
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Valorisation of lignocellulose and low concentration CO2 using a fractionation–photocatalysis–electrolysis process†
Santiago
Rodríguez-Jiménez‡
 ,
Erwin
Lam‡
,
Subhajit
Bhattacharjee
and
Erwin
Reisner
*
,
Erwin
Lam‡
,
Subhajit
Bhattacharjee
and
Erwin
Reisner
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, Cambridge, UK. E-mail: reisner@ch.cam.ac.uk
First published on 13th November 2023
Abstract
The simultaneous upcycling of all components in lignocellulosic biomass and the greenhouse gas CO2 presents an attractive opportunity to synthesise sustainable and valuable chemicals. However, this approach is challenging to realise due to the difficulty of implementing a solution process to convert a robust and complex solid (lignocellulose) together with a barely soluble and stable gas (CO2). Herein, we present the complete oxidative valorisation of lignocellulose coupled to the reduction of low concentration CO2 through a three-stage fractionation–photocatalysis–electrolysis process. Lignocellulose from white birch wood was first pre-treated using an acidic solution to generate predominantly cellulosic- and lignin-based fractions. The solid cellulosic-based fraction was solubilised using cellulase (a cellulose depolymerising enzyme), followed by photocatalytic oxidation to formate with concomitant reduction of CO2 to syngas (a gas mixture of CO and H2) using a phosphonate-containing cobalt(II) bis(terpyridine) catalyst immobilised onto TiO2 nanoparticles. Photocatalysis generated 27.9 ± 2.0 μmolCO gTiO2−1 (TONCO = 2.8 ± 0.2; 16% CO selectivity) and 147.7 ± 12.0 μmolformate gTiO2−1 after 24 h solar light irradiation under 20 vol% CO2 in N2. The soluble lignin-based fraction was oxidised in an electrolyser to the value-added chemicals vanillin (0.62 g kglignin−1) and syringaldehyde (1.65 g kglignin−1) at the anode, while diluted CO2 (20 vol%) was converted to CO (20.5 ± 0.2 μmolCO cm−2 in 4 h) at a Co(II) porphyrin catalyst modified cathode (TONCO = 707 ± 7; 78% CO selectivity) at an applied voltage of −3 V. We thus demonstrate the complete valorisation of solid and a gaseous waste stream in a liquid phase process by combining fractioning, photo- and electrocatalysis using molecular hybrid nanomaterials assembled from earth abundant elements.
Introduction
The renewable generation of valuable chemicals and fuels is a critical step towards a sustainable chemical industry.1,2 The valorisation of abundant waste resources, such as lignocellulosic biomass and the greenhouse gas CO2 offers great potential to achieve such an ambitious goal at the scale required to defossilise our industry. By harnessing the power of renewable energy sources, such as solar and wind, the conversion of non-edible biomass and CO2via photo- or electrochemical approaches presents an opportunity to produce sustainable fuels and chemicals.3Lignocellulosic biomass such as wood, is abundant and cheap and consists predominantly of three polymeric components: cellulose, hemicellulose and lignin.4,5 Cellulose and hemicellulose consist mainly of polysaccharide made from glucose, xylose, mannose and arabinose. Lignin is made of different polymerised aromatic units, and its utilisation remains challenging due to the robustness of its polyaromatic structure, which requires harsh conditions to break down the polymer (e.g., strong acids such as H2SO4).6,7 Unlike cellulose, lignin-to-chemical conversion technologies remain scarce.4,5,7,8 An important linkage in lignin, often found between its aromatic polymeric backbone, is the β-O-4 bond between two phenyl rings, which serves as an ideal target to depolymerise lignin into smaller aromatic fragments.9–11 The selective depolymerisation of all lignocellulose components and their subsequent chemical transformation would enable large-scale access to aliphatic and aromatic renewable feedstock chemicals.
Apart from ubiquitous biomass sources, the greenhouse gas CO2 can be used as an abundant carbon source to produce energy-rich chemicals such as CO, formate, hydrocarbons or alcohols.12 However, photo- and electroreduction of CO2 are predominantly performed in the presence of pure CO2, where concentrated CO2 streams have to be generated involving additional energy input.13–15 To alleviate the energy demand of the process, it is desirable to perform catalytic reactions at lower CO2 concentrations (e.g., ≤20% CO2).16–20 The challenge of using low concentration CO2 streams lies in maintaining high product selectivity and catalytic activity compared to reactions employing pure CO2 streams.17 Molecular CO2 reduction catalysts display an increased product selectivity compared to most heterogeneous electro- or photocatalysts.16,21,22
An attractive approach to utilise biomass and CO2 together is their simultaneous conversion in photo- or electrocatalytic processes.23–25 This strategy opens the possibility to couple biomass oxidation with CO2 reduction in a single process driven by sunlight or renewable electricity. The coupling of productive half-reactions thereby allows the conversion of a solid and a gaseous waste stream into valuable products such as CO, syngas, formate and aromatic chemicals, which can be more attractive than conventional systems performing overall water splitting to generate H2 and O2 from water. Additionally, the oxidation of biomass-derived substrates is thermodynamically less demanding than water oxidation, thereby facilitating the catalytic conversions, as well as the generation of value-added products.26–28 This combined approach also allows for the isolation of products in different phases and compartments, which can help with product separation. Conventional approaches such as water splitting generate explosive H2 and O2 mixtures in the reactor headspace.
The valorisation of cellulose and CO2 streams has recently been reported using a TiO2 nanoparticle with an immobilised CO2 reducing cobalt(II) bis(terpyridine) catalyst containing phosphonate anchors (CotpyP) (TiO2∣CotpyP). Photoexcitation of this hybrid TiO2∣CotpyP photocatalyst reduced aqueous CO2 to syngas, while cellulose-derived glucose was simultaneously oxidised to formate and arabinose. TiO2∣CotpyP could operate for 24 h and be recycled up to three times.29 However, only concentrated CO2 and pure cellulose have been used, and no strategy for lignin separation and utilisation were reported in this previous study.
Herein, we report the valorisation of all components in lignocellulose coupled to low concentration CO2 reduction, which has been achieved by employing molecular CO2 reduction hybrid nanomaterials (Fig. 1). First, lignocellulose is pre-treated and fractioned into predominantly cellulosic- and lignin-based components using acid hydrolysis. Second, the fractionated cellulosic solution was converted with low concentration CO2 using the TiO2∣CotpyP photocatalyst to HCOO− and syngas, respectively. Finally, an electrolysis process concomitantly converted the fractionated lignin solution on a carbon-based anode to vanillin and syringaldehyde, which find application in the food, pharma and cosmetics industries.10,30 Diluted CO2 is reduced to CO (with a single pass conversion yield close to 5% at both 10 vol% and 20 vol% CO2) with a molecular cobalt(II) porphyrin (CoPL) catalyst immobilised on a multiwall carbon nanotube (MWCNT) cathode. Thus, we demonstrate the complete valorisation of lignocellulose and low concentration CO2, which has been enabled by a precious-metal free fractionation–photocatalysis–electrolysis process.
 | ||
| Fig. 1 Schematic overview of a three-stage process for the complete valorisation of lignocellulose and diluted CO2. (1) Fractionation of lignocellulose from white birch into a predominant cellulose and lignin fraction (top and bottom, respectively), with the cellulose-based solid fraction being further incubated in cellulase to generate a soluble sugar (glucose and cellobiose) solution. Structures of xylose, mannose and arabinose are omitted for clarity. (2) Photocatalytic oxidation of cellulose-derived glucose and cellobiose to formate coupled with reduction of diluted CO2 to syngas (CO and H2) using TiO2∣CoptyP photocatalyst. (3) Electrolytic oxidation of lignin to aromatic aldehydes syringaldehyde and vanillin coupled with reduction of diluted CO2 to CO and H2 using CP∣MWCNT∣∣CP∣CoPL. | ||
Results and discussion
Fractionation of lignocellulose
The composition of dried and extracted white birch wood used in this study was determined by quantifying its sugar and lignin composition following standard characterisation procedures using H2SO4 hydrolysis:6 glucose (34.7 ± 1.6 wt%), xylose (20.3 ± 1.0 wt%), mannose (1.8 ± 0.1 wt%), arabinose (0.9 ± 0.1 wt%) and lignin (17.6 ± 1.4 wt%). Further details are provided in the Experimental section, Fig. 1, Fig. S1 and Tables S1 and S2.†To effectively utilise the different components of lignocellulose, their individual components are first separated by acid hydrolysis. Lignocellulose was pre-treated in a dioxane/HCl/HCOOH mixture at 80 °C for 3 h (500 mg in 5.9 mL) to obtain a liquid (or liquor) and a solid fraction.7 The dioxane/HCl/HCOOH mixture solubilises lignin and the liquid fraction predominantly consisted of lignin (41.2 ± 2.1 wt%) along with xylose (6.9 ± 1.3 wt%), glucose (2.4 ± 0.6 wt%), mannose (1.3 ± 0.3 wt%) and arabinose (2.5 ± 0.7 wt%). The second most abundant component xylose (derived from hemicellulose) is partially converted to furfural under these fractioning conditions (see Experimental section, and Fig. 1, Fig. S1 and Tables S1–S3† for further details).26 The isolated solid fraction consisted mainly of cellulosic components such as glucose (62.9 ± 1.9 wt%) and xylose (6.7 ± 1.6 wt%) with some lignin (5.4 ± 1.2 wt%).
Photocatalytic valorisation of cellulose and CO2
The solid fraction obtained from lignocellulose fractionation contained mainly cellulose and was used as a feedstock in photocatalysis following depolymerisation via cellulase pre-treatment.29 The depolymerised glucose and cellobiose are suitable electron donors in semiconductor suspension systems,31 and their valorisation has been previously reported.32,33 Specifically, the solid fraction was enzyme pre-treated with cellulase (0.05 mgcellulase mgsolid−1) to generate soluble sugars, predominantly glucose (39.3 ± 4.7 mM) and cellobiose (8.7 ± 2.1 mM) after 24 h incubation at 37 °C in an aqueous sodium acetate buffer solution (50 mM) at pH 5.29 With respect to white birch, 10.2 ± 0.4 wt% and 4.2 ± 0.6 wt% were converted to glucose and cellobiose, respectively (see Tables S1 and S2†).The lignocellulose-derived sugar solution was then utilised for CO2 reduction reactions using the TiO2∣CotpyP photocatalyst (Fig. 1).29 In a typical experiment, CotpyP (50 nmol) was added to a photoreactor containing a TiO2 suspension (5 mg, P25, particle diameter ∼20 nm) in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN:cellulase-treated solid fraction aqueous solution (3 mL). The photoreactor was sealed with a rubber septum and purged with 100% CO2 or 20% CO2 (balanced with N2) at a flow rate of 15 mL min−1 for 15 min. The sealed and stirred photoreactor was irradiated with a solar light simulator (100 mW cm−2, AM 1.5G, 25 °C, 600 rpm) for 24 h. The UV in the full solar spectrum is necessary to photoexcite electrons from the valence to the conduction band of TiO2.29 The gaseous products (H2 and CO) in the headspace (4.74 mL) were quantified by gas chromatography (GC), and HCOO− formed in the solution from glucose photooxidation was quantified by ion chromatography.
:1 MeCN:cellulase-treated solid fraction aqueous solution (3 mL). The photoreactor was sealed with a rubber septum and purged with 100% CO2 or 20% CO2 (balanced with N2) at a flow rate of 15 mL min−1 for 15 min. The sealed and stirred photoreactor was irradiated with a solar light simulator (100 mW cm−2, AM 1.5G, 25 °C, 600 rpm) for 24 h. The UV in the full solar spectrum is necessary to photoexcite electrons from the valence to the conduction band of TiO2.29 The gaseous products (H2 and CO) in the headspace (4.74 mL) were quantified by gas chromatography (GC), and HCOO− formed in the solution from glucose photooxidation was quantified by ion chromatography.
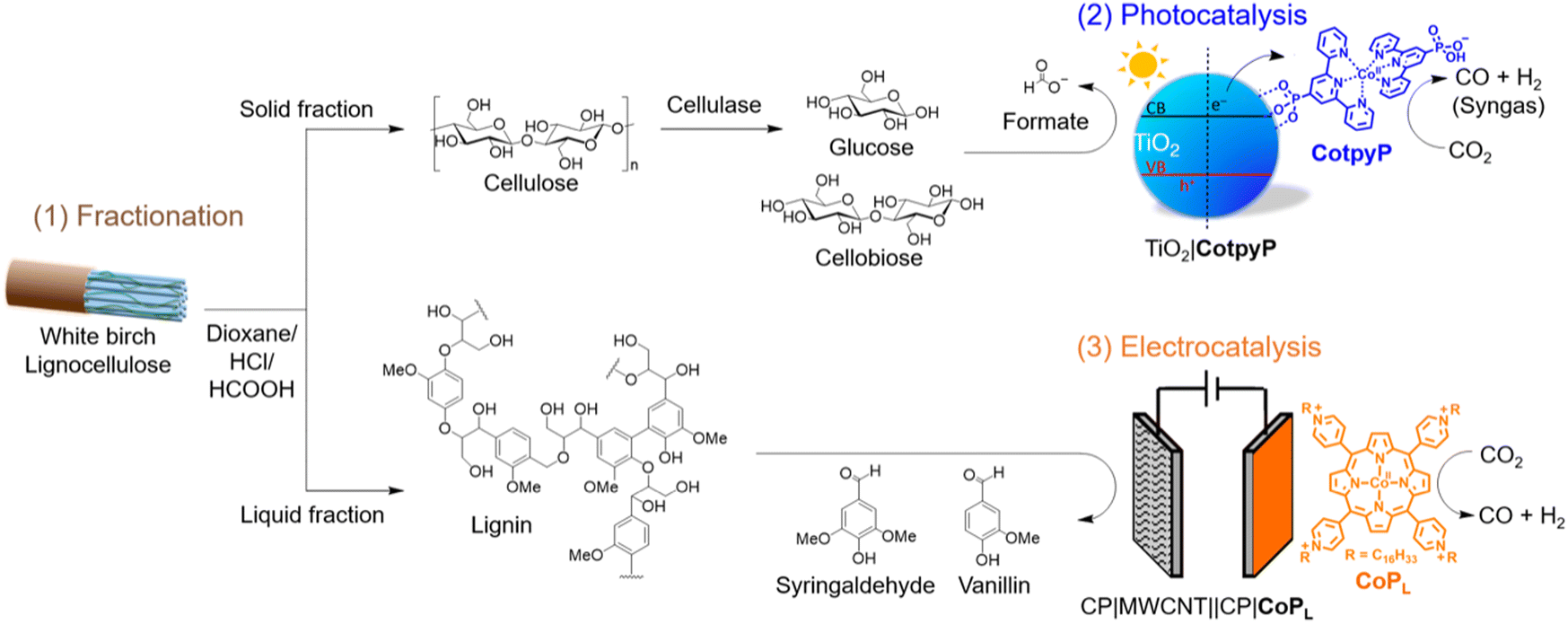
After 24 h of photocatalysis under 100 vol% CO2, 69.9 ± 4.0 μmolCO gTiO2−1, 109.8 ± 8.0 μmolH2 gTiO2−1 (i.e., 39% CO and 61% H2 selectivity for gaseous products) and 153.7 ± 4.0 μmolformate gTiO2−1 were formed. Under 20 vol% CO2, the formation yields were 27.9 ± 2.0 μmolCO gTiO2−1, 141.7 ± 27.9 μmolH2 gTiO2−1 (i.e., 16% CO and 84% H2 selectivities) and 147.7 ± 12.0 μmolformate gTiO2−1 (Fig. 2a and Table S4†). The obtained CO yields correspond to a CO2-to-CO conversion yield of ∼0.03% and ∼0.05% at 100 vol% and 20 vol% CO2, respectively. The reduction of protons (from water) to H2 and CO2 to CO by CotpyP as well as the oxidation of glucose/cellobiose by TiO2 to formate are two-electron processes with an expected 1:1 stoichiometric ratio for (H2 + CO):formate, which is close to the observed ratios.29,34–36
 | ||
| Fig. 2 (a) H2, CO and formate (blue, green and red, respectively) formation after 24 h of photocatalysis with TiO2∣CotpyP. (b) Transmission IR spectra with 13CO2 isotopic labelling of the headspace after 24 h of photocatalysis (sample, green curve), including 12CO (black) and 13CO (red) as reference. Reaction conditions: 5 mg TiO2 (P25); 50 nmol CotpyP; 3 mL 2:1 MeCN:pre-treated reaction solution (cellulose fraction pre-treated with cellulase); 25 °C, 100 mW cm−2, AM 1.5G. (a) 20 vol% CO2 was balanced with N2; (b) sample (green) was purged with 100% 13CO2. Experiments in (a) were performed in triplicates. | ||
The carbon source of the products was confirmed by isotopic labelling experiments. Experiments with 13CO2 and cellulase enzyme pre-treated cellulose were performed to confirm that the CO originates from CO2 reduction (for further details see Experimental section). Analysis of the gas headspace after photocatalysis by transmission IR spectroscopy reveals that 13CO produced by TiO2∣CotpyP was only formed when 13CO2 was used as the carbon source (Fig. 2b). Furthermore, in the case of formate, based on previous work using 13C6-glucose with TiO2∣CotpyP, 13C-formate is only formed through the photooxidation of 13C6-glucose, confirming formate's carbon source.29 Further mechanistic insights of the photooxidation of glucose to formate can be found in ref. 36.
These results demonstrate that the cellulose solid fraction, following cellulase pre-treatment, provides a source of suitable electron donors for photocatalytic CO2 reduction. The solar TiO2∣CotpyP reforming system was able to convert CO2-to-CO at concentrations of 20 vol% CO2, with an activity drop of only a factor of two despite the five-fold drop in CO2 concentration with respect to 100 vol% CO2 (Fig. 2a). Near stoichiometric amounts of HCOO− to CO/H2 were formed at both CO2 concentrations, demonstrating the effectiveness of TiO2∣CotpyP to photooxidise sugars to formate and concomitantly photoreduce CO2 and H2O to CO and H2.
Electrolytic valorisation of lignin and CO2
To optimise the simultaneous electrolysis of CO2 and lignin, the half-reactions of electroreduction of low CO2 concentration and electrooxidation of lignocellulose-derived lignin were first studied individually in a three-electrode setup. This was followed by the proof-of-concept coupling of both half reactions in a two-electrode electrolyser using the optimised cathode and anode.Low concentration CO2 electroreduction was performed on CoPL immobilised on MWCNT as a molecular catalyst (see Fig. 1). CoPL was chosen based on its known CO selectivity during electroreduction of pure CO2, and stability when immobilised on MWCNT via π–π stacking and its lipophilic alkyl chains (see ESI Note 1, and Fig. S2–S4†).28,37 We therefore further explored the electroreduction ability of CoPL supported on MWCNT under variable CO2 concentrations ranging 10, 20, 50 and 100 vol% (balanced with N2), using an electrochemical flow setup that allowed continuous purging of the electrolyte solution with a given gas composition.38
Cathodes containing CoPL were prepared, following a reported procedure (see Fig. S5†),28 by drop-casting a dimethylformamide (DMF) suspension containing 2.37 mg MWCNT mL−1 and 0.1 mM CoPL onto carbon paper (CP) (0.1 mLDMF cm−2, geometrical surface area = 1 cm2), which is denoted as CP∣CoPL. Electrocatalysis with CP∣CoPL was performed in a two-compartment electrochemical cell with a three-electrode setup. Pt foil was used as the counter electrode (CE) and Ag/AgCl (sat. KCl) as reference electrode (RE), a Nafion membrane separating the cathode and anode chambers, with the catholyte (0.1 M NaHCO3 in H2O) under a constant gas flow (9 mL min−1) of CO2 and N2 regulated by mass flow controllers. The generated gaseous products during electrochemical experiments (H2 and CO) were measured via online GC (schematically represented in Fig. S6†).38
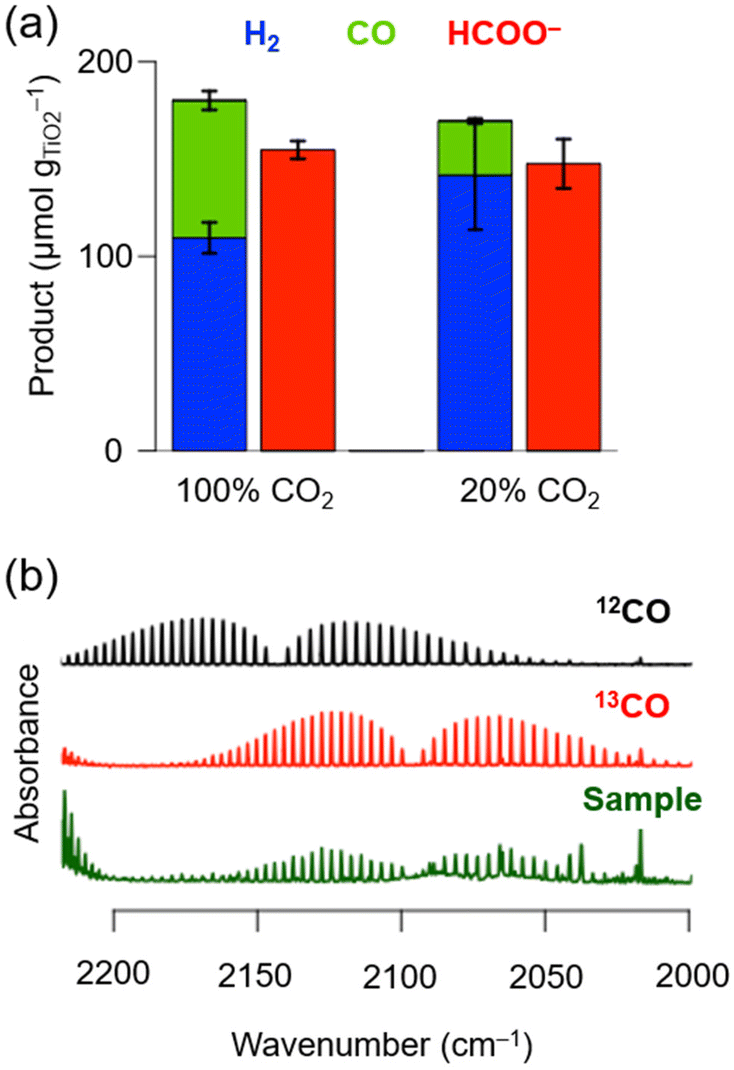
During chronoamperometry (CA) experiments, under an applied potential of −1.2 V vs. Ag/AgCl (sat. KCl) the catholyte chamber was continuously purged with pure N2 for 30 min, after which the CO2 concentration was gradually increased to 10, 20, 50 and 100 vol% every 45 min (Fig. 3a). Under pure N2, the current density was the lowest at approximately −0.5 mA cm−2 and around ∼0.08 μmolH2 min−1 evolved as the main gaseous product, with a minor CO background (<0.01 μmolCO min−1) likely caused by the chemical equilibrium between carbonic acid (H2CO3) and CO2. Upon increasing the CO2 concentration (in vol%), the current density gradually increased, from approximately −0.8 mA cm−2 (10 vol%), to −1.2 mA cm−2 (20 vol%), to −1.5 mA cm−2 (50 vol%) and to −1.8 mA cm−2 (100 vol%). The H2 formation rate remained constant at ∼0.06 μmolH2 min−1 at all CO2 concentration steps, whereas the CO formation rate and CO selectivity increased from ∼0.12 μmolCO min−1 and ∼65% (10 vol%), to ∼0.24 μmolCO min−1 and ∼80% (20 vol%), to ∼0.35 μmolCO min−1 and ∼90% (50 vol%) and to ∼0.50 μmolCO min−1 and ∼93% (100 vol%) (Fig. 3b). The low H2 formation rate across all studied CO2 concentrations (∼0.06 μmolH2 min−1) may be explained by the high affinity of CoPL to CO2.28 The carbon source of CO was previously confirmed for CoPL through 13CO2 isotopic labelling experiments.28,37
 | ||
| Fig. 3 (a) Chronoamperometry (CA) experiments as a function of concentration of CO2 and time during CO2 electrochemical reduction to CO using CP∣CoPL cathodes. (b) CO formation rate and selectivity during CA experiments. Experiments were performed in triplicates, and the shaded area represents the standard deviation. Reaction conditions: CA: 0.1 M NaHCO3 in H2O (pH = 6.7); Eapp = −1.2 V vs. Ag/AgCl for 3.5 h; WE: CP∣CoPL, CE: Pt foil, RE: Ag/AgCl (sat. KCl). Flow of CO2:N2 9 mL min−1. CO2 concentration was varied stepwise between 0 vol%, 10 vol%, 20 vol%, 50 vol% and 100 vol% and balanced with N2. | ||
Two main observations can be made by screening different CO2 gas flow concentrations: (1) CoPL exhibits a high product specificity with around ∼80% CO selectivity even under 20 vol% of substrate CO2. (2) The CO formation rate at 20 vol% CO2 (∼0.24 μmolCO min−1; CO turnover frequency (TOFCO) = 8.3 min−1; single-pass CO2 conversion yield39 = 4.7%) corresponds to roughly half the activity with respect to 100 vol% CO2 (∼0.50 μmolCO min−1; TOFCO = 17.2 min−1; single-pass CO2 conversion yield = 2.0%), thus indicating that decreasing five-fold the CO2 concentration only reduces two-fold the CO formation rate (Fig. 3b).
When comparing CP∣CoPL with TiO2∣CotpyP, both molecular hybrid systems exhibit similar CO formation rate trends, although CP∣CoPL maintains higher CO selectivity across all CO2 concentrations (see ESI Note 1 and Tables S4 and S5†). The origin of the observed trends for TiO2∣CotpyP and CP∣CoPL at different CO2 concentrations remains unclear but may be attributed to their molecular structure, which provides intrinsic affinity towards CO2. In comparison with state-of-the-art molecular systems, such as the rhenium bipyridine electrocatalyst [Re(4,4-dimethyl-2,2-dipyridyl)(CO)3(triethanolamine)], which operates in a DMF/triethanolamine solvent mixture at variable CO2 concentrations (1, 10 and 100%) under comparable flow conditions,22 CP∣CoPL is three orders of magnitude more active (i.e., TOFCO = 0.5 h−1 after 24 h at 10% CO2vs. 413 h−1 after 4 h at 20% CO2, respectively). Without taking into account that CO2 is more soluble in organic solvents than in water (e.g. ∼180 mM in DMF vs. ∼33 mM in water at 25 °C), these differences in performance could be tentatively associated to the catalytic mechanism of CO2 reduction, which enables cobalt porphyrins to achieve higher TOF than rhenium bipyridine electrocatalysts.22,37
The electrooxidation of the liquid fraction or liquor, containing predominantly lignin and obtained from pre-treating lignocellulose (250 mg), was studied in a two-compartment electrochemical cell with a three-electrode setup. For this purpose, the anodic conditions were initially optimised using the lignin model substrate 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol, which contains a β-O-4 linkage between two phenyl rings that mimics those ubiquitously found in lignin (see Fig. 1 and Fig. S7†).9–11
CP∣MWCNT anodes were fabricated by dropcasting a MWCNT suspension in ethanol (1.67 mg mL−1, 0.1 mL cm−2) containing Nafion 117 (2 vol% of a 5 wt% solution) to achieve a high surface area MWCNT layer with ∼20 μm thickness (see Fig. S8†). We found that MWCNT on hydrophilic carbon paper (CP∣MWCNT) acted as a suitable catalyst for the oxidation of the β-O-4 linkage in the model substrate (see Fig. S7 and ESI Note 2†). CP∣MWCNT (geometric surfaced area = 1 cm2) in the presence of 10 mM of lignin model substrate in 0.1 M Na2CO3 in 1:1 MeCN:H2O achieved high current densities (∼8 mA cm−2) at +1 V vs. Ag/AgCl (sat. KCl), while in absence of lignin, and under the same conditions, the current densities were lower (∼3 mA cm−2) (see Fig. S9 and S10†). MeCN was used to increase the solubility of the lignin model substrate and lignin. The generated oxidation product from the lignin model substrate during CA experiments, 3,4-dimethoxybenzaldehyde (3,4-MBA), was obtained in 36 ± 1% yield with a faradaic yield (FY) of 25 ± 1% assuming a two-electron oxidation (see Experimental details, Fig. S11 and Table S6†). The obtained 3,4-dimethoxybenzaldehyde was measured by 1H nuclear magnetic resonance (NMR) spectroscopy in CDCl3 with mesitylene as internal standard (see Fig. S12–S14†).
Having established the optimised conditions (see ESI Note 3, and Fig. S15–S20†) and the electrodes suitable for lignin oxidation and low concentration CO2 reduction, we aimed at coupling both redox half reactions in a single two-electrode electrolyser with the corresponding anolytes and catholytes separated by a bipolar membrane. Electrolysis was performed using a two-electrode setup (CP∣CoPL as WE and CP∣MWCNT as CE) with an applied voltage (Uapp) of −3 V for 4 h. The anolyte comprised of 0.1 M Na2CO3 in a 1:1 MeCN:H2O solvent mix containing lignin (obtained from pre-treating 250 mg lignocellulose), and the catholyte had 0.1 M NaHCO3 in H2O. The catholyte was constantly purged at 9 mL min−1 with 20 vol% CO2 (balanced with N2). The gaseous products on the cathodic side were monitored by online GC.38
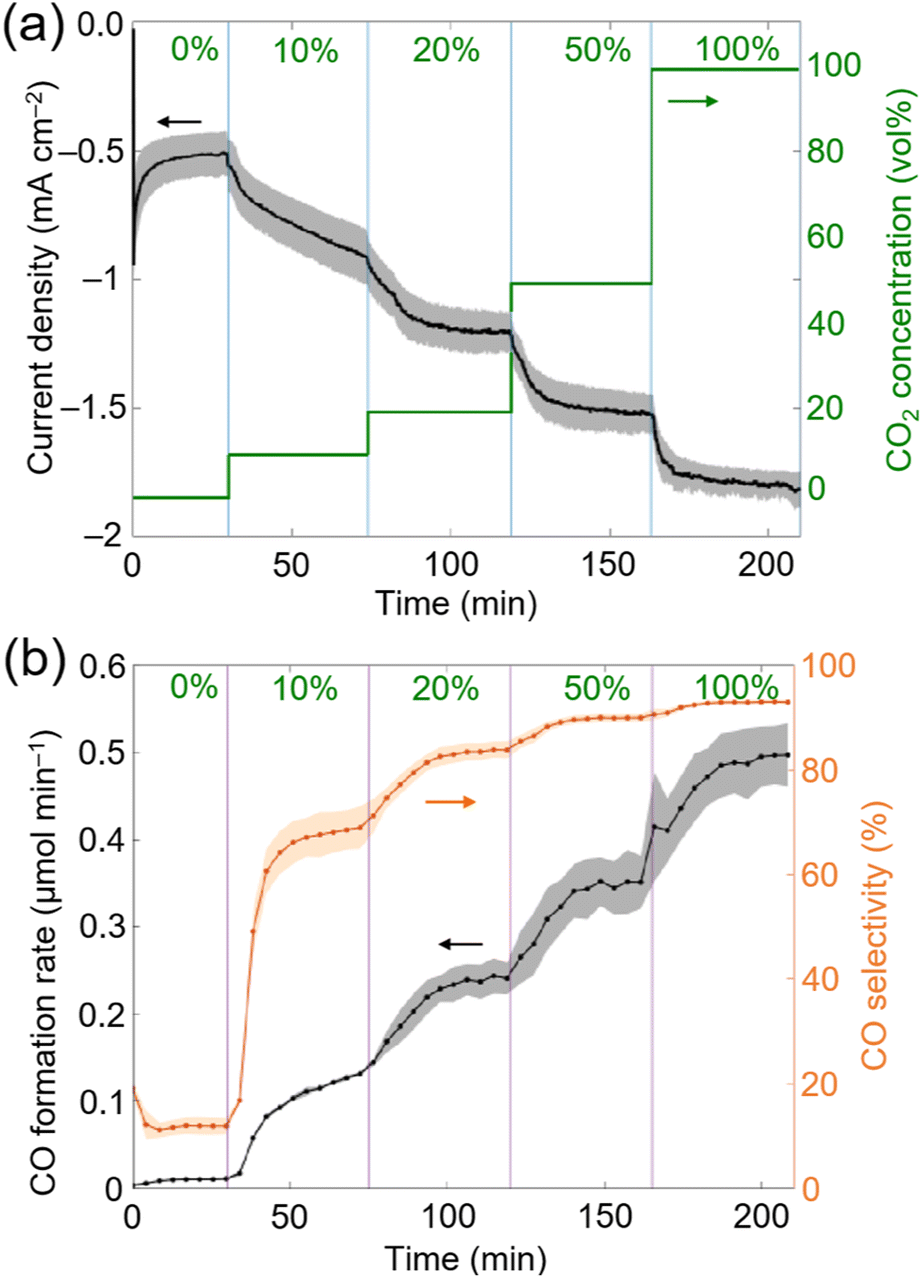
During 4 h electrolysis, the initial current density gradually decreased from approximately −1.4 mA cm−2 to around −0.4 mA cm−2 (Fig. S21†), and the initial maximum CO formation rate observed changed from ∼0.2 μmolCO min−1 to ∼0.05 μmolCO min−1 (Fig. 4a and b). After 4 h, 20.5 ± 0.2 μmolCO cm−2 was produced along with 5.8 ± 0.3 μmolH2 cm−2 (TONCO = 707 ± 7 and TONH2 = 200 ± 10) corresponding to a CO selectivity of 78 ± 2% and a FYCO+H2 of 59 ± 6% (Table S5†). Despite of the decrease in current density and CO formation rates, the CO selectivity remained stable.28
 | ||
| Fig. 4 Electrolysis of 20 vol% CO2 gas flow with CP∣CoPL at the cathode coupled to lignin conversion at the anode with CP∣MWCNT. (a) Current density as a function of time. (b) CO formation rate and selectivity during electrolysis at the cathode. (c) 1H NMR spectrum recorded in CDCl3 of the anolyte containing lignin after 4 h electrocatalysis showing the emergence of aldehydes and methoxy groups. Experiments were performed in triplicates, and the standard deviation are represented as shaded area (a, b). Reaction conditions: (a and b) Electrolysis (two electrode configuration) at Uapp = −3 V. Anolyte: 0.1 M Na2CO3, lignin fraction obtained from pre-treating 250 mg lignocellulose in 1:1 MeCN:H2O; anode: CP∣MWCNT. Catholyte: 0.1 M NaHCO3 in H2O with a flow of 20 vol% CO2 (balanced with N2) 9 mL min−1; cathode: CP∣CoPL; bipolar membrane; room temperature. (c) The asterisk (*) highlights the additional aldehyde signal formed from lignin electrooxidation. | ||
After electrolysis, the anolyte was worked-up (see Experimental section for details) and the crude product was analysed by 1H NMR spectroscopy in CDCl3. Control experiments, where the lignin fraction was stirred in the electrolyte solution for 4 h without applied bias, led to the formation of a small background level of aldehydes. Importantly, under an applied potential more aromatic aldehyde signals were formed (Fig. S20† and Fig. 4c), which shows that aromatic monomer formation is promoted by electrooxidation. The observed 1H NMR signals corresponded to syringaldehyde and vanillin (see Fig. 1 and 4c), which are aromatics that can be formed from lignin.8,10 After 4 h CA, 0.14 ± 0.03 μmol and 0.31 ± 0.03 μmol of vanillin and syringaldehyde were detected, respectively. These yields equated to 0.62 g of vanillin and 1.65 g of syringaldehyde per kilogram of lignin. Compared with oxygen evolved from water oxidation, vanillin and syringaldehyde have potential as bio-derived monomers in the polymer industry.40 The observed moderate yields for vanillin and syringaldehyde could be attributed to the acidic degradation of reaction intermediates, i.e., lignin-fragment oxidation decreased the local pH at the CP∣MWCNT anode surface below 11.5, and hence halt the formation of the targeted aromatics.40 In addition to the two identified aromatic compounds, other unidentified signals in the aromatic aldehyde (∼9.8 ppm) and methoxy regions (3.8–4.0 ppm) can be observed in the 1H NMR spectra (Fig. 4c).40
In comparison to CP∣MWCNT, the use of heterogeneous anodes for lignin oxidation based on metals/metal oxides, such as toxic Pb/PbO2, has been previously reported.41–43 For instance, these Pb/PbO2 anodes were able to generate different lignin-derived products, such as vanillin (5.83 g kglignin−1) and syringaldehyde (9.30 g kglignin−1), via electrooxidation/electrohydrogenation of bamboo-derived lignin when used with Cu cathodes in 1 M NaOH solution.41 Despite the difficulty to compare this previously reported system with our CP∣MWCNT∣∣CP∣CoPL system due to the different experimental conditions (i.e., electrode materials and surface, pH, temperature, substrate concentration, applied voltage and currents), the metal-free CP∣MWCNT anodes were able to generate yields of vanillin and syringaldehyde within the same order or one-order of magnitude lower than those reported for the metal/metal oxide based Pb/PbO2∣∣Cu system.
Previous work has shown that lignin valorisation can be coupled with hydrogen evolution or reduction of CO2 to formate using photoelectrochemical systems.44,45 In comparison, our work presents a unique and successful three-stage approach that shows that complete valorisation of lignocellulose is possible through the combination of fractionation and utilisation of photocatalysis and electrocatalysis to oxidise the resulting solid fraction to formate and liquid fraction to aromatics and reduce diluted CO2 and water to CO and H2. Although this system is a proof-of-concept demonstration, different factors would need consideration for practical implementation. These include optimisation of catalyst performance, scalability,46,47 integration of a carbon capture step,48 potential limitations such as catalyst stability, the engineering design of reactors49,50 for lignocellulose fractionation,46 photocatalysis51 and electrolysis,52 and the separation of products for further use.53,54
Conclusions
We report a combined fractionation-photocatalysis-electrolysis process for the complete valorisation of lignocellulose and low-concentration CO2. Fractioning lignocellulosic wood provides sugar-based solid and lignin-based soluble fractions that are photo- and electrocatalytically converted with immobilised molecular cobalt(II) catalysts to produce CO, formate, vanillin and syringaldehyde. The presented results set a precedent in integrating renewable processes for complete waste feedstock valorisation under ambient pressure and temperature. Moreover, our molecular hybrid systems were able to convert low CO2 concentrations (10–20 vol% CO2), which paves the way towards utilising CO2 concentrations nearing those found in flue gas (4–10 vol% CO2). However, to utilise actual flue gas additional CO2 purification steps are necessary to eliminate impurities,55 such as SO2, SO3, NOx, O2 and particulate matter depending on the type of fuel used. Learning from the use of low CO2 concentration also brings us a step closer towards operating directly from atmospheric CO2 (420 ppm).20,56Hence, this work demonstrates a proof-of-concept strategy to valorise challenging multicomponent waste streams simultaneously through solar-driven and electrochemical redox processes. This work further highlights the potential of molecularly engineered hybrid materials57 in the valorisation of waste streams, which can be expanded beyond CO2 reduction in future developments to perform chemistry in a more sustainable and circular manner. As biomass and CO2 emerge as the most scalable and readily available sustainable carbon sources to defossilise the chemical industry,58 this work aims to inspire new approaches in their practical valorisation.
Experimental section
Solvents and materials
Acetonitrile (MeCN, Fisher Chemicals), D2O (Sigma Aldrich), D(+)-glucose (Fisher Chemicals), D(+)-cellobiose (Acros), Cellulase from Trichoderma reesei ATCC 26921 (Sigma Aldrich), sodium acetate trihydrate (Fisher Chemicals), hydrochloric acid (34–37% Fisher Chemicals), 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol (lignin model substrate, FluoroChem), Vanillin (Thermo Scientific), syringaldehyde (Thermo Scientific), vanillic acid (Sigma Aldrich), syringic acid (Thermo Scientific), fufural (Sigma Aldrich), 2-methoxyphenoxyacetic acid (Alfa Aesar), 3,4-dimethoxybenzaldehyde (Acros), 4-hydroxybenzoic acid (Thermo Scientific), white birch (Merck), dioxane (Fisher Chemicals), formic acid (Fisher Chemicals), sulfuric acid (Fisher Chemicals), multiwalled carbon nanotubes (MWCNT 755117, Sigma Aldrich), carbon paper (Toray Paper 60, Fuel Cell Store), Nafion solution (5 wt% Sigma Aldrich), NaHCO3 (Merck), EtOAc (Fisher Chemicals), THF (Fisher Chemicals), Na2CO3 (Merck), MgSO4 (Fisher Chemicals) CDCl3 (Sigma Aldrich), 50 wt% H2SO4 (Fluka), Na2CO3 64 mM and NaHCO3 20 mM (ion chromatography eluent concentrate for Metrosep A Supp5; Sigma Aldrich) were purchased from commercial sources and used as received. MilliQ® grade H2O was used for all the experiments. TiO2 powder P25 (10–30 nm diameter; 50 m2 g−1) was obtained from Evonik; [Co(2,2′:6′,2′′-terpyridine-4′-phosphonic acid)2](BF4)2 (denoted as CotpyP)35 and CoPL37 were synthesised according to reported procedures. Reaction gases (CO2 and N2) were purchased from BOC. Ag/AgCl reference electrode was stored in a saturated NaCl solution (sat. NaCl; BasiMW-2030) and anion exchange membrane (Selemion, AGC Engineering), Nafion 117 membrane (Sigma Aldrich), bipolar membrane (Fumacep, Fuel Cell Store), were stored in MilliQ® grade H2O.
Physical characterisation
1H NMR spectroscopy was recorded on a Bruker DPX 400 MHz spectrometer with the chemical shifts (δ) of the 1H NMR spectrum being referenced against the residual solvent signal (D2O: δ = 4.79 ppm, CDCl3: δ = 7.26 ppm). Scanning electron microscopy was performed on a TESCAN MIRA3 FEG-SEM instrument. Before measuring the samples, they were sputtered with 10 nm of platinum.Product quantification
Gaseous H2 and CO under static conditions were analysed by a Shimadzu Tracera GC-2010 Plus with a barrier discharge ionization detector. The GC-2010 Plus was equipped with a ShinCarbon micro ST column (0.53 mm diameter) kept at 40 °C using helium carrier gas. The response factors for the gases were determined by calibration with known amounts of H2 and CO. Typically, 50 μL of headspace gas from the photoreactor was injected using an air-tight syringe (Hamilton, GASTIGHT). Formate was analysed by ion chromatography (IC) on a Metrohm 882 compact IC plus chromatography system equipped with a Metrosep A Supp 5 – 150/4.0 column using an aqueous Na2CO3 (3.2 mM) and NaHCO3 (1 mM) solution as eluent. The response factor of formate was determined by calibration with known amounts of aqueous formate solutions. Sugar concentrations (glucose, cellobiose, xylose, mannose and arabinose) were determined by high performance liquid chromatography (HPLC) on a Waters Breeze system equipped with a refractive index detector and a Rezex 8% Ca2+ Monosaccharide 300 × 7.80 mm HPLC column using 2.5 mM H2SO4 as the eluent with a flow rate of 0.5 mL min−1 (at 75 °C). The response factors for the sugars were determined by calibration of aqueous sugar solutions with known amounts of sugar.Klason lignin and sugar content determination
Klason lignin and sugar determination was performed following a reported procedure.6 For the compositional analysis of white birch, the sample was first dried at 105 °C overnight followed by cooling to room temperature under static vacuum in a desiccator containing CuSO4 as drying agent. The dried solid was extracted three times with an ethanol:water mixture (4:1 v/v and 20 mL g−1) followed by water (20 mL g−1) by sonication for 30 min. The sample was then dried at 105 °C overnight followed by cooling to room temperature under static vacuum in a desiccator containing CuSO4 as a drying agent.
To determine the Klason lignin content and sugar concentration, to 250 mg of the solid (dried and extracted white birch or solid after white birch pre-treatment in dioxane/HCl/HCOOH) was added 3.75 mL of a 72 wt% H2SO4 at room temperature. The suspension was stirred periodically (every 15 min with a glass rod) for 2 h and 145 mL of MilliQ® grade H2O was added, followed by refluxing the suspension at 120 °C for 4 h. The suspension was filtered on a tared frit. The solid was dried at 105 °C overnight and the mass of the filtered solid was determined gravimetrically to determine Klason Lignin content. The filtrate was diluted with H2O to 250 mL and concentrated to 50 mL at 80 °C. The solution was then analysed by HPLC to determine the sugar content (glucose, xylose, mannose, galactose and arabinose).
To determine the Klason Lignin and sugar content of the liquor from white Birch pre-treatment in dioxane/HCl/HCOOH, the liquor obtained from pre-treating 500 mg white Birch was used following the same procedure as with dried and extracted white Birch.
White Birch pre-treatment
Lignocellulose fractioning was performed following a reported procedure.7 To 500 mg of dried and extracted white birch was added dioxane (4.5 mL), conc. HCl (0.42 mL), formic acid (0.36 mL) and water (0.64 mL). The suspension was then stirred for 5 h at 80 °C and filtered and the residue was washed with dioxane until the solution became clear (solid/cellulose fraction). The combined filtrate was dried at 60 °C under vacuum and extracted with THF followed by drying at 40 °C to obtain a dark oil (liquid/lignin fraction).Enzyme hydrolysis of pre-treated white Birch
The obtained solid after treatment of white birch with dioxane/HCl/HCOOH was washed three times with H2O (20 mL g−1) to remove residual HCOOH and dried at 105 °C overnight. To the washed and dried solid fraction (150 mg) in an aqueous sodium acetate solution (50 mM, 3 mL) at pH 5 (adjusted by the addition of HCl) at 37 °C was added cellulase (15 mg in 0.75 mL) in a 50 mM aqueous sodium acetate solution at pH 5. The suspension was incubated at 37 °C for 24 h, followed by 15 min at 90 °C and filtration through a syringe filter (0.2 μm). The filtered solution was stored at −4 °C. The pre-treated cellulose fraction solution contained glucose (39.3 ± 4.7 mM) and cellobiose (8.7 ± 2.2 mM) as determined by HPLC. Control experiments with the recovered solid after treatment of white Birch with dioxane/HCl/HCOOH under the same condition in the absence of cellulase did not show the formation of glucose or cellobiose.CP∣MWCNT electrode preparation
A suspension containing MWCNT in ethanol (1.67 mg mL−1) and 2 vol% of a 5 wt% Nafion solution was sonicated for 15 minutes. The suspension was then dropcasted on a defined area of carbon paper (0.1 mL cm−2) that was masked with Teflon tape followed by overnight drying at room temperature. The electrode was then taped to a metal rod using a copper tape and the metal rod and copper tape were wrapped with Parafilm.CP∣CoPL electrode preparation
A suspension containing MWCNT in DMF (3.16 mg mL−1) was sonicated for 10 min. Subsequently, this ink was diluted 25% by adding 0.4 mM CoPL in DMF to achieve 2.37 mg MWCNT mL−1 and 0.1 mM CoPL. This ink was further sonicated for 10 min and then dropcasted on a defined area (masked with Teflon tape) of carbon paper (0.1 mL cm−2), followed by drying overnight at room temperature. The electrode was then taped to a metal rod using a copper tape and the metal rod and copper tape were wrapped with Parafilm.Photocatalytic experiments
In a typical experiment, to TiO2 (P25, 5 mg) was added 2 mL of MeCN and 1 mL of the solution obtained from cellulase pre-treatment in a glass photoreactor (7.74 mL total volume) equipped with a magnetic stir bar. 50 nmol of CotpyP (from a freshly prepared 2 mM solution in H2O; 0.025 mL) was added, and the photoreactor (3 mL solution with 4.74 mL headspace) was capped with a rubber septum and purged with CO2 (100 vol% or 20 vol% balanced with N2) controlled by mass flow controllers for 15 min at 15 mL min−1, followed by stirring for 15 min in the dark. The photoreactor (kept at 25 °C and stirred at 600 rpm) was then irradiated with a calibrated solar light simulator (Newport Oriel, 100 mW cm−2) equipped with an air mass 1.5 global (AM 1.5G) filter and a water filter to remove infrared radiation. The photocatalytic process was monitored by analysing the headspace after 6 and 24 h by gas chromatography to monitor H2 and CO formation. Formate in the solution (diluted in H2O (1:9 v:v photocatalysis solution:H2O)) was analysed at the end of the photocatalytic studies (after 24 h) by ion chromatography.
CO2-to-CO conversion yield
Photocatalytic CO2-to-CO conversion yield (%) of TiO2∣CotpyP was calculated by dividing the mol of CO produced after 24 h by the mol of CO2 in the photoreactor headspace and multiplying the product by 100. The mol of CO2 was obtained using the ideal gas law equation (pV = nRT), where p is 1 atm, V is the volume of the reactor headspace in L multiplied by the molar fraction of CO2, R is the ideal gas constant (0.082 atm L mol−1 K−1) and T is 298.15 K.Isotopic labelling experiments
To a suspension of cellulose (300 mg) in a 50 mM aqueous sodium acetate solution (6 mL) at pH 5 (adjusted by the addition of HCl) at 37 °C was added cellulase (15 mg in 1.5 mL) in a 50 mM aqueous sodium acetate solution at pH 5. The suspension was incubated at 37 °C for 24 h, followed by 15 min at 90 °C and filtration through a syringe filter (0.2 μm). The filtered solution was stored at −4 °C. The pre-treated cellulose solution contained glucose (53 ± 2 mM) and cellobiose (26 ± 1 mM) as determined by HPLC. To a glass photoreactor vial (7.74 mL total volume) equipped with a magnetic stir bar was added 5 mg of TiO2 which was suspended in 2.95 mL of 2:1 v:v MeCN:pre-treated cellulose solution. The molecular catalyst CotpyP (0.025 mL 50 nmol, 2 mM in H2O) and 0.025 mL H2O (to reach 3 mL) was added and the photoreactor was capped with a rubber septum. The photoreactor was then degassed for one min (vacuum at 10−2 mbar) after which 13CO2 (1 bar) was introduced. The photoreactor (kept at 25 °C and stirred at 600 rpm) was then irradiated (AM 1.5G, 100 mW cm−2). The headspace was then transferred to an air-tight evacuated IR cell (10 cm path length, equipped with KBr windows) and the background (IR cell under vacuum) corrected IR spectrum was recorded to detect 12CO and 13CO.
Flow CO2 electroreduction
Electrochemical experiments were performed on an Ivium Compactstat electrochemical analyser controlled by the Iviumsoft software. In a typical three-electrode setup experiment, a 0.1 M NaHCO3 solution in H2O was prepared and used as the anolyte and catholyte separated by a Nafion membrane. CA was performed with CP∣CoPL as working electrode, Pt foil as counter electrode and Ag/AgCl as reference electrode. The electrochemical cell was capped with rubber septa and the catholyte was purged for 30 minutes with N2 (for screening electrocatalytic performance of different gas composition) or the desired gas composition (for CA at a given gas composition (20 or 100 vol%) for 4 h) at a flow rate of 20 mL min−1 controlled by mass flow controllers (Brooks) to remove oxygen. Afterwards the flow rate was reduced to 9 mL min−1 and a potential of −1.2 V vs. Ag/AgCl was applied. CA was run at −1.2 V vs. Ag/AgCl for 4 h at constant gas flow of 20 or 100 vol% or during screening different gas composition, the electrocatalytic activity was measured for 30 min under N2 followed by increasing the CO2 concentration to 10, 20, 50 and 100 vol% every 45 min. Electrochemical experiments were carried out at room temperature. The formed gaseous products (H2 and CO) were measured by online GC measurement (injection every 4.5 min) using an Shimadzu Tracera GC-2010 Plus gas chromatograph equipped a barrier discharge ionization detector.38 Calibration was performed by determining the response factor by flowing a calibration gas with known CO and H2 composition under the same condition (9 mL min−1).Electrocatalytic lignin model substrate & lignin oxidation
Electrochemical experiments were performed in three-electrode configuration on a PalmSens MultiEMStat3+ potentiostat. In a typical experiment, 0.1 M Na2CO3 solution in MeCN:H2O (1:1 vol%) was used as the electrolyte. To the electrolyte was added 0.01 M of the lignin model substrate or the lignin fraction from pre-treating 250 mg white birch in dioxane/HCl/H2O. The solution was then used as anolyte (8 mL) in electrocatalysis with 0.1 M Na2CO3 solution in MeCN:H2O (1:1 vol%), and separated from the catholyte by a Selemion anion exchange membrane, and the electrochemical cell was capped with rubber septa. Cyclic voltammetry (CV) or CA were performed with CP∣MWCNT as working electrode, Pt foil as counter electrode and Ag/AgCl (sat. KCl) as reference electrode. CV scans were run from −0.2 to 1.0 V vs. Ag/AgCl followed by a backwards scan to −0.2 V vs. Ag/AgCl with a scan rate of 50 mV s−1. CA experiments were performed for 4 h at 1.0 V vs. Ag/AgCl. Electrochemical experiments were carried out at room temperature. To analyse the products after electrocatalysis, the anolyte was acidified to a pH of 3 with 0.1 M HCl, extracted with EtOAc (3 × 5 mL), dried over MgSO4, filtered and dried at 40 °C under vacuum to obtain a light brown solid. The solid was further analysed by 1H NMR spectroscopy in CDCl3 with mesitylene as internal standard. Control experiments were performed without an applied potential and stirring the anolyte containing the lignin model substrate or lignin fraction for 4 h followed work up and analysis by 1H NMR spectroscopy in CDCl3.
Electrocatalytic lignin oxidation coupled to CO2 reduction
Electrochemical experiments were performed in two-electrode configuration on an Ivium Compactstat electrochemical analyser controlled by the Iviumsoft software. In a typical experiment a 0.1 M Na2CO3 solution in MeCN:H2O (1:1 vol%) was prepared and used for the electrolyte. To the electrolyte was added 0.01 M of the lignin model substrate or the lignin fraction from pre-treating 250 mg white birch in dioxane/HCl/H2O. The solution was then used as anolyte (4 mL). A 0.1 M NaHCO3 solution in H2O was used as catholyte. The anolyte and catholyte were separated by a bipolar membrane. Electrolysis was performed with CP∣MWCNT and CP∣CoPL as anode and cathode, respectively. The electrochemical H-type cell was capped with two rubber septa and the catholyte was purged for 30 minutes with 20 vol% CO2 (balanced by N2) at a flow rate of 20 mL min−1 controlled by mass flow controllers (Brooks) to remove oxygen. Afterwards the flow rate was reduced to 9 mL min−1 and a potential of Uapp = −3 V was applied and run for 4 h. Electrochemical experiments were carried out at room temperature. The formed gaseous products (H2 and CO) on the cathode side were measured by online GC measurement (injection every 4.25 min) using Shimadzu Tracera GC-2010 Plus gas chromatograph equipped a barrier discharge ionization detector.38 Calibration was performed by determining the response factor by flowing a calibration gas with known CO and H2 composition under the same condition (9 mL min−1). To analyse the products, after electrocatalysis the anolyte was acidified to a pH of 3 with 0.1 M HCl, extracted with EtOAc (3 × 5 mL), dried over MgSO4, filtered and dried at 40 °C under vacuum to obtain a light brown solid. The solid was further analysed by 1H NMR in CDCl3 with mesitylene as internal standard. Control experiments were performed under the same conditions but without the lignin model substrate or lignin fraction dissolved in the anolyte.
Single-pass CO2 conversion yield
The electrochemical single-pass CO2 conversion yield (%) of CP∣CoPL was calculated by dividing the rate of CO formation (mol min−1) by the flow rate of CO2 (mol min−1) and multiplying the product by 100. The flow rate of CO2 was obtained by transforming mL min−1 to mol min−1 using the ideal gas law equation (pV = nRT), where p is 1 atm, V is the flow rate of CO2 in L min−1 (e.g., 1.8 × 10−3 L min−1 for 20 vol% CO2), R is the ideal gas constant (0.082 atm L mol−1 K−1) and T is 298.15 K.Data analysis
Experiments were performed in triplicates and the results are represented with the mean (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ) and standard deviation (σ) expressed as ± σ with
) and standard deviation (σ) expressed as ± σ with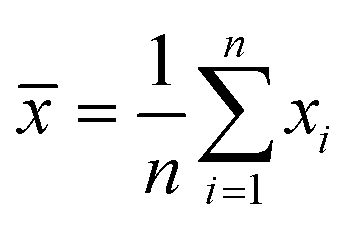
where n is the number of measurements and xi the individually determined value.
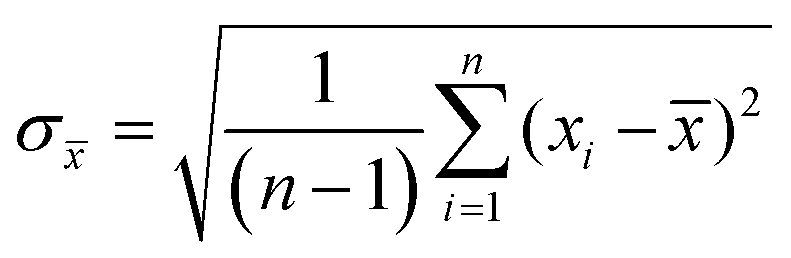
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the European Research Council (ERC) for a Proof of Concept Grant (SolReGen; to E. L. and E. R.), the European commission for a Horizon 2020 Marie Sklodowska-Curie individual Fellowship (GAN 891338, to S. R. J.), the Swiss National Science Foundation (Early Postdoc Fellowship: P2EZP2-191791 to E. L.), the Leverhulme Trust (P80336 to E. L. and E. R.), and the Cambridge Trust (HRH The Prince of Wales Commonwealth Scholarship to S. B.). The authors also thank Dr Nigel Howard at the University of Cambridge for performing elemental and ICP-OES analyses, and Dr Heather Greer at the University of Cambridge for assistance with the electron microscopy, as well as Dr Tessel Bouwens, Dr Bidyut B. Sarma and Dr Yongpeng Liu for useful feedback on the manuscript.References
- S. C. Peter, ACS Energy Lett., 2018, 3, 1557–1561 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- R. Schlögl, Green Chem., 2021, 23, 1584–1593 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- X. Liu, X. Duan, W. Wei, S. Wang and B.-J. Ni, Green Chem., 2019, 21, 4266–4289 RSC.
- TAPPI T 222 om-02, 2006 (Technical Association of the Pulp and Paper Industry, https://www.tappi.org/content/sarg/t222.pdf).
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- C. Zhang and F. Wang, Acc. Chem. Res., 2020, 53, 470–484 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- M. Rafiee, M. Alherech, S. D. Karlen and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- A. C. Forse and P. J. Milner, Chem. Sci., 2021, 12, 508–516 RSC.
- S. E. Renfrew, D. E. Starr and P. Strasser, ACS Catal., 2020, 10, 13058–13074 CrossRef CAS.
- E. Pérez-Gallent, C. Vankani, C. Sánchez-Martínez, A. Anastasopol and E. Goetheer, Ind. Eng. Chem. Res., 2021, 60, 4269–4278 CrossRef.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- A. Khurram, M. He and B. M. Gallant, Joule, 2018, 2, 2649–2666 CrossRef CAS.
- A. Khurram, L. Yan, Y. Yin, L. Zhao and B. M. Gallant, J. Phys. Chem. C, 2019, 123, 18222–18231 CrossRef CAS.
- S. Kar, M. Rahaman, V. Andrei, S. Bhattacharjee, S. Roy and E. Reisner, Joule, 2023, 7, 1496–1514 CrossRef CAS.
- A. Perazio, G. Lowe, R. Gobetto, J. Bonin and M. Robert, Coord. Chem. Rev., 2021, 443, 214018 CrossRef CAS.
- H. Kumagai, T. Nishikawa, H. Koizumi, T. Yatsu, G. Sahara, Y. Yamazaki, Y. Tamaki and O. Ishitani, Chem. Sci., 2019, 10, 1597–1606 RSC.
- X. Du, H. Zhang, K. P. Sullivan, P. Gogoi and Y. Deng, ChemSusChem, 2020, 13, 4318–4343 CrossRef CAS PubMed.
- C. Yang, S. Maldonado and C. R. J. Stephenson, ACS Catal., 2021, 11, 10104–10114 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- M. F. Kuehnel and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 3290–3296 CrossRef CAS PubMed.
- A. V. Puga, Coord. Chem. Rev., 2016, 315, 1–66 CrossRef CAS.
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Nat. Synth., 2023, 2, 182–192 CrossRef.
- E. Lam and E. Reisner, Angew. Chem., Int. Ed., 2021, 60, 23306–23312 CrossRef CAS PubMed.
- A. K. Sinha, U. K. Sharma and N. Sharma, Int. J. Food Sci. Nutr., 2008, 59, 299–326 CrossRef CAS PubMed.
- T. Kawai and T. Sakata, Nature, 1980, 286, 474–476 CrossRef CAS.
- J. C. Colmenares, A. Magdziarz and A. Bielejewska, Bioresour. Technol., 2011, 102, 11254–11257 CrossRef CAS PubMed.
- L. Da Vià, C. Recchi, E. O. Gonzalez-Yañez, T. E. Davies and J. A. Lopez-Sanchez, Appl. Catal., B, 2017, 202, 281–288 CrossRef.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354–365 CrossRef CAS.
- L. Lan, H. Daly, R. Sung, F. Tuna, N. Skillen, P. K. J. Robertson, C. Hardacre and X. Fan, ACS Catal., 2023, 13, 8574–8587 CrossRef CAS PubMed.
- S. Rodríguez-Jiménez, H. Song, E. Lam, D. Wright, A. Pannwitz, S. A. Bonke, J. J. Baumberg, S. Bonnet, L. Hammarström and E. Reisner, J. Am. Chem. Soc., 2022, 144, 9399–9412 CrossRef PubMed.
- C. D. Sahm, G. M. Ucoski, S. Roy and E. Reisner, ACS Catal., 2021, 11, 11266–11277 CrossRef CAS.
- C. P. O'Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- M. Liu, Y. Wen, J. Qi, S. Zhang and G. Li, ChemistrySelect, 2017, 2, 4956–4962 CrossRef CAS.
- P. Parpot, A. P. Bettencourt, A. M. Carvalho and E. M. Belgsir, J. Appl. Electrochem., 2000, 30, 727–731 CrossRef CAS.
- C. Lan, H. Fan, Y. Shang, D. Shen and G. Li, Sustainable Energy Fuels, 2020, 4, 1828–1836 RSC.
- J. King and S. S. C. Chuang, Catal. Commun., 2021, 149, 106219 CrossRef CAS.
- D. Wang, S. H. Lee, S. Han, J. Kim, N. V. T. Trang, K. Kim, E.-G. Choi, P. Boonmongkolras, Y. W. Lee, B. Shin, Y. H. Kim and C. B. Park, Green Chem., 2020, 22, 5151–5160 RSC.
- D. M. Alonso, S. H. Hakim, S. Zhou, W. Won, O. Hosseinaei, J. Tao, V. Garcia-Negron, A. H. Motagamwala, M. A. Mellmer, K. Huang, C. J. Houtman, N. Labbé, D. P. Harper, C. T. Maravelias, T. Runge and J. A. Dumesic, Sci. Adv., 2017, 3, e1603301 CrossRef PubMed.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- H. Ababneh, A. AlNouss and S. A. Al-Muhtaseb, Processes, 2022, 10, 2406 CrossRef CAS.
- C. Song, W. Pan, S. T. Srimat, J. Zheng, Y. Li, Y.-H. Wang, B.-Q. Xu and Q.-M. Zhu, in Studies in Surface Science and Catalysis, ed. S.-E. Park, J.-S. Chang and K.-W. Lee, Elsevier, 2004, vol. 153, pp. 315–322 Search PubMed.
- H. A. Ruiz, M. Conrad, S.-N. Sun, A. Sanchez, G. J. M. Rocha, A. Romaní, E. Castro, A. Torres, R. M. Rodríguez-Jasso, L. P. Andrade, I. Smirnova, R.-C. Sun and A. S. Meyer, Bioresour. Technol., 2020, 299, 122685 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- A. Caravaca, W. E. Garcia-Lorefice, S. Gil, A. de Lucas-Consuegra and P. Vernoux, Electrochem. Commun., 2019, 100, 43–47 CrossRef CAS.
- X. Ma, J. Albertsma, D. Gabriels, R. Horst, S. Polat, C. Snoeks, F. Kapteijn, H. B. Eral, D. A. Vermaas, B. Mei, S. de Beer and M. A. van der Veen, Chem. Soc. Rev., 2023, 52, 3741–3777 RSC.
- M. Peer, S. Mehdi Kamali, M. Mahdeyarfar and T. Mohammadi, Chem. Eng. Technol., 2007, 30, 1418–1425 CrossRef CAS.
- P. Wattanaphan, T. Sema, R. Idem, Z. Liang and P. Tontiwachwuthikul, Int. J. Greenhouse Gas Control, 2013, 19, 340–349 CrossRef CAS.
- S. J. Cobb, A. M. Dharani, A. R. Oliveira, I. A. C. Pereira and E. Reisner, Angew. Chem. Int. Ed., 2023, 62, e202218782 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, R. Bertrand, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- DECHEMA and FutureCamp, Roadmap Chemie 2050Verband der Chemischen Industrie e.V., https://www.vci.de/vci/downloads-vci/publikation/2019-10-09-studie-roadmap-chemie-2050-treibhausgasneutralitaet.pdf, 2023 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03258b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |