
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulated self-assembly of hcp topology MOFs of Zr/Hf and the extended 4,4′-(ethyne-1,2-diyl)dibenzoate linker†
Sophia S.
Boyadjieva
a,
Francesca C. N.
Firth
 b,
Mohammad R.
Alizadeh Kiapi
c,
David
Fairen-Jimenez
c,
Sanliang
Ling
d,
Matthew J.
Cliffe
*e and
Ross S.
Forgan
*a
b,
Mohammad R.
Alizadeh Kiapi
c,
David
Fairen-Jimenez
c,
Sanliang
Ling
d,
Matthew J.
Cliffe
*e and
Ross S.
Forgan
*a
aWestCHEM School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow G12 8QQ, UK. E-mail: ross.forgan@glasgow.ac.uk
bYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
cThe Adsorption & Advanced Materials Laboratory (A2ML), Department of Chemical Engineering & Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK
dAdvanced Materials Research Group, Faculty of Engineering, University of Nottingham, University Park, Nottingham NG7 2RD, UK
eSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: matthew.cliffe@nottingham.ac.uk
First published on 7th March 2023
Abstract
Careful control of synthetic conditions can enhance the structural diversity of metal–organic frameworks (MOFs) within individual metal-linker combinations. Herein, we show that hcp topology MOFs of both Zr(IV) and Hf(IV), linked by the extended (ethyne-1,2-diyl)dibenzoate linker, can be prepared by modulated self-assembly. The controlled addition of acetic acid and water to solvothermal syntheses is essential to generate these phase pure hcp topology materials, which are characterised experimentally and computationally. The central alkyne unit of the linker can be quantitatively brominated, but this results in partial degradation of the hcp phase, in contrast to the more stable fcu topology analogues. Nevertheless, the MOFs represent new members of the hcp topology isoreticular series showing high crystallinity and porosity, and demonstrate that new materials can be discovered in existing MOF phase spaces through judicious adjustment of key synthetic parameters.
Introduction
Metal–organic frameworks (MOFs) comprise metal ion or metal cluster nodes (also referred to as secondary building units, or SBUs) connected by multitopic organic ligands into multidimensional network structures.1 To date, it is estimated that over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOF structures have been deposited in the Cambridge Structural Database,2 with this huge number attributed to the chemical diversity in the choice of both metal SBU and organic ligand.3 In addition, it is possible to isolate multiple phases from the same metal–ligand combination—topological diversity—by careful control of reaction parameters.4 A pertinent example is that of trivalent metals linked by linear ditopic dicarboxylates, which can yield structurally rigid (MIL-101) and flexible (MIL-88 and MIL-53) phases.5 Furthermore, we have also previously shown that, in the specific case of Fe(III) MOFs with 1,4-benzenedicarboxylate (BDC2−), coordination modulation—the addition of reagents to MOF syntheses that can tune the coordination and pH equilibria during self-assembly6—is a viable strategy for kinetically selecting a specific material from a complex phase landscape.7
000 MOF structures have been deposited in the Cambridge Structural Database,2 with this huge number attributed to the chemical diversity in the choice of both metal SBU and organic ligand.3 In addition, it is possible to isolate multiple phases from the same metal–ligand combination—topological diversity—by careful control of reaction parameters.4 A pertinent example is that of trivalent metals linked by linear ditopic dicarboxylates, which can yield structurally rigid (MIL-101) and flexible (MIL-88 and MIL-53) phases.5 Furthermore, we have also previously shown that, in the specific case of Fe(III) MOFs with 1,4-benzenedicarboxylate (BDC2−), coordination modulation—the addition of reagents to MOF syntheses that can tune the coordination and pH equilibria during self-assembly6—is a viable strategy for kinetically selecting a specific material from a complex phase landscape.7
The multiple series of MOFs comprising Zr(IV) or Hf(IV) SBUs connected by linear ditopic dicarboxylate ligands present a particularly striking case study into the isolation of different MOF phases via synthetic control.8 Predominant among MOFs prepared from Zr(IV) or Hf(IV) and BDC2− or its derivatives is the UiO-66 series, in which [M6(μ3-O)4(μ3-OH)4(RCO2)12] metal clusters (M = Zr, Hf, Fig. 1a) are connected by twelve linear dicarboxylate linkers in a face-centred cubic (fcu) topology MOF (Fig. 1b). UiO-66(Zr) specifically exhibits the ideal formula [Zr6O4(OH)4(BDC)6],9 and a wide range of isoreticular derivatives of these materials, including interpenetrated UiO-66 topology phases, have been reported with both longer and functionalised linkers.9–15
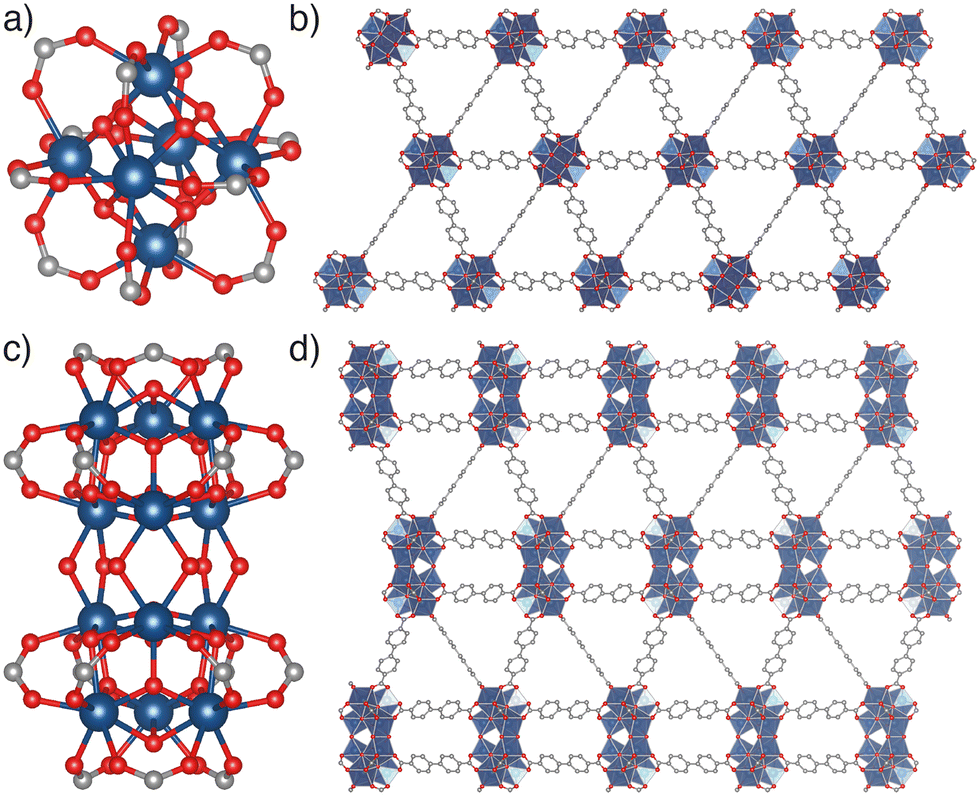 | ||
| Fig. 1 Comparison of the fcu and hcp topology MOFs adopted by tetravalent metals and linear dicarboxylate linkers, specifically focussing on those of Hf(IV) and biphenyl-4,4′-dicarboxylate (BPDC). a) The [Hf6(μ3-O)4(μ3-OH)4(RCO2)12] SBU. b) A section of the packing structure of fcu topology [Hf6O4(OH)4(BPDC)6], also known as UiO-67(Hf). c) The [Hf12(μ3-O)8(μ3-OH)8(μ2-OH)6(RCO2)18] SBU. d) A section of the packing structure of hcp topology [Hf12O8(OH)14(BPDC)9]. Hf: blue spheres (or, demonstrating Hf coordination environments in SBUs, polyhedra); C: grey; O: red. H atoms omitted for clarity. | ||
Varying the reaction composition can also affect the resultant MOF structure. For example, changing the metal source from ZrCl4 to Zr(iOPr)4 has led to the discovery of a polymorph of UiO-66 with a different connectivity and hex topology.16 Addition of monotopic carboxylic acids as coordination modulators has induced formation of “defect” phases where the modulators themselves are incorporated as charge-compensating defects. Such defect phases can form in MOFs with the linker 2,6-naphthalenedicarboxylate, where the connectivity of the hexanuclear SBU is reduced by the presence of charge-capping, monotopic acetate units.17 Modulated synthesis can even lower the connectivity of the structure in an ordered manner, as seen in the formation of reo UiO-66, which can be described as a missing cluster defect phase, where modulator incorporation (for example formic acid) leads to eight-connected SBUs that form well-defined nanodomains of the missing-cluster defect phase reo within the bulk of fcu UiO-66.18,19 Increasing reaction temperatures leads to the MIL-140 series of MOFs phase, which have an infinite one-dimensional chain SBU.20 MIL-140A, linked by BDC2−, is representative of the isoreticular series, having overall formula [ZrO(BDC)]. It is suggested that the presence of a condensed 1-D chain SBU and its isolation at higher reaction temperatures means that MIL-140A is the thermodynamic phase and UiO-66 is a kinetic product,20 although addition of water to syntheses promotes UiO-66 formation.21
Further careful synthetic control22 enables the synthesis of MOFs with higher nuclearity Zr(IV) or Hf(IV) SBUs linked by ditopic dicarboxylates.23–27 For example, MOFs containing the condensed [M12(μ3-O)8(μ3-OH)8(μ2-OH)6(RCO2)18] SBU (Fig. 1c) and adopting the hcp topology (Fig. 1d) have been reported with a range of linkers, including BDC2− and functionalised derivatives,28–33 1,4-naphthalenedicarboxylate,29 and longer linkers such as biphenyl-4,4′-dicarboxylate,23,30,34 1,1′:4′,1′′-terphenyl-4,4′′-dicarboxylate,35,36 4,4′-di(4-benzoato)-2,2′-bipyridine,37 and 5,15-di(p-benzoato)porphyrin.38 An example using poly(ethylene terephthalate) as the source of the BDC2− linker via direct synthesis has also been described.39 Individual systems have also been shown to undergo delamination by sonication, forming a hexagonal layered (hxl) phase and eventually hexagonal nanosheets (hns),23 which can also in specific cases be directly synthesised themselves.30
The hcp topology MOFs have been increasingly studied due to potential applications in luminescence,29 water remediation,31 heterogeneous catalysis,32,33,35 photocatalysis,37 nanomedicine,38 and molecular separations.39 Herein, we describe the synthesis and characterisation of Zr(IV) and Hf(IV) analogues of the hcp phase MOF linked by 4,4′-(ethyne-1,2-diyl)dibenzoate (EDB2−), which we have termed GUF-12 (GUF = Glasgow University Framework), by careful control of acetic acid and water content in solvothermal syntheses in DMF. We show the syntheses are scaleable, and that the MOFs have high porosity. Their metastability is demonstrated through efforts to postsynthetically modify them by bromination of their internal alkyne units, which, in the case of the Hf analogue, results in partial delamination or degradation.
Results and discussion
During attempts to prepare nanoparticulate versions of fcu topology Zr(IV) MOFs13,40 as part of our investigations into drug delivery,41 it was found that addition of high quantities of acetic acid and water to DMF-based solvothermal syntheses containing ZrCl4 and EDB-H2 resulted in formation of a new phase. This new MOF, termed GUF-12(Zr), was suspected to be the hcp analogue due to its characteristic hexagonal plate morphology and the nature of its powder X-ray diffractogram.The optimal conditions to isolate the hcp phases of both the Zr(IV) and Hf(IV) congeners of GUF-12 were identified via modulated solvothermal self-assembly. Specifically, 0.34 mmol each of MCl4 (M = Zr or Hf) and EDB-H2 (prepared according to a modified literature protocol13,42) were combined in 15 ml DMF, with addition of 75 μL deionised water (0.5% v/v) and 2.12 mL acetic acid (37.1 mmol, or 110 equiv. compared to the metal ion). The mixture was heated to 150 °C overnight in a PTFE-lined stainless steel autoclave, and the product isolated by centrifugation once cooled. Both the deliberate addition of water, and the use of a specific quantity of acetic acid as modulator, were key to ensure isolation of the hcp phases rather than the fcu analogue or other materials (see ESI,† Section S2). These observations tally with our previous mechanistic study that describes how the addition of large amounts of carboxylate modulator to syntheses is required to form the classical [M6O4(OH)4] clusters found in the fcu phases, while concomitant addition of water induces cluster merging to form the [M12O8(OH)14] units that comprise the hcp phases.22 Increasing water content in syntheses containing Hf(IV) and BPDC2− has previously led to the isolation of the hns phase over the hcp analogue,30 but with the EDB2− linker only hcp phase materials were formed when water content in syntheses was increased while retaining 110 equiv. of added acetic acid. In contrast, reducing the amount of acetic acid modulator in syntheses occasionally resulted in the failure to produce a MOF.
Even with careful optimisation of the synthesis conditions, single crystals large enough for characterisation by single crystal X-ray diffraction were unable to be grown for either GUF-12 analogue. SEM analysis showed formation of micron-scale hexagonal plates <100 nm in depth that are aggregated into a “desert rose” morphology, which is characteristic of MOF materials with this hcp topology (Fig. 2a and b).30 The structure of these materials was therefore determined by a combination of powder X-ray diffraction analysis and the use of model structures derived from density functional theory (DFT) calculations. Structural models of the hcp phase were constructed from the parent fcu phase, and then geometry-optimised by DFT calculations using the CP2K code (see ESI,† Section S3). These optimised structures confirmed the kinetic stability of the hcp topology models for the Zr(IV) and Hf(IV) analogues.
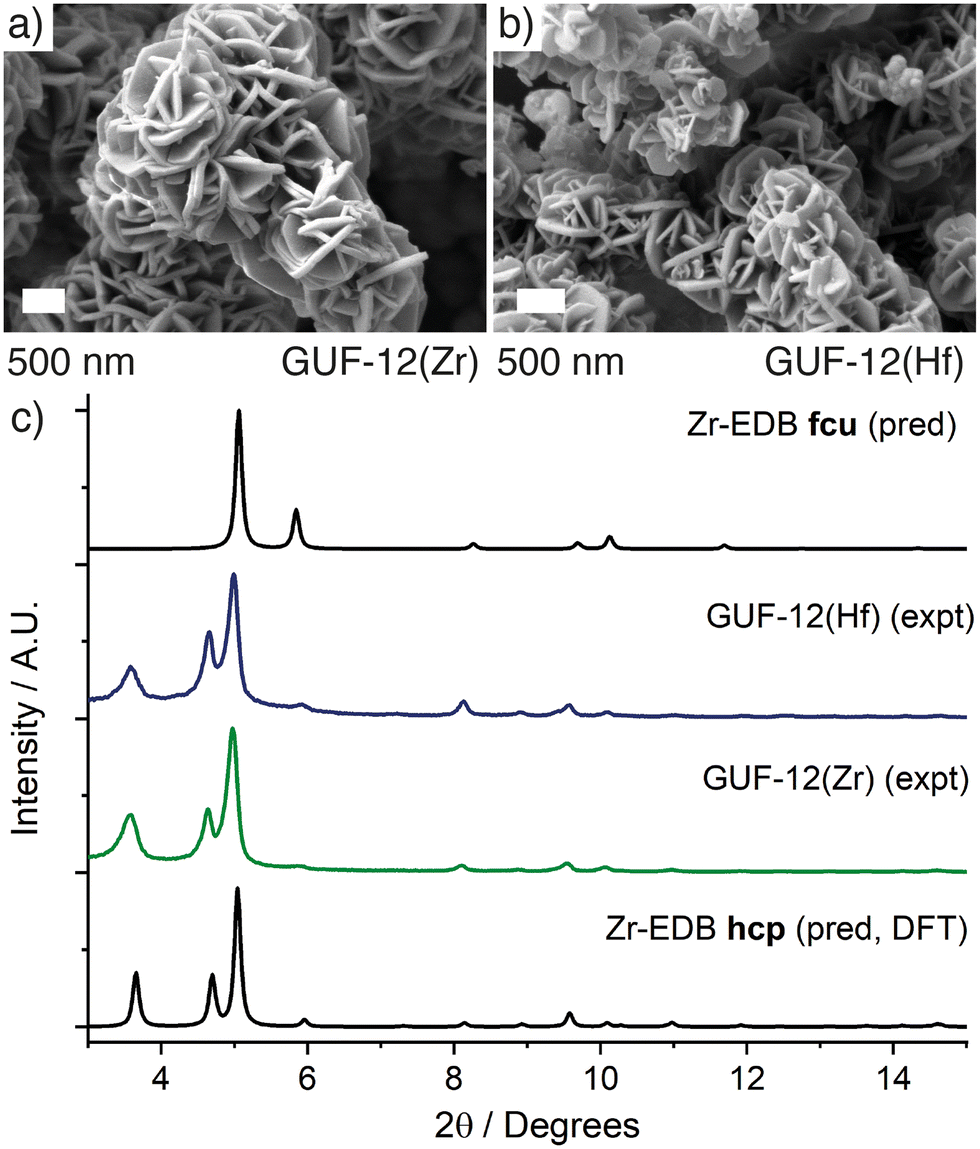 | ||
| Fig. 2 Scanning electron micrographs of the desert rose morphology of a) GUF-12(Zr) and b) GUF-12(Hf). c) Stacked powder X-ray diffractograms of GUF-12(Zr) and GUF-12(Hf) compared to patterns predicted for the hcp phase, derived from DFT model structures, and for the fcu phase, derived from the single crystal structures.13 | ||
Qualitative comparison of the powder X-ray diffractograms calculated from these DFT-derived hcp structures with those found experimentally (as well as those calculated from the crystal structures of the fcu phases) clearly indicate that GUF-12(Zr) and GUF-12(Hf) adopt the hcp topology (Fig. 2c). Quantitative Pawley fitting of the diffractograms (see ESI,† Section S4) in space group P63/mmc, using the Topas Academic 6.0 software package,43 confirmed that these materials were phase pure, and allowed accurate determination of the lattice parameters of these materials: GUF-12(Zr) a = 21.6121(31) Å, c = 48.014(12) Å; GUF-12(Hf) a = 21.5349(32) Å, c = 47.869(13) Å (Fig. 3). The DFT-optimised lattice parameters were within 0.6% of the experimental parameters. We also note that GUF-12(Hf) has a slightly smaller unit cell than GUF-12(Zr), due to slightly shorter Hf–O bonds.
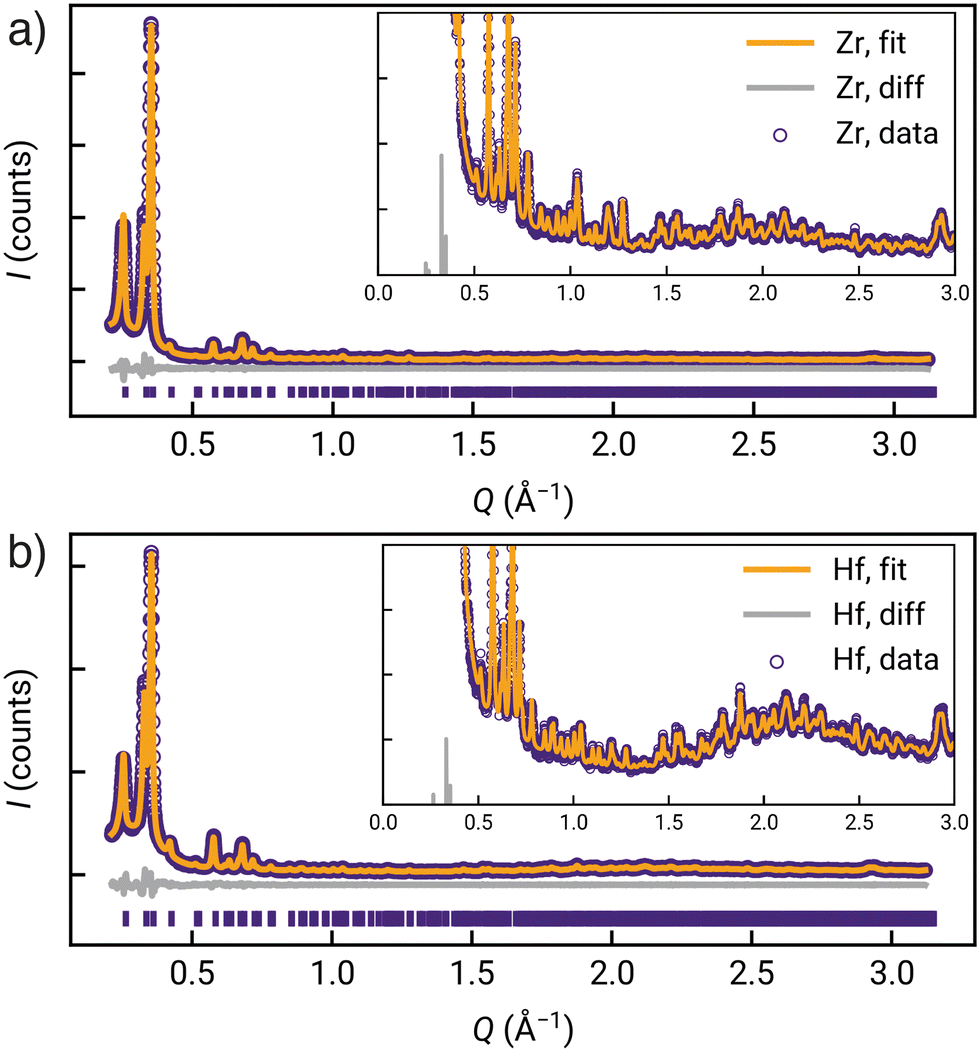 | ||
| Fig. 3 Quantitative Pawley fitting (with insets to show low intensity reflections) of the powder X-ray diffractograms of a) GUF-12(Zr) and b) GUF-12(Hf). | ||
Bulk composition was also assessed by thermogravimetric analysis (TGA) of the GUF-12 samples after activation at 120 °C for 20 h under turbomolecular pump vacuum to remove residual solvents (Fig. 4a). The metal oxide residue remaining after heating in air to 800 °C is indicative of metal content in the pristine MOF. For both GUF-12(Zr) and GUF-12(Hf), the residues were slightly higher than would be predicted for a pristine hcp phase; 42.1% wt for GUF-12(Zr) (theoretical: 40.8% wt) and 56.8% wt for GUF-12(Hf) (theoretical: 54.1% wt). These values suggest that the fcu phase is not significantly present in either case as the hcp phase has a higher overall metal weight percentage than the analogous fcu phase (theoretical residues of 34.9% wt and 47.8% wt would be expected for the pristine Zr(IV) and Hf(IV) fcu phases, respectively). Furthermore, these TGA results suggest that the hcp phases may exhibit some missing linker defects (replacement of EDB2− linkers by smaller charge-compensating acetates reduces the relative organic content in the MOF, increasing the metal weight percentage and subsequent TGA residue).41 This is commensurate with 1H NMR spectra of acid-digested samples (see ESI,† Section S5), which show the presence of acetate even after activation, indicating that the acetate groups are present as charge compensating defects rather than pore-bound solvents. Resonances that could be assigned to formate, produced by decomposition of DMF, were not observed in the materials.
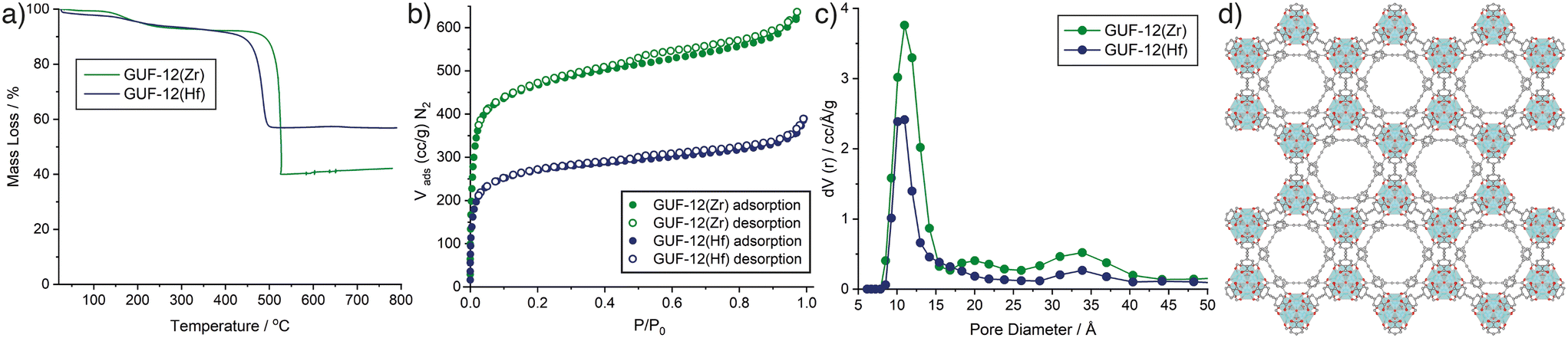 | ||
| Fig. 4 a) Thermogravimetric analyses of GUF-12(Zr) and GUF-12(Hf) in air. b) N2 adsorption desorption isotherms (77 K) of GUF-12(Zr) and GUF-12(Hf) with c) pore-size distributions (N2 at 77 K on carbon, slit pore, QSDFT, equilibrium model) calculated from the isotherms. d) Portion of the DFT model structure of GUF-12(Zr) viewed down the crystallographic c axis to visualise the hexagonal channel 11 Å in diameter. | ||
A pristine defect-free hcp structure would have formula [M12O8(OH)14(EDB)9]. 1H NMR spectroscopic analysis of activated, then acid-digested GUF-12(Zr) gives a 1:3.3 ratio of acetate to linker; assuming that one EDB2− is replaced by two acetates gives a formula of [Zr12O8(OH)14(EDB)7.8(CH3COO)2.4], which would leave a theoretical 42.5% wt ZrO2 residue after thermogravimetric analysis, matching well with the observed 42.1% wt. For GUF-12(Hf), the acetate content measured by 1H NMR spectroscopy is higher, at an approximate 1:2.5 acetate to EDB2− ratio. Similar levels of defectivity have been observed elsewhere for the hcp phase prepared from Zr(IV) and BDC2− using acetic acid modulated syntheses; these defects enhance its catalytic activity.33 Taking a similar approach, a formula of [Hf12O8(OH)14(EDB)7.5(CH3COO)3.0] for GUF-12(Hf) would correlate with the NMR spectroscopic data and leave a theoretical HfO2 residue of 56.3% wt, close to the experimental value of 56.8% wt. This greater acetate incorporation of the Hf(IV) congener may be reflected in its lower overall thermal decomposition temperature compared to the Zr(IV) analogue, as observed by TGA (Fig. 4a). The decomposition temperatures are broadly similar to the fcu analogues.13
The reliability of the synthetic method allowed us to upscale the synthesis of the hcp phases to quantities suitable for porosity analysis. N2 adsorption/desorption isotherms of GUF-12(Zr) and GUF-12(Hf) were collected at 77 K after activation of the samples at 120 °C for 20 h under vacuum (Fig. 4b). Both MOFs exhibit typical type I isotherms, associated with microporous materials, with small increases in uptake between 0.9 and 1.0 P/P0, indicating adsorption occurring in interparticle spacing or surface roughness. This can be explained by the aggregated “desert rose” morphology of the ∼100 nm thick hexagonal particles, which feature multiple surfaces and crevices where nitrogen can be adsorbed. Brunauer–Emmett–Teller (BET) areas were calculated from the experimental adsorption isotherms using BETSI, a publicly available software package that fully implements the extended Rouquerol criteria for an unambiguous BET area assignment (see ESI,† Section S6).44 The BET areas were found to be 1798 m2 g−1 and 1005 m2 g−1 for GUF-12(Zr) and GUF-12(Hf), respectively, with pore volumes of 1.03 cm3 g−1 and 0.59 cm3 g−1. As expected,30,35 these are lower than those reported for the fcu analogues (3280 m2 g−1 and 2000 m2 g−1, for the Zr and Hf congeners, respectively).13 Grand Canonical Monte Carlo (GCMC) simulations were performed to assess the potential total porosity of the two MOFs (see ESI,† Section S7). Simulated N2 adsorption isotherms for both GUF-12(Zr) and GUF-12(Hf) showed higher N2 uptakes and larger predicted BET areas (2765 m2 g−1 and 2127 m2 g−1, for the Zr and Hf congeners, respectively). GCMC simulations often overpredict porosity compared to experiment, but the magnitude of the difference between the experimental and simulated N2 uptakes suggest that the isolated MOFs may be partially amorphized or not fully activated.
In each isotherm, small type H4 hysteresis loops are discernible, which are indicative of minor levels of mesoporosity that could be a consequence of defectivity in the samples. Pore-size distributions (Fig. 4c), calculated from the experimental isotherms (N2 on carbon at 77 K, slit pore/QSDFD equilibrium) show a significant pore around 11 Å in diameter for each MOF, which correlates closely to the hexagonal pore evident along the crystallographic c axis (Fig. 4d), and is smaller than the major pore observed in the fcu analogues (∼12.5 Å).13 A broad feature is observed around 34 Å for each hcp MOF, which may be indicative of the defectivity implied by 1H NMR spectroscopy and TGA. This combination of high porosity and significant defectivity indicates the potential for application of the GUF-12 congeners in catalysis.33
We and others have previously shown that MOFs linked by ligands with internal alkene12,45–48 and alkyne12,13,49,50 subunits can be postsynthetically modified by halogenation. Specifically, we have demonstrated that the Zr(IV) and Hf(IV) fcu phases linked by EDB2− can be quantitatively brominated in a single-crystal to single-crystal manner.13 With two different linker environments in the hcp phases in this work, we wished to determine if (i) the linkers were accessible to bromination, and (ii) if this would result in a reactive delamination to form a nanosheet phase. Under conditions identical to those we have previously utilised for the fcu analogue,13 it was possible to quantitatively brominate the EDB2− ligands of the hcp materials, as assessed by 1H and 13C NMR spectroscopy of acid-digested samples. However, PXRD analysis of the brominated materials showed that stability was an issue when samples were scaled up, with inconsistent results. In the case of GUF-12(Hf), additional Bragg reflections were present that may represent degradation of the MOF or delamination, underlining the metastability of the hcp MOFs (see ESI,† Section S8).
Conclusions
We have shown that, by careful consideration of synthetic conditions, it is possible to synthesise Zr(IV) and Hf(IV) MOFs of EDB2− with the hcp topology to complement their established fcu analogues. The role of both water and acetic acid has been explored to optimise syntheses and reliably reproduce the materials on an increased scale, allowing characterisation of their porosity and possible defectivity, suggesting potential applications in heterogeneous catalysis. The hcp phases are likely kinetic products relative to the analogous fcu topology MOFs; moreover, the hcp materials are metastable, as demonstrated by partial degradation of GUF-12(Hf) during attempts to postsynthetically brominate the EDB2− linkers, in contrast to the stability of the fcu phase under identical conditions.13 Nevertheless, this work shows that careful control of modulated self-assembly allows access to new MOF materials exhibiting desirable physical properties within well-established phase spaces.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. F. thanks the Royal Society for receipt of a URF and the University of Glasgow for funding. The work has been supported by the European Research Council (ERC) under the European Union's Horizon 2020 Programme for Research and Innovation (R. S. F.: grant agreement no. 677289, SCoTMOF, ERC-2015-STG; D. F. J.: grant agreement no. 726380, NanoMOFdeli, ERC-2016-COG). M. J. C. thanks the School of Chemistry at the University of Nottingham for receipt of a Hobday Fellowship. M. R. A. K. acknowledges support from the Cambridge Trust Scholarship and the Trinity-Henry Barlow Scholarship. We acknowledge the use of the ARCHER2 supercomputer through membership of the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC grant no. EP/R029431. The data which underpin this submission are free to dowload at https://dx.doi.org/10.5525/gla.researchdata.1406.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- P. Z. Moghadam, A. Li, X.-W. Liu, R. Bueno-Perez, S.-D. Wang, S. B. Wiggin, P. A. Wood and D. Fairen-Jimenez, Chem. Sci., 2020, 11, 8373–8387 RSC.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- C. Ma, L. Zheng, G. Wang, J. Guo, L. Li, Q. He, Y. Chen and H. Zhang, Aggregate, 2022, 3, e145 CAS.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- D. Bara, E. Meekel, I. Pakamore, C. Wilson, S. Ling and R. S. Forgan, Mater. Horiz., 2021, 8, 3377–3386 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- A. Schaate, P. Roy, T. Preuße, S. J. Lohmeier, A. Godt and P. Behrens, Chem. – Eur. J., 2011, 17, 9320–9325 CrossRef CAS PubMed.
- J. Lippke, B. Brosent, T. von Zons, E. Virmani, S. Lilienthal, T. Preuße, M. Hülsmann, A. M. Schneider, S. Wuttke, P. Behrens and A. Godt, Inorg. Chem., 2017, 56, 748–761 CrossRef CAS PubMed.
- R. J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, Chem. – Eur. J., 2016, 22, 4870–4877 CrossRef CAS PubMed.
- R. J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, J. Am. Chem. Soc., 2015, 137, 9527–9530 CrossRef CAS PubMed.
- M. Schulz, N. Marquardt, M. Schäfer, D. P. Warwas, S. Zailskas and A. Schaate, Chem. – Eur. J., 2019, 25, 13598–13608 CrossRef CAS PubMed.
- X. Li, J. Xie, Z. Du, L. Jiang, G. Li, S. Ling and K. Zhu, Chem. Sci., 2022, 13, 6291–6296 RSC.
- M. Perfecto-Irigaray, G. Beobide, O. Castillo, I. da Silva, D. García-Lojo, A. Luque, A. Mendia and S. Pérez-Yáñez, Chem. Commun., 2019, 55, 5954–5957 RSC.
- V. Bon, I. Senkovska, M. S. Weiss and S. Kaskel, CrystEngComm, 2013, 15, 9572–9577 RSC.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef CAS PubMed.
- D. N. Johnstone, F. C. N. Firth, C. P. Grey, P. A. Midgley, M. J. Cliffe and S. M. Collins, J. Am. Chem. Soc., 2020, 142, 13081–13089 CrossRef CAS PubMed.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed.
- V. V. Butova, A. P. Budnyk, K. M. Charykov, K. S. Vetlitsyna-Novikova, C. Lamberti and A. V. Soldatov, Chem. Commun., 2019, 55, 901–904 RSC.
- F. C. N. Firth, M. W. Gaultois, Y. Wu, J. M. Stratford, D. S. Keeble, C. P. Grey and M. J. Cliffe, J. Am. Chem. Soc., 2021, 143, 19668–19683 CrossRef CAS PubMed.
- M. J. Cliffe, E. Castillo-Martínez, Y. Wu, J. Lee, A. C. Forse, F. C. N. Firth, P. Z. Moghadam, D. Fairen-Jimenez, M. W. Gaultois, J. A. Hill, O. V. Magdysyuk, B. Slater, A. L. Goodwin and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 5397–5404 CrossRef CAS PubMed.
- A. A. Bezrukov, K. W. Törnroos, E. Le Roux and P. D. C. Dietzel, Chem. Commun., 2018, 54, 2735–2738 RSC.
- S. Waitschat, H. Reinsch and N. Stock, Chem. Commun., 2016, 52, 12698–12701 RSC.
- S. Waitschat, H. Reinsch, M. Arpacioglu and N. Stock, CrystEngComm, 2018, 20, 5108–5111 RSC.
- S. Leubner, V. E. G. Bengtsson, K. Synnatschke, J. Gosch, A. Koch, H. Reinsch, H. Xu, C. Backes, X. Zou and N. Stock, J. Am. Chem. Soc., 2020, 142, 15995–16000 CrossRef CAS PubMed.
- M. Ermer, J. Mehler, M. Kriesten, Y. S. Avadhut, P. S. Schulz and M. Hartmann, Dalton Trans., 2018, 47, 14426–14430 RSC.
- L. Zhou, F. Liu, J. Wang, R. Chen and Y. Chen, CrystEngComm, 2021, 23, 2961–2967 RSC.
- F. C. N. Firth, M. J. Cliffe, D. Vulpe, M. Aragones-Anglada, P. Z. Moghadam, D. Fairen-Jimenez, B. Slater and C. P. Grey, J. Mater. Chem. A, 2019, 7, 7459–7469 RSC.
- B. Moll, T. Müller, C. Schlüsener, A. Schmitz, P. Brandt, S. Öztürk and C. Janiak, Mater. Adv., 2021, 2, 804–812 RSC.
- S. B. Peh, Y. Cheng, J. Zhang, Y. Wang, G. H. Chan, J. Wang and D. Zhao, Dalton Trans., 2019, 48, 7069–7073 RSC.
- X. Chen, Y. Lyu, Z. Wang, X. Qiao, B. C. Gates and D. Yang, ACS Catal., 2020, 10, 2906–2914 CrossRef CAS.
- R. Dai, F. Peng, P. Ji, K. Lu, C. Wang, J. Sun and W. Lin, Inorg. Chem., 2017, 56, 8128–8134 CrossRef CAS PubMed.
- P. Ji, K. Manna, Z. Lin, X. Feng, A. Urban, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 7004–7011 CrossRef CAS PubMed.
- M. R. Momeni and C. J. Cramer, Chem. Mater., 2018, 30, 4432–4439 CrossRef CAS.
- Y.-Y. Zhu, G. Lan, Y. Fan, S. S. Veroneau, Y. Song, D. Micheroni and W. Lin, Angew. Chem., Int. Ed., 2018, 57, 14090–14094 CrossRef CAS PubMed.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, G. Lan, H. Tang, C. Pelizzari, Y. X. Fu, M. T. Spiotto, R. R. Weichselbaum and W. Lin, Nat. Biomed. Eng., 2018, 2, 600–610 CrossRef CAS PubMed.
- L. Zhou, S. Wang, Y. Chen and C. Serre, Microporous Mesoporous Mater., 2019, 290, 109674 CrossRef CAS.
- X.-L. Lv, M. Tong, H. Huang, B. Wang, L. Gan, Q. Yang, C. Zhong and J.-R. Li, J. Solid State Chem., 2015, 223, 104–108 CrossRef CAS.
- I. Abánades Lázaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230–259 CrossRef.
- T. Gadzikwa, B.-S. Zeng, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2008, 3672–3674 RSC.
- A. A. Coelho, J. Evans, I. Evans, A. Kern and S. Parsons, Powder Diffr., 2011, 26, S22–S25 CrossRef CAS.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X.-H. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R.-B. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F.-X. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K.-I. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- C. A. Walshe, A. J. R. Thom, C. Wilson, S. Ling and R. S. Forgan, Chem. – Eur. J., 2022, 28, e202201364 CrossRef CAS PubMed.
- R. J. Marshall, T. Richards, C. L. Hobday, C. F. Murphie, C. Wilson, S. A. Moggach, T. D. Bennett and R. S. Forgan, Dalton Trans., 2016, 45, 4132–4135 RSC.
- S. C. Jones and C. A. Bauer, J. Am. Chem. Soc., 2009, 131, 12516–12517 CrossRef CAS PubMed.
- T. J. Azbell, R. M. Mandel, J.-H. Lee and P. J. Milner, ACS Appl. Mater. Interfaces, 2022, 14, 53928–53935 CrossRef CAS PubMed.
- D. Frahm, F. Hoffmann and M. Fröba, Cryst. Growth Des., 2014, 14, 1719–1725 CrossRef CAS.
- T. J. Matemb Ma Ntep, H. Reinsch, J. Liang and C. Janiak, Dalton Trans., 2019, 48, 15849–15855 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ce01529c |
| This journal is © The Royal Society of Chemistry 2023 |