
Modulating up-conversion and non-radiative deactivation to achieve efficient red thermally activated delayed fluorescence emitters†
Teng
Gao
ab,
Shaogang
Shen
ab,
Yuanyuan
Qin
ab,
Honglei
Gao
ab,
Xiangyu
Dong
ab,
Zhi
Pang
ab,
Pengfei
Wang
 ab,
Ying
Wang
*ab and
Xiaoxiao
Hu
*ac
ab,
Ying
Wang
*ab and
Xiaoxiao
Hu
*ac
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials and City U−CAS Joint Laboratory of Functional Materials and Devices, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wangy@mail.ipc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Chemistry and Chemical Engineering, Suzhou University, Suzhou 234000, China. E-mail: Huxchem@ahszu.edu.cn
First published on 4th November 2022
Abstract
The development of efficient red electroluminescent devices is one of the formidable challenges in organic light emitting diode (OLED) research. In this work, we reported three efficient red thermally activated delayed fluorescence (TADF) emitters based on the novel dibenzothioxanthene acceptor: 2SO-AD, 2SO-TBU and 2SO-F-TBU. The novel dibenzothioxanthene acceptor with large conjugation allows the energy of the locally excited triplet (3LE) state from the acceptor(A)-segment (3LEA) to be close to the charge transfer (CT) states in red emitters, thus enabling participation of the LE state to facilitate the reverse intersystem crossing (RISC) process. Furthermore, the bulky donor can effectively suppress the aggregation-caused quenching (ACQ), resulting in low non-radiative deactivation of 6.5 × 106 s−1. The red OLED devices based on 2SO-TBU and 2SO-F-TBU emitters achieved the maximum external quantum efficiency (EQEmax) of 16.27% and 14.47%, respectively. This work paves a new way towards efficient red TADF emitters.
Thermally activated delayed fluorescence (TADF) materials have attracted extensive attention in the application of organic light-emitting diodes (OLEDs) due to their high efficiency and noble metal-free organic framework.1–4 In general, TADF materials employ highly distorted D–A structures to reduce the energy gap between the singlet state (S1) and the triplet state (T1), enabling efficient up-conversion via thermally assisted reverse intersystem crossing (RISC), and thus nearly 100% internal quantum efficiency (IQE) can be theoretically achieved.5 After the pioneering work reporting close to 20% external quantum efficiency (EQE) in green TADF OLEDs,1 high-efficiency OLEDs have been achieved by several works with EQEs close to 40% in green and blue TADF devices.6–9 However, the development of red TADF OLEDs as one of the three primary colors is far from satisfactory.10,11
The relatively backward development of red TADF materials is mainly attributed to the following aspects: (1) red emission is usually achieved by employing a strong donor and acceptor with rigid and conjugated structures, which not only increases the probability of aggregation-caused quenching (ACQ), but also induces excited states to exhibit charge transfer (CT)-dominated properties.12 According to the El-Sayed rule, the same orbital properties between S1 and T1 will significantly reduce the spin–orbit coupling (SOC), which is detrimental to the RISC. (2) According to the energy gap law, the non-radiative deactivation rate constant increases exponentially with decreasing band gap,13,14 and therefore they are likely to suffer from lower photoluminescence quantum yields (PLQYs).15 As mentioned above, promoting the process of RISC from the triplet to singlet state and suppressing the non-radiative deactivation from the singlet to ground state will be the key to achieve high-efficiency red TADF materials.16–18 Many previous reports focused on employing multiple donors and acceptors to increase energy level degeneracy or manage different orbital types in singlet and triplet states to increase SOC for promoting the RISC process.19–22 Recently, Zhang's group brought a new perspective and found that the locally excited triplet states of currently employed donor (D)-segments (3LED) almost lie considerably higher than the 1CT and 3CT states of red emitters, and therefore cannot play a significant role in the RISC process. However, the energy levels of the locally excited triplet (3LE) states from these acceptor (A)-segments (3LEA) are close to those of the CT state in the red emitters, thus accelerating the RISC process.23 Generally, the energy level of the 3LE state from the A-segment (3LEA) can be reduced by designing a large conjugated structure, which not only facilitates the 3LE of the acceptor to contribute to the T1 excited state, but also allows the energy level of the 3LEA from the A-segment to be close to those of the CT states, which allows the 3LE to participate in up-conversion and thus promote the RISC process. However, this strategy inevitably increases the non-radiative deactivation of the molecules in the aggregated state.24 Fortunately, it has been demonstrated that non-radiative deactivation can be suppressed by employing a bulky donor that can effectively inhibit the ACQ derived from intermolecular aggregation, thereby enhancing the PLQY and improving the device performance.25,26
Herein, we designed and synthesized three red TADF emitters 2SO-AD, 2SO-TBU and 2SO-F-TBU with a novel dibenzothioxanthene acceptor. The rigidly conjugated acceptor unit not only provides a deeper LUMO for red emission, but also boosts the RISC process via managing the 3LEA. Additionally, the bulky tert-butylphenyl acridine donor can effectively inhibit the non-radiative deactivation caused by aggregation of the conjugated acceptor (Fig. 1a). Surprisingly, all molecules exhibit excellent AIE properties with high PLQY values of up to 58.3% in solid films. The red OLED devices based on 2SO-TBU and 2SO-F-TBU with emission peaks located at 608 and 612 nm exhibit EQEs of 16.27% and 14.47%, respectively.
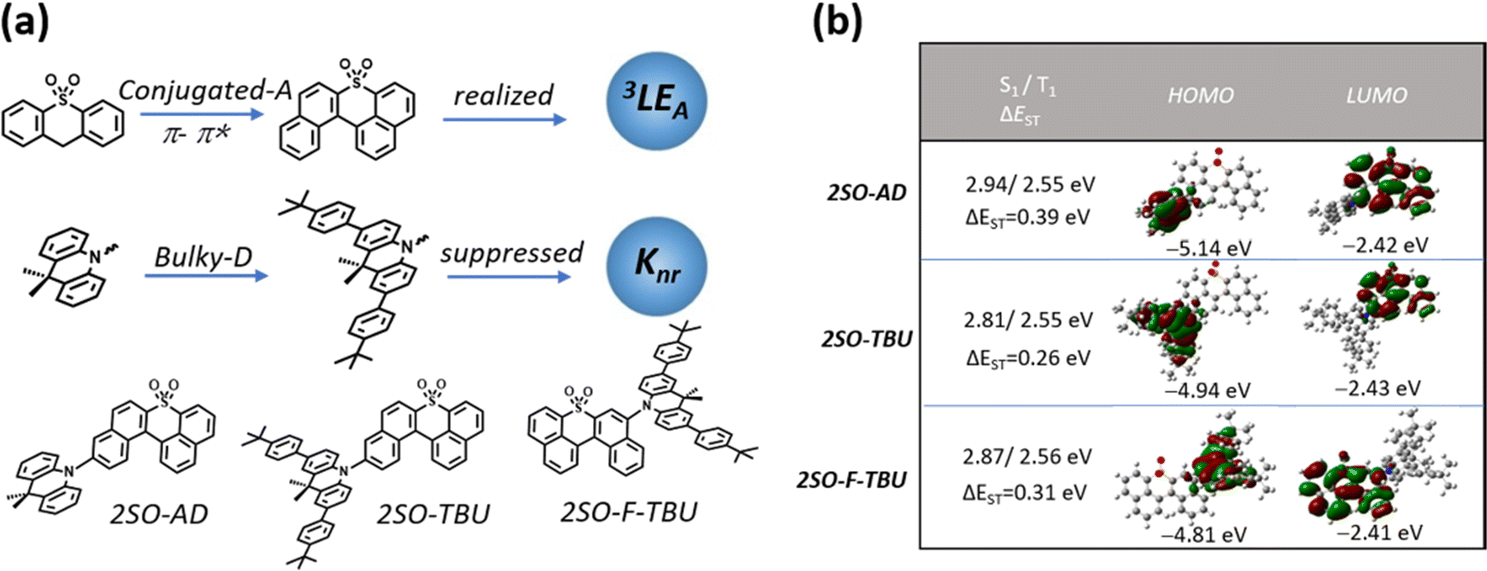 | ||
| Fig. 1 (a) Schematic diagram of the molecular design strategy; (b) HOMO and LUMO electronic distributions, and the lowest singlet (S1) and triplet (T1) energy levels for 2SO-AD, 2SO-TBU and 2SO-F-TBU calculated by DFT at the B3LYP/6-31g(d,p) level. | ||
The molecular synthesis and characterizations are shown in Schemes S1 and Fig. S1–S4 (ESI†). All compounds exhibited high thermal decomposition temperatures (Td) and glass transition temperatures (Tg): 411.7 °C/157.3 °C for 2SO-AD, 420.0 °C/219.1 °C for 2SO-TBU, and 421.6 °C/227.7 °C for 2SO-F-TBU, respectively (Fig. S5, ESI†). These results revealed their good thermal and morphological stability to aid in device fabrication and operation. Electrochemical test results (as shown in Fig. S6, ESI†) suggested a quasi-reversible, one-electron oxidation and reduction process mainly from the oxidation of the acridine donor and the reduction of the dibenzothioxanthene acceptor. By calculating the half-wave potentials in the reduction curves, it was found that 2SO-AD, 2SO-TBU and 2SO-F-TBU exhibited similar LUMO energy levels of −3.13, −3.10 and −3.11 eV, respectively. Differently, the HOMO energy levels are calculated to be −5.42 eV for 2SO-AD, −5.26 for 2SO-TBU and −5.28 eV for 2SO-F-TBU. The slight increase in HOMO energy levels for 2SO-TBU and 2SO-F-TBU were attributed to enhanced electron-donating properties from the tert-butylphenyl substituent on the donor. Correspondingly, the band gaps of molecules were achieved to be 2.29 eV, 2.16 eV and 2.17 eV, respectively.
Density functional theory (DFT) and time-dependent DFT (TDDFT) calculations were performed to evaluate the feasibility of the molecular design strategy. As shown in Fig. 1b, the LUMOs of all materials were distributed on the dibenzothioxanthene acceptor, while the HOMOs were located on the donor. The good HOMO–LUMO separation is beneficial to reduce ΔEST and thus achieve efficient RISC.27–30 The natural transition orbital (NTO) analysis demonstrated that all materials show the obvious CT-predominant nature of S1 states with the separated “holes” and “particles”. In contrast, their T1 states exhibited evident LE-predominant nature with a large overlap of “holes” and “particles”, which is contributed by the acceptor segment and this result is consistent with phosphorescence emission at 77 K (Fig. 2b). We further calculated the SOC matrix element values between different manifolds of these materials, the <S1|HSOC|T1> values of 2SO-AD, 2SO-TBU and 2SO-F-TBU are 1.061, 0.996 and 0.912 cm−1, respectively (Fig. 3d–f). Such high SOC values suggested that 3LEA can contribute to the fast RISC process and thus achieved efficient TADF emission.31
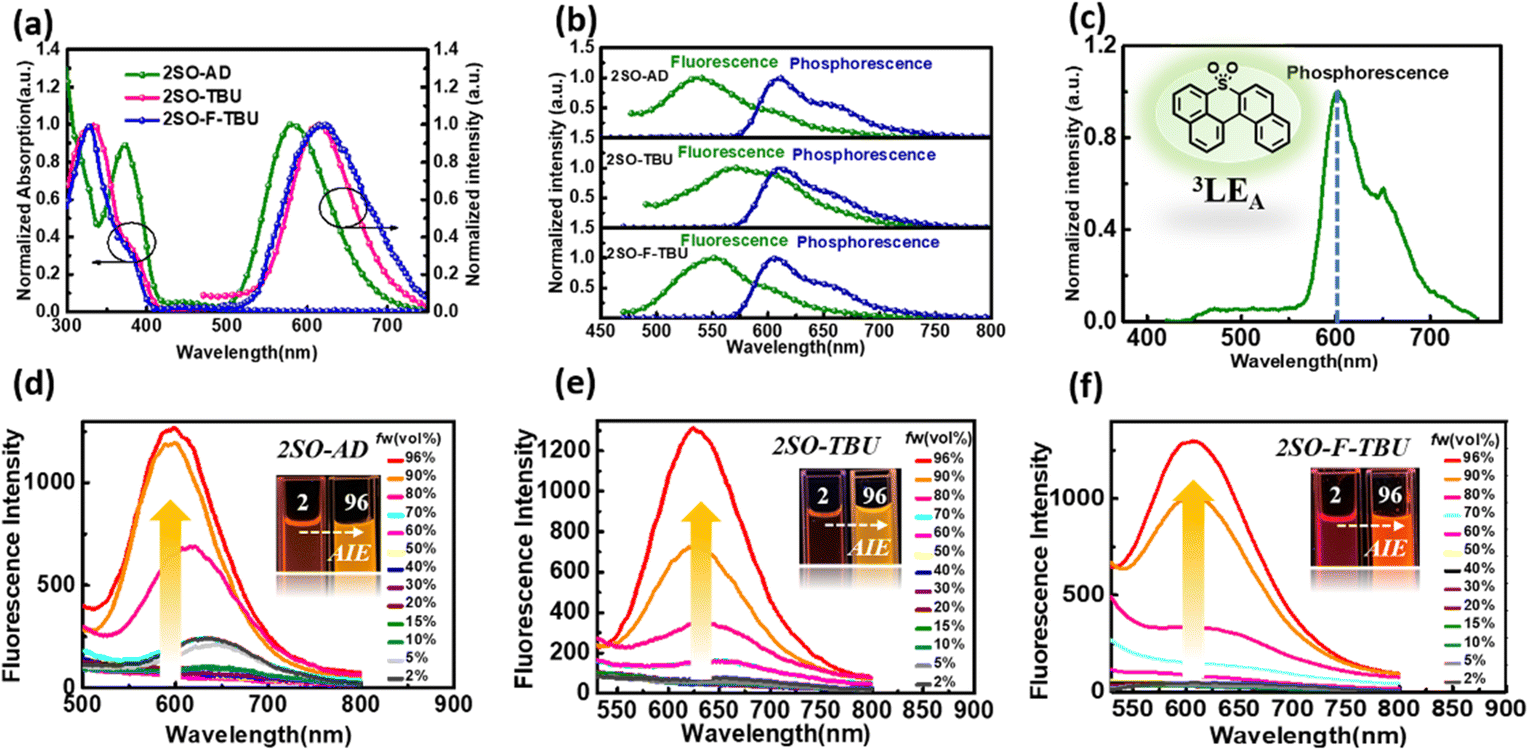 | ||
| Fig. 2 Photophysical characterizations of 2SO-AD, 2SO-TBU and 2SO-F-TBU: (a) UV-vis absorption and PL spectra in dilute toluene solutions (1 × 10−5 M) at room temperature; (b) fluorescence and phosphorescence spectra in 2-MeTHF at 77 K; (c) phosphorescence spectra of the dibenzothioxanthene acceptor in 2-MeTHF at 77 K; and the steady-state emission spectra in THF/H2O mixtures (1 × 10−5 M) at different fw (fw = 0 in pure THF solution) for (d) 2SO-AD, (e) 2SO-TBU and (f) 2SO-F-TBU. | ||
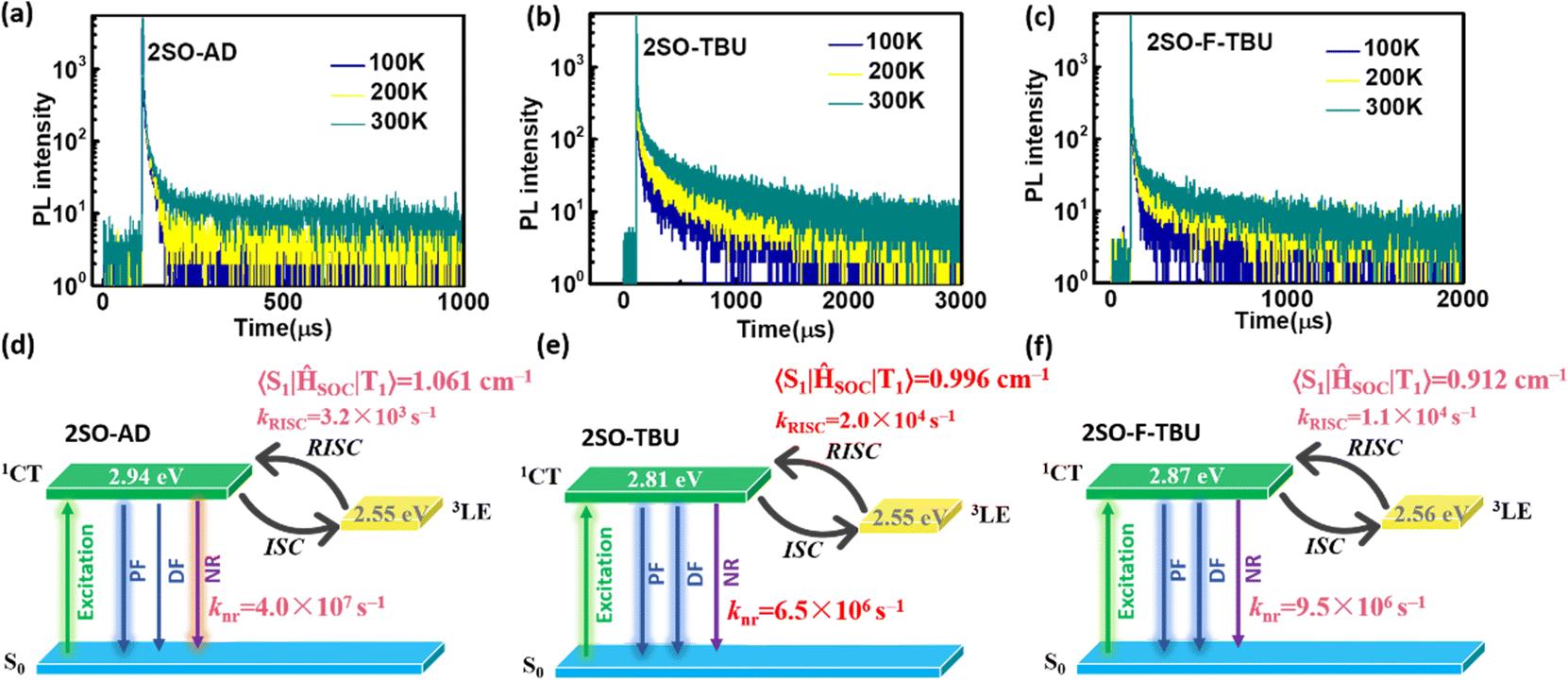 | ||
| Fig. 3 Temperature-dependent transient PL spectra in doped films (doping concentration: 10 wt%) for (a) 2SO-AD, (b) 2SO-TBU and (c) 2SO-F-TBU. TADF kinetic rate constants, electronic energies and SOC matrix element values were obtained by theoretical calculations for (d) 2SO-AD, (e) 2SO-TBU and (f) 2SO-F-TBU. | ||
The single crystals of 2SO-TBU were cultured by slow solvent vapor diffusion. As illustrated in Fig. S8 (ESI†), the dihedral angle between the donor and acceptor is nearly perpendicular with an angle of 81.0°. In addition, the molecule is fixed by short intermolecular contacts of C–H⋯O with 2.457–2.880 Å. Such a large dihedral angle and strong intermolecular contacts will increase the rigidity of the molecule in the aggregated state and reduce non-radiative deactivation due to molecular rotation or vibration.32–35 Interestingly, the shortest distance between adjacent molecules is 3.582 Å (Fig. S8, ESI†), and such a large stacking distance is expected to suppress concentration quenching and realize excellent emission in thin films according to Dexter energy transfer.36 Furthermore, theoretical calculations showed that the dihedral angles and molecular lengths of 2SO-AD, 2SO-TBU and 2SO-F-TBU are 88.43°/9.33 Å, 88.31°/21.88 Å and 88.24°/21.92 Å, respectively (Fig. S9, ESI†). The almost vertical molecular conformation and long molecular length for 2SO-TBU and 2SO-F-TBU theoretically supported their effective suppression of intermolecular aggregation and hence non-radiative deactivation.
The photophysical properties of these three compounds were investigated using ultraviolet-visible (UV-vis) and PL spectroscopy, and the results are summarized in Table S1 (ESI†). As shown in Fig. 2a, all materials in toluene exhibited strong absorption peaks around 340 nm, which were attributed to the π–π* transitions of the donor and acceptor, and the weak absorption peaks around 450 nm originated from the intramolecular charge transfer (ICT) from the donor to acceptor. The emission peaks of 2SO-AD, 2SO-TBU and 2SO-F-TBU in toluene were located at 580 nm, 614 nm and 618 nm, respectively. For 2SO-TBU and 2SO-F-TBU, the red shift of the emission peak can be ascribed to the enhanced electron-donating ability. Additionally, with the increase of solvent polarity, a significant redshift was observed in their emission spectra (Fig. S10–S12, ESI†). The positive solvation effects indicated that all compounds have significant CT characteristics, which is consistent with the results obtained from theoretical calculations. The ΔEST of 0.27 eV for 2SO-AD, 0.14 eV for 2SO-TBU and 0.20 eV for 2SO-F-TBU can be obtained according to the maximum emission peaks of the fluorescence and phosphorescence spectra at 77 K, which are comparable to those of reported TADF emitters. It is worth noting that the phosphorescence spectra of the three compounds showed clear LE characteristics with an energy level around 2.03 eV, which is consistent with our previous DFT calculations. These can be further verified by the phosphorescence spectra of the dibenzothioxanthene acceptor (Fig. 2c). This authenticates our strategy in achieving a locally excited triplet state on the acceptor (3LEA) fragment. To investigate the photophysical properties of compounds in the aggregated state, their phosphorescence spectra were examined in THF/water mixtures with different volume ratios. As shown in Fig. 2d–f, the luminescence intensity remained similar to and weaker than the initial intensity (I0) as fw increased from 2% to 70%. However, with the further increase of water content, the PL intensity is sharply enhanced and the emission spectrum is slightly blueshifted. Therefore, these results underline the obvious AIE behavior and will strongly promote the luminescence in solid films.
To further evaluate the TADF performances of the compounds in solid films, we dispersed them into 3,5-bis(3-(9H-carbazol-9-yl)phenyl)pyridine (35DCzPPY), a host material with high triplet energy level that can confine the excitons on guest molecules, to investigate their photophysical properties. As shown in the inset of Fig. S13 (ESI†), the photoluminescence spectra of all doped films showed red emission with peaks at 581 nm for 2SO-AD, 615 nm for 2SO-TBU and 591 nm for 2SO-F-TBU and photoluminescence quantum yields of 25.0%, 58.3% and 53.0%, respectively. The luminescence intensities in vacuum are significantly higher than those in air, and such oxygen sensitivity is a typical feature of TADF materials.37 The transient PL decay spectra of 2SO-AD, 2SO-TBU and 2SO-F-TBU at room temperature in a vacuum are presented in Fig. 3a–c. Both materials can be well fitted by a second-order exponential lifetime function, including a nanosecond-level prompt component and a microsecond-level delayed component. The prompt lifetimes of 2SO-AD, 2SO-TBU and 2SO-F-TBU are 10.4, 12.1 and 8.2 ns, respectively, which are attributed to the traditional S1 → S0 transition. The delayed lifetimes are 553.0, 272.1 and 577.5 μs, respectively, indicating that the delayed fluorescence via thermally activated RISC occurs. The proportion of the delay component increases with increasing temperature (Fig. 3a–c), and this trend follows the temperature-dependent characteristics of traditional TADF materials.1 To quantitatively evaluate the radiative and non-radiative deactivation processes of compounds, we calculated the related rate constants according to transient decay spectra, which are summarized in Table 1. As expected, the rate constants of singlet non-radiation decay (knrs) of 2SO-TBU with a large donor (6.5 × 106 s−1) effectively decreased by an order of magnitude compared to 2SO-AD without a large donor (4.0 × 107 s−1). This validates our strategy in suppressing non-radiative deactivation by introducing a bulky donor. The high singlet radiation decay constants (krs) of 1.3 × 107 s−1, 9.1 × 106 s−1 and 1.1 × 107 s−1 and reverse intersystem crossing constants (kRISC) of 3.2 × 103 s−1, 2.0 × 104 s−1 and 1.1 × 104 s−1 for 2SO-AD, 2SO-TBU and 2SO-F-TBU contribute positively to the efficient utilization of excitons. Among them, 2SO-TBU exhibited the fastest kRISC due to the synergism of its small ΔEST and strong SOC. Therefore, reduced non-radiative deactivation and fast RISC processes will help to achieve high-efficiency OLEDs.
| τ s (ns) | τ d (μs) | Φ Total (%) | Φ prompt (%) | Φ TADF (%) | Φ T (%) | k p (s−1) | k ISC(s−1) | k r s (s−1) | k nr s (s−1) | k RISC (s−1) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2SO-AD | 10.4 | 553.0 | 25.0 | 14.0 | 11.0 | 44.0 | 9.6 × 107 | 4.2 × 107 | 1.3 × 107 | 4.0 × 107 | 3.2 × 103 |
| 2SO-TBU | 12.1 | 272.1 | 58.3 | 11.0 | 47.3 | 81.2 | 8.3 × 107 | 6.7 × 107 | 9.1 × 106 | 6.5 × 106 | 2.0 × 104 |
| 2SO-F-TBU | 8.2 | 577.5 | 53.0 | 8.7 | 44.3 | 83.6 | 1.2 × 108 | 1.0 × 108 | 1.1 × 107 | 9.5 × 106 | 1.0 × 104 |
Multilayer OLEDs were fabricated to investigate the electrochemical characteristics with the device structure of ITO/1,1-bis[4-[N,N-di(p-tolyl)amino]phenyl]cyclohexane (TAPC) (35 nm)/1,3-Bis(carbazol-9-yl)benzene (mCP) (10 nm)/3,5-bis(3-(carbazol-9-yl)phenyl)pyridine (35DCzPPy):χ wt% TADF emitters (20 nm)/1,3,5-tri[(3-pyridyl)phen-3-yl]benzene (TmPYPB) (45 nm)/LiF (1 nm)/Al. The molecular structures and the energy levels of the device are shown in Fig. 4a. In these devices, 35DCzPPY is used as the host material, TAPC and mCP as the hole transport layer, TmPYPB as the electron transport layer, and LiF as the electron injection layer to construct a monochromatic optical device. The optimized doping concentrations (χ wt%) of 2SO-AD, 2SO-TBU and 2SO-F-TBU were 30 wt%, 5 wt% and 15 wt%, respectively. The characteristics of the devices are provided in Fig. 4b and c and the results are summarized in Table S2 (ESI†). The electroluminescence spectra of 2SO-AD, 2SO-TBU and 2SO-F-TBU cover red emission with peaks located at 599, 608 and 612 nm, and the turn-on voltages (Von) were 3.5, 3.5 and 3.0 V, respectively. No emission peak of the 35DCZPPY host was observed in the EL emission spectra, which confirms a complete confinement of the electrogenerated excitons on these three emitters during the EL process, and the energy transfer between the host and the guest is efficient. The devices based on 2SO-TBU and 2SO-F-TBU exhibited similar performance: maximum EQE of 16.27/14.47%, current efficiency (CE) of 26.07/26.31 cd A−1, and power efficiency (PE) of 27.55/20.44 lm W−1, respectively. The higher efficiency of the OLED based on 2SO-TBU is due to its reduced non-radiative deactivation and fast RISC process. However, it is also worth noting that the device based on 2SO-AD exhibited poor performance with a maximum EQE of 3.2%, which could be attributed to the lower PLQY mainly from serious non-radiative deactivation. Thus, modulating up-conversion and non-radiative deactivation by molecular engineering is crucial to achieve high-efficiency TADF emitters and devices.
 | ||
| Fig. 4 (a) Chemical structures of the materials used for the devices and the device diagram. (b) Current density–voltage–luminance characteristics; (c) external quantum efficiency versus luminance characteristics (inset: the EL spectra of the devices). | ||
Conclusions
In summary, we designed and synthesized three red TADF emitters, 2SO-AD, 2SO-TBU and 2SO-F-TBU, by employing a novel dibenzothioxanthene acceptor framework with large conjugation. The phosphorescence spectra and NTO theoretical calculations proved that this strategy not only provides a deeper LUMO for red emission, but also 3LEA from the A-segment to be close to those of the CT states, which allows the 3LE to participate in up-conversion and thus promote the RISC process. Furthermore, the molecular configuration with a bulky donor effectively inhibited intermolecular aggregation. Among them, 2SO-TBU and 2SO-F-TBU exhibited a faster reverse intersystem crossing process and suppressed non-radiative deactivation. The red OLED devices based on 2SO-TBU and 2SO-F-TBU with emission peaks located at 608 and 612 nm achieved high EQEs of 16.27% and 14.47%, respectively. Our work provides a new design strategy for high-performance red TADF emitters, which is instructive for the realization of efficient red OLEDs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21772209), International Partnership Program of Chinese Academy of Sciences (IPP) (1A1111KYSB20210028) and the National Program for Support of Top-notch Young Professionals.Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS
.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed
- D. Volz, J. Photonics Energy, 2016, 6, 020901 CrossRef
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed
- Y. Chen, X. Wei, Z. Li, Y. Liu, J. Liu, R. Wang, P. Wang, Y. Yamada-Takamurad and Y. Wang, J. Mater. Chem. C, 2017, 5, 8400–8407 RSC
- X. Cai, J. Xue, C. Li, B. Liang, A. Ying, Y. Tan, S. Gong and Y. Wang, Angew. Chem., Int. Ed., 2022, 134, e202200337 Search PubMed
- Y. Chen, D. Zhang, Y. Zhang, X. Zeng, T. Huang, Z. Liu, G. Li and L. Duan, Adv. Mater., 2021, 33, 2103293 CrossRef CAS PubMed
- R. Braveenth, H. Lee, J. D. Park, K. J. Yang, S. J. Hwang, K. R. Naveen, R. Lampande and J. H. Kwon, Adv. Funct. Mater., 2021, 31, 2105805 CrossRef CAS
- P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou and C. Yang, Adv. Mater., 2022, 34, 2106954 CrossRef CAS
- Z. Cai, X. Wu, H. Liu, J. Guo, D. Yang, D. Ma, Z. Zhao and B. Tang, Angew. Chem., Int. Ed., 2021, 60, 23635–23640 CrossRef CAS
- P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., Int. Ed., 2016, 55, 5739–5744 CrossRef CAS PubMed
- M. A. El Sayed, J. Chem. Phys., 1962, 36, 573–574 CrossRef CAS
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630 CrossRef CAS
- J. Liu, Z. Li, T. Hu, T. Gao, Y. Yi, P. Wang and Y. Wang, Adv. Opt. Mater., 2022, 10, 2102558 CrossRef CAS
- M. A. El Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS
- C. Li, C. Duan, C. Han and H. Xu, Adv. Mater., 2018, 30, 1804228 CrossRef
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050–5055 CrossRef CAS PubMed
- L.-S. Cui, A. J. Gillett, S. F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J.-L. Bredas, C. Adachi and R. H. Friend, Nat. Photonics, 2020, 14, 636–642 CrossRef CAS
- X. Hu, Y. Qin, Z. Li, H. Gao, T. Gao, G. Liu, X. Dong, N. Tian, X. Gu, C.-S. Lee, P. Wang and Y. Wang, Chin. Chem. Lett., 2022, 33, 4645–4648 CrossRef
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS
- D. Karthik, Y. H. Jung, H. Lee, S. Hwang, B.-M. Seo, J.-Y. Kim, C. W. Han and J. H. Kwon, Adv. Mater., 2021, 33, 2007724 CrossRef CAS
- J. Chen, Y. Xiao, K. Wang, D. Sun, X. Fan, X. Zhang, M. Zhang, Y. Shi, J. Yu, F. Geng, C. S. Lee and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2478–2484 CrossRef CAS
- J. Wang, J. Zhang, C. Jiang, C. Yao and X. Xi, ACS Appl. Mater. Interfaces, 2021, 13, 57713–57724 CrossRef CAS PubMed
- Y. Wada, K. Shizu, S. Kubo, K. Suzuki, H. Tanaka, C. Adachi and H. Kaji, Appl. Phys. Lett., 2015, 107, 183303 CrossRef
- W. Zeng, T. Zhou, W. Ning, C. Zhong, J. He, S. Gong, G. Xie and C. Yang, Adv. Mater., 2019, 31, 1901404 CrossRef
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS
- Y. Im, M. Kim, Y. J. Cho, J. A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS
- X. Wei, Y. Chen, R. Duan, J. Liu, R. Wang, Y. Liu, Z. Li, Y. Yi, Y. Yamada-Takamura, P. Wang and Y. Wang, J. Mater. Chem. C, 2017, 5, 12077–12084 RSC
- H. S. Kim, J. Y. Lee, S. Shin, W. Jeong, S. H. Lee, S. Kim, J. Lee, M. C. Suh and S. Yoo, Adv. Funct. Mater., 2021, 31, 2104646 CrossRef CAS
- T. Yang, Z. Cheng, Z. Li, J. Liang, Y. Xu, C. Li and Y. Wang, Adv. Funct. Mater., 2020, 30, 2002681 CrossRef CAS
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC
- Y. Shen, Z. Zhang, H. Liu, Y. Yan, S. Zhang, B. Yang and Y. Ma, J. Phys. Chem. C, 2019, 123, 13047–13056 CrossRef CAS
- S. K. Mohan Nalluri, J. Zhou, T. Cheng, Z. Liu, M. T. Nguyen, T. Chen, H. A. Patel, M. D. Krzyaniak, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1290–1303 CrossRef CAS
- J. Chen, W. Tao, W. Chen, Y. Xiao, K. Wang, C. Cao, J. Yu, S. Li, F. Geng, C. Adachi, C.-S. Lee and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14660–14665 CrossRef CAS PubMed
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311–11315 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2204277. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc04138c |
| This journal is © The Royal Society of Chemistry 2022 |