
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective electrochemical CO2 conversion with a hybrid polyoxometalate†
Satoshi
Kuramochi
a,
Jamie M.
Cameron
 b,
Tomoya
Fukui
a,
Kieran D.
Jones
b,
Stephen P.
Argent
c,
Shinpei
Kusaka
a,
Ryo
Shiraishi
a,
Yusuke
Tamaki
d,
Taiki
Yatsu
d,
Takuya
Shiga
a,
Osamu
Ishitani
de,
Hiroki
Oshio
*a and
Graham N.
Newton
*b
b,
Tomoya
Fukui
a,
Kieran D.
Jones
b,
Stephen P.
Argent
c,
Shinpei
Kusaka
a,
Ryo
Shiraishi
a,
Yusuke
Tamaki
d,
Taiki
Yatsu
d,
Takuya
Shiga
a,
Osamu
Ishitani
de,
Hiroki
Oshio
*a and
Graham N.
Newton
*b
aGraduate School of Pure and Applied Sciences, University of Tsukuba, Tennoudai 1-1-1, Tsukuba, Ibaraki 305-8571, Japan. E-mail: oshio@chem.tsukuba.ac.jp
bGSK Carbon Neutral Laboratories for Sustainable Chemistry, School of Chemistry, University of Nottingham, Nottingham, NG7 2TU, UK. E-mail: graham.newton@nottingham.ac.uk
cSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK
dDepartment of Chemistry, School of Science, Tokyo Institute of Technology, O-okayama, meguro-ku, Tokyo, 152-8550, Japan
eDepartment of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, Hiroshima 739 8526, Japan
First published on 9th August 2023
Abstract
A multi-component coordination compound, in which ruthenium antenna complexes are connected to a polyoxotungstate core is presented. This hybrid cluster effectively promotes the electrochemical conversion of CO2 to C1 feedstocks, the selectivity of which can be controlled by the acidity of the media.
Global energy demands continue to rise and the need for clean energy and effective methods of CO2 management has never been greater.1 In this regard, the sustainable photo- or electro-chemical reduction of CO2 to yield C1 feedstocks and fuels is a key target.2 This reduction can proceed via a proton-assisted multiple electron transfer pathway which can yield carbon monoxide (a 2e− reduction process), formate (2e−), formaldehyde (4e−), methanol (6e−) or methane (8e−).3 Despite the range of accessible products, the effective and selective reduction of CO2 remains an ongoing challenge due to the thermodynamic stability of CO2 and the substantial activation energy barrier to access the one electron-reduced radical anion CO2˙−.4
Transition metal complexes show promise as homogeneous electrocatalysts for the reductive conversion of CO2.5–7 The efficiency of transition metal based electrocatalysts is largely dependent how readily the catalyst is reduced, as well as the binding of the CO2 to the metal centre. Ruthenium complexes are a leading class of electrocatalysts in the field, with principle works by Tanaka, Ott, and Meyer, amongst others, illustrating the ways in which Ru centres can favourably interact with CO2.8–11 Furthermore, these reports demonstrate how the bidentate polypyridine ligand architecture (such as bpy = 2,2′-bipyridine and tpy = 2,2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6′,2′′-terpyridine) plays a significant role in the electrocatalytic reduction of CO2, with the Ru centre mediating electron transfer from bpy˙− and tpy˙− radical anions to the Ru-coordinated CO2 molecules.
:6′,2′′-terpyridine) plays a significant role in the electrocatalytic reduction of CO2, with the Ru centre mediating electron transfer from bpy˙− and tpy˙− radical anions to the Ru-coordinated CO2 molecules.
Polyoxometalates (POMs) on the other hand, are polyanionic metal oxide clusters with applications in energy storage,12–14 photocatalysis,15–17 and topically, electroreduction of CO2.18,19 POMs are structurally diverse, exhibit rich redox and photochemical properties, and can act as both electron and proton reservoirs; a desirable trait in the design of highly active or selective electrocatalysts.20–22 These versatile materials can be further modified to include organic moieties, permitting the design and synthesis of organic–inorganic hybrid systems.23–25 By tailoring the nature of the organic moiety in particular, the resulting hybrid species can demonstrate synergistic properties which can be finely tuned.26–30
Here, we show how targeted design of a hybrid POM cluster containing Ru-polypyridyl moieties as catalyst units can be employed to enhance the activity and dramatically alter the selectivity of the electroreduction of CO2. The new hybrid compound: (nBu4N)3H[(Ru(dmbpy)(ppt)Cl)2P2W17O57] (1) (nBu4N = tetrabutylammonium, dmbpy = 4,4′-dimethyl-2,2′-dipyridyl, ppt = 4′-(4-phosphonophenyl)-2,2′:6′,2′′-terpyridine), is shown to be an active species for both CO/H2 and formic acid (HCOOH) production, where the product distribution can be switched from CO/H2 to HCOOH by the addition of a weak proton donor due to the pre-association of protons to the reduced POM core.
The organic–inorganic hybrid bis-Ru POM complex (nBu4N)3H[(Ru(dmbpy)(ppt)Cl)2P2W17O57] (1) (Fig. 1) was prepared by condensation of the phosphonic acid derivatised mononuclear Ru2+ complex, [Ru(dmbpy)(ppt-H2)Cl]Cl,31,32 with the monolacunary Wells–Dawson polyanion, K10[P2W17O61], in N,N-dimethylacetamide (DMA) with nBu4NBr and HCl at 60 °C over 24 h. Hybrid POM 1 was isolated as a purple powder in good yield (85%), and characterised by elemental analysis, NMR, and ultraviolet-visible spectroscopies (see ESI†).
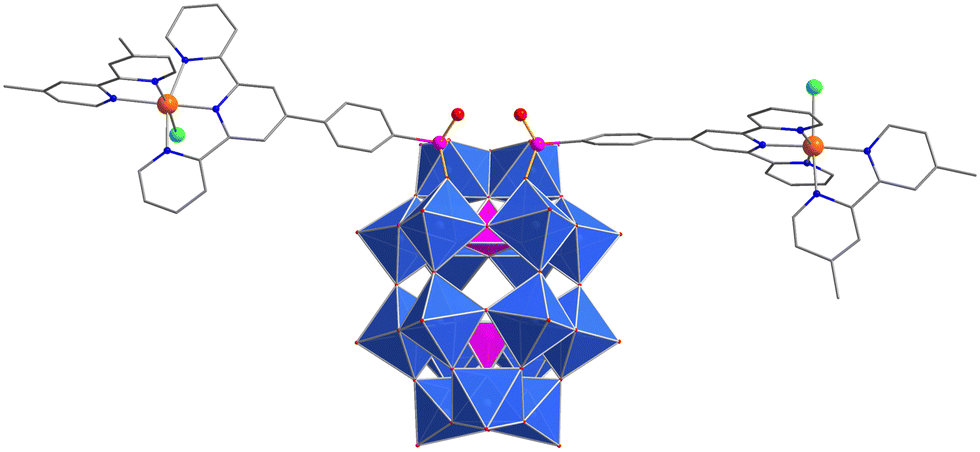 | ||
| Fig. 1 Single crystal X-ray structure of the hybrid-POM complex. Blue polyhedra = (WO6), pink polyhedra = (PO4), red spheres = O, pink spheres = P, grey spheres = C, green spheres = Cl, orange spheres = ruthenium. H-atoms, cations and solvent molecules are omitted. | ||
Crystals suitable for single crystal X-ray diffraction studies were prepared from a solution of 1 and tetramethylammonium tetrafluoroborate in N-methyl-2-pyrrolidone (NMP). Structural analysis showed that the phosphonate groups of two ppt ligands were embedded within the vacant site on the POM, while ruthenium ions occupied the tridentate terpyridine (tpy) binding sites. The distorted octahedral coordination environment of each Ru ion was completed by bidentate dmbpy ligands and a chloride ion, suggesting that the [Ru(dmbpy)(ppt)Cl]+ units had been structurally unaffected by the hybridization process.
Cyclic voltammetry (CV) measurements were conducted on 1 in a 1 mM NMP solution with nBu4NPF6 as the supporting electrolyte under N2. The CV showed seven quasi reversible redox processes corresponding to four one-electron POM-centred WVI → WV reductions (at Epc = −0.12, −0.54, −1.02 and −1.45 V vs. NHE); one-electron ppt/ppt˙− and dmbpy/dmbpy˙− processes on the antenna moieties at Epc = −1.23 V and −1.62 V, respectively; and a Ru2+/Ru3+ process at Epa = +1.06 V vs. NHE (Fig. 2 and see ESI† Fig. S4). The redox processes were further probed by controlled-potential UV-Vis-NIR absorption measurements under the same conditions (see ESI,† Fig. S6). The initial absorption spectrum showed overlaid absorption bands attributable to π–π* transitions on the organic moiety and O → W ligand-to-metal charge transfer (LMCT) bands in the UV/near-UV region and a metal-to-ligand charge transfer (MLCT) band at 528 nm originating from the Ru2+ ions. Scanning to negative potentials from 0.24 to −1.0 V vs. NHE induced the growth of WV → WVI intervalence charge transfer (IVCT) absorption bands above 600 nm. Beyond −1.0 V vs. NHE, the Ru2+ MLCT band decreased in intensity and appeared to shift to lower energy, due to the reduction of the ppt moieties to ppt˙−.
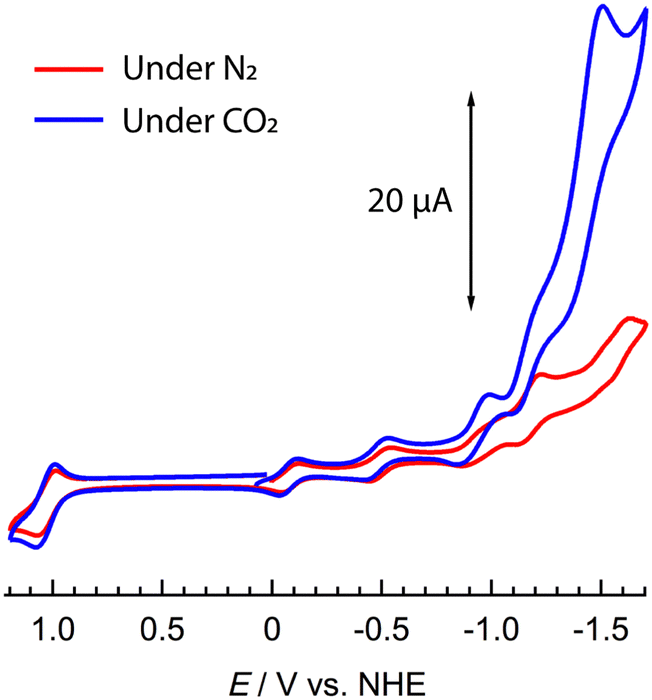 | ||
| Fig. 2 Cyclic voltammetry of 1 (1 mM) under N2 (red) or CO2 (Blue) in NMP, with 0.1 M nBu4NPF6 supporting electrolyte vs. NHE redox couple. Glassy carbon working electrode (d = 3 mm), Pt wire counter electrode, and a SCE reference (corrected to NHE, see ESI†). | ||
Increase of the negative potential caused the MLCT band to disappear as the dmbpy ligands were similarly reduced to radical anions, at which point the MLCT band was replaced with an intense absorption band attributable to π–π* transitions in the reduced ligands.33 Applying an oxidizing potential of +0.24 V led the absorption profile to return to its original state, indicating the good redox reversibility and stability of 1.
While there are few reports of POMs interacting with or binding CO2,34,35 in recent years, POMs have been applied towards photo- and electro-catalytic reduction of CO2 producing a variety of products.18,19,36–42 These POM catalysts are typically heterometallic clusters or electrostatically coordinated organometallic hybrid materials bearing transition metal active sites, with the POM often acting as an electron or proton reservoir. In 1, the antenna [Ru(dmbpy)(ppt)Cl]+ moieties are similar to the [(Ru(bpy)(tpy)Cl)]+ complex that was shown by Meyer et al. to catalyse the conversion of CO2 to CO and small amounts of CO2− and CO32− at −1.52 V vs. NHE.9,43
CV data collected under CO2 showed similar behaviour to those collected under N2 with three quasi-reversible waves indicating the stepwise reduction of the POM moiety from WVI17 to WVI14WV3. As the negative potential was increased beyond −1.00 V vs. NHE, however, the current rapidly increased with an onset value of ca. −1.08 V and a maximum at −1.51 V vs. NHE. The profile of the return scan echoed that collected under N2.
Additional controlled potential absorption measurements were conducted under CO2 between 0.24 and −1.76 V vs. NHE, to mirror the data obtained under N2 (see ESI,† Fig. S7). The absorption spectra initially behaved similarly, with the growth of IVCT (WV → WVI) bands above 600 nm as the potential was lowered to −1.0 V. In contrast to the spectra collected under N2, scanning the potential to −1.76 V vs. NHE caused no shift or reduction in intensity of the Ru2+ MLCT band at 528 nm and no apparent organic ligand reduction. This confirms that the organic anion radicals react rapidly in the presence of CO2, serving as the electron donors. Furthermore, multiple scans indicated that the Cl− ligand was lost over time (see ESI,† Fig. S8), where the peak associated with ppt ligand reduction shifts to a more positive potential in line with the literature.9
Controlled potential bulk electrolysis of 1 was employed in conjunction with gas chromatography (GC) and capillary electrophoresis to determine the gas phase products of the electrocatalytic CO2 reduction with 1 by headspace analysis in a sealed electrochemical cell. Under saturated CO2 conditions, controlled potential bulk electrolysis was performed at −1.56 V vs. NHE on a 0.03 mM solution of 1 in 0.1 M nBu4NPF6 MeCN at a carbon paper working electrode over a period of 1 h (Table 1). A mixture of hydrogen and carbon monoxide was formed with turnover numbers (TON) of 38 and 1.4 within 1 h, respectively (Table 1, entry 1), corresponding to a total faradaic efficiency (F.E.) of 94% for the evolution of H2 and CO. Hybrid POM connectivity was retained during electrolysis, as confirmed by 1H NMR spectroscopy and electrochemical analyses on the post-electrolysis solution (see ESI†, Fig. S9–S12). This is important as the phosphonate bridging unit in organic–inorganic hybrid POMs is known to be sensitive to hydrolytic cleavage.26,29,44
| Entry | Catalyst (mM) | Additive (equiv.) | TON-H2 | TON-CO | TON-HCOOH |
|---|---|---|---|---|---|
| a Measurements in MeCN at −1.56 V vs. NHE over 1 h. 0.1 M nBu4NPF6 supporting electrolyte, carbon paper working electrode, Pt wire counter electrode. GC and capillary electrophoresis analysis of headspace were used to determine products. b [Ru] (0.06 mM) and K6P2W18O62 (0.03 mM); 1 = (nBu4N)3H[(Ru(dmbpy)(ppt)Cl)2P2W17O57], [Ru] = [Ru(dmbpy)(ppt-Et2)Cl]Cl. | |||||
| 1 | 1 (0.03) | — | 38 | 1.4 | 0 |
| 2 | 1 (0.03) | H2O (10) | 33 | 2.4 | 4 |
| 3 | 1 (0.03) | AcOH (10) | <1 | <1 | 22 |
| 4 | [Ru] (0.06) | — | <1 | <1 | 0 |
| 5 | [Ru] (0.06) | AcOH (10) | 22 | <1 | 2 |
| 6b | [Ru]:K6P2W18O62 | AcOH (10) | <1 | <1 | <1 |
The electrolysis performance of hybrid POM 1 is distinct from that of the analogous Ru catalyst previously reported by Meyer et al., where comparable TONs for CO evolution are observed but no H2 is formed.9 For comparison, a control experiment using 2 equivalents (0.06 mM) of the diethylphosphonate [Ru(dmbpy)(ppt-Et2)Cl]Cl complex ([Ru]) shows slow evolution of H2 and CO over 1 h with TON less than 1 under the same conditions (Table 1, entry 4).
Polyoxoanion cluster hybridisation profoundly alters the overall reactivity, possibly due to the anchoring of the catalyst within the specific environment provided by the large POM anion outer coordination sphere. The affinity of the POM core for polar hydrogen-bonding substrates, and its Brønsted acidity, allows it to act as both a source of and a reservoir for protons (note that compound 1 itself is isolated as a semi-protonated salt). This may help to partly explain the high activity of 1 for the generation of H2 over CO. The proton source of the produced H2 is likely water impurities, and the introduction of CO2 should increase acidity of the solution. This is corroborated by the addition of a water to the electrolysis mixture, yielding similar number of turnovers for H2 and CO (Table 1, entry 2). Interestingly, modest levels of formic acid production can also be detected by capillary electrophoresis (TON-HCOOH (1 h) = 4), giving a total F.E. of 96%, suggesting that product selectivity may be tuneable by addition of different proton sources.
Addition of a proton source, or changes in local pH, or electrolyte composition are known to affect the efficacy and selectivity of CO2 reduction catalysts.45–51 Accordingly, we assessed how addition of a dedicated proton source influenced the electrocatalytic properties of 1. Cyclic voltammetry of 1 in the presence acetic acid under both N2 and CO2 atmospheres show that the compound remains stable up to the addition of at least 500 equivalents of AcOH (see ESI,† Fig. S13 and S14), where positive shifts in the half-wave potentials of the third and fourth POM reductions are observed on increasing concentrations of AcOH. Indeed, even upon addition of 1 equivalent, there is a substantial positive shift in the third POM reduction process, indicating that the acidic proton is associated with the anionic POM core. Under N2, addition of acid leads ultimately to the emergence of a sharp catalytic wave at −1.1 V vs. NHE corresponding to H2 evolution. Under CO2, the peak current of the catalytic wave at −1.52 V vs. NHE associated with CO2 reduction increases upon addition of AcOH before saturation after addition of 10 equivalents.
Following these observations, bulk electrolysis was repeated with 10 equivalents of AcOH added to CO2 saturated MeCN solutions of 1 (Table 1, entry 3), [Ru] (Table 1, entry 5), and a 2:1 mixture of [Ru] with [P2W18O62]6− (Table 1, entry 6), thereby modelling the ratio of components in the hybrid complex 1. Remarkably, the addition of AcOH was found to drastically alter the selectivity of 1 towards the reduction products. The evolution of H2 was almost completely halted upon addition of the weak acid (TON-H2 (1 h) = <1). Furthermore, CO evolution was similarly suppressed in favour of the production of formic acid as the major product (TON-HCOOH (1 h) = 22, F.E. = 67%). Comparatively, this unique selectivity is lost in the absence of the polyoxoanion component or division of the hybrid components, as [Ru] and the [Ru]: [P2W18O62]6− mixture favoured H2 evolution with minor formic acid and CO formation. To the best of our knowledge, such acid-mediated switching of the selectivity of these 2e− products in electrocatalytic CO2 reduction is unique to the hybrid POM complex 1, highlighting the synergetic properties of covalently hybridised organic–inorganic hybrid POMs.43
Typically, other Ru catalysts (such as Ru(bpy)(CO)2Cl2 and Ru(bpy)2(CO)22+) convert CO2 to formate under basic conditions, while acidic conditions favour isolation of H2 and CO.45,46,49–51 The impact of AcOH addition indicates that as the POM is reduced, protons are associated with the cluster, creating a highly protic local environment. These then interact with CO2 molecules undergoing reduction at the Ru sites and can facilitate proton-coupled electron-transfer (PCET), leading to HCOOH isolation. In the absence of acid, PCET is disfavoured, and solvent or electrolyte decomposition reactions can occur.
There is clearly a unique cooperative effect at play in the catalytic behaviour of 1, which cannot be replicated in the absence of direct hybridisation of the catalytic and polyoxoanion components into a single active complex. The protic local environment of the reduced hybrid system is likely a key factor in the enhancement of both the selectivity and efficiency of the electroreduction of CO2.
We have reported the synthesis of a bis-Ru-polypyridyl substituted hybrid polyoxometalate and investigated its solution-state electrochemical properties. The compound facilitates the selective electrochemical conversion of CO2 to H2/CO or to formic acid in the presence of a proton donor. The novelty of this approach suggests that the electron and proton buffering nature of hybrid POM systems may lead to the reversal of the expected trends in CO2 reduction. Crucially, this illustrates how multiple functional components can be combined to create a new class of hybrid material with unique electrocatalytic properties.
This work was supported by JSPS KAKENHI Grant Number JP16H06523 (Coordination Asymmetry), the EPSRC through grant EP/S031170/1, the University of Nottingham's Propulsion Futures Beacon of Excellence, a Grant-in-Aid for JSPS Research Fellow Grant Number J02555 (S. K.) and a JSPS Postdoctoral Fellowship for Foreign Researchers (J. M. C.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- IPCC, International panel on climate change 6th Assessment Report, http://www.ipcc.ch/.
- S. C. Peter, ACS Energy Lett., 2018, 3, 1557–1561 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- E. Fujita, Coord. Chem. Rev., 1999, 185–186, 373–384 CrossRef CAS.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- B. Das, A. Thapper, S. Ott and S. B. Colbran, Sustainable Energy Fuels, 2019, 3, 2159–2175 RSC.
- K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef CAS.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC.
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 CrossRef CAS PubMed.
- T. A. White, S. Maji and S. Ott, Dalton Trans., 2014, 43, 15028–15037 RSC.
- J.-J. Chen, L. Vilà-Nadal, A. Solé-Daura, G. Chisholm, T. Minato, C. Busche, T. Zhao, B. Kandasamy, A. Y. Ganin, R. M. Smith, I. Colliard, J. J. Carbó, J. M. Poblet, M. Nyman and L. Cronin, J. Am. Chem. Soc., 2022, 144, 8951–8960 CrossRef CAS PubMed.
- Y. Zhang, J. Liu, S.-L. Li, Z.-M. Su and Y.-Q. Lan, EnergyChem, 2019, 1, 100021 CrossRef.
- C. L. Peake, A. J. Kibler, G. N. Newton and D. A. Walsh, ACS Appl. Energy Mater., 2021, 4, 8765–8773 CrossRef CAS.
- N. Li, J. Liu, B.-X. Dong and Y.-Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 20779–20793 CrossRef CAS PubMed.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- S. Amthor, S. Knoll, M. Heiland, L. Zedler, C. Li, D. Nauroozi, W. Tobaschus, A. K. Mengele, M. Anjass, U. S. Schubert, B. Dietzek-Ivanšić, S. Rau and C. Streb, Nat. Chem., 2022, 14, 321–327 CrossRef CAS PubMed.
- Z. Lang, J. Miao, Y. Lan, J. Cheng, X. Xu and C. Cheng, APL Mater., 2020, 8, 120702 CrossRef CAS.
- Y. Cao, Q. Chen, C. Shen and L. He, Molecules, 2019, 24, 2069 CrossRef CAS PubMed.
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- J.-H. Kruse, M. Langer, I. Romanenko, I. Trentin, D. Hernández-Castillo, L. González, F. H. Schacher and C. Streb, Adv. Funct. Mater., 2022, 32, 2208428 CrossRef CAS.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- A. J. Kibler and G. N. Newton, Polyhedron, 2018, 154, 1–20 CrossRef CAS.
- G. Izzet, F. Volatron and A. Proust, Chem. Rec., 2017, 17, 250–266 CrossRef CAS PubMed.
- S. S. Amin, K. D. Jones, A. J. Kibler, H. A. Damian, J. M. Cameron, K. S. Butler, S. P. Argent, M. Winslow, D. Robinson, N. J. Mitchell, H. W. Lam and G. N. Newton, Angew. Chem., Int. Ed., 2023, 62, e202302446 CrossRef CAS PubMed.
- A. J. Kibler, N. Tsang, M. Winslow, S. P. Argent, H. W. Lam, D. Robinson and G. N. Newton, Inorg. Chem., 2023, 62, 3585–3591 CrossRef CAS PubMed.
- J. M. Cameron, S. Fujimoto, K. Kastner, R.-J. Wei, D. Robinson, V. Sans, G. N. Newton and H. H. Oshio, Chem. – Eur. J., 2017, 23, 47–50 CrossRef CAS PubMed.
- S. Fujimoto, J. M. Cameron, R.-J. Wei, K. Kastner, D. Robinson, V. Sans, G. N. Newton and H. Oshio, Inorg. Chem., 2017, 56, 12169–12177 CrossRef CAS PubMed.
- S. Amin, J. M. Cameron, J. A. Watts, D. A. Walsh, V. Sans and G. N. Newton, Mol. Syst. Des. Eng., 2019, 4, 995–999 RSC.
- V. Spampinato, N. Tuccitto, S. Quici, V. Calabrese, G. Marletta, A. Torrisi and A. Licciardello, Langmuir, 2010, 26, 8400–8406 CrossRef CAS PubMed.
- B. Jing, H. Zhang, M. Zhang, Z. Lu and T. Shen, J. Mater. Chem., 1998, 8, 2055–2060 RSC.
- V. Kalyanaraman, C. N. R. Rao and M. V. George, J. Chem. Soc. B, 1971, 2406–2409, 10.1039/J29710002406.
- G. Gao, F. Li, L. Xu, X. Liu and Y. Yang, J. Am. Chem. Soc., 2008, 130, 10838–10839 CrossRef CAS PubMed.
- B. Chen and R. Neumann, Eur. J. Inorg. Chem., 2018, 791–794 CrossRef CAS.
- M. Girardi, D. Platzer, S. Griveau, F. Bedioui, S. Alves, A. Proust and S. Blanchard, Eur. J. Inorg. Chem., 2018, 387–393 Search PubMed.
- D. Azaiza-Dabbah, C. Vogt, F. Wang, A. Masip-Sánchez, C. de Graaf, J. M. Poblet, E. Haviv and R. Neumann, Angew. Chem., Int. Ed., 2022, 61, e202112915 CrossRef CAS PubMed.
- M. Girardi, S. Blanchard, S. Griveau, P. Simon, M. Fontecave, F. Bedioui and A. Proust, Eur. J. Inorg. Chem., 2015, 3642–3648 CrossRef CAS.
- J. Du, Z.-L. Lang, Y.-Y. Ma, H.-Q. Tan, B.-L. Liu, Y.-H. Wang, Z.-H. Kang and Y.-G. Li, Chem. Sci., 2020, 11, 3007–3015 RSC.
- S.-X. Guo, F. Li, L. Chen, D. R. MacFarlane and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 12690–12697 CrossRef CAS PubMed.
- H. Yu, E. Haviv and R. Neumann, Angew. Chem., Int. Ed., 2020, 59, 6219–6223 CrossRef CAS PubMed.
- J. Gu, W. Chen, G. G. Shan, G. Li, C. Sun, X. L. Wang and Z. Su, Mater. Today Energy, 2021, 21, 100760 CrossRef CAS.
- See Table S2 (ESI†) for comparison of Ru-polypyridyl electrocatalysts for CO2 reduction. For a comprehensive review, please see: H. A. Younus, N. Ahmad, W. Ni, X. Wang, M. Al-Abri, Y. Zhang, F. Verpoort and S. Zhang, Coord. Chem. Rev., 2023, 493, 215318 CrossRef CAS.
- G. S. Kim, K. S. Hagen and C. L. Hill, Inorg. Chem., 1992, 31, 5316–5324 CrossRef CAS.
- H. Ishida, H. Tanaka, K. Tanaka and T. Tanaka, J. Chem. Soc., Chem. Commun., 1987, 131–132 RSC.
- Y. Tamaki and O. Ishitani, Faraday Discuss., 2017, 198, 319–335 RSC.
- M. Tomisaki, S. Kasahara, K. Natsui, N. Ikemiya and Y. Einaga, J. Am. Chem. Soc., 2019, 141, 7414–7420 CrossRef CAS PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS PubMed.
- A. Taheri, E. J. Thompson, J. C. Fettinger and L. A. Berben, ACS Catal., 2015, 5, 7140–7151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2283230. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02138f |
| This journal is © The Royal Society of Chemistry 2023 |