DOI:
10.1039/D2SC05827H
(Edge Article)
Chem. Sci., 2023,
14, 2289-2302
Selective chemical reagents to investigate the role of caspase 6 in apoptosis in acute leukemia T cells†
Received
20th October 2022
, Accepted 2nd January 2023
First published on 3rd January 2023
Abstract
Activated effector caspases 3, 6 and 7 are responsible for cleaving a number of target substrates, leading to the ultimate destruction of cells via apoptosis. The functions of caspases 3 and 7 in apoptosis execution have been widely studied over the years with multiple chemical probes for both of these enzymes. In contrast, caspase 6 seems to be largely neglected when compared to the heavily studied caspases 3 and 7. Therefore, the development of new small-molecule reagents for the selective detection and visualization of caspase 6 activity can improve our understanding of molecular circuits of apoptosis and shed new light on how they intertwine with other types of programmed cell death. In this study, we profiled caspase 6 substrate specificity at the P5 position and discovered that, similar to caspase 2, caspase 6 prefers pentapeptide substrates over tetrapeptides. Based on these data, we developed a set of chemical reagents for caspase 6 investigation, including coumarin-based fluorescent substrates, irreversible inhibitors and selective aggregation-induced emission luminogens (AIEgens). We showed that AIEgens are able to distinguish between caspase 3 and caspase 6 in vitro. Finally, we validated the efficiency and selectivity of the synthesized reagents by monitoring lamin A and PARP cleavage via mass cytometry and western blot analysis. We propose that our reagents may provide new research prospects for single-cell monitoring of caspase 6 activity to reveal its function in programmed cell death pathways.
Introduction
Caspases, a family of cysteine proteases, are mainly known for their indispensable roles in programmed cell death pathways (PCDs), such as apoptosis, pyroptosis and necroptosis.1 Apart from their fundamental roles in these processes, they have also been recognized as significant components in a variety of biochemical events, such as cell differentiation or protein secretion.2 Based on their functions, caspases can be assigned as apoptosis initiators (caspases 8, 9, and 10), apoptosis executioners (caspases 3, 6, and 7) and proinflammatory caspases involved in pyroptosis (caspases 1, 4, and 5 in humans; 1 and 11 in mice).3–5 Given the critical roles that caspases play under normal conditions, their catalytic activation must be tightly regulated. When a cell fails to accurately modulate caspase activity, numerous pathological processes can occur, such as carcinogenesis6 or neurodegeneration.7 Therefore, research efforts have been directed toward resolving the basis of caspase structures, activation mechanisms and catalytic preferences. Members of the caspase family share many structural and functional characteristics, which makes it challenging to distinguish them with small-molecule chemical reagents.
Caspase 6 (EC 3.4.22.59) is one of the executioner caspases and was first identified in 1995 in human Jurkat T cells using a PCR approach.8 In early years, it was named “mammalian Ced-3 homolog” (or Mch2) and identified as a proapoptotic protein activated by caspase 3.9 However, over the years, studies have shown that caspase 6 can operate before caspase 3 activation10 and even in the absence of caspase 3.11 Caspase 6 participates in both extrinsic and intrinsic apoptosis pathways and can be activated even in the absence of apical caspases, serving mostly as a feedback mechanism for upstream caspase activation.12 Moreover, it was recently demonstrated that caspase 6 plays a significant role in ZBP1-NLRP3 inflammasome activation by mediating innate immune responses.13 Caspase 6 promotes the activation of ZBP1-mediated cell death pathways, including pyroptosis, apoptosis and necroptosis, and plays an essential role in host defense against influenza A virus (IAV) infection. Moreover, this caspase has been shown to play a crucial role in coronavirus replication, underlying the importance of caspase 6 controlled inhibition.14 Taken together, there is multiple lines of evidence pointing to the role of caspase 6 in programmed cell death; however, we still suffer from the lack of selective chemical reagents to monitor its activity at the single-cell level.
Similar to other proteases from the caspase family, caspase 6 demonstrates rigorous requirements for aspartic acid at the P1 position of peptide substrates. Over the years, several endogenous caspase 6 substrates have been described and characterized. The first substrate, described in 1996, was lamin A, which is hydrolyzed by caspase 6 after the VEID sequence.15,16 Other examples include SATB1 cleaved after VEMD17 and huntingtin cleaved after IVLD.18 All these cleavage sites reflect the catalytic preferences of caspase 6 and were supported using synthetic substrates. The use of the positional scanning substrate combinatorial libraries (PS-SCL) approach revealed that caspase 6 indeed prefers substrate with the VEID, VEMD, and IVLD sequences, and its overall substrate specificity overlaps with caspase 8 and 9 pref. 3. A more detailed investigation of caspase 6 preferences with the use of combinatorial tetrapeptide substrate libraries containing unnatural amino acids (HyCoSuL approach) enabled the selection of caspase 6-selective peptides and partially distinguished this enzyme from other caspases.19 Moreover, caspase 6 was shown to also recognize pentapeptides, and this catalytic feature might be important for the development of selective chemical reagents.20
Chemical reagents for the visualization of caspase 6 activity include small-molecule substrates, covalent inhibitors and activity-based probes (ABPs), and over the years, these molecules have provided insight into caspase 6 activation mechanisms and biological functions.21,22 Nevertheless, such reagents suffer from suboptimal selectivity, as caspase 6 displays overlapping P4–P1 cleavage preferences with other apoptotic caspases. In our previous studies, we reported the synthesis and kinetic evaluation of a P5 combinatorial library toward caspase 2.23 The use of a broad range of unnatural amino acids enabled us to develop highly selective substrates and inhibitors for this caspase. Herein, we undertook the same approach and performed a broad screen of P5 preferences among apoptotic caspases, challenging the hypothesis that the P5 position may be a good discriminatory factor between these enzymes. Our screen revealed that caspase 6 highly prefers pentapeptides over tetrapeptides; thus, caspase 2 is not the only caspase displaying this kinetic characteristic. Since we found these preferences to be unusual within the caspase family, we decided to further explore this subject by performing in-depth kinetic characterizations with a set of fluorescent substrates and inhibitors. Moreover, we also developed first-in-class caspase 6 substrates with aggregation-induced emission (AIE) characteristics and used them for real-time imaging of caspase 6 activation in a leukemia T cell model. Our caspase 6-selective toolset, supported by western blotting and mass cytometry analysis, enabled us to dissect the kinetics of caspase 6 activation in apoptotic cells and revealed its kinetic cross-talk with other members of the caspase family.
Experimental section
Reagents and antibodies
All chemicals were obtained from commercial sources and were used without further purification. Fmoc- and Boc-protected amino acids were purchased from Iris Biotech GmbH (Marktredwitz, Germany), Sigma-Aldrich (Poznan, Poland), Bachem (Torrance, CA, USA), Creosalus (Louisville, KY, USA), and PE Biosciences Limited (Hong Kong, China). Fmoc-ACC-OH fluorescent dye was synthesized according to the procedure published previously by Maly et al.24 Rink amide AM resin (200–300 mesh, loading 0.74 mmol g−1), 2-chlorotrityl chloride resin (100–200 mesh, loading 1.59 mmol g−1), biotin, HBTU, HATU, piperidine (PAP), diisopropylcarbodiimide (DICI), 2,2,2-trifluoroethanol (TFE), and trifluoroacetic acid (TFA) were purchased from Iris Biotech GmbH. Anhydrous HOBt was purchased from Creosalus. 2,4,6-Collidine (2,4,6-trimethylpyridine), acetonitrile (ACN, HPLC gradient grade), triisopropylsilane (TIPS), hydrobromic acid solution (30% HBr wt. in acetic acid), N-methylmorpholine (NMM), tetrahydrofuran (THF, anhydrous), isobutylchloroformate (IBCF), and 2,6-dimethylbenzoic acid (2,6-DMBA) were purchased from Sigma-Aldrich. N,N′-Dimethylformamide (DMF, pure for analysis), methanol (MeOH), dichloromethane (DCM), acetic acid (AcOH), diethyl ether (Et2O), and phosphorus pentoxide (P2O5) were obtained from POCh (Gliwice, Poland). Diazomethane used for the synthesis of AOMK inhibitors was generated according to the Aldrich Technical Bulletin (AL-180) protocol. All compounds (peptides, ACC fluorescent substrates, AIE fluorescent substrates, and inhibitors) were purified by reverse-phase HPLC on a waters system (Waters M600 solvent delivery module and waters M2489 detector system) using a semipreparative Discovery® C8 column (particle size 10 μm). The purity of the compounds was confirmed on the above HPLC system using an analytical Discovery® C8 column (particle size 10 μm). The solvent composition was as follows: phase A (water/0.1% TFA) and phase B (ACN/0.1% TFA). For purification and compound analysis, the assay was run for 30 min in a linear gradient (from 5% phase B to 100% phase B). The molecular weight of each compound was confirmed on a WATERS LCT Premier XE High Resolution Mass Spectrometer with electrospray ionization (ESI) and a time of flight (TOF) module. Antibodies for western blot analysis were purchased from R&D Systems.
Synthesis of Ac-P5-LEHD-ACC and Ac-P5-DEVD-ACC substrate libraries and tetrapeptide fluorogenic substrates
The pentapeptide and tetrapeptide fluorogenic substrates were synthesized as described previously using the SPPS method.23 All substrates were purified and analyzed on an HPLC system and dissolved in DMSO to a concentration of 20 mM.
Synthesis of the P5 combinatorial substrate library
The synthesis of the P5 combinatorial substrate library with the general formula Ac-P5-Mix-Glu-Mix-Asp-ACC was described in our previous manuscript.23 This library was used to profile caspase preferences at the P5 position without further purification.
Preparation of recombinant caspases
The detailed protocol for the expression and purification of human apoptotic caspases was described elsewhere.25
Enzymatic kinetic studies
All kinetic studies were performed using an fMax fluorescence plate reader (Molecular Devices) operating in fluorescence kinetic mode using 96-well plates. The ACC fluorescence was monitored using 355 nm (excitation) and 460 nm (emission) wavelengths. Before kinetic analysis, all caspases were active site-titrated using the zVAD-fmk inhibitor (Cayman Chemical Company, Cat no. 14467) according to the protocol described by Stennicke and Salvesen.25 For all kinetic experiments, the caspase assay buffer consisted of 10% w/v sucrose, 20 mM PIPES, 10 mM NaCl, 1 mM EDTA, and 10 mM DTT (pH = 7.2–7.4). Additionally, the assay buffer for caspases 8, 9, and 10 was supplemented with 0.75 M sodium citrate to dimerize caspase monomers and ensure maximal enzymatic activity. Buffers were prepared at room temperature, and all kinetic assays were performed at 37 °C. All enzymes were preincubated in buffer for 15 min before kinetic assays were performed. All experiments (library screenings and substrate and inhibitor kinetics) were performed at least three times, and average values with SDs are presented. The kinetic data were analyzed using GraphPad Prism software.
Characterization of apoptotic caspase specificity at the P5 position
The P5 preferences of caspases 2, 3, and 8 determined using the Ac-P5-Mix-Glu-Mix-Asp-ACC library were described in our previous manuscript (16). Herein, we used the same approach to screen P5 preferences of other apoptotic caspases, including caspases 6, 7, 9, and 10. Each caspase was tested in the appropriate assay buffer (the buffer for caspases 9 and 10 was supplemented with sodium citrate). The P5 library was used at 100 μM, and the caspase concentrations were as follows: caspase 6, 50 nM; caspase 7, 20 nM; caspase 9, 50 nM; and caspase 10, 20 nM. All screenings were performed on 96-well plates in a 100 μl volume. The total assay time was 30 min, but only the linear portion of the fluorescence progress curve was taken for the analysis. For each caspase, the hydrolysis rate of the control tetrapeptide substrate (Ac-Mix-Glu-Mix-Asp-ACC) lacking the P5 position was set as 100% (or 1.0), and the P5 substrates were adjusted accordingly.
Synthesis and kinetic analysis of individual substrates
The procedure for the synthesis, purification, and kinetic analysis of the ACC-labeled substrates was described elsewhere.19
Synthesis of TPE-N3
Azide-functionalized tetraphenylethene (TPE-N3) was synthesized in two steps with an 80% total yield. (1) Synthesis of 1-(4-methylphenyl)-1,2,2-triphenylethene. To a 250 mL two-necked round-bottom flask equipped with a stirring bar, 5.05 g (30 mmol) of diphenylmethane was added and dissolved in 100 mL of anhydrous THF. Then, the mixture was cooled down to 0 °C, and 15 mL (2.5 M in hexane, 37.5 mmol) of n-butyllithium was slowly added by a syringe. The mixture was stirred at 0 °C for 1 h. Next, 4.91 g (25 mmol) of 4-methylbenzophenone was added into the flask, and the mixture was warmed to room temperature and stirred overnight. The reaction mixture was then quenched with saturated NH4Cl solution and subsequently extracted with DCM (three times). Organic layers were collected and concentrated on a rotary evaporator under reduced pressure. The crude product with 0.20 g of p-toluenesulfonic acid was dissolved in 100 mL of toluene. The mixture was heated and refluxed for 4 h. After cooling to room temperature, the mixture was extracted with DCM (three times). The organic layers were collected and concentrated. The crude product was analyzed with LC–MS (purity > 95%) and was used in the next step without purification (4.08 g, 95% yield). HR-MS (MALDI-TOF): m/z 346.1701 [(M)+, calc. 346.1722]. (2) Synthesis of 1-[(4-bromomethyl)phenyl]-1,2,2-triphenylethene. In a 250 mL round-bottom flask, a mixture of 3.04 g (8.77 mmol) of 1, 1.80 g (9.77 mmol) of freshly recrystallized N-bromosuccinimide, and 0.025 g of benzoyl peroxide in 60 mL of CCl4 was refluxed for 12 h. After this time, the mixture was extracted with water and DMC (3×). The organic layers were combined and dried over magnesium sulfate, and the DCM was removed under reduced pressure. The crude product was purified by silica-gel chromatography using hexane as the eluent to yield 2 as a white solid (2.42 g, 65% yield).
Synthesis of Fmoc-Lys-alkyne)-OH
Two grams (4.3 mmol) of Fmoc-Lys(Boc)-OH was dissolved in 5 mL of a TFA/DCM/TIPS (% v/v/v, 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:5) mixture and vigorously stirred for one hour. Next, the solution was concentrated, dissolved in DCM and dried in vacuo. The procedure was repeated six times to ensure TFA removal. The crude product (Fmoc-Lys-OH) was then dissolved in DMF, and subsequently, hex-5-ynoic acid-NHS (4.8 mmol, 1 g) with DIEA (14.4 mmol, 2.5 mL) was added. The reaction was stirred at room temperature for an hour. Next, the mixture was extracted with ethyl acetate, and the solvent was removed under reduced pressure. The crude product was dissolved in ACN:H2O (3:1), lyophilized and used in AIE substrate synthesis without purification.
:20:5) mixture and vigorously stirred for one hour. Next, the solution was concentrated, dissolved in DCM and dried in vacuo. The procedure was repeated six times to ensure TFA removal. The crude product (Fmoc-Lys-OH) was then dissolved in DMF, and subsequently, hex-5-ynoic acid-NHS (4.8 mmol, 1 g) with DIEA (14.4 mmol, 2.5 mL) was added. The reaction was stirred at room temperature for an hour. Next, the mixture was extracted with ethyl acetate, and the solvent was removed under reduced pressure. The crude product was dissolved in ACN:H2O (3:1), lyophilized and used in AIE substrate synthesis without purification.
Synthesis of AIE substrates
The synthesis of Ac-DEVDK-TPE, Ac-VEIDK-TPE, Ac-hPhe-VEIDK-TPE and Ac-Glu-VEIDK-TPE substrates involves both solution- and solid-phase chemistry. The alkyne-containing peptides were prepared by solid-phase peptide synthesis using the Fmoc strategy. In brief, Fmoc-protected Rink Amide resin was swollen for 30 minutes in DCM, followed by three washes with DMF. The Fmoc-protecting group was removed with 20% piperidine in DMF in three cycles (5 min, 5 min, 25 min), and the resin was washed six times with DMF. Next, an alkyne-functionalized lysine was attached to the resin with HOBt and DICI as coupling agents (3× excess, 3 hours, DMF, RT). Next, the mixture was filtered off, and the resin was washed four times with DMF. The coupling efficiency was confirmed with a ninhydrin test (5 min, 95 °C). Next, the resin was washed three times with DCM, three times with MeOH, dried over P2O5 and divided into four equal parts. Following the same SPPS procedure, Fmoc-protected amino acids were subsequently incorporated onto the resin to yield NH2-peptide-Lys-resin. Finally, the N-terminus of these peptides was capped with acetic acid, HOBt, and DIEPA in DMF (5× excess, 30 min). The products were washed with DMF (3×), DCM (3×) and MeOH (3×) and dried over P2O5 cleaved from the resin using a standard TFA procedure. Crude peptides were purified by HPLC and characterized with LC–MS. Subsequent coupling between TPE-N3 and azide-functionalized peptides via a Cu(I)-catalyzed “click” reaction was performed using CuSO4/sodium ascorbate as the catalyst and DMSO/water as the solvent. The synthesis yielded AIE substrates with >70% yield after HPLC purification. The purity and MW identity of each substrate were characterized by analytical HPLC and HR-MS.
Kinetic of AIE substrates
Caspases 3 and 6 were dissolved in assay buffer to a concentration of 10 nM or 50 nM (99 μL). After 15 min of preincubation, caspases were added to a 96-well plate containing AIE substrates dissolved in DMSO at various concentrations (1 μL; the final concentration of substrates in the well is presented in Fig. 5C). The gain of the fluorescence signal was monitored over a period of 30 min at 312 nm (excitation) and 450 nm (emission), and the relative rate of substrate hydrolysis was calculated from the linear part of the fluorescence curve. All experiments were performed at least three times, and the results are presented as average values with SDs. Unpaired, two-tailed t Test was performed to demonstrate the statistical significance of the results (*P < 0.05, **P < 0.01, ***P < 0.001).
Synthesis of AOMK inhibitors
The detailed protocol for the synthesis of P1 Asp inhibitors and probes with AOMK electrophile warheads has recently been described by Poreba et al.26 In brief, Boc-L-Asp(Bzl)-OH amino acids were transformed into Boc-L-Asp(Bzl)-CH2N2 through the use of diazomethane solution in diethyl ether. Next, the product was transformed into Boc-L-Asp(Bzl)-CH2Br with the use of a solution of HBr (30% wt. in acetic acid) and water. The crude product, a pale yellow oil, was then stirred with 2,6-dimethylbenzoic acid (2,6-DMBA) in the presence of KF in DMF to obtain Boc-L-Asp(Bzl)-AOMK. Then, the Boc group was removed using 50% TFA in DCM, and the final product (H2N-L-Asp(Bzl)-AOMK) was used for further synthesis without purification. In a separate synthesis, caspase-selective peptide fragments containing appropriate protecting groups (Ac-Glu(tBu)-Val-Glu(tBu)-Ile-COOH, Ac-hPhe-Val-Glu(tBu)-Ile-COOH and Ac-Val-Glu(tBu)-Ile-COOH) were synthesized on 2-chlorotrityl chloride resin and used without further purification. Next, the peptides were coupled with H2N-L-Asp(Bzl)-AOMK in DMF using HATU/DIPEA coupling reagents. The crude products were purified by HPLC, lyophilized and deprotected using 50% TFA in DCM, followed by removal of TFA/DCM under reduced pressure. Finally, the Bzl group from Asp was hydrolyzed via hydrogenolysis (Pd/C, H2, in DMF). The final products were purified by HPLC, lyophilized and dissolved in DMSO to a final concentration of 10 mM.
Determination of the kobs/I inhibition parameters for inhibitors
Caspases 3, 6, 7, 8, 9, and 10 were active site-titrated using the zVAD-fmk inhibitor. All enzymes were assayed in appropriate caspase buffer. The kobs/I parameter was measured under pseudo-first-order kinetic conditions. Inhibitors were diluted on a 96-well plate and mixed with the appropriate substrates (Ac-DEVD-ACC for caspases 3 and 7, Ac-VEID-ACC for caspase 6, and Ac-LEHD-ACC for caspases 8, 9, and 10; the final substrate concentration was 100 μM) and preincubated at 37 °C for 15 min. At the same time, in a separate tube, caspases were preactivated in assay buffer at 37 °C. After 15 min, enzymes were added to the wells, and fluorescence was monitored for 30 min, starting immediately. The second-order rate of inhibition (kobs/I) was determined in at least three independent experiments and is presented as average values with SDs.
Detection of proteins in apoptotic Jurkat T cells by immunofluorescence
Low-passage Jurkat T cells were seeded on a 12-well plate (100000 cells per well per 0.5 mL). Next, staurosporine (STS, 5 μM final concentration) was added to the cells and incubated for various times (0 min, 30 min, and 1, 2, 4, 6, and 8 hours). Next, cells were harvested, centrifuged at 500×g for 5 min, washed with 1× PBS, and centrifuged again at 500×g for 5 min. The cell pellet was then sonicated and centrifuged at 14000×g for 5 min. Then, PBS was aspirated, and cell pellets were suspended in 50 μL of 1× SDS/DTT and boiled for 5 min. Samples (20 μL) were subjected to SDS–PAGE (30 min, 200 V, 4–12% Bis-Tris 15-well gels), followed by transfer to a nitrocellulose membrane (60 min, 10 V) and blocking with 2% BSA in TBS-T (60 min, RT). Proteins were detected by probing the membrane with primary antibodies (anti-caspase 3 Cell Signaling #9662 1:1000; anti-PARP Cell Signaling #9542 1:1000; anti-caspase 6 Cell Signaling #9762 1:1000; anti-lamin A/C Cell Signaling #4777 1:1000; overnight, +4 °C) followed by incubation with fluorescently labeled secondary antibodies (IRDye 800CW donkey anti-goat, LI-COR; IRDye 680RD donkey anti-goat, LI-COR; 30 min, RT). The membranes were finally scanned in a LI-COR system (700 nm/red and 800 nm/green channels, Odyssey® CLx, Lincoln, NE, USA). The protein bands were quantified using Image Studio Lite software (Lincoln, NE, USA).
Real-time imaging of caspase activation in staurosporine-treated Jurkat T cells
Low-passage Jurkat T cells (100000 cells per well per 200 μL) in phenol red-free RPMI media were seeded on 4-well slides (Nunc Lab-Tek Chambered Coverglass, Thermo Fisher, #155361). Next, cells were treated with staurosporine (5 μM final concentration) and immediately treated with PI and selected TPE fluorogens (control, DEVD-KTPE for caspase 3/7, VEID-KTPE for caspase 3/6/7, and EVEID-KTPE for caspase 6, 100 μM final concentration). Slides were placed in the Leica TCS SP8 confocal microscope chamber in the presence of 5% CO2, and at selected time points (0, 30 min, 1, 2, 4, 6, 8 hours), images were taken. TPE fluorescence was detected as the UV channel (blue), whereas the PI signal was detected using a single-photon laser at 646 nm (red). Brightfield cell images were also taken using differential interference contrast (DIC).
Detection of proteins in apoptotic Jurkat T cells upon inhibitor treatment
Low-passage Jurkat T cells were seeded on a 12-well plate (100000 cells per well per 0.5 mL). Next, cells were preincubated with individual inhibitors (5 μM and 25 μM final concentration, 1% DMSO) or were left untreated (1% DMSO). After 2 hours of incubation, staurosporine (5 μM final concentration) was added to the cells. After 4 or 8 hours of incubation, cells were harvested and subjected to SDS-PAGE analysis and immunofluorescence protein visualization as described above.
Labeling antibodies with metal tags
Primary human monoclonal anti-caspase 6 (clone MCH2 14 1–190; # MA1-12558) and anti-lamin A (clone 133A2; # MA1-06101) antibodies were purchased from Thermo Fisher Scientific. The antibodies were labeled with 159Tb and 142Nd metals, respectively, according to a protocol described elsewhere (https://web.stanford.edu/group/nolan/protocols.html). All necessary reagents were purchased from Fluidigm. Other metal-tagged antibodies, anti-cleaved caspase 3 (172Yb, clone 5 A1E, # 3172023) and anti-cleaved PARP (143Nd, clone F21-852, # 3143011), were purchased from Fluidigm. Unless otherwise stated, all buffers and reagents for mass cytometry were purchased from Fluidigm.
Analysis of staurosporine-stimulated Jurkat T cells by mass cytometry
Jurkat T cells (1000000 cells per well per 0.5 mL) were seeded onto 12-well plates and preincubated with individual inhibitors (5 or 25 μM final concentration) for 2 hours. Next, the cells were stimulated with staurosporine (5 μM) for 4 hours. After this time, the cells were harvested, centrifuged at 500×g for 5 min, washed with 1 mL of 1× DPBS and centrifuged again. Next, the cell pellet with a residual amount of 1× DPBS (∼50 μL) was fixed with 1 mL of 4% PFA/DPBS for 20 min at 4 °C and washed again with DPBS. Following fixation, cells were permeabilized with Perm-S Buffer (Thermo Fisher Scientific), washed once with PBS and resuspended in Perm-S antibody cocktail containing anti-caspase 6 (159Tb), anti-lamin A (142Nd), anti-cleaved PARP (143Nd) and anti-cleaved caspase 3 (172Yb) antibodies. Cells were incubated with antibodies for 2 hours at RT, centrifuged at 900×g for 5 min, washed 1× with PBS, centrifuged 1× with permeabilization buffer, centrifuged, and stained with Ir191/193 DNA intercalator for 30 min at RT. Then, the samples were centrifuged at 900×g for 5 min, and the cell pellet was resuspended in 500 μL of CyFACS buffer and stored at 4 °C until CyTOF acquisition, but no longer than 12 h. Prior to acquisition, the cells were washed twice with CyFACS buffer and twice with Cell Acquisition Solution (Fluidigm). The results were analyzed with FlowJo and Cytobank software.
Results and discussion
Caspase screening at the P5 position
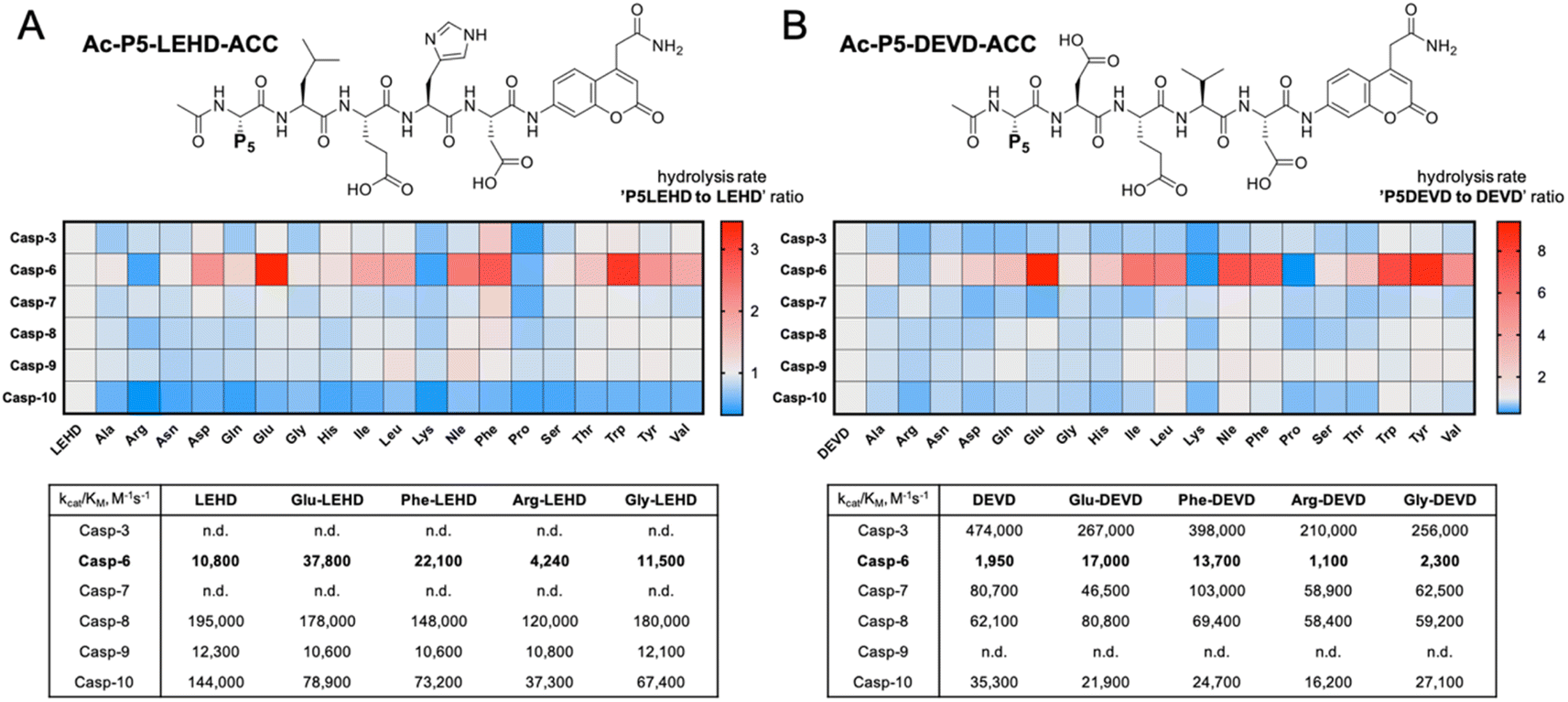
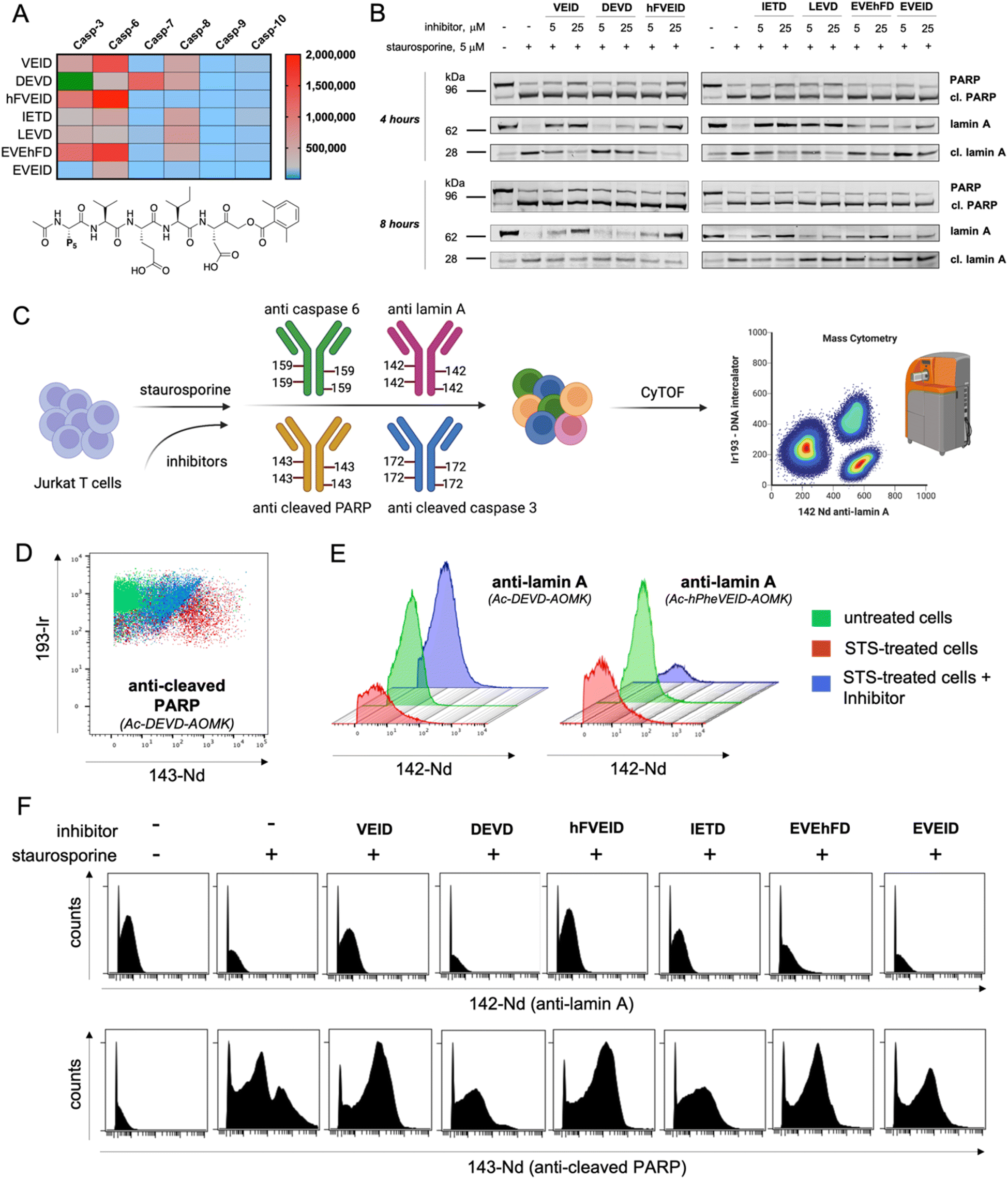
To evaluate caspase preferences toward pentapeptides, we synthesized two fluorescent substrate libraries with the general formulae DEVD and LEHD and a variety of natural amino acids at the P5 position. Each substrate was N-terminally acetylated and labeled with an ACC fluorescent tag at the C-terminus (Fig. 1). We selected DEVD and LEHD sequences to cover the specificity requirements of all apoptotic caspases, making these libraries useful to prime P5 preferences of six apoptotic caspases (DEVD for caspases 3, 7, 8, 10, and LEHD for caspases 6, 8, 9, 10). Based on our preliminary screening data, we concluded that similar to caspase 2, caspase 6 has a well-defined S5 pocket that facilitates efficient substrate turnover. Moreover, library screening enabled the selection of preferable pentapeptide substrates that contain both hydrophilic amino acid side chains (e.g., glutamic acid) and large hydrophobic side chains (phenylalanine, tryptophan and tyrosine) at the P5 position. Detailed kinetic analysis of selected substrates (kcat/KM), including varied residues at P5 (basic, acidic, aliphatic, and hydrophobic), revealed that caspase 6 indeed favors pentapeptide substrates. The kinetic data were consistent with the screening results, as glutamic acid was the most preferable residue at the P5 position.
 |
| | Fig. 1 Initial screening of apoptotic caspase preferences toward pentapeptide substrates. Two ACC-labeled substrate libraries (Ac-P5-LEHD-ACC and Ac-P5-DEVD-ACC) containing natural amino acids were used to determine the substrate specificity of six apoptotic caspases at the P5 position. Only caspase 6 was shown to preferably hydrolyze pentapeptide substrates from both libraries. (A) General structure of the pentapeptide Ac-P5-LEHD-ACC library. Kinetic parameters (kcat/KM) were determined for selected substrates with a variety of amino acid residues at P5. (B) General structure of the pentapeptide Ac-P5-DEVD-ACC library. Kinetic parameters (kcat/KM) were determined for selected substrates with a variety of amino acid residues at P5. The rate of pentapeptide substrate hydrolysis (expressed as RFU/s) by individual caspase was divided by the hydrolysis rate of control tetrapeptide substrates (LEHD or DEVD) and presented as a ratio. For better data presentation the hydrolysis rate of a control tetrapeptide substrate (LEHD and DEVD) for each caspase was set to 1.0 value, and the heat maps were scaled separately from 0 to the highest P5LEHD/LEHD (and P5DEVD/DEVD) ratio. The detailed kinetic analysis of selected substrates (kcat/KM) certifies the screening results. | |
P5 combinatorial library with unnatural amino acids
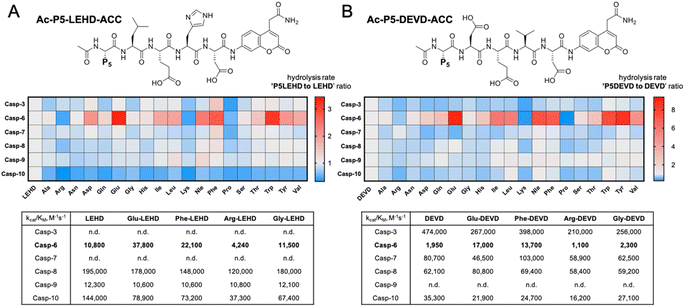
The initial screen uncovered unusual caspase 6 preferences toward pentapeptide substrates. Therefore, we decided to further explore caspase 6 P5 specificity using a P5 combinatorial substrate library containing over 100 unnatural amino acids. This library was successfully utilized to explore the chemical space in the caspase 2 S5 binding pocket, yielding a selective substrate and inhibitor.23 The general formula of the P5 library is Ac-P5-Mix-Glu-Mix-Asp-ACC (Fig. 2A), where P5 is a fixed natural or unnatural amino acid, Mix is an equimolar mixture of natural amino acids, and ACC is a fluorescent tag. As the library incorporates aspartic acid at the P1 position and glutamic acid at P3, it constitutes a convenient tool for the screening of P5 preferences of all caspases. The general caspase preferences at the P5 position are presented in Fig. 2B, and the whole specificity profiles can be found in Fig. S1.† The most favored amino acids in the caspase 6 S5 binding pocket are L-hPhe (homophenylalanine, L-hF) and L-Glu, as these substrates were hydrolyzed over 6 times faster than the reference tetrapeptide substrate lacking the P5 amino acid (Ac-Mix-Glu-Mix-Asp-ACC). Furthermore, we also identified several more amino acids at the P5 position that substantially enhanced the substrate hydrolysis rate, such as L-Gla (γ-carboxyglutamic acid), L-Igl (2-indanyl-glycine), L-Api ((homo)2-glutamic acid) and natural L-Trp. Given these results, we observed that caspase 6 displays the ability to accommodate two distinct types of amino acid side chains in the S5 pocket, namely, large hydrophobic (L-hPhe and its derivatives) and acidic (L-Glu and its derivatives) residues (Fig. 2C). Extensive P5 substrate specificity screening for apoptotic caspases enabled us to create an explicit outline of caspase substrate interactions. Therefore, we were able to extract amino acids that were further used to develop caspase 6-selective reagents.
 |
| | Fig. 2 Preferences of apoptotic caspases at the P5 position. (A) The P5 combinatorial library was designed based on general caspase preferences toward acidic amino acid residues at the P1 (Asp) and P3 (Glu) positions. The general architecture of the P5 combinatorial fluorogenic substrate library is Ac-P5-Mix-Glu-Mix-Asp-ACC. (B) Substrate specificity of seven apoptotic caspases at the P5 position. The activity ratio between pentapeptide and the control tetrapeptide substrate lacking a P5 amino acid is shown as a heatmap, where red indicates the best (most preferable) substrates and blue indicates the least preferable substrates. The results are arranged in accordance with caspase 6 preferences. All P5 specificity profiles are presented in the Supplemental Section; “none” – activity toward the corresponding tetrapeptide substrate (Ac-Mix-Glu-Mix-Asp-ACC). (C) The structures of the best amino acid residues for caspase 6 at the P5 position. | |
Kinetic analysis of caspase 6 pentapeptide substrates
After a broad analysis of caspase 6 P5 preferences, we aimed to precisely investigate the influence of P5 amino acids on the substrate hydrolysis rate, considering subsite cooperativity. To do so, we synthesized fluorescent pentapeptide substrates based on previously used peptide scaffolds (Ac-P5-LEHD-ACC, Ac-P5-DEVD-ACC), as well as also based on caspase 6 canonical tetrapeptides (Ac-P5-VEID-ACC, Ac-P5-VEHD-ACC), and we measured their kinetic parameters (kcat, KM, kcat/KM) toward this protease. P5 amino acids were selected to ensure the diversity of substrates; thus, we incorporated glycine, threonine, lysine, glutamic acid and homophenylalanine to decipher the potential subsite cooperativity of caspase 6. Fig. S2† presents the data collected for 22 different substrates. For almost all of them, we observed an inverse relationship between the kcat/KM and KM values. We found that substrates with VEHD and VEID tetrapeptide scaffolds are the most efficiently hydrolyzed, whereas substrates bearing LEHD and DEVD motifs are recognized much less efficiently by caspase 6. This observation is in line with the general P4–P2 preferences of caspase 6 determined by the PS-SCL approach. Importantly, regardless of the P4–P2 core scaffold, caspase 6 presents almost the same kinetic preferences within four sets of substrates. These results strongly indicate that caspase 6 does not display P5–P2 subsite cooperativity; therefore, the additional amino acid at the P5 position can be incorporated into caspase 6 peptides to improve their potency and selectivity.
AIEgens for caspase 6 investigation
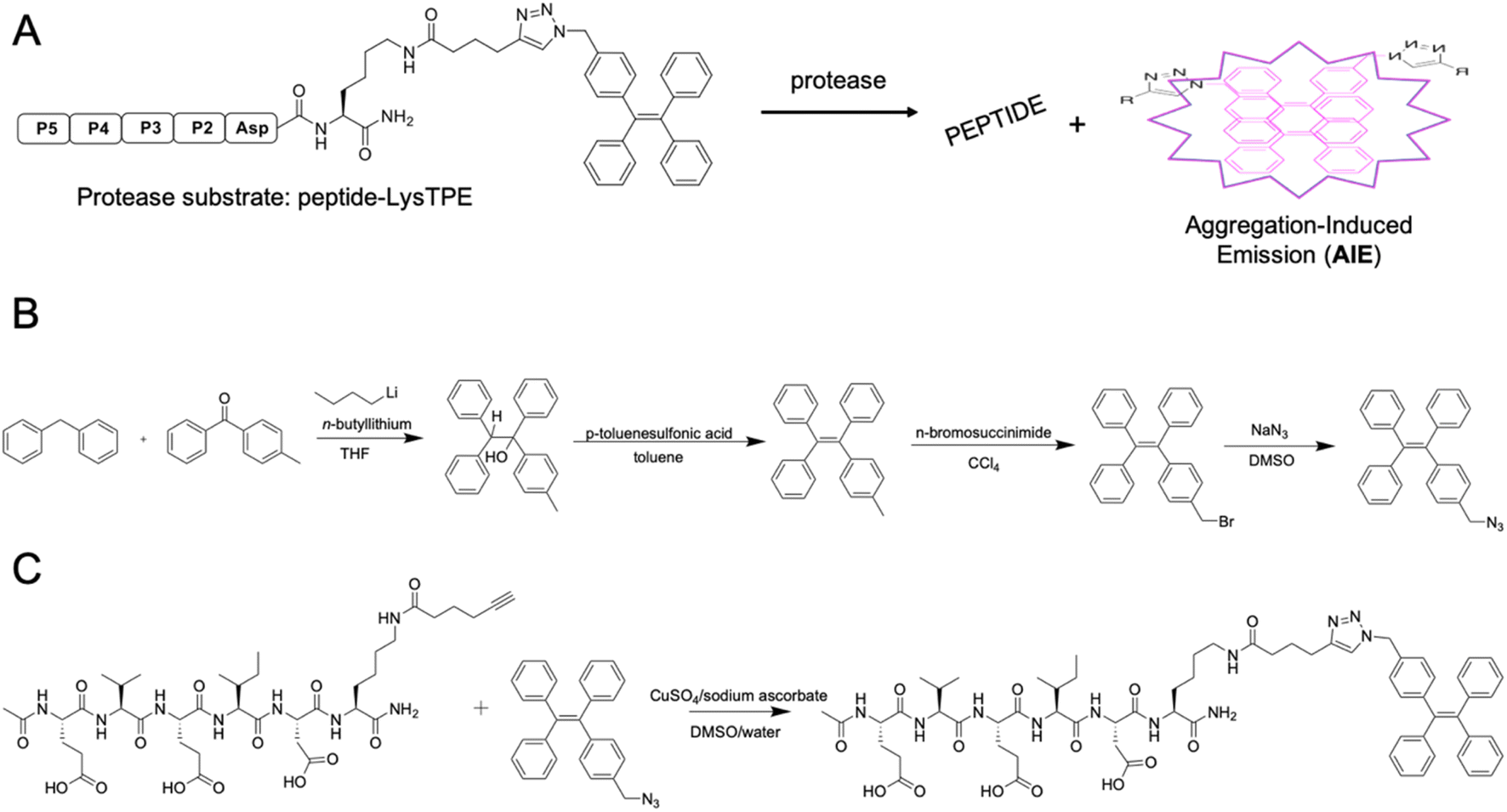
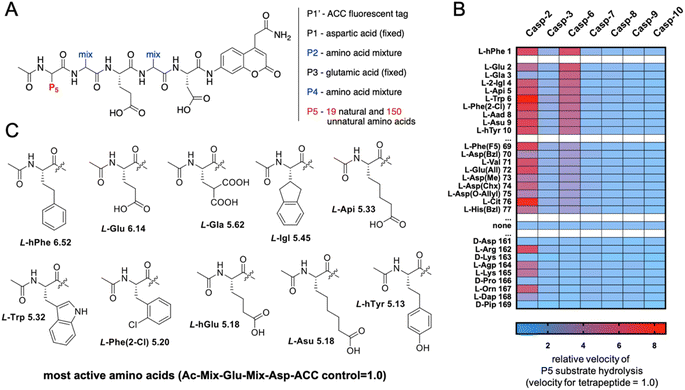
AMC- and ACC-labeled fluorescent substrates constitute one of the most convenient chemical tools for the profiling of protease substrate specificity and kinetic measurements in general.3,27 Unfortunately, their application is limited to simple in vitro experimental setups, as upon peptide bond hydrolysis, the fluorogenic reporter group diffuses away, hampering the localization of the active form of the enzyme at the single-cell level.28 Such limitations may be overcome with fluorescently labeled irreversible inhibitors that bind tightly to the enzyme active site; nevertheless, in the case of closely related enzymes such as caspases, strongly electrophilic warheads considerably attenuate inhibitor selectivity, causing off-target binding with other caspases.29 Given the above considerations, after almost three decades of caspase research, no truly selective reagents for caspase localization have been developed. A promising solution to this issue might be the use of substrates with aggregation-induced emission (AIE) characteristics, called AIEgens or AIE fluorogens.30 In these substrates, the standard fluorophore is replaced with an AIE fluorogen, which upon peptide bond hydrolysis undergoes fast and spontaneous aggregation leading to the emission of a strong fluorescence signal.31 Therefore, AIEgens exhibit all properties of regular substrates, including high selectivity and signal amplification. In addition, aggregated AIE fluorogens do not rapidly diffuse from the enzyme site, enabling the localization of the target protease. Therefore, AIE probes combine the advantages of fluorogenic substrates (selectivity) and irreversible inhibitors (localization). Such unique properties of AIE fluorogens offer an opportunity to develop specific light-up probes with many advantages, such as low background interference, superior photostability and a high signal-to-noise ratio.32 Given the above, and considering that no caspase 6-selective reagents have been developed, we utilized the unusual caspase 6 P5 catalytic preferences in the manufacturing of the first AIE-based substrates for this protease (Fig. 3A). In the first step, we synthesized a TPE-N3 (tetraphenylethene) fluorogen that displays AIE characteristics and subsequently coupled it through a lysine side chain to selected pentapeptides via a click chemistry approach (Fig. 3B and C).
 |
| | Fig. 3 AIE-based substrates for caspase 6. (A) Schematic representation of the activation of AIE-based protease substrates. The pentapeptide designed for caspase 6 was labeled with TPE fluorogen and treated with protease. After peptide bond hydrolysis, TPE tags are released and undergo spontaneous aggregation, yielding a strong fluorescence signal. (B) The synthesis route of TPE-N3 (TPE-azide) fluorogen. (C) Schematic representation of peptide-Lys-TPE synthesis. The caspase 6 peptide bearing alkyne-functionalized lysine (P1′ position) was synthesized and subsequently conjugated with the TPE-N3 moiety through click chemistry. | |
Kinetic analysis of caspase 6 AIE fluorogens
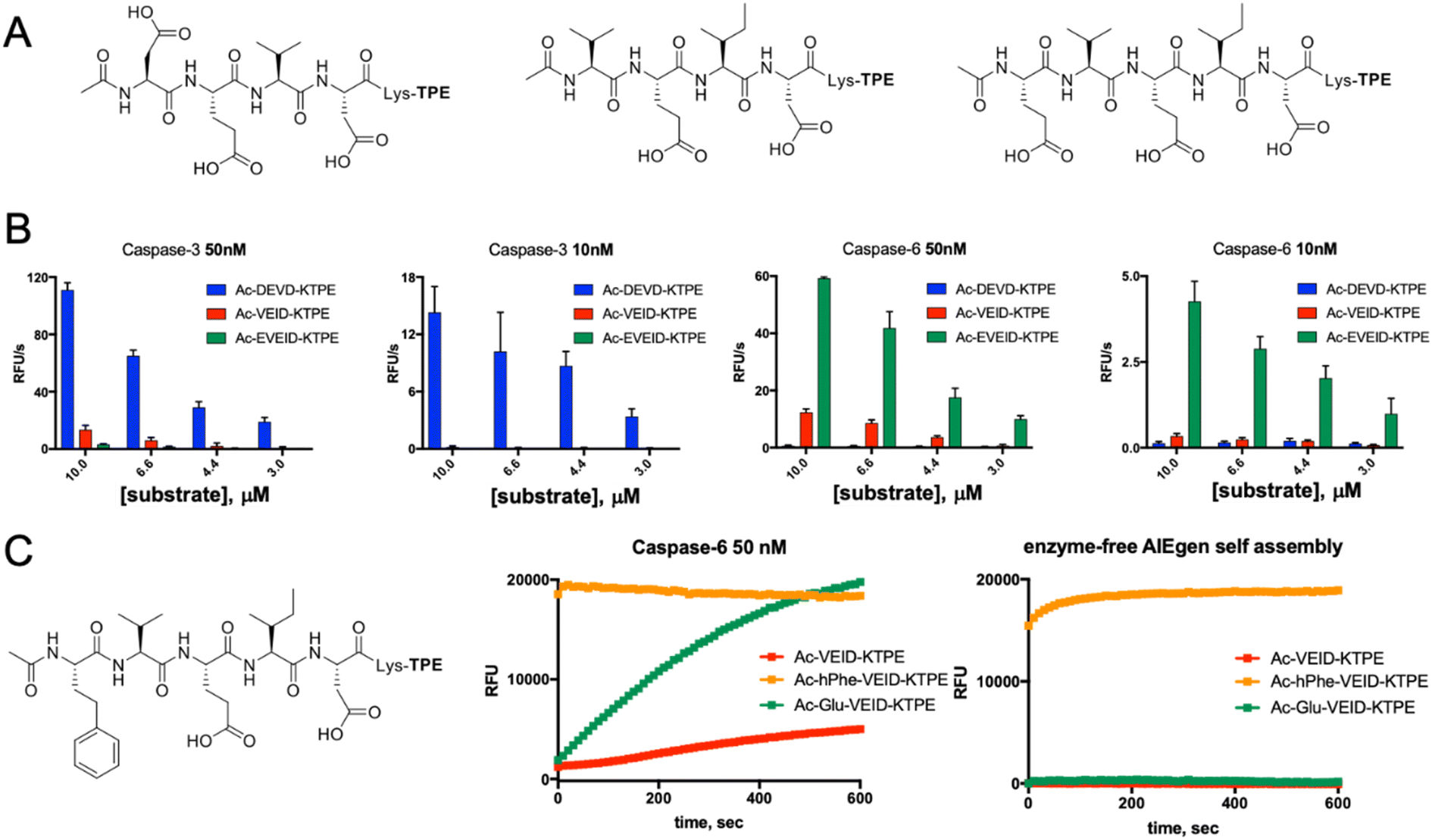
Along with caspases 3 and 7, caspase 6 orchestrates the execution phase of apoptosis. Very recently, this enzyme was also indicated to be a key factor in the activation of the NLRP3 inflammasome, the main pyroptotic machinery.13,33 Given the prominent role of caspase 6 in cell death, selective reagents for this enzyme would be of great importance. Therefore, we tested whether AIE fluorogens can discriminate between caspase 6 and caspases 3 and 7. To do so, taking advantage of P5 library screening, we synthesized two pentapeptide AIE substrates containing L-Glu or L-hPhe at the P5 position and the VEID motif at P4–P1. As a control, we also synthesized AIEgens with classical DEVD (caspase 3) and VEID (caspase 6) motifs (Fig. 4A). Kinetic analysis revealed that both tetrapeptide substrates are efficiently hydrolyzed by their target enzyme in a concentration-dependent manner (Fig. S3†). We demonstrated that although caspases 3 and 6 display overlapping substrate specificity, the tetrapeptide VEID-KTPE is very selective for caspase 6 over caspase 3. This result may be explained by the fact that caspases 6 and 3 differ significantly in their substrate preferences at the P1′ position. We previously demonstrated that caspase 3 recognizes only small amino acids at P1′, whereas caspase 6 displays broader specificity and can also accommodate other amino acids, such as lysine, which is used in AIE substrates for the incorporation of the TPE fluorogen.34 Notably, the introduction of glutamic acid at the P5 position significantly enhanced caspase 6 activity toward this AIE substrate (by approximately 4- to 5-fold) and, more importantly, improved its selectivity over caspase 3. The second caspase 6 pentapeptide AIE fluorogen included unnatural homophenylalanine (L-hPhe) at the P5 position (Fig. 4C, left). However, during kinetic measurements, we observed a very high background fluorescence signal generated by this substrate, and in particular, we detected no fluorescence increase after the addition of enzyme (Fig. 4C, middle). To further examine this phenomenon, we performed kinetic measurements in the absence of enzyme and found that this substrate undergoes rapid and spontaneous self-assembly when transferred from DMSO (stock) into the assay buffer (Fig. 4C, right). Therefore, we concluded that the hydrophobic P5 residue of L-hPhe might have caused rearrangements within the molecule and induced aggregate formation, even in the absence of enzyme. This feature strongly indicates that while developing new AIE substrates, a detailed kinetic analysis must be performed prior to cell imaging. Nevertheless, we showed that AIE-based compounds can be suitable for caspase kinetic assays, and by leveraging the peptide sequences, we can significantly improve their potency and selectivity toward individual caspases, as shown for caspase 6.
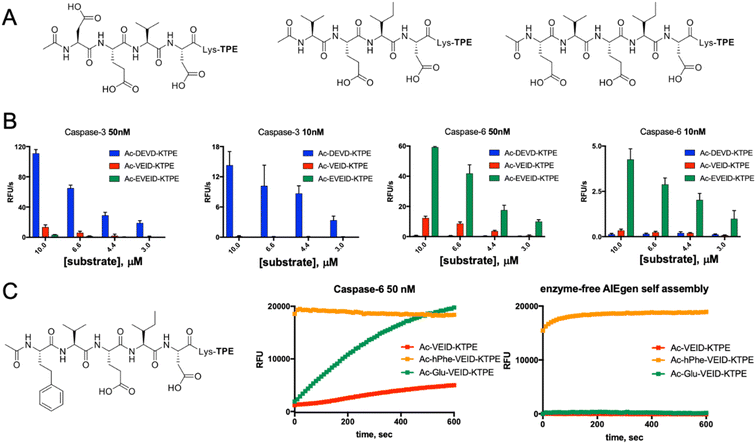 |
| | Fig. 4 Kinetic analysis of AIE-based substrates toward caspases 3 and 6. (A) Chemical structure of AIE-based substrates for caspases 3 and 6. (B) The efficiency of substrate hydrolysis by caspases 3 and 6. Tetrapeptide AIE substrates with classical sequences (DEVD for caspase 3 and VEID for caspase 6) are efficiently hydrolyzed as their target enzymes. The pentapeptide caspase 6 substrate Ac-EVEID-K-TPE displays very high selectivity over caspase 3. (C) The AIE-based substrate for caspase 6 with homophenylalanine at the P5 position is not suitable for kinetic studies, as it undergoes self-assembly due to high hydrophobicity and yields a strong fluorescent signal in the absence of the enzyme. | |
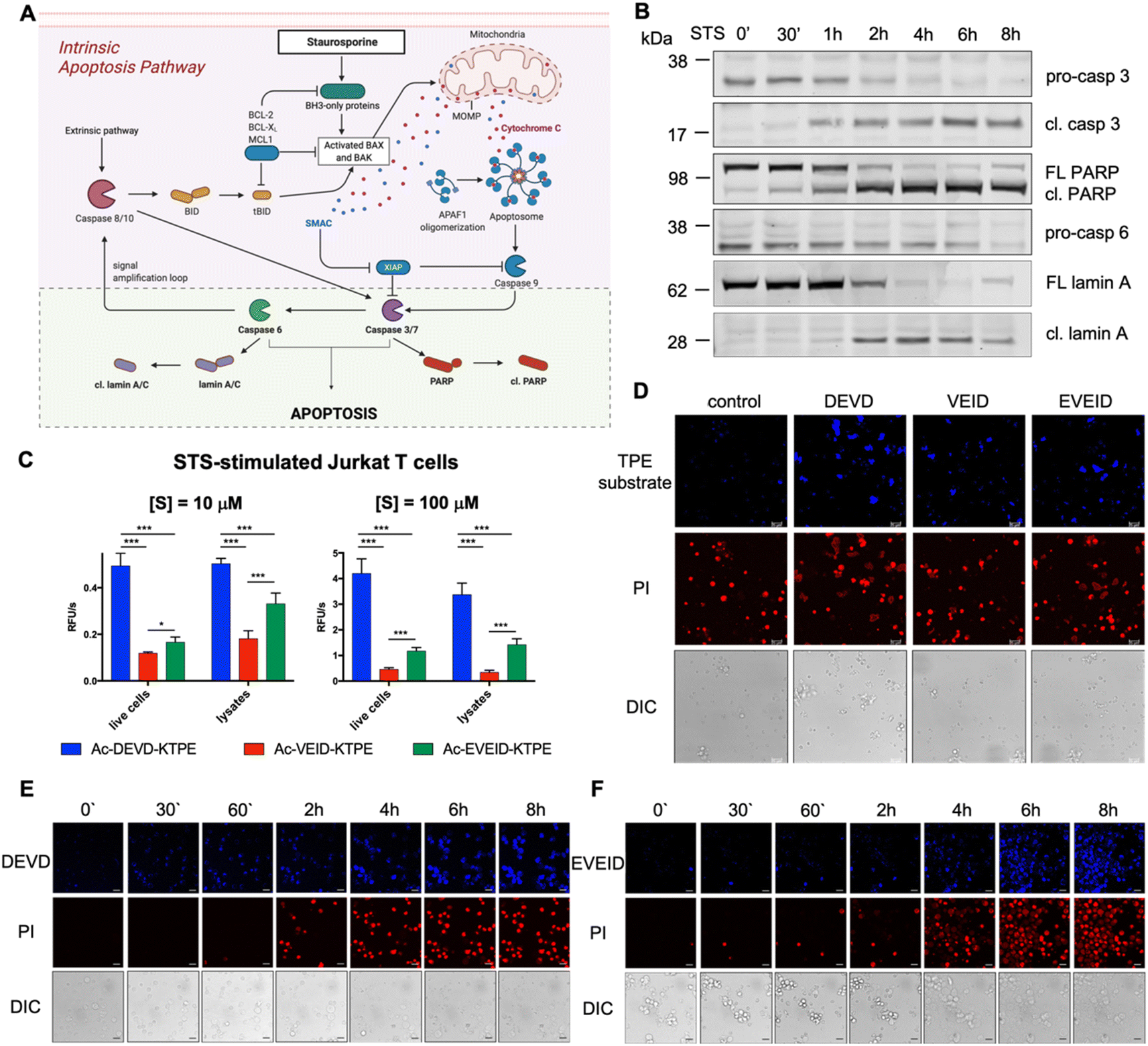
 |
| | Fig. 5 Caspase 6 activation in STS-treated Jurkat T cells. (A) Graphical representation of the intrinsic apoptosis pathway upon staurosporine treatment. The pale green background represents our working model to study hierarchical activation of executioner caspases and real-time imaging of caspase 6. (B) Activation of pro-caspase 3 (p32 → p17/p19) and pro-caspase 6 (p35) and processing of PARP (p116 → p89) and lamin A (p74 → p28) substrates demonstrate hierarchical activation of executioner caspases. (C and D) Detection of caspase 3/7 (DEVD), caspase 3/6/7 (VEID) and caspase 6 (EVEID) activity in STS-treated Jurkat T cells with AIE substrates. Cells were incubated with the appropriate AIE substrate for 2 hours, washed with PBS, stimulated with STS for 8 hours, and subjected to fluorescence readout (C) or confocal microscopy imaging (D). (E) Real-time imaging of caspase 3 activation in Jurkat T cells using Ac-DEVD-KTPE fluorogen. (F) Real-time imaging of caspase 6 activation in Jurkat T cells using Ac-EVEID-KTPE fluorogen. Panels E and F demonstrate hierarchical activation of caspase 3 followed by activation of caspase 6. An unpaired two-tailed T-test (P < 0.05) was performed to demonstrate the statistical significance of the results (C). | |
Real-time imaging of caspase 6 activation in Jurkat T cells
In the intrinsic pathway of apoptosis, pro-caspase 6 is activated by caspases 3 and 7, and then active caspase 6 cleaves apical pro-caspases 8 and 10, providing an amplification loop on the apoptotic signaling pathway.35 However, pro-caspase 6 can also undergo self-activation when it is directly upregulated by p53.36 To fully decipher the kinetics of caspase 6 activation, we decided to stimulate Jurkat T cells with staurosporine (STS), a protein kinase inhibitor that induces mitochondrial damage, cytochrome c release and subsequent activation of the caspase 9-dependent apoptosis pathway (Fig. 5A). Considering that in staurosporine-induced apoptosis, caspase 9 activates procaspase 3 but not procaspase 6,37 we aimed to investigate whether our selective AIE fluorogens would manage to distinguish the executioner caspase cascade in given time frames. First, we performed western blot analysis to monitor pro-caspase 3 and pro-caspase 6 activation and the processing of caspase substrates, i.e., PARP is cleaved by active caspase 3, and lamin A is processed by caspase 6 (Fig. 5B, S4†). We noticed that caspase 3 was fully activated within the first two hours of staurosporine treatment, while some portion of pro-caspase 6 remained for up to 6 hours. This hierarchical activation pattern is also reflected by the processing of downstream caspase 3 and 6 substrates, as PARP is cleaved before lamin A processing occurs. After establishing the kinetic pattern of executioner caspase activation, we used TPE substrates for the visualization of individual caspases. First, we incubated Jurkat T cells with selected AIE substrates (Ac-DEVD-TPE, Ac-VEID-TPE or Ac-EVEID-TPE at 10 μM and 100 μM), stimulated them with staurosporine for 8 hours and subjected them to fluorescence measurement (Fig. 5C). We demonstrated that the overall fluorescence signal from living cells and lysates was much stronger for DEVD than for EVEID and VEID fluorogens, highlighting the overwhelming role of caspase 3 over caspase 6 in apoptosis execution. Nevertheless, we were able to selectively detect caspase 6 activity. In the next experiments, we attempted to dissect the kinetics of activation of caspases 3 and 6 in cells. To do so, we incubated Jurkat T cells with AIE substrates for 2 hours and stimulated them with staurosporine for 8 hours. After this time, we imaged the cells with confocal fluorescence microscopy (Fig. 5D). As expected, the strongest signal was observed for the caspase 3/7 Ac-DEVD-TPE substrate, whereas the caspase 6-selective Ac-EVEID-TPE substrate produced lower fluorescence. Notably, we demonstrated that caspase 6 is sufficiently active to be selectively detected with AIE fluorogen. To further explore the caspase activation cascade, we simultaneously monitored PI uptake and AIE fluorogen hydrolysis in staurosporine-treated Jurkat T cells over time (Fig. 5E, F; S5, S6†). Real-time imaging revealed the sequential activation of caspase 3 (and caspase 7) followed by the downstream activation of caspase 6, demonstrating conserved hierarchical ordering in the execution phase of apoptosis. Importantly, the signal from caspase 6 activity is mostly observed in cells that are in the final stage of apoptosis execution, implying an important role of this protease in cell destruction.
Analysis of the intrinsic apoptosis pathway using diverse sets of inhibitors and mass cytometry
Using information about caspase 6 preferences at the P5 position, we synthesized covalent, irreversible inhibitors with an acyloxymethylketone (AOMK) moiety to test whether our substrate-based approach can be directly translated for the development of potent and selective caspase 6 inhibitors (Fig. S7†). By measuring their second-order rate of inhibition (kobs/I) toward recombinant caspases, we showed that inhibitors bearing VEID, hFVEID and EVEhFD sequences are more potent and selective toward caspase 6 than other caspases (Fig. 6A, S8†). Although our approach did not result in the development of entirely selective caspase 6 inhibitors, we created a panel of compounds with diverse potency toward individual caspases. Hence, we used them in a cell-based study to decipher the caspase activation cascade in staurosporine-treated Jurkat T cells, asking whether some of these inhibitors can distinguish between caspase 3 and caspase 6 and whether this simple approach can yield reliable information on the apoptotic machinery. As a readout, we first decided to analyze the results via western blotting. To do so, we preincubated Jurkat T cells with a panel of inhibitors (5 or 25 μM) followed by triggering intrinsic apoptosis with staurosporine (Fig. 6B, S9†). After 4 (or 8) hours, we collected cells and subjected them to western blotting analysis using a set of antibodies (anti-caspase 3, anti-caspase 6, anti-PARP and anti-lamin A). Surprisingly, the most potent caspase 3 inhibitor (Ac-DEVD-AOMK) only partially blocked PARP processing, indicating that negatively charged Asp and Glu residues hinder passive transport through the cell membrane. Similarly, highly potent caspase 6 inhibitors (Ac-EVEhFD-AOMK and Ac-EVEID-AOMK) due to the presence of three negatively charged amino acids were only partially effective in preventing lamin A cleavage in living cells. Conversely, more hydrophobic Ac-VEID-AOMK and Ac-hFVEID-AOMK largely prevent lamin A but not PARP processing, indicating their high in vitro selectivity toward caspase 6 over caspase 3. After prolonged stimulation of Jurkat T cells with staurosporine (8 hours), inhibitors are less effective, due to ongoing apoptosis. However, since the cell membrane is losing its integrity, even weakly cell-permeable inhibitors can be taken up and partially block lamin A processing. Therefore, to better assess the cellular potency of inhibitors toward individual caspases, only the early events of the apoptotic cascade have to be analyzed. To provide a closer insight into the cellular potency of the caspase 3 and 6 inhibitors we also performed a quantitative analysis of full-length and cleaved lamin A WB bands (Fig. S10†). Our data demonstrate that regardless of cell permeability characteristics all inhibitors retain their cellular selectivity between caspase 3 and 6. Moreover, our kinetic profiling indicated that Ac-hFVEID-AOMK, in contrast to VEID and IETD, is a poor inhibitor of apical caspases 8, 9, and 10; therefore, we speculate that this compound can be used as a potent and selective caspase 6 inhibitor in cell-based studies. The most distinctive features at the last phase of apoptosis execution include cell shrinkage, membrane blebbing and the formation of apoptotic bodies; hence, single-cell analysis is very challenging. To challenge this, we decided to test our inhibitors in staurosporine-treated Jurkat T cells in a mass cytometry format. Mass cytometry is a revolutionary technology that adopts atomic mass spectrometry in flow cytometry applications. The excellence of this method is that each metal isotope (used as an antibody tag) has its own peak on the mass spectrum, which eliminates the problem of signal overlap, thus allowing for simultaneous monitoring of more than 40 parameters at the single-cell level.38,39 Similar to flow cytometry, antibodies in mass cytometry are designed to target proteins that are differentially expressed across various cell populations (i.e., CD markers; Cluster of Differentiation) or undergo changes within cells (i.e., overexpression, phosphorylation or processing). Therefore, we decided to use two metal-tagged antibodies toward cleaved (active) caspase 3 (labeled with 172Yb) and cleaved PARP (labeled with 143Nd). This analysis should yield information regarding their cleavage status. However, since metal-tagged antibodies against cleaved caspase 6 or cleaved lamin A are not commercially available, we decided to perform in-house conjugation of anti-caspase 6 (159Tb) and anti-lamin A (142Nd) antibodies with the indicated metal tags. As antibodies can display differential binding affinity to various forms of target proteins, we tested whether our metal-tagged antibodies could distinguish between full-length and cleaved forms of caspase 6 and lamin A (Fig. 6C). To compare our data with western blotting analysis, we used the same experimental setup. Jurkat T cells were pretreated with inhibitors and then stimulated with staurosporine. Next, cells were collected, labeled with a panel of metal-tagged antibodies and subjected to mass cytometry analysis. As expected, we observed a considerable difference in the labeling of cleaved PARP between untreated and apoptotic cells (Fig. 6D). Moreover, the use of an Ac-DEVD-AOMK inhibitor partially reduced PARP cleavage, indicating direct caspase 3 inhibition. The same results were obtained for the labeling of cleaved caspase 3, where Ac-DEVD-AOMK only partially reduced the signal (Fig. S11†). These findings were in line with our western blotting analysis, where even a high concentration of Ac-DEVD-AOMK was not sufficient to fully block caspase 3 activation and subsequent PARP cleavage. More importantly, we also observed a considerable difference in the labeling of lamin A between untreated cells (high signal) and staurosporine-treated cells (low signal) (Fig. 6E). With this, we demonstrated that our anti-lamin A(142Nd) antibody binds more efficiently to the full-length than to the cleaved form of the protein, which can be explained by the fact that this antibody was raised against a peptide sequence that is partially removed by caspase 6, resulting in a weaker interaction with cleaved lamin A. Interestingly, the use of Ac-hFVEID-AOMK, but not Ac-DEVD-AOMK, significantly reduced the signal, indicating a direct inhibition of caspase 6 and high intracellular selectivity of this inhibitor. In the following experiments, we performed the same analysis using our diverse set of inhibitors (Fig. 6F, S10†). The data clearly demonstrate that the most potent and selective caspase 6 inhibitors bearing hFVEID, VEID and IETD efficiently block lamin A processing, while the only inhibitors affecting caspase 3 activity and subsequent PARP cleavage are Ac-DEVD-AOMK and Ac-IETD-AOMK. Our mass cytometry data are in very good agreement with the results obtained via western blotting, demonstrating that this combined approach might be useful to dissect cell death pathways and to decipher the individual roles of caspases.
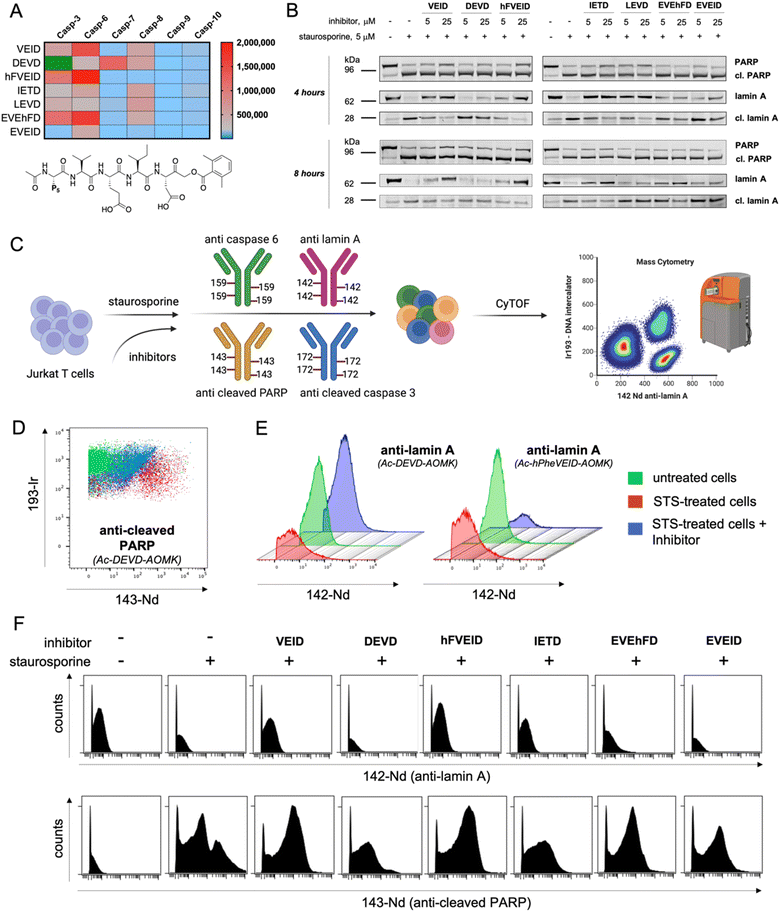 |
| | Fig. 6 Analysis of caspase 6 activity by covalent inhibitors and mass cytometry. To assess the selectivity and potency of the designed inhibitors, we performed a set of cell-based studies using western blotting and single-cell mass cytometry analysis. (A) The kinetic parameters (kobs/I, M−1 s−1) of tetra- and pentapeptide AOMK-based inhibitors toward recombinant apoptotic caspases. Inhibitors bearing VEID, hFVEID and EVEhFD peptides are the most potent inhibitors of caspase 6, whereas Ac-EVEID-AOMK is the most caspase 6-selective inhibitor. The kobs/I value of Ac-DEVD-AOMK for caspase-3 is presented in green as it is well of the scale (kobs/I = 6900000 M−1 s−1). (B) The influence of small-molecule inhibitors on PARP and lamin A processing by caspases 3 and 6 in staurosporine-treated Jurkat T cells. The most potent caspase 6 inhibitors almost completely blocked lamin A cleavage, whereas most caspase 3 inhibitors failed to prevent PARP processing. (C) Schematic representation of the experimental setup to analyze caspase activation in mass cytometry format. Jurkat T cells were preincubated with inhibitors, treated with staurosporine and labeled with a cocktail of metal-tagged antibodies to dissect the processing of individual caspases and their substrates. We used four metal-tagged antibodies: anti-lamin A (142Nd), anti-cleaved PARP (143Nd), anti-caspase 6 (159Tb), and anti-cleaved caspase 3 (172Yb). (D) Mass cytometry diagram demonstrating PARP processing in Jurkat T cells (untreated, treated with staurosporine, and treated with staurosporine in the presence of caspase 3/7 Ac-DEVD-AOMK inhibitor). The results show that the DEVD inhibitor attenuates but does not fully prevent PARP processing. (E) Mass cytometry histograms demonstrating the processing of lamin A in the presence of caspase 3 (DEVD) and caspase 6 (hFVEID) inhibitors. The data demonstrate that the hFVEID inhibitor, but not the DEVD inhibitor, prevents lamin A processing, which is in line with western blotting analysis. (F) Mass cytometry histograms demonstrating the labeling of caspase substrates lamin A (upper) and cleaved PARP (lower) in staurosporine-treated Jurkat T cells in the presence of various inhibitors. The data demonstrate that VEID, hFVEID and IETD inhibitors, targeting mainly caspase 6, block lamin A processing, whereas the DEVD inhibitor attenuates but does not fully prevent PARP processing. | |
Conclusions
Despite many research efforts that have been made over the last two decades, the exact role of caspase 6 in cell death processes remains to be fully elucidated. One of the well-defined substrates of caspase 6 is lamin A, which is cleaved after the VEID sequence,15 resulting in nuclear disassembly during the execution phase of apoptosis.40,41 Very recently, caspase 6 was demonstrated to play essential roles in pyroptosis and necroptosis processes, providing a great opportunity to develop selective reagents for studying the functions of this caspase. In this manuscript, we performed a large screen of caspase preferences at the P5 position and found that the incorporation of the P5 amino acid into the peptide backbone significantly increases the catalytic efficiency of caspase 6 and provides better selectivity of such substrates over other caspases. This in-depth analysis revealed that caspase 6 preferentially accommodates acidic and large hydrophobic residues at the P5 position. The most suitable amino acids in the S5 binding pocket were glutamic acid and unnatural homophenylalanine. Then, using individual pentapeptide substrate libraries with various P4–P2 scaffolds, we demonstrated that caspase 6 does not display subsite cooperativity, making it convenient to add various P5 amino acids to caspase 6-selective peptide motifs, which enhanced the substrate activity and selectivity. However, since ACC-labeled substrates are not convenient for cell-based studies, we decided to develop novel caspase 6 chemical reagents that display aggregation-induced emission phenomena. Such substrates enable the combination of the advantages of substrates and inhibitors, as AIEgens incorporated into the peptide backbone are nonemissive when dissolved in solution but form fluorescent aggregates after peptide bond hydrolysis. Therefore, these compounds enable real-time visualization of the activity and localization of enzymes. Moreover, they ensure high signal-to-noise ratios and show better performance than commercial coumarin-based fluorogenic probes. Accordingly, we synthesized TPE-labeled AIE substrates for caspase 6 and showed for the first time that such reagents indeed undergo rapid hydrolysis upon caspase 6 treatment and can be used in the real-time visualization of this enzyme in staurosporine-treated Jurkat T cells by fluorescence microscopy. Altogether, our work demonstrates that AIE substrates appear to be promising chemical reagents for the investigation of caspases. Moreover, we found that by combining selective peptide sequences with AIE fluorogens, it becomes possible to produce highly selective AIE substrates that can be further utilized to track caspase activity and localization in living cells. Finally, based on substrate screening, we also synthesized a diverse set of covalent inhibitors and used them in vitro to investigate intrinsic apoptosis in mass cytometry format. Our unique approach enables the multiparametric analysis of cell death mechanisms at the single-cell level. With this approach, we demonstrated that the Ac-hFVEID-AOMK inhibitor is highly potent and selective toward caspase 6, as it almost completely blocks this enzyme without cross-reacting with highly abundant caspase 3. We therefore propose that our caspase 6-selective reagents, Ac-EVEID-KTPE and Ac-hFVEID-AOMK, can be used to dissect the role of this enzyme in other cell-based studies that are not necessarily associated with apoptosis execution.
Data availability
All the supporting data are placed in the ESI.† Of course, we have multiple raw and unprocessed data, and we are happy to share them with other researchers per request.
Author contributions
Conceptualization and supervision MP. Funding acquisition KMG, MP. Investigation KMG, MK, JG, SK, MP. Methodology KMG, MK, SJS, MP. Resources KMG, SJS, MP. Validation and visualization KMG, MP. Writing – original draft KMG. Writing – review & editing MK, JG, SK, SJS, MP.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This project was supported by the National Science Centre in Poland (grant PRELUDIUM UMO-2017/25/N/ST5/02831 to KMG and grant OPUS UMO-2018/29/B/NZ1/02249 to MP) and by the National Institutes of Health USA (R01 grant GM099040 to SJS). KMG is a beneficiary of a START scholarship from the Foundation for Polish Science.
References
- D. R. McIlwain, T. Berger and T. W. Mak, Cold Spring Harbor Perspect. Biol., 2013, 5, a008656 CrossRef PubMed.
- S. Shalini, L. Dorstyn, S. Dawar and S. Kumar, Cell Death Differ., 2015, 22, 526–539 CrossRef CAS PubMed.
- N. A. Thornberry, T. A. Rano, E. P. Peterson, D. M. Rasper, T. Timkey, M. Garcia-Calvo, V. M. Houtzager, P. A. Nordstrom, S. Roy, J. P. Vaillancourt, K. T. Chapman and D. W. Nicholson, J. Biol. Chem., 1997, 272, 17907–17911 CrossRef CAS PubMed.
- N. Kayagaki, I. B. Stowe, B. L. Lee, K. O'Rourke, K. Anderson, S. Warming, T. Cuellar, B. Haley, M. Roose-Girma, Q. T. Phung, P. S. Liu, J. R. Lill, H. Li, J. Wu, S. Kummerfeld, J. Zhang, W. P. Lee, S. J. Snipas, G. S. Salvesen, L. X. Morris, L. Fitzgerald, Y. Zhang, E. M. Bertram, C. C. Goodnow and V. M. Dixit, Nature, 2015, 526, 666–671 CrossRef CAS PubMed.
- J. Shi, Y. Zhao, K. Wang, X. Shi, Y. Wang, H. Huang, Y. Zhuang, T. Cai, F. Wang and F. Shao, Nature, 2015, 526, 660–665 CrossRef CAS PubMed.
- C. E. Canman and M. B. Kastan, Semin. Cancer Biol., 1995, 6, 17–25 CrossRef CAS PubMed.
- S. Tacconi, R. Perri, E. Balestrieri, S. Grelli, S. Bernardini, R. Annichiarico, A. Mastino, C. Caltagirone and B. Macchi, Exp. Neurol., 2004, 190, 254–262 CrossRef CAS PubMed.
- M. Tewari, L. T. Quan, K. O'Rourke, S. Desnoyers, Z. Zeng, D. R. Beidler, G. G. Poirier, G. S. Salvesen and V. M. Dixit, Cell, 1995, 81, 801–809 CrossRef CAS PubMed.
- S. M. Srinivasula, T. Fernandes-Alnemri, J. Zangrilli, N. Robertson, R. C. Armstrong, L. Wang, J. A. Trapani, K. J. Tomaselli, G. Litwack and E. S. Alnemri, J. Biol. Chem., 1996, 271, 27099–27106 CrossRef CAS PubMed.
- T. E. Allsopp, J. McLuckie, L. E. Kerr, M. Macleod, J. Sharkey and J. S. Kelly, Cell Death Differ., 2000, 7, 984–993 CrossRef CAS PubMed.
- T. Miyashita, K. Nagao, S. Krajewski, G. S. Salvesen, J. C. Reed, T. Inoue and M. Yamada, Cell Death Differ., 1998, 5, 1034–1041 CrossRef CAS PubMed.
- A. Stepczynska, K. Lauber, I. H. Engels, O. Janssen, D. Kabelitz, S. Wesselborg and K. Schulze-Osthoff, Oncogene, 2001, 20, 1193–1202 CrossRef CAS PubMed.
- M. Zheng, R. Karki, P. Vogel and T. D. Kanneganti, Cell, 2020, 181, 674–687 CrossRef CAS PubMed.
- H. Chu, Y. Hou, D. Yang, L. Wen, H. Shuai, C. Yoon, J. Shi, Y. Chai, T. T. Yuen, B. Hu, C. Li, X. Zhao, Y. Wang, X. Huang, K.
S. Lee, C. Luo, J. P. Cai, V. K. Poon, C. C. Chan, A. J. Zhang, S. Yuan, K. Y. Sit, D. C. Foo, W. K. Au, K. K. Wong, J. Zhou, K. H. Kok, D. Y. Jin, J. F. Chan and K. Y. Yuen, Nature, 2022, 609, 785–792 CAS.
- A. Takahashi, E. S. Alnemri, Y. A. Lazebnik, T. Fernandes-Alnemri, G. Litwack, R. D. Moir, R. D. Goldman, G. G. Poirier, S. H. Kaufmann and W. C. Earnshaw, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8395–8400 CrossRef CAS PubMed.
- K. Orth, A. M. Chinnaiyan, M. Garg, C. J. Froelich and V. M. Dixit, J. Biol. Chem., 1996, 271, 16443–16446 CrossRef CAS PubMed.
- S. Galande, L. A. Dickinson, I. S. Mian, M. Sikorska and T. Kohwi-Shigematsu, Mol. Cell. Biol., 2001, 21, 5591–5604 CrossRef CAS PubMed.
- S. C. Warby, C. N. Doty, R. K. Graham, J. B. Carroll, Y. Z. Yang, R. R. Singaraja, C. M. Overall and M. R. Hayden, Hum. Mol. Genet., 2008, 17, 2390–2404 CrossRef CAS PubMed.
- M. Poreba, P. Kasperkiewicz, S. J. Snipas, D. Fasci, G. S. Salvesen and M. Drag, Cell Death Differ., 2014, 21, 1482–1492 CrossRef CAS.
- G. Fu, A. A. Chumanevich, J. Agniswamy, B. Fang, R. W. Harrison and I. T. Weber, Apoptosis, 2008, 13, 1291–1302 CrossRef CAS PubMed.
- L. E. Edgington, A. B. Berger, G. Blum, V. E. Albrow, M. G. Paulick, N. Lineberry and M. Bogyo, Nat. Med., 2009, 15, 967–973 CrossRef CAS PubMed.
- L. E. Edgington, B. J. van Raam, M. Verdoes, C. Wierschem, G. S. Salvesen and M. Bogyo, Chem. Biol., 2012, 19, 340–352 CrossRef CAS PubMed.
- M. Poreba, W. Rut, K. Groborz, S. J. Snipas, G. S. Salvesen and M. Drag, Cell Death Differ., 2019, 26, 2695–2709 CrossRef CAS PubMed.
- D. J. Maly, F. Leonetti, B. J. Backes, D. S. Dauber, J. L. Harris, C. S. Craik and J. A. Ellman, J. Org. Chem., 2002, 67, 910–915 CrossRef CAS PubMed.
- H. R. Stennicke and G. S. Salvesen, Methods, 1999, 17, 313–319 CrossRef CAS PubMed.
- M. Poreba, K. Groborz, M. Navarro, S. J. Snipas, M. Drag and G. S. Salvesen, Cell Death Differ., 2019, 26, 229–244 CrossRef CAS PubMed.
- Y. Choe, F. Leonetti, D. C. Greenbaum, F. Lecaille, M. Bogyo, D. Bromme, J. A. Ellman and C. S. Craik, J. Biol. Chem., 2006, 281, 12824–12832 CrossRef CAS PubMed.
- S. Grootjans, B. Hassannia, I. Delrue, V. Goossens, B. Wiernicki, Y. Dondelinger, M. J. Bertrand, D. V. Krysko, M. Vuylsteke, P. Vandenabeele and T. Vanden Berghe, Nat. Protoc., 2016, 11, 1444–1454 CrossRef PubMed.
- A. B. Berger, K. B. Sexton and M. Bogyo, Cell Response, 2006, 16, 961–963 CrossRef CAS PubMed.
- Y. Yuan, C. J. Zhang, R. T. K. Kwok, D. Mao, B. Z. Tang and B. Liu, Chem. Sci., 2017, 8, 2723–2728 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- R. T. Kwok, C. W. Leung, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC.
- M. Zheng, R. Karki, B. Kancharana, H. Berns, S. M. Pruett-Miller and T. D. Kanneganti, J. Biol. Chem., 2021, 297, 101379 CrossRef CAS PubMed.
- M. Poreba, A. Szalek, W. Rut, P. Kasperkiewicz, I. Rutkowska-Wlodarczyk, S. J. Snipas, Y. Itoh, D. Turk, B. Turk, C. M. Overall, L. Kaczmarek, G. S. Salvesen and M. Drag, Sci. Rep., 2017, 7, 43135 CrossRef CAS PubMed.
- S. Inoue, G. Browne, G. Melino and G. M. Cohen, Cell Death Differ., 2009, 16, 1053–1061 CrossRef CAS PubMed.
- T. K. MacLachlan and W. S. El-Deiry, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9492–9497 CrossRef CAS PubMed.
- I. V. Soni and J. A. Hardy, Biochemistry, 2021, 60, 2824–2835 CrossRef CAS PubMed.
- M. H. Spitzer and G. P. Nolan, Cell, 2016, 165, 780–791 CrossRef CAS PubMed.
- B. Bodenmiller, E. R. Zunder, R. Finck, T. J. Chen, E. S. Savig, R. V. Bruggner, E. F. Simonds, S. C. Bendall, K. Sachs, P. O. Krutzik and G. P. Nolan, Nat. Biotechnol., 2012, 30, 858–867 CrossRef CAS PubMed.
- E. A. Slee, C. Adrain and S. J. Martin, J. Biol. Chem., 2001, 276, 7320–7326 CrossRef CAS PubMed.
- Y. A. Lazebnik, A. Takahashi, R. D. Moir, R. D. Goldman, G. G. Poirier, S. H. Kaufmann and W. C. Earnshaw, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9042–9046 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a