
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnion carbide polymerization catalysts: from uncovering active site structures to designing molecularly-defined analogs†
David
Trummer‡
,
Anna Giorgia
Nobile‡
,
Pierre-Adrien
Payard‡
 ,
Anton
Ashuiev
,
Yuya
Kakiuchi
,
Daniel
Klose
,
Gunnar
Jeschke
* and
Christophe
Copéret
*
,
Anton
Ashuiev
,
Yuya
Kakiuchi
,
Daniel
Klose
,
Gunnar
Jeschke
* and
Christophe
Copéret
*
ETH Zürich Department of Chemistry and Applied Biosciences, Vladimir-Prelog-Weg 2, Zürich CH-8093, Switzerland. E-mail: ccoperet@ethz.ch; gunnar.jeschke@phys.chem.ethz.ch
First published on 15th September 2022
Abstract
The Union Carbide (UC) ethylene polymerization catalysts, based on chromocene dispersed on silica, show distinct features from the Phillips catalysts, but share the same heated debate regarding the structure of their active sites. Based on a combination of IR, EPR spectroscopies, labeling experiments, and DFT modeling, we identified monomeric surface-supported Cr(III) hydrides, (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)Cr(Cp)–H, as the active sites of the UC catalyst. These sites are formed in the presence of grafted and adsorbed chromocene as well as residual surface OH groups, only possible at high Cr loading, and involve a C–H activation of the Cp ring. These Cr-hydrides initiate polymerization, yielding Cr(III) alkyl species that insert ethylene through a Cossee–Arlman-type mechanism, as evidenced by spectroscopic studies. These insights inspired the design of a well-defined analog, CpCr(CH(SiMe3)2)2 grafted on partially dehydroxylated silica, that shows similar spectroscopic and polymer structure to the UC catalyst, further supporting the proposed active site structure.
SiO)Cr(Cp)–H, as the active sites of the UC catalyst. These sites are formed in the presence of grafted and adsorbed chromocene as well as residual surface OH groups, only possible at high Cr loading, and involve a C–H activation of the Cp ring. These Cr-hydrides initiate polymerization, yielding Cr(III) alkyl species that insert ethylene through a Cossee–Arlman-type mechanism, as evidenced by spectroscopic studies. These insights inspired the design of a well-defined analog, CpCr(CH(SiMe3)2)2 grafted on partially dehydroxylated silica, that shows similar spectroscopic and polymer structure to the UC catalyst, further supporting the proposed active site structure.
1 Introduction
Polyethylene (PE) is one of the most commonly used plastics worldwide.1 Since the 1950s, its production mostly relies on Ziegler–Natta or Phillips catalysts.1 In the early 1970s, an alternative Cr-based Union Carbide (UC) catalyst was developed based on silica-supported chromocene, [Cp2Cr(II)] (1).2–4 The UC catalyst is probably one of the first industrially used heterogeneous catalysts prepared from a silica supported organometallic reagent, thus pioneering the field of surface organometallic chemistry (SOMC).5,6 This catalyst shows high activity towards ethylene polymerization (EP) and its high response to H2 allows for chain length control.4,7–10Despite years of research and its unique features, its active sites and operating mechanism are unknown and still debated (Fig. 1A). The diversity of surface species, together with the low number of active sites (few %), have made the elucidation of the active site and mechanism challenging.9 Notably, the density of hydroxyl groups on the silica support influences the activity, which increases with decreasing OH density.11 A variety of Cr sites were previously reported: based on IR and UV-vis investigations, both mono-grafted 1, i.e. (SiO)CrCp and chemisorbed 1 (i.e. Cp2Cr⋯SiO2) at higher Cr loadings (Fig. 1A) were detected.4,7,12 The former was suggested to generate the corresponding monomeric Cr(IV) metallacycle (Fig. 1A, right) in the presence of ethylene.13 X-ray absorption spectroscopic (XAS) studies provided evidence for dimeric silica-supported Cr(III) species Cp2Cr2O4 before ethylene treatment.14 Dimeric half-sandwich Cr(II) sites on silica have also been identified, based on 1H MAS NMR investigations.8 The latter were proposed to react with C2H4 to yield dimeric Cr(III) alkyl silicates (Fig. 1A, right).13
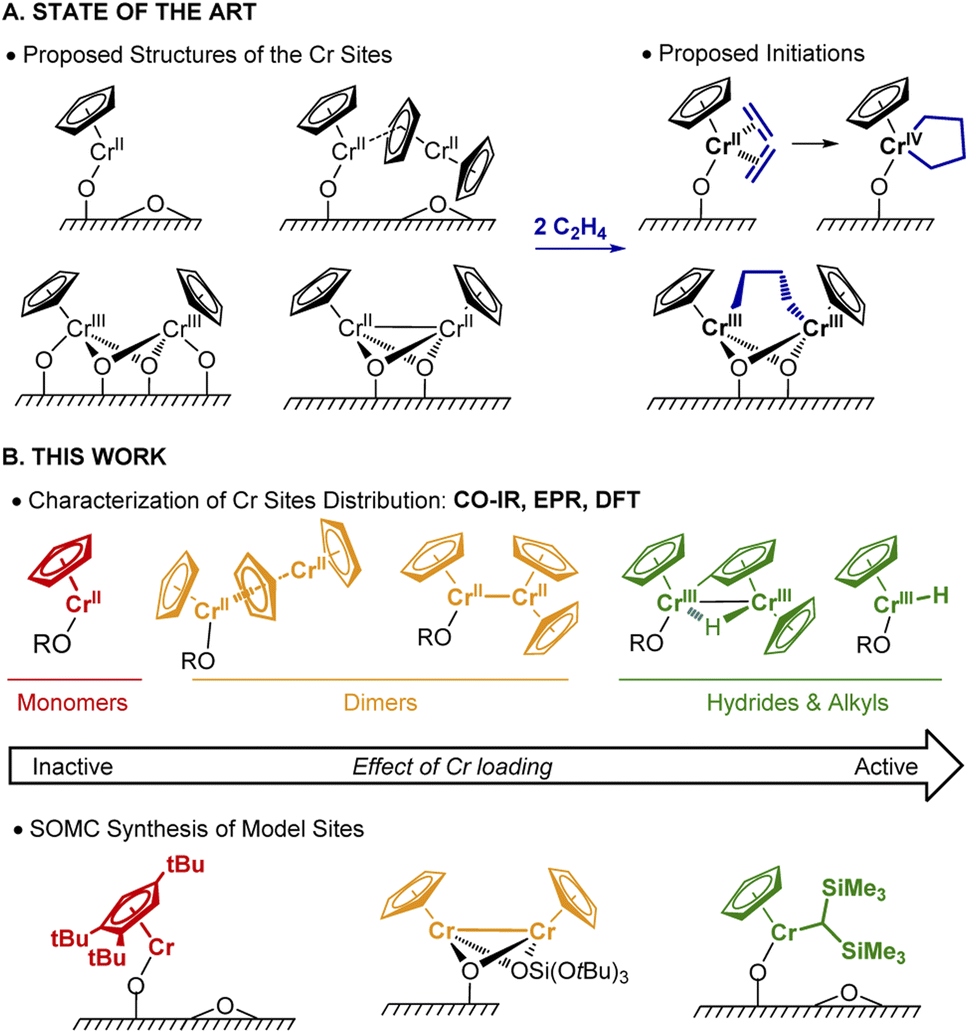 | ||
| Fig. 1 (A) Previously proposed surface site structures of the UC catalyst. (B) Illustration of Cr sites (above the arrow) investigated by CO-IR, EPR, and DFT modeling as a function of the Cr loading. The respective well-defined monomeric, dimeric Cr(II), and monomeric alkyl-Cr(III) sites, (below the arrow) are prepared via SOMC. | ||
In parallel, Theopold developed model systems based on molecular cyclopentadienyl Cr complexes, and showed that well-defined Cr(III) alkyls are able to insert ethylene, suggesting that the active sites of the UC catalyst could have similar electronic structure.15
In this work (Fig. 1B), we selectively prepare and characterize the Cr surface sites of the UC catalyst, using SOMC combined with spectroscopic and computational studies. In Part 1, using CO as a probe molecule for IR investigations, we identify various surface Cr sites as a function of Cr loadings and support our assignments by comparison with well-defined model systems prepared via SOMC. We evidence the presence of surface Cr hydrides in the UC catalysts at high loading, whose presence is confirmed by EPR spectroscopy. In Part 2, we investigate possible pathways for Cr–H formation using 2H labeling and DFT calculations. The CO vibrational frequencies of the proposed sites are calculated at the DFT level and compared with the experimental ones to validate the previously described assignments. Overall, we propose that two Cr moieties are cooperating to generate supported monomeric and dimeric Cr(III) hydrides via C–H activation of the Cp ring. In Part 3, using DFT calculations, we show that monomeric Cr(III) hydrides initiate EP via C2H4 insertion into the Cr(III)–H bond with the subsequent formation of Cr(III) alkyl species, enabling further polymerization. Finally, in Part 4, we develop a molecularly-defined analog of the proposed active site – namely (SiO)CrCp[CH(SiMe3)2]. This Cr(III)-alkyl initiates EP, as evidenced by EPR hyperfine spectroscopy and generates a similar polymer, further supporting that the active sites of the UC catalysts correspond to surface CpCr(III) hydrides/alkyls.
2 Results and discussion
2.1. Characterization of monomeric Cr(III)–H by CO-IR probe and EPR spectroscopies
While solving the structure of the active site of the UC catalyst is challenging, its identification should be possible, providing one detects a spectroscopic signature that correlates with polymerization activity.10 As reported earlier,2–4 the EP activity of the UC catalyst is critically dependent on the Cr loading. We therefore prepared and investigated four different catalysts varying in Cr loadings: 1-SiO2-0.25, 1-SiO2-0.5, 1-SiO2-1, and 1-SiO2-2 that contain 0.25, 0.5, 1.0, and 2.0 wt% of Cr, respectively (ESI, Section S2.1† for preparation). Contacting 1 with partially dehydroxylated silica at 700 °C (SiO2-700)6 led to a color change of the material, from white to dark blue/black and a decrease of the IR bands corresponding to the surface OH groups (Fig. S1).† According to elemental analysis and 1H NMR quantifications of the washing solutions (Section S2.1 and Table S1),† about one Cp/Cr is present on silica for 1-SiO2-0.25, suggesting the presence of mainly mono-grafted sites. The Cp/Cr ratio increases with increasing Cr loading, reaching almost 3 Cp/2 Cr in 1-SiO2-1, suggesting the presence of both grafted and strongly adsorbed 1 on silica. The EP activities for all the obtained materials show the following trends: while 1-SiO2-1 and 1-SiO2-2 are EP active, the low loading materials 1-SiO2-0.25 and 1-SiO2-0.5 show hardly any activity (Table S2).† Therefore, high Cr loadings (>1.0 wt% of Cr) favors the generation of the active sites in UC catalysts, consistent with previous findings.4IR spectroscopy using CO as a probe molecule6 was used to characterize the prepared UC catalysts (Fig. 2) and identify possible Cr surface sites (Section S1).† We assigned the various IR bands by comparison with the ones obtained for well-defined Cr surface species prepared by SOMC (Fig. 1B and 2, 2-SiO2 and 3-SiO2).
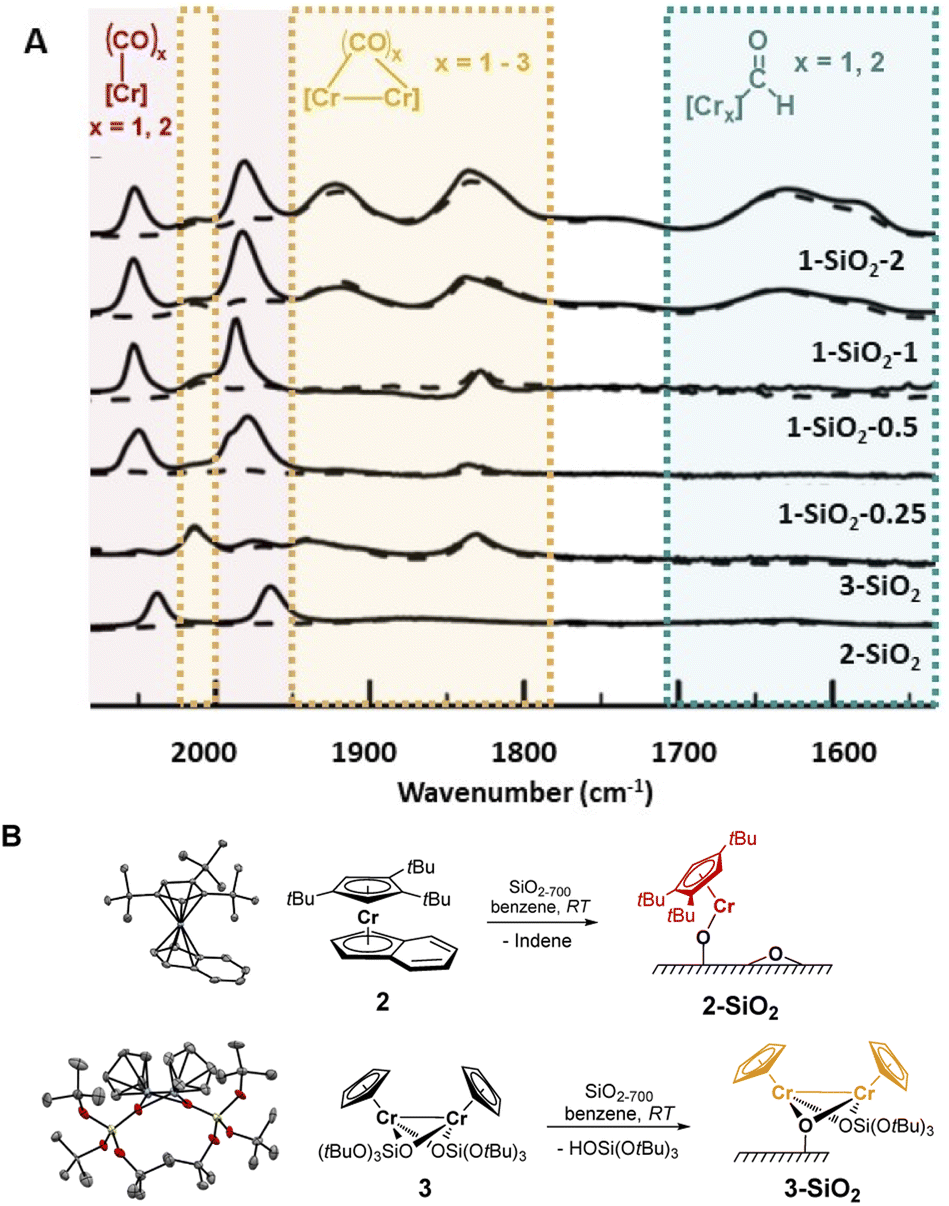 | ||
| Fig. 2 (A) Background-subtracted IR spectra (solid lines) of the UC catalysts with different Cr loadings and model systems 2-SiO2 and 3-SiO2 (Fig. 2B) after exposure to CO (25 mbar). Dashed lines indicate the IR spectra after the exposure to high vacuum (10−5 mbar). Reversible CO bands are marked in red (indicative of monomeric Cr(II) sites). Bands from irreversibly bound CO molecules are marked in orange (indicative of bridging CO between interacting Cr atoms). Low energy CO bands are marked in green (indicative of formyl groups). (B) Grafting of tailored monomeric and dimeric model complexes on SiO2-700. | ||
The low-loading material 1-SiO2-0.25 after exposition to CO shows only two predominant CO bands at 2061 cm−1 and 1981 cm−1 (Fig. 2A, red region). These bands disappear upon high vacuum (10−5 mbar; Fig. 2A, dashed lines), indicating the reversible and weak CO binding to these Cr surface sites. While increasing the Cr loading to 0.5 wt% (1-SiO2-0.5), three additional bands at 2015 cm−1, ∼1905 cm−1 (broad) and 1837 cm−1 arise (Fig. 2A, orange region). In contrast to the previously observed bands (Fig. 2A, red region), these bands are formed irreversibly, indicating a stronger CO binding to the Cr sites. Upon further increasing the Cr content to 1 wt% (1-SiO2-1), three additional broad CO bands appear at 1742 cm−1, 1630 cm−1, and 1579 cm−1 (Fig. 2A, green region).16 Notably, the appearance of the latter signals correlates with EP activity.
We then focused on the assignment of the CO-IR bands observed in the three groups (red, orange, green). The two reversible bands, together with a small shoulder at 1993 cm−1 (Fig. 2A, red) may be assigned to monomeric Cr surface sites. Indeed, the well-defined silica-supported monomeric Cr(II) [(SiO)Cr(tttCp)] 2-SiO2 (Fig. 2B, Section S2.2, and Fig. S3 for the IR spectra, S4† for XANES analysis) shows two reversible bands at 2035 cm−1 and 1960 cm−1 upon exposition to CO (Fig. 2A), as observed for 1-SiO2-0.25. Therefore, the two signals in the red region are assigned to the symmetric and antisymmetric CO stretching modes of di-carbonylic, monomeric Cr(II) sites.17 Notably, the well-defined Cr(II) monomeric species in 2-SiO2 show no EP activity at 10 bars and 40 °C, indicating that monomeric Cr(II) sites are not able to polymerize ethylene.
For 1-SiO2-0.5 (Fig. 2A, orange region), the irreversibility of CO adsorption could arise due to bridging CO molecules between two Cr centers, suggesting the presence of dimeric Cr surface sites. The well-defined dimeric Cr(II) surface site 3-SiO2 (Fig. 2B, ESI Section S2.3 & Fig. S6–S9†) shows four bands between 2012 cm−1 and 1830 cm−1 resulting from irreversibly bound CO molecules. These IR frequencies are consistent with the CO bands observed in 1-SiO2-0.5 (Fig. 2A, orange region). Therefore, we attribute these IR bands to the dimeric Cr(II) sites of the UC catalyst. Similar to 2-SiO2, 3-SiO2 shows no EP activity, indicating that the active sites of the UC catalyst do not correspond to monomeric or dimeric Cr(II) species.
This encouraged us to further investigate the surface species associated with the low energy CO bands (Fig. 2A, green region), which become significant only for the 1-SiO2-1 and 1-SiO2-2 materials (Fig. S10–11).† The CO vibrational frequencies down to 1550 cm−1 suggest the presence of formyl groups bound to Cr,18 that could result from the CO insertion into a Cr–H bond. While similar complexes, [Cp*CrPR3(CO)2CHO],18 have been formed upon the reaction of [Cp*Cr(PR3)(CO)3] with a hydride source, the formation of formyl intermediates via migratory insertion of CO is rare for early transition metals, but has been documented with zirconium hydride, a textbook example.19 Therefore, we propose that the UC catalysts with higher Cr loading are likely associated with the formation of surface-supported Cr hydrides. Since the IR bands of formyl grow in intensity for the UC catalysts active in EP (Fig. 2A), the proposed Cr–H could be the active or initiating sites, in analogy with previously described C2H4 polymerizing Ti hydrides.20
To inquire the possible presence of surface Cr hydrides as initiation sites of EP, we carried out 2D J-mediated heteronuclear multi-quantum correlation (HMQC)20 MAS NMR of 1-SiO2-1 after exposure to 13C2D4. The HMQC spectrum (Fig. S12B†) shows narrow and well-resolved correlations between the 13C signals at 29 and 31 ppm and the 1H signal at 1.5 ppm, indicating the presence of 1H in the obtained PE.21–23 The proton source has to originate from the surface OH groups or the Cp ligands. IR investigations of the same experiment show a weak band (Fig. S12A,† shown in green) at 2887 cm−1 related to a –CH stretching and an additional –CD2-stretching. This signals imply the presence of C–H vibrations, which could be assigned to the end –CD2H group resulting from multiple insertions of C2D4 into Cr hydrides as initiating sites.24,25 All together, these results support a hydride-mediated initiation mechanism.
The potential activity of Cr–H raises questions about the Cr oxidation state of these species. Previous work suggests the presence of Cr(III) sites in the UC system,14 we therefore performed EPR studies of the catalysts. We found that the CW EPR spectrum of 1-SiO2-1 (Cp2CrII on silica) contains paramagnetic species, even before contacting with C2H4 (Fig. S13A,† top), which are consistent with Cr(III) surface sites. The major fraction is represented by a nearly axial monomeric low-spin Cr(III) species. It appears that the EPR signature of this species changes upon contact with C2H4 (Fig. S13B†); the g tensor of this newly formed species is better fitted as being orthorhombic (Fig. S13A,† bottom) with an increased gx value, consistent with a change in the ligand surrounding of the LS Cr(III) sites after the contact with C2H4. This finding suggests that the observed Cr(III) species are the active sites of EP and likely a surface Cr hydride, as discussed above.
To summarize, while both mono- and dimeric Cr(II) surface sites are identified in the UC system, they are not active in EP. At higher Cr loadings, surface-supported Cr(III) hydride species are also formed; their presence correlates with EP activity and are thus likely associated with the active site of the UC catalyst.
2.2. Formation and characterization of surface Cr hydrides
In the next step, we investigated the possible pathways for the formation of these Cr(III) hydrides. In molecular systems, the formation of binuclear bridging Cr hydride complexes was previously reported,26–29 and in analogy to Cr, dinuclear bridging molybdocene–hydride complexes with activated Cp ligands are also known.30 Monomeric Cr hydrides are scarce,31 however, Sinema et al. reported a C–H activation step at the Cp ligand on ansa–chromocene complexes in the presence of [H(OEt2)2BAr4],32 and related activation pathways were also reported for other 3d metallocene-based complexes.33,34To elucidate whether the related C–H activation of the Cp ligand could take place on the surface, we grafted perdeuterated chromocene (1d10) on silica. If the C–D activation on the Cp ring is possible, D might exchange with surface OH groups, resulting in the appearance of an IR band corresponding to Si–OD groups. This IR band is indeed observed and suggests that C–H activation at the Cp ring is feasible (Fig. 3).
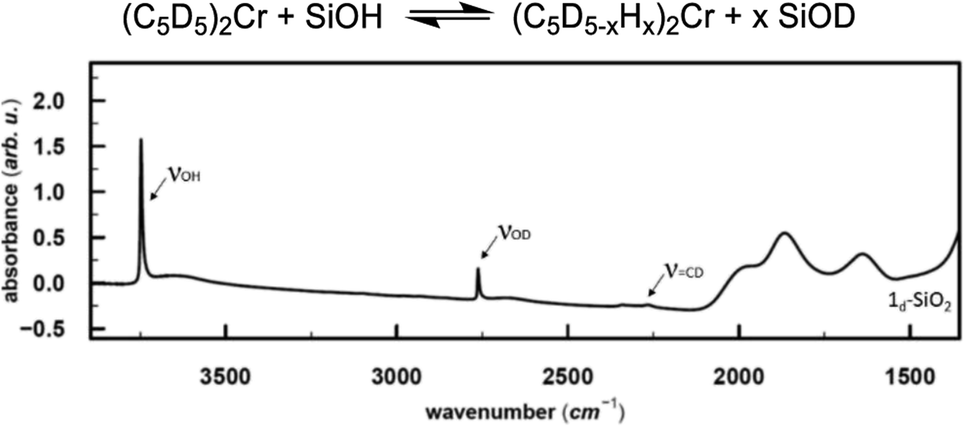 | ||
Fig. 3 Top: Proposed reaction for the interaction of 1d10 with surface OH groups, resulting in D exchange of deuterated Cp rings. Bottom: IR spectrum of D-labeled 1-SiO2-1. In addition to the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CD band, consistent with the deuterated Cp ring, the Si–OD band arises from the H/D exchange with the silica surface. CD band, consistent with the deuterated Cp ring, the Si–OD band arises from the H/D exchange with the silica surface. | ||
To elucidate how the C–H activation can take place, we performed subsequent grafting of non-labeled (1) and perdeuterated (1d10) chromocene and vice versa (Section S2.4 and Fig. S14†). When 1ll-d-SiO2-1 (0.25 wt% 1d10 & 0.75 wt% 1) is exposed to C2H4, only trace amounts of –CH2D end groups are observed in the IR spectrum (Fig. S15),† while after polymerization of C2D4, mainly –CD2H end groups are observed. This suggests that the C–H activation takes place on the chemisorbed 1 (see Fig. S15).† Congruent results are obtained when exposing 1hl-d-SiO2-1 (0.25 wt% 1 & 0.75 wt% 1d10) to labeled and non-labeled ethylene (Fig. S15†).35
We performed DFT studies to explore possible reaction pathways of the reaction of 1 with the silica surface (modeled by RO = –Si(OH)3). This simple model cannot account for secondary interactions with the surface, thus the computed pathways for the formation of the Cr-hydride species (Fig. 4) should be considered as providing a qualitative insight on the feasibility of the C–H activation process, but cannot be used to assess the relative stability of surface species. The structures of the Cr species (Tables S5 and S6† for the DFT-optimized structures) were optimized using unrestricted open-shell calculations with the B3LYP functional (see ESI and Table S4† for the computational details and optimization of the computational method) and the spin state with the lowest energy was selected (Table S7†). Dimeric species were modeled using the broken symmetry approach.36
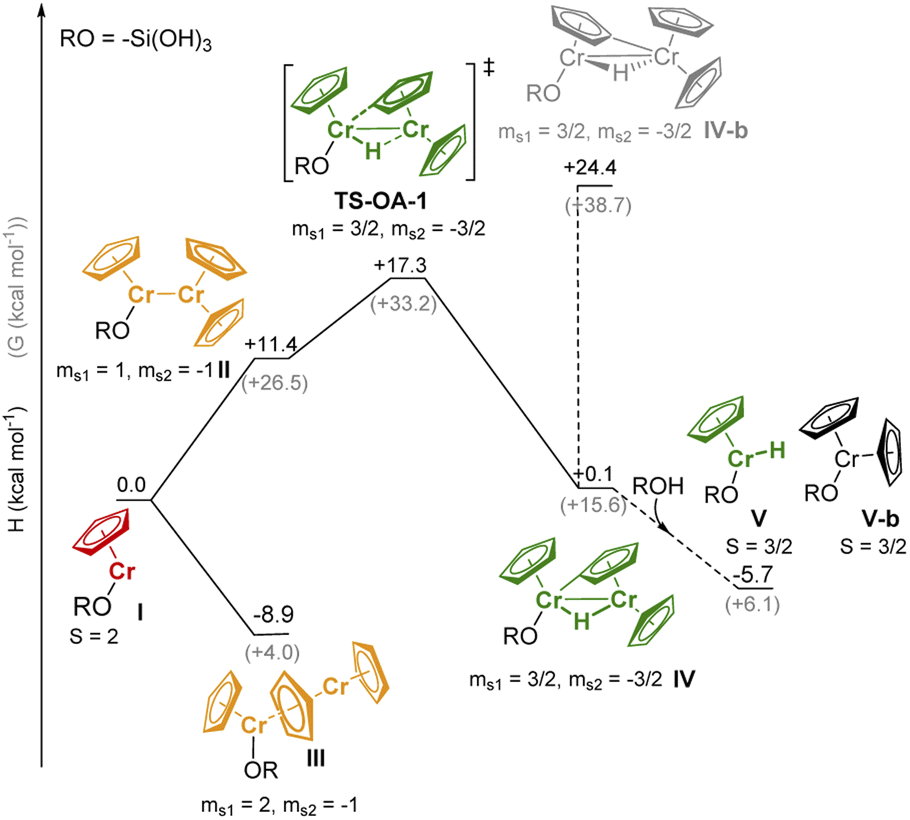 | ||
| Fig. 4 Proposed reaction pathways for the interaction of 1 on partially dehydroxylated silica. Enthalpies (H kcal mol−1) and free energies (G kcal mol−1) are calculated with respect to I and CrCp2. For monomeric sites, the spin quantum number is indicated with S; for dimeric sites, the local projection of the spin, as obtained from the Mulliken spin density on [CrCp] fragments I, is shown with msi. Since the formation of dimers is artificially disfavored given the approximations on the estimation of the entropy, only the enthalpy will be discussed in the text. | ||
The high-spin (S = 2) mono-grafted site I (see Fig. 4 and Table S7),† is coordinatively unsaturated and may interact with adjacent Si–O–Si bridges or surface OH groups. At higher Cr loading, site I can also interact with another molecule of 1, either to form stacked metallocene complex III (ΔH = −8.9 kcal mol−1), or complex II with a direct Cr–Cr bond (site II, ΔH = +11.4 kcal mol−1). Sites II can further reorganize through C–H activation at the Cp ring, yielding the surface site IV, a bridging Cr(III)–hydride complex (TS-OA-1, ΔH‡ = +17.3 kcal mol−1). The C–H activation at the other Cr moiety is energetically strongly disfavored (IV-b, ΔH = +24.4 kcal mol−1 with respect to I), consistent with the labeling studies (see Fig. S15).† The hydride in IV is expected to be located closer to the second Cr center, leading to two Cr(III) sites. The dimeric bridging Cr(III) hydride species (Fig. 4) are likely EPR silent due to a strong exchange between the neighboring Cr(III) centers. This would contradict the observation of the active Cr(III) sites, as evidenced by CW EPR (Fig. S13).† Therefore, we suggest that the dimeric bridging Cr(III) hydride species (IV) split apart to form monomeric Cr(III) sites upon reaction with residual surface OH groups to yield monomeric Cr(III) hydrides V and Cr(III) site V-b; this process would be energetically favored (ΔH = −5.8 kcal mol−1) as shown in Fig. 4.
All the proposed surface species I–V can coordinate CO molecules or allow CO insertion into the Cr–H bond, resulting in characteristic IR bands. We therefore calculated the CO vibrational frequencies (Section S3.2, Fig. S16 & Table S8† for computational details) of all possible Cr species after CO coordination to verify our previous assignments (see Part 1). The DFT calculated IR-CO frequencies obtained from the optimized structures are in good agreement with the assignments discussed above (Section S3.2†).
2.3. Reactivity of Cr(III) hydrides towards ethylene based on calculated reaction pathways
As outlined above, the EP activity of the UC catalyst correlates with the appearance of the dimeric (IV) and monomeric (V) Cr(III) hydrides (see Part 2), indicating that one of these hydrides could be the active site(s). We then calculated the energy barrier for the activation of ethylene and further chain propagation for sites IV and V (Fig. S31† and 5, respectively).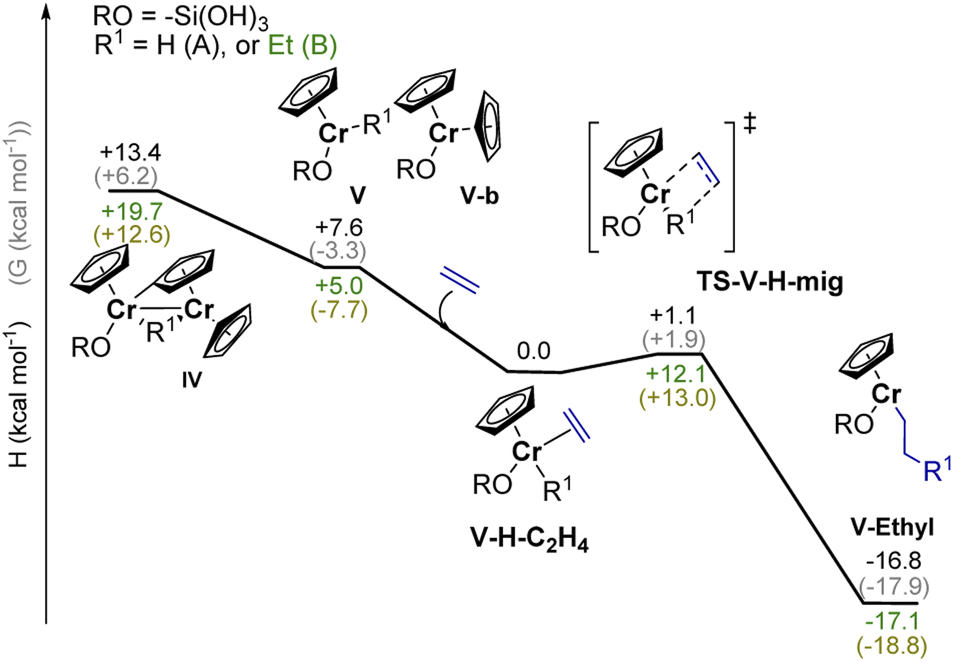 | ||
| Fig. 5 Calculated reaction enthalpies (H kcal mol−1) and free energies (G kcal mol−1) for potential ethylene activation pathways involving monomeric Cr(III) hydrides such as V (in black), or monomeric Cr(III) alkyl species such as V-ethyl (in green). Ethylene activation and chain propagation are both associated with reasonable energy costs. | ||
Ethylene coordination to the coordinatively unsaturated monomeric Cr(III) hydride V and further C2H4 insertion into the Cr–H bond is nearly barrier-less (ΔH† = 1.1 kcal mol−1, ΔG† = 1.9 kcal mol−1, Fig. 5). The resulting monomeric Cr(III) alkyl V-ethyl (ΔH = −16.3 kcal mol−1 for V-H-C2H4) can undergo chain growth via subsequent C2H4 insertions into the Cr–C bond, according to the Cossee–Arlman mechanism.20,37 This process is quite similar to what is described for the corresponding reaction of Ti(III) hydrides with C2H4.20 The obtained V-ethyl sites are thus highly active in EP, according to the “augmented” Cossee–Arlman mechanism previously described for the EP-active well-defined Ti(III) alkyls37 and for heterogeneous Ziegler–Natta catalysts.38 The subsequent C2H4 insertion has in fact a relatively low calculated TS barrier of ΔH = 12.1 kcal mol−1 (Fig. 5). Alternatively, ethylene can coordinate to the Cr atom grafted on the silica surface, while the initially bridging hydride is now localized on the C–H activated chromocene fragment in complex VI-a (Fig. S31).† The hydride can migrate to C2H4 while the first Cr–C bond is formed, leading to the formation of a dimeric Cr(III) alkyl complex. However, subsequent chain propagation is energetically prohibited in this case, further supporting that the active sites correspond to monomeric Cr(III) hydrides.
2.4. Synthesis and characterization of well-defined silica-supported Cr(III) alkyl analogs to the UC catalyst
The combination of experimental and computational studies suggest monomeric surface Cr(III) hydrides as the initiating active species. Inspired by the work of Theopold et al.,39,40 we developed CpCr[(CH(SiMe3)2)2] (4) (see Fig. 6A) by reacting [CpCrCl2]2 with two equivalents of LiCH(SiMe3)2 (ESI Section 4.1 & Fig. S33† for details on the synthesis and characterization).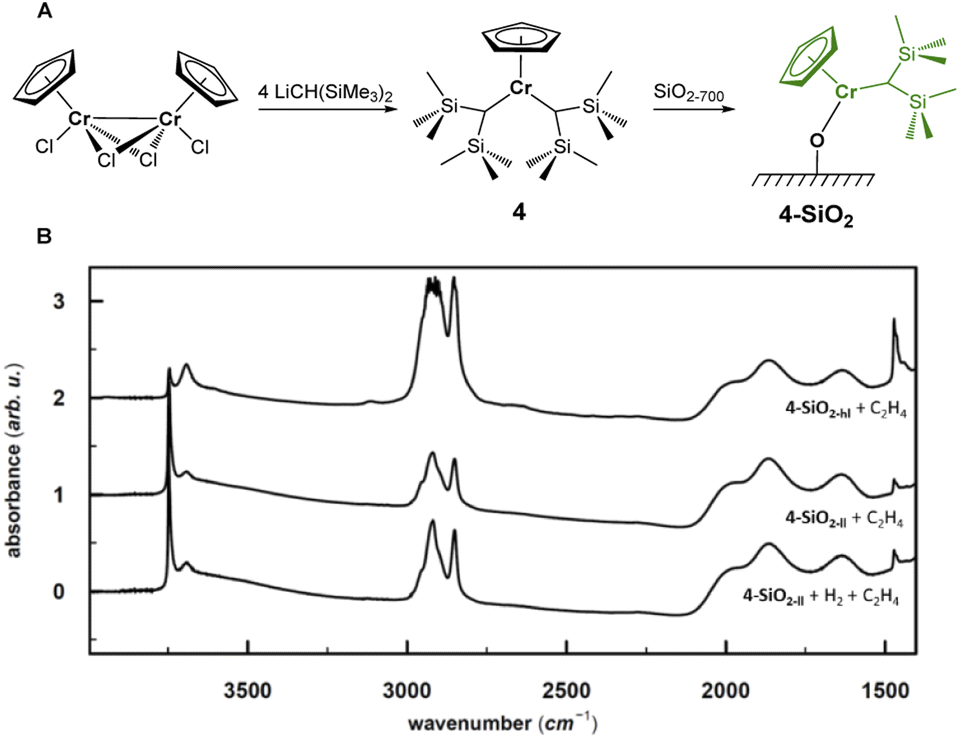 | ||
| Fig. 6 (A) Synthesis and surface chemistry of 4-SiO2. (B) IR spectra of 4-SiO2 with high Cr loading (0.8 wt%) after exposure to 25 mbar of C2H4 (top), and 4-SiO2 with low Cr loading (0.2 wt%) after exposure to C2H4 (middle) and H2 (50 mbar) prior to C2H4 (bottom). The higher intensity of C–H bands after exposure of the low Cr-loading material to H2 evidences for slightly higher EP activity. | ||
Upon contacting 4 with partially dehydroxylated silica (SiO2-700), CH2(SiMe3)2 is released. The IR spectrum of 4-SiO2 shows two intense bands at 3126 cm−1 and 2955 cm−1, indicating the presence of the [CH(SiMe3)2] ligand (Fig. S3 and S34†), consistent with the formation of surface alkyl species (SiO)CrCp[CH(SiMe3)2], whose structure is close to the proposed active sites, V-ethyl. Upon exposure to C2H4, 4-SiO2 rapidly polymerize ethylene, as evidenced by the strong C–H bands in the IR spectrum (see Fig. 6B). In fact, the average weight and number molar masses Mw and Mn respectively, as well as the Mw/Mn ratio of the produced PE is essentially the same as the one produced by the industrial-like UC catalyst (Table S15†), indicating that 4-SiO2 and the UC catalyst contain similar type of active sites.
The CW EPR spectrum41 of 4-SiO2 (Fig. 7A, black) shows the presence of HS (120–240 mT region) and a LS (300–400 mT region) Cr(III) species. Silica-supported Cr(III) HS species are characterized by a zero-field splitting (ZFS, described by parameters D and E) that exceeds Zeeman interaction at typical magnetic fields and by an effective g tensor (g⊥ ∼ 4, g∥ ∼ 2), limiting their assignment to structures based on the comparison of g tensor values or exact parameters of the ZFS tensor as the spectra only inform on the ZFS axiality (E/D).42 The major fraction is represented by a single line in the g ∼ 4 region, which suggests E/D values close to 0 and is thus indicative of nearly axial symmetry. In contrast, the LS species are best simulated with an orthorhombic g tensor (Fig. S35†). For 4-SiO2, the axially symmetric HS species can be assigned to the mono-grafted axially symmetric (SiO)CrCp[CH(SiMe3)2] surface site, while the LS species likely involve additional interactions of the Cr center with nearby Si–O–Si siloxane bridges. The g tensor principal values for this LS species are close to those obtained for the Cr(III) species of 1-SiO2-1 after its contact with C2H4 (Fig. S36†).
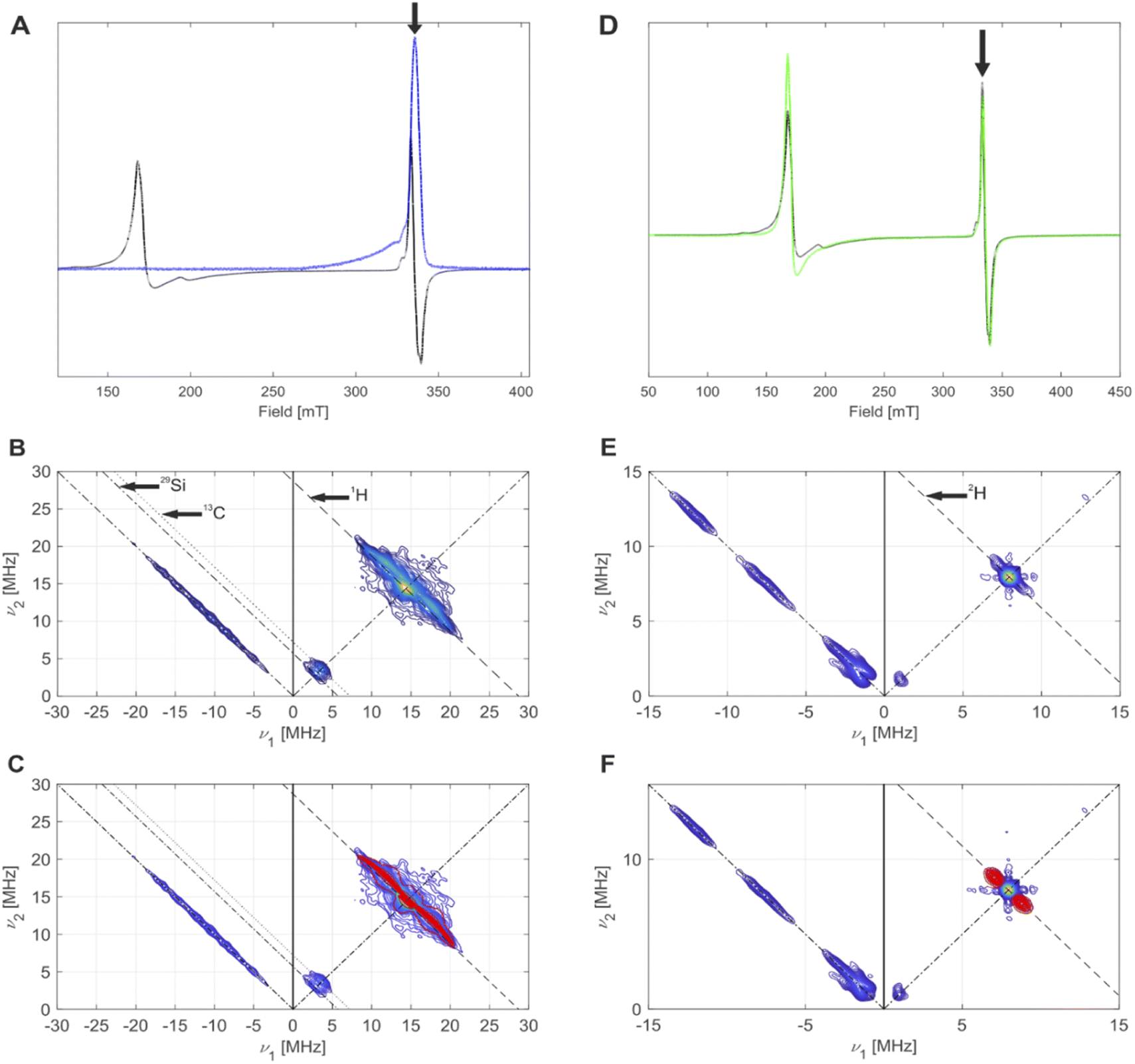 | ||
| Fig. 7 (A) CW EPR spectrum (black) and echo-detected EPR spectrum (blue) of 4-SiO2, measured at 5 K. The black arrow indicates the field position for HYSCORE. (B) HYSCORE spectrum (blue to yellow) of 4-SiO2, measured at 5 K. Antidiagonal lines indicate nuclear Zeeman frequencies of magnetically coupled nuclei that are present in this system. (C) Overlay of simulated (red) 2H hyperfine couplings with the experimental HYSCORE spectrum (blue to yellow) from (B) with an overall good agreement to the experimental spectrum. Details on the measurement conditions are outlined in the method Section of the ESI.† (D) CW EPR spectra of 4-SiO2 before (black) and after (green) EP. The black arrow indicates the field position used for HYSCORE. (E) Q-band HYSCORE spectrum (blue to yellow) of 4-SiO2 after the C2D4 exposure. (F) Overlay of simulated (red) 2H hyperfine couplings with the experimental HYSCORE spectrum (blue to yellow) from (E) with an overall good agreement to the experimental spectrum. | ||
We then focused on pulse EPR investigations of the LS species. As seen from the echo-detected EPR spectrum of 4-SiO2 (Fig. 7A), in pulse EPR spectroscopy at 5 K only the LS component is observable, enabling the selective characterization of these species by 2D hyperfine sublevel correlation (HYSCORE) spectroscopy.43 These spectra (Fig. 7B and C) reveal the presence of hydrogen atoms magnetically coupled to Cr(III) centers, and weak couplings to 29Si and 13C nuclei. The observation of 13C signals for 4-SiO2 in absence of 13C labeling is indicative of the presence of the Cp ligand within LS Cr(III) species. For 1H, spectral simulation of the observed hyperfine couplings (Fig. 7C) allows to extract the principal values of the 1H hyperfine coupling tensor (aiso = 3.0 MHz, adip = [−4.75–4.75 9.5] MHz), assuming axial symmetry. Using the point-dipole approximation,44 we determined a Cr(III)–H distance of 2.55 Å, suggesting α-H atoms of [CH(SiMe3)2] ligands in metal-alkyl complexes.37,45 This allows us to assign the observed S = 1/2 (g = [1.9916; 2.0028; 2.0157] species to (SiO)CrCp[CH(SiMe3)2] surface sites. Note the similar g tensor principal values to 1-SiO2-1 in presence of C2H4 (see Part 1), indicating that similar (SiO)CrCp–R species (R = –(CH2)n–CH3) are formed in the UC system during EP.
We further performed EPR studies of 4-SiO2 after its contact with C2D4 and observed unchanged principal g tensor values of the investigated LS species (Fig. 7D). In its Q-band HYSCORE spectrum 2H hyperfine couplings (Fig. 7E), with aiso = 1.7 MHz, adip = [−0.6–0.6 1.2] MHz and unresolved quadrupole coupling appear (Fig. 7F), indicating that 2H nuclei are in close proximity to the Cr(III) center, consistent with the C2D4 insertion into the Cr(III)–C bond according to the Cossee–Arlman mechanism.20,37 This is further confirmed by the disappearance of 1H hyperfine couplings in the X-band HYSCORE spectrum of 4-SiO2 after the C2D4 polymerization (Fig. S36†), consistent with its insertion into the Cr(III)–CH(SiMe3)2 bond of 4-SiO2. Therefore, we propose that the same Cossee–Arlman mechanism is involved in EP on both 4-SiO2 and the UC catalysts.
3 Conclusions
In this work, we characterized surface sites of the UC silica-supported Cr-based ethylene polymerization catalyst, using SOMC in combination with IR and EPR spectroscopic investigations, labeling experiments, and computational studies. We identified various surface sites depending on the Cr loading. Monomeric Cr(II) species are formed at very low Cr loading and show no polymerization activity. An increasing Cr loading leads to the formation of dimeric Cr(II) surface sites with various Cr–Cr interactions, that are inactive towards EP. At higher loadings, monomeric Cr(III) hydrides (SiO)CrCp–H are formed, their presence correlates with EP activity, and are thus proposed to be the active sites of the UC catalyst. These species are formed via C–H activation at the Cp ring and involvement of surface hydroxyl groups. The presence of a small fraction of adjacent surface OH groups is proposed to be involved in splitting the dimeric Cr(III) hydrides into the monomeric species, which are able to insert ethylene, thus corresponding to the active sites of the UC catalysts. Such structures inspired the preparation of an analog well-defined silica-supported system, namely (SiO)CrCp[CH(SiMe3)2] (4-SiO2). This material displays similar EPR signatures and EP activity as the UC catalysts and produces polymers of similar molecular weight distribution. Using hyperfine EPR spectroscopy, we also show that the well-defined Cr(III) surface alkyls perform ethylene polymerization via insertion into the Cr(III)–C bond, thus supporting the analog proposition for the EP mechanism for the active species of the UC catalysts. These findings open ways to rationally design novel heterogeneous EP catalysts.
Data availability
All data are provided in the ESI† and additional data can be available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This manuscript is dedicated to the memory of Prof. Richard A. Andersen. The authors also thank Dr Michael Wörle and Darryl Nater for assistance with crystallographic measurements; Dr Zachariah J. Berkson for the SS NMR measurements; Prof. Richard A. Andersen, Prof. Keith Searles and Dr Florian Allouche for scientific discussions. Dr Romain Berthoud and Dr John Severn are acknowledged for enabling HT-SEC measurements. D. T., A. N., and Y. K. are grateful to the Swiss National Foundation (SNF) for financial support of this work (grant no. 200021_169134, 200020B_192050, and 200020B_192050 respectively).Notes and references
- M. P. McDaniel, in Advances in Catalysis, B. C. Gates and H. Knözinger, eds., Academic Press, 2010, vol 53, pp. 123–606 Search PubMed.
- H. R. Sailors and J. P. Hogan, J. Macromol. Sci., Part A, 1981, 15, 1377–1402 Search PubMed.
- US Pat. 3,709,853, 1973.
- F. J. Karol, G. L. Karapinka, C. Wu, A. W. Dow, R. N. Johnson and W. L. Carrick, J. Polym. Sci., Part A: Polym. Chem., 1972, 10, 2621–2637 CrossRef CAS.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- A. Zecchina, G. Spoto and S. Bordiga, Faraday Discuss. Chem. Soc., 1989, 87, 149–160 RSC.
- M. Schnellbach, F. H. Köhler and J. Blümel, J. Organomet. Chem., 1996, 520, 227–230 CrossRef CAS.
- S. L. Fu and J. H. Lunsford, Langmuir, 1990, 6, 1774–1783 CrossRef CAS.
- F. J. Karol and C. Wu, J. Polym. Sci., Part A: Polym. Chem., 1974, 12, 1549–1558 CAS.
- F. J. Karol, C. Wu, W. T. Reichle and N. J. Maraschin, J. Catal., 1979, 60, 68–76 CrossRef CAS.
- S. L. Fu and J. H. Lunsford, Langmuir, 1990, 6, 1784–1792 CrossRef CAS.
- K. H. Theopold, R. A. Heintz, S. K. Noh and B. J. Thomas, in Homogeneous Transition Metal Catalyzed Reactions, American Chemical Society, 1992, vol 230, ch. 41, pp. 591–602 Search PubMed.
- P. J. Ellis, R. W. Joyner, T. Maschmeyer, A. F. Masters, D. A. Niles and A. K. Smith, J. Mol. Catal. A: Chem., 1996, 111, 297–305 CrossRef CAS.
- K. H. Theopold, Eur. J. Inorg. Chem., 1998, 1998, 15–24 CrossRef.
- A further increase in the Cr loading on silica (2 wt% Cr, 1-SiO2-2) does not lead to the appearance of new CO adsorption bands, though the peak at 1579 cm−1 gains in intensity.
- This was previously suggested and confirmed by carrying out the experiment with a 1/1 mixture of 12CO/13CO (see Fig. S5†) and is in agreement with previous assignment, see ref. 6.
- P. Leoni, A. Landi and M. Pasquali, J. Organomet. Chem., 1987, 321, 365–369 CrossRef CAS.
- P. T. Wolczanski and J. E. Bercaw, Acc. Chem. Res., 1980, 13, 121–127 CrossRef CAS.
- C. P. Gordon, S. Shirase, K. Yamamoto, R. A. Andersen, O. Eisenstein and C. Copéret, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5867–E5876 CAS.
- M. Pollard, K. Klimke, R. Graf, H. W. Spiess, M. Wilhelm, O. Sperber, C. Piel and W. Kaminsky, Macromolecules, 2004, 37, 813–825 CrossRef CAS.
- R. Kitamaru, F. Horii and K. Murayama, Macromolecules, 1986, 19, 636–643 CrossRef CAS.
- R. Blom, I. M. Dahl, A. Follestad and K. J. Jens, J. Mol. Catal. A: Chem., 1999, 138, 97–102 CrossRef CAS.
- S. Krimm, C. Y. Liang and G. B. B. M. Sutherland, J. Chem. Phys., 1956, 25, 549–562 CrossRef CAS.
- We also observed a weak band at 2887 cm−1, which implies the presence of C–H vibrations, and might be assigned to the end –CD2H group. This is further supported by the presence of two different types of –CD2– symmetric and antisymmetric stretching modes (see Fig. S12A†), indicating the existence of two different types of –CD2– moieties in the produced PE.
- S. Peitz, N. Peulecke, B. H. Müller, A. Spannenberg and U. Rosenthal, Acta Crystallogr., Sect. E:, 2010, 66, m296–m297 CrossRef CAS.
- K. A. Kreisel, G. P. A. Yap and K. H. Theopold, Eur. J. Inorg. Chem., 2012, 2012, 520–529 CrossRef CAS.
- L. A. MacAdams, G. P. Buffone, C. D. Incarvito, J. A. Golen, A. L. Rheingold and K. H. Theopold, Chem. Commun., 2003, 1164–1165, 10.1039/B301943H.
- W. H. Monillas, G. P. A. Yap and K. H. Theopold, Angew. Chem., Int. Ed., 2007, 46, 6692–6694 CrossRef CAS PubMed.
- J. ashkin, M. L. H. Green, M. L. Poveda and K. Prout, J. Chem. Soc., Dalton Trans., 1982, 2485–2494, 10.1039/DT9820002485.
- A. C. Filippou, S. Schneider and G. Schnakenburg, Angew. Chem., Int. Ed., 2003, 42, 4486–4489 CrossRef CAS.
- P.-J. Sinnema, P. J. Shapiro, D. M. J. Foo and B. Twamley, J. Am. Chem. Soc., 2002, 124, 10996–10997 CrossRef CAS PubMed.
- C. J. Harlan, T. Hascall, E. Fujita and J. R. Norton, J. Am. Chem. Soc., 1999, 121, 7274–7275 CrossRef CAS.
- R. Choukroun, C. Lorber and B. Donnadieu, Organometallics, 2004, 23, 1434–1437 CrossRef CAS.
- In contrast, the material 1hl-d-SiO2-1, shows –CH2D ending groups after polymerization of C2H4, indicating the formation of Cr–D bonds in case of the presence of chemisorbed 1d10.
- Metalloproteins: Methods and Protocols, ed. J. C. Fontecilla-Camps and N. Yvain, HumanaPress, Springer, 2014.
- A. Ashuiev, F. Allouche, N. Wili, K. Searles, D. Klose, C. Copéret and G. Jeschke, Chem. Sci., 2021, 12, 780–792 RSC.
- M. F. Delley, F. Núñez-Zarur, M. P. Conley, A. Comas-Vives, G. Siddiqi, S. Norsic, V. Monteil, O. V. Safonova and C. Copéret, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11624–11629 CrossRef CAS PubMed.
- R. A. Heintz, S. Leelasubcharoen, L. M. Liable-Sands, A. L. Rheingold and K. H. Theopold, Organometallics, 1998, 17, 5477–5485 CrossRef CAS.
- P. A. White, J. Calabrese and K. H. Theopold, Organometallics, 1996, 15, 5473–5475 CrossRef CAS.
- While both low-spin and high-spin Cr species are observ-able by CW EPR (see Fig. S13† as an example), pulse EPR investigations of Cr high-spin (S > 1/2) species, where zero-field splitting (ZFS) is often the dominant magnetic interaction, is often limited due to short T2 relaxation times, even at temperatures of around 2–20 K (see (a) S. Chabbra, D. M. Smith and B. E. Bode, Isolation of EPR spectra and estimation of spin-states in two-component mixtures of paramagnets, Dalton Trans., 2018, 47, 10473–10479 RSC; (b) N. A. Hirscher, C. H. Arnett, P. H. Oyala and T. Agapie, Characterization of Cr-Hydrocarbyl Species via Pulse EPR in the Study of Ethylene Tetramerization Catalysis, Organometallics, 2020, 39(24), 4420–4429 CrossRef CAS ). On the other hand, both high-spin (HS, S = 3/2) and low-spin (LS, S = 1/2) states were previously observed for Cr(III) alkyl complexes, depending on their ligand surrounding (ref. 29). Also, grafting of the Cr(N(SiMe3)2)3 complex on silica, followed by thermal treatment at 400 °C, provided Cr(III) species that are in a LS state, so that pulse EPR measure-ments were possible already at 110 K (ref. 30)..
- J. Telser, eMagRes, 2017, 207–234 CAS.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- D. M. Murphy, Proc. Natl. Acad. Sci. U. S. A., 2004, 79, 103 CAS.
- F. Allouche, D. Klose, C. P. Gordon, A. Ashuiev, M. Wörle, V. Kalendra, V. Mougel, C. Copéret and G. Jeschke, Angew. Chem., Int. Ed., 2018, 57, 14533–14537 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 21495472149548. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04235e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |