
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-electrocatalytic C–H hydroxyalkylation of N-heteroarenes with trifluoromethyl ketones†
Tianyu
He
,
Chaoqiang
Liang
and
Shenlin
Huang
 *
*
Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, International Innovation Center for Forest Chemicals and Materials, Nanjing Forestry University, Nanjing 210037, China. E-mail: shuang@njfu.edu.cn
First published on 21st November 2022
Abstract
Trifluoromethyl carbinols and N-heteroarenes are both prevalent in bioactive molecules. However, access to high-value pharmacophores combining these two functional groups still remains a challenge. Herein, we report an electro-chemical redox-neutral coupling for the synthesis of N-heteroaryl trifluoromethyl carbinols from readily available N-heteroarenes and trifluoromethyl ketones. The reaction starts with reversing the polarity of ketones to nucleophilic ketyl radicals through an electrocatalytic proton-coupled electron transfer (PCET), followed by radical addition to heteroarenes and rearomatization to afford tertiary alcohol products. Importantly, the merging of paired electrolysis and cobalt catalysis is crucial to this regioselective C–H hydroxyalkylation of heteroarenes, and thus avoids several known competing pathways including the spin-center shift (SCS) process. Collectively, this protocol provides straightforward access to heteroaryl trifluoromethyl carbinols, featuring ideal atom economy, excellent regioselectivity, and paired redox-neutral electrolysis.
Introduction
Both organofluorine compounds1 and N-heterocycles2 are prevalent in pharmaceuticals, agrochemicals, and other bioactive molecules. In particular, a number of therapeutic drugs contain CF3-substituted tertiary alcohols3 or N-heteroarenes,4 as illustrated in Scheme 1A. The combination of these two functional groups, trifluoromethyl carbinols and N-heteroarenes, is similarly impactful, expanding the chemical space for drug discovery.5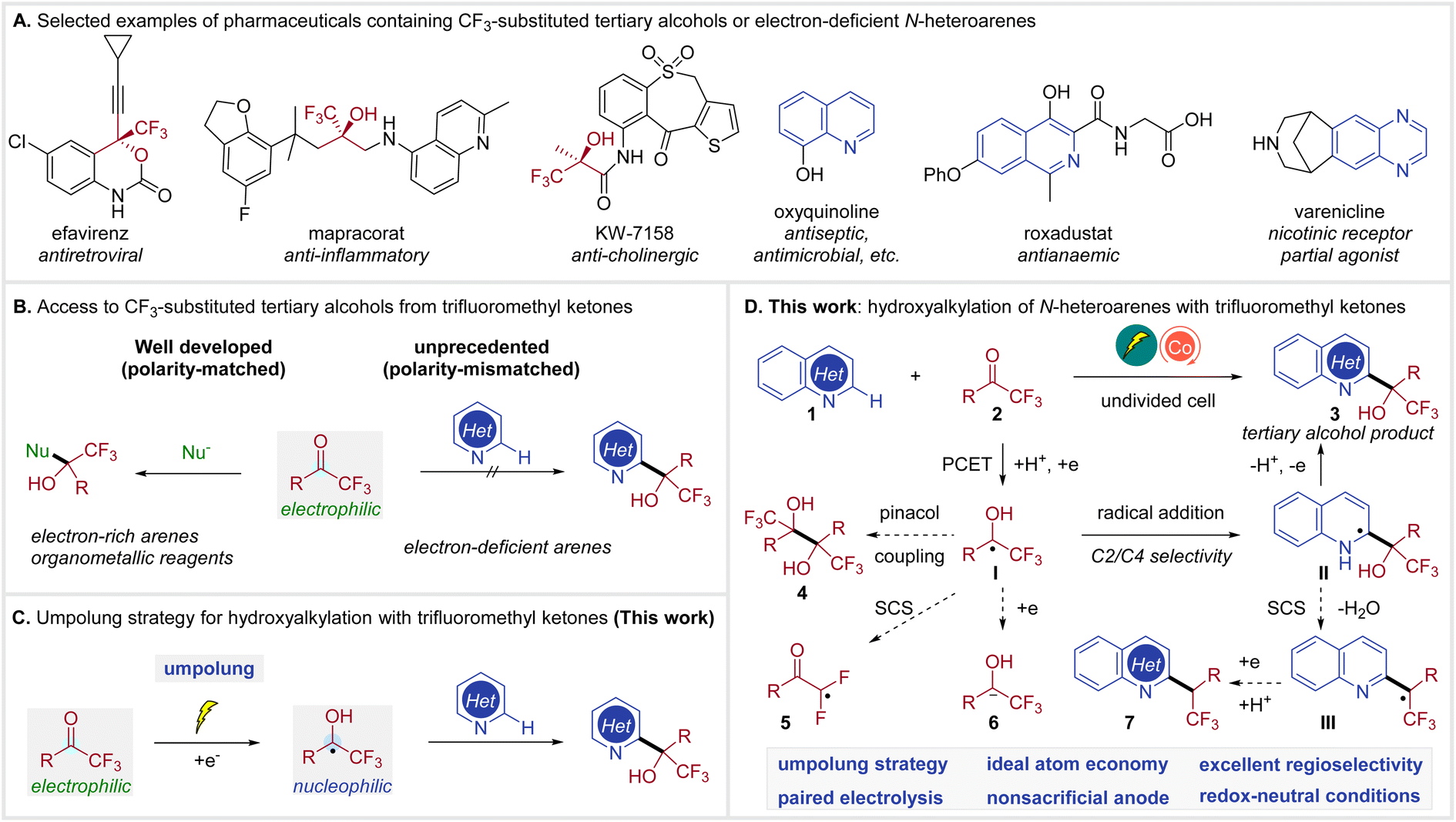 | ||
| Scheme 1 Context and strategy to access trifluoromethyl tertiary alcohols from trifluoromethyl ketones. | ||
While numerous methods have been developed to generate CF3-substituted tertiary alcohols,6 access to N-heteroaryl trifluoromethyl carbinols remains underdeveloped.5c,7 The prevailing approach to CF3-substituted tertiary alcohols involves nucleophilic addition of a nucleophile, such as organometallic reagents or electron-rich arenes, to the readily available trifluoromethyl ketones (Scheme 1B, left).6e,8 In contrast, the C–H hydroxyalkylation of electron-deficient N-heteroarenes with trifluoromethyl ketones remains unknown, probably due to their mismatched polarity (Scheme 1B, right).
The umpolung strategy, which converts trifluoromethyl ketones to the nucleophilic ketyl radicals,9 presents an attractive solution for this unprecedented coupling (Scheme 1C), but also raises formidable synthetic challenges. Currently, ketyl radical generation still relies primarily on stoichiometric amounts of Zn, Ti or SmI2.10 Alternatively, photoredox catalysis has been proved to be capable of generating ketyl radicals from ketones.11 However, this method typically requires the use of terminal reductants. Recently, Wang reported a photocatalytic C–H alkylation of heteroarenes with ketyl radicals from ketones.12 This reaction was achieved by the addition of ketyl radicals to heterarenes via a Minisci reaction13 pathway in combination with a spin-center shift (SCS) process,14 yielding alkylated products and not hydroxyalkylated adducts. Furthermore, organic electrochemistry15 has also been demonstrated as a sustainable method for the conversion of ketones to ketyl radicals.16 However, these protocols commonly require a divided cell setup or a sacrificial anode. Consequently, there remains no general method for accessing tertiary alcohols through the Minisci reaction of ketones and electron-deficient N-heteroarenes.
Herein, we report cobalt-electrocatalytic C–H hydroxyalkylation of N-heteroarenes with trifluoromethyl ketones for the synthesis of CF3-substituted tertiary alcohols (Scheme 1D). We envisioned that ketyl radical I could be generated from trifluoromethyl ketone 2via a proton-coupled electron transfer (PCET)17 under mild electroreductive18 conditions. The desired alcohol product 3 would be constructed through selective radical addition to heteroarene 1 and subsequent anodic oxidation of intermediate II. The following competing reaction pathways need to be suppressed. Firstly, a homocoupling of ketyl radical I is possible to deliver pinacol 4. Secondly, the CF3-substituted ketyl radical I may undergo a defluorinative spin-center shift giving radical 5.19 Alternatively, I may be further reduced to 6,20 which can undergo other side reactions. Moreover, the C2/C4 selectivity is also highly challenging.13c,21 Furthermore, intermediate II may be susceptible to SCS fragmentation12,22 to generate III, which could lead to alkylated side-product 7, since the α-hydroxy group proved to be active towards elimination of water. These challenges could be addressed via identification of a suitable paired electrocatalytic system.23 This electrocatalytic methodology enables a general, regioselective, and efficient synthesis of heteroaryl trifluoromethyl carbinols from trifluoromethyl ketones and N-heteroarenes.
Results and discussion
We commenced our investigation with the coupling of quinoline (1a) and 2,2,2-trifluoroacetophenone (2a) as the model reaction (Table 1). Pleasingly, the desired alcohol product 3a was isolated in 76% yield, when the electrolysis was conducted in a DMF/MeCN (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution of 1a and 2a at 50 °C in the presence of CoCl2·6H2O (5 mol%) as the catalyst, trifluoroacetic acid (TFA) as the acid, and nBu4NBr as the electrolyte with an undivided cell (entry 1). Notably, only C2-substituted quinoline product 3a was isolated, without the detection of a C4-substituted isomer. When a sacrificial zinc anode was used, no desired reaction was observed, highlighting the essential role of the paired electrolysis (entry 2). Other cathodes with a lower overpotential than stannum, for example, aluminum, were less suitable, indicating that a Sn cathode was necessary to avoid the competing proton reduction (entry 3).24 Further evaluation of various solvents revealed that the use of DMF/MeCN (7:1) was crucial for the efficient conversion of this coupling reaction. Reactions using other solvents, such as, DMF, MeCN, and DMSO, exhibited a dramatic decrease in reaction efficiency (entries 4–6). The choice of electrolyte also proved important to the success of the coupling, with nBu4NBr being optimal, while electrolytes with other counteranions, for example, nBu4NOAc, nBu4NI, and nBu4NCl, were less effective (entries 7–9). Replacing TFA with other acids, such as HCl and H2SO4, led to no formation of CF3-substituted tertiary alcohol 3a (entry 10). In addition, switching CoCl2·6H2O to SmI2, CoBr2, or Co(OAc)3 gave inferior yields (entries 11–13). Notably, the coupling reaction worked equally well at a lower current (10 mA), albeit with a longer reaction time (entry 14). When the electrolysis was conducted at room temperature, the isolated yield of 3a was modest (49%, entry 15). As expected, no conversion was observed without current (entry 16).
:1) solution of 1a and 2a at 50 °C in the presence of CoCl2·6H2O (5 mol%) as the catalyst, trifluoroacetic acid (TFA) as the acid, and nBu4NBr as the electrolyte with an undivided cell (entry 1). Notably, only C2-substituted quinoline product 3a was isolated, without the detection of a C4-substituted isomer. When a sacrificial zinc anode was used, no desired reaction was observed, highlighting the essential role of the paired electrolysis (entry 2). Other cathodes with a lower overpotential than stannum, for example, aluminum, were less suitable, indicating that a Sn cathode was necessary to avoid the competing proton reduction (entry 3).24 Further evaluation of various solvents revealed that the use of DMF/MeCN (7:1) was crucial for the efficient conversion of this coupling reaction. Reactions using other solvents, such as, DMF, MeCN, and DMSO, exhibited a dramatic decrease in reaction efficiency (entries 4–6). The choice of electrolyte also proved important to the success of the coupling, with nBu4NBr being optimal, while electrolytes with other counteranions, for example, nBu4NOAc, nBu4NI, and nBu4NCl, were less effective (entries 7–9). Replacing TFA with other acids, such as HCl and H2SO4, led to no formation of CF3-substituted tertiary alcohol 3a (entry 10). In addition, switching CoCl2·6H2O to SmI2, CoBr2, or Co(OAc)3 gave inferior yields (entries 11–13). Notably, the coupling reaction worked equally well at a lower current (10 mA), albeit with a longer reaction time (entry 14). When the electrolysis was conducted at room temperature, the isolated yield of 3a was modest (49%, entry 15). As expected, no conversion was observed without current (entry 16).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb |
| a Conditions: 1a (0.3 mmol), 2a (2 equiv.), CoCl2·6H2O (5 mol%), TFA (2 equiv.), nBu4NBr (2 equiv.), DMF (3.5 mL), MeCN (0.5 mL), graphite anode (1 cm × 1 cm × 0.2 cm), Sn cathode (1 cm × 1 cm × 0.1 cm), constant current (20 mA), undivided cell, 50 °C, 8 h. b Isolated yield. | ||
| 1 | None | 76% |
| 2 | Zn(+)/Sn(−) instead of C(+)/Sn(−) | 0% |
| 3 | C(+)/Al(−) instead of C(+)/Sn(−) | 34% |
| 4 | DMF as the solvent | 65% |
| 5 | MeCN as the solvent | 30% |
| 6 | DMSO as the solvent | 21% |
| 7 | nBu4NOAc instead of nBu4NBr | 19% |
| 8 | nBu4NI instead of nBu4NBr | 40% |
| 9 | nBu4NCl instead of nBu4NBr | 61% |
| 10 | Conc. HCl or H2SO4 instead of TFA | 0% |
| 11 | SmI2 instead of CoCl2·6H2O | 54% |
| 12 | CoBr2 instead of CoCl2·6H2O | 66% |
| 13 | Co(OAc)3 instead of CoCl2·6H2O | 52% |
| 14 | 10 mA, 36 h instead of 20 mA, 8 h | 70% |
| 15 | rt | 49% |
| 16 | Without current | 0% |
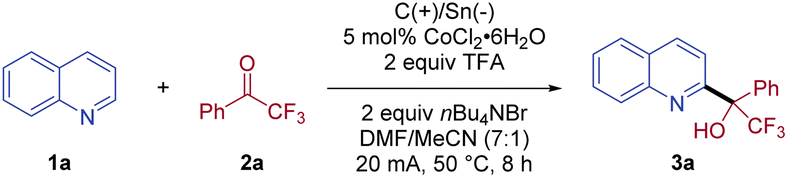
With these optimized conditions in hand, we then sought to evaluate the substrate scope with respect to trifluoromethyl ketones (Scheme 2). Trifluoromethyl aryl ketones bearing different substituents on the phenyl ring were well tolerated (3a–3k), including electron-donating groups (Me, tBu, MeS or MeO), phenyl, and electron-withdrawing groups (Cl, Br, or NO2). Aryl ketones with meta or para bromo-substituents exhibited good reactivity (3h and 3i), while ortho bromo-substituted aryl ketones delivered the desired product 3j in a lower yield, probably due to steric hindrance. 3,5-Dimethylphenyl and naphthyl alcohol products 3l and 3m were also obtained in synthetically useful yields. The heterocyclic substrate derived from piperonyl aldehyde was also effective, leading to tertiary alcohol 3n in 50% yield. Unfortunately, methyl 4-(2,2,2-trifluoroacetyl)benzoate with an ester group gave a complex mixture and 2,2,3,3,3-pentafluoro-1-phenylpropan-1-one with a longer perfluoroalkyl group was unreactive (see Scheme S1, in the ESI†).
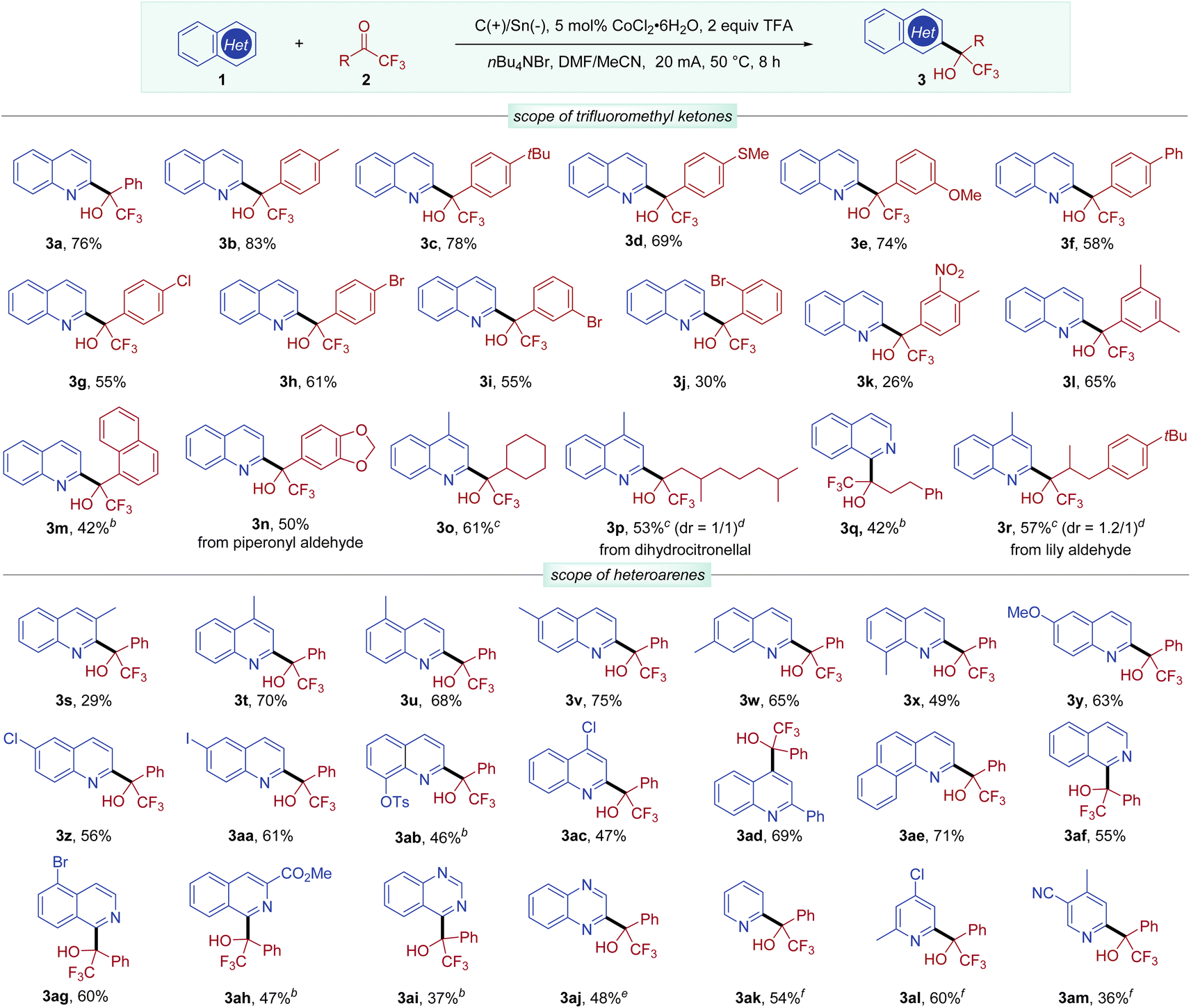 | ||
| Scheme 2 Substrate scope areaction conditions: undivided cell, graphite anode (1 cm × 1 cm × 0.2 cm), Sn cathode (1 cm × 1 cm × 0.1 cm), constant current (20 mA), 1 (0.3 mmol), 2 (2 equiv.), CoCl2·6H2O (5 mol%), TFA (2 equiv.), Bu4NBr (2 equiv.), DMF (3.5 mL), MeCN (0.5 mL), 50 °C, 8 h. Isolated yield. b12 h. cketones (3 equiv.). dDetermined by 1H NMR. eTFA (3 equiv.). f6,6′-dimethyl-2,2′-bipyridine (10 mol%) was used. | ||
Besides aryl ketones, aliphatic trifluoromethyl ketones were also examined in this transformation. A cycloalkyl trifluoromethyl ketone was converted to the corresponding alcohol product 3o in 61% yield. Trifluoromethyl ketones with linear alkyl substituents also underwent efficient Minisci-type reactions to afford alcohols (3p–3r). Specifically, trifluoromethyl ketones derived from natural products, including dihydrocitronellal and lily aldehyde, were well transformed into the desired alcohols 3p and 3r.
We next investigated the scope of the heteroarene component using 2,2,2-trifluoroacetophenone (2a). An initial evaluation of the substituent effect at different positions of quinolines demonstrated that the reactivity was mainly determined by steric effects, with 3-methylquinoline delivering 3s in 29% yield. Quinolines bearing a methyl group at other positions afforded alcohol products 3t–3x in good yields. Simple quinolines bearing MeO, Cl, and I were suitable substrates, regioselectively providing C2-substituted products 3y–3aa in good yields. Interestingly, an 8-hydroxyquinoline derivative could be functionalized to provide 3ab without incident, showing potential for pharmacological applications.4b 4-Chloroquinoline underwent coupling with 2a exclusively at the C2-position (3ac), while 2-phenylquinoline was selectively hydroxyalkylated at the C4-position (3ad). Benzo[h]quinoline was also an effective coupling partner leading to 3ae in 71% yield. Moreover, simple isoquinoline and isoquinolines bearing a Br or ester group reacted well in this coupling reaction at the C1 site (3af–3ah). It is important to note that quinazoline and quinoxaline readily participated in this electrocatalytic protocol, affording 3ai and 3aj, respectively. Notably, simple pyridine and pyridines bearing methyl, chloro, or cyano substituents were also found to be amenable to this coupling reaction, providing 3ak–3am in 36–60% yields, when 6,6′-dimethyl-2,2′-bipyridine was employed as a ligand. Moreover, selective mono-alkylation could be achieved with 2,6-unsubstituted pyridine substrates.
To gain further insight into the mechanism of this redox-neutral coupling of N-heteroarenes with trifluoromethyl ketones, we conducted several control experiments (Scheme 3). A ketyl radical pathway was indicated by the radical-trapping experiment, as no desired reaction was observed in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) and TEMPO-adduct 8 could be detected by HRMS (Scheme 3A). In the absence of TFA, no reactivity was obtained with only the recovery of starting materials 1a and 2a (Scheme 3B), suggesting that a PCET process might be involved. Evidence that electrolysis of ketone 2a delivers the ketyl radical A is supported by the formation of pinacol product 4a from the reaction carried out in the absence of a N-heteroarene under standard conditions (Scheme 3C). Without using a cobalt catalyst, the reaction of quinoline 1a with 2a underwent inefficiently to provide C2-substituted product 3a in 40% yield, along with 12% of C2 and C4 disubstituted product 9 (Scheme 3D), highlighting the importance of the cobalt catalyst for both reactivity and regioselectivity. During the solvent screening, we did not observe any significant influence of solvents on C2/C4 regioselectivity. Therefore, the coordination of Co with substrates was proposed to explain regioselectivity.13c,21
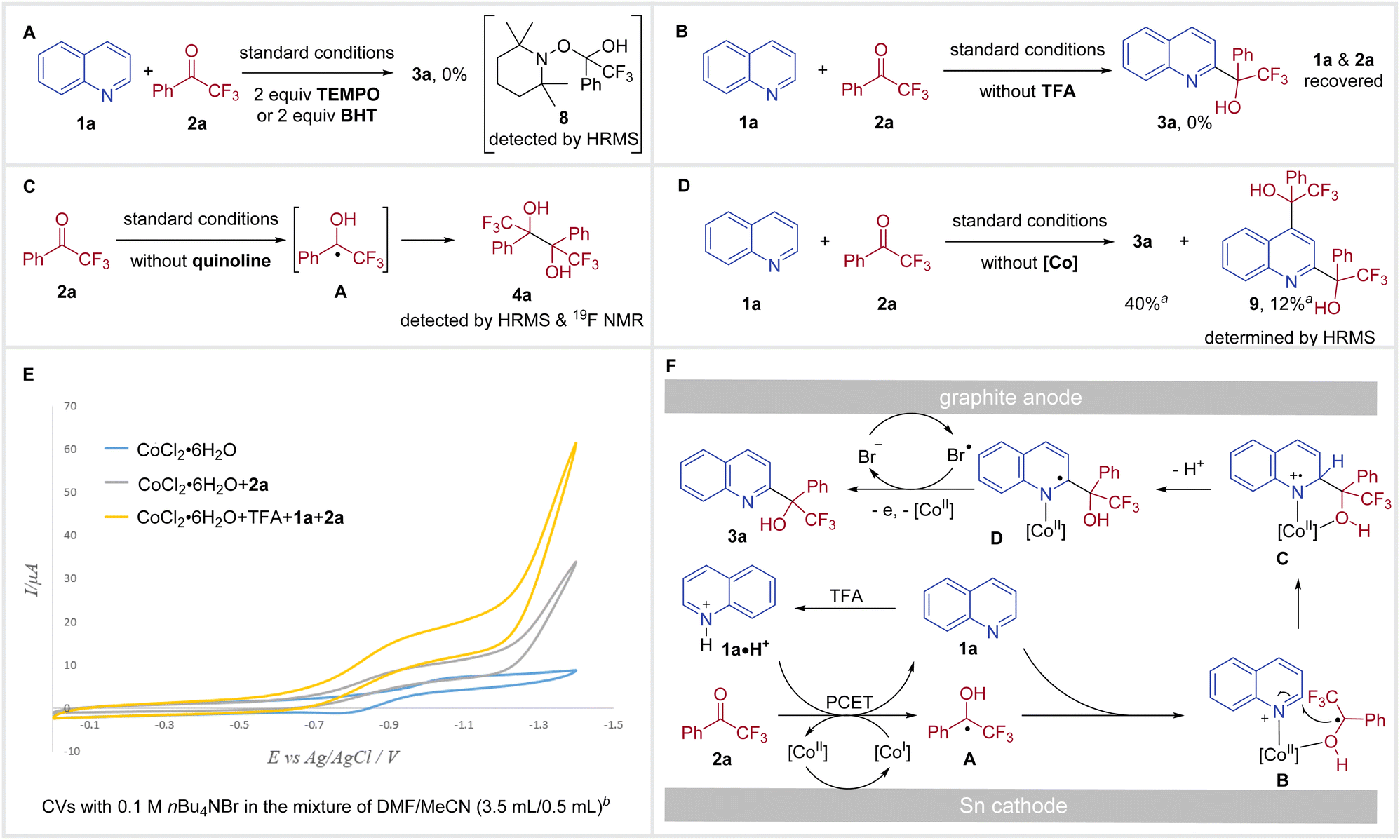 | ||
| Scheme 3 Mechanistic experiments and proposal ayields were determined by 19F NMR analysis using (trifluoromethyl)benzene as an internal standard. bCVs of a 0.01 M solution of CoCl2·6H2O (blue trace), the mixture of 0.01 M CoCl2·6H2O and 0.1 M 2a (gray trace), the mixture of 0.01 M CoCl2·6H2O, 0.1 M 2a, 0.1 M TFA and 0.1 M 1a (yellow trace) with 0.1 M TBAB at 100 mV s−1 in the mixture of DMF/MeCN (3.5 mL/0.5 mL). | ||
In order to reveal the essential role of CoCl2·6H2O, we next performed the cyclic voltammetry (CV) experiments. Examination of CoCl2·6H2O showed a reversible reduction peak at E = −1.02 V vs. Ag/AgCl, which was assigned to the reduction of Co(II) to Co(I).25 Addition of 2a to the CV solution of CoCl2·6H2O resulted in increased reductive current and loss of reversibility (Scheme 3E, gray trace).26 When 1a, TFA, and 2a were added to the CV solution of CoCl2·6H2O, a more significant increase in catalytic current was observed (Scheme 3E, yellow trace). Moreover, the catalytic current increased as a function of the ketone concentration (see Fig. S7 in the ESI†). Taken together, these results suggest that a PCET process takes place between Co(I) and 2a forming a ketyl radical and the resulting Co(II) is reduced to Co(I) at the cathode.
On the basis of these mechanistic experiments, a plausible reaction pathway for our redox-neutral coupling of N-heteroarenes with trifluoromethyl ketones is outlined in Scheme 3F. Initially, reduction of Co(II) at the cathode forms the Co(I) catalyst, which facilitates a homogeneous PCET with 2a to give ketyl radical A. At this stage, the ketyl radical A and 1a could be coordinated onto the Co(II) catalyst, forming the intermediate B. Subsequently, intramolecular radical addition delivers C, which then loses a proton to afford D. Meanwhile, nBu4NBr performs a dual role as supporting electrolyte and a redox mediator. Thus, bromine radicals (Br˙) could be generated from the oxidation of bromide ions (Br−) at the anode (E = +0.84 V vs. Ag/AgCl, see Fig. S8 in the ESI†).27 In the final step, Br˙ accomplishes a single electron oxidation of intermediate D,28 and the subsequent dissociation of Co(II) delivers the desired tertiary alcohol product 3a.
Conclusions
In summary, we have established the cobalt-electrocatalytic C–H hydroxyalkylation of N-heteroarenes with trifluoromethyl ketones, featuring broad substrate scope, ideal atom economy, and excellent regioselectivity. This redox-neutral method involves carbonyl umpolung via electrocatalytic proton-coupled electron transfer, radical addition to heteroarenes, and rearomatization. By merging paired electrolysis and cobalt catalysis, this regioselective C–H hydroxyalkylation avoids the known competing spin-center shift process and offers an efficient access to high-value pharmacophores, N-heteroaryl trifluoromethyl carbinols.Data availability
The ESI† contains method description, product characterization data, and NMR spectra.Author contributions
T. H. and C. L. performed the experiments, obtained all data, and analyzed the results. S. H. designed the project, directed the study, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support provided by the Natural Science Foundation of China (32171724) is warmly acknowledged. We are also grateful for the support from the advanced analysis and testing center of Nanjing Forestry University.Notes and references
-
(a) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS
; (b) H. Mei, J. Han, K. D. Klika, K. Izawa, T. Sato, N. A. Meanwell and V. A. Soloshonok, Eur. J. Med. Chem., 2020, 186, 111826 CrossRef CAS PubMed
-
(a) C. Prandi and E. G. Occhiato, Pest Manage. Sci., 2019, 75, 2385 CrossRef CAS
-
(a) L. A. Solt, N. Kumar, P. Nuhant, Y. Wang, J. L. Lauer, J. Liu, M. A. Istrate, T. M. Kamenecka, W. R. Roush, D. Vidovic, S. C. Schurer, J. Xu, G. Wagoner, P. D. Drew, P. R. Griffin and T. P. Burris, Nature, 2011, 472, 491 CrossRef CAS
-
(a) V. Yadav, J. Reang, V. Sharma, J. Majeed, P. C. Sharma, K. Sharma, N. Giri, A. Kumar and R. K. Tonk, Chem. Biol. Drug Des., 2022, 100, 389 CrossRef CAS PubMed
-
(a) M. Awale, R. Visini, D. Probst, J. Arus-Pous and J. L. Reymond, Chimia, 2017, 71, 661 CrossRef CAS
-
(a) K. Domino, C. Veryser, B. A. Wahlqvist, C. Gaardbo, K. T. Neumann, K. Daasbjerg, W. M. De Borggraeve and T. Skrydstrup, Angew. Chem., Int. Ed., 2018, 57, 6858 CrossRef CAS
- W. J. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 17198 CrossRef CAS PubMed
- C. B. Kelly, M. A. Mercadante and N. E. Leadbeater, Chem. Commun., 2013, 49, 11133 RSC
- X. Xu, Q. Q. Min, N. Li and F. Liu, Chem. Commun., 2018, 54, 11017 RSC
- A. Peter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349 RSC
- Q. Xia, J. Dong, H. Song and Q. Wang, Chem.–Eur. J., 2019, 25, 2949 CAS
- J. Dong, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Sci. Adv., 2019, 5, eaax9955 CrossRef CAS PubMed
-
(a) W. Meng, K. Xu, B. Guo and C. Zeng, Chin. J. Org. Chem., 2021, 41, 2621 CrossRef
-
(a) F. L. Zhang, B. Li, K. N. Houk and Y. F. Wang, JACS Au, 2022, 2, 1032 CrossRef CAS PubMed
-
(a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415 CrossRef CAS PubMed
-
(a) P. Hu, B. K. Peters, C. A. Malapit, J. C. Vantourout, P. Wang, J. Li, L. Mele, P. G. Echeverria, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 20979 CrossRef CAS
-
(a) P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017 CrossRef CAS
-
(a) J. Derosa, P. Garrido-Barros and J. C. Peters, Inorg. Chem., 2022, 61, 6672 CrossRef CAS
- Y. J. Yu, F. L. Zhang, T. Y. Peng, C. L. Wang, J. Cheng, C. Chen, K. N. Houk and Y. F. Wang, Science, 2021, 371, 1232 CrossRef CAS
- J. R. Box, A. P. Atkins and A. J. J. Lennox, Chem. Sci., 2021, 12, 10252 RSC
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 CrossRef CAS PubMed
- J. Jin and D. W. MacMillan, Nature, 2015, 525, 87 CrossRef CAS
-
(a) S. Zhang and M. Findlater, Chem.–Eur. J., 2022, e202201152 CAS
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866 CrossRef CAS PubMed
- D. P. Hickey, C. Sandford, Z. Rhodes, T. Gensch, L. R. Fries, M. S. Sigman and S. D. Minteer, J. Am. Chem. Soc., 2019, 141, 1382 CrossRef CAS PubMed
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404 RSC
- L.-S. Kang, M.-H. Luo, C. M. Lam, L.-M. Hu, R. D. Little and C.-C. Zeng, Green Chem., 2016, 18, 3767 RSC
- P. Xu, P. Y. Chen and H. C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. See DOI: https://doi.org/10.1039/d2sc05198b |
| This journal is © The Royal Society of Chemistry 2023 |