
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-switchable anion binding and catalysis with a visible light responsive halogen bonding receptor†
Aidan
Kerckhoffs
,
Isabelle
Moss
and
Matthew J.
Langton
 *
*
Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: matthew.langton@chem.ox.ac.uk
First published on 23rd November 2022
Abstract
Photo-switchable receptors allow for photo-control over guest binding and release with spatial and temporal precision. Here we report the first halogen bonding photo-switchable anion receptors in which chloride binding may be reversibly modulated by irradiation with red and blue light, with over a 50-fold enhancement in chloride binding affinity observed for the Z isomer. We demonstrate that this switchable binding enables unprecedented photo-controlled catalysis of XB-mediated halide abstractions and a Mukaiyama Aldol reaction.
The recognition of anions by synthetic supramolecular receptors is a rapidly expanding field of research, driven by applications in sensing, catalysis and transmembrane transport.1,2 Amongst the array of Lewis acidic intermolecular interactions available for anion recognition, halogen bonding has emerged in recent years as a highly effective interaction for strong and selective anion binding.3 Halogen bonding (XB) is the attractive intermolecular interaction between the electrophilic region of a halogen atom and a Lewis base, and has found wide-ranging applications in supramolecular chemistry,4 including catalysis,5–7 self-assembly,8,9 anion recognition10–13 and anion transport across lipid bilayer membranes.14–17 The stringent linear interaction and resistance to solvent effects18 are particularly beneficial for the design of potent anion receptors.
Photo-switchable receptors, in which guest binding is modulated by photo-isomerization of the host in order to switch between strongly and weakly binding states, enable control over when and where guests are bound and released. Since Shinkai's work on potassium hosts, the field of photo-responsive cation receptors has become relatively well established.19 However, photo-responsive receptors for anions remain rare,20 with only a handful of systems reported to date based on azobenzene foldamers21,22 and receptors,23–27 azopyrazoles,28 acylhydrazones29 or stilbene photoswitches,30–33 all exploiting hydrogen bonding interactions for anion recognition, and apart from two exceptions reported by us,24,25 all responding to UV light, which has poor biocompatibility and limits applications in biological anion sensing or transport.
Here we report the first example of a halogen bonding, visible light responsive anion receptor, by functionalizing a red-shifted ortho-chloro azobenzene with electron deficient iodo-triazole motifs. The receptor can be switched from a weakly binding state as the E isomer to a strongly binding state as the Z isomer using red light, and this process may be reversed using blue light. We also demonstrate that the charged iodo-triazolium analogue acts as an unprecedented XB-mediated photo-switchable catalyst, in which the Z isomer is a more potent catalyst for a variety of model XB- catalyzed reactions.
The target halogen bonding photo-switchable receptor is shown in Fig. 1, which incorporates an ortho-chloro-functionalized azobenzene. Recent work has demonstrated that ortho-substitution of azobenzenes with heteroatoms leads to significant red shifting of the n–π* band of the E isomer, separating it from the Z n–π* transition, and enabling visible light, bi-directional photo-switching with orthogonal wavelengths.34–39 Chloro-substitution enables red light switching, and affords long Z isomer lifetimes (half-life of ∼1 week),25 which is particularly advantageous for switchable binding applications, where thermal relaxation would lead to un-controllable ion release. The central azobenzene core is functionalised with electron deficient aryl-iodotriazoles, which are potent XB donors.12,40,41
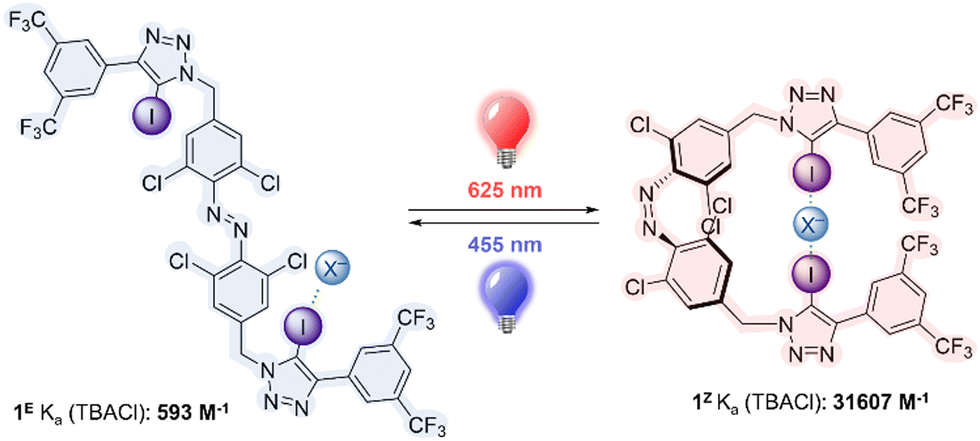 | ||
| Fig. 1 Photoswitchable anion receptor 1. | ||
Receptor 1 was prepared from commercially available carboxylic acid 2, which was reduced and subsequently protected to afford aniline 4. Azobenzene 5 was generated in good yield with in situ generated DBU-Cl+ in the presence of 4 (Scheme 1).42 After deprotection, alcohol 6 was tosylated and subsequently converted to the azide 8, which was subjected to Cu-catalyzed ‘click’ conditions with iodoalkyne 9 to afford receptor 1 in good yields.
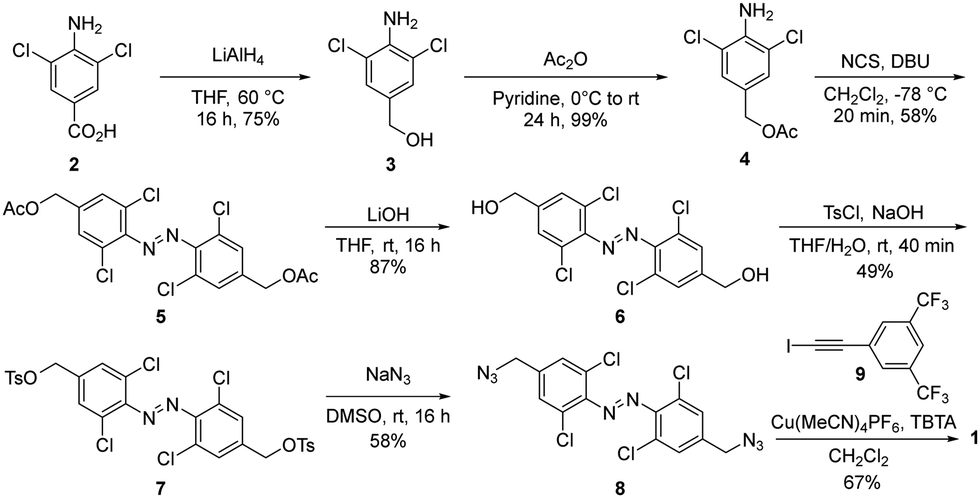 | ||
| Scheme 1 Synthesis of red-shifted azobenzene 1. | ||
We first investigated the photo-switching properties of 1. Irradiation with red light from an LED (625 nm, ∼0.9 W) afforded high conversion to the Z isomer with a photo-stationary state (PSS) distribution of 83% Z in acetone as determined by 1H NMR experiments (Fig. S49, ESI†). Irradiation with blue light (455 nm, ∼1.1 W) triggered Z to E isomerisation (87% E). It was also possible to generate near-quantitative amounts of 1E by heating to 60 °C in the oven for 1 h (> 98% E). The UV-vis spectra of 1 are shown in Fig. 2A. As observed in related tetra ortho-chloro azobenzenes, the n–π* transitions are well separated, as reflected in the high PSS in both switching directions. The switching process is fully reversible and could be repeated multiple times without detectable fatigue (Fig. 2B).
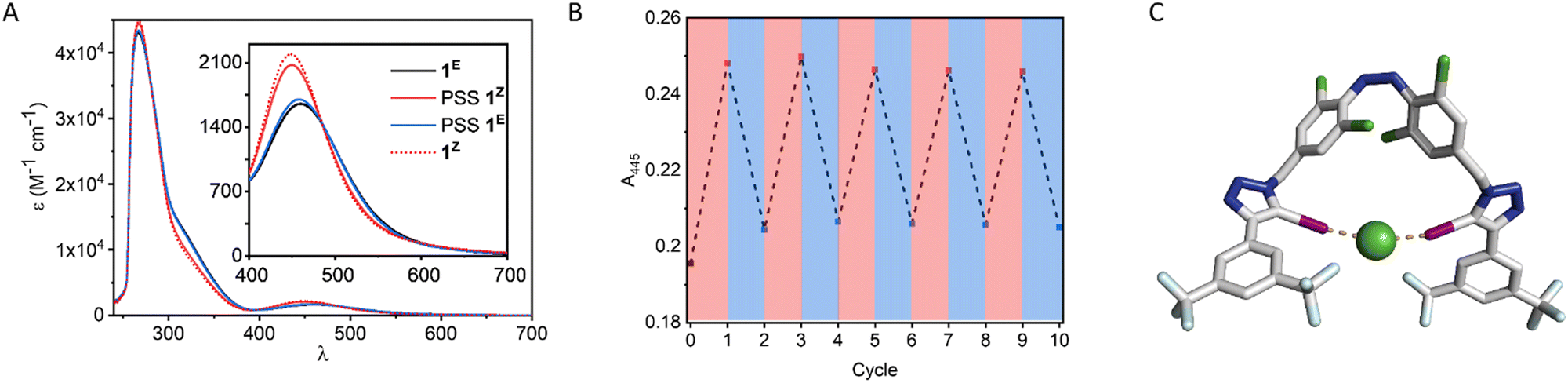 | ||
| Fig. 2 (A) Absorption spectra of 1 as the E and Z isomer. Inset: n → π* bands and composition of the PSS mixtures upon irradiation with red light (625 nm, determined by NMR integration). Pure Z-isomer spectrum was calculated from the spectra of pure 1E and the PSS mixture of known composition. (B) Absorption at 445 nm after successive cycles of irradiation with 625/455 nm light (acetone). (C) Geometry optimised molecular mechanics calculated structure of 1ZCl. | ||
Molecular mechanics calculations of 1Z as a complex with chloride suggested that both iodotriazole XB donors are capable of forming a convergent bidentate interaction with the anion (Fig. 2C). We therefore investigated the anion binding capability of the receptor by conducting 1H NMR titration experiments in d6-acetone by addition of aliquots of the anion as the tetrabutylammonium salt. The data were fitted to the appropriate binding isotherms using Dynafit,43 and the association constants are shown in Table 1. K1,E is the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding constant for anion binding by 1E and K1,Z is the corresponding 1:1 binding constant for 1Z, which was determined by generating the Z-rich PSS in the NMR tube with red light (83% Z), and accounting for binding to the minor population of 1E in the analysis.
:1 binding constant for anion binding by 1E and K1,Z is the corresponding 1:1 binding constant for 1Z, which was determined by generating the Z-rich PSS in the NMR tube with red light (83% Z), and accounting for binding to the minor population of 1E in the analysis.
| Aniona | K E 1/M−1 | K E 2/M−1 | K Z 1/M−1 | EM/mM | K refEM |
|---|---|---|---|---|---|
| a Anions added as the tetra butyl ammonium salt in d6-acetone, 298K. b No binding. Errors at the 95% confidence limit. | |||||
| Cl− | 593 ± 70 | 148 | 31607 ± 52 | 360 | 107 |
| Br− | 311 ± 2 | 78 | 4616 ± 45 | 188 | 30 |
| I− | 108 ± 5 | 27 | 677 ± 3 | 232 | 13 |
| NO3− | NBb | NBb | 67 ± 3 | — | — |
| HSO4− | NBb | NBb | 62 ± 9 | — | — |
Chloride binding to 1E was statistical (binding to one iodotriazole XB donor is independent of that to the other).33 For statistical binding, K1 = 4K2 by accounting for the statistical factors, and K1 is 2Kref, where Kref is the binding affinity to an individual iodo-triazole binding site. We have previously determined this single site binding affinity to be 197 ± 14 M−1 in a related 3,5-bistrifluoromethyl aryl-iodotriazole, in which the pendent azobenzene in 1 is replaced by a hexyl group.15 In this case the binding affinity to one site of 1E is 296 M−1. This suggests that the adjacent azobenzene leads to a modest enhancement of chloride affinity, further evidenced by chemical shift perturbations of the adjacent H atoms on the azobenzene aryl rings (Fig. S59–S64, ESI†). Binding of the halides to 1E decreased in the order Cl− > Br− > I−; and no binding of either nitrate or hydrogen sulfate was observed.
The affinity of the corresponding Z isomer 1Z to the halides was much enhanced compared to the E isomer. In the case of chloride this enhancement was significant, with KZ/KE = 53, the highest reported to date for a switchable anion receptor incorporating halogen bonding interactions,33 and amongst the highest known across all anion receptor types.20 This provides strong evidence that both iodotriazole XB donors are involved in a bidentate chelating cooperative interaction with the anion.
We quantified the chelate cooperativity in the system through the effective molarity, EM, which defines the concentration at which simple intermolecular interactions compete with cooperative intramolecular interaction (Table 1 and eqn (1)).44
| EM = KobsZ/Kref2 | (1) |
We also prepared the iodotriazolium receptor 1-Me-BarF by methylation and subsequent anion exchange of 1 (Fig. 3A, see ESI† for synthesis and characterization). As expected, the affinity of the dicationic host for chloride was enhanced relative to 1, necessitating the use of the more competitive solvent DMSO-d6 for titration experiments for the triflate derivative. The switchable anion affinity was maintained in this system, albeit significantly diminished (K1E = 349 M−1, K1Z = 945 M−1, KZ/KE = 2.7, DMSO-d6). This may be due to charge repulsion in the Z-isomer where both binding sites are in close proximity.
 | ||
| Fig. 3 (A) Structure of iodotriazolium receptor and control compound. (B and C) Friedel–Crafts alkylation reaction performed with activator 1-Me-BArF4. Data shown in presence and absence of activator for each isomer (black points) and fit to first order rate equation (lines). (D) Mukaiyama aldol reaction with activator 1-Me-BArF4. | ||
The ability of the photo-switchable halogen bonding anion receptors to catalyze a Friedel–Crafts alkylation by bromide abstraction was then explored (Fig. 3B).45 Both isomers of 1-Me-BArF were able to efficiently catalyze this benchmark reaction at low concentrations (1.7 mM), with notably a ∼3-fold rate enhancement for 1Z-Me-BArF4 (Fig. 3B). However, both isomers of the neutral analogue 1 were unable to catalyse the desired reaction (Fig. S72–S73, ESI†), suggesting that charge-assisted halogen bonding is essential for this reaction to take place; in line with Huber's work.45 We observed significantly diminished rates for the reaction in the presence of 2 equivalents of control compound 10, and no background activity in the absence of an activator (Fig S71–S80, ESI†).
Encouraged by this result, we screened a variety of other benchmark XB-mediated reactions. Pleasingly, our system efficiently catalyzed the analogous chloride abstraction reaction (Fig. 3C and Fig. S81–S88, ESI†), with a ∼2-fold enhancement in rate for the Z-isomer relative to the E-isomer. 1-Me-BArF4 was also capable of catalyzing a Mukaiyama aldol reaction at 0.5 mol% catalyst loadings (Fig. 3D and Fig. S89–S96, ESI†),46 with the Z-isomer affording approximately twice as much product relative to the E-isomer within 7 hours. Finally, 1-Me-BArF4 efficiently catalyzed a Michael addition of 1-methylindole and crotonophenone at 10 mol % loading (Fig. S97–S104, ESI†),47 although there was no significant discrimination between the isomers. This is perhaps due to the steric demands of the enone within the relatively small binding site of 1Z-Me-BArF4, leading to monodentate binding modes. In all benchmark reactions, two equivalents of control compound 10 relative to 1-Me-BArF4 resulted in sluggish reactions, suggesting that the dicationic nature of 1-Me-BArF4 enhances catalysis rates.
In conclusion, we report the first visible light switchable anion receptor and catalyst utilising halogen bonding interactions. The neutral bis-iodo-triazole functionalised red-shifted azobenzene can be switched between a weakly binding E isomer and the strongly binding Z isomer using blue and red light, with over a 50-fold enhancement in binding affinity observed. The analogous dicationic triazolium system enables unprecedented photo-regulated XB-mediated halide abstraction catalysis of a Friedel-Crafts alkylation and Mukaiyama aldol reaction, in which the stronger binding Z isomers acts as the more potent catalytic species. These results demonstrate that combining molecular photoswitches with potent halogen bond donors could open up new applications of photo-responsive receptors for achieving spatio-temporal control over both molecular recognition and catalytic processes using light.
A. K. acknowledges the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship (EP/L015838/1), generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. M. J. L. acknowledges funding from the Royal Society. M. J. L. is a Royal Society University Research Fellow.
Conflicts of interest
There are no conflicts to declare.References
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed
.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS
- J. Y. C. Lim and P. D. Beer, Chemistry, 2018, 4, 731–783 CrossRef CAS
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS
- C. Xu, V. U. B. Rao, J. Weigen and C. C. J. Loh, Nat. Commun., 2020, 11, 4911 CrossRef PubMed
- O. Dumele, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 12339–12344 CrossRef CAS PubMed
- C. J. Serpell, N. L. Kilah, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2010, 49, 5322–5326 CrossRef CAS PubMed
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, Angew. Chem., Int. Ed., 2019, 58, 13823–13827 CrossRef CAS
- M. G. Sarwar, B. Dragisic, S. Sagoo and M. S. Taylor, Angew. Chem., Int. Ed., 2010, 49, 1674–1677 CrossRef CAS PubMed
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS
-
A. Docker and P. D. Beer, Halogen Bonding in Solution, John Wiley & Sons, Ltd, 2021, pp. 83–120 Search PubMed
- A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef PubMed
- L. E. Bickerton, A. J. Sterling, P. D. Beer, F. Duarte and M. J. Langton, Chem. Sci., 2020, 11, 4722–4729 RSC
- L. E. Bickerton, A. Docker, A. J. Sterling, H. Kuhn, F. Duarte, P. D. Beer and M. J. Langton, Eur. J. Chem., 2021, 27, 11738–11745 CrossRef CAS PubMed
- A. Singh, A. Torres-Huerta, T. Vanderlinden, N. Renier, L. Martínez-Crespo, N. Tumanov, J. Wouters, K. Bartik, I. Jabin and H. Valkenier, Chem. Commun., 2022, 58, 6255–6258 RSC
- C. C. Robertson, R. N. Perutz, L. Brammer and C. A. Hunter, Chem. Sci., 2014, 5, 4179–4183 RSC
- S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu and O. Manabe, J. Am. Chem. Soc., 1981, 103, 111–115 CrossRef CAS
- S. Lee and A. H. Flood, J. Phys. Org. Chem., 2013, 26, 79–86 CrossRef CAS
- Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838–12840 CrossRef CAS PubMed
- F. C. Parks, Y. Liu, S. Debnath, S. R. Stutsman, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 17711–17723 CrossRef CAS PubMed
- K. Dąbrowa, P. Niedbała and J. Jurczak, Chem. Commun., 2014, 50, 15748–15751 RSC
- A. Kerckhoffs, Z. Bo, S. E. Penty, F. Duarte and M. J. Langton, Org. Biomol. Chem., 2021, 19, 9058–9067 RSC
- A. Kerckhoffs and M. J. Langton, Chem. Sci., 2020, 11, 6325–6331 RSC
- Y. R. Choi, G. C. Kim, H.-G. Jeon, J. Park, W. Namkung and K.-S. Jeong, Chem. Commun., 2014, 50, 15305–15308 RSC
- S. T. J. Ryan, J. del Barrio, R. Suardíaz, D. F. Ryan, E. Rosta and O. A. Scherman, Angew. Chem., Int. Ed., 2016, 55, 16096–16100 CrossRef CAS PubMed
- S. Lv, X. Li, L. Yang, X. Wang, J. Zhang, G. Zhang and J. Jiang, J. Phys. Chem. A, 2020, 124, 9692–9697 CrossRef CAS PubMed
- Z. Kokan and M. J. Chmielewski, J. Am. Chem. Soc., 2018, 140, 16010–16014 CrossRef CAS
- S. J. Wezenberg, M. Vlatković, J. C. M. Kistemaker and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 16784–16787 CrossRef CAS PubMed
- S. J. Wezenberg, L.-J. Chen, J. E. Bos, B. L. Feringa, E. N. W. Howe, X. Wu, M. A. Siegler and P. A. Gale, J. Am. Chem. Soc., 2022, 144, 331–338 CrossRef CAS PubMed
- D. Villarón, M. A. Siegler and S. J. Wezenberg, Chem. Sci., 2021, 12, 3188–3193 RSC
- A. Docker, X. Shang, D. Yuan, H. Kuhn, Z. Zhang, J. J. Davis, P. D. Beer and M. J. Langton, Angew. Chem., Int. Ed., 2021, 60, 19442–19450 CrossRef CAS
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS
- D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538–6547 CrossRef CAS
- A. Kerckhoffs, K. Christensen and M. J. Langton, Chem. Sci., 2022, 13, 11551–11559 RSC
- D. Mungalpara, S. Stegmüller and S. Kubik, Chem. Commun., 2017, 53, 5095–5098 RSC
- F. Ostler, D. G. Piekarski, T. Danelzik, M. S. Taylor and O. García Mancheño, Eur. J. Chem., 2021, 27, 2315–2320 CrossRef CAS
- A. Antoine John and Q. Lin, J. Org. Chem., 2017, 82, 9873–9876 CrossRef CAS
- P. Kuzmič, Anal. Biochem., 1996, 237, 260–273 CrossRef
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS
- A. Dreger, E. Engelage, B. Mallick, P. D. Beer and S. M. Huber, Chem. Commun., 2018, 54, 4013–4016 RSC
- F. Heinen, D. Reinhard, E. Engelage and S. Huber, Angew. Chem., Int. Ed., 2021, 60, 5069–5073 CrossRef CAS PubMed
- R. Sutar, N. Erochock and S. Huber, Org. Biomol. Chem., 2021, 19, 770–774 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05199k |
| This journal is © The Royal Society of Chemistry 2023 |