
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic approaches towards highly durable proton exchange membrane fuel cells with minimized Pt use
Hee-Eun
Kim
 ,
Jaehoon
Kwon
and
Hyunjoo
Lee
*
,
Jaehoon
Kwon
and
Hyunjoo
Lee
*
Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon 34141, South Korea. E-mail: azhyun@kaist.ac.kr
First published on 4th May 2022
Abstract
Proton exchange membrane fuel cells (PEMFCs) produce electricity from H2 without carbon emission, and they are considered as environmentally benign energy conversion devices. Although PEMFCs are mature enough to find themselves in a few commercial automobiles such as Hyundai Nexo and Toyota Mirai, their durability should be enhanced, especially under transient conditions, and Pt use should be reduced significantly to expand their market. Herein, we introduce examples of how catalysts can contribute to enhancing the durability of PEMFCs while minimizing Pt use. Numerous electrocatalysts have been reported claiming superior activity in a half-cell setup, but they often fail to show the same enhancement in a single cell setup due to various transfer problems, impurity poisoning, etc. This perspective focuses on catalysts tested in a membrane-electrode-assembly (MEA) setup. As examples to obtain durability under transient conditions, catalysts used in reversal-tolerant anodes (RTAs) and selective anodes are explained. RTAs can endure sudden H2 starvation, and selective anodes can operate properly when O2 is unexpectedly mixed with H2 in the anode. As examples with high durability in long-term operation, Pt-based nanoparticle catalysts encapsulated with carbon shells are explained. Interestingly, PtCo nanoparticles supported on Co–N–C or PtFe nanoparticles encapsulated with a carbon shell presented a superior cell performance in spite of <1/10 Pt use in an MEA setup. Non-Pt group metal (PGM) catalysts used in an MEA setup are also briefly explained. With these highly durable catalysts which can respond properly under transient conditions with minimum Pt use, PEMFC technology can bring about a more sustainable society.
 Hee-Eun Kim | Hee-Eun Kim received her B. S. degree from Yonsei University (2017) and M. S. degree from Korea Advanced Institute of Science and Technology (KAIST) (2019). She is currently a PhD candidate at KAIST under the supervision of Prof. Hyunjoo Lee. Her research interests focus on electrocatalysts for PEMFC applications. |
 Jaehoon Kwon | Jaehoon Kwon received his B. Eng. degree from the University of Edinburgh (2019) and his M. Eng. degree from the University of Toronto (2020). He is currently a graduate student at KAIST under the supervision of Prof. Hyunjoo Lee. His research focuses on electrocatalysts for PEMFC applications. |
 Hyunjoo Lee | Prof. Hyunjoo Lee received her B. S. and M. S. degree from Seoul National University and PhD degree from California Institute of Technology. She is currently a full professor at KAIST. Her research interest is in heterogeneous atomic catalysts with structure control and their applications for various gas-phase and electrochemical reactions. |
1. Introduction
The H2 economy has emerged as a promising step towards a carbon-neutral society.1 Fuel cells that convert H2 into electricity without emitting pollutants are expected to play a pivotal role in the H2 economy. Proton exchange membrane fuel cells (PEMFCs) have the features of low operating temperature (60–200 °C), high efficiency, and zero-emission, and they are applied in power generation and transportation.2,3 However, an excessive amount of Pt must be used, which leads to up to 43% of the total stack cost.4 Extensive efforts are being put into enhancing the catalytic activity to minimize the use of Pt catalysts.5–7 Also, PEMFCs used in automobiles are constantly exposed to various harsh conditions on the road. The current status of fuel cell durability is still far below the targets set by the U.S. Department of Energy (DOE).8 To design highly active and durable catalysts for PEMFCs, it is critical to understand the reactions occurring at the electrode under various transient conditions.Pt loading used in Mirai, which is a fuel cell electric vehicle (FCEV) produced by Toyota, was 0.05 mgPt cm−2 for the anode and 0.315 mgPt cm−2 for the cathode.9 These values are very high compared to the DOE target, which is 0.025 mgPt cm−2 at the anode and 0.1 mgPt cm−2 at the cathode.10 The specifications of FCEVs currently available on the market are shown in Table 1. The Pt content used for FCEVs is in the range of 10–80 g. Internal combustion engine vehicles (ICEVs) also use Pt group metals (PGM) such as Pt, Pd, and Rh to convert pollutants of NOx, CO, and HxCy into N2, CO2, and H2O, and the typical PGM amount used in ICEVs is known to be 2–4 g.11 FCEVs would be much more cost-competitive if the Pt loading could be reduced by an order of magnitude.
PEMFCs can encounter changes in power demand and gas distribution at the electrodes. To ensure their stable operation in PEMFC vehicles, various systematic approaches have been sought: local current densities of the segmented cell can be monitored,15 outlet gas composition can be monitored,16 or fuel supply can be regulated using a proportional-integral-differential (PID) controller.17 To remove air from the anode compartment, in which only H2 is supposed to flow, high-flow gas purging can be employed before and after the start-up/shut-down of the engine.18 However, these methods are both time and energy-consuming. Receiving feedback about the fuel supply might not be swift enough, and installing extra sensors would lower the system efficiency. Catalytic approaches to ensure durability even under transient operating conditions can be more promising.
Although remarkable progress has been made within these few decades to develop better fuel cell catalysts, those efforts did not result in better PEMFC performances. Electrocatalysts are typically tested in a half-cell setup, which consists of a working electrode, a reference electrode, and a counter electrode. Very small amounts (<50 μg) of the catalyst are deposited on the tip of the working electrode. Intrinsic activity can be measured excluding the effect of transfer limitations. However, many studies reporting high activity in a half-cell setup failed to show good performance in a single cell.19,20 A PEMFC single cell consists of a membrane-electrode-assembly (MEA). In the MEA, the current–voltage (I–V) polarization curve can be divided into three parts: (1) the low current density region corresponding to catalyst activation, (2) the linear region attributed to ohmic losses, and (3) the high current density region corresponding to mass transport of reactants and products.21 The activation overpotential can be reduced by enhancing the intrinsic activity or increasing the surface area of the catalyst. The ohmic and mass transfer overpotential can be reduced by modifying ionomer distribution or the porous structure of the electrodes. Particularly, the three-phase boundary (TPB) where electrons, protons, and reactants meet together should be formed efficiently; otherwise, the electrode reaction would not occur efficiently despite the high intrinsic activity.
In this perspective, we will introduce how catalytic approaches can enhance PEMFC performances, and especially focus on the studies reporting MEA cell data. Fig. 1 summarizes the conditions which fuel cell catalysts should satisfy for efficient MEA operation. For the anode, various transient conditions that potentially degrade the cell performance will be discussed, and how the durability under such transient conditions can be improved by modifying the catalyst will also be explained. For the cathode, catalysts that showed better activity and durability in a single cell will be discussed. Particularly, recent reports showing ultra-low Pt loading with high activity and durability in a single cell will be explained. Non-PGM catalysts and their applications in a single cell (if they exist) will also be addressed.
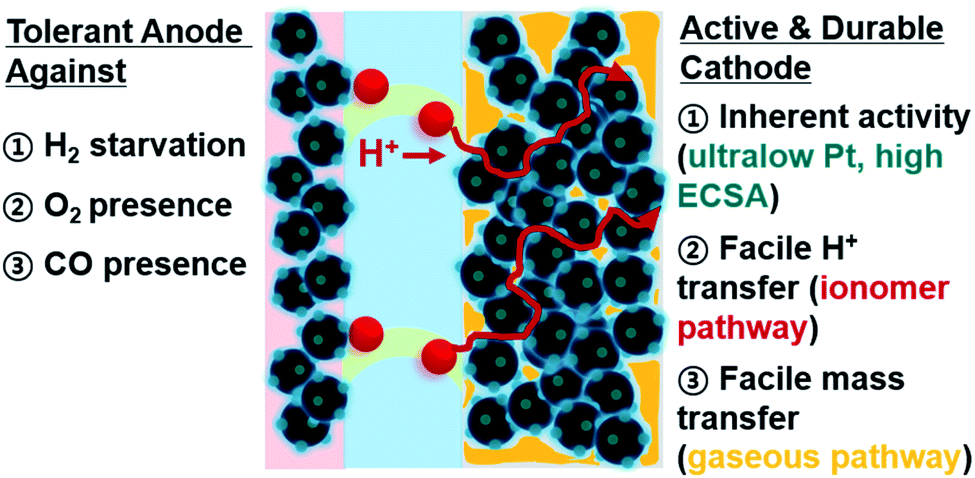 | ||
| Fig. 1 Illustrative description that fuel cell catalysts should satisfy for MEA operation. | ||
2. Durable anode catalysts under transient conditions
Fast kinetics of the hydrogen oxidation reaction (HOR: H2 → 2H+ + 2e−) at the anode enables facile reaction even with a small amount of Pt. Therefore, cathode catalysts have mainly been studied to enhance the sluggish kinetics of the oxygen reduction reaction (ORR: O2 + 4H+ + 4e− → 2H2O). However, PEMFCs can suffer from transient operating conditions such as frequent load changing, idling, start-up/shut-down, and cold start, especially in automotive applications,22,23 and many problems can occur at the anode under these conditions. This section will focus on a strategy to design durable anode catalysts under various transient conditions.2.1 Reversal-tolerant anodes under H2 starvation
Fig. 2a illustrates the reactions occurring at each electrode during PEMFC operation. Under normal operating conditions, the anode potential stays near the reversible hydrogen electrode (RHE) potential due to the very small HOR overpotential. However, when the H2 supply is not sufficient to meet the requirements during load changes or if there is uneven H2 distribution, H2 starvation occurs at the anode. To provide protons needed at the cathode, the anode potential readily rises to reach the potential for the carbon oxidation reaction (COR: C + 2H2O → CO2 + 4H+ + 4e−).22,24 Due to the very high overpotential of the COR, the cell voltage is reversed. Carbon in the catalyst layer or gas diffusion layer is corroded by the reaction with water causing pore structure collapse,25 Pt catalyst loss,26 and even pinhole formation on the MEA.27–29 The pinholes can be particularly dangerous because gas mixing of H2 and O2 can lead to an explosion. This irreversible damage by cell voltage reversal should be avoided. Various efforts have been made to develop a reversal-tolerant anode (RTA).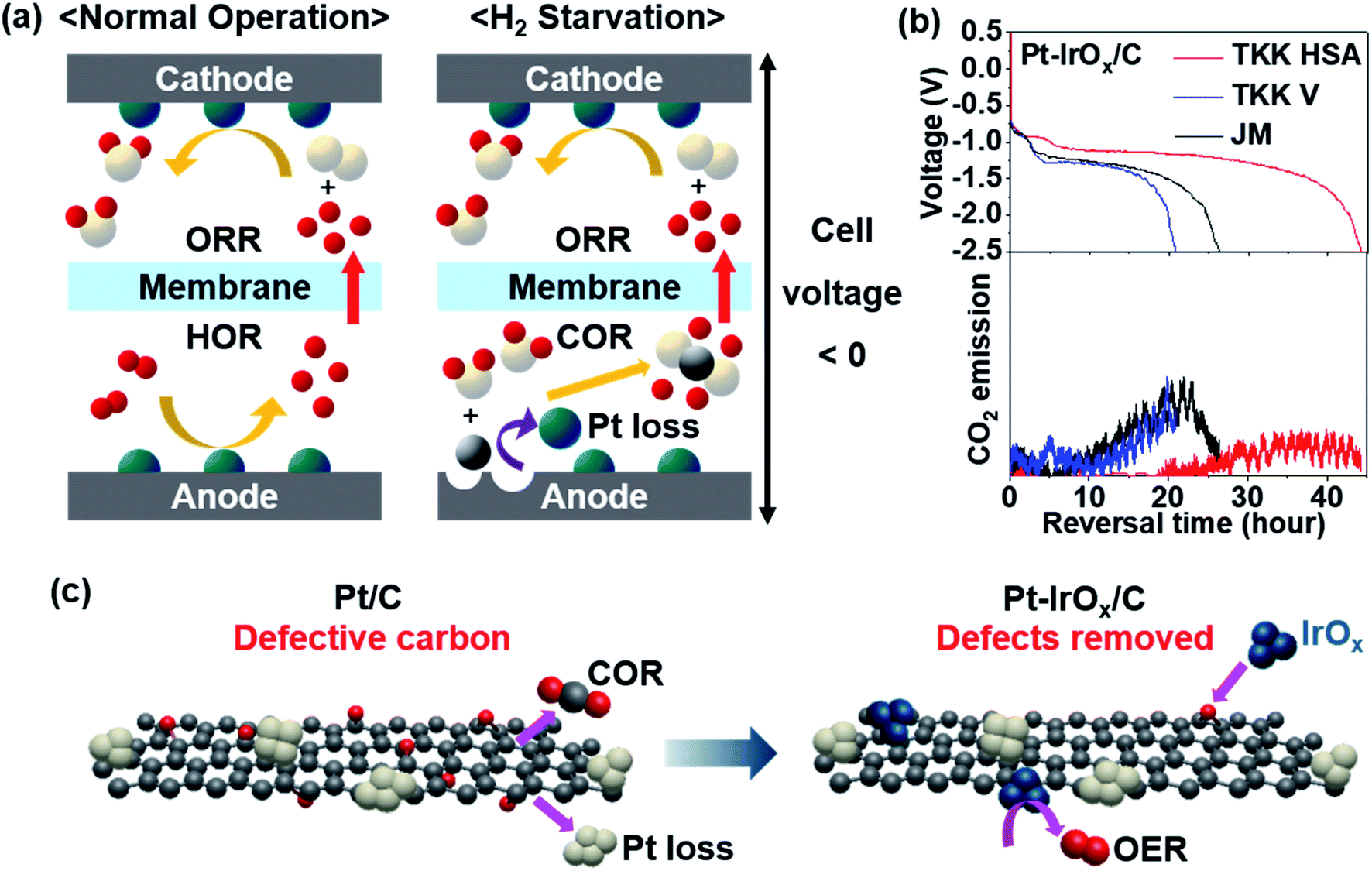 | ||
| Fig. 2 (a) Schematic illustration of electrode reactions occurring in normal operation (left) and under H2-starved conditions (right). The white, red, grey and turquoise spheres represent O, H, C, and Pt atoms, respectively. (b) Reversal tolerance test results of Pt–IrOx supported on different types of carbon (top) and CO2 emission monitored concurrently (bottom). Commercial Pt/C (TKK high surface area (HSA), TKK Vulcan (V), Johnson Matthey (JM)) was used to immobilize IrOx clusters. Longer reversal time indicates better durability. (c) Schemes of Pt/C and Pt–IrOx/C under H2-starved conditions. Reproduced from ref. 30 with permission, copyright 2021, Elsevier. | ||
Corrosion-tolerant supports were introduced to enhance the durability of the anode under cell reversal. Highly graphitized carbon showed better reversal tolerance.31 Metal oxides32 and carbides33 were used as supports instead of carbon. To protect the carbon from reacting with water to be oxidized, a hydrophobic coating such as polytetrafluoroethylene (PTFE) was applied to repel water.23 In addition, an oxygen evolution reaction (OER: 2H2O → O2 + 4H+ + 4e−) catalyst was added onto the anode so that water dissociation dominates carbon oxidation.28,30,34,35 As one way to estimate the reversal tolerance of the catalyst, chronopotentiometry is performed at 0.2 A cm−2 with Ar flow instead of H2 flow to simulate H2-starved conditions.22,31 The time taken for the cell voltage to reach −2.5 V is denoted as the reversal time. Litster's group prepared an RTA by physically mixing commercial Pt/C with IrO2 synthesized by the Adams fusion method.34 When tested at a relative humidity (RH) of 68%, which is typical for automotive PEMFCs, the reversal time was 76 min. The reversal time was only 12 min at a higher RH of 82% due to severe carbon loss. We previously prepared monodisperse IrOx and deposited them onto commercial Pt/C with a reversal time of 240 min at RH 100%.28 The oxidation state and dispersion of Ir species were found to be the key factors affecting the reversal tolerance. The oxidic Ir species was more active for the OER with better reversal tolerance. When monodisperse Ir species were finely distributed on the carbon surface, carbon corrosion could be prohibited more efficiently.28 Particularly, a highly defective carbon support was more advantageous to immobilize IrOx clusters uniformly with better reversal tolerance.30 While defective carbon is prone to the COR, small IrOx clusters could be anchored onto those defects better, facilitating the OER (Fig. 2c). As a result, the reversal time could be significantly lengthened up to 44 h at RH 100% (Fig. 2b). Litster's group adopted a Ti layer between Pt black and the gas diffusion layer (GDL).36 Even when a carbon-free catalyst such as Pt black is used at the anode, carbon corrosion still occurs on the microporous layer side of the GDL. By using a Ti protective layer, GDL degradation was prevented and the cell reversal tolerance was improved. Non-Pt catalysts were reported to have high activity for both HOR and OER in an RTA. Pak's group reported an IrRu2 catalyst supported on graphitized carbon.35 They prepared an IrRu2/C catalyst with a strong electronic interaction between Ir and Ru and showed a reversal time of ∼7 h at RH 50%. The IrRu/C series with various compositions showed similar or higher cell performance due to the excellent HOR activity. The test conditions to evaluate the reversal tolerance should be refined further to provide a general guideline.37
2.2 Selective anodes in the presence of O2
Automotive PEMFCs also suffer from frequent start-up/shut-down (SU/SD) cycles. After shut-down, atmospheric air inevitably flows into an anode gas chamber through an exhaust line.38 When H2 is replenished to the anode upon starting up, the remaining air and H2 can be mixed. Also, O2 crossover can occur from the cathode to the anode through a thin membrane.39 Pt-based catalysts are commonly used in the anode because of their high HOR activity. Since Pt catalysts are also highly active in the ORR, the presence of air in the anode can lead to the ORR at the anode, particularly during the SU/SD. Then the high potential imposed at the cathode causes the COR, severely damaging the catalyst layer (Fig. 3a).40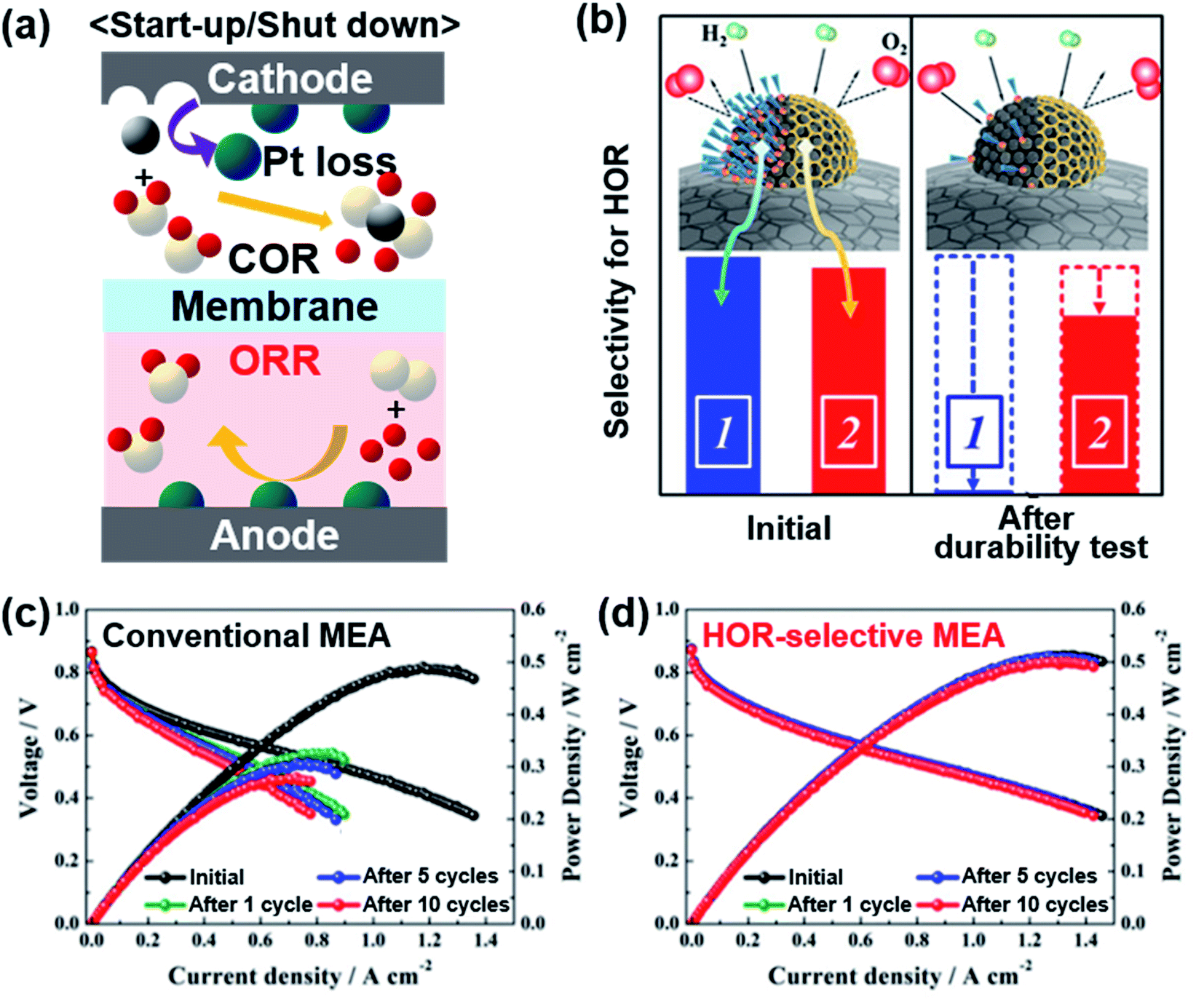 | ||
| Fig. 3 (a) Schematic illustration of electrode reactions occurring under start-up/shut-down conditions. (b) Comparison of the oleylamine-attached Pt catalyst and carbon-encapsulated Pt catalyst for selective HOR. Changes in the single cell performance of the (c) conventional MEA and (d) HOR-selective MEA during start-up/shut-down cycles. Reproduced from ref. 41 with permission, copyright 2019, American Chemical Society. | ||
A selective anode, which catalyzes the HOR while hindering the ORR, can minimize the degradation during the SU/SD cycles. Whereas the σ-bond of H2 can be activated atop a Pt atom, O2 has a π-bond that is dissociated effectively on Pt ensemble sites with a high coordination number.42 To selectively promote the HOR and impede the ORR, various efforts were put into covering the Pt ensemble sites. Markovic's group reported calix[4]arene-modified Pt(111) to be tolerant against the ORR and active towards the HOR.43 They also applied calix[4]arene on a 3 M nanostructured thin film (NSTF) and TKK 5 nm Pt/C with selective HOR activity, but these studies remained to be half-cell tests.44 Kim's group blocked Pt sites on commercial Pt/C with 1-dodecanethiol and this chemically modified catalyst presented less activity loss than bare Pt after 10 cycles of SU/SD test in a single cell.45 Jung and Yoo's groups were able to form graphitized carbon shells on Pt nanoparticles, which selectively allow H2 to pass but block O2 like a molecular sieve (Fig. 3b).41 Carbon-encapsulated Pt was formed by treating oleylamine-attached Pt/C at 900 °C. The oleylamine was easily detached after long-term reaction recovering the original ORR activity, but the carbon shell could block the ORR stably, enabling the HOR only. While bare Pt/C presented significant degradation after SU/SD cycles, the Pt/C encapsulated with a carbon shell experienced very little change during the single cell test (Fig. 3c and d). Stühmeier's group also showed that the Pt/TiOx/C catalyst, in which TiOx layers encapsulate Pt nanoparticles by a strong metal–support interaction (SMSI), had high HOR activity with hindered ORR.46
Blocking the surface Pt sites of Pt nanoparticles would waste many Pt atoms occluded inside the nanoparticles. Single-atomic Pt catalysts can maximize Pt utilization while catalyzing the HOR only. Pt single-atom catalysts (SACs) can hinder the ORR. Hashimoto and Nakanishi's groups prepared single-atomic Pt supported on covalent triazine frameworks (Pt-CTF) and showed ORR-tolerance of isolated Pt atoms.47 2.8 wt% Pt-CTF presented a cell performance comparable to that of commercial 20 wt% Pt/C. Liao's group prepared highly dispersed Pt on TiN with low ORR activity, but their HOR activity was not good enough compared to commercial Pt/C even in a half-cell setup.48 Additionally, the ORR on the Pt SAC can induce a two-electron pathway producing H2O2.49 The H2O2 formation is detrimental to PEMFC durability, because it forms hydroxyl or hydroperoxyl (˙OH or ˙OOH) radicals, which chemically degrade the perfluorosulfonic acid ionomer or membrane.50
The electrical conductivity of metal oxide semiconductors can be changed under H2 or air, and this feature was used to develop efficient selective anodes. Kakinuma and Uchida's groups showed that Pt/Ta–TiO2 had an order of magnitude higher conductivity in H2 than in air.51 The loss of electrochemically active surface area (ECSA) in the cathode was much less in Pt/Ti0.9Ta0.1O2−δ compared to the value in Pt/C after 1000 cycles of the start-up. Kim's group reported Pt/HxWO3 that converts itself into an insulator when exposed to O2 while regaining its conductivity under H2.52 After 10 cycles of SU/SD test by switching the anode gas from H2 to air, the cathode catalyst layer thickness decreased by only 10% and 71% of the maximum power density was retained in the Pt/m-HxWO3 anode, whereas the cathode catalyst layer thickness decreased by 65% and only 30% of the initial maximum power density was retained in the commercial Pt/C anode. However, these metal oxides typically lack surface area and Pt dispersion, and electrical conductivity is often insufficient. Non-Pt catalysts such as Ir/C, which shows HOR activity comparable to that of Pt/C but hardly shows ORR activity, can be used.53
3. Durable cathode catalysts with minimum Pt use
A vast amount of effort is being put into developing highly active and durable cathode catalysts. In this section, we will focus on how cathode catalysts should be designed to have better PEMFC performance with a smaller amount of Pt use. Unlike many ORR catalysts tested only in a half-cell setup, cathode catalysts should work properly in a single cell setup and the conditions that cathode catalysts should satisfy will be enumerated. Recently, Pt catalysts encapsulated with carbon shells were reported to have impressive durability in a single cell, and these studies will be explained as means to enhance the durability. Catalysts with ultra-low Pt contents were successfully used in a single cell, showing cell performances similar to or even better than that of commercial Pt/C despite <1/10 Pt use. These studies will be introduced as examples to minimize Pt use.3.1 Engineering catalyst layers in an MEA setup
There are many studies reporting high activity in electrochemical ORR, but they were usually tested in a half-cell setup using a rotating disk electrode (RDE). As shown in Fig. 4a, there is a significant discrepancy between the mass activities obtained in a half-cell and a single cell.54–63 In a half-cell test using an RDE, reactants are continuously supplied to the surface while the electrode is rotating (Fig. 4b). The reaction takes place at the interface between the liquid-phase electrolyte and the catalyst on the electrode. Mass transport is possible only by diffusion and convection vertically to the working electrode, which is facilitated by electrode rotation. If the activity is measured in the low current density region, typically at 0.9 V, the reaction kinetics can be investigated solely.64,65 This method was devised to evaluate the intrinsic activity, but it does not necessarily represent the activity in the high current density region. In a single cell test using an MEA, the cell performance is determined by combining activation, ohmic, and mass transfer overpotentials.21 The activation overpotential depends on the intrinsic activity and ECSA of catalysts. Not only should catalysts have high intrinsic activity, but they should also have a high ECSA to minimize the activation overpotential. The triple-phase boundary (TPB) is the region where gaseous O2 meets electrons and protons as shown in Fig. 4c, and it is important to form this TPB efficiently to obtain a high ECSA.66 The ohmic and mass transfer overpotentials can be reduced by securing good conductivity and a highly porous network within the electrode layer. Minimizing these overpotentials becomes more important as the current density increases. Typically, a thinner catalyst layer is more advantageous to achieving more facile mass transfer.67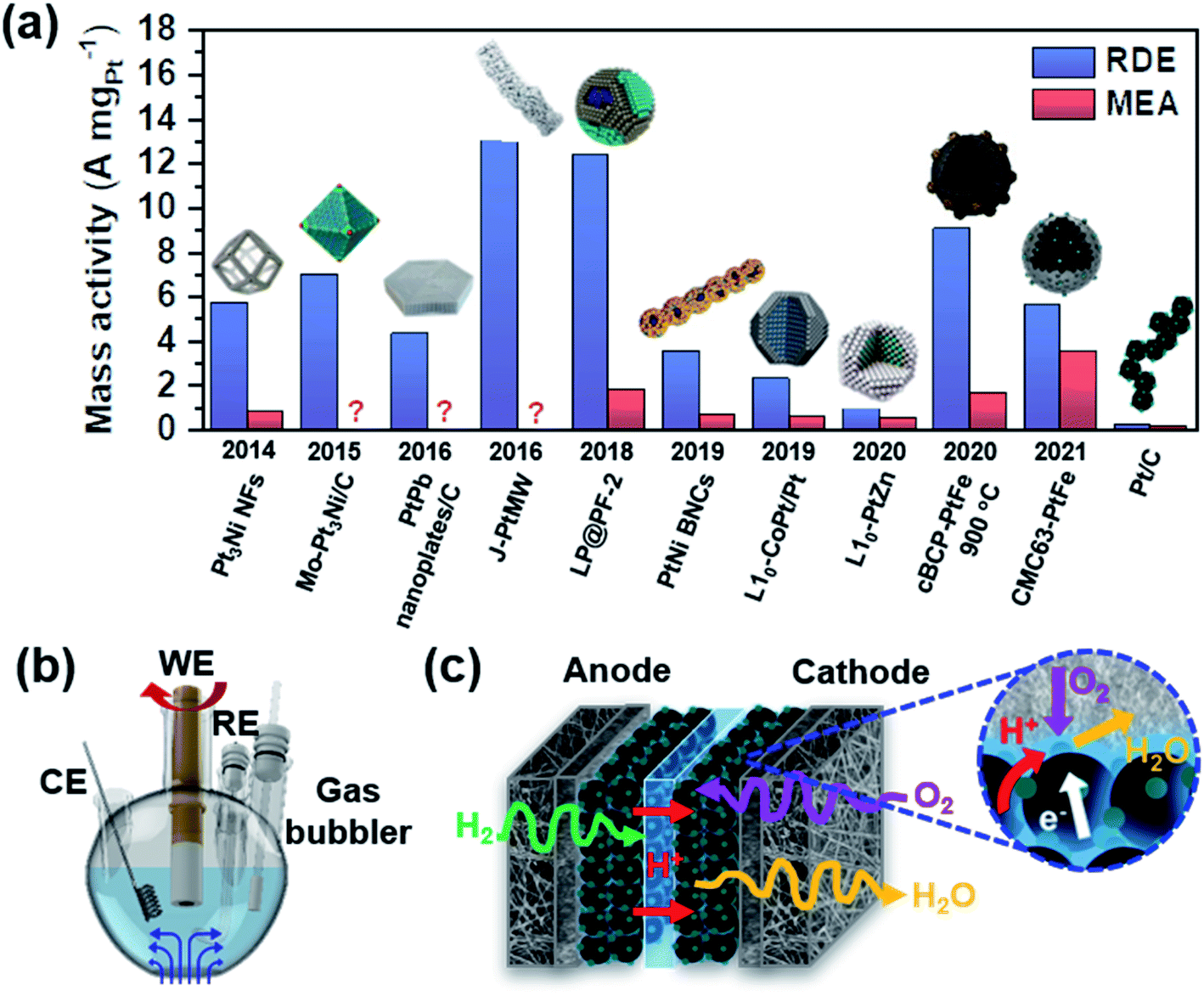 | ||
| Fig. 4 (a) Comparison of the catalytic activity measured in the RDE and MEA setup (Pt3Ni NFs,54 Mo–Pt3Ni/C,55 PtPb nanoplates/C,56 J-PtMW,57 LP@PF-2,58 PtNi BNCs,59 L10-CoPt/Pt,60 L10-PtZn,61 cBCP-PtFe 900 °C,62 CMC63-PtFe,63 and Pt/C63). This figure was modified from ref. 68 with permission, copyright 2021, Nature Publishing Group. Schematic illustration of (b) a half-cell setup (WE: working electrode, CE: counter electrode, and RE: reference electrode) and (c) MEA configurations. The inset shows the triple-phase boundary in the cathode layer, in which O2, H+, and e− meet together. | ||
Alloying Pt with 3d transition metals such as Co or Ni was reported to enhance the activity in a single cell.69,70 The Pt electronic structure could be modulated by alloying, tuning the adsorption energy of oxygen species, leading to higher ORR activity.71–73 However, transition metals are easily dissolved and leached out, contaminating the ionomer and membrane, which all leads to poor cell durability.74,75 Catalysts were coated with a silica or carbon shell to prevent metal dissolution and agglomeration, but these shells also induced poorer conductivity or less active sites, leading to decreases in the cell performance.76,77
The ionomer network that provides proton pathways in the MEA is often not homogeneous throughout the catalyst layer. Pt sites that are not in close proximity to Nafion can only proceed reaction with the aid of water transporting the protons. Functionalizing the support surface, modifying the catalyst ink composition, and controlling the support morphology have been tried to improve ionomer distribution. But direct contact of the ionomer with Pt atoms can poison the Pt active sites with –SO3− or –O− groups present in the Nafion ionomer.78,79 When the amount of ionomer is excessive, the micropores can be easily clogged and O2 permeation would not occur through the thick ionomer layer. 30–40 wt% of Nafion in the catalyst layer is known to be the optimal ionomer content for the PEMFC performance.80 Chandezon and Gebel's groups reported a 3D morphology of Nafion, showing that the average Nafion layer thickness was ∼7.2 nm when the Nafion content was 33%.81 Suzuki's group reported an average ionomer thickness ranging from 4.0 to 14.9 nm with Nafion content varying from 14 to 50%.80 The optimal cell performance was obtained with 33% of the ionomer content with an ionomer thickness of 9.0 nm. However, it is noteworthy that the ionomer thickness can vary from 4 to 20 nm depending on the temperature and relative humidity conditions.82 Orfanidi and Strasser's groups used the coulombic interaction between sp2 N-groups on the carbon and ionomer to form a thin ionomer layer uniformly.66 Fujigaya's group coated the carbon support with polybenzimidazole (PBI) to have positive charges, and then the negatively charged Nafion ionomer could be distributed more evenly.83
Controlling the pore structure is also very important to facilitate mass transfer. Although the importance of accessible pores, which can protect Pt from direct contact with the ionomer but allow access to protons and O2, was recently emphasized,84 micropores are usually not beneficial to forming an efficient TPB because the ionomer cannot form the proton pathway inside these small pores.85 Water flooding is the major reason for degraded cell performance, especially in the high current density region, blocking active sites and O2 transport.86 Evenly distributed macropores would be beneficial to relieve water flooding. We recently could control the pore size of the carbon support from 13 nm to 63 nm using block copolymers and showed that 63 nm was the best with minimum mass transfer overpotential. The detailed discussion will be provided in Section 3.3. Macropores throughout the electrode with additional accessible pores seem to be ideal. Cathode catalysts should be designed not only to have high intrinsic activity but also to form a TPB efficiently with minimized ohmic and mass transfer overpotentials.
3.2 Enhanced durability by carbon shell encapsulation
As a distinct method to enhance durability for long-term PEMFC operation, using carbon shell-encapsulated Pt catalysts (Pt@C) has been reported. Sung and Hyeon's groups prepared ordered face-centered tetragonal (fct)-PtFe nanoparticles coated with N-doped carbon shells.87 The PtFe nanoparticles were coated with polydopamine, and then annealed at 700 °C to allow the formation of N-doped carbon shells. After 100 h of MEA operation, the maximum power density decreased by 3.4% for the encapsulated catalyst, whereas for the commercial Pt/C, it decreased by 27%. Wang's group also reported an fct-PtFe catalyst encapsulated with porous carbon shells.88 Fe/C was first synthesized by pyrolyzing C2H2 and Fe(CO)5 together, and then the Pt precursor was added and annealed at 900 °C. Although this catalyst showed better durability for the ORR performed in a half-cell test, its durability in a single cell was not assessed. Lim, Sung, and Kwon's groups synthesized Pt nanoparticles encapsulated by N-containing carbon shells using aniline.89 The Pt–aniline complex deposited on carbon nanofibers was annealed at 900 °C. After 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in a single cell test, the maximum power density barely changed for the encapsulated catalyst but decreased by ∼30% for commercial Pt/C.
000 cycles in a single cell test, the maximum power density barely changed for the encapsulated catalyst but decreased by ∼30% for commercial Pt/C.
Jung and Yoo's groups reported carbon-encapsulated PtFe nanoparticles with various extents of Pt surface atom exposure.90 Pt and Fe precursors containing organic ligands were pyrolyzed, and then carbon atoms absorbed inside the metal alloys diffused to the surface, forming a carbon shell. Although the carbon shell was too thick to enable the reaction initially, H2 treatment could remove the carbon shell partially; 5% H2 could preserve the carbon shell while enabling the reaction with high durability, whereas 20% H2 removed the carbon shell completely (Fig. 5a). This work nicely showed a trade-off relation between activity and durability, which depended on the carbon shell thickness. A thicker carbon shell presented better durability, but the maximum power density was smaller due to the smaller ECSA, and a thinner carbon shell showed poorer durability, but the maximum power density was higher due to more exposed Pt atoms (Fig. 5b–d). Because Pt-based nanoparticles are typically sintered during annealing for carbon shell formation, preserving the small nanoparticle size is important to avoid sacrificing the activity. Controlling the thickness and extent of porosity in the carbon shell is highly desired to enhance both activity and durability.
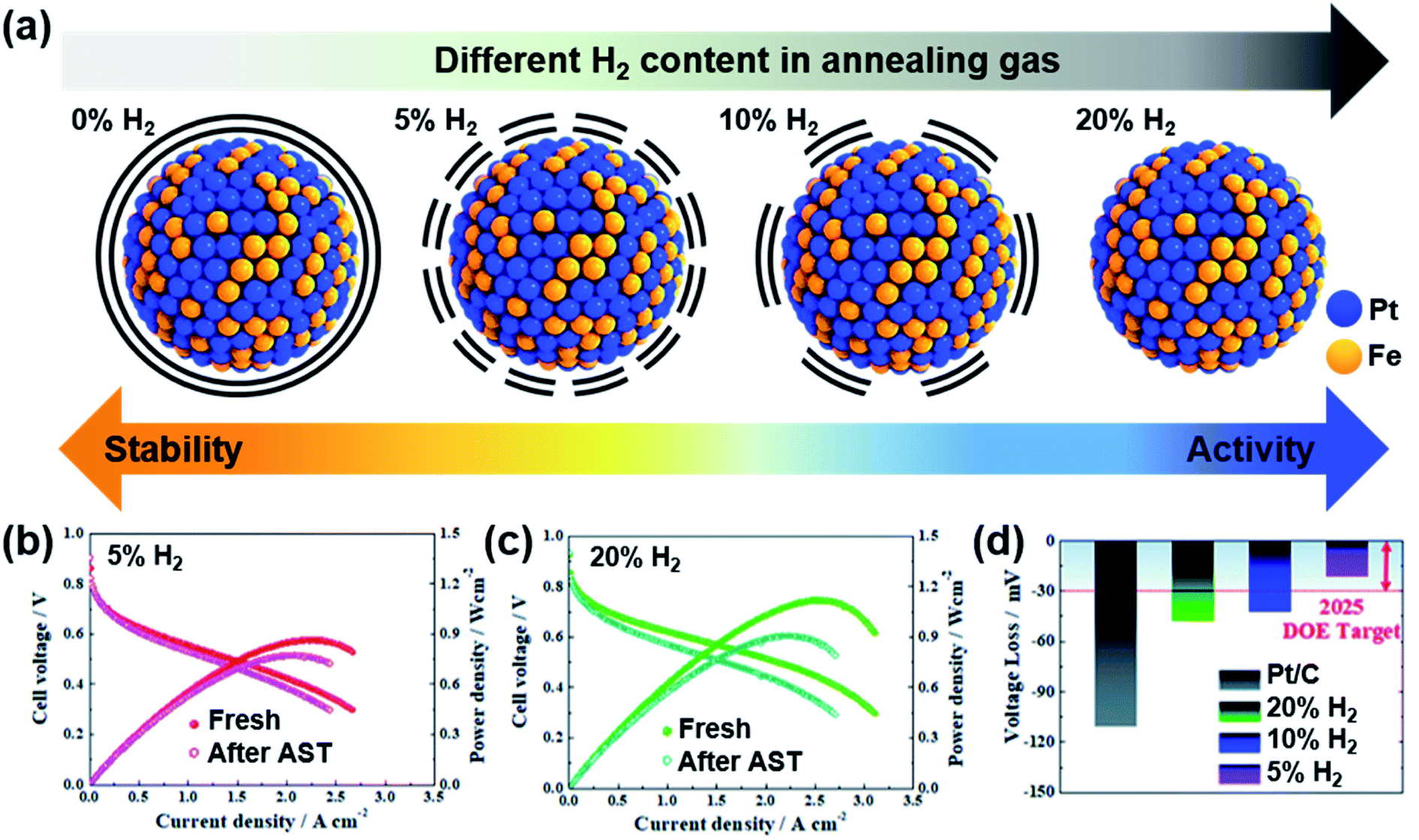 | ||
| Fig. 5 (a) Difference in the carbon shell depending on annealing conditions; various percentages of H2 flow were used at 700 °C. I–V curves before and after accelerated stress tests (ASTs) performed under (b) 5% H2 and (c) 20% H2. (d) Voltage loss at 0.8 A cm−2 after ASTs. Reproduced from ref. 90 with permission, copyright 2021, the Royal Society of Chemistry. | ||
3.3 Ultra-low Pt catalysts
High ORR activity in a half-cell does not guarantee good performance in a single cell. Although the mass activity at 0.9 V is typically used to compare the intrinsic activity in a single cell, the current density is often undetected at 0.9 V, probably because the TPB is not formed efficiently enough. Additionally, even when the current density at 0.9 V is high, the current density might drop suddenly in the intermediate voltage range due to severe ohmic/mass-transfer resistance. This behavior is often observed for ultra-low Pt loading catalysts. The catalyst layer becomes much thicker due to low Pt loading, and then transfer resistances become a hindrance to achieving high current density as the voltage decreases, resulting in much lower power density.67,91 High intrinsic activity and facile transport should be obtained together to reduce Pt amounts significantly.Pt catalysts combined with transition metals embedded in nitrogen–carbon composites (M–N–C) have shown high activity and durability in a single cell setup. Shui, Zheng, and Yu's groups reported Pt single atoms grafted onto Fe–N–C.92 This catalyst showed better stability than Fe–N–C alone because Pt single atoms prevented the Fe center from catalyzing the Fenton reaction generating hydroxyl radicals, which was the main reason for the poor durability of Fe–N–C catalysts. Liu's group reported LP@PF catalysts in which PtCo nanoparticles, Co@graphene, and Co–N–C co-existed.58 Despite the ultra-low Pt loading of 0.035 mgPt cm−2, this catalyst presented a much higher maximum power density of ∼1.4 W cm−2 than commercial Pt/C with a loading of 0.35 mgPt cm−2 with ∼1.2 W cm−2 in H2/O2 flow. The mass activity at 0.9 V after 30000 cycles of durability test in a single cell was 0.26 A mgPt−1, which meets the DOE 2025 target of 0.26 A mgPt−1 for mass activity after an accelerated stress test (AST).93 Myers, Xie, Spendelow, and Wu's groups reported that Pt3Co nanoparticles dispersed on Fe–N–C showed a high initial mass activity of 0.72 A mgPt−1 at 0.9 V with a Pt loading of 0.1 mgPt cm−2.94 The mass activity at 0.9 V decreased by 38% after 30000 cycles of durability test, which still exceeds the DOE target. Wang and Liu's groups reported Pt alloy nanoparticles, which consist of an ordered intermetallic Pt3M (M = Fe and Zn) core and a Pt shell, supported on Fe–N–C.95 The mass activity at 0.9 V was 0.45 A mgPt−1 with 0.13 mgPt cm−2 of cathode loading. The maximum power density was 1.31 W cm−2, whereas commercial Pt/C with the same Pt loading showed 0.92 W cm−2.
We recently reported PtFe nanoparticles encapsulated with carbon shells synthesized using a block copolymer-based mesoporous carbon support.62 With an ultra-low Pt loading of 0.01 mgPt cm−2, a high maximum power density of 0.96 W cm−2 was obtained, while the value for the commercial Pt/C was 0.98 W cm−2 with a Pt loading of 0.2 mgPt cm−2 in H2/O2 flow. The mass activity at 0.9 V after 30000 cycles of durability test in a single cell was 0.81 A mgPt−1. In a follow-up study, we were able to control the diameter of pore channels from 13 nm to 63 nm (Fig. 6a).63 The mesoporous carbon support was synthesized by selectively carbonizing styrene domains of poly(styrene-block-dimethylsiloxane) sphere particles. When the molecular weights of these block copolymers became larger, the diameter of the pore channels became larger. Despite 1/20 Pt use, catalysts with 63 nm diameter showed even higher maximum power density than commercial Pt/C in H2/air flow (Fig. 6b). Distinctly, this catalyst also showed very high durability, probably due to the presence of a carbon shell structure (Fig. 6c). We confirmed that there were no N species on the catalyst surface, excluding the effect of Fe–N–C species. The effect of pore diameter was evaluated by estimating the kinetic overpotential, electron migration overpotential estimated from high frequency resistance (HFR), proton migration overpotential, and mass transfer overpotential separately at various O2 concentrations (Fig. 6d). The mass transfer overpotentials decreased as the diameter became larger, enhancing the cell performance greatly. Further study is still required to elucidate at which sites the surface reactions occur.
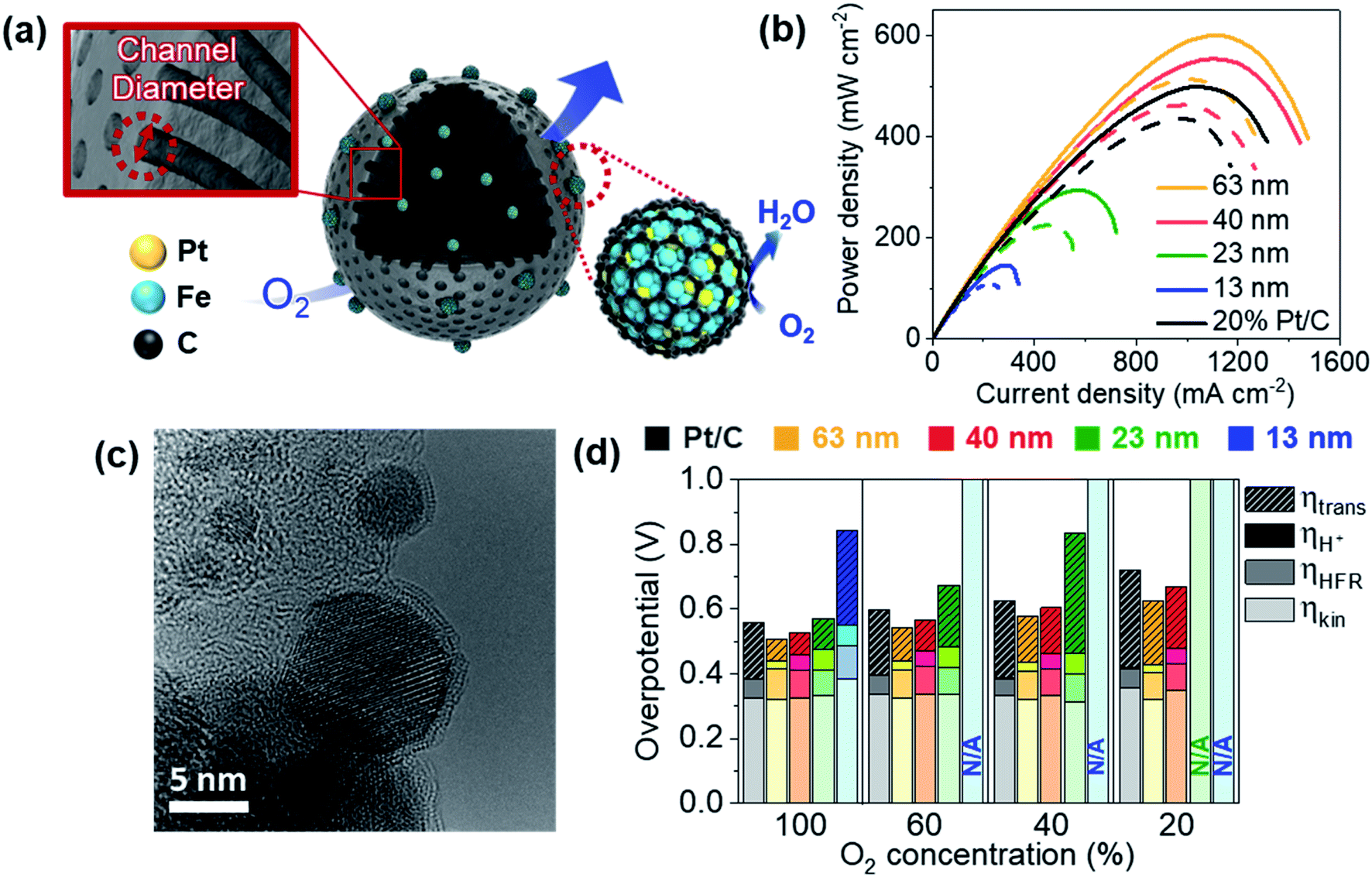 | ||
| Fig. 6 (a) PtFe@C catalyst with various channel diameters. (b) Polarization curves under H2/air flow. The solid lines indicate the initial performance and the dotted lines indicate the performance after 30000 cycles of the durability test in a single cell. The cathode Pt loading was 0.01 mgPt cm−2 for PtFe@C and 0.2 mgPt cm−2 for Pt/C. (c) HR-TEM image of the PtFe@C catalyst (63 nm) after the durability test. (d) Kinetic, high frequency resistance (HFR), proton transfer, and mass transfer overpotentials (ηkin, ηHFR, ηH+, and ηtrans, respectively) estimated at 1.1 A cm−2 at various O2 concentrations (N/A indicates ‘not available’ due to too large overpotentials). Reproduced from ref. 63 with permission, copyright 2021, Wiley-VCH. | ||
4. Non-PGM electrode catalysts
Non-PGM catalysts have been extensively studied for the HOR and ORR, but most of these studies are based on the half-cell test. For the anode, non-PGM catalysts with a performance similar to that of commercial Pt/C in a single cell could hardly be found. Because PGM loading is typically very low, as small as 0.05 mgPt cm−2 in the anode,96 replacing Pt with a non-PGM catalyst might not be urgent. Instead, non-PGM HOR catalysts can be more durable against impurities such as CO unlike Pt catalysts. While the current PEMFCs require high purity H2 (>99.97 mol%),97 non-PGM catalysts might be able to alleviate this demand, which can potentially reduce H2 prices significantly. We will introduce CO-tolerant HOR catalysts here. For the cathode, Fe–N–C or Co–N–C catalysts have been actively investigated, and they actually showed similar performances to the commercial Pt/C in a single cell test. However, the catalyst layer is usually much thicker, resulting in poor performance in the high current density region. Recent examples addressing this issue will be introduced.4.1 Non-PGM anode catalysts
Ni- or Co-doped metal oxides and metal carbides have been developed for the HOR but they are easily dissolved under acidic conditions.98–100 Burstein's group reported that carbon-encapsulated Ni crystals are stable in highly acidic solution with a good HOR activity of 0.12 mA cm−2 at 0.27 V (vs. SHE).101 Koel's group reported that hafnium oxynitride (HfNxOy) has high activity and durability for the HER and HOR under acidic conditions.102 However, these catalysts have barely shown promising performances in a single cell. They might have some advantages as CO-tolerant HOR catalysts. H2 is currently produced from steam reforming of fossil fuel or biomass, producing CO inevitably. Because a very small amount of CO can poison Pt catalyst surfaces, CO should be completely removed from H2 by using additional reactions and adsorption processes of water-gas shift reaction, preferential oxidation, pressure-swing adsorption, etc.97,103 CO-tolerant HOR catalysts might enable the use of cheap impure H2. Burstein's group showed that tungsten carbide (WC) is less susceptible to CO poisoning in the HOR.104 Jousselme and Artero's groups reported a Ni complex with biomimetic organic ligands immobilized on carbon nanotubes for the HER, showing no degradation in the current density upon CO (50 ppm) exposure.105 Nagai's group showed that metal carbides (Co–MoC) can behave as an anode catalyst in a single cell, but the maximum power density was very low at ∼19 mW cm−2.99 When the CO concentration in H2 increased from 0 to 600 ppm, the Co–MoC presented less increase in the overpotentials compared to Pt/C.4.2 Non-PGM cathode catalysts
Fe–N–C catalysts have been most extensively studied for the ORR as non-PGM catalysts. Many good review papers can be found,106–109 so we will focus on their MEA performance instead of providing full description. The density of Fe–N–C active sites is not high enough, which leads to a thick catalyst layer, resulting in poor cell performance (Fig. 7a). Metal–organic framework (MOF)-based carbon materials have been developed to increase the density of active sites.110–112 Chang and Wu's groups introduced (Fe, Co)–N–C, in which Fe–Co dual sites were embedded on N-doped porous carbon.113 Fe3+ ions were adsorbed onto a Zn/Co bimetallic MOF, and then reduced forming Fe–Co dual sites. The maximum power density was 0.98 W cm−2 in H2/O2 flow and 505 mW cm−2 in H2/air flow, which was 76% of the value obtained with commercial Pt/C. Litster's group studied the effect of ionomer distribution on Fe–N–C catalysts synthesized from Fe-MOFs.110 By using 100 nm-sized particles, an efficient ionomer pathway could be obtained with a maximum power density of 1.14 mW cm−2 in H2/O2 flow and 610 mW cm−2 in H2/air flow. Smaller or larger particles rather hindered the proton transfer. Wang and Zhou's groups reported a very high maximum power density of 1.33 W cm−2 in H2/O2 flow using Fe–N–C catalysts derived from zeolitic imidazolate frameworks (ZIF-8) as shown in Fig. 7b and c.112 The performance in the high current density region could be enhanced further by enlarging the pores of Fe–N–C catalysts using mild CO2 etching.114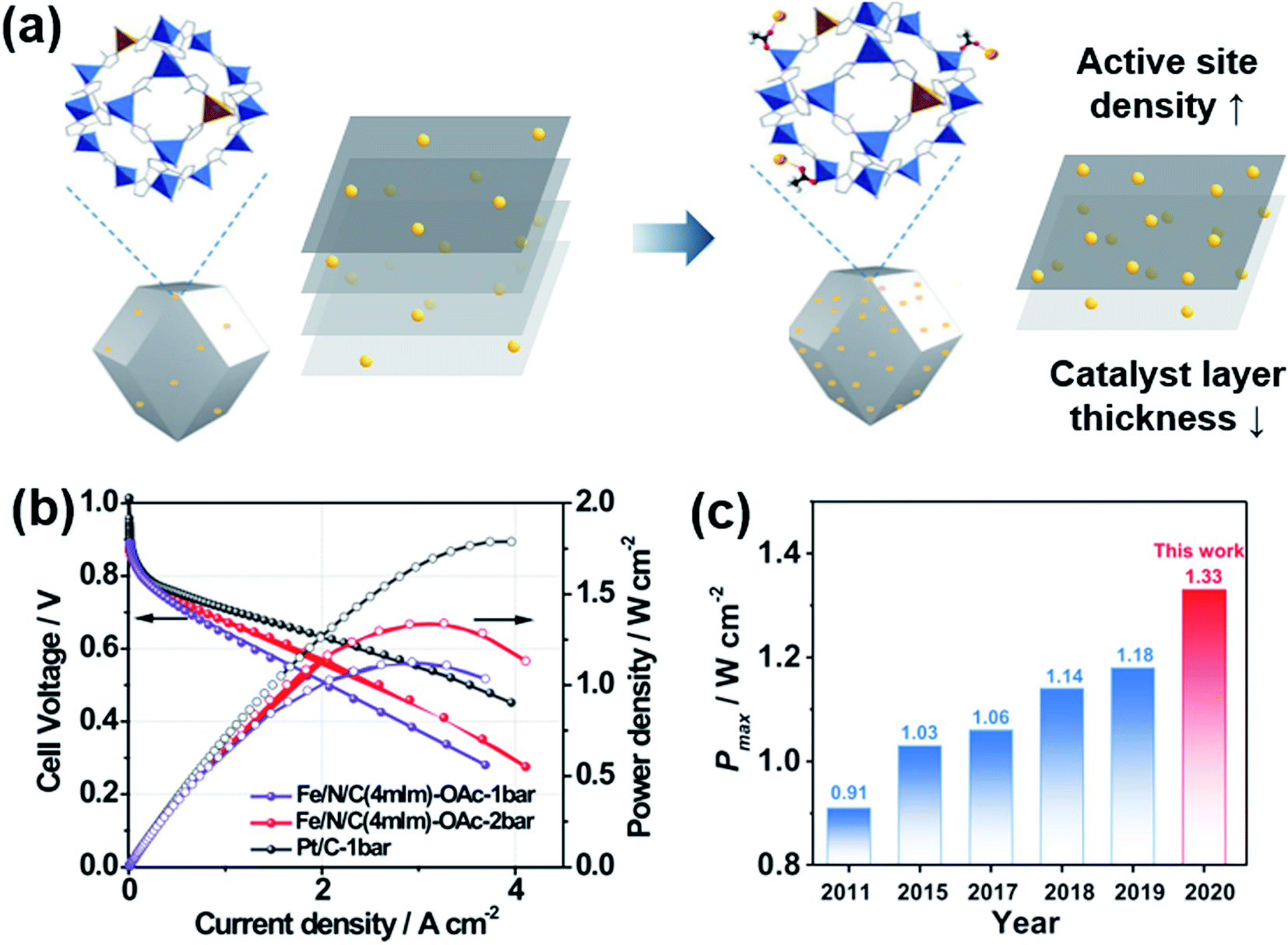 | ||
| Fig. 7 (a) Schematic diagram of Fe-doping on ZIF-8 with acetates (OAc−). (b) Polarization and power density curves of Fe/N/C and Pt/C under H2/O2 flow with 1 or 2 bar of backpressure. (c) Maximum power density (Pmax) of Fe–N–C catalysts in recent years. Reproduced from ref. 112 with permission, copyright 2021, Wiley-VCH. | ||
Fe–N–C catalysts suffer from poor durability due to Fenton reactions, producing H2O2, which degrades the cell significantly.115 As an alternative, Wang, Litster, and Wu's groups prepared Co–N–C catalysts derived from Co-doped ZIF-8 capped with Pluronic F127 surfactants.111 The surfactant layers eventually formed carbon shells after pyrolysis. The Co–N–C catalyst showed a high maximum power density of 0.87 W cm−2 in H2/O2 flow. The durability was tested at 0.7 V in an MEA for 100 h in H2/air flow, and significant initial performance loss was observed. Very recently, Shahbazian-Yassar, and Shao's groups adopted Ta–TiOx nanoparticles as additives to Fe–N–C as scavengers of ˙OH radicals and H2O2.116 The performance loss was as small as 3% at 0.9 ViR-free after the accelerated degradation test, whereas the degradation was 33% without the scavengers. The durability test results for the M–N–C catalysts, as typically obtained for Pt-based catalysts, are barely found in literature.
5. Summary and outlook
PEMFCs provide a promising way for energy conversion without carbon emission. However, the real application of PEMFCs requires durability under various operating conditions, such as H2 starvation and O2 crossover. Additionally, the use of Pt catalysts in PEMFCs is still too much, and the minimization of Pt use is essential to expand the PEMFC market further. Among the various methods to address these issues, we focused on catalytic approaches and how electrode catalysts have been developed to enhance the durability under harsh operating conditions and to minimize Pt use. Particularly, we focused on catalysts applied in an MEA setup. Although many electrocatalysts have been reported with high activity and durability in a half-cell setup, they often have various problems, leading to them not showing good performance in an MEA setup.More specifically, we introduced reversal-tolerant anode and selective anode catalysts for enhancing the durability under abnormal operating conditions. H2 starvation can induce carbon corrosion in the anode, causing voltage reversal. Water splitting reactions in the anode can prevent carbon corrosion in the unexpected absence of H2 fuel, so the reversal-tolerant anode typically consists of Pt/C and OER catalysts such as Ir-based catalysts. Uniform dispersion of fine IrOx catalysts on carbon supports could be an effective way to prevent voltage reversal. H2 fuel can be mixed with O2 in the anode, and selective anodes which catalyze the HOR only without the ORR can prevent the voltage loss. Because Pt ensemble sites are required to catalyze the ORR, attempts were made to block the ensemble sites by capping the Pt surface with organic moieties or by using Pt single-atom structures. Modulating the conductivity of supports has also been used for selective anodes; while the conductivity is good enough under reducing conditions with H2 only, the exposure to O2 decreases the conductivity significantly, blocking the electron transfer.
Massive amounts of Pt are still required to catalyze the sluggish ORR in the cathode. While numerous novel structures have been proposed with superior half-cell activities, only a limited number of catalysts demonstrate enhanced performance in an MEA setup. Particularly, when Pt use in the cathode is minimized, long-term durability becomes much worse. Innovative catalysts with minimum Pt use, but superior durability, are highly required. Recently, Pt catalysts encapsulated with carbon shells presented impressive durability for long-term operation. Unique catalysts with PtCo nanoparticles supported on Co–N–C or PtFe nanoparticles encapsulated with carbon shells exhibited very high performance while using only less than 1/10 of Pt compared to commercial Pt/C.
Non-PGM catalysts have been actively investigated for PEMFCs. While transition metals inherently suffer from poor stability under acidic conditions, Fe–N–C catalysts have recently become promising candidates as ORR catalysts in an MEA setup for PEMFCs because of their excellent catalytic activity. However, because the electrode thickness is typically much thicker, the Fe–N–C catalysts suffer from mass transfer resistance, showing poorer performance in H2/air flow than in H2/O2 flow. The durability for long-term reaction should also be guaranteed further. Various non-PGM catalysts are also studied for developing impurity-tolerant HOR catalysts, but most of them remain as half-cell research.
Catalytic approaches are surely promising to address the problems which the current PEMFC technology is faced with. It cannot be emphasized enough that PEMFC catalysts should be developed in an MEA setup. Understanding only the surface reaction itself is not enough to promote the overall performance. Catalysts can suffer from various transfer problems of poor conductivity, ion transfer, and mass transfer while being adapted in an MEA setup, and these issues should be addressed together. With highly durable PEMFC catalysts using a minimum amount of Pt, PEMFCs can become the most realistic engine to drive the current fossil-fuel-based society to a more sustainable one.
Author contributions
H.-E. Kim and H. Lee organized the manuscript and all authors wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Research Foundation of Korea (2021R1A3B1076715, 2019M3D1A1079306, 2017M3D1A1040692, and 2016R1A5A1009592).References
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- S. Satyapal, FC EXPO 2018, U.S. Department of Energy Hydrogen and Fuel Cell Technology Overview, Tokyo, Japan, March, 2018, https://www.energy.gov/sites/default/files/2018/03/f49/fcto_doe_h2_fc_overview_satyapal_fc_expo_2018_0.pdf, accessed October 2021 Search PubMed.
- A. Serov, A. D. Shum, X. Xiao, V. De Andrade, K. Artyushkova, I. V. Zenyuk and P. Atanassov, Appl. Catal., B, 2018, 237, 1139–1147 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- E. Padgett and G. Kleen, Automotive Fuel Cell Targets and Status, U.S. Department of Energy, 2020, https://www.hydrogen.energy.gov/pdfs/20005-automotive-fuel-cell-targets-status.pdf, accessed October, 2021 Search PubMed.
- Y. Wang, H. Yuan, A. Martinez, P. Hong, H. Xu and F. R. Bockmiller, Adv. Appl. Energy, 2021, 2, 100011 CrossRef.
- D. Papageorgopoulos, Fuel Cell R&D Overview: 2019 Annual Merit Review and Peer Evaluation Meeting, U.S. Department of Energy, United States, April, 2019, https://www.hydrogen.energy.gov/pdfs/review19/plenary_fuel_cell_papageorgopoulos_2019.pdf, accessed September 2021 Search PubMed.
- T. A. M. Suter, K. Smith, J. Hack, L. Rasha, Z. Rana, G. M. A. Angel, P. R. Shearing, T. S. Miller and D. J. L. Brett, Adv. Energy Mater., 2021, 11, 2101025 CrossRef CAS.
- B. K. Hong, S. H. Kim and C. M. Kim, Johnson Matthey Technol. Rev., 2020, 64, 236–251 Search PubMed.
- Reuters, https://www.reuters.com/article/us-platinum-week-bosch-fuelcells-exclusi-idUSKCN1SJ0FG, accessed October, 2021.
- Autonews, https://www.autonews.com/article/20160314/OEM05/303149972/honda-gm-set-targets-for-2020-fuel-cell-vehicle, accessed January, 2022.
- H. Schulenburg, B. Schwanitz, N. Linse, G. G. Scherer, A. Wokaun, J. Krbanjevic, R. Grothausmann and I. Manke, J. Phys. Chem. C, 2011, 115, 14236–14243 CrossRef CAS.
- E. Brightman and G. Hinds, J. Power Sources, 2014, 267, 160–170 CrossRef CAS.
- C. Lebreton, C. Damour, M. Benne, B. Grondin-Perez and J. P. Chabriat, Int. J. Hydrogen Energy, 2016, 41, 15615–15621 CrossRef CAS.
- P. T. Yu and F. T. Wagner, US Pat., 007270904B2, 2007 Search PubMed.
- V. Yarlagadda, S. E. McKinney, C. L. Keary, L. Thompson, B. Zulevi and A. Kongkanand, J. Electrochem. Soc., 2017, 164, F845–F849 CrossRef CAS.
- F. Zhu, L. Luo, A. Wu, C. Wang, X. Cheng, S. Shen, C. Ke, H. Yang and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 26076–26083 CrossRef CAS PubMed.
- J. Marie, R. Chenitz, M. Chatenet, S. Berthon-Fabry, N. Cornet and P. Achard, J. Power Sources, 2009, 190, 423–434 CrossRef CAS.
- T. R. Ralph and M. P. Hogarth, Platinum Met. Rev., 2002, 46, 117–135 CAS.
- S. D. Knights, K. M. Colbow, J. St-Pierre and D. P. Wilkinson, J. Power Sources, 2004, 127, 127–134 CrossRef CAS.
- T. Joo, L. M. Hu, B. K. Hong, J. G. Oh and S. Litster, J. Power Sources, 2020, 472, 228439 CrossRef CAS.
- S. Ghosh, H. Ohashi, H. Tabata, Y. Hashimasa and T. Yamaguchi, J. Power Sources, 2017, 362, 291–298 CrossRef CAS.
- J. Wang, G. Yin, Y. Shao, S. Zhang, Z. Wang and Y. Gao, J. Power Sources, 2007, 171, 331–339 CrossRef CAS.
- F. Nandjou, J. P. Poirot-Crouvezier, M. Chandesris, J. F. Blachot, C. Bonnaud and Y. Bultel, J. Power Sources, 2016, 326, 182–192 CrossRef CAS.
- C. W. Roh, H. E. Kim, J. Choi, J. Lim and H. Lee, J. Power Sources, 2019, 443, 227270 CrossRef CAS.
- B. T. Huang, Y. Chatillon, C. Bonnet, F. Lapicque, S. Leclerc, M. Hinaje and S. Rael, Int. J. Hydrogen Energy, 2013, 38, 543–550 CrossRef CAS.
- H. E. Kim, S. Shin and H. Lee, J. Catal., 2021, 395, 404–411 CrossRef CAS.
- T. R. Ralph, S. Hudson and D. P. Wilkinson, ECS Trans., 2006, 1, 67–84 CrossRef CAS.
- H. Chhina, S. Campbell and O. Kesler, J. Power Sources, 2006, 161, 893–900 CrossRef CAS.
- H. Chhina, S. Campbell and O. Kesler, J. Power Sources, 2008, 179, 50–59 CrossRef CAS.
- B. K. Hong, P. Mandal, J. G. Oh and S. Litster, J. Power Sources, 2016, 328, 280–288 CrossRef CAS.
- S. W. Lee, B. Lee, C. Baik, T. Y. Kim and C. Pak, J. Mater. Sci. Technol., 2021, 60, 105–112 CrossRef CAS.
- L. Hu, B. K. Hong, J.-G. Oh and S. Litster, ACS Appl. Energy Mater., 2021, 4, 119–127 CrossRef CAS.
- C. E. Moore, J. Eastcott, M. Cimenti, N. Kremliakova and E. L. Gyenge, J. Power Sources, 2019, 417, 53–60 CrossRef CAS.
- Y. Yu, H. Li, H. Wang, X.-Z. Yuan, G. Wang and M. Pan, J. Power Sources, 2012, 205, 10–23 CrossRef CAS.
- K. D. Baik, B. K. Hong and M. S. Kim, Int. J. Hydrogen Energy, 2013, 38, 8927–8933 CrossRef CAS.
- T. Mittermeier, A. Weiß, F. Hasché, G. Hübner and H. A. Gasteiger, J. Electrochem. Soc., 2016, 164, F127–F137 CrossRef.
- J. Jang, M. Sharma, D. Choi, Y. S. Kang, Y. Kim, J. Min, H. Sung, N. Jung and S. J. Yoo, ACS Appl. Mater. Interfaces, 2019, 11, 27735–27742 CrossRef CAS PubMed.
- R. A. van Santen, M. Neurock and S. G. Shetty, Chem. Rev., 2010, 110, 2005–2048 CrossRef CAS PubMed.
- B. Genorio, D. Strmcnik, R. Subbaraman, D. Tripkovic, G. Karapetrov, V. R. Stamenkovic, S. Pejovnik and N. M. Markovic, Nat. Mater., 2010, 9, 998–1003 CrossRef CAS PubMed.
- B. Genorio, R. Subbaraman, D. Strmcnik, D. Tripkovic, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2011, 50, 5468–5472 CrossRef CAS PubMed.
- S. W. Yun, S. A. Park, T. J. Kim, J. H. Kim, G. W. Pak and Y. T. Kim, ChemSusChem, 2017, 10, 489–493 CrossRef CAS PubMed.
- B. M. Stühmeier, S. Selve, M. U. M. Patel, T. N. Geppert, H. A. Gasteiger and H. A. El-Sayed, ACS Appl. Energy Mater., 2019, 2, 5534–5539 CrossRef.
- R. Kamai, K. Kamiya, K. Hashimoto and S. Nakanishi, Angew. Chem., Int. Ed., 2016, 55, 13184–13188 CrossRef CAS PubMed.
- J. Luo, H. Tang, X. Tian, S. Hou, X. Li, L. Du and S. Liao, ACS Appl. Mater. Interfaces, 2018, 10, 3530–3537 CrossRef CAS PubMed.
- H.-E. Kim, I. H. Lee, J. Cho, S. Shin, H. C. Ham, J. Y. Kim and H. Lee, ChemElectroChem, 2019, 6, 4757–4764 CrossRef CAS.
- V. A. Sethuraman, J. W. Weidner, A. T. Haug, S. Motupally and L. V. Protsailo, J. Electrochem. Soc., 2008, 155, B50 CrossRef CAS.
- H. Shintani, Y. Kojima, K. Kakinuma, M. Watanabe and M. Uchida, J. Power Sources, 2015, 294, 292–298 CrossRef CAS.
- S.-M. Jung, S.-W. Yun, J.-H. Kim, S.-H. You, J. Park, S. Lee, S. H. Chang, S. C. Chae, S. H. Joo, Y. Jung, J. Lee, J. Son, J. Snyder, V. Stamenkovic, N. M. Markovic and Y.-T. Kim, Nat. Catal., 2020, 3, 639–648 CrossRef CAS.
- J. Durst, A. Orfanidi, P. J. Rheinlander, F. Hasche, C. Eickes, P. Suchsland, M. Binder and H. A. Gasteiger, ECS Trans., 2015, 69, 67–76 CrossRef CAS.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D. J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- X. Tian, X. Zhao, Y. Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. D. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu and S. Sun, Joule, 2019, 3, 124–135 CrossRef CAS.
- J. Liang, Z. Zhao, N. Li, X. Wang, S. Li, X. Liu, T. Wang, G. Lu, D. Wang, B.-J. Hwang, Y. Huang, D. Su and Q. Li, Adv. Energy Mater., 2020, 10, 2000179 CrossRef CAS.
- J. Choi, Y. J. Lee, D. Park, H. Jeong, S. Shin, H. Yun, J. Lim, J. Han, E. J. Kim, S. S. Jeon, Y. Jung, H. Lee and B. J. Kim, Energy Environ. Sci., 2020, 13, 4921–4929 RSC.
- Y. J. Lee, H. E. Kim, E. Lee, J. Lee, S. Shin, H. Yun, E. J. Kim, H. Jung, H. C. Ham, B. J. Kim and H. Lee, Adv. Energy Mater., 2021, 11, 2102970 CrossRef CAS.
- Z. Jia, G. Yin and J. Zhang, in Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, ed. W. Xing, G. Yin and J. Zhang, Elsevier, Amsterdam, 2014, vol. 6, pp. 199–229 Search PubMed.
- S. Martens, L. Asen, G. Ercolano, F. Dionigi, C. Zalitis, A. Hawkins, A. Martinez Bonastre, L. Seidl, A. C. Knoll, J. Sharman, P. Strasser, D. Jones and O. Schneider, J. Power Sources, 2018, 392, 274–284 CrossRef CAS.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hubner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- L. Xia, M. Ni, Q. Xu, H. Xu and K. Zheng, Appl. Energy, 2021, 294, 117012 CrossRef CAS.
- J. T. Fan, M. Chen, Z. L. Zhao, Z. Zhang, S. Y. Ye, S. Y. Xu, H. J. Wang and H. Li, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- M. Ammam and E. B. Easton, J. Power Sources, 2013, 236, 311–320 CrossRef CAS.
- S. Mukerjee and S. Srinivasan, J. Electroanal. Chem., 1993, 357, 201–224 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- L. Dubau, M. Lopez-Haro, L. Castanheira, J. Durst, M. Chatenet, P. Bayle-Guillemaud, L. Guétaz, N. Caqué, E. Rossinot and F. Maillard, Appl. Catal., B, 2013, 142–143, 801–808 CrossRef CAS.
- D. D. Papadias, R. K. Ahluwalia, N. Kariuki, D. Myers, K. L. More, D. A. Cullen, B. T. Sneed, K. C. Neyerlin, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F3166–F3177 CrossRef CAS.
- T. Y. Yoo, J. M. Yoo, A. K. Sinha, M. S. Bootharaju, E. Jung, H. S. Lee, B. H. Lee, J. Kim, W. H. Antink, Y. M. Kim, J. Lee, E. Lee, D. W. Lee, S. P. Cho, S. J. Yoo, Y. E. Sung and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 14190–14200 CrossRef CAS PubMed.
- S. Takenaka, H. Miyamoto, Y. Utsunomiya, H. Matsune and M. Kishida, J. Phys. Chem. C, 2014, 118, 774–783 CrossRef CAS.
- R. Subbaraman, D. Strmcnik, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, ChemPhysChem, 2010, 11, 2825–2833 CrossRef CAS PubMed.
- C. Liu, T. Uchiyama, N. Nagata, M. Arao, K. Yamamoto, T. Watanabe, X. Gao, H. Imai, S. Katayama, S. Sugawara, K. Shinohara, K. Oshima, S. Sakurai and Y. Uchimoto, ACS Appl. Energy Mater., 2021, 4, 1143–1149 CrossRef CAS.
- A. Suzuki, U. Sen, T. Hattori, R. Miura, R. Nagumo, H. Tsuboi, N. Hatakeyama, A. Endou, H. Takaba, M. C. Williams and A. Miyamoto, Int. J. Hydrogen Energy, 2011, 36, 2221–2229 CrossRef CAS.
- M. Lopez-Haro, L. Guétaz, T. Printemps, A. Morin, S. Escribano, P. H. Jouneau, P. Bayle-Guillemaud, F. Chandezon and G. Gebel, Nat. Commun., 2014, 5, 5229 CrossRef CAS PubMed.
- T. Morawietz, M. Handl, C. Oldani, K. A. Friedrich and R. Hiesgen, ACS Appl. Mater. Interfaces, 2016, 8, 27044–27054 CrossRef CAS PubMed.
- S. M. Jayawickrama, D. Wu, R. Nakayama, S. Ishikawa, X. Liu, G. Inoue and T. Fujigaya, J. Power Sources, 2021, 496, 229855 CrossRef CAS.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner, W. B. Gu, L. Thompson and A. Kongkanand, ACS Energy Lett., 2018, 3, 618–621 CrossRef CAS.
- N. Ramaswamy and S. Kumaraguru, ECS Trans., 2018, 85, 835–842 CrossRef CAS.
- L. Cindrella, A. M. Kannan, J. F. Lin, K. Saminathan, Y. Ho, C. W. Lin and J. Wertz, J. Power Sources, 2009, 194, 146–160 CrossRef CAS.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y. H. Chung, H. Kim, B. S. Mun, K. S. Lee, N. S. Lee, S. J. Yoo, D. H. Lim, K. Kang, Y. E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 CrossRef CAS PubMed.
- X. X. Du, Y. He, X. X. Wang and J. N. Wang, Energy Environ. Sci., 2016, 9, 2623–2632 RSC.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J. H. Jang, Y. H. Cho, T. Lim, Y. E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- Y. Kim, J.-H. Jang, J. Min, A. A. Jeffery, S. Lee, S. S. Chougule, M. Kim, N. Jung and S. J. Yoo, J. Mater. Chem. A, 2021, 9, 24480–24487 RSC.
- D. Banham, J. X. Zou, S. Mukerjee, Z. H. Liu, D. Yang, Y. Zhang, Y. Peng and A. G. Dong, J. Power Sources, 2021, 490 CrossRef CAS.
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef.
- U.S. DRIVE Partnership, Fuel Cell Technical Team Roadmap, Nov. 2017, https://www.energy.gov/sites/default/files/2017/11/f46/FCTT_Roadmap_Nov_2017_FINAL.pdf, accessed March 2022 Search PubMed.
- Z. Qiao, C. Y. Wang, C. Z. Li, Y. C. Zeng, S. Hwang, B. Y. Li, S. Karakalos, J. Park, A. J. Kropf, E. C. Wegener, Q. Gong, H. Xu, G. F. Wang, D. J. Myers, J. Xie, J. S. Spendelow and G. Wu, Energy Environ. Sci., 2021, 14, 4948–4960 RSC.
- X. Ao, W. Zhang, B. T. Zhao, Y. Ding, G. Nam, L. Soule, A. Abdelhafiz, C. D. Wang and M. L. Liu, Energy Environ. Sci., 2020, 13, 3032–3040 RSC.
- H. A. Gasteiger, J. E. Panels and S. G. Yan, J. Power Sources, 2004, 127, 162–171 CrossRef CAS.
- Z. Du, C. Liu, J. Zhai, X. Guo, Y. Xiong, W. Su and G. He, Catalysts, 2021, 11, 393 CrossRef CAS.
- M. Nagai, M. Yoshida and H. Tominaga, Electrochim. Acta, 2007, 52, 5430–5436 CrossRef CAS.
- S. Izhar and M. Nagai, J. Power Sources, 2008, 182, 52–60 CrossRef CAS.
- S. Izhar, M. Yoshida and M. Nagai, Electrochim. Acta, 2009, 54, 1255–1262 CrossRef CAS.
- G. E. Haslam, X. Y. Chin and G. T. Burstein, Phys. Chem. Chem. Phys., 2011, 13, 12968–12974 RSC.
- X. Yang, F. Zhao, Y.-W. Yeh, R. S. Selinsky, Z. Chen, N. Yao, C. G. Tully, Y. Ju and B. E. Koel, Nat. Commun., 2019, 10, 1543 CrossRef PubMed.
- G. W. Roberts, P. Chin, X. Sun and J. J. Spivey, Appl. Catal., B, 2003, 46, 601–611 CrossRef CAS.
- D. R. McIntyre, G. T. Burstein and A. Vossen, J. Power Sources, 2002, 107, 67–73 CrossRef CAS.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- Y. Shao, J.-P. Dodelet, G. Wu and P. Zelenay, Adv. Mater., 2019, 31, 1807615 CrossRef PubMed.
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Adv. Mater., 2019, 31, 1806545 CrossRef PubMed.
- T. Asset and P. Atanassov, Joule, 2020, 4, 33–44 CrossRef CAS.
- A. Uddin, L. Dunsmore, H. Zhang, L. Hu, G. Wu and S. Litster, ACS Appl. Mater. Interfaces, 2020, 12, 2216–2224 CrossRef CAS PubMed.
- Y. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. Chen, D. Myers, D. Su, K. L. More, G. Wang, S. Litster and G. Wu, Energy Environ. Sci., 2019, 12, 250–260 RSC.
- Y. Li, P. Zhang, L. Wan, Y. Zheng, X. Qu, H. Zhang, Y. Wang, K. Zaghib, J. Yuan, S. Sun, Y. Wang, Z. Zhou and S. Sun, Adv. Funct. Mater., 2021, 31, 2009645 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- L. Wan, W. Chen, H. Xu, Y. Wang, J. Yuan, Z. Zhou and S. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 45661–45669 CrossRef CAS PubMed.
- F. D. Speck, J. H. Kim, G. Bae, S. H. Joo, K. J. J. Mayrhofer, C. H. Choi and S. Cherevko, JACS Au, 2021, 1, 1086–1100 CrossRef CAS PubMed.
- H. Xie, X. Xie, G. Hu, V. Prabhakaran, S. Saha, L. Gonzalez-Lopez, A. H. Phakatkar, M. Hong, M. Wu, R. Shahbazian-Yassar, V. Ramani, M. I. Al-Sheikhly, D.-e. Jiang, Y. Shao and L. Hu, Nat. Energy, 2022, 7, 281–289 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |