
In vivo targeting of a tumor-antigen encoded DNA vaccine to dendritic cells in combination with tumor-selective chemotherapy eradicates established mouse melanoma†‡
Sugata
Barui§¶
 *a,
Soumen
Saha§||
ab,
Yakati
Venu**
ab,
Gopi Krishna
Moku
abc and
Arabinda
Chaudhuri
*abd
*a,
Soumen
Saha§||
ab,
Yakati
Venu**
ab,
Gopi Krishna
Moku
abc and
Arabinda
Chaudhuri
*abd
aBiomaterials Group, CSIR-Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad-500007, Telangana, India. E-mail: arabinda.chaudhuri@iiserkol.ac.in; barui.sugata@gmail.com
bAcademy of Scientific & Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh-201002, India
cDepartment of Physical Sciences, Kakatiya Institute of Technology and Science, Yerragattu Gutta, Warangal 506 015, Telangana, India
dDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia-74126, West Bengal, India
First published on 26th July 2023
Abstract
Despite remarkable progress during the past decade, eradication of established tumors by targeted cancer therapy and cancer immunotherapy remains an uphill task. Herein, we report on a combination approach for eradicating established mouse melanoma. Our approach employs the use of tumor selective chemotherapy in combination with in vivo dendritic cell (DC) targeted DNA vaccination. Liposomes of a newly synthesized lipopeptide containing a previously reported tumor-targeting CGKRK-ligand covalently grafted in its polar head-group region were used for tumor selective delivery of cancer therapeutics. Liposomally co-loaded STAT3siRNA and WP1066 (a commercially available inhibitor of the JAK2/STAT3 pathway) were used as cancer therapeutics. In vivo targeting of a melanoma antigen (MART-1) encoded DNA vaccine (p-CMV-MART1) to dendritic cells was accomplished by complexing it with a previously reported mannose-receptor selective in vivo DC-targeting liposome. Liposomes of the CGKRK-lipopeptide containing encapsulated FITC-labeled siRNA, upon intravenous administration in B16F10 melanoma bearing mice, showed remarkably higher accumulation in tumors 24 h post i.v. treatment, compared to their degree of accumulation in other body tissues including the lungs, liver, kidneys, spleen and heart. Importantly, the findings in tumor growth inhibition studies revealed that only in vivo DC-targeted genetic immunization or only tumor-selective chemotherapy using the presently described systems failed to eradicate the established mouse melanoma. The presently described combination approach is expected to find future applications in combating various malignancies (with well-defined surface antigens).
Introduction
In cancer immunotherapy – arguably the most rapidly emerging cancer therapeutic modality – the power of body's immune cells is harnessed to kill cancer cells. Ever since their discovery in 1973,1 dendritic cells (DCs) have been shown to play a key role in activating our adaptive immune systems in cancer immunotherapy. The distinguishing abilities of DCs in capturing and processing tumor antigens and in presenting the processed antigenic fragments to the T cells in tumor-draining lymph nodes result in the generation of tumor-specific Cytotoxic T Lymphocytes (CTLs) that kill tumor cells.2–6 DCs are capable not only of presenting extracellular antigen fragments in complexation with the major histocompatibility complex (MHC) class II molecules to CD4+ T helper (TH) cells but also of cross-presenting the antigen fragments on MHC class I molecules to CD8+ T cells.7 Such cross-presentation capability of DCs is crucial in mounting anti-tumor immune responses.7 These potent antigen presentation and T cell activation abilities of DCs are finding widespread uses in designing effective anticancer vaccines.3,8–11In clinical studies involving DC-based cancer vaccines, classically autologous monocytes are first isolated from the patient's body using an apheresis technique, and ex vivo differentiation of DCs is effected by exposing the isolated DCs to tumor associated/specific antigens (TAAs/TSAs) in the presence of maturation-inducing agents. Such ex vivo matured DCs are finally reinfused back into the patient's body.9,12–15 DNA vaccination (genetic immunization) is another promising therapeutic modality for mounting anti-tumor immune responses in cancer immunotherapy.16 In this promising therapeutic modality, autologous DCs are first ex vivo transfected with the tumor antigen encoded DNA. Ex vivo transfected DCs then process the expressed antigenic proteins (using their proteosome complexes) into small peptide fragments and present the resulting antigen fragments in complexation with MHC-I and MHC-II to CD8+ and CD4+ T-lymphocytes, respectively.17,18 Effective ex vivo DC transfection-based DNA vaccinations for mounting immune responses have been accomplished in the past through the use of numerous DNA vaccine carriers including cationic microparticles,19,20 cationic peptides,21 cationic liposomes,22–27 gold nanoparticles,28etc. More recent studies reported the development of mannose-receptor selective in vivo DC-targeting liposomal29 and gold nanoparticle30-based DNA vaccine carriers for inducing effective and lasting anti-tumor immune responses in mice against melanoma under prophylactic settings (i.e. in preventive mode).
Although the above-mentioned priorly reported DNA vaccine carriers could protect mice from post-immunization tumor challenge under preventive settings, they failed to eradicate established mouse tumors under therapeutic settings. This is why DC-transfection-based DNA vaccination strategies are now being combined with other effective therapeutic modalities including chemotherapy, radiotherapy, etc. in DC-based cancer immunotherapy.31–37 To this end, we have begun exploring the therapeutic promises of in vivo DC-targeted DNA vaccination in combination with tumor-selective chemotherapy for inducing effective anti-tumor immune responses. Using previously designed in vivo DC-targeting cationic liposomes prepared from cationic amphiphile containing both transfection-enhancing guanidine functionality and DC-targeting mannose-mimicking shikimoyl functionality29 and two newly designed BBB-crossing brain-tumor targeting cationic liposomes, we showed that in vivo DC-targeted DNA vaccination (containing brain tumor antigen encoded DNA vaccines) in combination with brain-tumor selective chemotherapy remarkably enhanced the overall survivability of mice bearing orthotopically established mouse glioblastoma.38,39 Although by using such a combination approach in DC-based cancer immunotherapy, we could accomplish 300–500% enhancement of overall survivability of mice bearing orthotopically established brain tumors, we failed to eradicate the established brain tumor.38,39
Herein, we report on the first example of applying our above-mentioned unique combination approach38,39 in DC-based cancer immunotherapy for eradicating established mouse tumors. In this study, for the tumor-selective chemotherapy component in our combination approach, we used liposomes of a new cationic lipopeptide (CLP, Fig. 1) containing a previously reported,40 and subsequently, widely exploited41–44 tumor and tumor endothelial cell targeting CGKRK-ligand in its polar head-group region. The liposomes of CLP contained STAT3siRNA (small interfering RNA against signal transducer and activator of transcription 3) encapsulated in its aqueous core and WP1066 (a commercially available hydrophobic JAK (Janus Kinase)/STAT inhibitor) solubilized in its lipid bilayer region. STAT3, aberrantly activated in many cancer cells, causes continuous transcription of cell growth factors and anti-apoptotic molecules, thereby playing a crucial role in maintaining cancer cell growth and survival.45 However, since STAT3 is a cytoplasmic transcription factor, it is often difficult to target it directly. To this end, JAKs, the upstream activators of STATs, are being used in both preclinical and clinical settings as potential therapeutic targets for decreasing STAT activation in cancer cells.46 Cytokines and growth factor ligands provide the initial stimulus for activating the JAK/STAT pathway.47 For instance, binding of cytokines with their corresponding transmembrane receptors leads to multimerization with other subunits and close physical association of receptor-associated JAKs.48 Thereafter, the receptor-associated JAKs become activated via trans-phosphorylation.48 The phosphorylated (activated) JAKs then phosphorylate the tyrosine residues on the cytoplasmic region of the cytokine receptor to provide docking sites for the Src Homology 2 (SH2) domain of STAT proteins. Binding of STAT proteins to the phosphorylated intracellular domains of the cytokine receptors eventually leads to activated-JAK-catalyzed phosphorylation of the bound STAT proteins.49 The phosphorylated (activated) STATs become dimerized, and thereafter, the dimerized STATs are translocated into the nucleus to execute their functions as transcription factors, namely, inducing the expression of genes that regulate cellular proliferation, survival, and invasion, as well as the host immune response.48,50–52
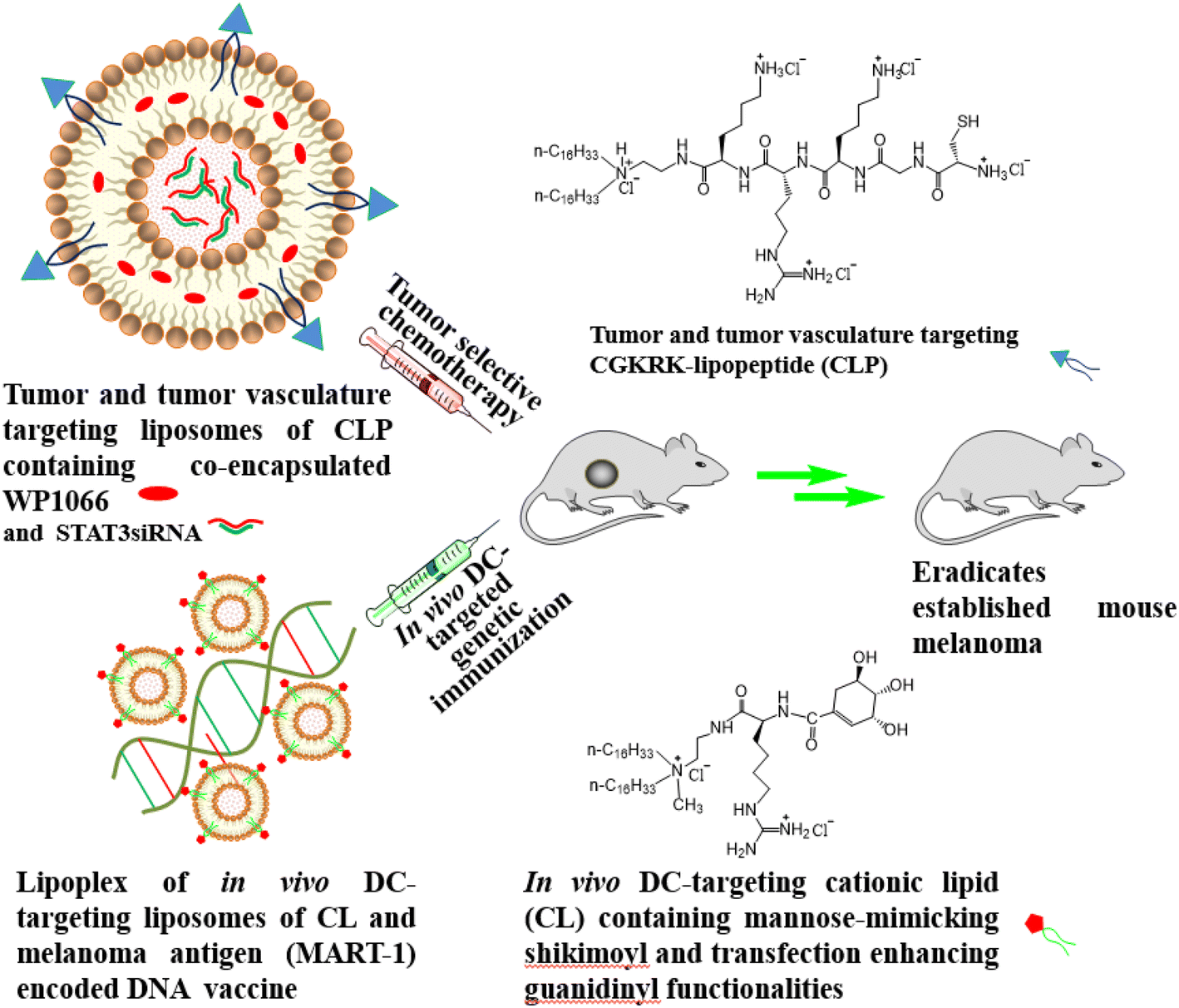 | ||
| Fig. 1 Cartoon for the presently described combination approach for eradicating the established mouse melanoma tumor. The sizes of the DC-targeting liposomes and the MART-1 encoded DNA vaccine shown in the cartoon are not drawn on the same scale. | ||
This is why many small molecule-based JAK inhibitors are finding widespread uses in inhibiting tumor growth. By inhibiting JAKs, they inhibit the activation (phosphorylation) of STAT proteins, and thereby, inhibit the above-mentioned subsequent downstream JAK–STAT signal transduction processes, eventually arresting the proliferation of cancer cells.46
Activation of the transcription factor STAT3 produces several immunosuppressive factors such as VEGF, TGFβ, IL-6 and IL-10, which, in turn, negatively affect the functional maturation of DCs.53,54 More specifically, these immunosuppressive factors inhibit DC maturation by inhibiting expressions of MHC class II, co-stimulatory molecules CD80 and CD86 and immune-stimulating molecules, such as tumor-necrosis factor (TNF) and IL-12.55 These factors were our rationale for simultaneously targeting both the widely used JAK2 inhibitor WP106638,39,46,56–60 and STAT3siRNA using tumor-targeting liposomes of the CGKRK-lipopeptide (CLP, Fig. 1). In the in vivo DC-targeted genetic immunization component of our combination approach, we used a previously reported in vivo DC-targeting liposome of a cationic lipid (CL, Fig. 1) containing both transfection-enhancing guanidine and DC-targeting mannose-mimicking shikimoyl head-groups. This in vivo DC-targeting cationic liposome was electrostatically complexed with a DNA vaccine p-CMV-MART1 encoding melanoma tumor antigen MART1 (Fig. 1). Selection of p-CMV-MART1 in the present study was based on our prior experience in working with this efficient DNA vaccine.25–30 Exploiting the abovementioned present systems, herein we show that the combined use of tumor-selective chemotherapy and in vivo DC-targeted genetic immunization using tumor antigen encoded DNA vaccines eradicates established mouse melanoma. Such a combination mode may find future uses for the treatment of many cancers where the antigenic markers of the tumor are well characterized.
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF (1:4, v/v, 10 mL, 4 min, 2 times) at room temperature. Following the same Fmoc strategy, sequential couplings of Fmoc-Lys(BOC)-OH, Fmoc-Gly-OH and BOC-Cys(Trt)-OH (2 equiv. each) using HATU (2 equiv.) and DIPEA (4 equiv.) in DMF at room temperature for 1.5 h for each amino acid afforded the resin-associated penta-peptide intermediate 3. The resin-bound intermediate 3 was taken out of the reaction vessel with excess DCM, washed thoroughly with DCM (5 × 10 mL) and dried well. The resulting dried resin bound intermediate was treated with 0.5% TFA in DCM (70 mL) for 2 h at 0 °C to obtain a protected penta-peptide intermediate 4 (0.058 g, 58%). N,N-Di-n-hexadecyl-N-2-aminoethylamine (23 mg, 0.046 mmol) was then dissolved in dry DCM (3 mL) and the solution was added to an ice cold reaction mixture (which had been under stirring conditions for 30 min) containing EDCI (8.2 mg, 0.042 mmol), HOBT (6.4 mg, 0.042 mmol) and the protected penta-peptide intermediate 4 (0.058 g, 0.042 mmol) in dry DCM (5 mL). The resulting solution was left under stirring at room temperature for 12 h. Then the solvent was evaporated using a rotary evaporator at 30 °C and the residue was dried completely under high vacuum. The dried intermediate was treated with TFA–thioanisole–ethanedithiol–anisole (90:5:3:2 v/v, 2 mL) for 3 h at 0 °C and washed with TFA:DCM (1:9, v/v, 8 mL). The acid washings were concentrated to about 1 mL and Et2O was added until a white precipitate separated. The precipitate upon chloride ion exchange chromatography over Amberlyst IRA-400 resin afforded the target CGKRK-lipopeptide (CPL, Fig. 1) as a white, fluffy solid (25 mg, 55% based on intermediate 4). The purified CGKRK-lipopeptide was found to be essentially insoluble in chloroform and could be dissolved in 3:1 (v/v) methanol:chloroform. The 1H NMR spectra of the pure CGKRK-lipopeptide was thus recorded in CD3OD/CDCl3 (3/1, v/v) mixed solvent (Fig. S1A, ESI‡). The final CGKRK-lipopeptide was characterized by the molecular ion peak (C in ESI-MS) (Fig. S1B, ESI‡) and the purity was confirmed by reversed phase analytical HPLC using two different mobile phases (Fig. S2, ESI‡).
:DMF (1:4, v/v, 10 mL, 4 min, 2 times) at room temperature. Following the same Fmoc strategy, sequential couplings of Fmoc-Lys(BOC)-OH, Fmoc-Gly-OH and BOC-Cys(Trt)-OH (2 equiv. each) using HATU (2 equiv.) and DIPEA (4 equiv.) in DMF at room temperature for 1.5 h for each amino acid afforded the resin-associated penta-peptide intermediate 3. The resin-bound intermediate 3 was taken out of the reaction vessel with excess DCM, washed thoroughly with DCM (5 × 10 mL) and dried well. The resulting dried resin bound intermediate was treated with 0.5% TFA in DCM (70 mL) for 2 h at 0 °C to obtain a protected penta-peptide intermediate 4 (0.058 g, 58%). N,N-Di-n-hexadecyl-N-2-aminoethylamine (23 mg, 0.046 mmol) was then dissolved in dry DCM (3 mL) and the solution was added to an ice cold reaction mixture (which had been under stirring conditions for 30 min) containing EDCI (8.2 mg, 0.042 mmol), HOBT (6.4 mg, 0.042 mmol) and the protected penta-peptide intermediate 4 (0.058 g, 0.042 mmol) in dry DCM (5 mL). The resulting solution was left under stirring at room temperature for 12 h. Then the solvent was evaporated using a rotary evaporator at 30 °C and the residue was dried completely under high vacuum. The dried intermediate was treated with TFA–thioanisole–ethanedithiol–anisole (90:5:3:2 v/v, 2 mL) for 3 h at 0 °C and washed with TFA:DCM (1:9, v/v, 8 mL). The acid washings were concentrated to about 1 mL and Et2O was added until a white precipitate separated. The precipitate upon chloride ion exchange chromatography over Amberlyst IRA-400 resin afforded the target CGKRK-lipopeptide (CPL, Fig. 1) as a white, fluffy solid (25 mg, 55% based on intermediate 4). The purified CGKRK-lipopeptide was found to be essentially insoluble in chloroform and could be dissolved in 3:1 (v/v) methanol:chloroform. The 1H NMR spectra of the pure CGKRK-lipopeptide was thus recorded in CD3OD/CDCl3 (3/1, v/v) mixed solvent (Fig. S1A, ESI‡). The final CGKRK-lipopeptide was characterized by the molecular ion peak (C in ESI-MS) (Fig. S1B, ESI‡) and the purity was confirmed by reversed phase analytical HPLC using two different mobile phases (Fig. S2, ESI‡).
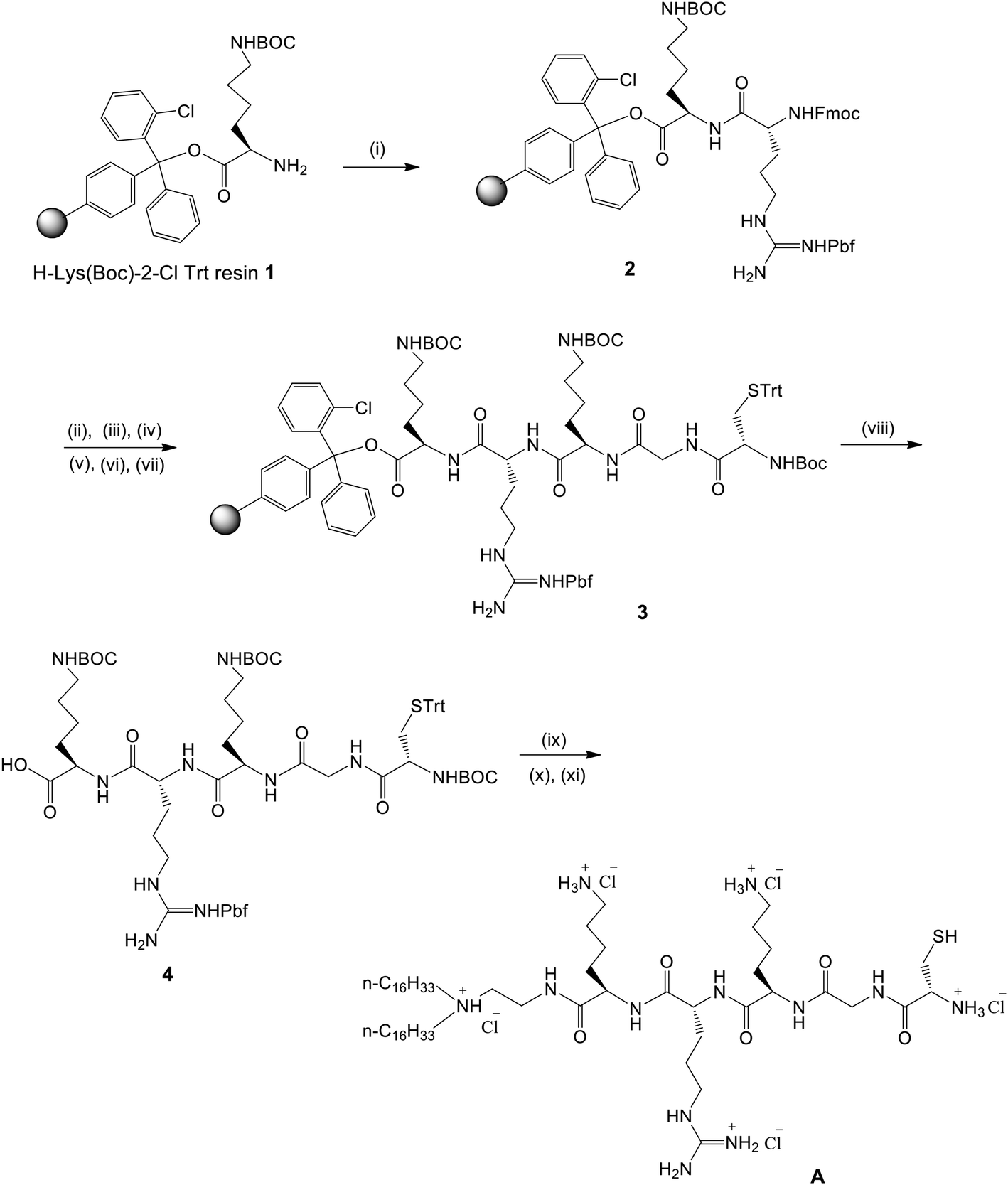 | ||
| Fig. 2 Solid phase synthesis of the CGKRK-lipopeptide (CLP). Reagents: (i) Fmoc-Arg(Pbf)-OH (2 equiv.), HATU (2 equiv.), DIPEA (4 equiv.), rt, 1 × 30 min, and then 1 × 1 h (ii) piperidine-DMF (1:4), rt, 4 min, (×2); (iii) Fmoc-Lys(BOC)-OH (2 equiv.), HATU (2 equiv.), DIPEA (4 equiv.), rt, 1 × 30 min, and then 1 × 1 h (iv) piperidine-DMF (1:4), rt, 4 min, (×2); (v) Fmoc-Gly-OH (2 equiv.), HATU (2 equiv.), DIPEA (4 equiv.), rt, 1 × 30 min, and then 1 × 1 h; (vi) piperidine-DMF (1:4), rt, 4 min, (×2); (vii) BOC-Cys(Trt)-OH (2 equiv.), HATU (2 equiv.), DIPEA (4 equiv.), rt, 1 × 30 min, and then 1 × 1 h; (viii) TFA-Dry DCM (0.5%), 2 h, 0 °C; (ix) N,N-di-n-hexadecyl-N-2-aminoethylamine, dry DCM, EDCI, HOBt, 12 h; (x) TFA–thioanisole–ethanedithiol–anisole (90:5:3:2), 3 h, 0 °C; and (xi) Cl− ion exchange (Amberlyst resin). | ||
1H NMR (200 MHz, CDCl3 + CD3OD): δ/ppm = 0.9 [t, 6H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3–(CH2)15–]; 1.1–1.5 [bs, 56H, –(C2)14–; m, 6H, Lys Cβ2 + Arg Cβ2]; 1.5–2 [m, 8H, Lys Cγ2 + Lys Cδ2; m, 2H, Arg Cγ2; m]; 2.5–3.2 [m, 4H, –N(–C2–CH2–)2; m, 2H, –N–C2–CH2– NH–CO; m, 4H, Lys Cω2; m, 2H, Arg Cδ2; m, 2H, Cys Cβ2]; 3.3–3.8 [m, 2H, –N–CH2–C2–NH–CO–; m, 2H, LysCα; m, 1H, Cys Cα; m, 1H, Arg Cα]; 4.4–4.5 [m, 2H, Gly Cα2]. ESI-MS: m/z = 1082 [M]+.
3–(CH2)15–]; 1.1–1.5 [bs, 56H, –(C2)14–; m, 6H, Lys Cβ2 + Arg Cβ2]; 1.5–2 [m, 8H, Lys Cγ2 + Lys Cδ2; m, 2H, Arg Cγ2; m]; 2.5–3.2 [m, 4H, –N(–C2–CH2–)2; m, 2H, –N–C2–CH2– NH–CO; m, 4H, Lys Cω2; m, 2H, Arg Cδ2; m, 2H, Cys Cβ2]; 3.3–3.8 [m, 2H, –N–CH2–C2–NH–CO–; m, 2H, LysCα; m, 1H, Cys Cα; m, 1H, Arg Cα]; 4.4–4.5 [m, 2H, Gly Cα2]. ESI-MS: m/z = 1082 [M]+.
:dicationic amphiphile:cholesterol mole ratios of 0.25:1:0.5. To prepare liposomal formulations containing WP1066, the stock solution of WP1066 was prepared by dissolving it in chloroform (5 mg mL−1). The final total lipid:WP1066 ratio (w/w) used in preparing the liposomal formulations containing only WP1066 and formulations containing both WP1066 and STAT3 siRNA was 10:1 for both in vitro and in vivo experiments. The appropriate lipid mixtures were dissolved in a mixture of chloroform and methanol (3:1, v/v) in a glass vial and the solvent was removed with a thin flow of moisture-free nitrogen gas. The dried lipid film was then kept under high vacuum for 8 h and hydrated overnight either in autoclaved water to prepare liposomal formulations containing only WP1066 or in nuclease free water containing a siRNA–protamine complex (total lipid:siRNA w/w 25:1, siRNA:protamine w/w 1:10) to prepare liposomal formulations containing either only siRNA or both siRNA and WP1066. The hydrated lipid film was first vortexed for 1–2 minutes at room temperature to produce multi-lamellar vesicles (MLVs). For liposomes having only WP1066, MLVs were then sonicated in an ice bath until clarity using a Branson 450 sonifier at 100% duty cycle and 25 W output power to produce small unilamellar vesicles (SUVs). For liposomes having siRNA, MLVs were bath-sonicated for 1 min to produce small unilamellar vesicles (SUVs) and then frozen and thawed 16–20 times at −78 °C to achieve equilibrium transmembrane solute distributions. Unentrapped siRNA was eluted by using Amicon Ultra centrifugal filter units (10 kD), and the liposomes were finally concentrated using Amicon Ultra centrifugation to prepare a final lipid concentration of 1 mM for the in vitro experiments or 5 mM for the in vivo experiments. In the case of FITC labeled siRNA, the concentration of siRNA entrapped in liposomes was measured by lysing the liposomes with 1% Triton-X and using fluorescence measurement from a standard graph of FITC labeled siRNA.
Liposomes of the in vivo DC-targeting cationic lipid (CL, Fig. 1) were prepared using DOPE as a co-lipid maintaining DOPE:CL mole ratios of 1:1. The appropriate lipid mixtures were dissolved in a mixture of chloroform and methanol (3:1, v/v) in a glass vial and the solvent was removed with a thin flow of moisture free nitrogen gas. The dried lipid film was then kept under high vacuum for 8 h and hydrated overnight in autoclaved water. The hydrated lipid film was first vortexed for 1–2 min at room temperature to produce multi-lamellar vesicles (MLVs). MLVs were then sonicated in an ice bath until clarity using a Branson 450 sonifier at 100% duty cycle and 25 W output power to produce small unilamellar vesicles (SUVs).
000 cells per well in a 96-well plate for 18–24 h and Rho-PE labeled liposomes containing the CGKRK-lipopeptide (CLP, Fig. 1) were added to each well. The total volume in each well was made up to 100 μL with complete medium. After 3 h of treatment, cells were washed with phosphate buffered saline and the live B16F10 cells were viewed using an epifluorescence microscope. To analyze the cellular uptake of FITC-labeled siRNA encapsulated in the liposomes of the CGKRK-lipopeptide (CLP, Fig. 1), HUVEC and B16F10 cells were seeded at a density of ∼10000 cells per well in a 96-well plate for 18–24 h before the treatment and liposomally encapsulated FITC-siRNA (30 nM) was added to each well. The total volume in each well was made up to 100 μL with serum-free medium. After 3 h of treatment, cells were washed with phosphate buffered saline and the live cells were viewed using an epifluorescence microscope.
:1000 dilutions (in 10 mL 0.05% PBS-T) overnight at 4 °C. After washing with PBS-T (3 × 10 mL, 10 min each), the membranes were incubated with a goat anti-rabbit secondary antibody conjugated to alkaline phosphatase (in 10 mL 0.05% TBS-T with 1:5000 dilution) for 2 h. Protein bands were developed using a BCIP/NBT chromogen solution (Calbiochem) and the expression levels were normalized to β-actin.
000 rpm for 20 minutes at 4 °C and 100 μL of the supernatants were taken in each well of a 96-well plate in doublet. Concentrations of FITC labeled siRNA present in the samples were measured by fluorescence measurements from a standard graph of fluorescence vs. the FITC labeled siRNA concentration. Background fluorescence from the tissue extracts, if any, were corrected by subtracting the fluorescence of the tissue extracts from the untreated control mice.
:1 lipid:DNA charge ratio for each mouse). Both 5th and 6th groups were immunized (s.c.) with the lipoplexes of p-CMV-MART1 and the liposomes of lysinylated cationic amphiphiles with guanidine and mannose-mimicking shikimoyl head-groups (cationic amphiphile 2) on days 15, 17 and 19 (using 200 μL of 5% glucose solution containing 15 μg of DNA, 4:1 lipid:DNA charge ratio for each mouse). Tumor volumes (V = ½ab2, where a = maximum length of the tumor and b = minimum length of the tumor measured perpendicular to each other) were measured using a slide calipers.
Statistical analysis. Error bars represent mean values ± SEM. The statistical significance of the experiments was determined by a two-tailed Student's test. *P < 0.05 was considered statistically significant.
:100 dilution) in PBS for 30 min and then with a goat anti-rabbit HRP-conjugated secondary antibody (at 1:100 dilution) for 15 min to mark the tumor endothelial microvessels. Tumor slides stained with CD31 were observed in a bright field using an inverted microscope (×20 magnification).
:1 to 100:1) in each well of U-bottomed 96-well plates for 6 h at 37 °C under 5% CO2, and then the lactate dehydrogenase (LDH) levels in the cell culture supernatants were measured according to the manufacturer's protocol (Promega, USA).
Results and discussion
Chemistry
CGKRK-lipopeptide 1 (CLP, Fig. 1) was synthesized by using a conventional Fmoc strategy-based solid phase peptide synthetic route as shown schematically in Fig. 2. The details of the synthetic schemes, procedures, nuclear magnetic resonance (NMR), and mass spectral data for CLP as well as the high performance liquid chromatography (HPLC) profiles of the purified lipopeptide in the two different mobile phases are provided in the ESI (Fig. S1A, S1B and S2,‡ respectively). The in vivo DC-targeting lipid containing both guanidine and mannose-mimicking shikimoyl head-groups (CL, Fig. 1) was synthesized as described previously.29Hydrodynamic sizes and zeta potentials (ξ) of liposomes
Hydrodynamic diameters and the global surface charges (zeta potentials) of the liposomal formulations containing only STAT3 siRNA, only WP1066 and both STAT3 siRNA and WP1066 were measured by photon correlation spectroscopy and electrophoretic mobility using a Zeta sizer 3000HSA (Malvern, UK). The liposomal sizes and surface potentials for all these three formulations were found to be within the range of 180–210 nm and 4–6 mV, respectively (Table S1, ESI‡).Apoptosis of endothelial and tumor cells
Toward probing the synergistic effects of the presently described liposomal formulations of STAT3 siRNA and WP1066 in inducing apoptosis in endothelial cells, we studied the conventional Annexin V/propidium iodide (PI) binding-based flow cytometric apoptosis assay. Incubation of HUVEC cells with liposomal formulations of CLP containing both STAT3 siRNA and WP1066 induced significantly enhanced accumulation of late apoptotic cells compared to the late apoptotic populations in HUVEC cells treated with liposomal formulations containing only WP1066 and only STAT3 siRNA (Fig. 3A). Importantly, scrambled siRNA encapsulated in the liposomes of CGKRK-lipopeptide 1 was found to be incompetent to induce apoptosis in endothelial cells (Fig. 3A). Consistently, similar synergistic effects of liposomally co-loaded STAT3 siRNA and WP1066 in inducing enhanced apoptosis were also observed in the tumor (B16F10) cells (Fig. S3, ESI‡).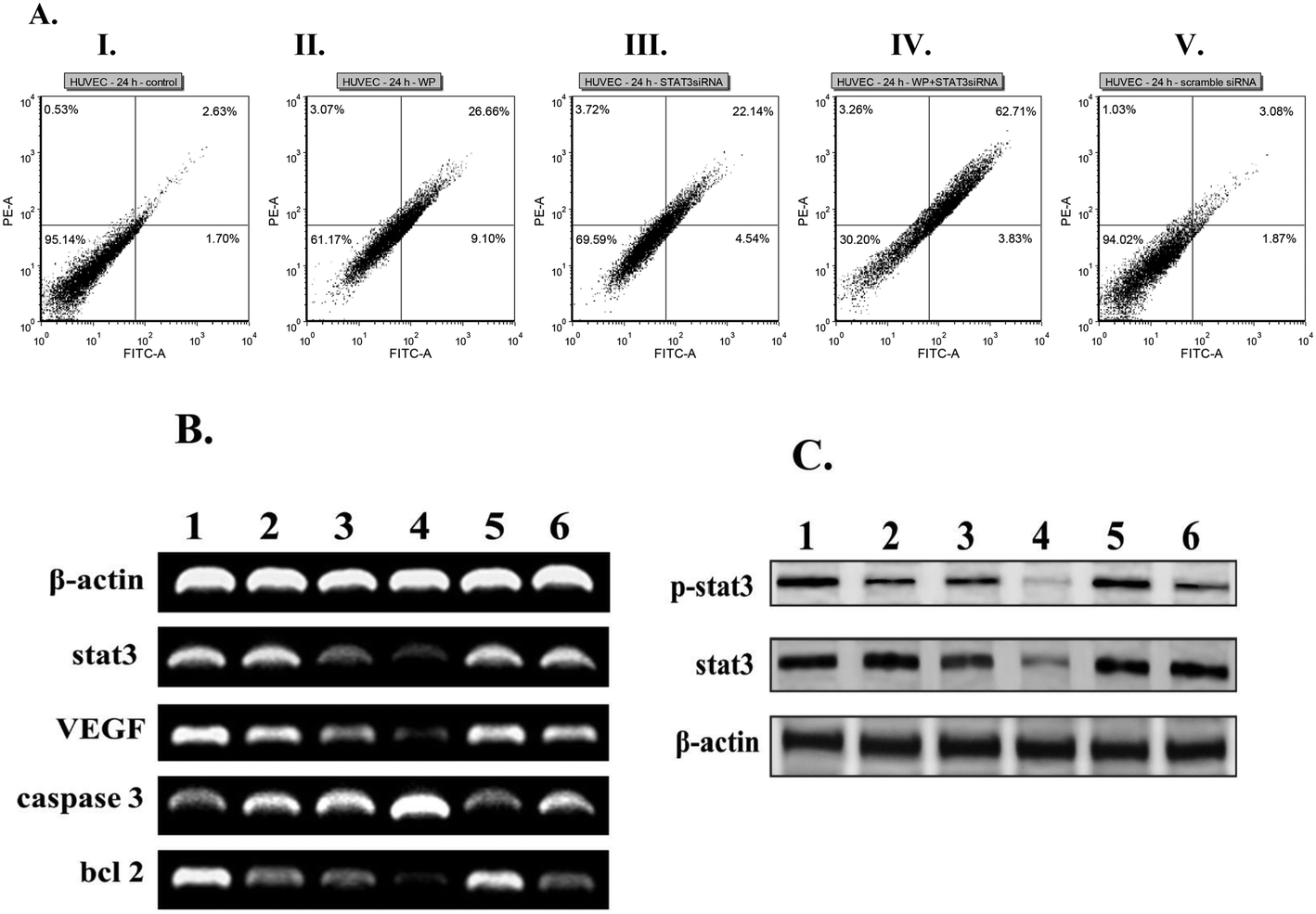 | ||
| Fig. 3 Synergistic effects of STAT3 siRNA and WP1066 co-loaded liposomes of CLP in inducing apoptosis and inhibiting STAT3 activation in endothelial cells. (A) Untreated control HUVEC (I); HUVEC treated with only WP1066-loaded liposomes of CLP (II); HUVEC treated with only STAT3 siRNA-loaded liposomes of CLP (III); HUVEC treated with STAT3 siRNA and WP1066 co-loaded liposomes of CLP (IV); and HUVEC treated with scrambled siRNA loaded liposomes of CLP (V). Both untreated (I) and treated cells were stained with FITC-Annexin V and propidium iodide (PI) for the flow cytometric analysis. The horizontal and vertical axes represent cells labeled with FITC-Annexin V and PI, respectively in the dot plot. Dots in the upper right quadrant represent late apoptotic cells (positive for both Annexin V and PI). (B and C) mRNA levels (B) of the indicated genes involved in the STAT3 signalling pathway; the levels of STAT3 and p-STAT3 protein expressions (C) were measured by RT-PCR and western blotting, respectively. Lane 1, untreated cells; lane 2, cells treated with the targeted liposomal WP1066; lane 3, cells treated with the targeted liposome containing STAT3 siRNA; lane 4, cells treated with the targeted liposome containing both WP1066 and STAT3 siRNA; lane 5, cells treated with the targeted liposome containing scrambled siRNA; and lane 6, cells treated with the targeted liposome containing both WP1066 and scrambled siRNA. | ||
RT-PCR and western blot analysis
With a view to analyze the effects of combined therapeutics co-loaded in CLP liposomes on the mRNA expressions of genes involved in the STAT3 signaling pathway, RT-PCR experiments were performed. The decrease in the expressions of anti-apoptotic genes such as VEGF, STAT3, Bcl2, and BclXL genes as well as the increase in expression of the pro-apoptotic caspase-3 gene were observed when compared with the loading control β-actin in both HUVEC (Fig. 3B, the original gel images used for drawing Fig. 3B are provided in Fig. S4‡) and B16F10 (Fig. S5A, ESI, the original gel images used for drawing Fig. S5A are provided in Fig. S6‡) cells. Collectively, the findings in the RT-PCR experiments (Fig. 3B and Fig. S5A, ESI‡) showed that liposomally entrapped combined therapeutics (STAT3 siRNA + WP1066) were more potent in downregulating the transcription of anti-apoptotic genes and upregulating the transcription of pro-apoptotic genes compared to their levels in cells treated with liposomally entrapped individual therapeutics (either STAT3 siRNA or WP1066). Importantly, liposomally encapsulated scrambled siRNA showed no effect on genes involved in the STAT3 signaling pathway (Fig. 3B and Fig. S5A, ESI‡). To examine the possible inhibition of STAT3 phosphorylation, HUVEC and B16F10 cells were treated with liposomal formulations containing chemotherapeutics, and cell lysates (proteins) were subjected to western blot analysis. Liposomal formulations of combined therapeutics (STAT3 siRNA and WP1066) were found to be more efficient in downregulating the expressions of STAT3 and p-STAT3 protein levels compared to their levels in the cells treated with liposomally entrapped individual therapeutics (either STAT3 siRNA or WP1066) in both endothelial and tumor cells (Fig. 3C and Fig. S5B, ESI‡). An important observation needs further clarification at this point of discussion. As mentioned above, activated (phosphorylated) JAKs catalyze phosphorylation of the STAT proteins bound to the phosphorylated intracellular domains of the cytokine receptors.46,49 Thus, WP1066, the inhibitor of JAK2, decreases the level of pSTAT3 protein in cells treated with liposomal formulations containing only WP1066 (compared to the level of pSTAT3 protein in the untreated control cells). Since lesser pSTAT3 would be produced in JAK2-catalyzed phosphorylation of STAT3 proteins in the cells treated with liposomal formulations containing only WP1066, in a quantitative study, one may observe a somewhat increased level of STAT3 proteins (not a decreased level) after such treatment of cells. However, in qualitative PCR and western blot experiments (Fig. 3B and C), depending on the degree of JAK2 inhibition by WP1066, we may not always be able to see the enhanced levels of STAT3 proteins in cells treated with liposomal formulations containing only WP1066. Such a possibility could play a role in the decreased pSTAT3 level (compared to the pSTAT3 level in the untreated cells, lane 2, Fig. 3C) and no significant changes in the STAT3 protein levels (lane 2, Fig. 3B and C) observed in the cells treated with liposomal formulations containing only WP1066.Targeting FITC labeled siRNA to tumor vasculature
With a view to evaluate the efficiency of liposomes of CLP to deliver encapsulated siRNA in tumor vasculature, we intravenously administered FITC-labeled siRNA encapsulated in the liposomes of CLP in C57BL/6J mice bearing aggressive B16F10 tumors. Immunohistochemical staining of fixed tumor cryosections (prepared 24 h post the liposome injection) using antibodies against vWF (tumor vasculature marker) revealed co-localization of FITC (green) and tumor endothelial cells (Fig. S7, ESI‡). Thus, the findings summarized in Fig. S7‡ indicate that the liposomes of CLP deliver its entrapped siRNA cargo to tumor endothelial cells.Biodistribution of liposomally encapsulated FITC-labeled siRNA
Toward examining the tissue distribution profiles of the presently described liposomal systems, FITC-labeled siRNA encapsulated in the liposomes of CLP was intravenously administered in mice bearing B16F10 melanoma tumors. After 24 h of treatment, tumors and tissue samples were obtained to quantify the concentration of FITC-labeled siRNA in each sample. Importantly, the concentration of FITC labeled siRNA in the tumor was found to be remarkably higher than those in the other body tissues including the lungs, liver, kidneys, spleen and heart (Fig. S8, ESI‡). Autofluorescence intensities for each organ of the untreated control mice were subtracted from the corresponding measured values in the organs of the treated mice for constructing the biodistribution profile (Fig. S8‡). The fluorescence and autofluorescence intensities measured for each organ used for drawing Fig. S8‡ are provided in an Excel Sheet format in Table S2, ESI.‡Tumor growth inhibition study using tumor-selective CLP liposomes
With a view to evaluate the therapeutic potential of the synergistic use of WP1066 and STAT3 siRNA encapsulated in the presently described liposomes of CLP, C57BL/6J mice bearing aggressive B16F10 tumors were separately administered (i.v.) with liposomal formulations of CLP containing only WP1066, only STAT3 siRNA and both WP1066 and STAT3 siRNA. Pronounced tumor growth inhibition was observed in mice treated with formulations containing both the therapeutics (Fig. 4A and B, the original colored images of the tumors shown in Fig. 4B are provided in Fig. S9, ESI‡). Comparatively, tumor growth inhibition was found to be 2–3 fold less in mice treated with formulations containing only WP1066 or only STAT3 siRNA (Fig. 4A and B). Mice intravenously administered with vehicle alone (in 5% aqueous glucose solution) developed large tumors on day 23 (Fig. 4A and B) and were sacrificed on day 24 post tumor inoculation.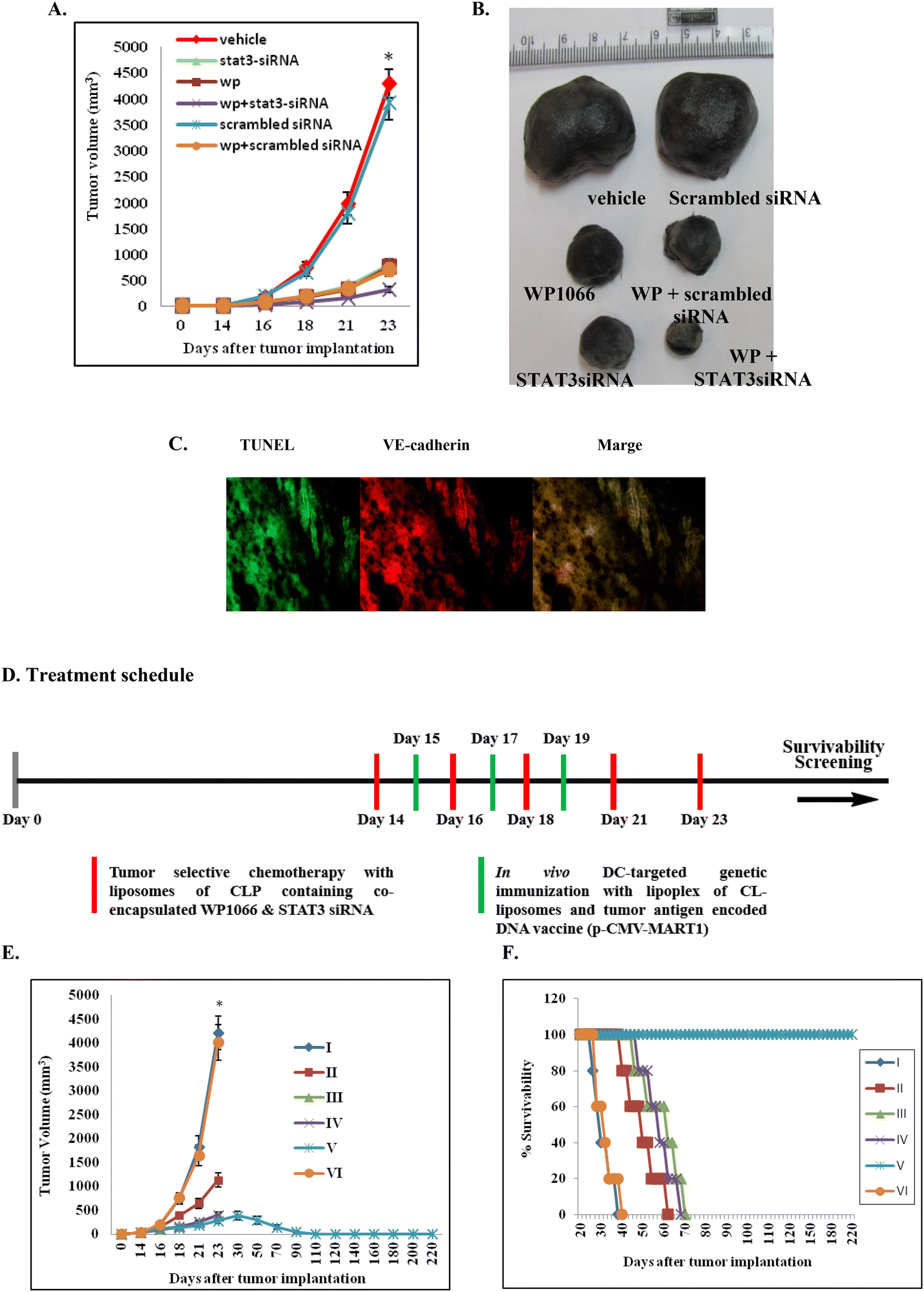 | ||
| Fig. 4 Tumor selective chemotherapy with CLP-liposomes containing co-encapsulated WP1066 and STAT3 siRNA in combination with in vivo DC-targeted genetic immunization with lipoplexes of CL liposomes and a melanoma antigen (MART-1) encoded DNA vaccine results in the eradication of the established mouse melanoma. (A) Relative tumor growth inhibition in the mouse groups (n = 5) treated with CLP liposomes containing only WP1066, CLP liposomes containing only STAT3 siRNA, and CLP liposomes containing only scrambled siRNA; targeted liposomal formulation containing both WP1066 and scrambled siRNA; and targeted liposomal formulation containing both WP1066 and STAT3 siRNA. (B) Representative tumor sizes in each group on day 24 post tumor inoculation. (C) On day 24 post tumor inoculation, the fixed tumor cryosections from the mice treated with the targeted liposomal formulation containing both WP1066 and STAT3 siRNA were immunostained using an anti-VE-cadherin antibody (markers of tumor endothelial cells) and a TUNEL assay kit (markers for apoptotic cells, green). All the images were taken at ×10 magnification. (D) Treatment schedule. (E) Relative tumor growth inhibition observed in the mouse groups i.v. treated with only the vehicle control 5% glucose (I), only in vivo DC-targeting CL-liposomes in complexation with the melanoma antigen (MART-1) encoded DNA vaccine (Lipo-MART1, II), only (STAT3 siRNA and WP1066) co-loaded CLP liposomes (III), simultaneously with the control non-specific lipoplexes of the p-CMV-β-gal plasmid and in vivo DC-targeting CL liposomes and (WP1066 + STAT3 siRNA) co-loaded CLP liposomes ((Lipo-(WP + STAT3 siRNA) + Lipo-β-gal), IV), simultaneously with lipoplexes of the melanoma antigen (MART-1) encoded DNA vaccine (p-CMV-MART-1) and in vivo DC-targeting CL liposomes and (WP1066 + STAT3 siRNA) co-loaded CLP liposomes (Lipo-(WP + STAT3 siRNA) + Lipo-MART1, V), and empty CLP liposomes (VI). (F) Day 25 onwards, mice were subjected to a survival study. *P < 0.05 with respect to the tumor sizes for the 5% glucose treatment. | ||
Apoptosis in tumor vasculature
Since liposomes of CLP were found to be efficient in targeting siRNA to tumor vasculatures (Fig. S8, ESI‡), we envisaged that the observed tumor growth inhibition (Fig. 3A and B) presumably resulted from the apoptosis of tumor endothelial cells. To confirm this, C57BL/6J mice bearing aggressive murine B16F10 melanoma tumors were administered liposomal formulations having both WP1066 and STAT3 siRNA. On day 24, mice were sacrificed, and tumors were cryosectioned, fixed and immunostained for VE-cadherin (another endothelial cell marker). The same tumor cryosections were also stained using TUNEL assay kits for labeling apoptotic cells. The colocalization of the TUNEL-positive and VE-cadherin-positive cells in the tumor cryosections (Fig. 4C, the original snapshots are provided in Fig. S10 and S11, ESI‡) was consistent with the proposition that the remarkable tumor growth inhibition properties of liposomal formulations of CLP containing co-loaded WP1066 and STAT3 siRNA are presumably mediated through apoptosis of tumor endothelial cells. Toward confirming the efficiencies of the presently described liposomal formulations of the CGKRK-lipopeptide containing WP1066 and STAT3 siRNA in inhibiting the formation of microvessels around tumor tissues, tumor cryosections were immunostained with CD31 (one of the most widely used markers of tumor endothelial cells) antibodies. The findings in these immunohistochemical staining experiments clearly showed least tumor microvessel densities in the tumor cryosections from mice treated with liposomal formulations containing both WP1066 and STAT3 siRNA (Fig. S12‡).Inhibition of STAT3 phosphorylation in vivo
To examine the possible inhibition of STAT3 phosphorylation under in vivo conditions, B16F10 tumor cells were isolated from mice treated with chemotherapeutics encapsulated in liposomes of CLP. Most significant decrease in STAT3 activation at the protein level was observed in B16F10 cells isolated from tumors treated with STAT3 siRNA and WP1066 co-encapsulated in the liposomes of CGKRK-lipopeptide 1 (Fig. S13, ESI‡). Importantly, liposomally encapsulated scrambled siRNA showed no effect on the STAT3 and p-STAT3 protein levels in vivo (Fig. S13, ESI‡).Combined effects of tumor-selective chemotherapy and in vivo DC-targeted genetic immunization on tumor growth inhibition
Toward evaluating the therapeutic promise of the present combination approach in inhibiting the established mouse melanoma, mice were immunized (s.c.) after two weeks of tumor inoculation with lipoplexes of a melanoma antigen encoded DNA vaccine (p-CMV-MART1) and direct in vivo DC-targeting liposomes of CL (Fig. 1) on days 15, 17 and 19 post tumor inoculation. Tumor growth inhibition studies (Fig. 4A) suggest that only genetic immunization (without using any tumor-selective chemotherapy) is not enough to inhibit tumor growth. Most notably, complete regression of the established melanoma tumor was observed only when established melanoma-bearing mice were treated with five WP1066 and STAT3 siRNA co-loaded tumor-selective CLP liposomes in combination with three s.c. genetic immunization with melanoma antigen (MART1) encoded p-CMV-MART1 DNA vaccines complexed with in vivo DC-targeting liposomes of CL (Fig. 4E and F) following the treatment schedules shown in Fig. 4D.Melanoma-specific cellular immune response
Toward examining the role of cellular immune responses in the presently described combination approach, we conducted a cytotoxicity (CTL) assay and measured the amounts of secreted IFN-γ and IL-4 when effector cells (splenocytes isolated from immunized mice) were incubated with target B16F10 melanoma cells. Primed splenocytes were collected and used for lysing the target melanoma cells across the effector:target cell ratios of 10:1–100:1. The effector splenocytes isolated from mice receiving the combined treatment were significantly more efficient in lysing the target melanoma cells than those isolated from mice treated (s.c.) with only lipoplexes of p-CMV-MART1 and CL or with only STAT3 siRNA and WP1066 co-loaded liposomes of CLP (Fig. S14A, ESI‡). Thus, the findings summarized in Fig. S14A‡ demonstrate that the use of genetic immunization in combination with targeted chemotherapy is most efficient to induce formations of anti-B16F10 specific CTLs. Using an ELISA-based protocol, we measured the relative amounts of IFN-γ and IL-4 in the supernatant of the overnight co-culture of the target cells B16F10 and splenocytes of the treated mice. Consistently, results in the cytokine secretion assays revealed remarkably higher amounts of IFN-γ and IL-4 secreted by the activated T cells in mice receiving the combined treatment (Fig. S14B and C, ESI‡). Thus, the findings summarized in Fig. S12A–C‡ indicate that heightened cellular immune responses may play crucial roles in the observed eradication of the established melanoma in mice receiving the presently described combination therapy.
The observed synergistic therapeutic outcome in the combination approach described herein presumably originates from enhanced DC maturation (and hence more T-cell mediated killing of tumor cells) resulting from the inhibition of STAT3 activation by WP1066-loaded liposomes of CLP. In other words, in the initial phases of the treatment schedules, the concentrations of immunosuppressive factors in the tumor microenvironment were likely to be at much higher concentrations than their concentrations at the end of the treatment schedules. Presumably, during the initial phase of the treatment schedule, immunosuppressive factors dominated the race between immunosuppression-related tumor growth vs. T-cell mediated tumor cell killing and the trend was reversed at the end of the treatment schedule. After the end of the treatment schedule, the pronounced decrease in the concentrations of the immunosuppressive factors in the tumor microenvironment might have significantly enhanced DC maturation, eventually leading to T-cell mediated eradication of the established mouse melanoma. Such a mechanistic possibility might play a key role in the observed slow increase of the tumor volume, followed by a gradual decrease of the tumor volume and eventual eradication of the established mouse melanoma in group V mice (Fig. 4E). Prior reports also showed that targeting small molecule inhibitors of the JAK/STAT pathway selectively to the tumor site is capable of inducing antitumor immune responses in the tumor microenvironment.61,62 Besides cell-mediated immunity mounted by T-cells, natural killer (NK) cells are primary effectors of innate immunity against transformed tumor cells.63,64 Bellucci et al. demonstrated increased susceptibility of tumor cells to NK cells upon treatment with tyrosine kinase inhibitors.65 Such enhanced activities of NK cells may also play an important role in the presently observed synergistic therapeutic outcome. Clearly, in-depth mechanistic studies need to be carried out in future to decipher the origin of the observed synergy between tumor-selective chemotherapy and in vivo DC-targeted genetic immunization. Whether or not other potent commercially available JAK-STAT inhibitors (about 10 available)46 would be equally efficient for use in our present combination approach needs to be studied in future. Similarly, whether the present combination approach works efficiently for other melanoma antigen encoded DNA vaccines66 (e.g. DNA vaccines encoding melanoma antigens gp100, tyrosinase related protein1, etc.) needs to be examined in future. The preclinical findings described herein are expected to attract interest among the physician scientists for undertaking clinical studies aimed at exploring the therapeutic promise of the present combination approach in cancer patients.
Conclusions
We have shown that the use of tumor-selective chemotherapy in combination with in vivo dendritic cell (DC) targeted DNA vaccination eradicates established mouse melanoma. Liposomes of a newly synthesized lipopeptide containing a previously reported tumor-targeting CGKRK-ligand were used for tumor selective delivery of liposomally co-loaded cancer therapeutics STAT3siRNA and WP1066 (a commercially available inhibitor of the JAK2/STAT3 pathway). In vivo targeting of a melanoma antigen (MART-1) encoded DNA vaccine (p-CMV-MART1) to dendritic cells was accomplished by complexing it with a previously reported mannose-receptor selective in vivo DC-targeting cationic liposome. Liposomes of the CGKRK-lipopeptide containing encapsulated FITC-labeled siRNA, upon intravenous administration in B16F10 melanoma bearing mice, showed remarkably higher accumulation in tumors 24 h post i.v. treatment, compared to their degree of accumulation in other body tissues including the lungs, liver, kidneys, spleen and heart. Importantly, findings in tumor growth inhibition studies revealed that only in vivo DC-targeted genetic immunization or only tumor-selective chemotherapy using the presently described systems failed to eradicate the established mouse melanoma. Since the presently described combination therapy avoids the need for painstaking isolation of DCs or T-cells, and their ex vivo transduction and subsequent reinfusion into the recipient's body, this unique combination approach is expected to find future uses in combating established melanoma and possibly other solid tumors containing distinguishing tumor antigens.Author contributions
A. C. and S. B. conceived this project and designed the experiments; S. B. and S. S. carried out the major work of the experimental parts reported in this manuscript; Y. V. and G. K. M. contributed in both in vitro and in vivo experiments and designed the experiments; and A. C. and S. B. wrote the manuscript with inputs from all the other authors.Conflicts of interest
S. B., S. S. and A. C. are inventors of US Patent No. 9944676B2, issued on April 17, 2018.Acknowledgements
A. C. thanks the Council of Scientific and Industrial Research (CSIR), Government of India for funding this work (project codes: CSC0302 and BSC0123) and the Indian Council of Medical Research (ICMR), Government of India, for awarding him an ICMR Emeritus Scientist position. S. B. and G. K. M. thank CSIR, Government of India, for their doctoral fellowships, Y. V. thanks the University Grant Commission (UGC), Government of India, New Delhi, for his doctoral fellowship and S. S. thanks CSIR, Government of India for awarding him the Shyama Prasad Mukherjee Fellowship for pursuing doctoral research.References
- R. M. Steinman and Z. A. Cohn, J. Exp. Med., 1973, 137, 1142–1162 CrossRef CAS PubMed.
- G. J. Randolph, J. Exp. Med., 2021, 218, e20202077 CrossRef PubMed.
- A. Harari, M. Graciotti, M. Bassani-Sternberg and L. E. Kandalaft, Nat. Rev. Drug Discovery, 2020, 19, 635–652 CrossRef CAS PubMed.
- S. Nava, D. Lisini, S. Frigerio, S. Pogliani, S. Pellegatta, L. Gatti, G. Finocchiaro, A. Bersano and E. A. Parati, Pharmaceutics, 2020, 12, 215–227 CrossRef CAS PubMed.
- S. Jhunjhunwala, C. Hammer and L. Delamarre, Nat. Rev. Cancer, 2021, 21, 298–312 CrossRef CAS PubMed.
- S. C. Eisenbarth, Nat. Rev. Immunol., 2019, 19, 89–103 CrossRef CAS PubMed.
- O. P. Joffre, E. Segura, A. Savina and S. Amigorena, Nat. Rev. Immunol., 2012, 12, 557–569 CrossRef CAS PubMed.
- Y. Lu, Y. Shi and J. You, J. Controlled Release, 2022, 341, 184–205 CrossRef CAS.
- M. Sadeghzadeh, S. Bornehdeli, H. Mohahammadrezakhani, M. Abolghasemi, E. Poursaei, M. Asadi, V. Zafari, L. Aghebati-Maleki and D. Shanehbandi, Life Sci., 2020, 254, 117580 CrossRef CAS PubMed.
- A. Gardner, A. P. de Mingo and B. Ruffell, Front. Immunol., 2020, 11, 924 CrossRef CAS PubMed.
- S. K. Wculek, F. J. Cueto, A. M. Mujal, I. Melero, M. F. Krummel and D. Sancho, Nat. Rev. Immunol., 2020, 20, 7–24 CrossRef CAS PubMed.
- G. Y-Z. X. Zhao and X.-R. Song, Acta Pharmacol. Sin., 2020, 41, 959–969 CrossRef PubMed.
- P. Han, D. Hanlon, O. Sobolev, R. Chaudhury and R. L. Edelson, Int. Rev. Cell Mol. Biol., 2019, 349, 251–307 CAS.
- B. Giri, P. Sharma, T. Jain, A. Ferrantella, U. Vaish, S. Mehra, B. Garg, S. Iyer, V. Sethi and Z. Malchiodi, et al. , OncoImmunology, 2021, 10, 1976952 CrossRef PubMed.
- C. I. DeVette, H. Gundlapalli, S.-C. A. Lai, C. P. McMurtrey, A. R. Hoover, H. R. Gurung, W. R. Chen, A. L. Welm and W. H. Hildebrand, Oncoimmunology, 2020, 9, 1685300 CrossRef PubMed.
- J. Rice, C. H. Ottensmeier and F. K. Stevenson, DNA vaccines: precision tools for activating effective immunity against cancer, Nat. Rev. Cancer, 2008, 8, 108–120 CrossRef CAS.
- O. Akbari, N. Panjwani, S. Garcia, R. Tascon, D. Lowrie and B. Stockinger, J. Exp. Med., 1999, 189, 169–178 CrossRef CAS PubMed.
- J. Banchereau and R. M. Steinman, Nature, 1998, 392, 245–252 CrossRef CAS PubMed.
- M. L. Hedley, J. Curley and R. Urban, Nat. Med., 1998, 4, 365–368 CrossRef CAS PubMed.
- M. Singh, M. Briones, G. Ott and D. O'Hagan, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 811–816 CrossRef CAS PubMed.
- A. S. Irvine, P. K. Trinder, D. L. Laughton, H. Ketteringham, R. H. McDermott, C. Reid, A. M. Haines, A. Amir, R. Husain, R. Doshi, L. L. S. Young and A. Mountain, Nat. Biotechnol., 2000, 18, 1273–1278 CrossRef CAS PubMed.
- Y. Hattori, S. Kawakami, S. Suzuki, F. Yamashita and M. Hashida, Biochem. Biophys. Res. Commun., 2004, 317, 992–999 CrossRef CAS PubMed.
- W. Wijagkanalan, S. Kawakami, M. Takenaga, R. Igarashi, F. Yamashita and M. Hashida, J. Controlled Release, 2008, 125, 121–130 CrossRef CAS PubMed.
- Y. Lu, S. Kawakami, F. Yamashita and M. Hashida, Biomaterials, 2007, 28, 3255–3262 CrossRef CAS PubMed.
- R. Srinivas, P. P. Karmali, D. Pramanik, A. Garu, Y. Mahidhar, B. K. Majeti, S. Ramakrishna, G. Srinivas and A. Chaudhuri, J. Med. Chem., 2010, 53, 1387–1391 CrossRef CAS PubMed.
- R. Srinivas, A. Garu, G. Moku, S. Agawane and A. Chaudhuri, Biomaterials, 2012, 33, 6220–6229 CrossRef CAS PubMed.
- C. Voshavar, R. C. Meka, S. Samanta, S. Marepally and A. Chaudhuri, J. Med. Chem., 2017, 60, 1605–1610 CrossRef CAS PubMed.
- R. C. Reddy, S. Mukherjee, C. R. Patra and A. Chaudhuri, Nanoscale, 2019, 11, 7931–7943 RSC.
- A. Garu, G. Moku, S. K. Gulla and A. Chaudhuri, Mol. Ther., 2016, 24, 385–397 CrossRef CAS PubMed.
- S. K. Gulla, B. R. Rao, G. Moku, S. Jinka, N. V. Nimmu, S. Khalid, C. R. Patra and A. Chaudhuri, Biomater. Sci., 2019, 7, 773–788 RSC.
- M. V. Gulijk, F. Dammeijer, J. G. J. V. Aerts and H. Vroman, Front. Immunol., 2018, 9, 2759 CrossRef PubMed.
- G.-Y. Jang, Y. S. Kim, S. E. Lee, J. W. Lee, H. D. Han, T. H. Kang and Y.-M. Park, Cancer Immunol., Immunother., 2021, 70, 1075–1088 CrossRef CAS PubMed.
- S. P. Lau, N. van Montfoort, P. Kinderman, M. Lukkes, L. Klaase, M. van Nimwegen, M. van Gulijk, J. Dumas, D. Mustafa and S. L. A. Lievense, et al. , J. Immunother. Cancer, 2020, 8, e000772 CrossRef PubMed.
- T. Oba, K. Makino, R. Kajihara, T. Yokoi, R. Araki, M. Abe, H. Minderman, A. E. Chang, K. Odunsi and F. Ito, J. Immunother. Cancer, 2021, 9, e002432 CrossRef PubMed.
- J. Guo, E. Muse, A. J. Christians, S. J. Swanson and E. Davila, Cancer Immunol. Res., 2019, 7, 1523–1534 CrossRef CAS PubMed.
- M. Bassani-Sternberg, A. Digklia, F. Huber, D. Wagner, C. Sempoux, B. J. Stevenson, A.-C. Thierry, J. Michaux, H. Pak and J. Racle, et al. , Front. Immunol., 2019, 10, 1832 CrossRef CAS PubMed.
- R. Ramanathan, H. Choudry, H. Jones, M. Girgis, W. Gooding, P. Kalinski and D. L. Bartlett, Ann. Surg. Oncol., 2021, 28, 4637–4646 CrossRef PubMed.
- S. Bhunia, V. Vangala, D. Bhattacharya, H. G. Ravuri, M. Kuncha, S. Chakravarty, S. Ramakrishna and A. Chaudhuri, Mol. Pharm., 2017, 14, 3834–3847 CrossRef CAS PubMed.
- S. Saha, V. Yakati, D. Bhattacharya, S. D. Kompella, M. Kuncha, S. Chakravarty, S. Ramakrishna and A. Chaudhuri, Adv. Biosyst., 2017, 1, 1600009 CrossRef PubMed.
- J. A. Hoffman, E. Giraudo, M. Singh, L. Zhang, M. Inoue and K. Porkka, et al. , Cancer Cell, 2003, 4, 383–391 CrossRef CAS PubMed.
- L. Agemy, D. Friedmann-Morvinski, V. R. Kotamraju, L. Roth, K. N. Sugahara and O. M. Girard, et al. , Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17450–17455 CrossRef CAS PubMed.
- X. L. Yao, Y. Yoshioka, G. X. Ruan, Y. Z. Chen, H. Mizuguchi and Y. Mukai, et al. , Biomacromolecules, 2012, 13, 2402–2409 CrossRef CAS PubMed.
- Q. Hu, X. Gao, T. Kang, X. Feng, D. Jiang, Y. Tu, Q. Song, L. Yao, X. Jiang, H. Chen and J. Chen, Biomaterials, 2013, 34, 9496–9508 CrossRef CAS PubMed.
- S. K. Gulla, R. Kotcherlakota, S. Nimushakavi, N. V. Nimmu, S. Khalid, C. R. Patra and A. Chaudhuri, ACS Omega, 2018, 3, 8663–8676 CrossRef CAS PubMed.
- M. Tolomeo and A. Cascio, Int. J. Mol. Sci., 2021, 22, 603 CrossRef CAS PubMed.
- Z. Qureshy, D. E. Johnson and J. R. Grandis, J. Cancer Metastasis Treat., 2020, 6, 27 CAS.
- R. Morris, N. J. Kershaw and J. J. Babon, Protein Sci., 2018, 27, 1984–2009 CrossRef CAS PubMed.
- J. S. Rawlings, K. M. Rosler and D. A. Harrison, J. Cell Sci., 2004, 117, 1281–1283 CrossRef CAS PubMed.
- C. P. Lim and X. Cao, Mol. BioSyst., 2006, 2, 536–550 RSC.
- J. V. Alvarez, D. A. Frank, J. Alvarez and F. Lab, Cancer Biol. Ther., 2004, 3, 1045–1050 CrossRef CAS PubMed.
- W. Vainchenker and S. N. Constantinescu, Oncogene, 2013, 32, 2601–2613 CrossRef CAS PubMed.
- Y. Wang, Y. Shen, S. Wang, Q. Shen and X. Zhou, Cancer Lett., 2018, 415, 117–128 CrossRef CAS PubMed.
- H. Yu, M. Kortylewski and D. Pardoll, Nat. Rev. Immunol., 2007, 7, 41–51 CrossRef CAS PubMed.
- H. Yu, D. Pardoll and R. Jove, Nat. Rev. Cancer, 2009, 9, 798–809 CrossRef CAS PubMed.
- D. Gabrilovich, Nat. Rev. Immunol., 2004, 4, 941–952 CrossRef CAS PubMed.
- A. Ferrajoli, S. Faderl, Q. Van, P. Koch and D. Harris, et al. , Cancer Res., 2007, 67, 11291–11299 CrossRef CAS PubMed.
- A. Horiguchi, T. Asano, K. Kuroda, A. Sato and J. Asakuma, et al. , Br. J. Cancer, 2010, 102, 1592–1599 CrossRef CAS PubMed.
- X. Zhou, Y. Ren, A. Liu, L. Han and K. Zhang, et al. , Oncol. Rep., 2014, 31, 2173–2180 CrossRef CAS PubMed.
- L. Kong, K. A. Mohamed, W. Jun and A. Chakraborty, et al. , Clin. Cancer Res., 2008, 14, 5759–5768 CrossRef CAS PubMed.
- V. Vangala, N. V. Nimmu, S. Khalid, M. Kuncha, R. Sistla, R. Banerjee and A. Chaudhuri, Mol. Pharm., 2020, 17, 1859–1874 CrossRef CAS PubMed.
- W. Zou, Nat. Rev. Cancer, 2005, 5, 263–274 CrossRef CAS PubMed.
- T. Wang, G. Niu, M. Kortylewski, L. Burdelya, K. Shain, S. Zhang, R. Bhattacharya, D. Gabrilovich, R. Heller, D. Coppola, W. Dalton, R. Jove, D. Pardoll and H. Yu, Nat. Med., 2004, 10, 48–54 CrossRef CAS PubMed.
- M. A. Caligiu, Blood, 2008, 112, 461–469 CrossRef PubMed.
- E. Vivier, D. H. Raulet, A. Moretta, M. A. Caligiuri, L. Zitvogel, L. L. Lanier, W. M. Yokoyama and A. S. Ugolini, Science, 2011, 331, 44–49 CrossRef CAS PubMed.
- R. Bellucci, H. N. Nguyen, A. Martin, S. Heinrichs, A. C. Schinzel, W. C. Hahn and J. Ritz, J. Clin. Invest., 2012, 122, 2369–2383 CrossRef CAS PubMed.
- T. Rezaei, E. Davoudian, S. Khalili, A. M. Amini and M. Hejazi, et al. , Pigm. Cell Melanoma Res., 2021, 34, 869–891 CrossRef CAS PubMed.
Footnotes |
| † CSIR-IICT Manuscript No. IICT/Pubs./2023/149. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00702b |
| § These authors contributed equally to this work. |
| ¶ Present address: Enveda Therapeutics India Private Limited, Genome Valley, Hyderabad-500101, Telangana, India. |
| || Present address: Department of Biomedical Engineering, Duke University, Durham, NC 27708-0281, USA. |
| ** Present address: Department of Chemical and Biological Engineering, The University of Alabama, Tuscaloosa, AL 35487, USA. |
| This journal is © The Royal Society of Chemistry 2023 |