
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling catalyst-free electro-photochemical reactivity of aryl diazoesters and facile synthesis of oxazoles, imide-fused pyrroles and tetrahydro-epoxy-pyridines via carbene radical anions†
Debajit
Maiti
 a,
Argha
Saha
b,
Srimanta
Guin
b,
Debabrata
Maiti
b and
Subhabrata
Sen
*a
a,
Argha
Saha
b,
Srimanta
Guin
b,
Debabrata
Maiti
b and
Subhabrata
Sen
*a
aDepartment of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence Deemed to be University, Chithera, Dadri, Gautam Buddha Nagar, UP 201314, India. E-mail: Subhabrata.sen@snu.edu.in
bDepartment of Chemistry, IIT-Bombay, Powai, Mumbai 400076, MH, India
First published on 18th April 2023
Abstract
Herein, we report a reagent-less (devoid of catalyst, supporting electrolyte, oxidant and reductant) electro-photochemical (EPC) reaction [electricity (50 μA) and blue LED (5 W)] of aryl diazoesters to generate radical anions which are subsequently reacted with acetonitrile or propionitrile and maleimides to generate diversely substituted oxazoles, diastereo-selective imide-fused pyrroles and tetrahydroepoxy-pyridines in good to excellent yield. Thorough mechanistic investigation including a ‘biphasic e-cell’ experiment supports the reaction mechanism involving a carbene radical anion. The tetrahydroepoxy-pyridines could be fluently converted to fused pyridines resembling vitamin B6 derivatives. The source of the electric current in the EPC reaction could be a simple cell phone charger. The reaction was efficiently scaled up to the gram level. Crystal structure, 1D, 2D NMRs and HRMS data confirmed the product structures. This report demonstrates a unique generation of radical anions via electro-photochemistry and their direct applications in the synthesis of important heterocycles.
Introduction
In recent years, electrochemistry has constituted a major pathway in steering numerous organic transformations.1 By virtue of being generated via various renewable energy sources, it has successfully evolved as an ecologically sustainable and economical energy source to promote organic reactions.2 Seminal reviews and perspective articles on electrochemistry have ignited the “electro-curiosity” of researchers to investigate electrochemical organic reactions.3 Similar to electrochemistry, photochemistry has been extensively utilized as an appropriate energy source for various organic transformations for many years. It has gained tremendous attention from the community of synthetic organic chemists, especially after the evolution of light-emitting diodes (LED) during the last decade.4In recent years, these two apparently related but orthogonal techniques have been coupled together to facilitate and modulate organic transformations driven by single electron-redox pathways.5 As a concept, electro-photochemistry (EPC) was first reported by Moutet and Reverdy in 1979 through electrochemical generation of phenothiazine radical cations and their subsequent photoexcitation.6 Immediately after that Rustling illustrated a photoexcited and electrochemically generated anthracene radical anion that reduced 4-chlorobiphenyl.7 Despite such ground-breaking disclosures it was not until 2019, that EPC was first used in facilitating an organic reaction.5,8
In recent times, thermal and photochemical decomposition of diazoalkane and diazoester with and without metal catalysts have been extensively explored and widely used in synthetic organic chemistry.9 Among them, aryl diazoesters are one of the most unique carbene precursors where the electrophilicity of carbene can be tuned precisely through varying substitution in the donor as well as in the acceptor domain attached to it. Electrophilic carbenes generated from aryl and other diazoesters are employed in numerous chemical transformations including C–H, N–H, O–H insertions, cyclopropanation, ylide generation etc.10 However, electro-photochemical reactivity of aryl diazoesters remained unexplored so far. In one recent example, Huang et al. used diazoesters in an electrochemical reaction but the diazoester was not electrochemically activated or explored in that work.11
Careful curation of the literature revealed that there are only a few reports to date demonstrating the generation of carbene anion radicals. The McDonald research group (1986) and later Parker and Bethell et al. (1989) reported the electrochemical generation of the diazoalkane radical followed by thermal loss of dinitrogen to form the carbene anion radical.12 Bierbaum et al. (2008) reported mechanistic insight for halogen substituted carbene radical anion in the gas phase.13 At the beginning of the last decade, Betrand et al. (2013) documented the genesis of this research in an important review article which summarised the research based on main group radicals and radical ions that are stabilised by carbenes.14 Bruin and co-workers reported metal carbenoid radical-mediated reactions like HAT, radical C–C coupling and cyclopropanation at electron-deficient olefins.15 However, there are no reports so far on electro-photochemical generation of carbene anion radicals and their direct synthetic application.
In an effort to bolster the concept of EPC and to explore the electro-photochemical reactivity of diazoesters, herein we report a hitherto unknown transformation involving activation of aryl diazoesters using electricity and a blue LED to a radical anion and their subsequent reaction with acetonitrile or propionitrile and maleimide to afford diversely substituted N-heterocycles such as oxazoles, imide fused pyrroles and tetrahydro-epoxy-pyridines with excellent atom-economy, yield and diastereoselectivity (Scheme 1). The reaction occurs at a constant current as low as 50 μA and does not require any supporting electrolyte, electromediated photoredox catalyst and photoelectrodes. The reaction is successfully scaled up to the gram level. The synthesised tetrahydro-epoxy-pyridines were easily transformed into diversely substituted fused pyridines (resembling vitamin B6) with fluorescence-sensing properties in different solvents. Discovering an efficient industry-compliant strategy to synthesise this diverse class of heterocyclic compounds has always remained as the most popular research goal for a synthetic chemist. Control experiments, cyclic voltammetry and a uniquely designed bi-phasic e-cell experiment assisted in deciphering the mechanism of this novel EPC reaction.
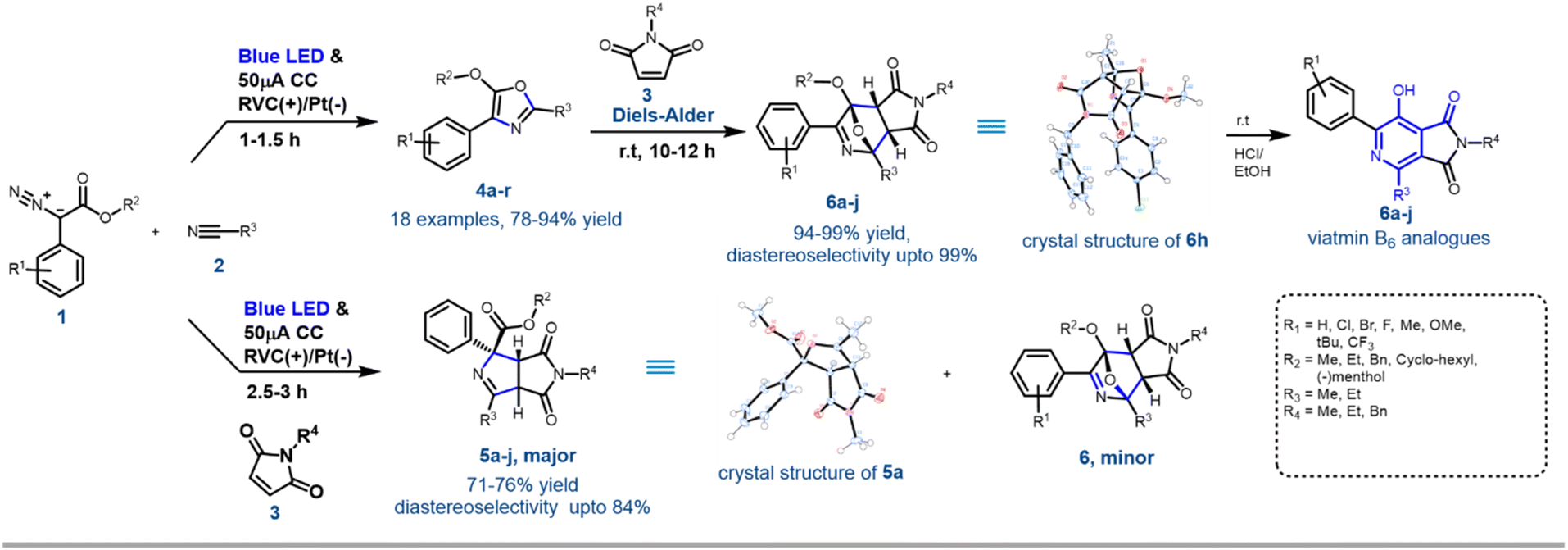 | ||
| Scheme 1 General scheme of the EPC reaction and further synthetic application. | ||
Results and discussion
Optimisation of the EPC protocol to synthesise oxazole
At the outset, the EPC reaction for the synthesis of oxazole was optimized with phenyl diazoester, 1a and acetonitrile, 2a. The reaction parameters were carefully modulated to standardise the reaction conditions. These observations are summarised in Table 1. The electrodes that were screened for the purpose were RVC, graphite, platinum and copper (Table 1, entry 1–5). The best outcome was observed when RVC was used as an anode and platinum as a cathode (Table 1, entry 1). The electrochemical reaction when performed in the dark (without a blue LED) afforded no product (Table 1, entry 6). The photochemical reaction under the blue LED and without electricity resulted in only diazo decomposition (Table 1, entry 7). These preliminary reactions suggested that both blue LEDs and electric current are required to facilitate the reaction. The conventional EPC reactions require a supporting electrolyte to allow adequate electricity to pass through the solvents by decreasing the resistance of the reaction solution. Accordingly, tetra-butyl ammonium hexafluorophosphate (NBu4PF6) was used as the supporting electrolyte. The yield here was comparable to the best effort (Table 1, entry 1). But the reaction completion time was prolonged (Table 1, entry 8). Next, the electrical parameters were optimized. With 20 μA constant current, the reaction prolonged to 6 h with nearly the same yield as with 50 μA (Table 1, entry 9), whereas with 100 μA constant current within 1 h the diazoester 1a was completely consumed but 4a was obtained in only 52% yield (Table 1, entry 10). The reaction under constant voltage of 1.5 and 2.0 V (Table 1, entry 11 and 12) was more time-consuming and low yielding than the reaction with constant current @ 50 μA (Table 1, entry 1). After a thorough investigation of these observations, it was concluded that our EPC reaction with 1a and 2a works best when conducted under a blue LED with a constant current of 50 μA, using RVC as the anode and platinum (Pt) as the cathode. All the reactions were carried out under a nitrogen atmosphere using dry and degassed acetonitrile.|
|
|||
|---|---|---|---|
| Entry | Deviation from the above conditions | Yieldb | Comments |
| a Reaction conducted with 50 mg of 1a. b Isolated yield. | |||
| 1 | No deviation | 92% | 1.5 h reaction time |
| 2 | RVC (−), platinum (+) | 74% | 4.5 h reaction time |
| 3 | Graphite (+), platinum (−) | 90% | 4.5 h reaction time |
| 4 | Copper (−) | 30% | 1a decomposition prevalent |
| 5 | Graphite (−) | 65% | 6 h reaction time |
| 6 | Dark | 0% | Partial 1a decomposition |
| 7 | No electricity | 0% | 1a decomposition after 3 h |
| 8 | NBu4PF6 as supporting electrolyte | 88% | 4 h reaction time |
| 9 | Constant current @ 20 μA | 90% | 6 h reaction time |
| 10 | Constant current @ 100 μA | 52% | In <1 h all 1a consumed |
| 11 | Constant voltage @ 1.5 V | 88% | 2.5 h reaction time |
| 12 | Constant voltage @ 2 V | 78% | 1.5 h reaction time |
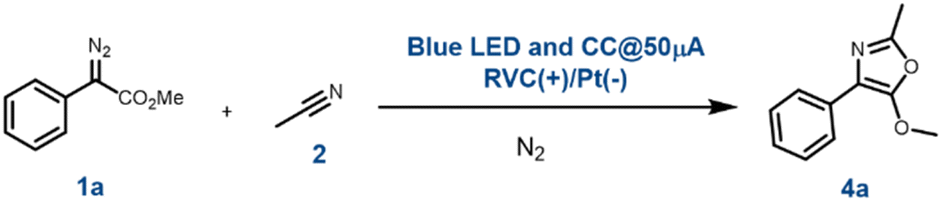
Oxazole synthesis with the EPC reaction
With the optimised reaction conditions in hand, next, the scope of the reaction was explored with various substituted aryl diazoesters [1a–m: methyl ester derivative 1a–j, cyclohexyl methyl ester 1l, benzo[d][1,3]dioxol-5-ylmethyl ester 1m and (−)menthol ester 1n] along with acetonitrile 2a and propionitrile 2b to afford the desired oxazoles in good to excellent yield (Schemes 2 and S1 [refer to the ESI†]). In general, for the methyl diazo esters, all the substitutions at the phenyl ring were tolerated affording the desired oxazoles 4a–r in excellent yield (Scheme 2). However, the reaction with aryl diazo acetates containing electron donating substituents such as 1f, 1h, 1j, 1l, and 1n occurred at a sluggish rate, thereby providing the desired compounds 4f, 4h, 4j, 4n, 4p, 4r, respectively, in a slightly lesser yield (80 to 90%) compared to their electron withdrawing counterparts (1b, e and g → 4b, 4e, 4g and 4l) (92 to 94%). This could be attributed to the formation of the radical anion C (Scheme 4) as a key intermediate in the reaction pathway, which was more stabilised by the electron withdrawing groups. This is discussed in the later part of the manuscript. The optimised reaction conditions were also amenable towards propionitrile 2b and with different diazoesters such as 1a, e and l, thereby affording the desired compounds 4k–o in 81 to 93% yield (Scheme 2). It is noteworthy that with natural (−)-menthol derivative p-chlorophenyl diazo ester 1n, the resulting oxazole 4r was obtained in 88% yield. Reaction with 3-thiophene methyl diazo acetate [1k, Scheme S1 (ESI†)] and 2a failed to generate the desired product. Hence, the EPC reaction could successfully convert acetonitrile 2a and propionitrile 2b to various oxazoles when reacted with differently substituted aryl diazo esters 1a–o. To demonstrate the scalability of this EPC reaction compound 4a was synthesized on a 0.95 g scale from 1 g of 1a and 5 ml of CH3CN under the optimized conditions. The reaction went smoothly in 3.5 h to afford the desired product in 88% yield. A careful survey of the literature suggests that there are few protocols reported so far to synthesise oxazoles from diazo compounds; however, none of those had utilized the electro-photochemical reactivity of aryl-diazoesters as we have demonstrated here.16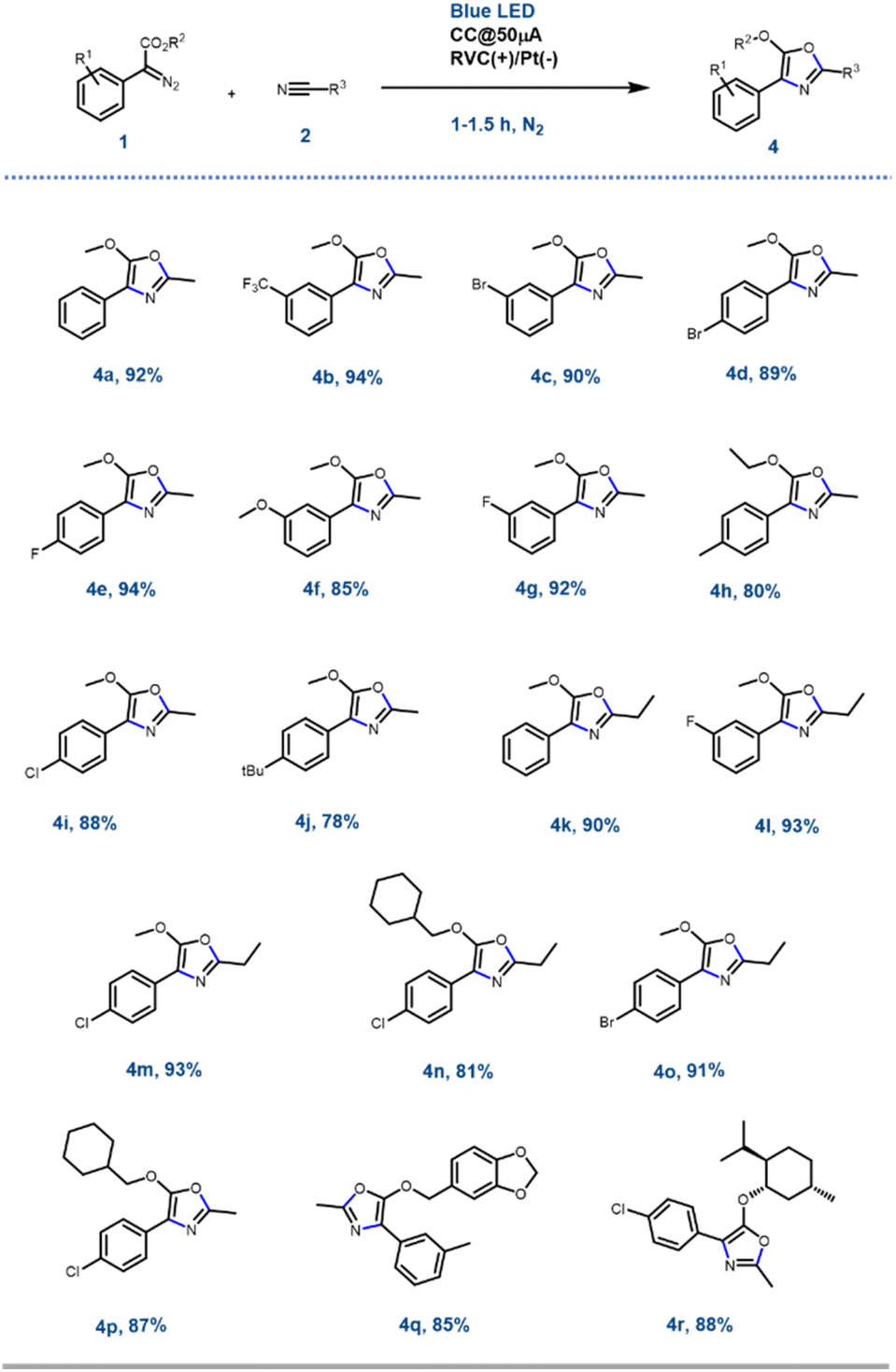 | ||
| Scheme 2 different oxazoles through the EPC reaction. | ||
Further synthetic application of the EPC reaction using N-protected maleimides
To demonstrate the further utility of the EPC strategy, in situ cycloaddition reactions of oxazole 4 with appropriate dipolarophiles were envisioned. The efficiency of N-substituted maleimides and acrylates as dipolarophiles in various [3 + 2] cycloaddition reactions has been demonstrated.10f Accordingly, when maleimide 3a was added to the reaction mixture at the beginning of the electro-photochemical (EPC) reaction, two different maleimide adducts, 5a and 6a were obtained in ∼75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 ratio with nearly 100% conversion (Scheme 3). HRMS, NMR and single crystal XRD data (5a and 6h) confirmed the structures of 5 and 6 (Scheme 1, Fig. S5 and S6 in ESI†). Hence as a generic reaction strategy, numerous aryl diazoacetates, 1a, d and f were reacted with 2a/b and 3a–c to afford both 5a–j as the major product with yields ranging from 71–76% (Scheme 3) and 6 as the minor products (enlisted in Scheme 4 separately). It was anticipated that maleimides 3 reacted with an intermediate as well as product 4 to afford 5 and 6. This was clarified later while deciphering the mechanism of the reaction (Scheme 3A).
:25 ratio with nearly 100% conversion (Scheme 3). HRMS, NMR and single crystal XRD data (5a and 6h) confirmed the structures of 5 and 6 (Scheme 1, Fig. S5 and S6 in ESI†). Hence as a generic reaction strategy, numerous aryl diazoacetates, 1a, d and f were reacted with 2a/b and 3a–c to afford both 5a–j as the major product with yields ranging from 71–76% (Scheme 3) and 6 as the minor products (enlisted in Scheme 4 separately). It was anticipated that maleimides 3 reacted with an intermediate as well as product 4 to afford 5 and 6. This was clarified later while deciphering the mechanism of the reaction (Scheme 3A).
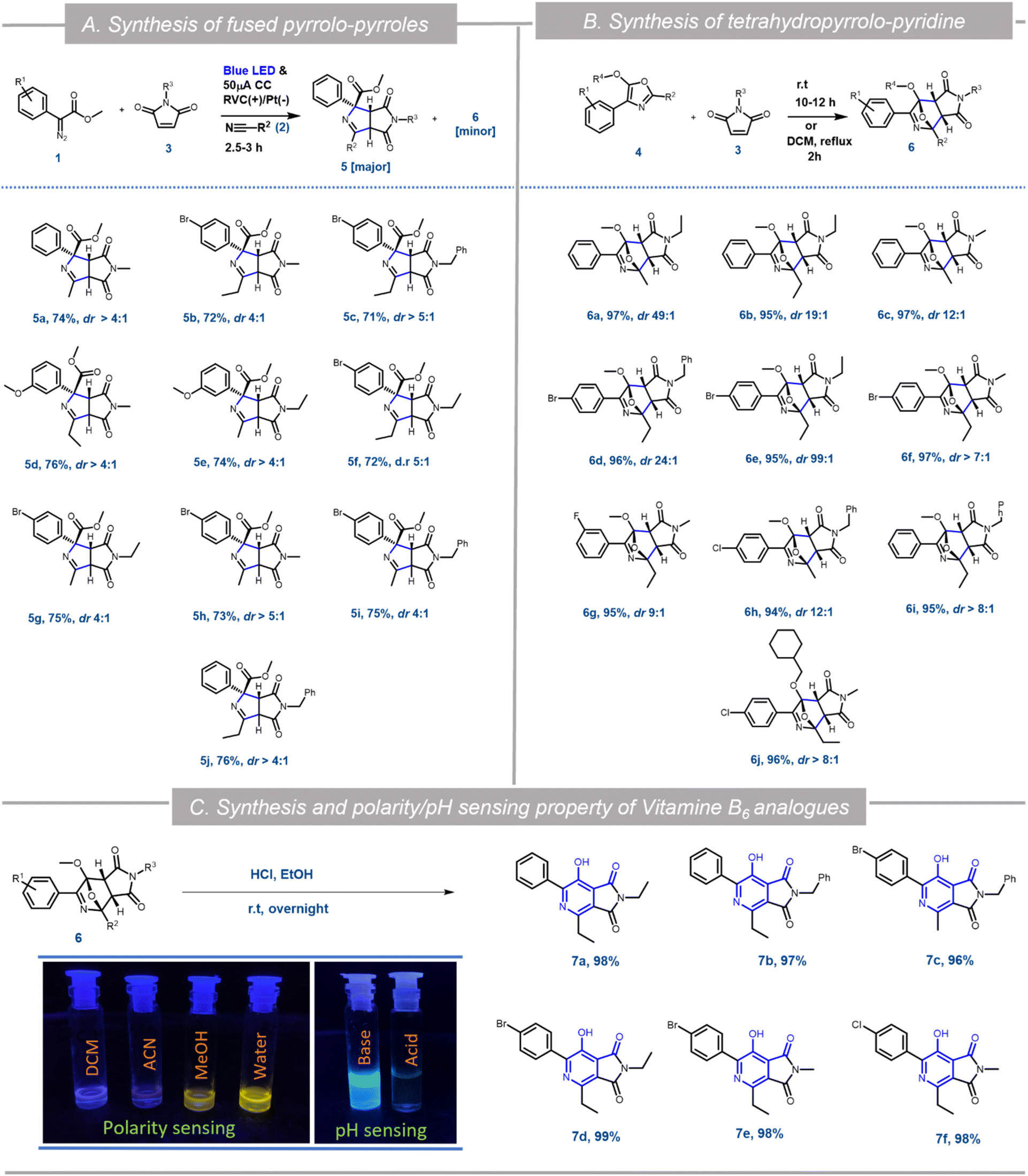 | ||
| Scheme 3 Further application of the EPC reaction: (A) synthesis of pyrrolo-pyrroles through the EPC reaction, (B) synthesis of tetra-hydro-pyrrolo pyridine through the Diels–Alder reaction from oxazole; (C) synthesis and polarity and pH sensing properties of vitamin B6 analogues [diastereomeric ratio (dr) determined through NMR analysis]. | ||
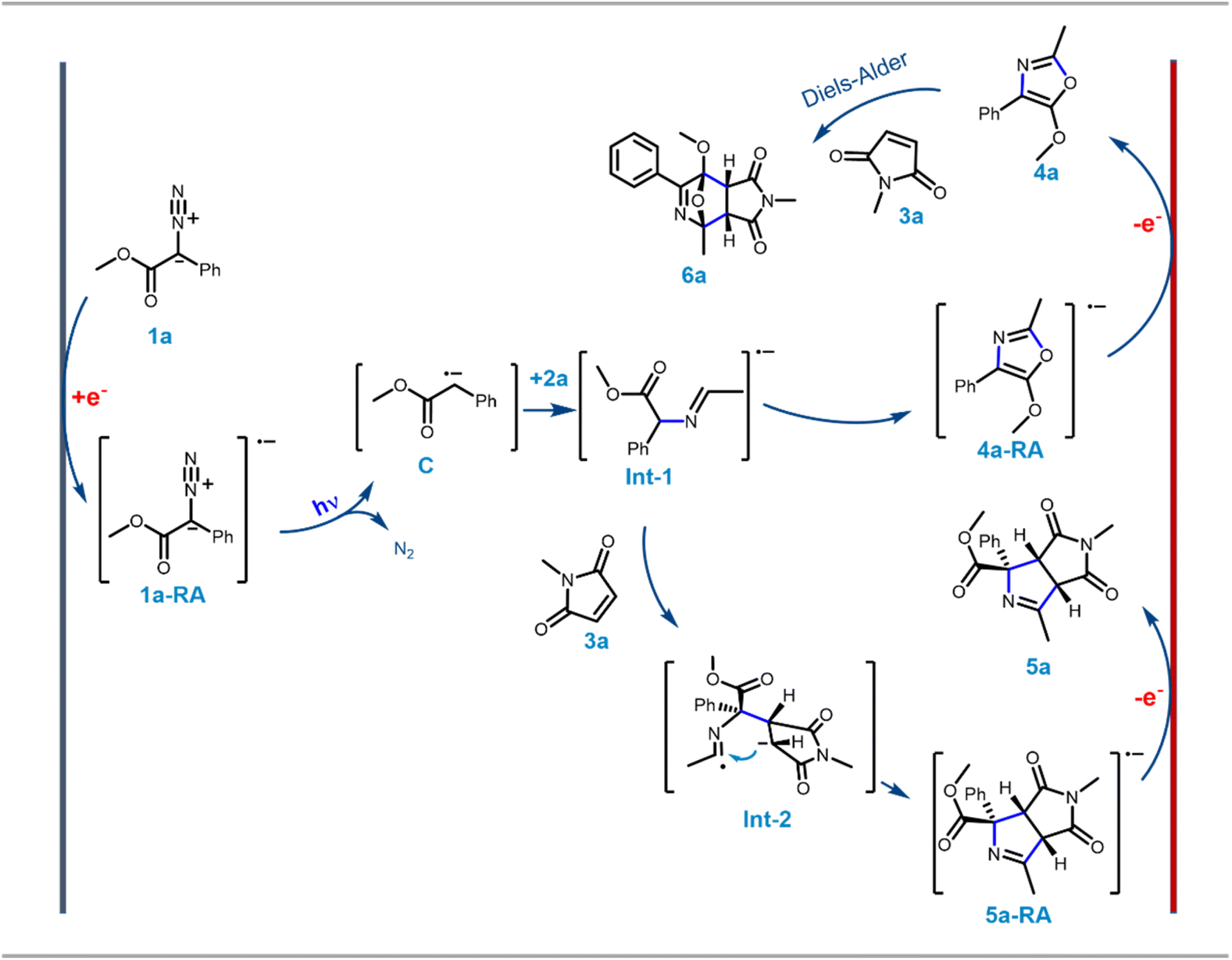 | ||
| Scheme 4 Possible reaction mechanism based upon control experiments. Structure of the isomer obtained in the crystal structure used here as a representative entity. | ||
Since the single crystal X-ray of 6a stipulated that it is the [4 + 2] cycloaddition reaction product between the oxazole 4a and maleimide 3a, in a bid to generate 6 exclusively, the N-methyl maleimide 3a was added in the reaction mixture after the complete formation of 4a in the EPC reaction of 1a and 2a. The blue LED and the electric current were turned off. Reaction 6a was obtained as the exclusive product in 97% yield (Scheme 3). To further optimize the conditions for the formation of 6, compound 4a (isolated) and maleimide 3a were reacted in dichloromethane under three different conditions: (i) under reflux conditions (ii) under a blue LED in an ice-bath and (iii) in the dark in an ice bath. We observed complete conversion and ∼100% yield of 6a under reflux within 2 h. However, under conditions (ii) and (iii) there was no product formation. The scope of the [4 + 2] cycloaddition reaction was demonstrated by generating ten derivatives 6a–j (Scheme 3) by reacting 1a, 1d, 1g and 1i with 2a, 2b and 3a–c (N-methyl/ethyl/benzyl maleimides respectively). By using either of the protocols i.e., with in situ addition of 3 at r.t. after the formation of 4 or with isolated 4 under reflux the yield of the reactions was nearly quantitative with excellent diastereoselectivity of up to 99% (Scheme 3B).
[4 + 2] Cycloaddition reactions between oxazoles and alkenes and their subsequent aromatization are one of the key strategies for the synthesis of pyridine derivatives. The pyridine derivatives are important because of their ubiquitous presence in natural products including Vitamin B6 (pyridoxine) and its analogues.17 To demonstrate the synthetic application of 6 in the synthesis of fused pyridines (with the structural motif resembling vitamin B6 derivatives), compounds 6b, 6d–f, 6i and 6j were treated with 1 N HCl in ethanol in r.t. overnight. To our expectation, 7a–f were generated in excellent yield. Interestingly, these analogues as demonstrated by compound 7b, displayed different fluorescence in different polar solvents (Scheme 3C). More interestingly, the aqueous basic solution of 7b in 1 N NaOH exhibited bright fluorescence which was diminished in the aqueous acidic solution of 7b in 1 N HCl (Scheme 3C). These observations indicated that such fused pyridines could potentially be utilised as polarity and pH sensing probes for biological studies. This would form the basis of a separate endeavour as further investigation is ongoing in our laboratory.
Mechanistic studies and the possible mechanism
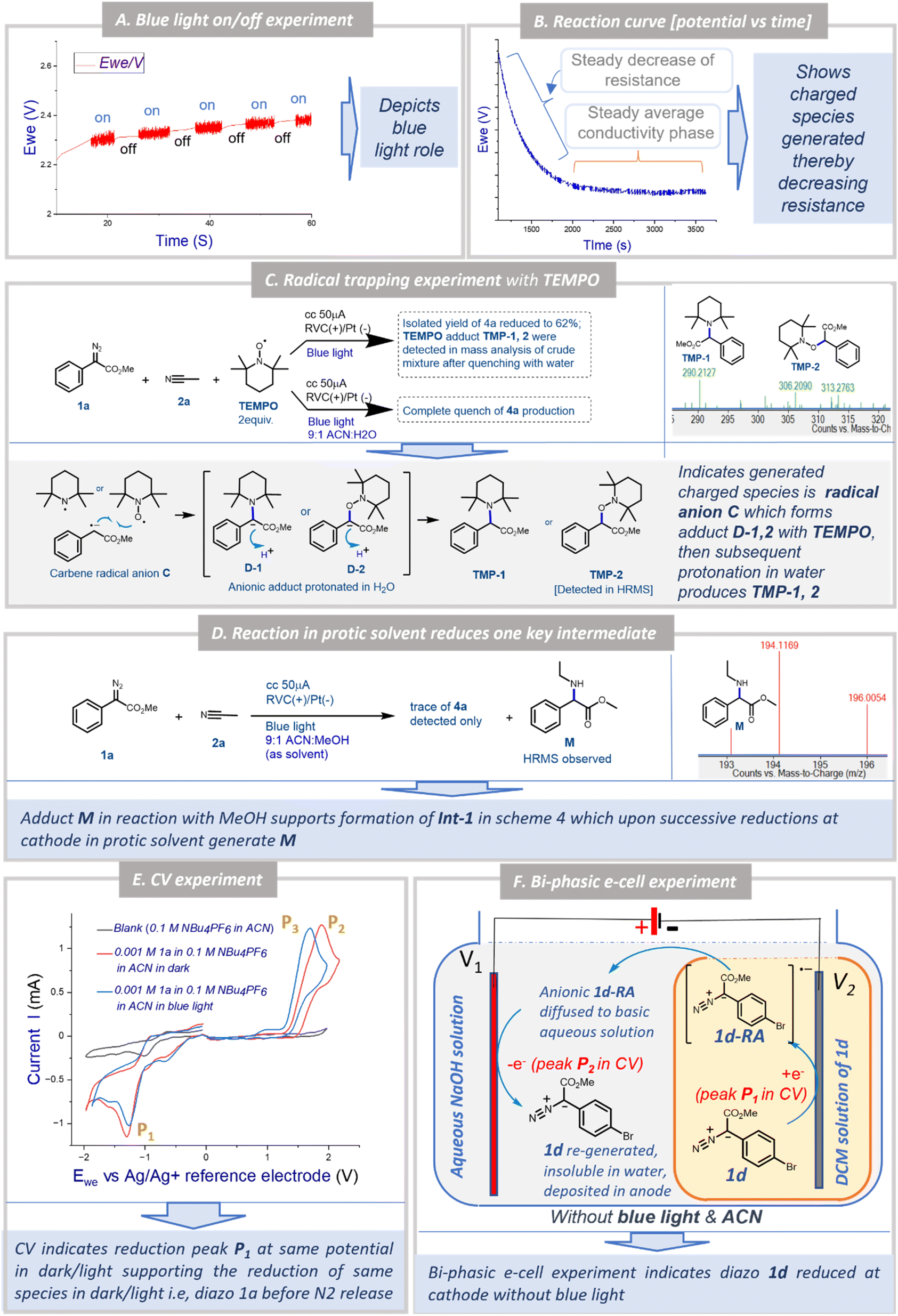
To understand the possible mechanism of the EPC reaction, we performed several control experiments. Based on literature reports and our earlier observation, we initially could think of three possible mechanistic proposals for the transformation: Pathway [A]: under a blue LED, carbene was generated which could form the nitrogen ylide with acetonitrile followed by cycloaddition to afford oxazole 4a where electricity had no role to play; Pathway [B]: the aryl diazoester 1a could be reduced at the cathode to generate the anion radical which could liberate di-nitrogen under a blue LED to generate the carbene radical anion which could attack acetonitrile to generate Int-1 (Scheme 4) and subsequent cyclisation and oxidation at the anode could lead to the formation of 4a; Pathway [C]: at the outset the aryl diazoester could release dinitrogen under a blue LED to generate carbene and subsequently it is reduced at the cathode to generate the carbene radical anion which could attack acetonitrile to generate Int-1 which then cyclise and get oxidised at the anode to generate 4a.To realise the possible mechanism, the observations from the optimisation experiments were analysed and several control experiments were performed. In the first place, it was observed that during the optimisation reaction of 1a and acetonitrile 2a, without the blue LED (with all the other parameters unchanged) there was no reaction (the reaction was monitored over a 3 h period) (Table 1, entry 6). Next, the blue LED was turned on and off at a regular interval of ∼5 s and the potential was recorded against the time at a constant current. A distinct fluctuation of potential (Ewe) was observed when the blue LED was ‘on’ and the plot was ∼linear when the blue LED was turned off (Fig. 1A). This indicated that during the reaction under the blue LED the electron flow to/from the potentiostat varied and that the reaction system could be experiencing a change in electron transfer from the cathode to the reaction solution and from the reaction solution to the anode. Since the reaction is free from any supporting electrolyte, additive and mediator, this change in electron transfer could be happening directly to/from the substrates. Again, in the optimisation study, the reaction under the blue LED and without any electricity failed to afford the formation of the desired product 4a within 3 h of reaction time. Instead, partial diazo decomposition was observed (monitored through TLC) (Table 1, entry 7). Earlier Jurberg and Davies18 reported that the reaction of phenyl diazoester with acetonitrile under a 15 W blue LED and overnight formed a product which could be an oxazole (based on the characterisation data) possibly through nitrile ylide. There are also other reports of the formation of nitrile ylide under a blue LED in the reaction with nitrile and diazoesters.19 It is noteworthy that in all those cases higher intensity of the blue LED was used for a longer duration. But our EPC reaction protocol generated 4a in 92% yield within 1.5 h reaction time and in the presence of a 5 W blue LED (Table 1, entry 1). As mentioned earlier, in this short time span and with a milder LED source no formation of oxazole was observed until electricity was applied (as mentioned in Table 1, entry 7). This observation and literature reports furthermore indicated that the mechanism in our case is not “Pathway [A]”.
 | ||
| Fig. 1 Mechanistic investigation. | ||
Next, when the reaction kinetics was explored through a potential vs. time plot (Fig. 1B), two distinct phases of the overall reaction were observed. In the initial phase of the reaction at a constant current, the potential (Ewe) decreased steadily to a certain level and then in the remaining phase of the reaction it remained constant. This implicated that there must be a formation of charged species during the reaction which reduced the resistance of the solution. After reaching a certain concentration of the charged species (equilibrium), the reaction at a constant current behaved like that at a constant voltage. This continued until the completion of the reaction after which the conductivity decreased and the potential increased to maintain the constant current possibly due to the consumption of all charged species towards the formation of a neutral product (Figs. 1B and S2 [ESI†]). These observations support two possibilities: (a) electricity is playing an important role during the reaction of oxazole 4a formation and (b) there must be formation of a charged species which ultimately led to the formation of the product.
To understand the nature of the charged species and whether the reaction involved the radical mechanism, the EPC reaction of 1a and 2a was performed in the presence of TEMPO (2 equiv.) (Fig. 1C) in two different ways. In the first case, the reaction without water led to a decrease in the isolated yield of 4a to 62%. The HRMS analysis of the reaction mixture after quenching with water whilst the reaction was still ongoing indicated the formation of TEMPO adducts TMP-1 and TMP-2. Next, when the same reaction was performed with water (10% v/v), the formation of 4a was completely quenched. This observation indicates that TEMPO formed anionic adducts D-1 and D-2 by reacting with anion radical C (Fig. 1C) which was then protonated in water to form TMP-1 and TMP-2. Since the adducts D-1 and D-2 formed via reactions between C and TEMPO, the reaction towards the formation of 4a was retarded. And that is why in the next case no formation of 4a was observed when water was there to protonate both the anionic TEMPO adducts D-1 and D-2, thereby driving the reaction more towards the formation of TMP-1 and TMP-2 as well as the direct quenching of C (Fig. 1C). These two complementary experiments outline the crucial formation of carbene radical anion C during the EPC reaction.
Furthermore, to identify any other possible intermediates, the experiment was performed using 9:1 CH3CN:MeOH as solvent (all other standard parameters unchanged). Interestingly, a trace of product 4a and an adduct M was detected in HRMS analysis of the reaction mixture (Fig. 1D). This supported the formation of Int-1 in the reaction between C and acetonitrile (in Scheme 4) which after protonation and successive reductions at the cathode might form M (unable to generate 4a in the presence of methanol). In both TEMPO + water and ACN/MeOH experiments, no adduct of ‘–OH’ or ‘–OMe’ was observed which could eliminate the formation of any cationic intermediate.
Hitherto, the experiments and subsequent observations eliminated the possibility of mechanistic Pathway [A] (i.e. ylide mediated pathway) and supported the formation of carbene radical anion C. Now, C can be generated in two ways: (a) direct one-electron reduction of 1a at the cathode generates diazo anion radical 1a-RA which liberates di-nitrogen under blue light to generate C, or (b) carbene, after generation from 1a under blue light undergoes one electron reduction at the cathode to generate C. In a bid to determine the immediate precursor of C we performed two experiments: cyclic voltammetry and the biphasic e-cell experiment. In cyclic voltammetry of 1a in acetonitrile the reduction peak P1 was observed both in the dark and under a blue LED (Fig. 1E). However, under the blue LED oxidation peak P3 was observed instead of peak P2 in the dark. This indicated that the same reduction is happening both under LED and dark conditions which could possibly correspond to the reduction of 1a to generate 1a-RA (Scheme 4). Peak P2 possibly corresponds to the oxidation of 1a-RA to re-generate 1a under dark conditions which had been further supported by the bi-phasic e-cell experiment explained below.
To demonstrate the reduction of methyl aryl diazoester in the dark as indicated by CV recording, a unique bi-phasic e-cell was devised in our lab (Figs. 1F, S3A and B†). The e-cell consisted of a 20 ml reaction vial, V1 containing 1 M NaOH solution, and a smaller 5 ml vial, V2 containing aryl diazoester 1d as a solution in DCM. As DCM is immiscible with and heavier than water, V2 could be easily immersed in the aqueous solution without mixing the two solutions. Next, a RVC (as a cathode) was immersed in V2 and a RVC (as an anode) was immersed in V1, and the electrolysis was performed at 2 V constant voltage in the absence of a blue LED. Interestingly, after some time yellow solid of 1d was seen floating over the surface of the aqueous solution (Fig. S3C–E†). After about 60 minutes of electrolysis, the yellow deposit of 1d was also observed over the anode (Fig. S3F†). This observation strongly stipulated the reduction of 1d at the cathode (∼corresponds to peak P1 @ −1.29 V, Fig. 1E) followed by the formation of a radical anion and oxidation at the anode (∼corresponds to peak P2 @ +1.89 V, Fig. 1E) to regenerate aryl diazoester under standard electrochemical conditions in the absence of blue light. At the cathode, the reduction of 1d initially generates radical anion 1d-RA, which traversed through a basic aqueous solution to the anode, where it got oxidised to re-generate 1d. Since 1d is insoluble in water it is either deposited on the anode as a solid or floats over the surface of the aqueous layer after regenerating at the anode. It is noteworthy that there was no other way for 1d to reach the anode from the DCM solution in V2 through an aqueous barrier except in the form of an anion (since it had originated at the cathodic chamber). There could be a possibility that in an aqueous solution the anion radical was protonated to an extent. This was anticipated and the basic aqueous solution was used to minimise it. Had an acidic solution or water been used then chances of protonation could have been higher and regeneration of diazo through oxidation could have been minimal or nothing. 1d was considered for this experiment as it is a bright yellow coloured flaky solid at room temperature so that either buoyancy or deposition could be easily visible. Acetonitrile was not used in this experiment as the objective was to demonstrate the reduction of 1d and not the formation of oxazole. Since acetonitrile is miscible with water the distinct two phases (water/DCM) could not have been constructed with acetonitrile.
In summary, the control experiments inferred that: (a) optimisation study and blue LED on/off experiments highlighted the necessity of blue LEDs for the transformation; (b) potential vs. time plot exhibited the generation of charged species during the EPC reaction; (c) the TEMPO experiment indicated that the charged species is carbene radical anion C; (d) the acetonitrile: MeOH experiment indicated the possible structure of Int-1; (e) CV and the bi-phasic e-cell experiment supported that aryl diazoester (1a/1d) was first reduced to generate 1a-RA/1d-RA, subsequently nitrogen release under a blue LED generated the carbene radical anion C. All of these observations supported possible mechanism Pathway [B] over [A] or [C] which has been depicted in Scheme 4.
Considering 1a, 2a and 3a as the representative participants, the reaction could have begun with the formation of radical anion 1a-RA (Scheme 4) which could be generated through single electron reduction at the cathode from 1a. Radical anion 1a-RA then released di-nitrogen under a blue LED to afford the carbene radical anion C. The anion radical or “radical anion” is unique chemical species where in general the LUMO (lowest unoccupied antibonding molecular orbital) of a neutral molecule becomes the SOMO (singly occupied molecular orbital) of the radical by addition of one electron to the neutral molecule.20 Herein the anionic charge of C could have been delocalised to the adjacent ester carbonyl which could have made the SOMO lying over C (carbene carbon) to be electrophilic enough to attack the N-terminus of nitrile to generate Int-1. This could undergo cyclisation to afford the oxazole radical anion 4a-RA. 4a-RA was oxidised leaving an electron at the anode to form oxazole 4a (possibly corresponds to peak P3 in CV). When the reaction occurred in the presence of 3a, it was the intermediate Int-1 and the product 4a that participated in the reaction with maleimide 3a to afford 5a and 6a as a mixture of ∼75:25, respectively. When 3a was added after the complete formation of 4a and kept stirring overnight in r.t without electricity and blue light, 6a was obtained as the exclusive [4 + 2] cycloaddition product as there was no Int-1 left to react with 3a (Scheme 4).
Manual set up
Finally, to demonstrate the simplicity of handling and cost-effectiveness of our photo-electrochemical reaction set-up, a customised apparatus was assembled in our lab with readily available electronic chips and components which included a 5V–1A (AC to DC) mobile adapter, 1 K ohm resistors, LEDs, connecting wires, a breadboard, 5 watt blue LED strip (430–450 nm) with 12 V AC to DC converter, a basic multimeter (to measure potential and current) (Fig. S4†). The total cost of all these components is ∼1600 INR or 20 USD and could be purchased from a local electrical shop. A schematic of this setup is depicted in Fig. S4.† The reaction between 1a and 2a was performed using this set up and to our utmost gratification compound 4a was obtained in 90% yield in 2.5 h.Conclusion
In summary, herein we have unveiled a practical strategic amalgamation of electrochemistry and photochemistry to exploit aryl diazoesters in the generation of radical anions and their application in the synthesis of meaningful heterocycles like oxazoles 4, imide-fused pyrroles 5 (from the anion radical) and tetrahydro-epoxypyrrolo-pyridines, 6 (from oxazole) from acetonitrile/propionitrile and maleimide. Compounds 6 could be easily transformed to diversely substituted fused pyridines 7 in excellent yield, resembling vitamin B6 analogues. Compound 7 demonstrated fluorescence in various solvents and at different pH subject to further investigation. A plausible mechanism based on control experiments and a uniquely designed bi-phasic e-cell experiment reveals a hitherto unexplored chemistry of carbene anion radicals generated from aryl-diazoesters under electro-photochemical conditions which facilitated the organic transformations. This EPC strategy devoid of any photoelectrodes, photo-redox catalysts, mediator and supporting electrolytes demonstrated a highly efficient organic transformation that could be scalable to the gram level. A cost-effective and practical reactor system was designed (Fig. S4†) using a simple cell phone charger as a source of electricity where the reaction occurred readily to generate the oxazole in excellent yield. Thus, this efficient synthetic process holds great promise to explore more organic transformations utilising the EPC protocol with diazoesters. Additionally, acetonitrile is a toxic solvent which gets metabolized to hydrogen cyanide (the source of its toxicity).21 Sustainable transformation of acetonitrile to useful heterocyclic compounds as demonstrated in this work is a value addition and could be envisioned as a strategy to utilise industry excess acetonitrile (as by-product) towards the synthesis of widely used pharmaceutical building blocks.22 Hence, it is worth mentioning, that our all-new EPC strategy could direct a new way to generate and utilize carbene anion radical in the field of electro-photochemistry.Data availability
ESI† containing experimental and characterisation data are provided alongwith crystal data.Author contributions
Mr DM conceptualized the project and synthesized the compounds. Mr AD and Mr SG assisted in the synthesis of compounds. Prof. DM and Prof. SBS managed the project. Prof. SBS supervised the project and wrote the manuscript with the help of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mr Debajit Maiti is thankful to Shiv Nadar IoE for the PhD scholarship. We are thankful to Shiv Nadar IoE for their support and Dr Santosh K. Singh, for his valuable guidance and suggestions during setting up of electrochemical reactions. Prof. Maiti, Mr Saha and Dr Guin are thankful to IIT Bombay for their instrumental facilities.Notes and references
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- C. Zhu, N. W. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- (a) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed; (b) R. D. Little, J. Org. Chem., 2020, 85, 13375–13390 CrossRef CAS PubMed; (c) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata, P. S. Baran, M. Yan, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2019, 53, 72–83 CrossRef PubMed; (d) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117(21), 13230–13319 CrossRef CAS PubMed; (e) M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., 2018, 57, 4149–4155 CrossRef CAS PubMed; (f) C. Zhu, N. W. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7(3), 415–431 CrossRef CAS PubMed.
- (a) B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef; (b) N. S. Lewis and D. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed; (c) J. L. Schwarz, R. Kleinmans, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2020, 142, 2168–2174 CrossRef CAS PubMed; (d) R. Davenport, M. Silvi, A. Noble, Z. Hosni, N. Fey and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 6525–6528 CrossRef CAS PubMed; (e) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed; (f) A. F. Prusinowski, R. K. Twumasi, A. Wappes and D. A. Nagib, J. Am. Chem. Soc., 2020, 142, 5429–5438 CrossRef CAS PubMed; (g) J. V. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. Macmillan, Nat. Rev. Chem., 2017, 1, 52 CrossRef CAS; (h) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed; (i) K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. H. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 CrossRef CAS PubMed; (j) B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed; (k) M. R. Becker, A. D. Richard- son and S. Schindler, Nat. Commun., 2019, 10, 5095 CrossRef PubMed; (l) R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203–1207 CrossRef CAS PubMed.
- (a) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci, 2020, 6, 1317–1340 CrossRef CAS PubMed; (b) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- J. Moutet and G. Reverdy, Tetrahedron Lett., 1979, 20, 2389–2393 CrossRef.
- S. Shukla and J. Rusling, J. Phys. Chem., 1985, 89, 3353–3358 CrossRef CAS.
- (a) Y. Yu, P. Guo, J. Zhong, Y. Yuan and K. Ye, Org. Chem. Front., 2020, 7, 131–135 RSC; (b) F. Wang and S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed; (c) H. Yan, Z.-W. Hou and H. C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS PubMed; (d) W. Zhang, K. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS; (e) H. Huang, Z. Strater, M. Rauch, J. Shee, T. Sisto, C. Nuckolls and T. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed.
- (a) G. T. Gurmessa and G. S. Singh, Res. Chem. Intermed., 2017, 43, 432–448 CrossRef; (b) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC; (c) J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9(27), 8895–8918 CrossRef CAS.
- (a) R. M. Koenigs, C. Empel, S. Jana and C. Pei, Angew. Chem., Int. Ed., 2020, 60, 13271–13279 Search PubMed; (b) D. Maiti, R. Das and S. Sen, J. Org. Chem., 2021, 86(3), 2522–2533 CrossRef CAS PubMed; (c) M. L. Stivananin, A. A. G. Fernandes, A. F. D. Silva, C. Y. Okada and I. D. Jurberg, Adv. Synth. Catal., 2019, 362, 1106–1111 CrossRef; (d) R. M. Koenigs, C. Empel, S. Jana, C. Pei and T. V. Nguyen, Org. Lett., 2020, 22, 7225–7229 CrossRef PubMed; (e) D. Maiti, R. Das and S. Sen, Green Chem., 2021, 29, 5893 Search PubMed; (f) J. Day, B. McKeever-Abbas and J. Dowden, Angew. Chem., Int. Ed., 2016, 55, 5809–5813 CrossRef CAS PubMed.
- Z. He, W. Zhao, Y. Li, Y. Yu and F. Huang, Org. Biomol. Chem., 2022, 20, 8078 RSC.
- (a) D. Bethell and V. D. Parker, J. Am. Chem. Soc., 1986, 108(23), 7194–7200 CrossRef CAS; (b) R. N. McDonald, Tetrahedron, 1989, 45(13), 3993–4015 CrossRef CAS.
- S. M. Villano, N. Eyet, W. C. Lineberger and V. M. Bierbaum, J. Am. Chem. Soc., 2008, 130(23), 7214–7215 CrossRef CAS PubMed.
- C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4(8), 3020–3030 RSC.
- W. I. Dzik, X. P. Zhang and B. D. Bruin, Inorg. Chem., 2011, 50, 9896–9903 CrossRef CAS PubMed.
- (a) T. Ibata, T. Yamashita, M. Kashiuchi, S. Nakano and H. Nakawa, Bull. Chem. Soc. Jpn., 1984, 57, 2450 CrossRef CAS; (b) T. Ibata and Y. Isogami, Bull. Chem. Soc. Jpn., 1989, 62, 618 CrossRef CAS; (c) R. Liu, Q. Liu, H. Meng, H. Ding, J. Hao, Z. Ji, H. Yue and W. Wei, Org. Chem. Front., 2021, 8, 1970 RSC.
- (a) G. V. Suárez-Moreno, E. González-Zamora and F. Méndez, Org. Lett., 2011, 13, 6358–6361 CrossRef PubMed; (b) T. Ibata, S. Nakano, H. Nakawa, J. Toyoda and Y. Isogami, Bull. Chem. Soc. Jpn., 1986, 59, 433 CrossRef CAS.
- I. D. Jurberg and H. M. L Davies, Chem. Sci., 2018, 9, 5112–5118 RSC.
- (a) H. Li, X. Wu, W. Hao, H. Li, Y. Zhao, Y. Wang, P. Lian, Y. Zheng, X. Bao and X. Wan, Org. Lett., 2018, 20, 5224–5227 CrossRef CAS PubMed; (b) B. Cai, Y. Bao, C. Pei, Q. Li, L. Li, R. M. Koenigs and J. Xuan, Chem. Sci., 2022, 13, 13141 RSC; (c) K. Zhu, M. Cao, G. Zhao, J. Zhao and P. Li, Org. Lett., 2022, 24, 5855–5859 CrossRef CAS PubMed; (d) B. Cai, W. Yao, L. Li and J. Xuan, Org. Lett., 2022, 24, 6647–6652 CrossRef CAS PubMed.
- N. L. Holy and J. D. Marcum, Angew. Chem., Int. Ed., 1971, 10, 115–124 CrossRef CAS.
- (a) U. C. Pozzani, C. P. Carpenter, P. E. Palm, C. S. Weil and J. H. Nair, J. Occup. Med., 1959, 1(12), 634–642V CrossRef CAS PubMed; (b) K. A. Z. U. O. Hashimoto, Jpn. J. Ind. Health, 1991, 33(6), 463–474 CAS; (c) V. Chaban and O. V. Prezhdo, J. Phys. Chem. Lett., 2011, 2(19), 2499–2503 CrossRef CAS.
- R. Xia, D. Tian, S. Kattel, B. Hasa, H. Shin, X. Ma, J. G. Chen and F. Jiao, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2176481 and 2179729. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00089c |
| This journal is © The Royal Society of Chemistry 2023 |