
The effects of intercalated environmental gas molecules on carrier dynamics in WSe2/WS2 heterostructures†
Yanxue
Zhang‡
a,
Hongsheng
Liu‡
 b,
Yanyan
Zhao
a,
Jiaqi
Lin
*c,
Yizhen
Bai
b,
Jijun
Zhao
*ab and
Junfeng
Gao
*ab
b,
Yanyan
Zhao
a,
Jiaqi
Lin
*c,
Yizhen
Bai
b,
Jijun
Zhao
*ab and
Junfeng
Gao
*ab
aState Key Laboratory of Structural Analysis, Optimization and CAE Software for Industrial Equipment (Dalian University of Technology), Ministry of Education, Dalian, 116024, China. E-mail: gaojf@dlut.edu.cn; zhaojj@dlut.edu.cn
bKey laboratory of Materials Modification by Laser, Ion and Electron Beams (Dalian University of Technology), Ministry of Education, Dalian, 116024, China
cThe School of Bioengineering, Dalian University of Technology, Dalian, 116024, China. E-mail: jqlin@dlut.edu.cn
First published on 5th April 2023
Abstract
Effective tuning of carrier dynamics in two-dimensional (2D) materials is significant for multi-scene device applications. Using first-principles and ab initio nonadiabatic molecular dynamics calculations, the kinetics of O2, H2O, and N2 intercalation into 2D WSe2/WS2 van der Waals heterostructures and its effect on carrier dynamics have been comprehensively explored. It is found that the O2 molecule prefers to dissociate into atomic O atoms spontaneously after intercalation of WSe2/WS2 heterostructures, whereas H2O and N2 molecules remain intact. O2 intercalation significantly speeds up the electron separation process, while H2O intercalation largely speeds up the hole separation process. The lifetime of excited carriers can be prolonged by O2 or H2O or N2 intercalations. These intriguing phenomena can be attributed to the effect of interlayer coupling, and the underlying physical mechanism for tuning the carrier dynamics is fully discussed. Our results provide useful guidance for the experimental design of 2D heterostructures for optoelectronic applications in photocatalysts and solar energy cells.
New conceptsThe 2D van der Waals transition metal dichalcogenide (TMD) heterostructures with type-II band alignment are promising for applications in optoelectronics. During the process of fabrication and transfer, gas molecules can be easily intercalated into the interlayer space of the heterostructure. However, knowledge about the effect of gas molecule intercalation on the photo-generated carrier dynamics of heterostructures is still lacking. Herein, we systematically explore the carrier dynamics of environmental gas molecule (O2/H2O/N2) intercalation in WSe2/WS2 heterostructure using the advanced TDDFT method. Intercalation of either O2 or H2O or N2 reduces the band gap of pristine WSe2/WS2 heterostructure. The type-II band alignment is robust after O2/H2O/N2 intercalation. Importantly, intercalation suppresses the electron–hole recombination and thus prolongs the lifetime of excited carriers. Our exploration provides new ideas for using gas molecule intercalation to modulate the carrier dynamics of van der Waals TMD heterojunctions. |
Introduction
The van der Waals (vdW) heterostructures constructed by vertically stacking different 2D materials bring new physical properties and enrich the family of 2D materials.1–5 Compared with pristine 2D materials, vdW heterostructures show unique electronic and optical properties that can be used for transistors and photodetectors.6–9 Recent theoretical and experimental studies have demonstrated the feasibility of band alignment engineering by stacking different 2D monolayer materials.7,10–13 Indeed, ultrafast electron transfer in transition metal dichalcogenide (TMD) vdW heterostructures, such as MoS2/MoSe2, WSe2/MoSe2, and MoS2/WS2, has been confirmed.14–19 In particular, the unique type-II band alignment promotes the interlayer exciton formation20–22 and effectively separates the photo-generated electron–hole pairs, which holds promise in the fields of photocatalysts and solar cells.23–26 Due to the robust type-II band alignment, WSe2/WS2 vdW heterostructure has attracted considerable attention.27–38In vertical heterostructures, the vdW gap between monolayer components offers space for the intercalation of ions and molecules, thereby giving rise to new opportunities for property modulation. It has been evidenced that ion intercalation into TMD heterostructures has a significant impact on the chemical and physical properties and leads to broad applications in energy storage.39–41 For example, Liang et al. proposed a strategy of water and oxygen incorporation to tune the interlayer space and hydrophilicity of MoS2 and achieved high Zn2+ storage capacity.42 Zhang et al. significantly enhanced the photoluminescence and photocurrent of TMD materials through tailoring the interlayer interaction by soft plasma intercalation.43 Recently, Qian et al. successfully synthesized a chiral molecule-intercalated TaS2 or TiS2 superlattice, which provides a platform for studying the chiral-induced spin selectivity effect.44 Beyond, Liu et al. successfully synthesized Cu atom intercalated NbS2, which improves the electrical conductivity and suppresses the transition temperature and volume fraction of the superconductor.45
In the laboratory, the vdW heterostructures can be synthesized by mechanically-assembled stacking, chemical vapor deposition, and physical epitaxy method.46,47 In the process of fabrication and transfer, intercalation of the ambient gas molecules into the interlayer space is inevitable.48 On the one hand, similar to the effects of ion intercalation, molecule intercalation could also have a significant influence on the electronic and optoelectronic properties of TMD heterostructures,40,43 which should be carefully considered. On the other hand, intercalation of ambient gas molecules could be an easy but powerful way to modify the structure, stacking mode, and related properties of 2D TMD materials.49 Small molecule (such as O2, H2O) intercalation in interlayer space has been extensively observed experimentally.50–57 To our best knowledge, however, the intercalation behavior of ambient gas molecules such as O2, H2O, and N2 and their influence on the carrier dynamics of vdW heterostructures has been rarely studied.
In this study, taking the WSe2/WS2 vdW heterostructure as an example, the intercalation effect of O2, H2O, and N2 molecules on the carrier dynamic was carefully investigated by using the state-of-the-art Hefei-NAMD58 code (more details in ESI†), which can well describe the dynamic behavior of excited state carriers at time, space, momentum, and energy scales.59–64 We find that O2, H2O, and N2 intercalation reduces the band gap of the system but would not change the type-II band alignment of the pristine heterostructure. Interestingly, O2 intercalation, H2O intercalation, and N2 intercalation can effectively modulate carrier dynamics in WSe2/WS2 vdW heterostructures. Our work provides a new perspective into the carrier dynamics modulation in WSe2/WS2 vdW heterostructures by molecule intercalation.
Results and discussions
The equilibrium lattice constants of monolayer WS2 and WSe2 are 3.183 Å and 3.317 Å, respectively, in agreement with previous theoretical results.65–68 According to previous study, the WSe2/WS2 vdW heterostructure with C7 stacking is the most stable configuration.35,37,38,65,69,70 Thus, we focus on C7 configuration in this study, while our test calculations on other geometries like T stacking demonstrate that molecule intercalation would not change the sequence of energies for the bilayer heterostructures. In order to reduce the lattice mismatch, the lattice constant of the WSe2/WS2 heterostructure is set to be the average value of the lattice constants of WSe2 and WS2. The WSe2 layer is compressed by 2%, and the WS2 layer is stretched by 2%. To check the strain effect on the band gap, we calculated the band structures of WSe2 and WS2 monolayers with different strain values, as shown in Fig. S1 (ESI†). The results show that a strain of 4% has a significant effect on the band gap. Fortunately, a strain of 2% has only a small effect on the band gap. Therefore, the deformation of both layers is a good choice to maintain the band gap, consistent with the previous theoretical work.67,71 According to the projected density of states (DOS) in Fig. 1(e) and the orbital projected band structures in Fig. S2 (ESI†) for pristine WSe2/WS2 vdW heterostructure, the valence band maximum (VBM) mainly originates from WSe2 and the conduction band minimum (CBM) mainly comes from WS2. Therefore, WSe2/WS2 vdW heterostructures exhibit typical type-II band alignment, which is promising for photocatalysts and solar cells due to the easy separation of photo-generated electron–hole pairs. Using the Perdew–Burke–Ernzerhof functional (PBE) functional, the theoretical band gap of WSe2/WS2 vdW heterostructure is 0.81 eV, which is rather close to the previous reports (0.82 eV).65 The band gap is largely reduced in the heterostructure compared with monolayer WS2 (1.80 eV) and WSe2 (1.54 eV),66,72 as shown in Fig. S1 (ESI†).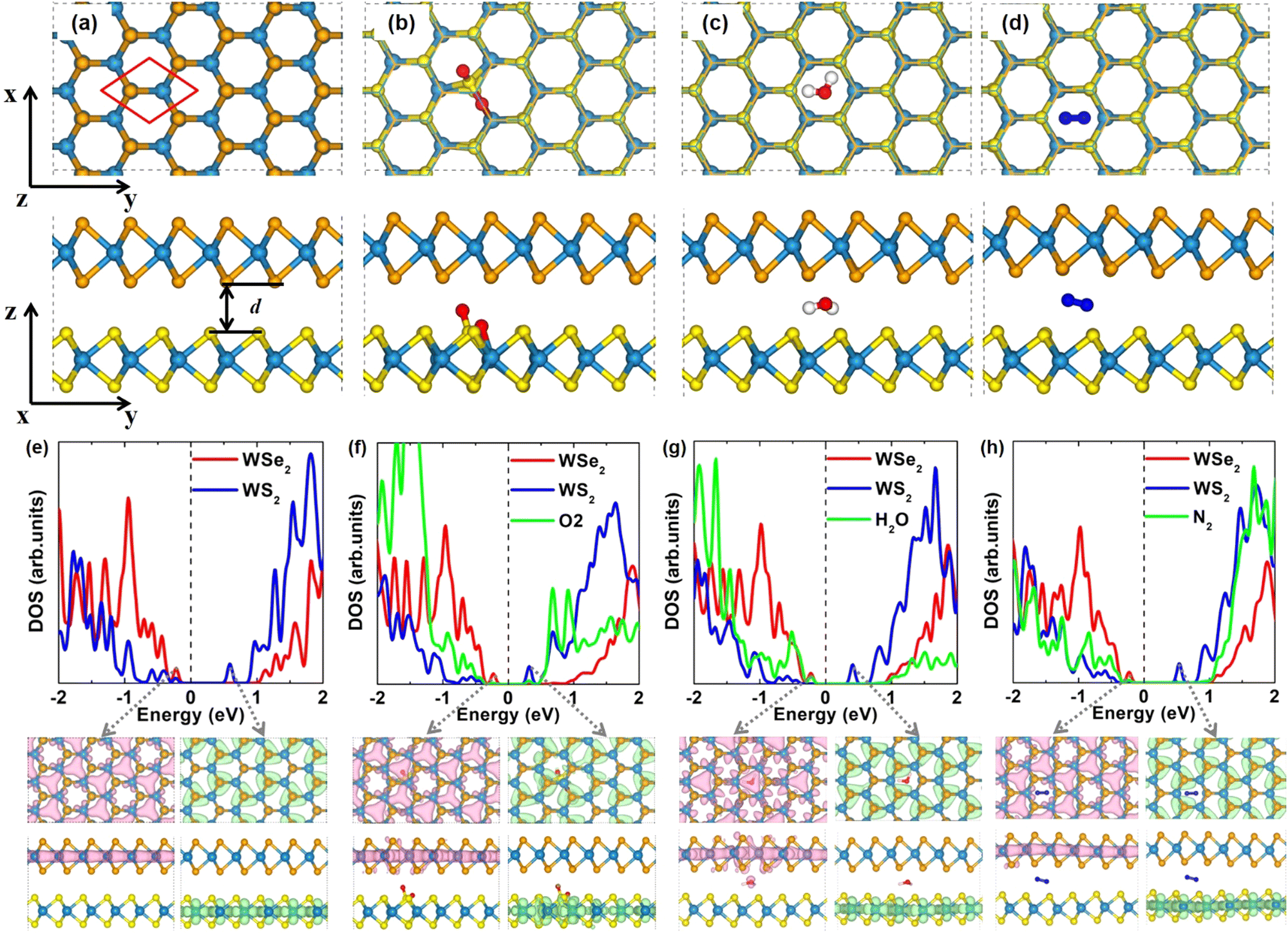 | ||
| Fig. 1 Optimized structure, projected density of states (DOS), and charge density of (a), (e) pristine, (b), (f) O2 and (c), (g) H2O, and (d), (h) N2 intercalated WSe2/WS2 vdW heterostructures. The white, red, dark blue, yellow, orange, and blue balls represent H, O, N, S, Se, and W atoms, respectively. To visualize the structure of molecules in the interlayer space, the WSe2 layer is shown in thin-stick style in (b)–(d). The pink and green colors represent the charge density of VBM and CBM, respectively. The isosurface value is set as 0.0008 |e| Bohr−3. | ||
As shown in Fig. 1(a)–(d), we build a 3 × 3 × 1 orthogonal supercell for WSe2/WS2 vdW heterostructure and insert one O2 or H2O or N2 molecule per supercell to investigate the intercalation effect. The lateral dimensions of the supercell are 9.75 Å and 16.89 Å, which are sufficiently large to avoid the intercalated molecule and its periodic replicas. Different intercalation sites for O2 molecules were tested (see Fig. S3, ESI†). O2 molecules are more likely adsorbed and dissociated on the WS2 layer. Notably, O2 intercalation did not change the stacking geometry of C7. Upon intercalation, the O2 molecule will spontaneously dissociate. As depicted in Fig. 1(b), one S–W bond is broken, and a new W–O–S–O bond geometry is formed. The detailed bond lengths are summarized in Table S1 (ESI†). Intercalation of the O2 molecule increases the interlayer spacing d by about 0.07 Å. (see Table S1, ESI†). Fig. S4 (ESI†) shows that the dissociation barrier of the O2 molecule in the interlayer of the WSe2/WS2 heterostructure is 0.18 eV. In contrast, the O2 molecule prefers physisorption on pristine monolayer WS2.72,73 Therefore, the confinement environment does activate O2 molecules, which can be understood by analyzing the charge transfer. The charge density difference (Δρ) was calculated as follows,
| Δρ = ρtotal − ρWSe2 − ρWS2 − ρO2/H2O/N2 | (1) |
Different intercalation sites for H2O molecules have also been tested (Fig. S6, ESI†), and the most stable configuration is presented in Fig. 1(c). Again, the stacking pattern of C7 is not changed by H2O intercalation. In contrast to the case of O2 intercalation, the intercalated H2O molecule would not dissociate. The H2O molecule prefers to locate at the hollow site independent of initial configurations. The two H atoms of the H2O molecule tend to be closer to the S atoms of the WS2 layer. Intercalation of the H2O molecule enlarges the interlayer distance by 0.13 Å. The details of the interlayer distances of H2O intercalated heterostructures can be found in Table S2 of ESI.† Compared to the H2O molecule in vacuum, the intercalated one has a longer O–H bond and a narrower H–O–H bond angle, indicating that the confinement environment also activates the H2O molecule. The charge density difference (Fig. S5c, ESI†) reveals the electron transfer between the heterostructures and the H2O molecule.
Possible intercalation sites for the N2 molecule are examined as shown in Fig. S7 (ESI†), and the most stable configuration is shown in Fig. 1(d). N2 molecule intercalation also did not change the stacking pattern of C7, and the N2 molecule did not dissociate. N2 molecule prefers to locate at the hollow site after optimization independent of initial configurations. Intercalation of the N2 molecule increases the larger interlayer distance by about 0.32 Å. The details of the bond lengths and interlayer distances of N2 intercalated heterostructures are shown in Table S3 of ESI.† Furthermore, the charge density difference of the N2 intercalated heterostructure is calculated as shown in Fig. S5d (ESI†), from which we can see that the interface charge is reduced compared with the pristine heterostructure (Fig. S5a, ESI†).
It is noted that the pristine WSe2/WS2 heterostructure is built by deforming both layers. Whether it is the strain that causes O2 dissociation is unclear. To examine this, the O2, H2O, and N2 intercalation in an unstrained bilayer WS2 layer with 2H stacking was investigated, as shown in Fig. S8 (ESI†).68,74 Similar to the case of the intercalation in WSe2/WS2 heterostructure, in the bilayer WS2, O2 tends to dissociate while H2O and N2 remain intact. Therefore, the strain effect on the dissociation of O2 molecules can be excluded. In experiments, small molecule intercalation in the interface of graphene and substrate has been extensively observed.50–52,56 For example, O2 and H2O intercalation in the interface of graphene/Cu(111) surface leads to surface oxidation and promotes the decoupling of graphene from the substrate.56 Besides, the surface and catalysis are strongly influenced by a 2D cover, and the interface between adjacent 2D layers or 2D overlayers and metal substrates can be interesting 2D nanocontainers as well as 2D nanoreactors.53 According to the above analysis, the dissociation of the O2 molecule is expected to be the result of the space confinement effect.
According to the projected density of states (DOS) and charge densities in Fig. 1(e)–(h), intercalation of O2 or H2O or N2 does not change the feature of type-II band alignment of the WSe2/WS2 vdW heterostructure, i.e., the VBM mainly from WSe2 and the CBM mainly from WS2, respectively. However, molecule intercalation can prominently reduce the band gap of the system. The band gaps of O2, H2O, and N2 intercalated heterostructures are 0.54 eV, 0.63 eV, and 0.76 eV, respectively. As shown in Fig. 1(e), the VBM and CBM of pristine heterostructure are located at −0.276 eV and 0.538 eV, respectively. It can be seen in Fig. 1(f) that the decreased band gap in O2 intercalated heterostructure is caused by the downward movement of conduction bands of the WS2 layer (CBM: 0.304 eV) and slightly upward shift of valence bands of the WSe2 layer (VBM: −0.233 eV) after O2 intercalation. The downward movement of conduction bands near CBM can be used as an intermediate state of electron transfer from the WSe2 layer to the WS2 layer and is expected to promote the fast charge transfer from the WSe2 layer to the WS2 layer. To clearly see the energy positions of O-related defect levels in the electronic structures, the projected density of states of molecules in Fig. 1 is magnified by a factor of one hundred. From Fig. 1(f), we can see that O defect levels do not contribute to the VBM and CBM. Impurity energy levels induced by O2 dissociation locate between the CBM of the WS2 layer and the CBM of the WSe2 layer in the conduction band region. Therefore, during the electron transfer process, the donor state is not changed. O defect levels can be regarded as intermediate states of electron transfer from the WSe2 layer to the WS2 layer. In the valence band area, the valence bands of the WS2 layer are shifted down, the coupling of the WSe2 layer and WS2 layer is reduced after O2 intercalation, and a small part of O impurity energy levels exist between the VBM of the WS2 layer and VBM of WSe2 layer. As shown in Fig. 1(g), the reduced band gap in H2O intercalated heterostructure is due to the shift down of the conduction bands of the WS2 layer (CBM: 0.407 eV) and mildly upward shift of VBM of the WSe2 layer (VBM: −0.223 eV). The conduction bands near the CBM of the WS2 layer are moving down, which can also serve as an intermediate state of electron transfer between the WSe2 and WS2 layers. In the valence band area, the valence bands of the WS2 layer are shifted down and the coupling of WSe2 and WS2 is reduced, similar to O2 intercalation. To clearly show the effect of the H2O molecule, the project DOS of the H2O molecule is magnified by 100 times in Fig. 1(g). From this, we can see that the projected DOS of the H2O molecule located between the VBM of the WS2 layer and VBM of the WSe2 layer, which can be an intermediated state between the WS2 layer and the WSe2 layer and promote hole transfer. The charge density of VBM is composed of H2O molecule and WSe2 layer, indicating that the highest occupied molecular orbital of the H2O molecule is elevated and can be a hole acceptor. As shown in Fig. 1(h), in the conduction band area, N2 intercalation also induced the conduction bands of the WS2 layer (CBM: 0.513 eV) and VBM of the WSe2 layer (VBM: −0.248 eV) to move down and up slightly. The conduction band near CBM is also moved downward, which can be as an intermediate state between the CBM of the WSe2 layer and the CBM of WS2 layer and promote electron transfer. In the VBM, the coupling of WSe2 and WS2 is also reduced. The projected DOS of the N2 molecule is also magnified 100 times for clarity, which has less effect on the VBM than the H2O molecule.
The large supercell-induced band folding containing seven irreducible K points is shown in Fig. S9 (ESI†). In order to clearly show the changes in band edge position, the band structures of pristine, O2 intercalated, H2O intercalated, and N2 intercalated WSe2/WS2 heterostructures are unfolded as shown in Fig. S10b, d, f and h (ESI†), respectively.75 Compared with the pristine heterostructure, the CBM band offset ΔEC between the CBM at K of WS2 (CB-WS2@K) state and the CBM at K of WSe2 (CB-WSe2@K) state increased after molecule intercalation. The VBM at Γ of WSe2 (VB-WSe2@Γ) state moved down after molecule intercalation. The VBM at K of WS2 (VB-WS2@K) state also shifted down. Correspondingly, the VBM band offset ΔEV between the VB-WS2@K state and the VB-WSe2@K state increased after molecule intercalation. The ΔEC and ΔEV of pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures are summarized in Table S4 (ESI†).
As illustrated in Fig. 2(a), we focus on three processes about carrier dynamic behavior of the heterostructures: (I) transfer of photo-generated electrons from WSe2 to WS2, (II) transfer of photo-generated holes from WS2 to WSe2, (III) recombination of the photo-generated electrons and holes.
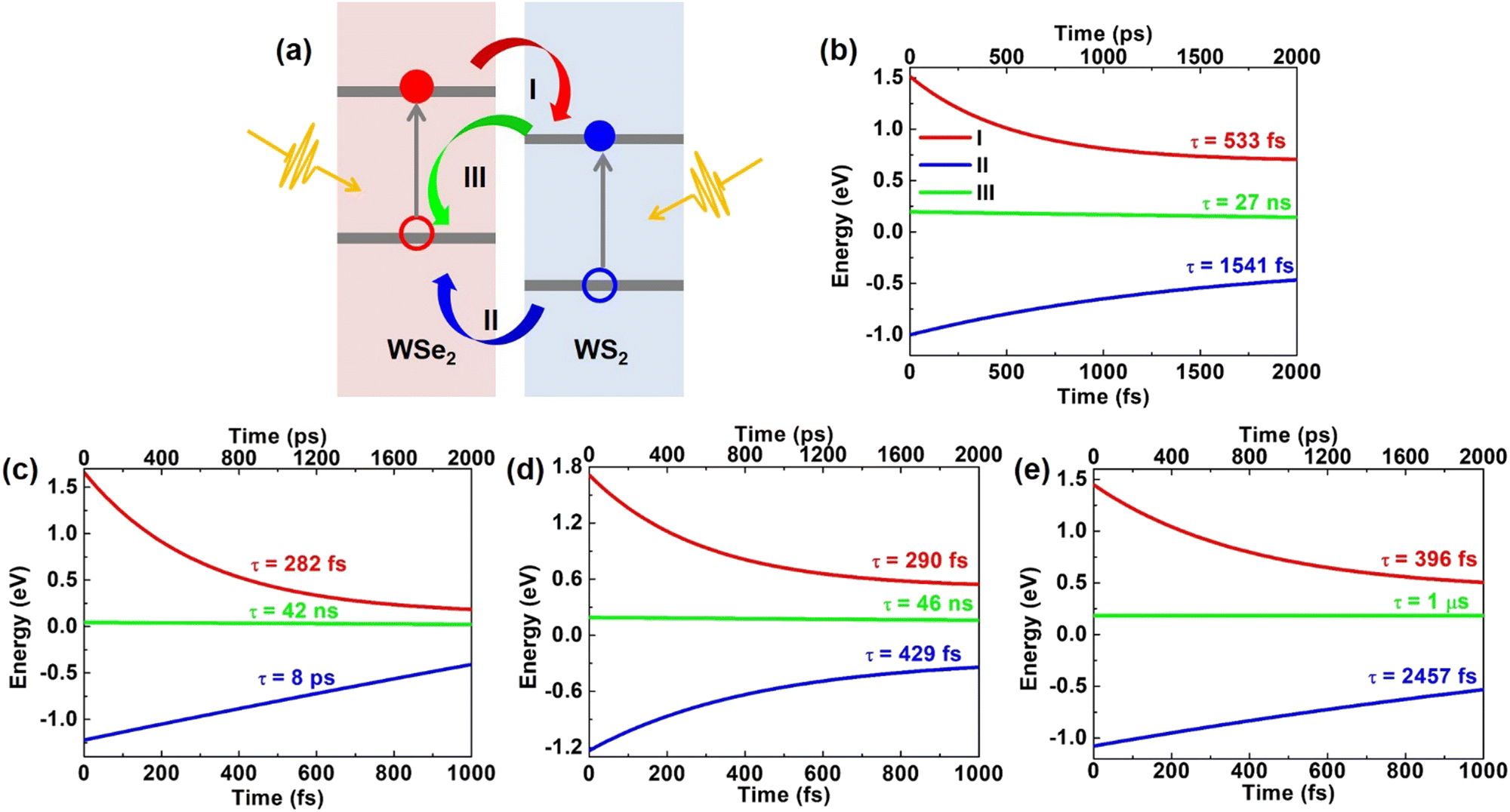 | ||
| Fig. 2 (a) Schematic plots of (I) electron transfer, (II) hole transfer, and (III) electron and hole recombination processes. (b–e) show the fitting curves of time-dependent carrier energy change of three processes in (a) for pristine, O2 intercalated, H2O intercalated, and N2 intercalated WSe2/WSe2 vdW heterostructures. | ||
The time-dependent electron localization behavior of process (I) for pristine, O2 intercalated, H2O intercalated, and N2 intercalated vdW heterostructures at 300 K has been carefully investigated. (for details, see Fig. S11a–d of ESI†). For pristine heterostructure, the initial electron distribution on the WSe2 and WS2 layers is 29% and 71%, respectively. After 12 fs, the spatial electron population on the WSe2 layer decreases to 11%. Our finding agrees well with a previous theoretical study, which suggested ultrafast electron transfer from the WSe2 layer to the WS2 layer. In addition, recent experimental transient absorption spectroscopy has verified the ultrafast electron transfer from WSe2 to WS2 (∼40 fs).65,76 In this process, the adiabatic (AD) mechanism makes the main contribution, indicating that the process is sensitive to phonon excitation. We will discuss the dominant phonon modes in the charge transfer process later. For O2 intercalated heterostructures, the initial electron population on the WSe2 layer is 39%. After 18 fs, the electron population on WSe2 decreases to 19%. At 463 fs, the electron population on the WSe2 layer completely diminishes to 0%. In this process, both AD and nonadiabatic (NA) mechanisms contribute to ultrafast electron transfer. The NA mechanism is related to direct charge hopping. For H2O intercalated heterostructures, the initial electron distribution on the WSe2 layer is 34%. After 18 fs, the electron population on WSe2 decreased to 22%. At 1 ps, the electron population on the WSe2 layer decreases to 2%. In this process, the AD mechanism plays a key role. For the N2 intercalated heterostructure, the initial electron distribution on the WSe2 layer is 70%. After 18 fs, the electron population on WSe2 decreased to 21%. AD mechanism dominates the fast charge transfer. Then, the electron population on the WSe2 layer decreased and converged to 2% within 1 ps. In this process, AD and NA mechanism plays a key role. By fitting the average electron energy with exponential decay function [A × exp(−t/τ)], the electron transfer time for pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures is 533 fs, 282 fs, 290 fs, and 396 fs, respectively, as shown in Fig. 2(b)–(e) (red lines). Therefore, O2, H2O, and N2 intercalation can speed up the intervalley electron transfer process effectively. Details of the time-dependent electron energy profile of process (I) are shown in Fig. S11e–h (ESI†). The electron transfer pathway in momentum spaces is CB-WSe2@K → CB-WS2@3 → CB-WS2@K state for pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures.
For pristine heterostructure, the initial hole distribution on the WS2 layer is 94%. Then, holes migrate from the WS2 layer to the WSe2 layer quickly. Within 40 fs, the hole population on the WS2 layer decreases to 57%. Subsequently, at 1 ps, the hole in the WSe2 layer gradually increases and converges to 76%. From Fig. S12e (ESI†), we can see that within 2 ps, the hole transfer pathway in momentum spaces is VB-WS2@K → VB-WSe2@5 → VB-WSe2@K state. The average hole energy reached the VB-WSe2@5 state within 1 ps. For O2 intercalated heterostructures, the initial hole distribution on the WS2 layer is 97%. After 25 fs, the hole population on the WS2 layer decreases to 76%. At 73 fs, the hole population on the WS2 layer decreases to 57%. In this process, the AD mechanism dominates. Subsequently, there is a slow hole transfer until 1 ps, where the hole population on the WSe2 layer converges to 74%. In this process, both AD and NA mechanisms take effect. As shown in Fig. S12f (ESI†), the hole transfer pathway in momentum spaces is VB-WS2@K → VB-WSe2@Γ → VB-WSe2@K state and the average hole energy reached the VB-WSe2@Γ state within 1 ps. For H2O intercalated heterostructures, the initial hole population on the WS2 layer is 90%. After 18 fs, the hole population on the WS2 layer decreases to 52%. In this process, the AD mechanism dominates. Then, the hole population on the WSe2 layer slowly increased and converged to 74%. In this process, AD and NA mechanisms together promote hole transfer. As shown in Fig. S12g (ESI†), the hole transfer pathway in momentum spaces is VB-WS2@K → VB-WSe2@Γ → VB-WSe2@K state and the average hole energy approaches the VB-WSe2@K state within 1 ps. For N2 intercalated heterostructures, the initial hole population on the WS2 layer is 98%. The hole from the WS2 layer transfers to the WSe2 layer, and the hole population on the WS2 layer decreases to 39% within 60 fs. AD mechanism contributes to the fast hole transfer. Then, the hole localization on the WS2 layer oscillated until 230 fs. In this process, the AD mechanism makes the main contribution. Subsequently, the hole population on the WSe2 layer slowly increased and converged to 66%. In this process, AD and NA mechanisms together promote hole transfer. As shown in Fig. S12h (ESI†), the hole transfer pathway in momentum spaces is VB-WS2@K → VB-WSe2@Γ → VB-WSe2@K state and the average hole energy approaches the VB-WSe2@Γ state within 1 ps. Details of the time-dependent hole localization of process (II) are given in Fig. S12a–d (ESI†). By fitting the average hole energy with an exponential decay function [A × exp(−t/τ)], we derive the hole transfer time as 1541 fs, 8062 fs, 429 fs, and 2457 fs for pristine, O2 intercalated, H2O intercalated and N2 intercalated heterostructures, respectively, as shown in Fig. 2(b)–(e) (blue lines). Therefore, H2O intercalation can promote ultrafast hole transfer from the VB-WS2@K state to the VB-WSe2@K state, while O2 and N2 intercalation cannot promote hole transfer. Details of the time-dependent hole energy of process (II) are shown in Fig. S12e–h (ESI†).
To investigate the electron–hole recombination, i.e., the process (III) in Fig. 2(a), the time-dependent electron populations on the VBM of heterostructures have also been calculated for pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures at 300 K. (details in Fig. S13a–d, ESI†). The initial electron population on the WSe2 layer (VBM of the heterostructures) is 0%. For pristine heterostructure, after 2 ns, the electron population on the VBM rises to 7.6%. For O2 intercalated heterostructures, after 2 ns, the electron population on the VBM grows to 4.9%. For the case of H2O intercalation, after 2 ns, the electron population on the VBM rises to 4.5%. For the N2 intercalated heterostructure, the electron population on the VBM is only 0.2% after 2 ns. By fitting the population of the electron with exponential decay: exp(−t/τ) ≈ 1 − t/τ, the electron–hole recombination time is calculated to be 27 ns, 42 ns, 46 ns, and 1 μs for pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures, respectively, as shown in Fig. 2(b)–(e) (green lines). Therefore, molecular intercalation can effectively slow down the recombination. In the process of recombination, the NA mechanism dominates because there is no crossing between the donor state and the acceptor state.
Furthermore, we consider the effect of temperature on the carrier dynamics by performing NAMD simulations at 78 K. Our results show that for both pristine and molecule intercalated heterostructures, the decrease in temperature slightly slows down the electron transfer process but greatly slows down the hole transfer process and the recombination process. The detailed discussion can be found in the ESI† (Fig. S11–S19).
The time dependent energy level evolution of pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures at 300 K is shown in Fig. 3(a)–(d), respectively. From this, we can see that the donor state (CB-WSe2@K) is strongly orbital hybridized and mixed with the intermediate state during the electron transfer process. These hybrid states confirm that the AD mechanism dominates the fast electron transfer process. Especially, the CBM + 3 level and CBM + 4 level cross in intercalated heterostructures, which promotes fast electron transfer. O2 intercalation introduces a large number of intermediate states, which further promotes the rapid transfer of electrons. In the hole transfer process, the hybrid VBM-2 level in pristine heterostructure becomes flat, localized, and gapped with VBM-1 level and VBM-3 level after O2 intercalation, which suppresses the hole transfer. Similarly, the VBM-2 level is gapped with the VBM-1 level in N2 intercalated heterostructure, which inhibits the hole transfer. For H2O intercalated heterostructure, the donor state (VB-WS2@K) is mixed with the intermediate states, which promotes rapid hole transfer.
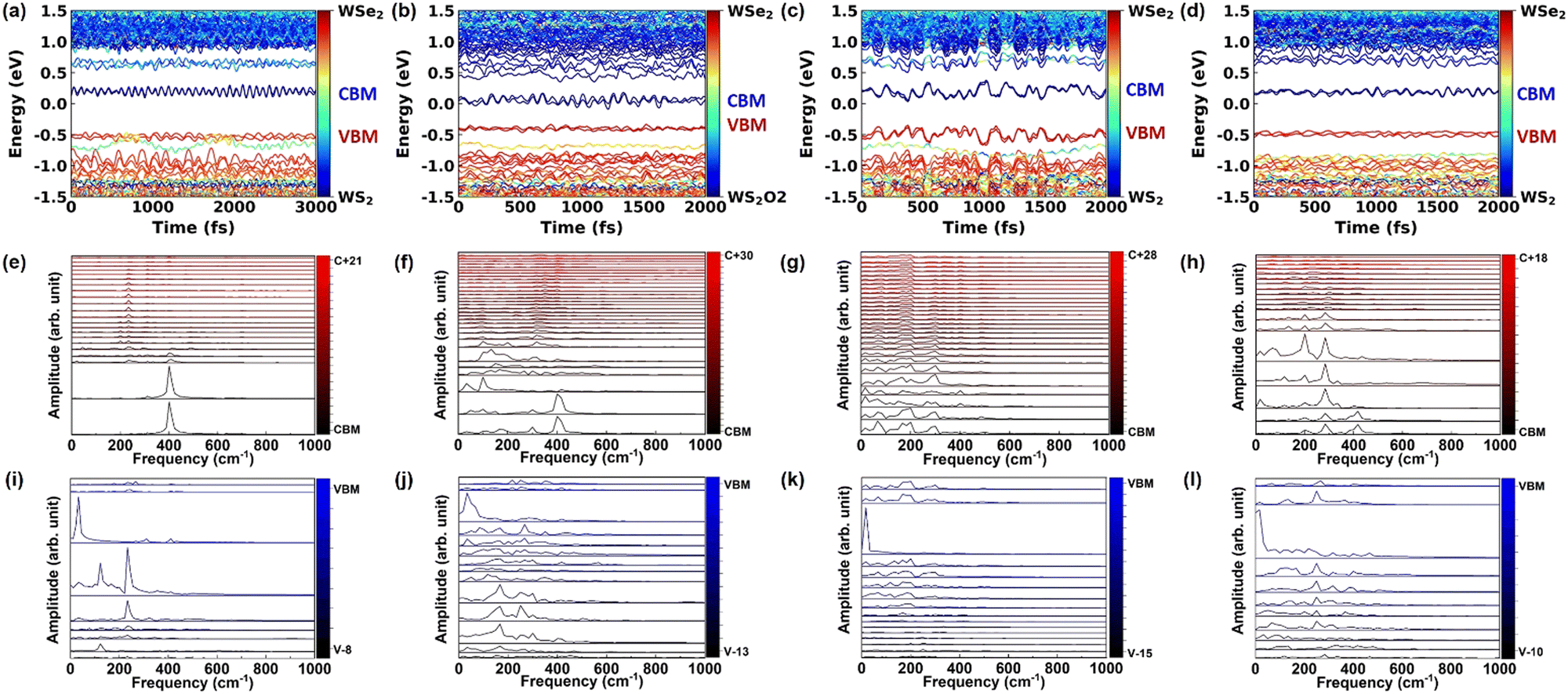 | ||
| Fig. 3 The time-dependent energy evolution at Γ point of (a) pristine, (b) O2 intercalated, (c) H2O intercalated, and (d) N2 intercalated heterostructures at 300 K, respectively. The color bar in (a)–(d) represents the contribution from WSe2 layer (red part) and WS2 layer (blue part), respectively. Fourier transforms of the donor and relevant acceptor energy states during the process of electron transfer and hole transfer in (e), (i) pristine, (f), (j) O2 intercalated, (g), (k) H2O intercalated, and (h), (l) N2 intercalated WSe2/WS2 vdW heterostructures at 300 K, respectively. The color bar in (e)–(l) represent the band number. | ||
Electron–phonon coupling during charge transfer can be obtained from the Fourier transform of the time-dependent energy level evolution. The electron–phonon coupling during charge transfer in pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures at 300 K and 78 K is shown in Fig. 3(e)–(l) and Fig. S14(e)–(l) (ESI†), respectively. First, we discuss the phonon coupling in the pristine heterostructure. In the electron transfer process, the donor state (CBM + 21), intermediate states (CBM + 1–CBM + 20), and acceptor state (CBM) mainly couple with phonons located at 200 cm−1, 233 cm−1, and 400 cm−1 (Fig. 3(e)). In the hole transfer process, the donor state (VBM-8), intermediate state (VBM-7–VBM-1), and acceptor state (VBM) mainly couples with phonons located at 33 cm−1, 122 cm−1, 178 cm−1, 233 cm−1, 267 cm−1, and 411 cm−1 (Fig. 3(i)). Compared with the pristine heterostructure, molecule intercalation induces many low-frequency phonon modes participating in the electron and hole transfer process, as shown in Fig. 3(f)–(h) and (j)–(l), respectively. The phonon spectra77 of pristine, O2 intercalated, H2O intercalated, and N2 intercalated WSe2/WS2 heterostructures are shown in Fig. S15a–d (ESI†). It can be seen that molecule intercalation induces high-frequency phonons, but phonons with a frequency larger than 1000 cm−1 do not participate in band edge coupling, as shown in Fig. 3(e)–(l) and Fig. S14e–l (ESI†).
Furthermore, we investigate the origin of different carrier dynamic behavior in pristine and intercalated WSe2/WS2 vdW heterostructures. The transfer probability of charge carriers is determined by the value of nonadiabatic coupling (NAC), which is described as:78,79
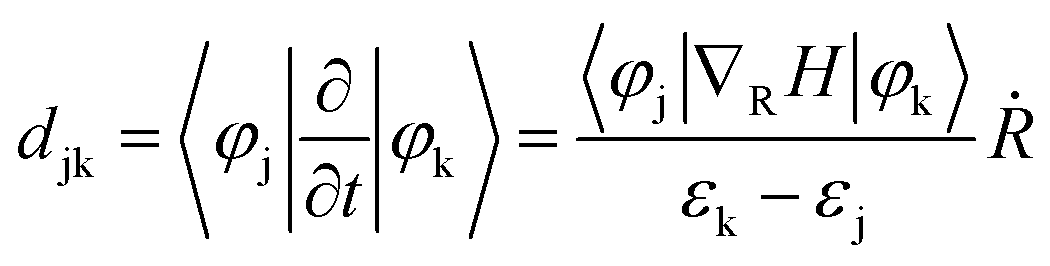 | (2) |
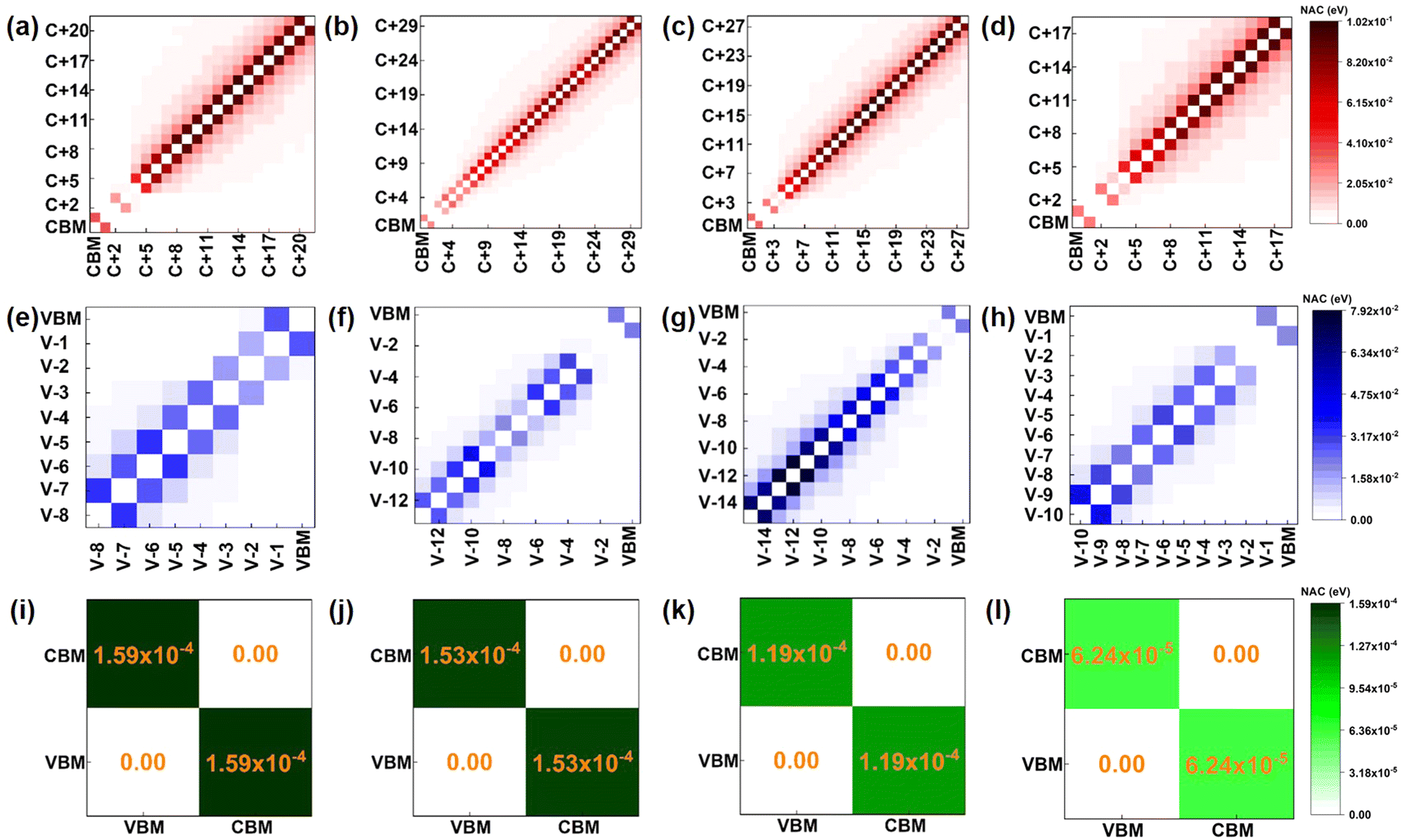 | ||
| Fig. 4 The averaged NAC values between the orbitals in the process of electron transfer, hole transfer, and recombination in (a), (e), (i) pristine, (b), (f), (j) O2 intercalated, (c), (g), (k) H2O intercalated, and (d), (h), (l) N2 intercalated WSe2/WS2 vdW heterostructures at 300 K, respectively. Here, C+1, C+2, … means CBM+1, CBM+2,… level, respectively; V-1, V-2, …means VBM-1, VBM-2,… energy level, respectively. | ||
In the process of electron transfer (process (I) in Fig. 2(a)), the averaged NAC values between different electronic states for pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures are plotted in Fig. 4(a)–(d). In Fig. 4, each square represents the average NAC value between two different orbitals. The average NAC value is indicated by the color of the square. The color ruler is shown on the right. The darker the color, the higher the NAC value. The NAC value for two identical orbitals is zero, which is represented by white squares on the diagonal. The size of the square is meaningless. In general, according to eqn (2), the NAC value between adjacent energy levels is larger than that between distant energy levels. For example, in the pristine heterostructure, the averaged NAC value between CBM + 21 and CBM + 20 is 0.087 eV, while the NAC between CBM + 21 and CBM + 19 is 0.025 eV. Therefore, photo-excited electrons prefer to relax to the energetically adjacent states. The average NAC values between conduction states and the VBM are very small, as seen in Fig. 4(i)–(l). This accounts for the difficulty of electron–hole recombination, i.e., process (III) in Fig. 2(a). The average NAC values between CBM and VBM are 1.59 × 10−4, 1.53 × 10−4, 1.19 × 10−4, and 6.24 × 10−5 for pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructure, respectively. Therefore, the recombination of electron and hole is the most difficult in N2 intercalated heterostructure.
The average NAC values of O2, H2O, and N2 intercalated heterostructures are smaller than the pristine heterostructure, which agrees with the result of Fig. S13a–d (ESI†). Within 2 ns, pristine heterostructure has a fast recombination rate, followed by O2 intercalation, H2O intercalation, and N2 intercalation. As shown in Fig. S17 (ESI†), we plot the band gap distribution during MD simulation, and the average band gap of pristine, O2, H2O, and N2 intercalated heterostructures are reduced compared with the static computation. The average band gaps of pristine, H2O, and N2 intercalated heterostructures are 0.69 eV, 0.67 eV, and 0.64 eV, respectively. In contrast, O2 intercalation induced a small average energy gap of 0.42 eV. A small band gap is expected to lead to large NAC values and promote fast electron–hole recombination in O2 intercalated heterostructure. Thus, the energy level difference is not the main factor for NAC. As mentioned above, except for energy level difference, electron–phonon coupling element 〈φj|∇RH|φk〉 and nuclear velocity Ṙ also play an important role in determining NAC values. The larger the energy level oscillations, the more phonons are excited and the stronger the electron phonon coupling.58 As shown in Fig. S18a (ESI†), the relative phonon amplitude of CBM and VBM states of pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures at 300 K are plotted. It can be seen that after molecule intercalation, the phonon amplitudes of CBM state and VBM state are reduced, indicating the smaller phonon occupation and weaker electron–phonon coupling element. Furthermore, we calculated the root mean square displacement75,80 (RMSD) of pristine, O2 intercalated, H2O intercalated, and N2 intercalated heterostructures using MD trajectories, which is defined as:
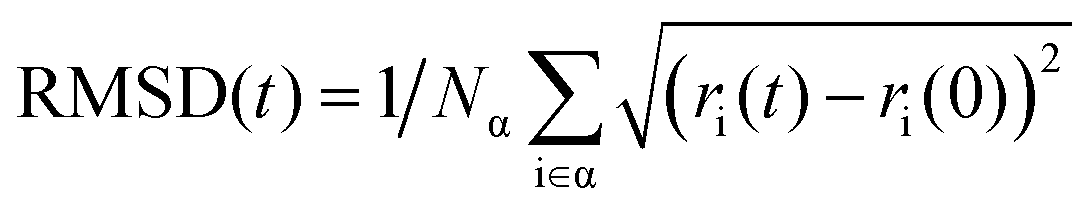 | (3) |
In summary, in O2 intercalated heterostructures, the downward shifted conduction band and the O impurity energy level induce more intermediate states between donor and acceptor, resulting in fast electron transfer from the WSe2 layer to the WS2 layer. In H2O intercalated and N2 intercalated heterostructures, the downward shifted conduction band increasing intermediate states also promote the fast intervalley electron transfer from CB-WSe2@K state to CB-WS2@K state at 300 K. H2O intercalation gives rise to fast hole transfer from VB-WS2@K state to VB-WSe2@K state, while O2 and N2 intercalation do not promote hole transfer. Molecule intercalation alters the hole transfer pathway compared to pristine heterostructure. More importantly, molecule intercalation generally suppresses the recombination of electrons and holes. Overall, O2, H2O, and N2 intercalation can effectively tune the carrier dynamic of WSe2/WS2 vdW heterostructures at a certain temperature.
Conclusions
The equilibrium structure and carrier dynamics of pristine, O2 intercalated, H2O intercalated, and N2 intercalated WSe2/WS2 vdW heterostructures have been systematically investigated by first-principles and ab initio nonadiabatic molecular dynamics calculations. Our results show that at the interface of WSe2/WS2 vdW heterostructure, O2 molecules will spontaneously dissociate, whereas H2O and N2 molecules will not. Either O2 or H2O or N2 intercalation reduces the band gap of the heterostructure but will not change the type-II band alignment. At 300 K, O2 or H2O, or N2 intercalation significantly speeds up the electron separation process from the CB-WSe2@K state to the CB-WS2@K state. At 300 K, H2O intercalation largely accelerates the hole separation process. All three molecules suppress the recombination of electrons and hole due to the reduced electron–phonon coupling element. Our results reveal a new atomistic perspective of modulating the carrier dynamics in WSe2/WS2 vdW heterostructures by molecule intercalation and provide valuable guidance for the experimental design of functional 2D heterostructures.Author contributions
Y. Zhang initialized the project, carried out the calculations, and wrote the draft. H. L., Y. Z., J. L., Y. B., J. G. and J. Z. helped with the calculation process and provided inspiring suggestions for improvement. J. G. and J. Z. supervised the whole project. All authors contributed to the discussion of the results and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 12074053, 91961204, 12004064), the Fundamental Research Funds for the Central Universities (DUT21LAB112, DUT22ZD103), and National Foreign Expert Project (G2022127004L). The authors also acknowledge Computational support from Shanghai Supercomputer Center, DUT supercomputing center, and Tianhe supercomputer of Tianjin center.References
- M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584–2586 RSC.
- M.-Y. Li, C.-H. Chen, Y. Shi and L.-J. Li, Mater. Today, 2016, 19, 322–335 CrossRef CAS.
- Y. Liu, N. O. Weiss, X. Duan, H.-C. Cheng, Y. Huang and X. Duan, Nat. Rev. Mater., 2016, 1, 16042 CrossRef CAS.
- Y. Liu, Y. Huang and X. Duan, Nature, 2019, 567, 323–333 CrossRef CAS PubMed.
- Y. Liu, S. Zhang, J. He, Z. M. Wang and Z. Liu, Nano-Micro Lett., 2019, 11, 13 CrossRef CAS PubMed.
- Q. Zeng and Z. Liu, Adv. Energy Mater., 2018, 4, 1700335 Search PubMed.
- R. Liu, F. Wang, L. Liu, X. He, J. Chen, Y. Li and T. Zhai, Small Struct., 2020, 2, 2000136 CrossRef.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- S. J. Liang, B. Cheng, X. Cui and F. Miao, Adv. Mater., 2020, 32, 1903800 CAS.
- K. Kośmider and J. Fernández-Rossier, Phys. Rev. B, 2013, 87, 075451 CrossRef.
- C. Zhang, C. Gong, Y. Nie, K.-A. Min, C. Liang, Y. J. Oh, H. Zhang, W. Wang, S. Hong, L. Colombo, R. M. Wallace and K. Cho, 2D Mater., 2016, 4, 015026 CrossRef.
- T. Kang, Z. Jin, X. Han, Y. Liu, J. You, H. Wong, H. Liu, J. Pan, Z. Liu, T. W. Tang, K. Zhang, J. Wang, J. Yu, D. Li, A. Pan, D. Pan, J. Wang, Y. Liu and Z. Luo, Small, 2022, 18, 2202229 CrossRef CAS PubMed.
- C. Quan, C. Lu, C. He, X. Xu, Y. Huang, Q. Zhao and X. Xu, Adv. Mater. Interfaces, 2019, 6, 1801733 CrossRef.
- Q. Zheng, W. A. Saidi, Y. Xie, Z. Lan, O. V. Prezhdo, H. Petek and J. Zhao, Nano Lett., 2017, 17, 6435–6442 CrossRef CAS PubMed.
- Q. Zheng, Y. Xie, Z. Lan, O. V. Prezhdo, W. A. Saidi and J. Zhao, Phys. Rev. B, 2018, 97, 205417 CrossRef CAS.
- Y. Tian, Q. Zheng and J. Zhao, J. Phys. Chem. Lett., 2020, 11, 586–590 CrossRef CAS PubMed.
- Z. Hu, X. Liu, P. L. Hernández-Martínez, S. Zhang, P. Gu, W. Du, W. Xu, H. V. Demir, H. Liu and Q. Xiong, InfoMat, 2022, 4, e12290 CAS.
- F. Ceballos, M. Z. Bellus, H.-Y. Chiu and H. Zhao, ACS Nano, 2014, 8, 12717–12724 CrossRef CAS PubMed.
- J. E. Zimmermann, Y. D. Kim, J. C. Hone, U. Hofer and G. Mette, Nanoscale Horiz., 2020, 5, 1603–1609 RSC.
- Y. Jiang, S. Chen, W. Zheng, B. Zheng and A. Pan, Light: Sci. Appl., 2021, 10, 72 CrossRef CAS PubMed.
- A. Tartakovskii, Nat. Rev. Phys., 2020, 2, 8–9 CrossRef.
- N. P. Wilson, W. Yao, J. Shan and X. Xu, Nature, 2021, 599, 383–392 CrossRef CAS PubMed.
- D. Unuchek, A. Ciarrocchi, A. Avsar, K. Watanabe, T. Taniguchi and A. Kis, Nature, 2018, 560, 340–344 CrossRef CAS PubMed.
- C. H. Lee, G. H. Lee, A. M. van der Zande, W. Chen, Y. Li, M. Han, X. Cui, G. Arefe, C. Nuckolls, T. F. Heinz, J. Guo, J. Hone and P. Kim, Nat. Nanotechnol., 2014, 9, 676–681 CrossRef CAS PubMed.
- J. S. Ross, P. Rivera, J. Schaibley, E. Lee-Wong, H. Yu, T. Taniguchi, K. Watanabe, J. Yan, D. Mandrus, D. Cobden, W. Yao and X. Xu, Nano Lett., 2017, 17, 638–643 CrossRef CAS PubMed.
- M. M. Furchi, A. Pospischil, F. Libisch, J. Burgdorfer and T. Mueller, Nano Lett., 2014, 14, 4785–4791 CrossRef CAS PubMed.
- C. Jin, J. Kim, M. I. B. Utama, E. C. Regan, H. Kleemann, H. Cai, Y. Shen, M. J. Shinner, A. Sengupta and K. Watanabe, Science, 2018, 360, 893–896 CrossRef CAS PubMed.
- E. C. Regan, D. Wang, C. Jin, M. I. Bakti Utama, B. Gao, X. Wei, S. Zhao, W. Zhao, Z. Zhang, K. Yumigeta, M. Blei, J. D. Carlstrom, K. Watanabe, T. Taniguchi, S. Tongay, M. Crommie, A. Zettl and F. Wang, Nature, 2020, 579, 359–363 CrossRef CAS PubMed.
- Y. Tang, L. Li, T. Li, Y. Xu, S. Liu, K. Barmak, K. Watanabe, T. Taniguchi, A. H. MacDonald, J. Shan and K. F. Mak, Nature, 2020, 579, 353–358 CrossRef CAS PubMed.
- L. Yuan, B. Zheng, J. Kunstmann, T. Brumme, A. B. Kuc, C. Ma, S. Deng, D. Blach, A. Pan and L. Huang, Nat. Mater., 2020, 19, 617–623 CrossRef CAS PubMed.
- C. Jin, Z. Tao, T. Li, Y. Xu, Y. Tang, J. Zhu, S. Liu, K. Watanabe, T. Taniguchi, J. C. Hone, L. Fu, J. Shan and K. F. Mak, Nat. Mater., 2021, 20, 940–944 CrossRef CAS PubMed.
- S. Miao, T. Wang, X. Huang, D. Chen, Z. Lian, C. Wang, M. Blei, T. Taniguchi, K. Watanabe, S. Tongay, Z. Wang, D. Xiao, Y. T. Cui and S. F. Shi, Nat. Commun., 2021, 12, 3608 CrossRef CAS PubMed.
- W. Zhao, E. C. Regan, D. Wang, C. Jin, S. Hsieh, Z. Wang, J. Wang, Z. Wang, K. Yumigeta, M. Blei, K. Watanabe, T. Taniguchi, S. Tongay, N. Y. Yao and F. Wang, Nano Lett., 2021, 21, 8910–8916 CrossRef CAS PubMed.
- H. J. Chuang, M. Phillips, K. M. McCreary, D. Wickramaratne, M. R. Rosenberger, V. P. Oleshko, N. V. Proscia, M. Lohmann, D. J. O'Hara, P. D. Cunningham, C. S. Hellberg and B. T. Jonker, ACS Nano, 2022, 16, 16260–16270 CrossRef CAS PubMed.
- S. Rahman, X. Sun, Y. Zhu and Y. Lu, ACS Nano, 2022, 16, 21505–21517 CrossRef CAS PubMed.
- A. Sood, J. B. Haber, J. Carlström, E. A. Peterson, E. Barre, J. D. Georgaras, A. H. Reid, X. Shen, M. E. Zajac and E. C. Regan, Nat. Nanotechnol., 2023, 18, 29–35 CrossRef CAS PubMed.
- M. Zhu, Z. Zhang, T. Zhang, D. Liu, H. Zhang, Z. Zhang, Z. Li, Y. Cheng and W. Huang, Nano Lett., 2022, 22, 4528–4534 CrossRef CAS PubMed.
- K. Wang, B. Huang, M. Tian, F. Ceballos, M. W. Lin, M. Mahjouri-Samani, A. Boulesbaa, A. A. Puretzky, C. M. Rouleau, M. Yoon, H. Zhao, K. Xiao, G. Duscher and D. B. Geohegan, ACS Nano, 2016, 10, 6612–6622 CrossRef CAS PubMed.
- Y. Li, J. Zhang, Q. Chen, X. Xia and M. Chen, Adv. Mater., 2021, 33, 2100855 CrossRef CAS PubMed.
- Y. Jung, Y. Zhou and J. J. Cha, Inorg. Chem. Front., 2016, 3, 452–463 RSC.
- A. H. Biby, B. A. Ali and N. K. Allam, Mater. Adv., 2021, 2, 5052–5056 RSC.
- H. Liang, Z. Cao, F. Ming, W. Zhang, D. H. Anjum, Y. Cui, L. Cavallo and H. N. Alshareef, Nano Lett., 2019, 19, 3199–3206 CrossRef CAS PubMed.
- L. Zhang, H. Nan, X. Zhang, Q. Liang, A. Du, Z. Ni, X. Gu, K. K. Ostrikov and S. Xiao, Nat. Commun., 2020, 11, 5960 CrossRef CAS PubMed.
- Q. Qian, H. Ren, J. Zhou, Z. Wan, J. Zhou, X. Yan, J. Cai, P. Wang, B. Li, Z. Sofer, B. Li, X. Duan, X. Pan, Y. Huang and X. Duan, Nature, 2022, 606, 902–908 CrossRef CAS PubMed.
- X.-C. Liu, Sci. Adv., 2020, 6, eaay4092 CrossRef CAS PubMed.
- K. S. Novoselov, A. Mishchenko, A. Carvalho and A. H. Castro Neto, Science, 2016, 353, aac9439 CrossRef CAS PubMed.
- J. Li, J. Liang, X. Yang, X. Li, B. Zhao, B. Li and X. Duan, Small, 2022, 18, 2107059 CrossRef CAS PubMed.
- S. Zhang, X. Deng, Y. Wu, Y. Wang, S. Ke, S. Zhang, K. Liu, R. Lv, Z. Li, Q. Xiong and C. Wang, Chem. Soc. Rev., 2022, 51, 4000–4022 RSC.
- J. Wu, J. Peng, H. Sun, Y. Guo, H. Liu, C. Wu and Y. Xie, Adv. Mater., 2022, 34, 2200425 CrossRef CAS PubMed.
- P. Sutter, J. T. Sadowski and E. A. Sutter, J. Am. Chem. Soc., 2010, 132, 8175–8179 CrossRef CAS PubMed.
- E. Granas, J. Knudsen, U. A. Schröder, T. Gerber, C. Busse, M. A. Arman, K. Schulte, J. N. Andersen and T. Michely, ACS Nano, 2012, 6, 9951–9963 CrossRef PubMed.
- R. Larciprete, S. Ulstrup, P. Lacovig, M. Dalmiglio, M. Bianchi, F. Mazzola, L. Hornekær, F. Orlando, A. Baraldi and P. Hofmann, ACS Nano, 2012, 6, 9551–9558 CrossRef CAS PubMed.
- Q. Fu and X. Bao, Chem. Soc. Rev., 2017, 46, 1842–1874 RSC.
- C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen, X. Sun, S. Siahrostami, K. Chan and H. Wang, Nat. Catal., 2020, 3, 125–134 CrossRef CAS.
- L. Zhou, X. Wang, H. J. Shin, J. Wang, R. Tai, X. Zhang, H. Fang, W. Xiao, L. Wang, C. Wang, X. Gao, J. Hu and L. Zhang, J. Am. Chem. Soc., 2020, 142, 5583–5593 CrossRef CAS PubMed.
- P. R. Kidambi, B. C. Bayer, R. Blume, Z. J. Wang, C. Baehtz, R. S. Weatherup, M. G. Willinger, R. Schloegl and S. Hofmann, Nano Lett., 2013, 13, 4769–4778 CrossRef CAS PubMed.
- J. W. Tyrrell and P. Attard, Phys. Rev. Lett., 2001, 87, 176104 CrossRef CAS PubMed.
- Q. Zheng, W. Chu, C. Zhao, L. Zhang, H. Guo, Y. Wang, X. Jiang and J. Zhao, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1411 CAS.
- S. Tan, A. Argondizzo, J. Ren, L. Liu, J. Zhao and H. Petek, Nat. Photonics, 2017, 11, 806–812 CrossRef CAS.
- S. Tan, Y. Dai, S. Zhang, L. Liu, J. Zhao and H. Petek, Phys. Rev. Lett., 2018, 120, 126801 CrossRef CAS PubMed.
- L. Zhang, Q. Zheng, Y. Xie, Z. Lan, O. V. Prezhdo, W. A. Saidi and J. Zhao, Nano Lett., 2018, 18, 1592–1599 CrossRef CAS PubMed.
- W. Chu, Q. Zheng, O. V. Prezhdo and J. Zhao, J. Am. Chem. Soc., 2020, 142, 3214–3221 CrossRef CAS PubMed.
- W. Chu, Q. Zheng, O. V. Prezhdo, J. Zhao and W. A. Saidi, Sci. Adv., 2020, 6, eaaw7453 CrossRef CAS PubMed.
- X. Jiang, Q. Zheng, Z. Lan, W. A. Saidi, X. Ren and J. Zhao, Sci. Adv., 2021, 7, eabf3759 CrossRef PubMed.
- H. Zeng, X. Liu, H. Zhang and X. Cheng, Phys. Chem. Chem. Phys., 2021, 23, 694–701 RSC.
- D. Muoi, N. N. Hieu, H. T. T. Phung, H. V. Phuc, B. Amin, B. D. Hoi, N. V. Hieu, L. C. Nhan, C. V. Nguyen and P. T. T. Le, Chem. Phys., 2019, 519, 69–73 CrossRef CAS.
- K. Zollner, P. E. F. Junior and J. Fabian, Phys. Rev. B, 2019, 100, 195126 CrossRef CAS.
- H.-g Kim and H. J. Choi, Phys. Rev. B, 2021, 103, 085404 CrossRef CAS.
- J. Zhang, J. Wang, P. Chen, Y. Sun, S. Wu, Z. Jia, X. Lu, H. Yu, W. Chen, J. Zhu, G. Xie, R. Yang, D. Shi, X. Xu, J. Xiang, K. Liu and G. Zhang, Adv. Mater., 2016, 28, 1950–1956 CrossRef CAS PubMed.
- X. Wu, X. Wang, H. Li, Z. Zeng, B. Zheng, D. Zhang, F. Li, X. Zhu, Y. Jiang and A. Pan, Nano Res., 2019, 12, 3123–3128 CrossRef CAS.
- L. Debbichi, O. Eriksson and S. Lebègue, Phys. Rev. B, 2014, 89, 205311 CrossRef.
- V. Q. Bui, T. T. Pham, D. A. Le, C. M. Thi and H. M. Le, J. Phys.: Condens. Matter, 2015, 27, 305005 CrossRef PubMed.
- H. Liu, N. Han and J. Zhao, RSC Adv., 2015, 5, 17572–17581 RSC.
- A. Ramasubramaniam, D. Naveh and E. Towe, Phys. Rev. B, 2011, 84, 205325 CrossRef.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- H. Zhou, C. Sun, W. Xin, Y. Li, Y. Chen and H. Zhu, Nano Lett., 2022, 22, 2547–2553 CrossRef CAS PubMed.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- J. C. Wong, L. Li and Y. Kanai, J. Phys. Chem. C, 2018, 122, 29526–29536 CrossRef CAS.
- X. Niu, Y. Li, Y. Zhang, Q. Zheng, J. Zhao and J. Wang, J. Mater. Chem. C, 2019, 7, 1864–1870 RSC.
- F. Zhang, X. Wang, W. Gao and J. Zhao, Phys. Rev. Appl., 2022, 17, 064016 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Computational methods, band structures, O2/H2O/N2 intercalation model, bond lengths, bond angle and energy comparison, charge density difference, time-dependent energy state evolution, electron and hole population, phonon spectra, average NAC values, band gap distribution, RMSD. See DOI: https://doi.org/10.1039/d3mh00420a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2023 |