
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of anti-Kasha photoemission from a biazulene core embedded in an antiaromatic/aromatic hybrid†
Aitor
Diaz-Andres
 a,
Jose
Marín-Beloqui
c,
Junting
Wang
b,
Junzhi
Liu
b,
Juan
Casado
c and
David
Casanova
*ad
a,
Jose
Marín-Beloqui
c,
Junting
Wang
b,
Junzhi
Liu
b,
Juan
Casado
c and
David
Casanova
*ad
aDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain. E-mail: david.casanova@dipc.org
bDepartment of Chemistry, State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
cDepartment of Physical Chemistry, University of Malaga, Campus de Teatinos s/n, 29071 Malaga, Spain
dIKERBASQUE – Basque Foundation for Science, 48009 Bilbao, Euskadi, Spain
First published on 22nd May 2023
Abstract
The violation of the Kasha photoemission rule in organic molecules has intrigued chemists since their discovery, being always of relevance given its connection with unique electronic properties of molecules. However, an understanding of the molecular structure–anti-Kasha property relationship in organic materials has not been well-established, possibly because of the few existing cases available, limiting their prospective exploration and ad hoc design. Here we introduce a novel strategy to design organic emitters from high excited states combining intramolecular J-coupling of anti-Kasha chromophores with the hindering of vibrationally-induced non-radiative decay channels by enforcing molecular rigidity. We apply our approach to the integration of two antiparallel azulene units bridged with one heptalene all inserted into a polycyclic conjugated hydrocarbon (PCH). With the help of quantum chemistry calculations, we identify a suitable PCH embedding structure and predict its anti-Kasha emission from the third high energy excited singlet state. Finally, steady fluorescence and transient absorption spectroscopy studies corroborate the photophysical properties in a recently synthesized chemical derivative with this pre-designed structure.
Introduction
Light emission (fluorescence or phosphorescence) in organic molecules, in the vast majority of cases, proceeds from the lowest energy excited state irrespective of the excitation energy used.1–3 This is a consequence of the energy gap law based on the fact that closely packed excited states, that is, with small energy separations, always lie with strong vibronic overlaps promoting efficient Sn → S1 non-radiative processes, i.e., internal conversion (IC). Contrarily, when the interstate gaps are large, radiative transitions can emerge as the most efficient relaxation mechanisms, as it is usually the case for the S1 → S0 decay. This is known as the Kasha's rule, which states that most of the molecules are emissive from the lowest energy, same (ground state) spin multiplicity, S1 excited state.4 Since it was claimed by Michael Kasha in 1950, few cases with emission from higher excited states have appeared, also known as anti-Kasha emitters.5–10 Azulene, the non-alternant isomer of naphthalene, and its derivatives form a paradigmatic family of anti-Kasha emitters.5,8–14 Azulene exhibits fluorescent emission from the second excited singlet state (S2), mainly triggered by: (i) the large energy separation with S1 (preventing efficient S2 → S1 IC), and (ii) the larger oscillator strength of S2 (S2 → S0) compared to S1 (S1 → S0). These two factors allow S2-fluorescence to compete with IC.A potential strategy to expand the pool of molecules endowed with anti-Kasha luminescence is via integration of azulene-like fragments in polycyclic conjugated hydrocarbons (PCHs). PCHs with non-alternant and antiaromatic topologies have constantly attracted the curiosity of chemists15–17 owing to their unique electronic,18–20 optical21,22 and structural features23–28 in comparison with their respective polycyclic aromatic/benzenoid hydrocarbons (PAHs).
Recently, non-alternant PCHs have engaged renewed interest due to their role as chemical defects,29i.e., Stone–Wales, in nanographene topological insulators and semiconductors,30,31 graphene quantum dots,32,33 or as spin qubits.30 Modular bottom-up organic synthesis has constructed several PCHs containing one or multiple azulene units,34 showing exotic photophysical and electronic properties.19,22,23,25,26,35–40 However, most of the known PCHs with azulene fragments in expanded forms do not show the intrinsic azulene-like anti-Kasha fluorescence, since, among other factors, benzenoid rings surrounding the azulene core and included for thermodynamic stabilization increase the local aromatic character what adversely bleaches anti-Kasha conditions.38,41 The task to encounter molecules with a fine balance between aromatic/non-alternant composition able to be stable and simultaneously conserve the anti-Kasha anomaly is certainly challenging. A comprehensive understanding of the modes of manipulation of light absorption and emission over broader wavelength ranges of the electromagnetic spectrum, to which anti-Kasha emitters can contribute, is a key aspect for the design of next-generations organic fluorophores able to serve as more versatile and efficient energy conversion substrates.
To design new molecules with anti-Kasha fluorescence, two aspects should be considered: (i) to selectively increase the optical activity of a given high-energy singlet excited state from which fluorescence will detune; and (ii) to promote the conditions for inefficient IC towards lowest-lying excited singlets. For this twofold purpose, we herein conceive a design strategy for a bright S3 state based on the coupling of two equivalent non-Kasha emitters, e.g., azulene. Firstly, to enhance the optical activity of the high-energy excited state, we borrow the J-coupling concept from supramolecular photophysics, in which “head-to-tail” stacked dimers induce an augment of radiative decay rates (larger oscillator strengths).42 In this sense, we apply the Kasha's excitonic coupling model,43,44 based on the interaction of transition dipole moments, to the coupling of the S2 state of azulene in order to design new dimeric azulene compounds with the suitable disposition between the two S2 transition dipole moments (Fig. 1). Secondly, to hinder IC from the bright state to lower spin-singlet states, we intend to preserve a large energy gap with respect to same-spin states. For that, it is desirable to have a (J-like) weak coupling of the two local S2 transitions, and simultaneously a vanishing interaction between S1 states of the building monomers. Finally, we explore the structural embedding of the target dimer in various PCHs in order to ensure antiparallel planar disposition of the two azulenes and enforce structural rigidity to further hinder non-radiative decay pathways.
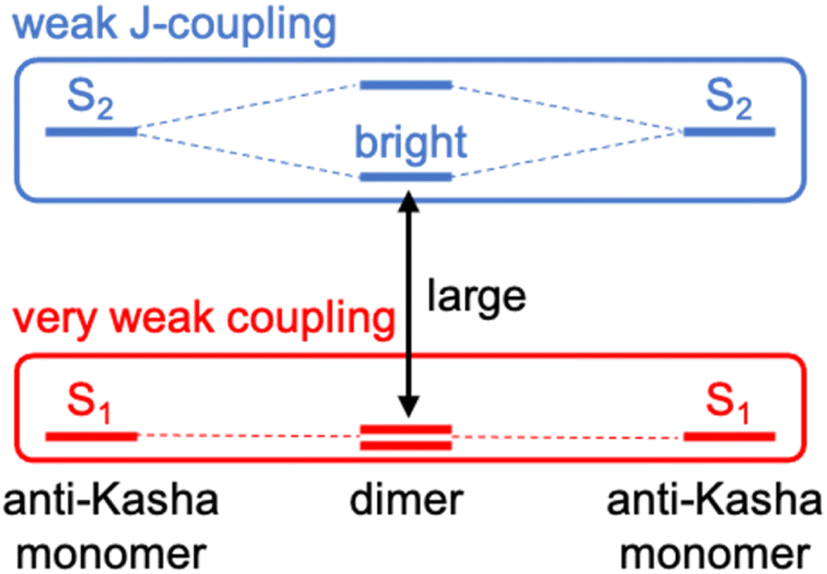 | ||
| Fig. 1 Molecular design strategy based on the coupling of non-Kasha emitters with weak J-coupling between high-energy (anti-Kasha) states (in blue) and very weak interaction between lowest excited states (in red). | ||
Our study relies on the results from quantum chemical calculations of potentially suitable dimeric azulene structures, i.e., biazulenes. In this article, we find out that a J-type parallel coupling of two azulene units through the lateral positions of five member rings gathers the conditions for anti-Kasha emission from a bright S3 excited state. Moreover, we identify a proper PCH embedding for the optimal biazulene core that preserves the requirements for high-excited state emission. Finally, the predicted photophysical properties are experimentally corroborated in a recently synthesized derivative of the computationally designed anti-Kasha emitter.
Experimental details
Electronic structure calculations
Hückel molecular orbital energies were obtained using the orbital parameters α (the energy of an electron in a pz-orbital) and β (the interaction energy between two vicinal orbitals) from the reported literature values.45 Ground-state molecular geometries have been optimized at the density functional theory (DFT) level with the CAM-B3LYP functional46 and the cc-pVDZ basis set. Vertical electronic transitions and excited state minima were obtained with the time-dependent version of DFT (TDDFT), by using a larger basis set (cc-pVTZ) and with the rCAM-B3LYP functional, which has shown to minimize many-electron self-interaction error in largely conjugated systems.47 Nucleus-independent chemical shift (NICS)48 calculations have been done at the same computational level. All calculations have been performed with the Q-Chem package.49Fluorescence and transient absorption measurements
Femtosecond transient absorption spectroscopy characterization was carried out with a Helios equipment from Ultrafast Systems, equipped with an amplified femtosecond Spectra-Physics Solstice-100F laser (with a 128 fs pulse width and 1 kHz repetition rate) coupled with a Spectra-Physics TOPAS Prime F optical parametric amplifier (195–22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nm). Samples were studied in CH2Cl2 solutions. The excitation power was modified using neutral density filters to match the desired power.
000 nm). Samples were studied in CH2Cl2 solutions. The excitation power was modified using neutral density filters to match the desired power.
Results and discussion
Optimal covalent arrangement for S3 anti-Kasha emission
Firstly, we explore possible linking modes in covalent dimers of azulene in order to generate the proper interstate couplings and a state energy diagram similar to the one envisaged in Fig. 1. For that, we devise five symmetrically connected dimers, biA1–biA6 (Fig. 2a). Interestingly, the optoelectronic and charge-transport properties of some of these symmetric biazulenes (biA1 and biA6) have been recently characterized.50 Asymmetric biazulene arrangements, i.e., connected by different azulene positions, are disregarded, since they should be prone to induce exciton localization, recovering (at most) the emission properties of the monomer. In other words, the bright excited state (S3) should delocalize over the two non-Kasha moieties, which require the two monomers to be equivalent. Therefore, only symmetrical dimers, i.e., connected by the same azulene positions, are considered. Besides, we are interested in orientations of the two local transition dipole moments of S2 of azulene (aligned along the long molecular axis) resulting in J-type interactions, so the syn-forms of biazulenes, expected to trigger H-coupling, are not taken into account here.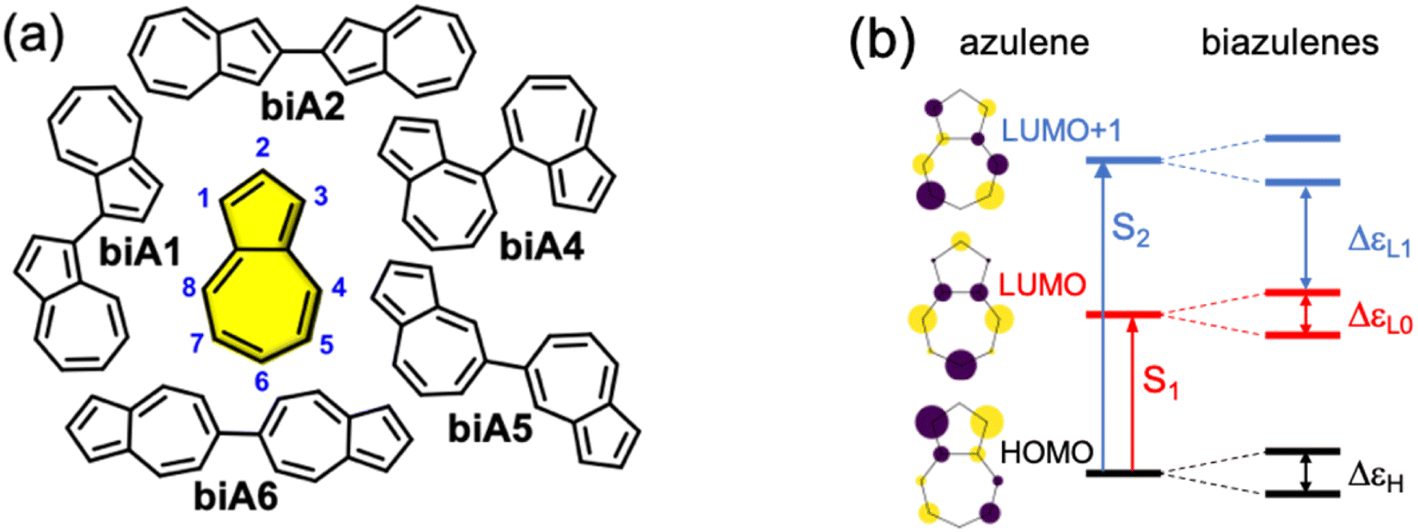 | ||
| Fig. 2 (a) Molecular structures of azulene (yellow) and symmetrical biazulenes (biA1–biA6). (b) Hückel-type molecular orbital diagram for azulene and biazulenes. Vertical arrows indicate orbital transitions for S1 and S2 states in azulene. Double arrows indicate orbital gaps in biazulenes. | ||
To achieve a bright S3 excited state, we aim to establish simple guidelines for biazulene violating Kasha's rule based on the magnitude of orbital couplings. The Hückel molecular orbitals of azulene and the general orbital distribution in biazulenes are presented in Fig. 2b. The two lowest excited singlet states of azulene correspond to the HOMO-to-LUMO (S1) and HOMO-to-LUMO+1 (S2) electronic promotions (Fig. 2b). Within the one-electron picture, we can associate the S2/S3 gap in biazulene with the energy difference between LUMO+1 and LUMO+2 (ΔεL1 in Fig. 2b). Strong interaction between the two LUMOs of azulene would result in large destabilization of the LUMO+1 of the dimer, which will shrink ΔεL1 and accelerate IC decay from S3. Hence, a small LUMO/LUMO+1 gap in the dimer (ΔεL0) is desirable, which requires a weak coupling between LUMOs of each azulene. On the other hand, degeneracy between the two highest occupied orbitals (ΔεH = 0) might induce an increase of the density of states, e.g., low-energy HOMO−1 → LUMO transition, favoring non-radiative processes. Additionally, to avoid orbital mixing with the π-system of the peripheral conjugated rings when embedded into a PCH (see below), a high energy HOMO on the biazulene is desired, which requires a large ΔεH, i.e., a strong coupling between HOMOs of each azulene. Therefore, considering all these conditions, we predict that anti-Kasha emission from S3 will be facilitated in those dimers with: large ΔεL1 and ΔεH, and small ΔεL0.
The molecular orbital energy differences for the five symmetric biazulenes are presented in Fig. 3a as obtained by the Hückel model. The biA1 dimer (antiparallel azulenes linked at the 1,1-position) presents the largest ΔεL1 and ΔεH, and a tiny ΔεL0, hence, according to the requirements discussed above, biA1 is identified as the most suitable arrangement for (S3) non-Kasha emission. These results can be rationally understood by the π-electron density at the connecting positions, which are optimal in position 1 (Fig. 3b): large in the HOMO (strong coupling), vanishing in the LUMO (no interaction), and small in LUMO+1 (weak coupling). It is also important to notice that the 1,1-linkage in biA1 triggers antibonding (bonding) interaction in the HOMO (LUMO+2) and ensures that the HOMO → LUMO+2 excitation corresponds indeed to a “head-to-tail” (like) orientation of the transition dipole moments on each azulene, predicting a bright S3 state (J-coupling in Fig. 1).
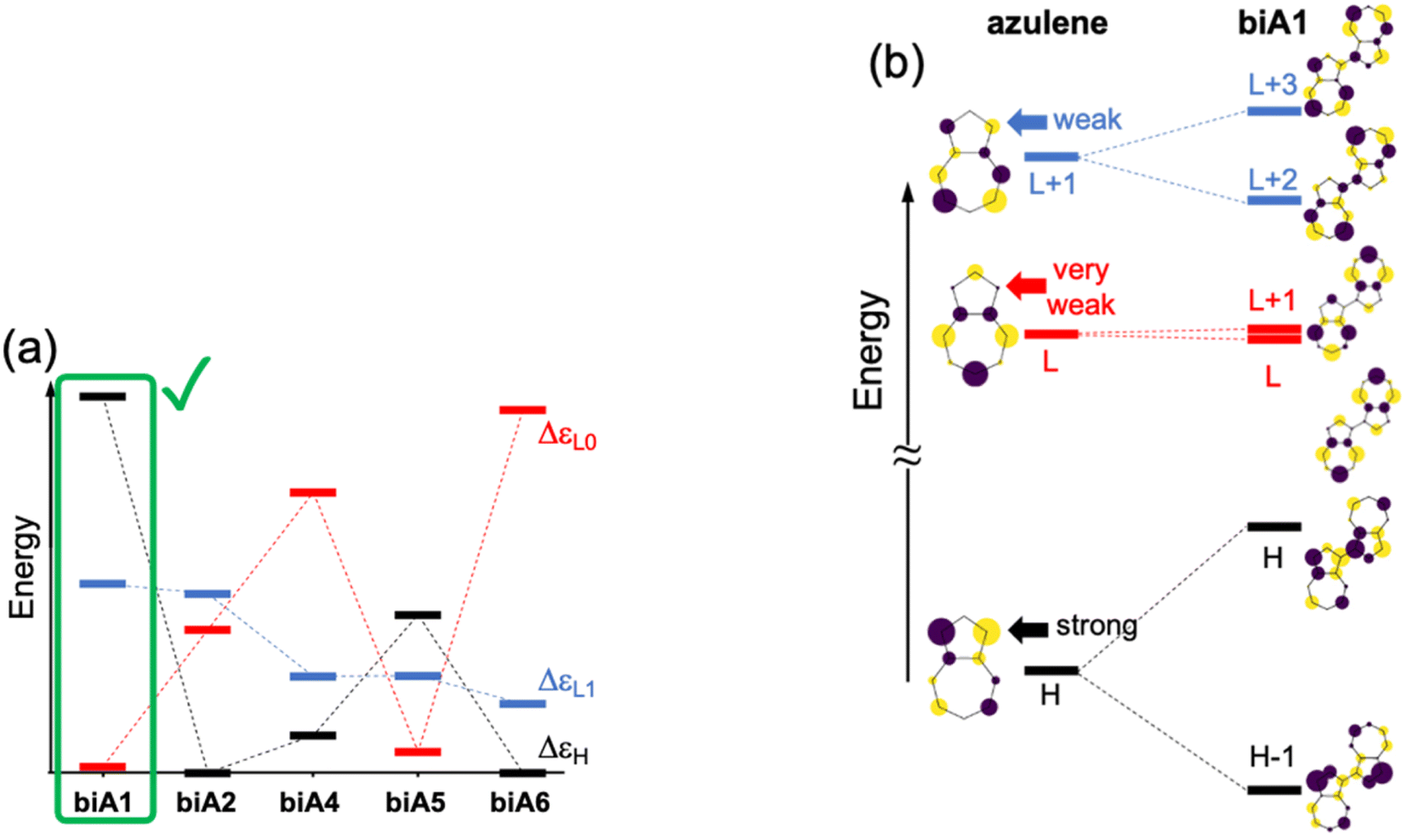 | ||
| Fig. 3 (a) Orbital energy differences (ΔεH, ΔεH, and ΔεL1) for symmetrical biazulenes (biA1–biA6). (b) Molecular orbital diagrams of azulene and biA1. Participation of pz-orbital in position 1 of azulene highlighted. Energy gaps and orbitals obtained with the Hückel model. H = HOMO; L = LUMO. | ||
Incorporation into a polycyclic conjugated hydrocarbon
Once we have identified the best link mode for efficient S3 fluorescence in biazulenes, we explore different forms to embed it into a PCH while preserving its structure. The motivation for surrounding the biA1 unit with conjugated rings is twofold: (i) ensure the antiparallel planar disposition of the two azulenes by blocking inter-azulene rotations, and (ii) enforce molecular rigidity to hinder secondary non-radiative decay pathways. Moreover, to maintain the predicted properties for biA1, the desired extended form must preserve the symmetry between the two azulene moieties and avoid orbital mixing, i.e., ensure that frontier molecular orbitals (FMOs) are localized on the biazulene unit.Based on biA1, we can bridge the two azulenes with six-membered or seven-membered rings, which are generally used and easily synthesized. However, when the two azulenes are bridged with two six-membered rings, the FMOs delocalize beyond the biazulene unit and eventually disrupt the properties of biA1 (Fig. S1–S4 and Tables S1–S4†). Then, we propose two seven-membered rings bridged molecules with different number of benzene rings symmetrically fused in order to preserve equivalency between the two azulenes (biA1a–biA1d in Fig. 4a).
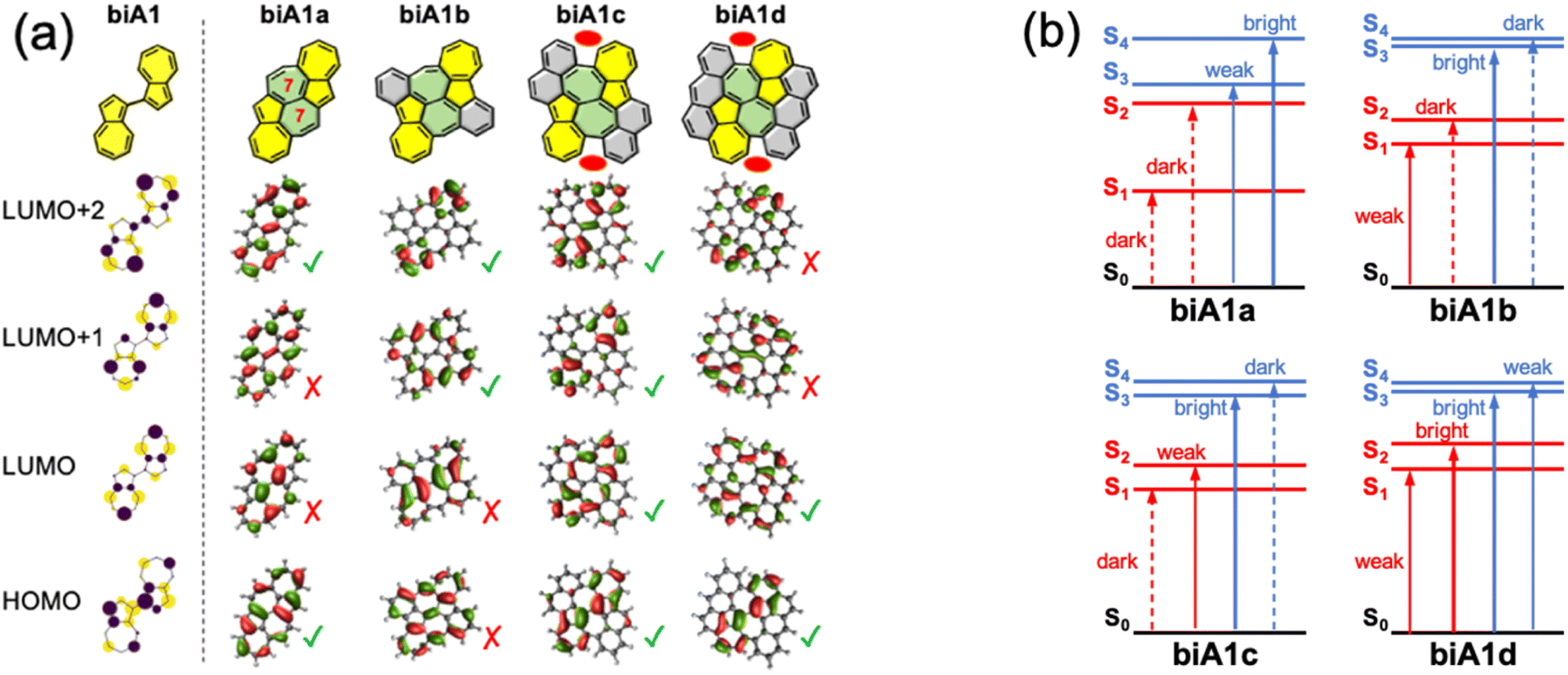 | ||
| Fig. 4 FMOs (a) and state energy diagrams (b) of compounds biA1a–d computed at the rCAM-B3LYP/cc-pVDZ level. Molecular orbitals similar with the (Hückel) biA1 model distribution (left) marked with green checkmarks, otherwise marked with red crosses. Red ovals in structures biA1c and biA1d indicate steric hindrance. | ||
The presence of the additional fused rings notably alters the nature of FMOs for biA1a and biA1b with respect to the biA1 model. These changes can be related to the interaction of the biazulene with the peripheral pz-orbitals, resulting in orbital hybridization and delocalization. Consequently, computed transitions for biA1a (Fig. 4b) discard it as a potentially efficient backbone (weak emission properties of S3 and small S2/S3 gap). On the other hand, despite the different orbital distribution with respect to biA1, biA1b shows good potential capabilities as a high state emitter, with large S2/S3 gap and oscillator strength. To our interest and delight, the FMOs of biA1c are notably localized on the biazulene core and recover the biA1 model orbital distribution. TDDFT computed low-lying states of biA1c predict good anti-Kasha emission properties from S3, with a rather large energy difference with respect to S2 and an oscillator strength notably larger than in azulene. It is also worth noticing that the additional benzene rings in biA1c with respect to biA1b, introduce steric hindrance at the armchair region (red ovals in Fig. 4a), triggering the loss of planarity through molecular torsion, and inducing a decoupling of the biazulene core to the π-system of the external naphthalene moieties, which helps to the localization of FMOs. Extending the peripheral conjugation in biA1d stabilizes the bonding interaction of azulene LUMO+1s (LUMO+2 in biA1 model), swapping the LUMO+1/LUMO+2 energy ordering with respect to biA1c. Consequently, biA1d exhibits the same state distribution as biA1c, but with a much-reduced S2/S3 gap, induced by the orbital energies of LUMO+1 and LUMO+2. From these results, we conclude that molecular structure based on biA1c can be a good candidate as anti-Kasha (S3) emitter.
The addition of benzenoid rings surrounding the biazulene core in biA1b–d with respect to biA1a decreases the local antiaromatic character of the seven-membered rings. Concretely, NICSzz(1) results indicate a large release of antiaromaticity with the two more external seven-membered rings becoming non-aromatic (Fig. S11†).
Experimental realization of the anti-Kasha emitter
To prove our strategy, we explore the photophysical properties of the recently synthesized compound 1 (Fig. 5),51 corresponding to the optimal biA1c backbone decorated with two tert-butylphenyl groups symmetrically distributed. For the sake of completeness, we will compare the photophysical properties of 1 with those of its isoelectronic homologue bischrysene (2), only containing six-membered rings.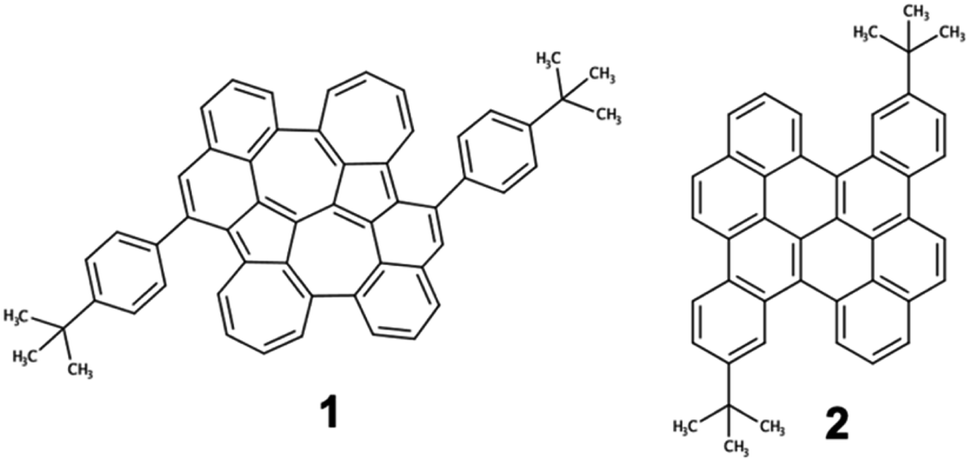 | ||
| Fig. 5 Molecular structures of diazuleno[2,1,8-efg:2′,1′,8′-kla]heptalene PCH 1 and substituted bischrysene (2). | ||
The ground state optimized geometry of 1 is noticeably non-planar with bond length alternation along the two embedded azulene units (Table S17†), in very good agreement with the X-ray molecular structure.51 FMOs of 1 (Fig. 6a) are largely localized on the biazulene unit with the HOMO built as the in-phase (with respect to inversion) coupling of two monomeric HOMOs on each of the two azulenes, the LUMO (LUMO+1) is the in-phase (out-of-phase) combination of azulene LUMOs, and the LUMO+2 is obtained as the out-of-phase interaction of azulene's LUMO+1. As a result, S1 and S2 states of 1 can be interpreted as emerging from the + and − combinations of excitations localized on each azulene moiety, respectively (Fig. 6b). The weak inter-azulene coupling results in small S1/S2 energy gap (E(S1) = 2.13 eV, E(S2) = 2.37 eV). Due to the relative disposition of the two azulenes in 1, transition dipole moments in S1 cancel each other out, while they present a weak intensity in S2 (f = 0.068). Interestingly, excitation to S3, obtained as the HOMO → LUMO+2 promotion, appears at much larger energy (E(S3) = 3.20 eV). Moreover, the parallel disposition of local transition dipole moments in the third excited singlet triggers a rather strong oscillator strength (f = 0.175). Higher singlet state excitations present much larger delocalization degree over the entire PCH (Fig. S10 and Table S11†). Therefore, the state distribution exhibited by 1 at the Franck–Condon region is in perfect accordance with the simple designed principles stablished from biA1, and the nature of molecular orbitals and low-lying states are in excellent agreement with the biA1c model system (Fig. 4b). On the other hand, the lowest singlet–singlet transition in 2 corresponds to the electronic excitation between the π-delocalized HOMO and LUMO (Fig. 6c). The vertical excitation to S1 is computed at 3.07 eV with a large oscillator strength (f = 0.442), in agreement with the conventional behavior of other PCHs. Overall, these results are in perfect agreement with the experimental absorption profiles of 1 and 2.51
 | ||
| Fig. 6 (a) Molecular orbitals involved in the lowest singlet excited states of 1; (b) biazulene (core) model employed to characterize S1–S3 states of 1; and (c) molecular orbitals involved in the lowest singlet excited state of 2. | ||
Recorded steady-state fluorescence emission spectra of 2 displays one red-shifted band with respect to the S0 → S1 absorption, which is mirror-like image with the excitation spectrum with a small Stokes shift, indicating the rigidity of its PCH skeleton (Fig. 7a). 2 strictly follows Kasha's rule, that is, independently of the frequency of absorbed photons, highly efficient Sn → S1 IC yields strong S1 population and posterior emission. On the other hand, 1 discloses completely different photophysics. When 1 is excited at either one of the two low-energy absorption bands, i.e., 600–800 nm and 430–500 nm, no fluorescence emission is observed either at room temperature or upon temperature lowering at 80 K. Excitation on the high energy group of bands (370 nm) of 1 gives rise to no neat emission, whereas a well-vibronically resolved emission band is clearly detected upon cooling at 80 K, with vibrational peaks at 410, 434 and 463 nm (Fig. 7b), and associated lifetime of 2–3 ns. The excitation spectrum recorded for this emission is also a specular image of its emission, also with vanishing Stokes shift. However, the excitation spectrum does not perfectly fit the absorption profile though the main excitation peaks (at 361, 380 and 405 nm) lie within the limits of its corresponding absorption band (340–410 nm). Though the compound is prepared with a high degree of purity,51 the presence of trace amounts of impurities might give rise to anomalous emissions and wrong anti-Kasha assignments. However, here, the presence of a turning on of the fluorescence at cryogenic conditions points out towards the existence of an intramolecular process ruling out the presence of other molecular entities. TDDFT calculations characterize this emission as the S3 → S0 electronic deexcitation (Tables S14 and S15†) and, according to its vibronic resolution, it belongs to a rigidified S3 state of 1 highly prompted to fluoresce.
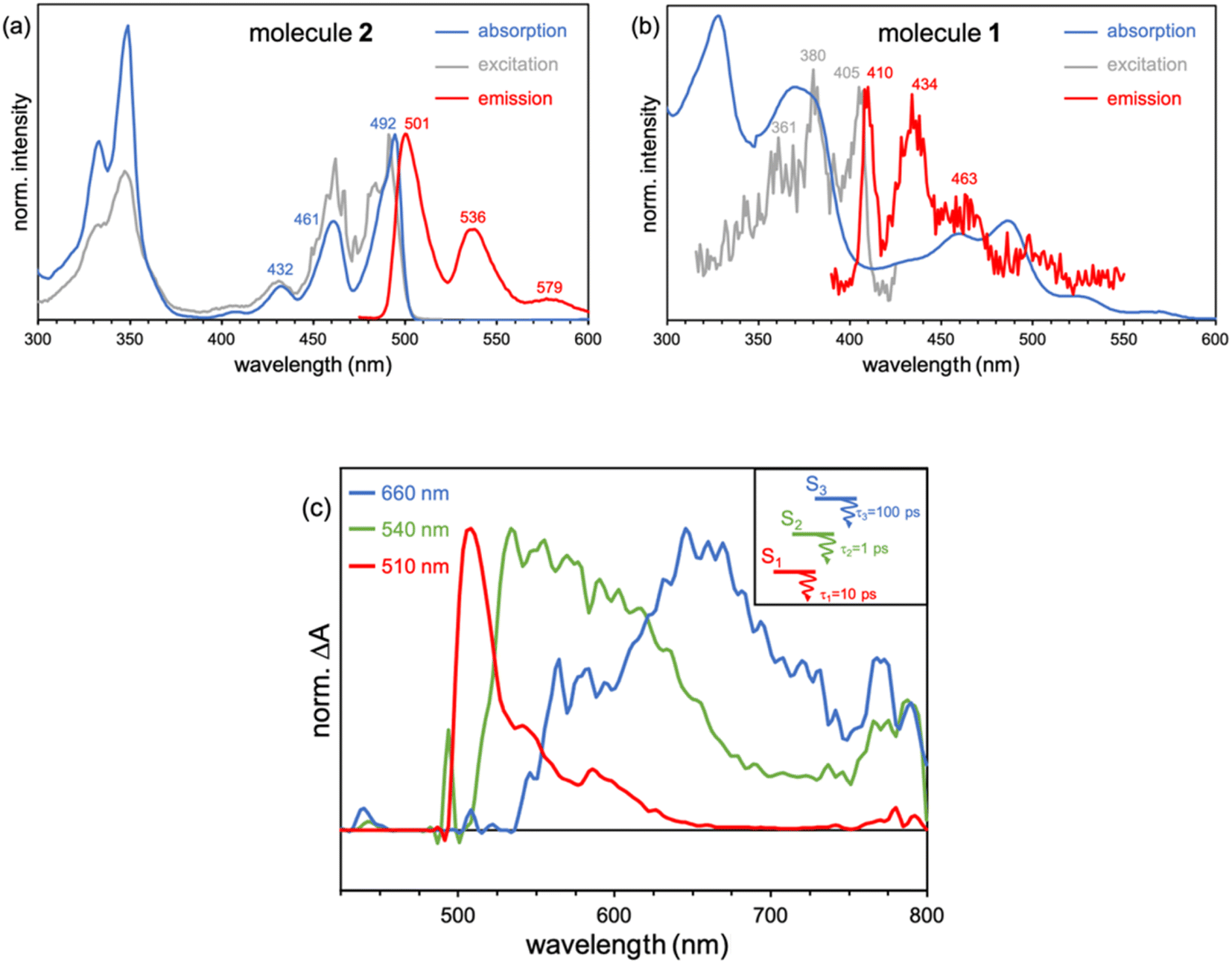 | ||
| Fig. 7 (a) Absorption (black), excitation (gray) and emission (red line) (exciting at 360 nm) spectra of 2 in methyl THF at 80 K. (b) Absorption (black) of 1 together with its emission spectrum (red, exciting at 360 nm) and the corresponding excitation (grey) spectrum of this emission at 80 K. (c) Femtosecond excited state transient absorption spectra (ESA bands) of the species formed upon excitation at 370 nm in methyl THF solution at room temperature together with the excited state diagram and lifetimes deduced from the experimental photophysical data for 1. | ||
Femtosecond transient absorption spectroscopy (fs-TAS) of 1 (Fig. 7c) has been carried out to further characterize the manifold of excited states detected in the steady-state emission. Upon pumping at 370 nm, we detect the initial formation of three different excited state absorption bands (ESA) with wavelength absorbance maxima at 660, 540 and 510 nm (Fig. S12†). The band component at 540 nm disappears in 1 ps followed by the ESA of the 510 nm band in 10 ps. Lastly, the 660 nm band gets to zero absorbance in 100 ps. On the other hand, by photoexcitation at 480 nm, only two components in the band time evolution are observed corresponding to the ESA features at 540 and 510 nm. The formation of the ESA feature at 660 nm is only under 370 nm excitation, which, according to TDDFT calculations and assignment, might indicate that corresponds to an absorption from the S3 high energy state, i.e., S3 → Sn, whereas those ESA bands at 540 and 510 nm emerge as transient absorptions from the two lowest energy lying excited states, i.e., S2 → Sn and S1 → Sn, respectively. The observed 660 → 540 → 510 nm blue-shift also supports the excited state energy ordering as: S3 (660 nm) > S2 (540 nm) > S1 (510 nm). On the other hand, the larger relaxation time for the higher energy S3 (660 nm) excited state fully agrees with the detection of its fluorescence in the steady state experiment and by electronic structure calculations (see above). On the other hand, the fs-TAS characterization of 2 gave very different results (Fig. S13†). As seen, upon low energy excitation (485 nm, matching the low energy absorption band), a band centered at 710 nm is formed right upon the technique time detection limit. This band is assigned to an excited singlet state that decays to ground state with a lifetime of 800 ps, in accordance with the obtained fluorescence lifetime for 2 (Fig. S14†).
In order to rationalize the high energy anti-Kasha emission of 1, we computationally search for local energy minima on the excited state manifold. Relaxation on the PES of the S1 (dark) state substantially reduces the vertical gap to S0 (Table S12†), facilitating the non-radiative decay to the ground state. Minimization of the S2 energy preserves its electronic character with a weak oscillator strength (Table S13†). Moreover, at the S2 minimum, there is a rather small S2/S1 vertical gap, suggesting fast IC to S1 with a subsequent (non-radiative) transit to the ground state. These results are consistent with the lack of fluorescent emission when the molecule is photoactivated with low-energy photons, and with the fast decay of the ESA bands at 540 and 510 nm. This behavior can be rationalized by: (i) weak absorption to low-lying excited states (S1 and S2); (ii) fast IC to the S1 triggered by the small energy gap; and (iii) reduced S1/S0 optical gap favoring non-radiative decay to the ground state over fluorescent emission.
On the other hand, our TDDFT calculations identify a minimum on the PES of the S3 (bright) state (Table S14†), with a molecular geometry that slightly deviate from the Franck–Condon structure (Table S17†). The computed energy at the excited state structure is compatible with the anti-Kasha fluorescence registered at 410 nm and shows large oscillator strength, facilitating their radiative decay. Moreover, it presents a quite large energy gap with lower states of the same multiplicity, i.e., S2 and S1. In particular, S3/S2 vertical gap at the S3 minimum is considerably large (∼0.8 eV), inhibiting IC to S2 (and S1).
From these results, we can stablish that the anti-Kasha emission in 1 has its origin in the photophysical properties of azulene. Concretely, the nature of the low-lying singlet states of 1 are largely dictated by the interaction of the two azulene moieties in the biazulene core, resulting in two dark (or weakly emitting) singlets reminiscent of the lowest excited singlet in azulene, and a higher (S3) state with important optical activity (related to the second singlet of azulene). Although in 1 the gap of the S3 state with the lower singlets is reduced with respect to the S1/S2 energy difference in pristine azulene (Table S15†), it remains sufficiently large for the emission purpose. On the other hand, the parallel and constructive J-like disposition of the two local (azulene-like) transition dipole moments composing the S3 → S0 transition, triggers a notably stronger oscillator strength as compared to the non-Kasha state in azulene. Moreover, the molecular structure rigidity imposed by the embedding of the biazulene core in a PCH hinders vibrational distortions potentially assisting the deactivation of the high energy singlets.
Conclusions
In summary, we propose a molecular strategy for intense anti-Kasha emission referring to the intramolecular J-coupling concept. The key strategy is to optimize FMO coupling and energy level alignment to selectively increase the photoactivity of higher-order excited states (S3), while rationally controlling energy gaps between different excited states to suppress IC processes. To validate our strategy, we have explored the emission properties of the recently synthetized diazuleno[2,1,8-efg:2′,1′,8′-kla]heptalene embedded in a PCH holding the optimal connection mode of biazulene and the surrounding fused units as computationally predicted. Contrary to its benzenoid isomer 2 only exhibiting luminescence following Kasha's rule, PCH 1 shows fluorescence from the S3 state in the range of 410–470 nm upon excitation at 370 nm, which was well verified by fs-TAS, as corresponding to high energy excited state absorption bands at 660 nm. Our work reported herein provides a new way to rationally design anti-Kasha emitters and encourages the exploration of improving the competition of emission against nonradiative decay at high-energy states of photoluminescent materials. New strategies of manipulation of light–matter interaction in organic molecules revealed from these sort of in-depth multidisciplinary analysis paves the unique way for rational design of new chromophores and energy converter in all-carbon-based molecular systems.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
A. D.-A. performed all the electronic structure calculations. A. D.-A. and D. C. analyzed the computational results. J. M.-B., J. W., J. L. and J. C. performed experiments for molecules 1 and 2. J. M.-B. and J. C. carried out transient and steady-state spectroscopic measurements and discuss the interpretation. J. L., J. C. and D. C. conceived the work and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MINECO/FEDER of the Spanish Government (PGC2018-098533-B-100, PID2019-109555GB-I00, PID2021-127127NB-I00 and RED2018-102815-T), the Junta de Andalucía (UMA18FEDERJA057 and PROYEXCEL-00328 PY21), the Eusko Jaurlaritza (PIBA19-0004), the Hong Kong Research Grants Council (27301720, 17304021), and the National Natural Science Foundation of China (22122114). F. G. and A. D.-A. acknowledge the FPI program of the Spanish Government for predoctoral fellowships. J. L. is grateful for the funding from The University of Hong Kong (HKU) and ITC to the SKL. We thank the UGC funding administered by HKU for supporting the Time-of-Flight Mass Spectrometry Facilities under the Support for Interdisciplinary Research in Chemical Science. We acknowledge the computer cluster (HPC2021) of HKU and the Donostia International Physics Center (DIPC) Computational Center for generous allocations of compute resources.References
- M. Kasha and S. P. McGlynn, Molecular Electronic Spectroscopy, Annu. Rev. Phys. Chem., 1956, 7, 403–424 CrossRef CAS.
- P. Klán and J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice, Wiley, 2009 Search PubMed.
- N. J. Turro, V. Ramamurthy, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, 2009 Search PubMed.
- M. Kasha, Characterization of electronic transitions in complex molecules, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- M. Beer and H. C. Longuet-Higgins, Anomalous Light Emission of Azulene, J. Chem. Phys., 1955, 23, 1390–1391 CrossRef CAS.
- P. A. Geldof, R. P. H. Rettschnick and G. J. Hoytink, Fluorescence from the second excited singlets of pyrene and 3,4-benzpyrene, Chem. Phys. Lett., 1969, 4, 59–61 CrossRef CAS.
- S. Z. Levine, A. R. Knight and R. P. Steer, Fluorescence from the second excited singlet state of thiophosgene vapour, Chem. Phys. Lett., 1974, 29, 73–76 CrossRef CAS.
- T. Itoh, Fluorescence and Phosphorescence from Higher Excited States of Organic Molecules, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS PubMed.
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Breaking the Kasha Rule for More Efficient Photochemistry, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- D. Malpicci, E. Lucenti, C. Giannini, A. Forni, C. Botta and E. Cariati, Prompt and Long-Lived Anti-Kasha Emission from Organic Dyes, Molecules, 2021, 26, 6999 CrossRef CAS PubMed.
- G. Binsch, E. Heilbronner, R. Jankow and D. Schmidt, On the fluorescence anomaly of azulene, Chem. Phys. Lett., 1967, 1, 135–138 CrossRef CAS.
- H. J. Griesser and U. P. Wild, The fluorescence lifetimes of carbonyl derivatives of azulene showing dual emission, J. Photochem., 1980, 13, 309–318 CrossRef CAS.
- H. Yamaguchi, M. Higashi and K. Fujimori, Anomalous fluorescence spectra of azuleno[1,2-b]furan, Spectrochim. Acta, Part A, 1987, 43, 1377–1378 CrossRef.
- R. P. Steer, Photophysics of molecules containing multiples of the azulene carbon framework, J. Photochem. Photobiol., C, 2019, 40, 68–80 CrossRef CAS.
- S. H. Pun and Q. Miao, Toward Negatively Curved Carbons, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed.
- A. Konishi and M. Yasuda, Breathing New Life into Nonalternant Hydrocarbon Chemistry: Syntheses and Properties of Polycyclic Hydrocarbons Containing Azulene, Pentalene, and Heptalene Frameworks, Chem. Lett., 2020, 50, 195–212 CrossRef.
- Chaolumen, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Construction of Heptagon-Containing Molecular Nanocarbons, Angew. Chem., Int. Ed., 2021, 60, 23508–23532 CrossRef CAS PubMed.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, A grossly warped nanographene and the consequences of multiple odd-membered-ring defects, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- A. Konishi, K. Horii, D. Shiomi, K. Sato, T. Takui and M. Yasuda, Open-Shell and Antiaromatic Character Induced by the Highly Symmetric Geometry of the Planar Heptalene Structure: Synthesis and Characterization of a Nonalternant Isomer of Bisanthene, J. Am. Chem. Soc., 2019, 141, 10165–10170 CrossRef CAS PubMed.
- Y. Ni, T. Y. Gopalakrishna, H. Phan, T. Kim, T. S. Herng, Y. Han, T. Tao, J. Ding, D. Kim and J. Wu, 3D global aromaticity in a fully conjugated diradicaloid cage at different oxidation states, Nat. Chem., 2020, 12, 242–248 CrossRef CAS PubMed.
- C. M. Cruz, I. R. Márquez, I. F. A. Mariz, V. Blanco, C. Sánchez-Sánchez, J. M. Sobrado, J. A. Martín-Gago, J. M. Cuerva, E. Maçôas and A. G. Campaña, Enantiopure distorted ribbon-shaped nanographene combining two-photon absorption-based upconversion and circularly polarized luminescence, Chem. Sci., 2018, 9, 3917–3924 RSC.
- X.-S. Zhang, Y.-Y. Huang, J. Zhang, W. Meng, Q. Peng, R. Kong, Z. Xiao, J. Liu, M. Huang, Y. Yi, L. Chen, Q. Fan, G. Lin, Z. Liu, G. Zhang, L. Jiang and D. Zhang, Dicyclohepta[ijkl,uvwx]rubicene with Two Pentagons and Two Heptagons as a Stable and Planar Non-benzenoid Nanographene, Angew. Chem., Int. Ed., 2020, 59, 3529–3533 CrossRef CAS PubMed.
- H. Reel and E. Vogel, Dicyclohepta[cd,gh]pentalene—A New Pyrene Isomer, Angew. Chem., Int. Ed., 1972, 11, 1013–1014 CrossRef CAS.
- S. H. Pun, Y. Wang, M. Chu, C. K. Chan, Y. Li, Z. Liu and Q. Miao. Synthesis, Structures, and Properties of Heptabenzo[7]circulene and Octabenzo[8]circulene, J. Am. Chem. Soc., 2019, 141, 9680–9686 CrossRef CAS PubMed.
- N. Ogawa, Y. Yamaoka, H. Takikawa, K.-i. Yamada and K. Takasu, Helical Nanographenes Embedded with Contiguous Azulene Units, J. Am. Chem. Soc., 2020, 142, 13322–13327 CrossRef CAS PubMed.
- T. Kirschbaum, F. Rominger and M. Mastalerz, A Chiral Polycyclic Aromatic Hydrocarbon Monkey Saddle, Angew. Chem., Int. Ed., 2020, 59, 270–274 CrossRef CAS PubMed.
- K. Y. Cheung, K. Watanabe, Y. Segawa and K. Itami, Synthesis of a zigzag carbon nanobelt, Nat. Chem., 2021, 13, 255–259 CrossRef CAS PubMed.
- Y. Fei, Y. Fu, X. Bai, L. Du, Z. Li, H. Komber, K.-H. Low, S. Zhou, D. L. Phillips, X. Feng and J. Liu, Defective Nanographenes Containing Seven-Five-Seven (7–5–7)-Membered Rings, J. Am. Chem. Soc., 2021, 143, 2353–2360 CrossRef CAS PubMed.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, Structural Defects in Graphene, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- O. V. Yazyev and S. G. Louie, Electronic transport in polycrystalline graphene, Nat. Mater., 2010, 9, 806–809 CrossRef CAS PubMed.
- J. Lahiri, Y. Lin, P. Bozkurt, I. I. Oleynik and M. Batzill, An extended defect in graphene as a metallic wire, Nat. Nanotechnol., 2010, 5, 326–329 CrossRef CAS PubMed.
- D. Geng, H. Wang and G. Yu, Graphene Single Crystals: Size and Morphology Engineering, Adv. Mater., 2015, 27, 2821–2837 CrossRef CAS PubMed.
- X.-Y. Wang, A. Narita and K. Müllen, Precision synthesis versus bulk-scale fabrication of graphenes, Nat. Rev. Chem., 2017, 2, 0100 CrossRef.
- Y. Fei and J. Liu, Synthesis of Defective Nanographenes Containing Joined Pentagons and Heptagons, Adv. Sci., 2022, 9, 2201000 CrossRef CAS PubMed.
- J. Ma, Y. Fu, E. Dmitrieva, F. Liu, H. Komber, F. Hennersdorf, A. A. Popov, J. J. Weigand, J. Liu and X. Feng, Helical Nanographenes Containing an Azulene Unit: Synthesis, Crystal Structures, and Properties, Angew. Chem., Int. Ed., 2020, 59, 5637–5642 CrossRef CAS PubMed.
- H. Xin, C. Ge, X. Jiao, X. Yang, K. Rundel, C. R. McNeill and X. Gao, Incorporation of 2,6-Connected Azulene Units into the Backbone of Conjugated Polymers: Towards High-Performance Organic Optoelectronic Materials, Angew. Chem., Int. Ed., 2018, 57, 1322–1326 CrossRef CAS PubMed.
- K. Uehara, P. Mei, T. Murayama, F. Tani, H. Hayashi, M. Suzuki, N. Aratani and H. Yamada, An Anomalous Antiaromaticity That Arises from the Cycloheptatrienyl Anion Equivalent, Eur. J. Org. Chem., 2018, 2018, 4508–4511 CrossRef CAS.
- Y. Han, Z. Xue, G. Li, Y. Gu, Y. Ni, S. Dong and C. Chi, Formation of Azulene-Embedded Nanographene: Naphthalene to Azulene Rearrangement During the Scholl Reaction, Angew. Chem., Int. Ed., 2020, 59, 9026–9031 CrossRef CAS PubMed.
- Y. Yamaguchi, K. Ogawa, K.-i. Nakayama, Y. Ohba and H. Katagiri, Terazulene: A High-Performance n-Type Organic Field-Effect Transistor Based on Molecular Orbital Distribution Control, J. Am. Chem. Soc., 2013, 135, 19095–19098 CrossRef CAS PubMed.
- Y. Yamaguchi, M. Takubo, K. Ogawa, K.-i. Nakayama, T. Koganezawa and H. Katagiri, Terazulene Isomers: Polarity Change of OFETs through Molecular Orbital Distribution Contrast, J. Am. Chem. Soc., 2016, 138, 11335–11343 CrossRef CAS PubMed.
- A. Ong, T. Tao, Q. Jiang, Y. Han, Y. Ou, K.-W. Huang and C. Chi, Azulene-Fused Acenes, Angew. Chem., Int. Ed., 2022, 61, e202209286 CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- M. Kasha, Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates, Radiat. Res., 1963, 20, 55–70 CrossRef CAS PubMed.
- R. M. Hochstrasser and M. Kasha, Application of the exciton model to mono-molecular lamellar systems, Photochem. Photobiol., 1964, 3, 317–331 CrossRef CAS.
- F. A. Van-Catledge, A Pariser-Parr-Pople-based set of Hueckel molecular orbital parameters, J. Org. Chem., 1980, 45, 4801–4802 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Development of exchange-correlation functionals with minimal many-electron self-interaction error, J. Chem. Phys., 2007, 126, 191109 CrossRef PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, Á. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, Á. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince, R. A. DiStasio, A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the frontiers of quantum chemistry: an overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- Y. Shibuya, K. Aonuma, T. Kimura, T. Kaneko, W. Fujiwara, Y. Yamaguchi, D. Kumaki, S. Tokito and H. Katagiri, Linear Biazulene Isomers: Effects of Molecular and Packing Structure on Optoelectronic and Charge-Transport Properties, J. Phys. Chem. C, 2020, 124, 4738–4746 CrossRef CAS.
- J. Wang, F. Gordillo, J. M. Beloqui, A. Diaz-Andres, X. Miao, D. Casanova, J. Casado and J. Liu, Synthesis of a Dicyclohepta[a,g]heptalene-Containing Polycyclic Conjugated Hydrocarbon and the Impact of Non-Alternant Topologies, Angew. Chem., Int. Ed., 2023, 62, e202217124 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00405h |
| This journal is © The Royal Society of Chemistry 2023 |