
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast relaxing red and near-IR switchable azobenzenes with chalcogen and halogen substituents: periodic trends, tuneable thermal half-lives and chalcogen bonding†
Aidan
Kerckhoffs
 ,
Kirsten E.
Christensen
and
Matthew J.
Langton
*
,
Kirsten E.
Christensen
and
Matthew J.
Langton
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: matthew.langton@chem.ox.ac.uk
First published on 20th September 2022
Abstract
Molecular photoswitches operating in the red to near-IR region with controllable thermal relaxation rates are attractive components for photo-regulating biological processes. Herein, we report the synthesis of red-shifted azobenzenes functionalised with the heavier chalcogens and halogens that meet these requirements for biological application; namely fatigue-resistant photo-switching with red and near IR light and functional handles for further functionalisation for application. We report robust periodic trends for the chalcogen and halogen azobenzene series, and exploit intramolecular chalcogen bonding to tune and redshift the absorption maxima, supported by photo-physical measurements and solid-state structural analysis. Remarkably, the rate of the Z → E thermal isomerisation can be tuned over timescales spanning 107 s by judicious choice of chalcogen and halogen substituents.
Introduction
Azobenzenes (ABs) are attractive photoswitches due to their robust photo-switching, large conformational changes, low rate of photobleaching, and ease of synthesis and derivatization.1 Azobenzene motifs have thus been successfully employed in a plethora of applications, including molecular machines,2,3 smart materials,4–6 crystal engineering,7 transmembrane ion transport8–10 and non-invasive manipulation of biological systems, in particular within the emerging field of photo-pharmacology.11–13 Within the last decade, there have been significant efforts towards developing ABs,14–17 as well as related photoswitches such as diazocine,18 azonium19 and azo-BF2 derivatives,20 that can be isomerised using light in the biocompatible red to near-infrared region with controlled thermal relaxation rates. This is primarily driven by potential applications within biological contexts: longer wavelengths of light (>600 nm) exhibit significantly higher tissue penetrance,21 are absorbed less by hemoglobin,22 and do not display any toxic or mutagenic effects observed with high energy UV light that are typically required to isomerise ABs and other photoswitches such as stilbene.23,24Tetra ortho-substitution of azobenzenes with the lighter chalcogens and halogens (Fig. 1) is an established approach for red-shifting trans-ABs, pioneered by Woolley (X = Cl, Br, OMe, SEt)15,25,26 and Hecht (X = F).14 In these systems E → Z isomerisation is triggered using green or red light, exciting into the red-shifted n → π* transitions, whereas Z → E isomerisation occurs using higher energy blue or violet LEDs, with photo-stationary states (PSS) ranging from ∼85–95%. Thermal relaxation of the Z isomer of these derivatives is generally slow (half-lives of hours to years). Developing faster relaxing derivatives of these systems, whilst maintaining the efficient switching with visible light (and ideally with red/near IR wavelengths), is an attractive goal, because generation of the Z isomer can be achieved using bio-compatible wavelengths of light whilst relying only on thermal relaxation to quantitatively regenerate the E isomer. Typically, it is more convenient for the Z isomer to be the active ‘on’ form of the photoswitch, because this allows for the activation of systems that switch ‘off’ over time in the absence of light, without requiring a second excitation wavelength.11
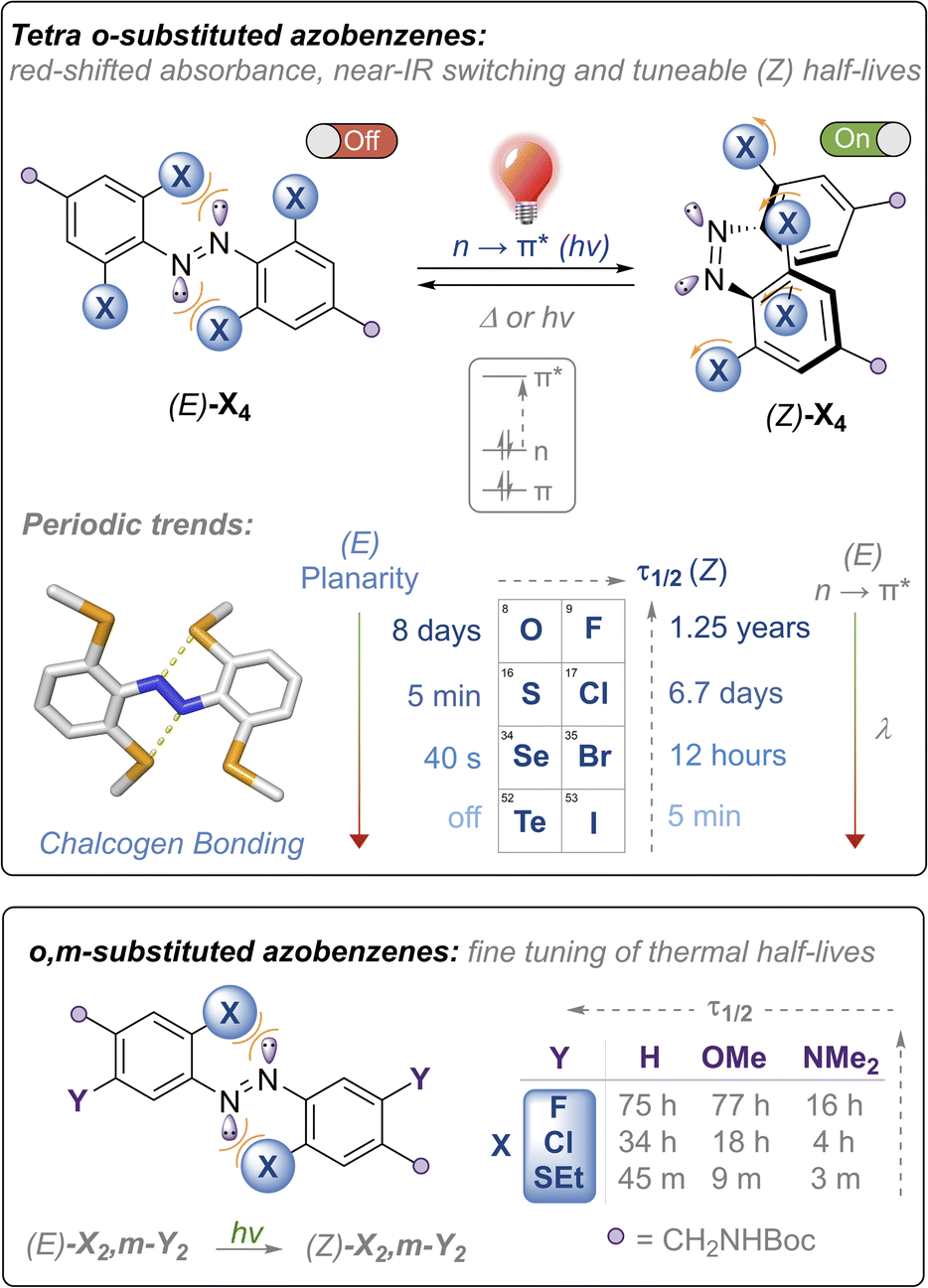 | ||
| Fig. 1 Heavy chalcogen and halogen substituted azobenzenes described in this article: periodic trends, thermal half-life tuning and near-IR photo-switching. No photo-switching was detected at room temperature for the tetra-telluride Te4. | ||
Access to a tuneable arsenal of structurally similar azobenzenes activated with biocompatible wavelengths of light, and exhibiting varying thermal relaxation rates, is also desirable to match the timescales of various biological processes. For example, Z → E thermal relaxation kinetics in the timeframe of minutes is suitable for photocontrol of transcription factors,25 whereas photochemical manipulation of ion channels has been demonstrated using azobenzenes that relax within seconds.27 It is therefore appealing to design redshifted photoswitches that thermally relax in the timescale of seconds to minutes, where relaxation does not outcompete the influx of light and photoisomerisation.28,29 To date, only a handful of studies guiding the design of redshifted azobenzenes exist, and scope for further functionalisation is limited.28–30 Furthermore, the vast majority of fast-relaxing azobenzenes are asymmetric push–pull systems, and not structurally analogous to the highly red-shifted and thermally stable ortho-substituted azobenzenes, preventing interchange of the photo-switch in applications where systematic modulation of thermal half-lives are desired.31
Herein, we report the first examples of the heavy chalcogen and halogen tetra-ortho functionalised azobenzenes (where X = SeMe, TeMe and I), and demonstrate how steric clash and intramolecular chalcogen bonding may be used to tune and red-shift the absorption maxima for azobenzene isomerisation, achieving photo-switching in the unprecedented near IR region for the tetra-ortho-iodo derivative (Fig. 1, top). We comprehensively examine the substituent effects and periodic trends down the chalcogen and halogen series, and show that thermal half-lives may be tuned over seven orders of magnitude within the family of structurally related ABs, ranging from essentially bistable derivatives (half-lives of days to years) through to those that relax on the timescale of seconds, all photo-switched using visible or near-IR light. We also explore the impact of di-ortho/di-meta substituted scaffolds on photo-switching properties and show that this substitution pattern allows for fine-tuning of the thermal half-lives (Fig. 1, bottom). Importantly, all scaffolds are appended with functional handles for further post-synthetic modification that is required for downstream application.
Results and discussion
Approach
The nature of the ortho-substituent directly influences the n orbital energies of ABs. In the case of (E)-Cl4 and (E)-(OMe)4, repulsive interactions between the diazo lone pairs and ortho substituent has been shown to destabilise the n molecular orbital, leading to red-shifted n → π* transitions in the E isomer (Fig. 1, top).26,29 The tetra-chloro derivative (E)-Cl4 has been proposed to be conformationally flexible, exhibiting n → π* (S0 → S1) transitions with extended red-shifted tails, because the minor populations of planar conformations raise the n orbital energy.29 Upon photo-switching to the Z-isomer, this repulsion is reduced, leading to large separations of the n → π* bands of the E and Z isomers, enabling high photo-stationary state ratios to be accessed. It is desirable to photo-isomerise ABs by exciting n → π* bands, as these typically proceed with higher quantum yields and longer wavelengths than by exciting the π → π* transitions.16With these previous results in mind, our initial strategy to access highly red-shifted, fast relaxing ABs was to extend the known tetra-ortho azobenzene derivatives X4 to the unknown heavier congeners (Fig. 1, top). We anticipated that the larger, heavier atoms (Se, Te, I) would lead to more redshifted n → π* tails due to increased destabilising clash with the diazo N lone pairs. Furthermore, we sought to explore whether, in the case of the chalcogens, intramolecular chalcogen bonds between the diazo lone pair and the chalcogen atom sigma hole would lead to higher populations of planar (and thus more red-shifted) conformations. Analogous to the well-known halogen bonds,32 chalcogen bonds are potent non-covalent interactions between a polarised chalcogen atom and a Lewis base, with applications in crystal engineering,33 construction of complex hierarchal supramolecular assemblies,34 anion recognition35–37 and transport.38–40
Our second strategy was to explore the di-ortho-substituted analogues (E)-X2,m-Y2, anticipating that with only two ortho heteroatoms the azobenzenes would retain a planar conformation whilst red-shifting the n → π* transition.26 We reasoned that a meta electron-donating substituent would shift absorbances to higher wavelengths due to the rule of auxochromes, whilst meta electron donating groups have previously been shown to significantly reduce the half-life of ortho-substituted azobenzenes to the (milli)second range when X = OMe.41 We therefore systematically explored combinations of X = F, Cl, OMe, SEt, pyrrolidine and phosphonate ester, and Y = H, OMe, NMe2, whilst retaining functional handles for derivatisation (see ESI† for full experimental procedures and characterisation).
Our synthetic strategy is summarised in Fig. 2 and all synthesised derivatives are tabulated in Table 1 (see ESI† for full experimental details and optimisation of synthetic routes). In general, ortho-halogenated and methoxy derivatives were accessed via oxidative dimerisation of the corresponding aniline, with yields typically decreasing with increasing steric bulk. Preparation of tetra-thio derivatives via this strategy generally proceeded with extremely low yields (∼2%),25 presumably due to competitive oxidation of the thioether units and the inherent challenges of forming the extremely sterically hindered bond. Thus, a strategy using nucleophilic aromatic substitution with ortho-fluoro derivatives was employed, providing yields up to 90%. This strategy was also exploited to access the unprecedented ortho-seleno and telluro derivatives. The photo-switches are equipped with functional handles (p-CH2NHBoc, p-CO2Et, p-CH2N3, p-CH2OH) for further derivatisation.
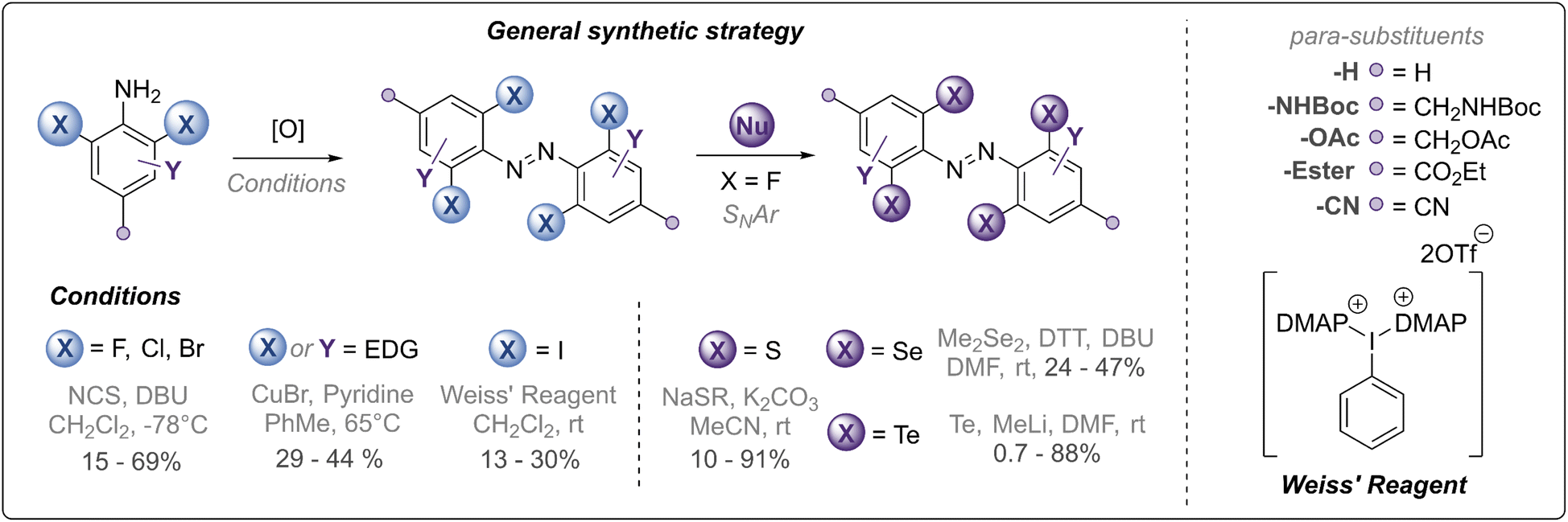 | ||
| Fig. 2 Synthetic strategy towards chalcogen and halogen substituted azobenzene derivatives (see Fig. 1 and Table 1 for structures). | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AB | X1, X2a | Y | Para | Conditions | Yield | τ 1/2 | λ excite (nm) | λ max (nm) | PSS | |
| a X1 = X2 if one entry. b Not determined. c Decomposed under irradiation with light. d No photo-switching observed. e Emission maximum of the LED used for photo-switching. f λ max of n → π* transition. g Switching possible up to 660 nm (43% Z PSS). h Switching possible up to 730 nm. i Thermal half-life too fast to determine the PSS distribution. All half-life and photo-switching experiments conducted in DMSO at 298 K. | ||||||||||
| 1 | F4-H | F | H | H | KMnO4/FeSO4 | 28% | ||||
| 2 | F4-NHBoc | F | H | CH2NHBoc | NCS/DBU | 69% | 1.25y | 530 | 454 | 84% |
| 3 | F4-Ester | F | H | CO2Et | NCS/DBU | 92% | ||||
| 4 | Cl4-NHBoc | Cl | H | CH2NHBoc | NCS/DBU | 43% | 160 h | 625 | 462.6 | 77% |
| 5 | Br4-NHBoc | Br | H | CH2NHBoc | NCS/DBU | 15% | 12 h | 625 | 465.9 | 66% |
| 6 | I4-OAc | I | H | CH2OAc | Weiss' Reagent | 13% | 2 m | 625 | 501.3 | |
| 7 | I4-OH | I | H | CH2OH | From entry 6 | — | 500.3 | |||
| 8 | I4-N3 | I | H | CH2N3 | From entry 6 | — | 3 m | 625 | 514.4 | |
| 9 | I4-NPhth | I | H | CH2NPhth | From entry 6 | — | 3 m | 625g | 511.8 | 50% |
| 10 | I4-NHBoc | I | H | CH2NHBoc | From entry 6 | — | 5 m | 660 | 514.3 | |
| 11 | I4-CN | I | H | CN | Weiss' Reagent | 26% | 20 s | 660 | 520.8 | |
| 12 | I4-ester | I | H | CO2Et | Weiss' Reagent | 30% | 46 s | 660h | 519.6 | |
| 13 | (OMe)4-NHBoc | OMe | H | CH2NHBoc | CuBr/pyridine | 38% | 8.2 d | 625 | 459.8 | 95% |
| 14 | (OMe)2F2,m-OMe2-NHBoc | OMe, F | OMe | CH2NHBoc | CuBr/pyridine | 3% | 530 | 425.4 | ||
| 15 | (SMe)4-H | SMe | H | H | SNAr from 1 | 91% | 5 m | 625 | 514.2 | |
| 16 | (SEt)4-NHBoc | SEt | H | CH2NHBoc | SNAr from 2 | 72% | 3 m | 625 | 514.5 | 46% |
| 17 | (SEt)4-ester | SEt | H | CO2Et | SNAr from 3 | 44% | 52 s | 660 | 590.0 | |
| 18 | (SiPr)4-NHBoc | SiPr | H | CH2NHBoc | SNAr from 2 | 35% | 3 m | 590 | 500.4 | |
| 19 | (SEt)4,m-OMe2-NHBoc | SEt | OMe | CH2NHBoc | SNAr | 36% | 590 | 512.9 | ||
| 20 | (SeMe)4-H | SeMe | H | H | SNAr from 1 | 47% | 80 s | 625 | 533.9 | |
| 21 | (SeMe)4-NHBoc | SeMe | H | CH2NHBoc | SNAr from 2 | 24% | 34 s | 625 | 534.3 | |
| 22 | (SeMe)4-ester | SeMe | H | CO2Et | SNAr from 3 | 34% | 2.5 s | 660 | 588.8 | |
| 23 | Te2F2-H | TeMe, F | H | H | SNAr from 1 | 88% | n/a | 543 | ||
| 24 | (TeMe)4-H | TeMe | H | H | SNAr from 23 | 0.7% | n/a | 518.5 | ||
| 25 | F2,m-H2 | F | H | CH2NHBoc | NCS/DBU | 36% | 75 h | 530 | 452.5 | 50% |
| 26 | F2,m-OMe2 | F | OMe | CH2NHBoc | CuBr/pyridine | 14% | 77 h | 530 | 387.1 | 50% |
| 27 | F2,m-(NMe2)2 | F | NMe2 | CH2NHBoc | CuBr/pyridine | 38% | 16 h | 530 | 405.5 | 48% |
| 28 | Cl2,m-H2 | Cl | H | CH2NHBoc | NCS/DBU | 10% | 34 h | 530 | 471.0 | 50% |
| 29 | Cl2,m-OMe2 | Cl | OMe | CH2NHBoc | CuBr/pyridine | 14% | 18 h | 530 | 475.0 | 77% |
| 30 | Cl2,m-(NMe2)2 | Cl | NMe2 | CH2NHBoc | CuBr/pyridine | 44% | 4 h | 530 | 432.1 | 50% |
| 31 | OMe2,m-OMe2 | OMe | OMe | CH2NHBoc | CuBr/pyridine | 17% | 530 | 426.6 | ||
| 33 | (SEt)2,m-H2 | SEt | H | CH2NHBoc | SNAr from 25 | 10% | 44 m | 590 | 431.3 | 50% |
| 34 | (SEt)2,m-OMe2 | SEt | OMe | CH2NHBoc | SNAr from 26 | 21% | 9 m | 590 | 464.1 | |
| 35 | (SEt)2,m-(NMe2)2 | SEt | NMe2 | CH2NHBoc | SNAr from 27 | 10% | 140 s | 590 | 543.0 | |
| 36 | (N(CH2)4)2,m-H | N(CH2)4 | H | CH2NHBoc | CuBr/pyridine | 29% | 530 | 530.2 | ||
| 37 | (PO(OEt)2)2,m-H | PO(OEt)2 | H | H | Cross coupling | 51% | 530 | 458.2 | ||
Optical, electronic and structural analysis
The UV-vis spectra of the (E)-X4 derivatives and (E)-X2,m-Y2 are shown in Fig. 3 (analogous spectra for the Z isomers, and deconvoluted spectra are available in the ESI†). Tetra-ortho substitution proved to be a reliable method of red-shifting the n → π* absorbance: within each group, increasing atomic size leads to red-shifting of the n → π* absorption maximum. The heavier atoms exhibit deeply redshifted tails (>650 nm for X = I and >700 nm for X = Te), attributed to increased steric clash between the larger element with the n orbital in planar conformations (Fig. 3b). In contrast, for (E)-X2,m-Y2, (E)-Cl2,m-Y2 and (E)-(SEt)2,m-Y2 derivatives, it was found that although the meta auxochrome (Y = OMe and NMe2) broadened and increased the absorbance of the n → π* transition, the tail did not generally extend much further than the R = H derivatives; with absorptions not exceeding 600 nm (Fig. 3a). The n → π* transition maximum, λmax, typically followed the order meta-H > –NMe2 > –OMe and ortho-S > Cl > F. It is clear from these experiments that the four ortho X substituents in the X4 derivatives are essential for achieving the deeply red-shifted tails which allows for red/near-IR triggered photo-isomerisation.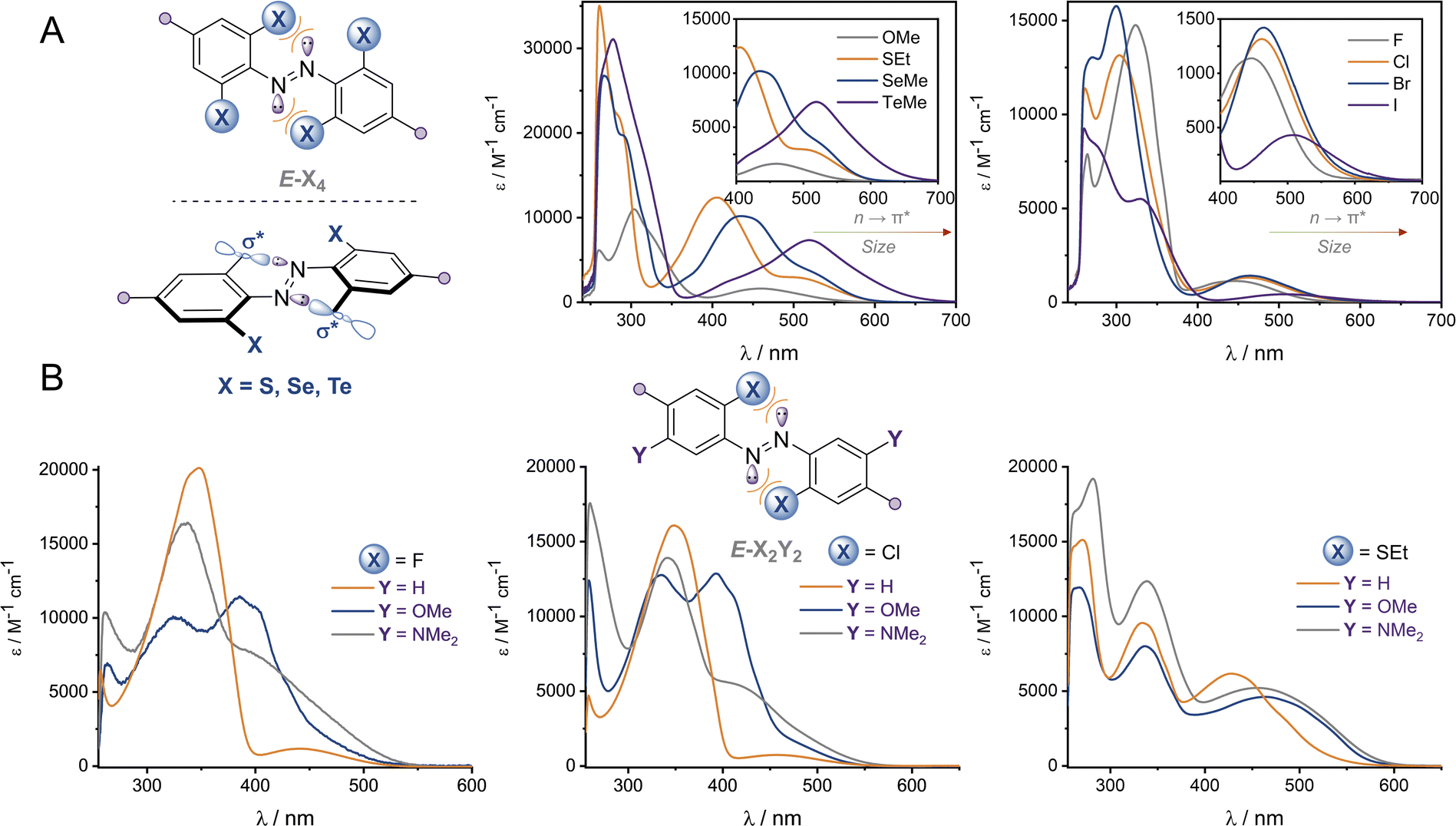 | ||
| Fig. 3 UV-vis spectra of (A) (E)-X4 derivatives and (B) (E)-X2,m-Y2 in DMSO at 25 °C. | ||
To explore the role of the ortho-substituents on the azobenzene structure, we grew single crystals of the novel tetra-ortho derivatives and analysed their structures by X-ray crystallography (Fig. 4 – see ESI† for experimental procedures and diffraction data). Structures for the known F4,42 Cl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 and (OMe)415 derivatives were accessed from the Cambridge Crystallographic Data Centre (CCDC).
29 and (OMe)415 derivatives were accessed from the Cambridge Crystallographic Data Centre (CCDC).
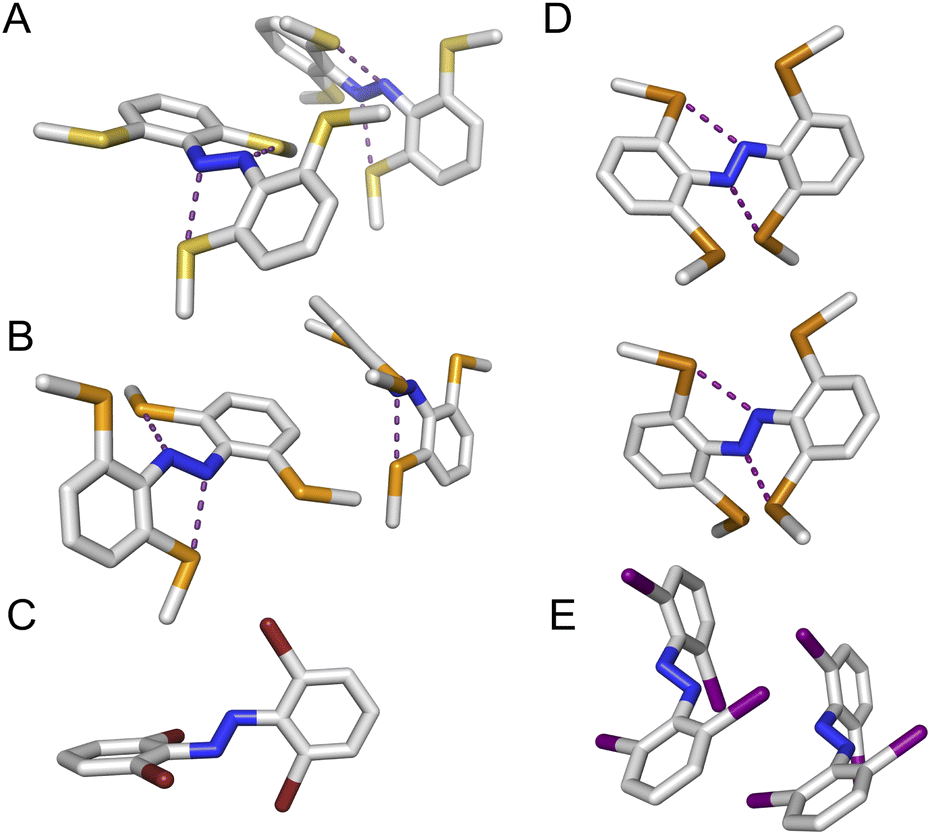 | ||
| Fig. 4 Crystal structures of novel tetra-ortho substituted azobenzene scaffolds (para substituents not shown for clarity). (A) (SMe)4-H (B) (SeMe)4-H (C) Br4-NHBoc (D) (TeMe)4-H (E) I4-N3. Intramolecular chalcogen bonds forming 5-membered rings between the chalcogen and azo N atoms represented with purple dotted lines. For A, B, D and E two conformations are present within the unit cell. | ||
We observed multiple conformations within the same unit cell for tetra -thio, -seleno, -telluro and -iodo derivatives, as has been observed previously by Trauner and co-workers for the tetra chloro azobenzenes.29 This provides evidence for the high flexibility of the E isomers, which are able to sample a range of conformations including the more planar, redshifted geometries. The geometries of the various structures across the tetra-ortho series are compared in Fig. 5. In general, the diazo bond is more twisted for the (heavier) chalcogens (Fig. 5a), perhaps to accommodate the intra-molecular 5-membered ring chalcogen bond. Increased N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N distortion and longer N–N bonds leads to more redshifted wavelengths for the tetra-chloro derivatives, based on previous theoretical calculations,29 which presumably contribute here to the high absorbance at longer wavelengths of the chalcogen derivatives.
N distortion and longer N–N bonds leads to more redshifted wavelengths for the tetra-chloro derivatives, based on previous theoretical calculations,29 which presumably contribute here to the high absorbance at longer wavelengths of the chalcogen derivatives.
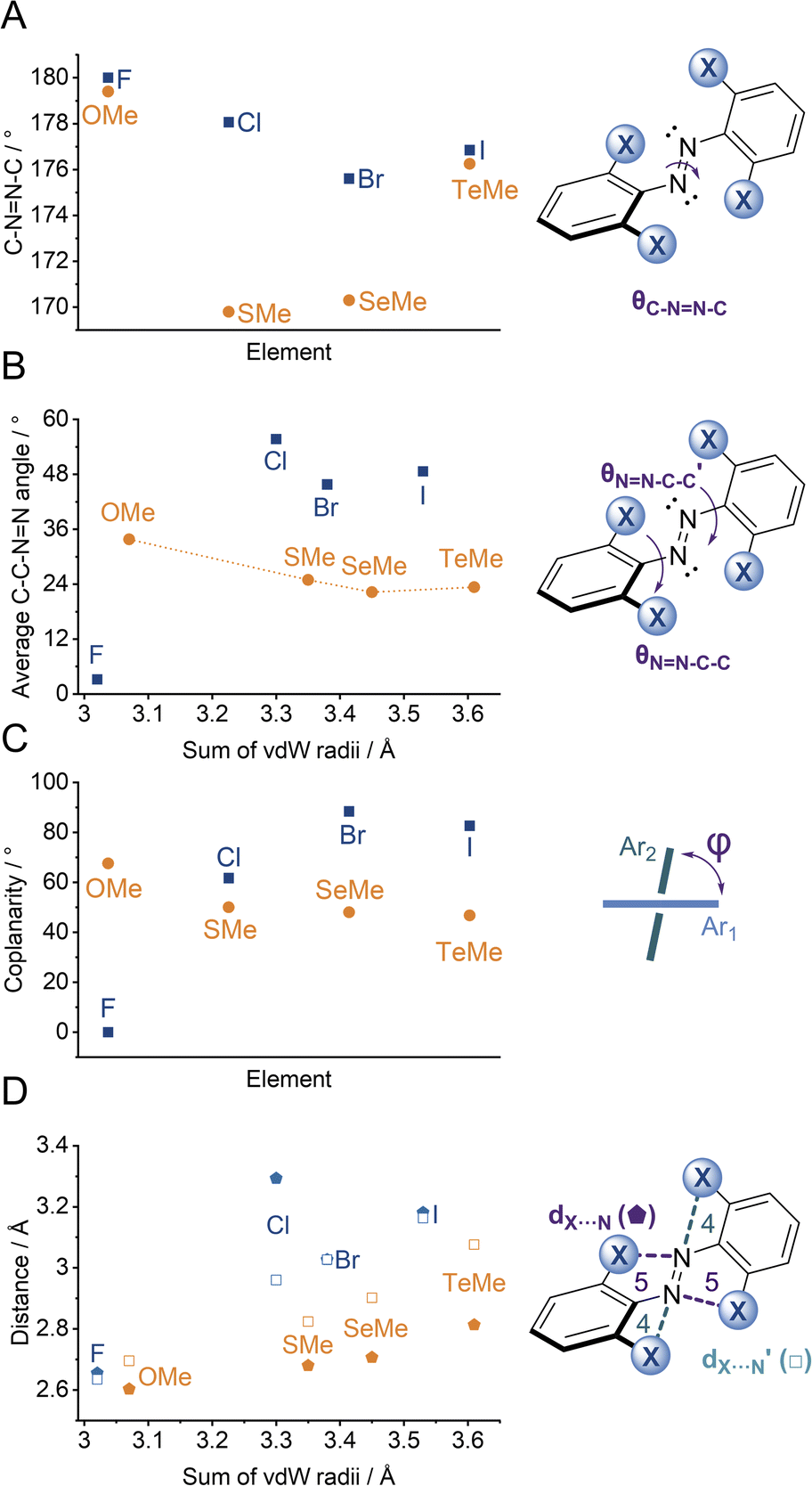 | ||
Fig. 5 Geometric parameters from single-crystal data for (E)-X4 (A) C–NN–C dihedral, (B) average NN–C–C dihedral, (C) coplanarity of rings, (D) distances between diazo lone pair and X-atom (formation of intramolecular 5 (![[pentagon filled]](https://www.rsc.org/images/entities/char_e13d.gif) ) and 4 (□) membered rings). ) and 4 (□) membered rings). | ||
Generally, each aryl ring is independently twisted away from the diazo bond (dihedral NN–C–C ≠ NN–C–C′), in line with the proposed conformational flexibility of the E isomers. The average NN–C–C dihedral and coplanarity of the aryl rings generally decreases down the chalcogen group despite the increasing atomic size, whereas in contrast, it broadly increases down the halogen group (Fig. 5b and c). This is consistent with the chalcogens enforcing planar character via an intramolecular chalcogen bond. Further evidence for this X⋯N chalcogen bonding interaction can be inferred from the inherent preference for 5-membered intramolecular X⋯N interactions over 4-membered (dX⋯N < dX⋯N′), where the highly directional chalcogen bond overlap is more feasible (Fig. 5d).43 Conversely, the halogens have no preference between 4- or 5-membered interactions because the halogen centred σ-hole is on the pole of the halogen atom and therefore cannot interact with the azo N via a linear halogen bond. Conformations where steric clash is minimised are therefore favoured (dX⋯N ≈ dX⋯N′).
The di-, tri- and tetra-telluro derivatives (Te2F2-H, Te3F-H and Te4-H) were prepared via an unprecedented nucleophilic aromatic substitution of the methyl-telluride on the corresponding fluoro-azobenzene derivative (Fig. 6A). UV-vis spectra of di, tri and tetra ortho tellurides reveal that with increasing number of ortho-TeMe substituents, the n → π* λmax is blue-shifted, but the tail of the absorption extended further into the red/near IR region (Fig. 6B). The solid-state structure of Te2F2-H (Fig. 6C), revealed that this di-telluride is completely planar, owing to the strong 5-membered Te⋯N chalcogen bond (2.72 Å), well within the sum of the N–Te van der Waals radii (3.61 Å; 75%). When Te is exchanged to Cl (Cl2F2-H)26 or H (F2H2-NHBoc), the 5-membered interaction between the azo-N atoms and the ortho-substituents is biased to the smallest substituent to minimise repulsion with the lone pairs. Notably, Cl2F2-H adopts a more twisted geometry than Te2F2-H despite the smaller size of Cl relative to Te (Fig. 6C); serving as further evidence for strong intramolecular chalcogen bonding within the ortho-telluro ABs.
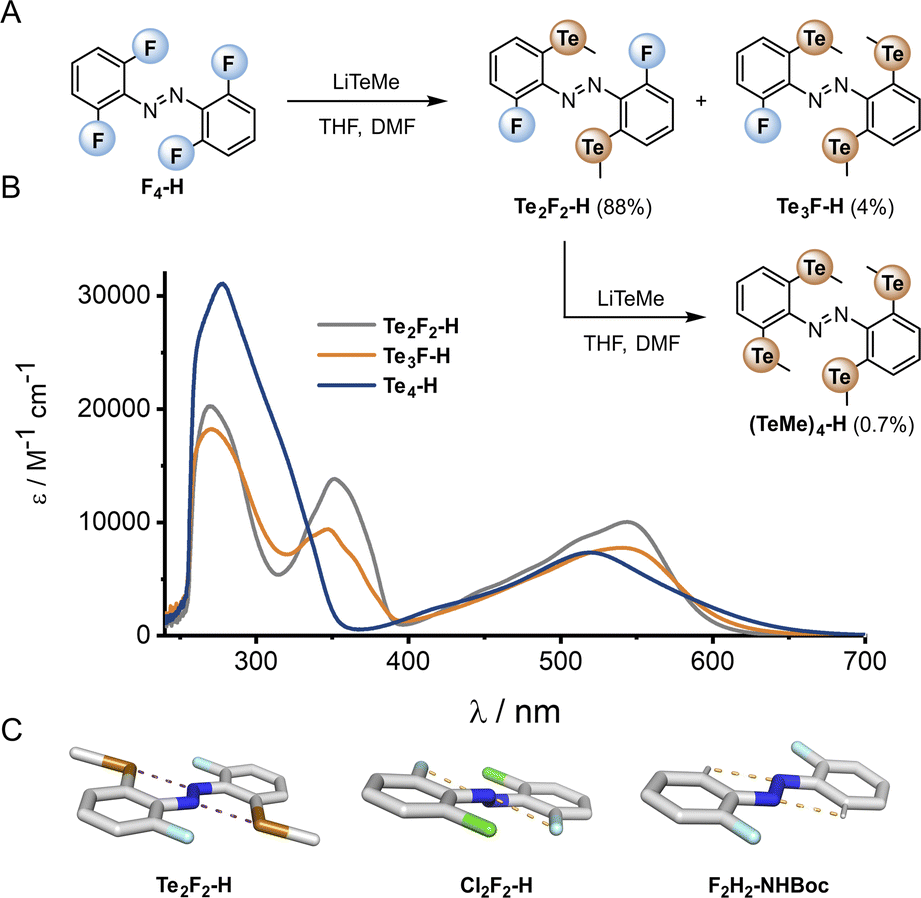 | ||
| Fig. 6 (A) Preparation of tellurium-substituted azobenzenes via an SNAr reaction of the corresponding fluoro-derivative. (B) UV-vis spectra of Te derivatives. (C) Crystal structures: Cl2F2 structure accessed from CCDC,29F2H2-NHBoc determined in-house (para substituent omitted for clarity). | ||
Analysis of the UV-vis spectra and data from the crystal structures suggests that the di-telluride Te2F2-H exhibits an optimal balance of intra-molecular chalcogen bonds and steric bulk to enforce planarity, because it can form two strong chalcogen bonds without any destabilising steric interactions arising from extra tellurium atoms. This leads to Te2F2-H exhibiting the most red-shifted n → π* λmax. However, as with the other heavy ortho main group ABs, the derivatives with additional Te atoms also appear to have minor populations of planar conformations, leading to a deeply red-shifted tail of the n → π* transition.
Photo-isomerisation and glutathione stability
The E → Z photo-isomerisation and half-life for the Z → E thermal isomerisation was evaluated for all derivatives (Fig. 7 and Table 1, see ESI† for original data). To our surprise, the half-lives for (Z)-X2,m-Y2 derivatives were in the order of hours to days for X = F and Cl: the effect of the meta group was not as drastic as anticipated. We typically observed a ∼4 fold decrease in half-life by substituting H with OMe, and a further 3-fold drop from OMe to NMe2 for X = F, Cl and SEt. The nature of the ortho-substituent appeared to be the main influence on τ1/2 (F > Cl ≫ SEt). However, these predictable trends observed for the meta-substitution allow for precise tuning of thermal half-lives. The photo-stationary states (PSSs) of these derivatives ranged from 48–77% Z isomer (Table 1). We also prepared the di-ortho, mono-meta-methoxy derivative, OMe2,m-OMe2, however this was found to be unstable to light (ESI Fig. S359†).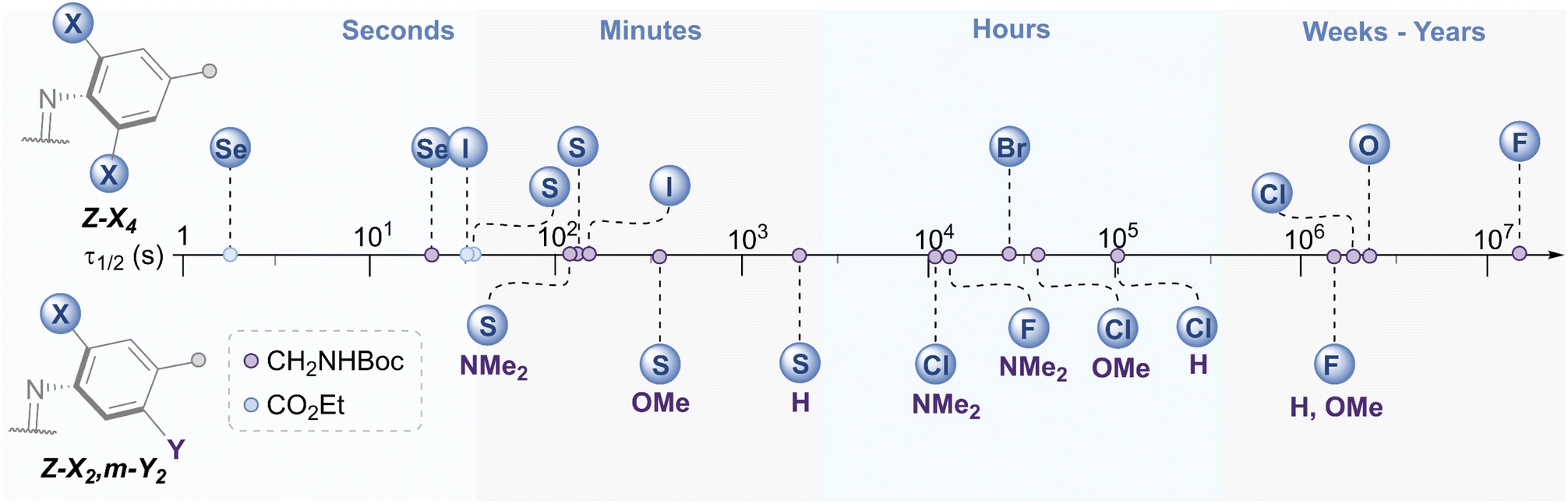 | ||
| Fig. 7 Schematic timeline showing the range of half-lives for the Z → E thermal isomerisation of the azobenzene derivatives. | ||
The X4 derivatives exhibited more interesting photo-switching properties, with heavier elements displaying robust, reversible switching behaviour using solely red light and thermal decay, with half-lives for the Z → E thermal isomerisation of the order of seconds to minutes (Fig. 8A and B). Remarkably, it was possible to photo-switch the tetra-iodo derivative I4-ester using near-IR light (730 nm) (Fig. 8C). Notably, these thermal half-lives can be tuned over seven orders of magnitude within this isostructural series and each derivative is amenable to further functionalisation. Photo-isomerisation was not detected for the ortho-telluro derivatives. This is consistent with findings by Haberhauer and co-workers, who propose that the strong chalcogen bonds in the E isomer of a PhTe-substituted azobenzene lead to dramatic energy differences between the E and Z isomers, which suppresses photoswitching.43 The photo-stationary states of the halogen X4 derivatives range from 50–84% (Table 1) and could also be determined for the slower relaxing chalcogen derivatives (PSS 46–95%, see Fig. S308–S323† for original data).
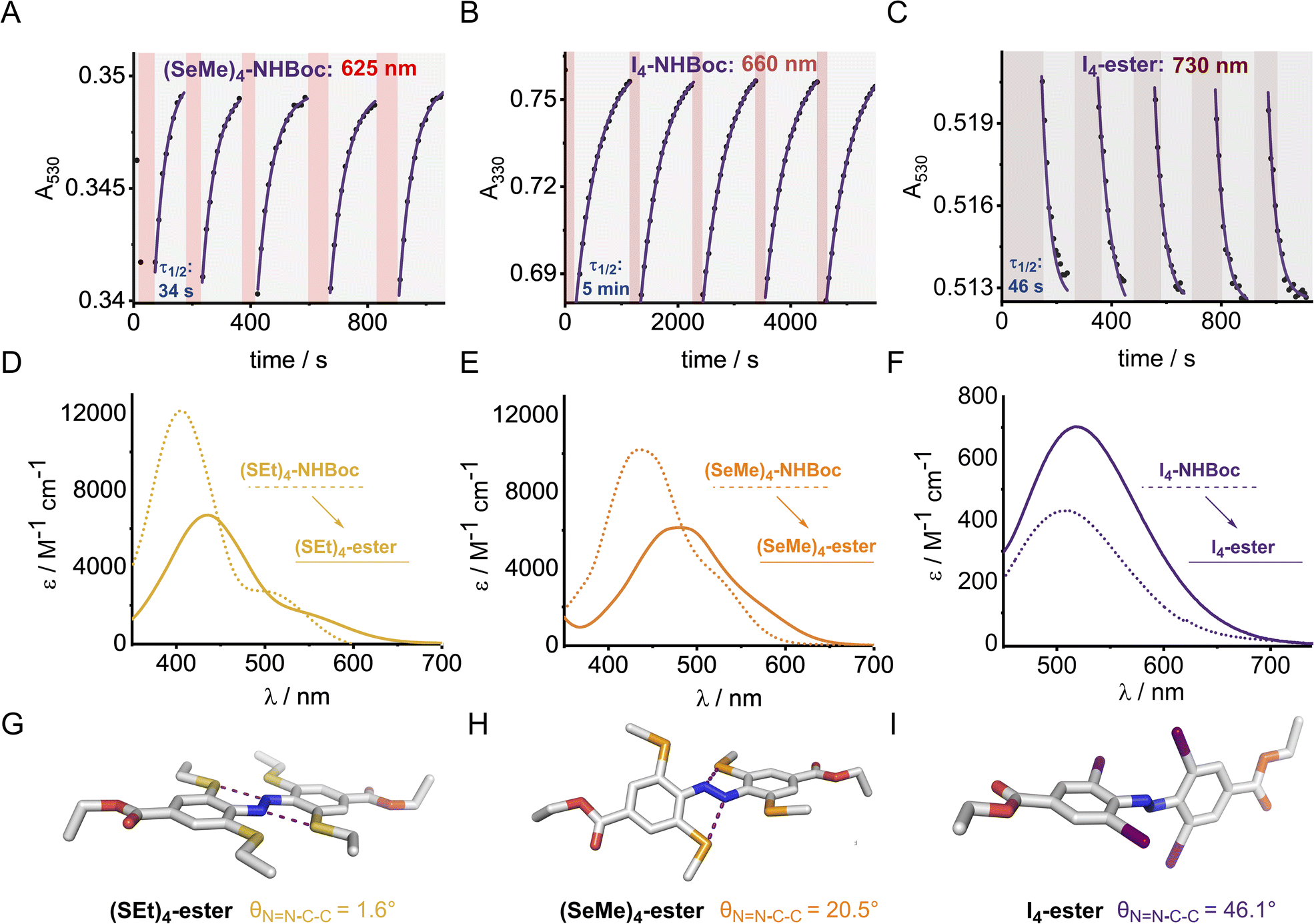 | ||
| Fig. 8 Reversible photo-switching for tetra ortho-substituted azobenzene derivatives: (A) (SeMe)4-NHBoc with 625 nm, (B) I4-NHBoc with 660 nm and (C) I4-ester with 730 nm irradiation. Data point: absorbance at the indicated wavelength, solid line: 1st order exponential fit. Red shaded region indicates time period of irradiation with an LED of the indicated wavelength. Samples of ∼100 μM were irradiated with LEDs (625 nm, ∼0.9 W; 660 nm, ∼1.1 W, 730 nm ∼0.7 W). (D–F) Stacked UV-vis spectra of S, Se and I derivatives (G–I) Solid-state structures of para-ester derivatives with average aryl-NN dihedral angles. | ||
The half-lives of the (Z)-X4 derivatives decrease down each group (Table 1, entries 2, 4, 5, 10 and 13, 16, 21, 24), consistent with electronegative substituents relieving the NN lone pair repulsion and stabilising the overall Z isomer energy.14 For X = F, anomalously high Z isomer stability has been previously suggested to arise from intramolecular C⋯F interactions.44 Increasing the steric bulk of the alkyl group appended to sulfur from ethyl to iso-propyl blueshifts the switching transition, presumably due to steric clash that distorts from planarity outweighing the chalcogen bonding interactions (entry 16 → 18). This structural change does not affect the thermal half-life of the Z-isomer.
In general, switching from a para-H substituent to a methylene group lowers the half-life around 2-fold, most likely due to increased electron density of the azobenzene.45 Switching to an ester group significantly lowers the half-life for each derivative (∼4–10-fold), consistent with previously reported systems, and rationalised by π-acceptors stabilising the linear Ar–N–N–Ar transition state during thermal isomerisation.16 Furthermore, the ester group typically allows switching with more deeply red-shifted wavelengths and acts as a potential functional handle for further derivatisation (Fig. 8D–F). These deeply red-shifted tails may be attributed to increased average planarity to maximise conjugation between the diazo bond and ester. Indeed, for the S4 derivatives, the ester motif has a dramatic effect on planarity relative to (SMe4)-H in the solid state (Fig. 8G), with an essentially planar structure observed. For the seleno and iodo derivatives, the decrease in planarity is less dramatic in the solid-state crystal structures, presumably due to competing steric effects with the larger atoms (Fig. 8H–I). Combining tetra-ortho substitution with a meta-methoxy substituent lead to dyes that were unstable to light (entry 14, 19 see ESI S68 and S363†). ABs with ortho-phospho and amino groups were also found to decompose in the presence of light, and thus are not suitable candidates for photo-switching applications (entry 36–37, S364–S365).†
The (SeMe)4, (SEt)4, and I4 derivatives display thermal half-lives in the order useful for spatial targeting applications (second to minute range). Crucially, these photo-switches may be reversibly switched between both isomers using only red wavelengths of light (E → Z) and thermal isomerisation (Z → E). Furthermore, there is scope for selectively switching one scaffold in the presence of another with orthogonal wavelengths. For instance, (SeMe)4-NHBoc does not switch with 660 nm light, whereas I4-NHBoc or I4-CO2Et derivatives readily isomerise under these conditions.
Finally, the stability of the X4 derivatives were assessed by exposing them to the highest expected intracellular concentration of glutathione (GSH, 10 mM).28 This was achieved by treating the Boc-protected amino-methyl derivatives X4-NHBoc (X = F, Cl, Br, I, OMe, SMe and SeMe) with TFA to remove the protecting groups. The resulting ammonium-appended AB derivative was dissolved in N2-degassed 100 mM phosphate buffer at pH 7 containing 10 mM GSH, and the changes in absorbance were monitored over an extended period. Generally, the larger, less electron withdrawing derivatives are more stable under these conditions, which is expected based on the substituent's ability to polarise the diazo bond towards nucleophilic attack, and steric arguments. Strong σ-donating substituents are also proposed to aid diazo lone pair protonation, and subsequent GSH attack.25 Notably, the seleno derivatives exhibit stability for days, advantageous for applications in cellular environments, whereas the lighter elements generally decay in the order of hours (Table 2). The I4 derivative displays similar stability to the F4 analogue.
Conclusion
We report the synthesis of heavy main group element substituted azobenzenes, revealing robust periodic trends in absorption and thermal half-lives within the tetra-ortho-chalcogen and halogen substituted series. We observe that the unprecedented azobenzenes substituted with the heavier (less electronegative) elements exhibit shorter half-lives for the thermal Z to E isomerisation and more redshifted n → π* bands. We provide a uniquely comprehensive study on the ortho-substituent effects in these systems, and demonstrate that introduction of meta-electron donating groups could be used to fine-tune the thermal half-lives, whilst para-electron withdrawing groups further red-shift the absorption, and decrease the thermal stability of the Z isomer. In particular, the sulfur, seleno and iodo derivatives display short (second to minute) half-lives, and are reversibly switched using red light, with no evidence of photobleaching. The corresponding tetra-ortho-iodo, para-ester derivative could be reversibly switched using near-IR irradiation and thermal relaxation. The red-shifted seleno derivative also exhibits excellent stability to the cellular reducing agent glutathione. This work provides a comprehensive point of reference for red-shifted azobenzene design, and we anticipate these tuneable, robust and biocompatible photo-switches will find utility within the field of photo-pharmacology, and other applications in which the precise tuning of photo-switching and thermal relaxation, within otherwise isostructural molecular switches, is desired.Author contributions
A. K. carried out the experimental work. K. E. C. performed the X-ray crystallography experiments. M. J. L. conceived and directed the project. All authors contributed to the discussion and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. acknowledges the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship (EP/L015838/1), generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. We gratefully acknowledge the EPSRC for a Strategic Equipment Grant (EP/V028995/1). M. J. L. acknowledges funding from the Royal Society. M. J. L. is a Royal Society University Research Fellow.Notes and references
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- L. W. Giles, C. F. J. Faul and R. F. Tabor, Mater. Adv., 2021, 2, 4152–4164 RSC.
- X. Pang, J. Lv, C. Zhu, L. Qin and Y. Yu, Adv. Mater., 2019, 31, 1904224 CrossRef CAS PubMed.
- J. Vapaavuori, C. G. Bazuin and A. Priimagi, J. Mater. Chem. C, 2018, 6, 2168–2188 RSC.
- O. S. Bushuyev, A. Tomberg, T. Friščić and C. J. Barrett, J. Am. Chem. Soc., 2013, 135, 12556–12559 CrossRef CAS PubMed.
- Y. Rin Choi, G. Chan Kim, H.-G. Jeon, J. Park, W. Namkung and K.-S. Jeong, Chem. Commun., 2014, 50, 15305–15308 RSC.
- A. Kerckhoffs and M. J. Langton, Chem. Sci., 2020, 11, 6325–6331 RSC.
- A. Kerckhoffs, Z. Bo, S. E. Penty, F. Duarte and M. J. Langton, Org. Biomol. Chem., 2021, 19, 9058–9067 RSC.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem.–Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–51595 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, M. J. Hansen, L. Kálmán and G. A. Woolley, Chem. Commun., 2015, 51, 12981–12984 RSC.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 13190–13193 CrossRef CAS PubMed.
- W. F. Cheong, S. A. Prahl and A. J. Welch, IEEE J. Quantum Electron., 1990, 26, 2166–2185 CrossRef.
- S. Samanta, A. Babalhavaeji, M. Dong and G. A. Woolley, Angew. Chem., Int. Ed., 2013, 52, 14127–14130 CrossRef CAS.
- T. R. Dunkern, G. Fritz and B. Kaina, Oncogene, 2001, 20, 6026–6038 CrossRef CAS.
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef.
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos and G. A. Woolley, Chem. Commun., 2013, 49, 10314 RSC.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS.
- A. Mourot, M. A. Kienzler, M. R. Banghart, T. Fehrentz, F. M. E. Huber, M. Stein, R. H. Kramer and D. Trauner, ACS Chem. Neurosci., 2011, 2, 536–543 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS PubMed.
- D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538–6547 CrossRef CAS.
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS.
- A. Docker, A. J. M. Martínez, H. Kuhn and P. D. Beer, Chem. Commun., 2022, 58, 3318–3321 RSC.
- G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef CAS PubMed.
- A. Borissov, I. Marques, J. Y. C. Lim, V. Félix, M. D. Smith and P. D. Beer, J. Am. Chem. Soc., 2019, 141, 4119–4129 CrossRef CAS.
- A. Docker, C. H. Guthrie, H. Kuhn and P. D. Beer, Angew. Chem., Int. Ed., 2021, 60, 21973–21978 CrossRef CAS PubMed.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS PubMed.
- G. Park and F. P. Gabbaï, Chem. Sci., 2020, 11, 10107–10112 RSC.
- L. E. Bickerton, A. Docker, A. J. Sterling, H. Kuhn, F. Duarte, P. D. Beer and M. J. Langton, Chem.–Eur. J., 2021, 27, 11738–11745 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, M. J. Hansen, L. Kálmán and G. A. Woolley, Chem. Commun., 2015, 51, 12981–12984 RSC.
- J. Broichhagen, D. H. Woodmansee, D. Trauner and P. Mayer, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o459–o460 CrossRef CAS PubMed.
- S. Mehrparvar, Z. N. Scheller, C. Wölper and G. Haberhauer, J. Am. Chem. Soc., 2021, 143, 19856–19864 CrossRef CAS PubMed.
- K. Aggarwal, T. P. Kuka, M. Banik, B. P. Medellin, C. Q. Ngo, D. Xie, Y. Fernandes, T. L. Dangerfield, E. Ye, B. Bouley, K. A. Johnson, Y. J. Zhang, J. K. Eberhart and E. L. Que, J. Am. Chem. Soc., 2020, 142, 14522–14531 CrossRef CAS PubMed.
- M. A. Kienzler, A. Reiner, E. Trautman, S. Yoo, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2013, 135, 17683–17686 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2196083–2196092. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04601f |
| This journal is © The Royal Society of Chemistry 2022 |