
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of (−)-scabrolide A and (−)-yonarolide†
Nicholas J.
Hafeman‡
 ,
Steven A.
Loskot‡
,
Christopher E.
Reimann
,
Beau P.
Pritchett
,
Scott C.
Virgil
and
Brian M.
Stoltz
*
,
Steven A.
Loskot‡
,
Christopher E.
Reimann
,
Beau P.
Pritchett
,
Scott C.
Virgil
and
Brian M.
Stoltz
*
Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu
First published on 1st April 2023
Abstract
The complete account of the total syntheses of scabrolide A and yonarolide is disclosed. This article describes an initial approach involving a bio-inspired macrocyclization/transannular Diels–Alder cascade, which ultimately failed due to undesired reactivity during macrocycle construction. Next, the evolution of a second and third strategy, which both involve an initial intramolecular Diels–Alder reaction followed by a late-stage closure of the seven-membered ring of scabrolide A are detailed. The third strategy was first validated on a simplified system, but problems were encountered during a key [2 + 2] photocycloaddition on the fully elaborated system. An olefin protection strategy was employed to circumvent this problem, ultimately leading to the completion of the first total synthesis of scabrolide A and the closely related natural product yonarolide.
Introduction
The polycyclic furanobutenolide-derived norcembranoids are a small family of structurally diverse, highly oxygenated C19 norditerpenoids, which are isolated exclusively from soft corals of the genus Sinularia (Fig. 1).1 Since the first reports of their isolation over two decades ago, these molecules have captivated synthetic chemists owing to their unique and synthetically challenging structural features, as well as the range of biological effects they have been shown to elicit. Consequently, the polycyclic furanobutenolide-derived norcembranoids have garnered considerable attention from the synthetic community, and several attempted total syntheses have been reported to date.2,3 | ||
| Fig. 1 Polycyclic furanobutenolide-derived norcembranoids. | ||
All known polycyclic furanobutenolide-derived norcembranoids are believed to originate from the macrocyclic norcembranoids sinuleptolide and 5-episinuleptolide (9, Scheme 1), which are thought to be in equilibrium in vivo. Through a series of divergent, transannular reaction cascades, these two progenitors give rise to the diverse set of carbocyclic skeletons that make up the furanobutenolide-derived polycyclic norcembranoid family. For example, scabrolide A (1) is proposed to arise from 5-episinuleptolide (9) following a series of two transannular Michael additions: a 7-exo-trig cyclization between C(5) and C(13), and a 5-exo-trig cyclization between C(7) and C(11), which produce isomeric sinulochmodin C (3). β-Elimination of the C(5) tertiary ether then generates 12 (the structure originally and erroneously assigned as scabrolide B), which undergoes olefin isomerization to scabrolide A (1). This biosynthetic hypothesis is supported by the fact that these natural products are often isolated together, and alongside their macrocyclic counterparts.1 Futhermore, Pattenden's groundbreaking biomimetic semisyntheses of sinulochmodin C (3) and ineleganolide (6) from the same naturally-derived precursor (9) beautifully demonstrated the feasibility of this type of chemistry.4
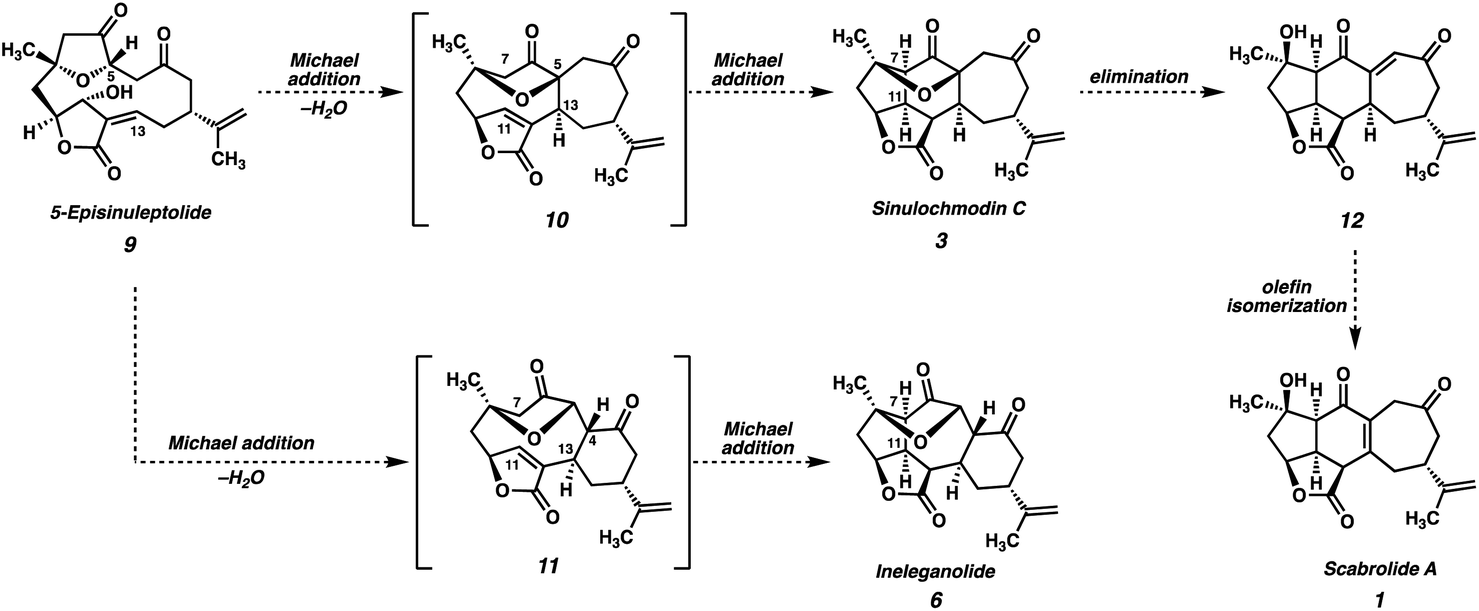 | ||
| Scheme 1 Proposed biosynthesis of sinulochmodin C (3), scabrolide A (1), and ineleganolide (6). | ||
Efforts have been reported toward the total synthesis of several of the polycyclic furanobutenolide-derived norcembranoid diterpenoids. Ineleganolide (6) has a long history of thwarted attempted syntheses. Disclosures by Vanderwal2d as well as our group2e detailed the syntheses of highly advanced intermediates en route to this target, which could not be carried forward to the natural product itself. Regarding the [5-6-7] fused carbocyclic core of the yonarane-type norcembranoids (scabrolide A, yonarolide, sinulochmodin C), two recent publications5 demonstrated strategies for the synthesis of the fused [5-6] portion of this framework. However, these reports did not detail the formation of the 7-membered carbocycle. Additionally, pioneering research by Barriault,6 Ito,3 and Mehta7 demonstrated approaches to this ring system, but again without further advancement to any natural product targets.
Given our laboratory's interest in the total synthesis of structurally complex natural products, we have had a longstanding fascination with the polycyclic furanobutenolide-derived norcembranoid diterpenoids. Beginning with our efforts toward ineleganolide (6), which culminated in the enantioselective synthesis of several isomeric “ineleganoloids”,2e we have continued our pursuit of this family of natural products, shifting our focus to (−)-scabrolide A (1). Our efforts resulted in the first total syntheses of (−)-scabrolide A (1),8 as well as the related natural product yonarolide (4), representing the first successful total synthesis of any member of the polycyclic furanobutenolide-derived norcembranoid class.
Since our initial disclosure of this research, several completed syntheses of polycyclic furanobutenolide-derived norcembranoids and cembranoids (the C20 counterparts) have been reported. The Fürstner group published an elegant total synthesis of both scabrolide A (1) and the (incorrectly) reported structure of scabrolide B (12).9 Additionally, the Wood group very recently reported the first successful total syntheses of ineleganolide (8) and sinulochmodin C (3),10 a ground-breaking achievement within this field. Finally, recent publications by the Romo group as well as our group detailed the successful syntheses of the closely related C20 cembranoids ramesewaralide11 and havellockate,12 respectively.
This article provides a complete account of our total syntheses of scabrolide A (1) and yonarolide (4). Our efforts toward these targets, which built heavily upon our laboratory's experience with ineleganolide (6), involved several pivots in the strategic approach. In this manuscript we hope to present the challenges faced in pursuit of scabrolide A (1) and yonarolide (4), and to show how these challenges informed the development of the route that ultimately proved successful in delivering these targets.
First-generation approach
At the outset, we planned to exploit a bioinspired approach to scabrolide A (1), in which the core ring system would be assembled via a transannular Diels–Alder reaction, which retrosynthetically delivers macrolactone 13 (Fig. 2). Given the fact that the central six-membered ring of the natural product is believed to arise from a transannular double-Michael cascade (i.e., a formal Diels–Alder) in vivo, we hypothesized that this approach might be feasible provided we could access macrocycle 13. We anticipated that a ring-closing metathesis (RCM) of an intermediate such as ester 14 might generate the requisite macrocycle, given the longstanding precedent for the use of this reaction for the formation of large and medium-sized rings.13 Ester 14, in turn, would be formed by a convergent esterification of two enantioenriched fragments: dihydroxyvinylcyclopentene 15 and ynoic acid 16.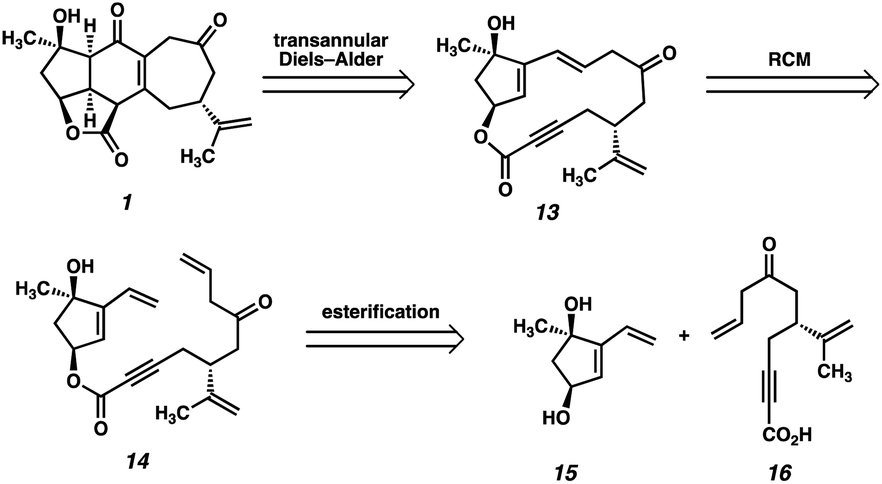 | ||
| Fig. 2 First-generation retrosynthetic analysis of scabrolide A (1). | ||
The synthesis of dihydroxyvinylcyclopentene ent-15 had been previously reported by our laboratory during our efforts toward ineleganolide (6).14,2e It should be noted that, at the outset of that study, the absolute stereochemistry of ineleganolide (6)15 and of several of the other polycyclic furanobutenolide-derived norcembranoids16 was not known (and had been erroneously reported as the ent-series). It was later shown that these molecules possess the opposite absolute stereochemistry, shown correctly in Fig. 1.17 Thus, unbeknownst to us at the time, our initial efforts targeted the ent-series of these natural products.
Our original synthesis of dihydroxyvinylcyclopentene ent-15, outlined in Scheme 2, commences with the advancement of 1,1-dimethoxycyclohexane (17) and Tris HCl (18) to TES enol ether 19, achieved in 6 steps and 28% overall yield. Enol ether 19 then serves as a substrate for a Pd-catalyzed asymmetric allylic alkylation, delivering enantioenriched tertiary ether 20 in 82% yield and 92% ee. Following an oxidative bromination/Wittig-type cyclization, enone 21 is obtained. A diastereoselective reduction followed by protecting group manipulations results in the formation of monoprotected triol 22, which may then undergo oxidation, Wittig methenylation, and deprotection to deliver the key fragment ent-15.
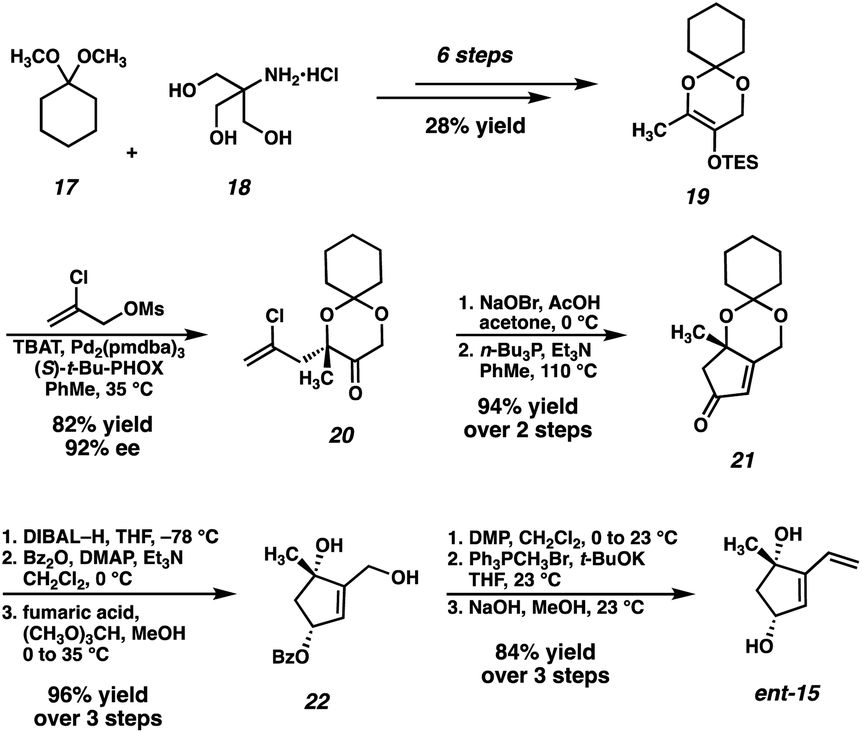 | ||
| Scheme 2 First-generation synthesis of vinylcyclopenteneone ent-15. | ||
The second coupling partner is accessed from (S)-carvone (23) beginning with a known sequence18 which delivers Weinreb amide ent-24 in 4 steps (Scheme 3). Seyferth–Gilbert homologation19 affords terminal alkyne ent-25, and subsequent monoaddition of allyl magnesium bromide furnishes ketone 26, which is then protected as the corresponding dioxolane (27). Deprotonation of 27 with n-BuLi and quenching with CO2 delivers the desired ynoic acid, which is carried forward crude to the convergent esterification. Gratifyingly, we found that the two fragments are smoothly coupled under Yamaguchi conditions,20 delivering ester 28 in 44% yield over the two steps, setting the stage for the key macrocyclization reaction.
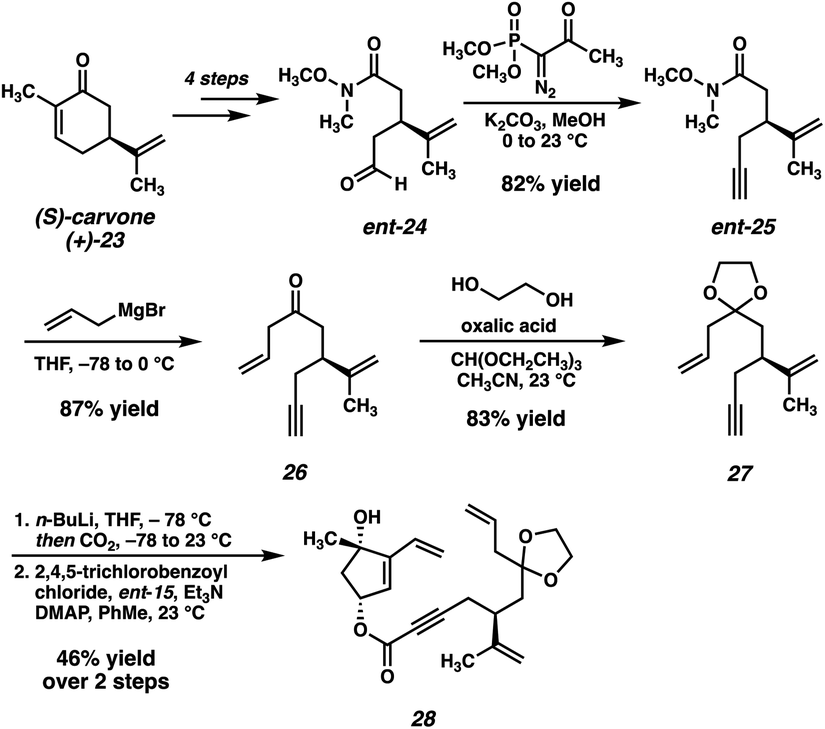 | ||
| Scheme 3 Synthesis of RCM substrate 28. | ||
Despite ample precedent for RCM-mediated macrocyclization, ester 28 failed to undergo the desired RCM, instead delivering undesired cyclohexene 29 as the predominant product (Scheme 4A). Even after examining a variety of known RCM catalysts, macrocycle 30 was never observed, thus thwarting this synthetic plan. Although substrate 28 possesses two monosubstituted olefins (the vinyl group and the allyl group), it is likely that the electronic deactivation of the conjugated vinyl group causes the allyl olefin to be the preferential site of initial reactivity.21 The resulting Ru-carbenoid is then poised to undergo a facile 6-membered ring closure, resulting in undesired RCM product 29.
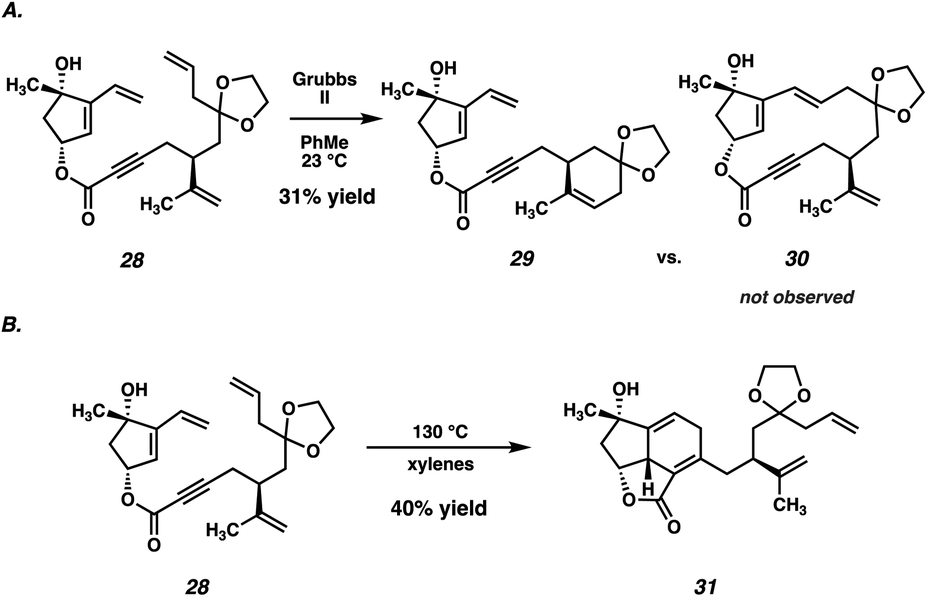 | ||
| Scheme 4 (A) Failed RCM macrocyclization. (B) Diels–Alder of ester 28. | ||
Although disappointed in the failure of our key macrocyclization, we recognized that access to 28 still represented an important milestone in the synthesis, and we proceeded to probe its reactivity further. Specifically, we viewed this as an opportunity to test whether an intramolecular Diels–Alder reaction of a compound such as 28 would be feasible. To that end, heating of ester 28 to 130 °C in xylenes resulted in a facile Diels–Alder cycloaddition, delivering tricycle 31 in 40% yield as a single diastereomer (Scheme 4B). Although adduct 31 would not serve as a viable intermediate in our total synthesis given the incorrect substitution on the carvone-derived tether, the success of this reaction served to validate our hypothesis that the central six-membered ring of scabrolide A (1) could be formed by an intramolecular Diels–Alder reaction. Thus, we opted to revise our retrosynthetic strategy around this significant result.
Second-generation approach
Our second-generation retrosynthetic analysis (Fig. 3) relied upon a late-stage closure of the eastern seven-membered carbocycle of scabrolide A (1) from a precursor such as enone 32. We envisioned access to 32 from Diels–Alder adduct 33, which, in turn, could be furnished from ester 34. Ester 34 would be prepared by a similar convergent esterification of dihydroxyvinylcyclopentene 15 and a new ynoic acid, 35, appended with a methyl ketone.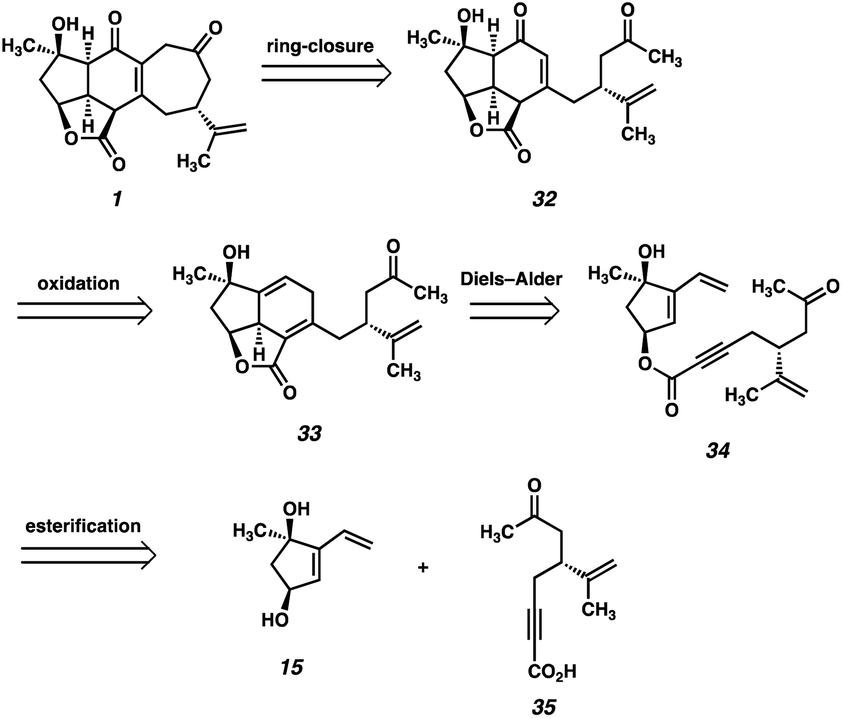 | ||
| Fig. 3 Second-generation retrosynthetic analysis of scabrolide A (1). | ||
At this point, we also became interested in a more expedient route to dihydroxyvinylcyclopentene 15. Inspired by a report from Maimone and coworkers detailing the synthesis of enone 37 (Scheme 5) in two steps from (R)-linalool (36),22 we hypothesized that we could exploit this building block for the preparation of 15. Our second-generation synthesis of this fragment commences with a conjugate addition of vinyl cuprate to enone 37, and, following a dehydrogenation protocol, vinyl enone 40 is furnished in good yield. A diastereoselective Luche reduction then establishes the final stereocenter, delivering a TBS-protected variant of the desired fragment (41). Treatment with TBAF at elevated temperature then delivers dihydroxyvinylcyclopentene 15 in 20% yield over 7 steps (vs. 15), and in the correct enantiomeric series.
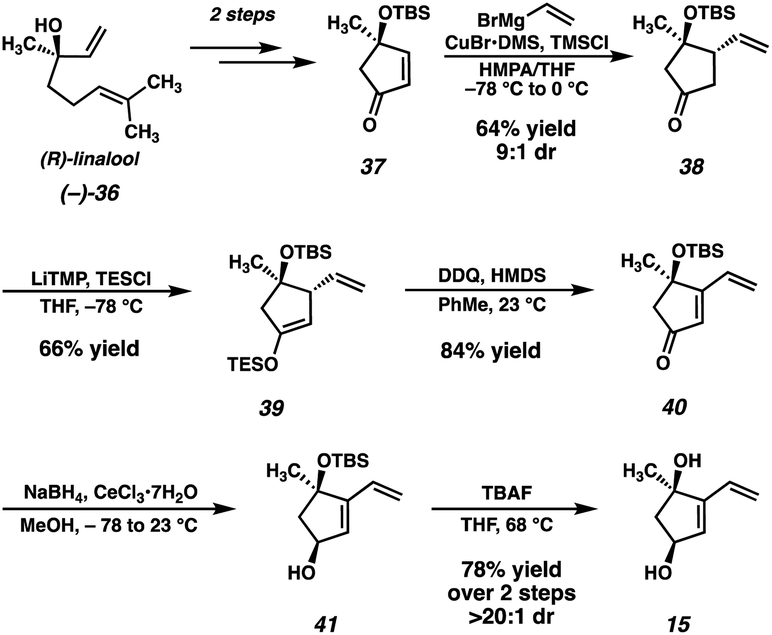 | ||
| Scheme 5 Second-generation synthesis of vinylcyclopentenone 15. | ||
The new ynoic acid coupling partner (35) is prepared from alkyne 25 (Scheme 6), intercepted from the route to the ynoic acid used previously (now starting from (R)-carvone to match the enantiomer of dihydroxyvinylcyclopentene 15). Monoaddition of methyl magnesium bromide installs the requisite methyl ketone in the form of intermediate 42. Pd-catalyzed oxidative carboxylation23 then affords methyl ester 43, which, following saponification, is coupled with TBS-protected dihydroxyvinylcyclopentene 41 under Steglich conditions24 to afford Diels–Alder precursor 44.
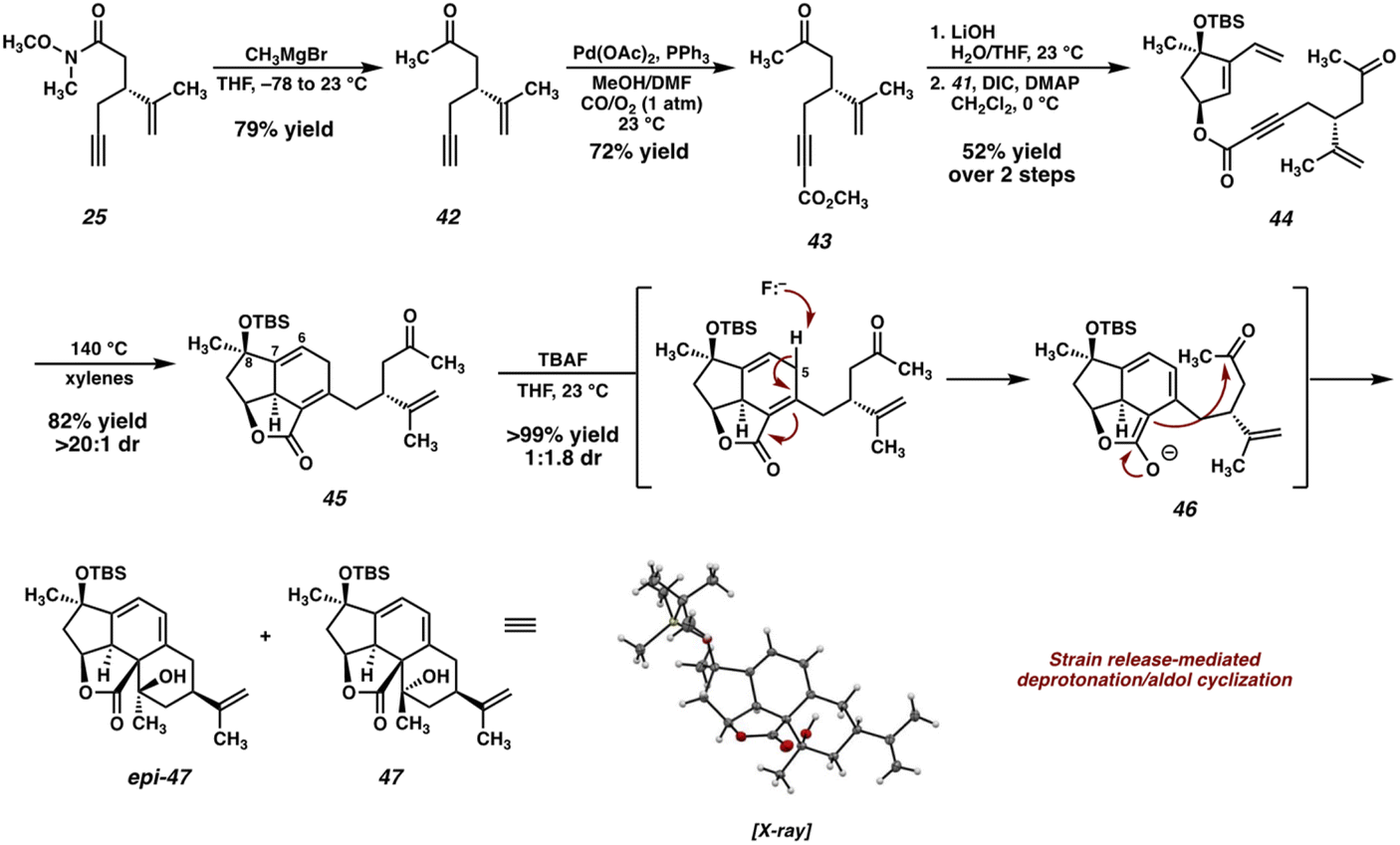 | ||
| Scheme 6 Synthesis of ester 44, Diels–Alder cycloaddition, and undesired aldol cyclization. | ||
As before, heating ester 44 to 140 °C in xylenes smoothly triggers the desired [4 + 2] cycloaddition, delivering tricycle 45 in good yield as a single diastereomer. At this stage, oxidation of the Δ6,7 olefin is required in order to install the C(6) ketone present in the natural product. Initially, we planned to accomplish this via a directed epoxidation and subsequent Meinwald rearrangement. To this end, we attempted the deprotection of the C(8) alcohol by treatment of 42 with TBAF, however, we were surprised to isolate tetracycles 47 and epi-47 instead of the anticipated deprotected alcohol. We hypothesize that these unexpected products are the result of an aldol cyclization between an extended enolate (46) generated by deprotonation of 45 at C(5) with TBAF. Although TBAF is typically far too weak of a base to deprotonate γ-protons of an α,β-unsaturated lactone, we reason that the strain imparted on tricycle 45 significantly reduces the pKa of these protons, thus facilitating deprotonation and subsequent cyclization. Interestingly, a compound possessing a ring system very similar to 47 was recently observed by Gaich and coworkers during their total synthesis of norcembrene 5 (a macrocyclic norcembranoid) resulting from an unexpected [4 + 2] cycloaddition.25 The authors propose that this ring system may correspond to a yet-undiscovered subclass of the polycyclic norcembranoid natural products.
In light of this undesired reactivity, we simply opted to utilize dihydroxyvinylcyclopentene 15 instead of its TBS-protected congener (Scheme 7). Not surprisingly, the Diels–Alder reaction of ester 48 proceeds smoothly, delivering tricycle 49 in good yield as a single diastereomer. The now free hydroxyl group can then direct a VO(acac)2-catalyzed epoxidation, which furnishes epoxide 50 in excellent yield. Unfortunately, the planned Meinwald rearrangement failed under a variety of Lewis acid-mediated conditions forcing us to settle on a two-step approach to enone 32.26 Performing a Ti-mediated reductive opening27 of epoxide 50 furnishes diol 51. However, despite surveying a wide variety of oxidation conditions, ketone 52 (or enone 32) was never isolated. Instead, decomposition, presumably through the aforementioned aldol pathway, was observed under several oxidation conditions (i.e., Swern, DMP, Cr(VI)), while other conditions (i.e., TPAP/NMO, CuOTf/ABNO/O2)28,29 failed to induce any reactivity whatsoever. The failure of this oxidation ultimately thwarted the advancement of intermediate 51 to enone 32.
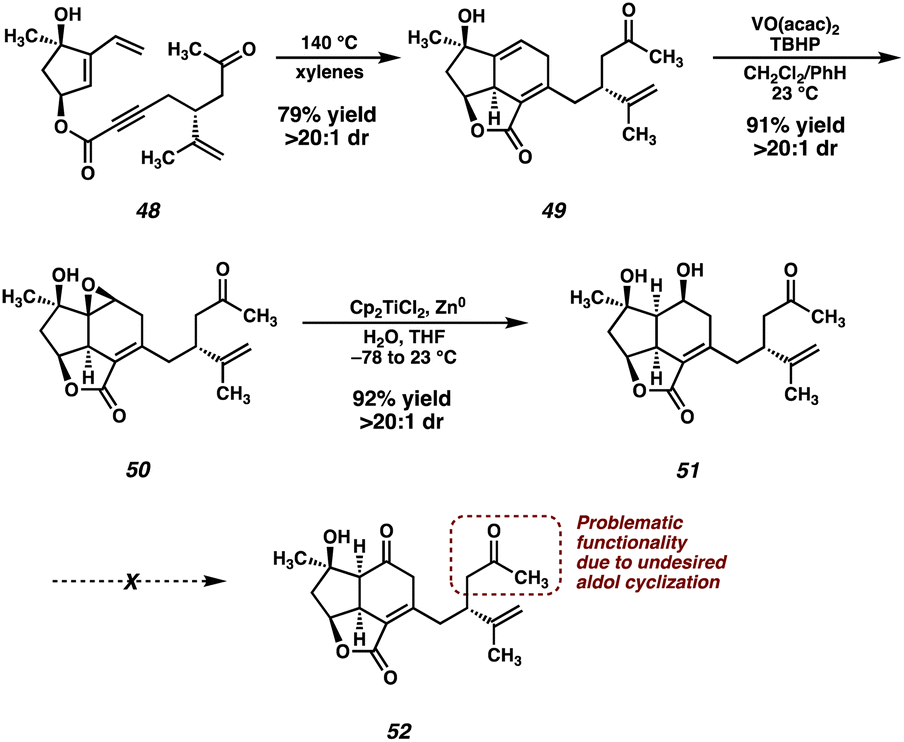 | ||
| Scheme 7 Diels–Alder reaction of 48, directed epoxidation, reductive epoxide opening and failed oxidation of alcohol 51. | ||
Photocycloaddition strategy: model studies
Although surprised by the difficulty of this transformation, we immediately identified the pendant ketone as the problematic functionality in diol 51, which was hampering the advancement of this intermediate further in the synthesis. As such, we wondered whether the replacement of the ketone with a non-electrophilic surrogate might mitigate the undesired reactivity and facilitate the completion of the synthesis. Identifying a vinyl silane as a suitable choice, which would be converted to the ketone via a Tamao–Fleming30 oxidation at a later point in the synthesis, we redesigned our route once again. When considering the proposed intermediate (i.e., 55) we identified the vinyl silane/enone system as a well suited substrate for a [2 + 2] photocycloaddition,31 which might prove successful in forging the final C–C bond (C(4)–C(5)) of the natural product. With these facts in mind, we ultimately developed the retrosynthetic plan outlined in Fig. 4 as a suitable path forward.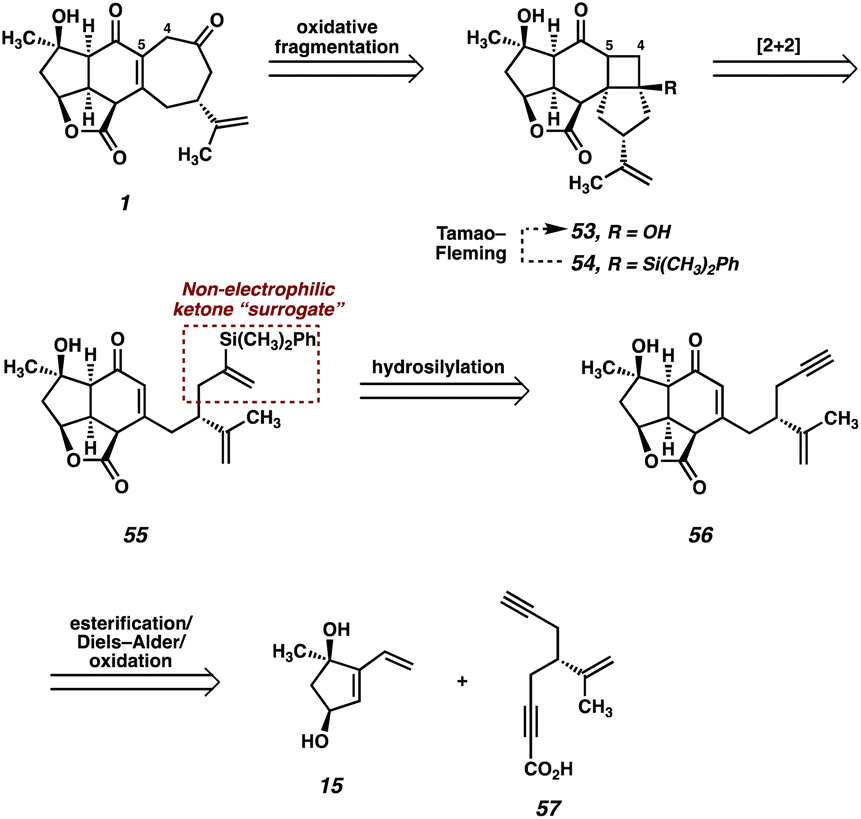 | ||
| Fig. 4 Third-generation retrosynthetic analysis of scabrolide A (1). | ||
In this approach the seven-membered carbocycle would be introduced at a late stage via an oxidative fragmentation32 of a fused [4-5] ring system such as 53, which would be accessed by Tamao–Fleming oxidation of tertiary silane 54. Pentacycle 54 was envisioned as the product of an intramolecular enone–olefin photocycloaddtion, retrosynthetically delivering vinyl silane 55, which would be accessed via a hydrosilylation of alkyne 56. Notably, we hoped that replacement of the pendant methyl ketone present in prior intermediates with a relatively inert alkyne might allow access to the previously intractable enone (in this case 56) by suppression of problematic decomposition pathways (i.e., aldol). In order to first test this new strategy, we opted to utilize a simplified system which omits the isopropenyl group. Removal of this distal stereocenter obviates the need for an enantiospecific synthesis of the carboxylic acid coupling partner in the esterification reaction, which allows expedient access to this fragment. We reasoned that, once a route to the core of the natural product was established, we would then go back and modify the route to include the isopropenyl group from an early stage.
In the event, known ester 58,33 available in one step from commercially available 1,6-heptadiyne, is saponified and coupled with dihydroxyvinylcyclopentene 15 (Scheme 8). The previously established Diels–Alder/epoxidation/reductive epoxide opening sequence then delivers diol 60. With the problematic ketone now replaced with a relatively inert terminal alkyne, the oxidation of diol 60 proceeds under the action of DMP, delivering enone 61 after migration of the olefin into conjugation with the newly formed ketone. However, the outcome of this transformation proved to be highly variable, with a maximum yield of 47%, although yields as low as 20% were also observed. Specifically, this oxidation tended to proceed very slowly, and often completely stalled even after addition of excess DMP and heating. Despite this irreproducibility, we were still able to access sufficient quantities of crucial intermediate 61 to explore the feasibility of the remainder of our new strategy. To this end, a Ru-catalyzed hydrosilylation34 delivers vinyl silane 62, setting the stage for the exploration of the crucial [2 + 2] photocycloaddition.
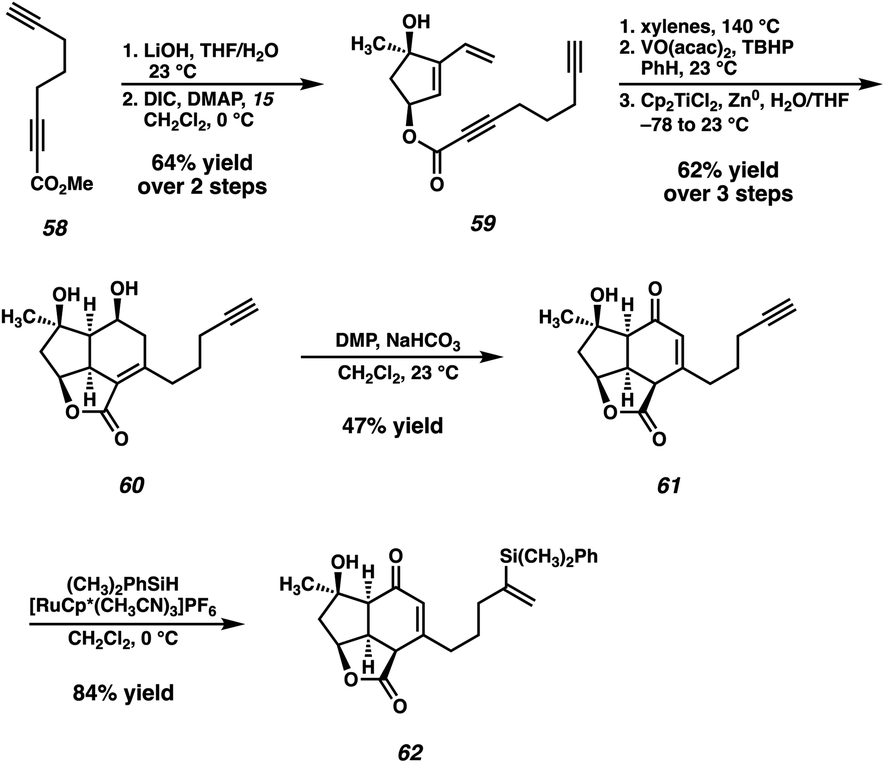 | ||
| Scheme 8 Synthesis of simplified [2 + 2] substrate 62. | ||
To our delight, irradiation of vinyl silane 62 at 350 nm in benzene smoothly initiates the enone–olefin cycloaddition, delivering cyclobutanes 63 and 64 as a separable, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers, which were characterized unambiguously by X-ray diffraction (Scheme 9). Notably, diastereomer 63, the major product of this reaction, displays trans fusion at the [6-4] ring juncture, which is an uncommon stereochemical outcome of [2 + 2] photocycloadditions.35 Additionally, both products 63 and 64 maintain conserved stereochemistry at C(5). Based upon these results, we can infer the mechanistic scenario outlined in Scheme 9.36
:1 mixture of diastereomers, which were characterized unambiguously by X-ray diffraction (Scheme 9). Notably, diastereomer 63, the major product of this reaction, displays trans fusion at the [6-4] ring juncture, which is an uncommon stereochemical outcome of [2 + 2] photocycloadditions.35 Additionally, both products 63 and 64 maintain conserved stereochemistry at C(5). Based upon these results, we can infer the mechanistic scenario outlined in Scheme 9.36
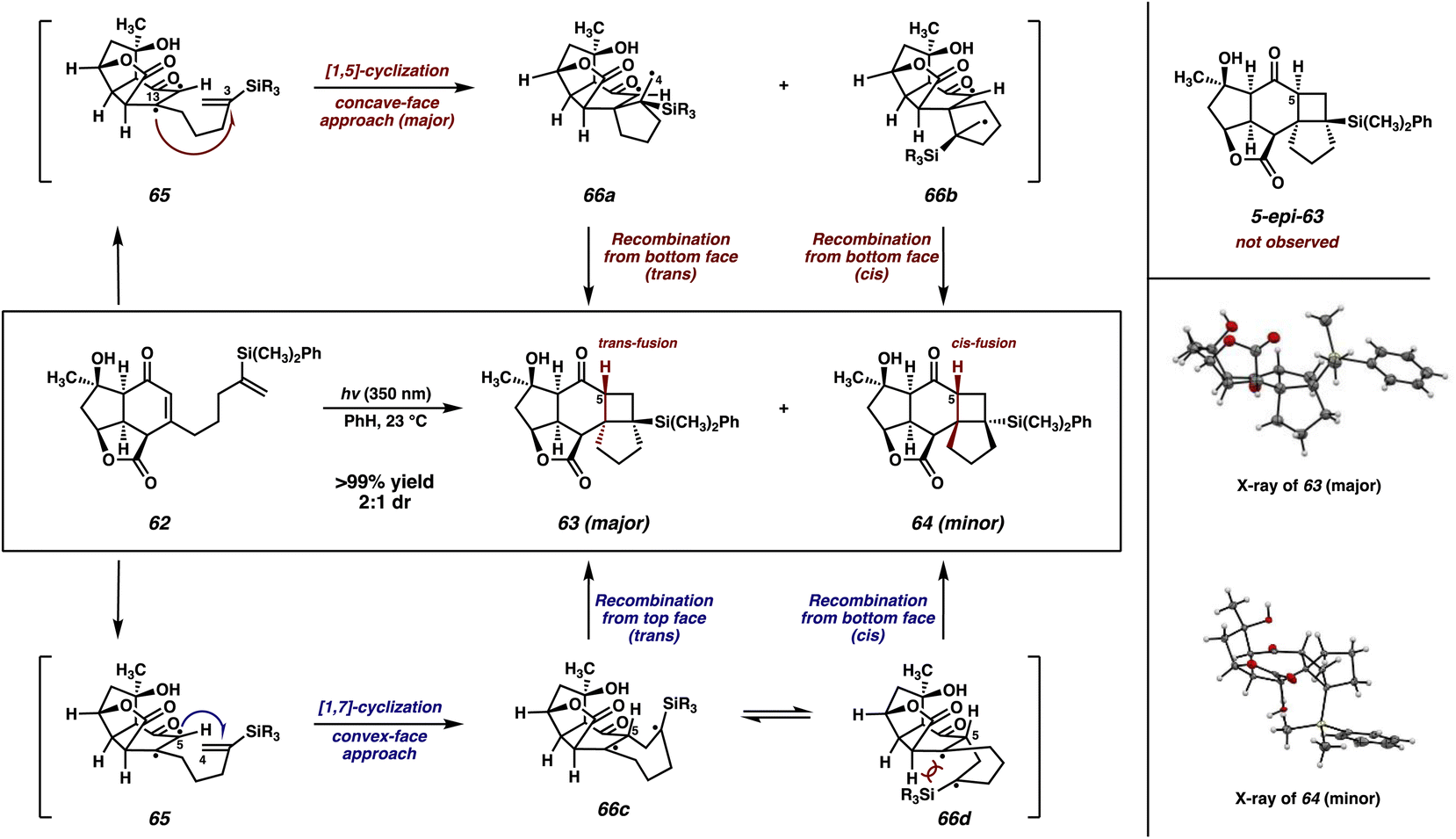 | ||
| Scheme 9 [2 + 2] photocycloaddition of enone 62. | ||
Initially, photoexcited enone 65 can undergo either a 1,5-cyclization (Scheme 9, top pathway, red) or a 1,7-cyclization (bottom pathway, blue). Considering first the top pathway, this initial 1,5-cyclization between C(3) and C(13) would need to proceed with a dr of 2:1, favoring attack from the concave face of the enone, to align with the experimentally observed result. Subsequently, diradicals 66a and 66b would be formed. Minor product 64 could form in a straightforward fashion via radical recombination from the same face as the initial attack to form the cis-fused cyclobutane. However, in order to generate major product 63, diradical 66a, already seemingly poised to undergo recombination from the concave face to form 5-epi-63 (not observed), would instead need to undergo an unlikely conformational shift in order for the C(4)–C(5) bond to be formed from the opposite, convex face.
Alternatively, photoexcited enone 65 could instead undergo a 1,7-cyclization between C(4) and C(5). If this proceeded with high diastereoselectivity from the convex face, as could be reasonably expected, it would explain the conservation of stereochemistry at C(5) between the two observed photocycloadducts (63 and 64). This 1,7-cyclization would lead to the formation of a 1,4-diradical, which could then undergo radical recombination from either the concave (66c) or convex (66d) face of the molecule. Inspection of the latter reveals a steric interaction between the bulky silyl group and the underside of the cyclohexanone ring. Thus, preferential (albeit not exclusive) recombination occurs from the concave face leading to the formation of trans-fused cyclobutane 63 as the major product.
Although it is typically understood in intramolecular enone–olefin photocycloadditions that triplet-state enones preferentially form 5-membered rings when possible,37 exceptions to this tendency do exist, and the prevailing mechanism in each case is believed to be highly substrate-dependent. Specifically, cases in which the reacting ground-state olefin is substituted at the internal position (e.g., 1,1-disubstituted olefins) this type of reactivity is often observed.38 This is presumably due to steric repulsion between the enone and the fully substituted olefinic carbon, and in the case of a bulky Ph(CH3)2Si substituent, it would be expected that this steric effect would be even more pronounced. Thus, we propose the mechanism involving 1,7-cyclization followed by 1,4-radical recombination for this particular transformation. Evidence for this type of mechanism operating in our cycloaddition includes the preferential formation of the trans-fused cyclobutane, as well as the conservation of stereochemistry at C(5) between both the trans- and cis-fused cyclobutane products. However, we do not have any definitive evidence to rule out the alternative pathway.
Gratified that our simplified system successfully undergoes the key [2 + 2] photocycloaddition, we set out to explore the remainder of our new strategy to access the carbocyclic core of scabrolide A. The next task would be the Tamao–Fleming oxidation, which would convert the tertiary silane to an oxygen atom that would eventually become the C(3) ketone of the natural product. To our delight, trans-fused silylated cyclobutane 63 readily undergoes Hg-mediated Tamao–Fleming oxidation, affording tertiary alcohol 67 in 30% yield (Scheme 10). Interestingly, the cis-fused diastereomer 64 fails to engage in the same reaction either in the presence of Hg(OAc)2 or more reactive Hg(TFA)2,39 returning only starting material. This may be due to the inaccessibility of the tertiary silane in 64, which is shielded by the bulk of the molecule, preventing access to activation/nucleophilic attack. Efforts to perform this reaction at elevated temperatures simply result in the nonspecific decomposition of silane 64. Fortuitously, the reactive diastereomer happens to be the major product of the photocycloaddition, and thus material throughput is not significantly hampered by this unexpected divergence in reactivity.
 | ||
| Scheme 10 Tamao–Fleming oxidation of 63 and failed Tamao–Fleming oxidation of 64. | ||
With cyclobutanol 67 in hand, we were presented with the opportunity to examine the final phase of our strategy on this model system. Specifically, an oxidative fragmentation of the cyclobutanol moiety present in intermediate 67 to generate the cycloheptenone was all that remained for the synthesis of the carbocyclic framework of scabrolide A (1).
We were intrigued by a report from Qi and Zhang40 which details the Ag-catalyzed oxidative fragmentation of a variety of cyclobutanol substrates to the corresponding γ-chloroketones. When we subjected model cyclobutanol 67 to these conditions, we were initially pleased to observe signs of selective reactivity. Upon purification, however, we were surprised to isolate pentacycle 72 duct of this reaction (Scheme 11), rather than the expected γ-chloroketone or enone. We believe that 72, the structure of which was determined unambiguously by X-ray diffraction, arises from chlorohydrin formation/oxa-Michael addition of enone 71, after initial oxidative cyclobutanol fragmentation and chloride elimination. Although not the expected product, we were delighted to note that 72 is adorned with the requisite 7-membered carbocycle, and thus possesses the full carbocyclic framework of scabrolide A (1).
 | ||
| Scheme 11 Oxidative ring expansion of cyclobutanol 67 to ether 72. | ||
Photocycloaddition strategy: complete system
Given these results, we were confident that our newly designed strategy would be successful in delivering the natural product if we introduced the isopropenyl group at an early stage and carried it through the remainder of the synthesis. As such, we targeted ynoic acid 57 (Scheme 12) as a coupling partner for the convergent esterification. Returning to (R)-carvone ((−)-23) as our chiral-pool starting material, we designed a synthesis of the requisite fragment (57) starting from known monoprotected dialdehyde 73.41 This is accomplished using a series of two Corey–Fuchs homologations,42 which enables the sequential installation of both alkynyl fragments while maintaining the stereochemical fidelity of the isopropenyl stereogenic center. | ||
| Scheme 12 Synthesis of isopropenyl-bearing acid 57. | ||
 | ||
| Scheme 13 Esterification of ynoic acid 57 and Diels–Alder/oxidation sequence. | ||
With acid 57 in hand, the convergent esterification is performed between diol 15 and ynoic acid 57, and, following the intramolecular Diels–Alder reaction, tricycle 78 is obtained (Scheme 13). This adduct is then carried through the same epoxidation/reductive opening sequence, this time utilizing a catalytic quantity of Ti for the epoxide opening.43 At this stage, we recognized the need to address the challenging oxidation of diol 80, and turned our attention toward a model system for this optimization (Table 1).
|
|
|||
|---|---|---|---|
| Entry | Conditions | Substrate | Result |
| 1 | DMP, CH2Cl2, 23 °C | 81 and 60 | Inconsistent yield/incomplete conversion |
| 2 | PCC, CH2Cl2, 23 °C | 81 | 35% yield |
| 3 | TPAP, NMO, 3 Å MS, CH2Cl2, 23 °C | 81 | No reaction |
| 4 | Cu(CH3CN)4OTf, MeOppy, ABNO, NMI, CH3CN, O2, 23 °C | 81 | No reaction |
| 5 | TEMPO, PIDA, CH2Cl2, 23 °C | 81 | No reaction |
| 6 | IBX, DMSO, 23 °C | 81 | 43% yield |
| 7 | IBX, CH3CN, 80 °C | 81 | 71% yield |
| 8 | IBX, CH3CN, 50 °C | 80 | 72% yield |
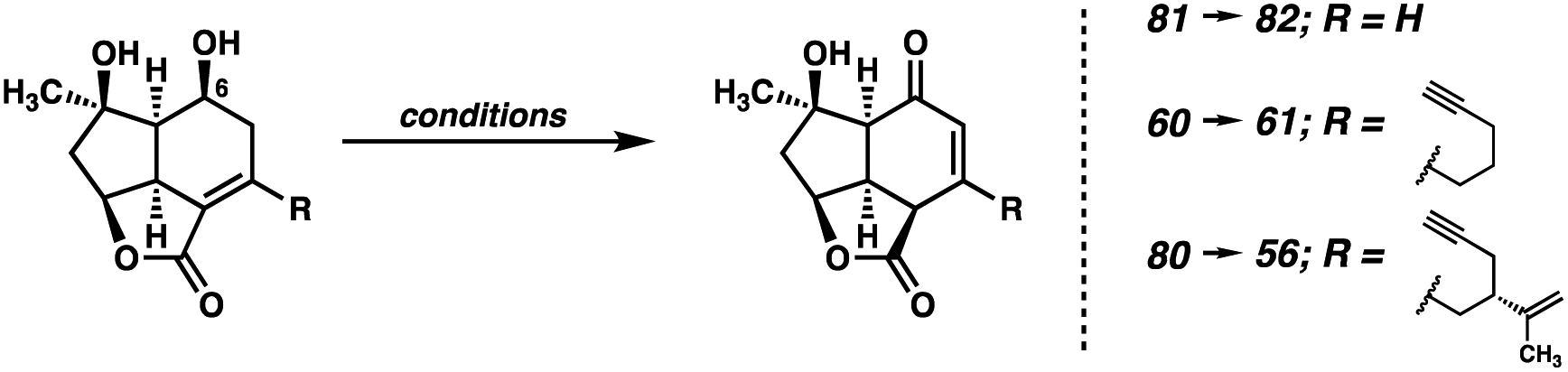
As noted earlier, buffered DMP could be employed for this oxidation in previous systems, however, these results were highly irreproducible. Specifically, the degree of conversion varied widely between batches both on the same and different scales. Initial experiments with Cr(VI) oxidants proved somewhat promising, inducing the desired reactivity, however large amounts of nonspecific decomposition were observed, and the yield was never optimized past 35% (entry 2). Interestingly, TPAP/NMO, Cu(OTf)/ABNO/O2, and TEMPO systems failed to produce any meaningful levels of reactivity, returning only starting material (entries 3–5). We were pleased to see oxidation when utilizing IBX in DMSO, however, in this solvent the formation of several unidentified byproducts was observed (entry 6). Gratifyingly, switching the solvent to MeCN and elevating the temperature suppressed the undesired side reactivity, delivering model enone 82 in 71% yield (entry 7). To our delight, after slight modification, this IBX/MeCN system proved effective on the full system, delivering enone 56 in 72% yield (entry 8).
Failed photocycloaddition and olefin protection
With a robust route to enone 56 now established, we were poised to examine the key [2 + 2] photocycloaddition with the now fully elaborated system (Scheme 14). Hydrosilylation of enone 56 delivers vinyl silane 55 in good yield, setting the stage for the key [2 + 2] photocycloaddition, this time on the fully elaborated system. To our dismay, however, upon irradiation of 55 at 350 nm in benzene, the expected fused [6-4-5] product (i.e., 54) was not observed. Instead, we isolated fused [6-4-4] product 83, the major product of this photocycloaddition, which arises from the undesired reaction between the enone and the isopropenyl olefin rather than the vinyl silane. While the presence of two olefins on our [2 + 2] substrate was always a concern, we reasoned that, due to the highly strained nature of 83, that the desired product 54 would likely be formed with at least some degree of selectivity. Consequently, we were astonished to discover that this transformation was exquisitely selective for the [6-4-4] fused system, with no signs of the isopropenyl vinyl proton signals observed in the crude 1H NMR. | ||
| Scheme 14 Photocyclization of fully elaborated substrate 55 to form undesired product 83. | ||
As outlined in Fig. 5, we believe that the observed regioselectivity arises from steric interactions imposed by the bulky Ph(CH3)2Si group. In the desired reactive conformation of photoexcited enone 84 (i.e., for 1,7-cyclization leading ultimately to 54), the large silyl group is required to come into close proximity with the tricyclic core of the intermediate (Fig. 5, left). We believe that this causes the favorability of a conformation such as 84′, where the silyl group is held at a distance, thus bringing the isopropenyl olefin into closer proximity to the triplet-state enone. Subsequent 1,6–cyclization44 results in the formation of a 1,4–diradical, which then undergoes radical recombination to the observed [6-4-4] product (83).
 | ||
| Fig. 5 Mechanistic rationale for the undesired photocycloaddition outcome. | ||
Given the overwhelming preference for the formation of 83 in the photocycloaddition, we reasoned that it would likely prove fruitless to attempt overturning selectivity for the desired product. We instead opted to utilize a protecting group strategy to overcome this undesired reactivity, where temporary “masking” of the isopropenyl olefin could allow for the correct regioselectivity in the [2 + 2]. In practice, this would require a functional group that could be easily installed on the olefin in question, and more importantly, could be removed at a later stage in the synthesis. After surveying several options, we decided that an epoxide could meet both of these criteria. To this end, treatment of enone 56 with m-CPBA delivers epoxide 85 as an inseparable mixture of epoxide epimers at C(15), which can then undergo hydrosilylation to furnish vinyl silane 86 (Scheme 15). To our delight, with the reactive isopropenyl group now suitably “protected” as the corresponding epoxide, this intermediate smoothly undergoes the [2 + 2] cycloaddition at the intended olefin, delivering the desired trans-fused [6-4-5] product 87 in 71% yield.
 | ||
| Scheme 15 Epoxidation of 56, successful [2 + 2] photocycloaddition of protected substrate 86, and failed Tamao–Fleming oxidation. | ||
Initial attempts at performing the Tamao–Fleming oxidation on silane 87 proved unsuccessful. Treatment with Hg(OAc)2/AcOOH, KBr/AcOOH, and HBF4/H2O2 all result in rapid decomposition, presumably due to the instability of the reactive epoxide under these strongly acidic conditions. Indeed, control experiments revealed that, even in the absence of activating agents, 87 quickly decomposes in solution with AcOOH/AcOH, rendering the desired Tamao–Fleming oxidation infeasible. This being the case, we chose to circumvent this issue by converting the problematic epoxide to the corresponding primary alcohol via a second Ti-catalyzed epoxide opening, which also serves as the first of two steps for the removal of the olefin protecting group (Scheme 16). Silane 89 and its C(15) epimer are obtained in excellent yield from this reaction, and are readily separable by column chromatography at this stage. Gratifyingly, we found that, in the absence of the epoxide, silane 89 smoothly undergoes a Tamao–Fleming oxidation to deliver cyclobutanol 90 (epi-89 is also competent in this transformation and can thus be carried through the remainder of the synthesis in an analogous fashion to produce epi-90).
 | ||
| Scheme 16 Reductive opening of epoxide 87, and successful Tamao–Fleming oxidation of 89. | ||
Oxidative fragmentation and attempted dehydration
With cyclobutanol 90 in hand, we were poised to investigate the critical fragmentation reaction on our fully-elaborated system. To our delight, under our previously employed Ag-catalyzed conditions, cyclobutanol 90 undergoes fragmentation to cycloheptenone 91 (Scheme 17). Presumably the formation of 91 proceeds through the intermediacy of the corresponding γ-chloroketone or a THF-bridged species analogous to 72 (an intermediate product can be observed and isolated that is unstable to chromatography and thus has eluded characterization). However, this intermediate undergoes elimination of chloride during purification by preparative TLC, or stirring in a suspension of TLC-grade SiO2 in MeOH, to deliver the desired enone (91) directly. With this key transformation complete and the carbocyclic core of the natural product now established on a fully elaborated system, the final remaining task was the elimination of the C(16) alcohol to regenerate the isopropenyl group and complete the total synthesis.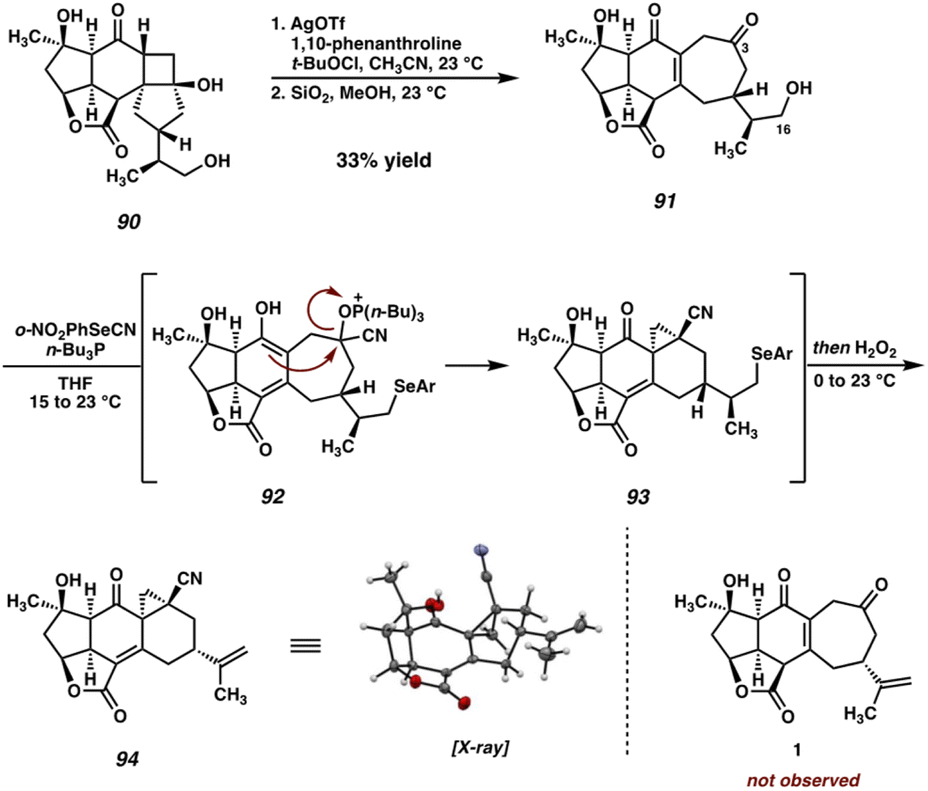 | ||
| Scheme 17 Oxidative ring-expansion of cyclobutanol 90 and unexpected cyclization of 91 during Grieco dehydration. | ||
Given its frequent use in total synthesis, particularly in late-stage, complex settings, we turned to the venerable Grieco elimination45 for this critical step (Scheme 17). To our surprise, however, exposure of alcohol 91 to the canonical Grieco conditions did not afford any trace of scabrolide A (1). After several attempts, we were able to isolate and characterize pentacycle 94 as the primary product of this reaction.
While initially puzzling, we recognized that this product might arise if, under the reaction conditions, a cyanohydrin is formed at the C(3) ketone by HCN generated during the Grieco reaction. Activation of the cyanohydrin hydroxyl group by tetrabutyl phosphonium selenide (an intermediate in the Grieco reaction) could precede cyclization either via a cationic intermediate or by direct displacement to close the three-membered ring. Following the usual oxidation/elimination of the selenide at C(16), pentacycle 94 would be formed. Upon closer analysis (LCMS) no trace of the diketone-selenide is detected during the course of the reaction, suggesting that cyanohydrin formation is faster than alcohol displacement under these conditions, and thus that this undesired cyclization would be difficult to suppress. In light of this unexpected result, we turned our attention to alternative alcohol elimination protocols (Scheme 18). Mesylate 95 is readily formed using standard conditions, however, all attempts at performing the final elimination proved fruitless. Instead, nonspecific decomposition was observed in all cases, presumably a consequence of the base sensitivity of the substrate. Alternatively, thiocarbamate 96 could be prepared, which has been shown to undergo Chugaev-type eliminations at elevated temperatures.46 In our hands, however, this intermediate failed to undergo the required elimination, instead decomposing when heated to high temperatures.
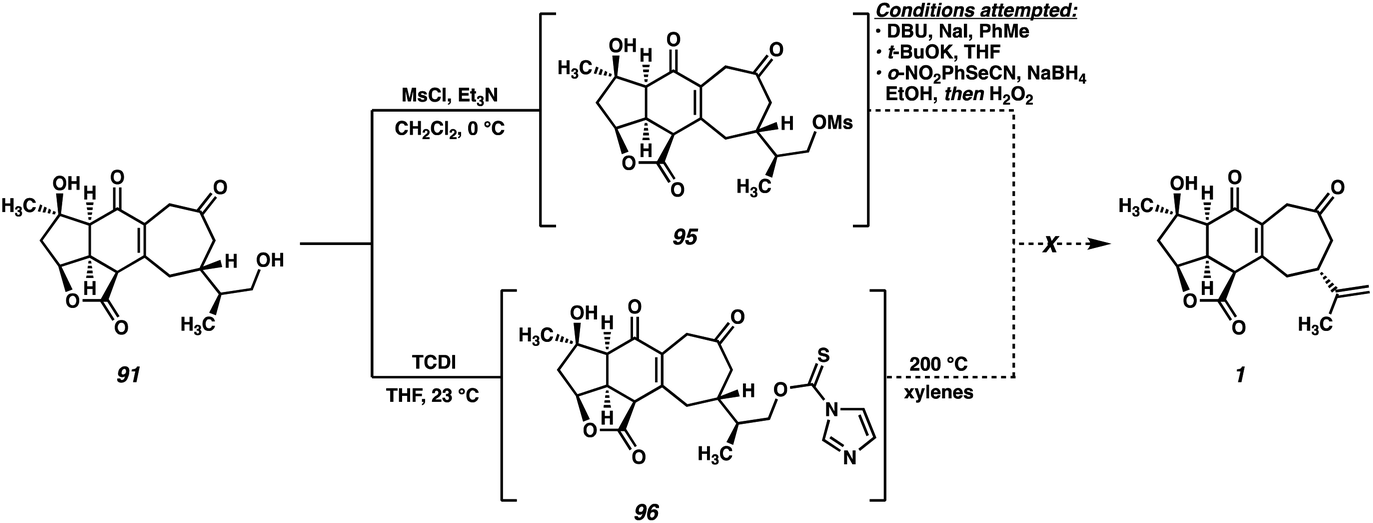 | ||
| Scheme 18 Failed dehydration attempts of alcohol 91. | ||
Upon examining intermediate 91, the presence of the relatively reactive vinylogous β-ketoester is evident (Fig. 6, red), which we hypothesize may be responsible for the undesired reactivity observed in this system. Indeed, armed with the knowledge that, under the Grieco conditions, this system does have the capability of undergoing transannular enolate chemistry across the 7-membered ring, we reasoned that the presence of this reactive functional group might be hindering our efforts toward elimination of the primary alcohol at C(16). With this in mind, we concluded that an intermediate in which the vinylogous relationship between the C(6) and C(19) carbonyls had not yet been established might be better suited for this elimination reaction. As such, we identified triol 90 as a suitable substrate for the key dehydration.
 | ||
| Fig. 6 Rationale for failure of dehydration attempts of enone 91, and switching to a different substrate (90). | ||
Completion of the total synthesis
To our delight, both 90 and its C(15) epimer smoothly undergo dehydration under Grieco conditions to deliver cyclobutanol 53 in good yield, converging these two diastereomeric intermediates to a single product (Scheme 19). With the isopropenyl group effectively deprotected, all that remained at this stage was the oxidative fragmentation of the cyclobutanol to generate the cycloheptenone and complete the synthesis. However, with the isopropenyl olefin now fully unmasked, this transformation was complicated by the potential for undesired oxidation of the newly revealed olefin. Indeed, exposure of cyclobutanol 53 to the Ag-catalyzed conditions previously established for this ring expansion generated a complex mixture of polychlorinated products (detected by LCMS). This can be attributed to the strongly oxidizing t-BuOCl, which is known to chlorinate both olefins and allylic systems in many cases.47 | ||
| Scheme 19 Successful Grieco dehydration of triol 90, NIS-mediated ring-expansion of cyclobutanol 53 to complete the total synthesis of scabrolide A (1), and dehydration of scabrolide A (1) to yonarolide (4). | ||
In search of milder conditions for the oxidative ring expansion, we turned our attention to previously reported protocols for this type of transformation that employ weaker oxidants. To our delight, we found that the NIS/CuI-mediated protocol reported by Takasu and Ihara48 proved to be selective for the desired transformation, furnishing scabrolide A (1) in 61% yield after oxidative ring-expansion of the cyclobutanol (Scheme 19).
Under these conditions, it is proposed that reaction between the tertiary cyclobutanol (53) and NIS generates a hypoiodite (97), which then undergoes thermal homolysis to generate an O-centered radical. This high-energy intermediate then undergoes a β-scission of the C(3)–C(13) bond with subsequent recombination of the resultant tertiary C-centered radical (98) with iodine. The resultant iodide (99), situated at the β-position of both the C(6) ketone and the C(19) lactone, then undergoes facile elimination spontaneously under the reaction conditions to deliver scabrolide A (1). We were also able to convert scabrolide A (1) to a closely related natural product, yonarolide (4), after dehydration with Burgess reagent. This research represents the first total synthesis of either of these two natural products.8,9
Conclusions
Disclosed herein is the full account of our total synthesis of (−)-scabrolide A (1) and yonarolide (4). An initial bioinspired macrocyclization/transannular cycloaddition strategy was thwarted by a failed RCM, but revealed a possible sequential ring-forming strategy which ultimately proved successful. As such, an approach consisting of convergent esterification of two chiral pool-derived fragments followed by West-to-East ring formation was designed. This involved an intramolecular [4 + 2] cycloaddition for the construction of the central 6-membered ring of the natural product, followed by a [2 + 2] cycloaddition/cyclobutane fragmentation sequence to generate the 7-membered ring. This strategy was initially realized in a des-isopropenyl model system. Translation to the full system revealed an unexpected regioselectivity issue in the [2 + 2] step, which was overcome using an unconventional olefin-protection strategy (i.e., masking of the olefin as an epoxide). Finally, efforts to perform a late-stage elimination of a primary alcohol revealed unexpected and unprecedented reactivity, which was overcome by a slight modification in the synthetic sequence. Our route provided access to both scabrolide A (1) as well as closely related yonarolide (4), neither of which had been previously prepared by total synthesis.Since the publication of our initial report in 2020, several key advancements have been made in both the total synthesis and isolation/characterization of the polycyclic furanobutenolide-derived norcembranoids. As mentioned in the introduction, the past three years have seen several elegant syntheses of both the Sinularia-derived polycyclic cembranoid and norcembranoid natural products.9–12,49 In addition to total syntheses, a number of new members of the polycyclic furanobutenolide-derived norcembranoid family have been isolated1d,e and characterized (including the structural reassignment of flagship member scabrolide B). Despite the fact that these molecules have been known for decades, the recent explosion in total syntheses and the continuing isolation of new members indicates that this class of natural products remains a relevant and exciting area of research.
Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compounds 1, 47, 63, 64, 72, 83, 90, epi-90, and 94 have been deposited in the Cambridge Crystallographic Data Center under accession numbers CCDC 1988002 (1), 2239956 (47), 2239963 (63), 2239959 (64), 2239957 (72), 1987997 (83), 1987989 (90), 1987999 (epi)-90 and 2239966 (94).Author contributions
N. J. H., S. A. L., C. E. R., B. P. P., and S. C. V. conceived of and performed experiments. B. M. S. supported and supervised the research. N. J. H. wrote the original draft of the manuscript, which was edited by all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors wish to thank NSF (CHE1800511), NIH (R35GM 145239), and the Heritage Medical Research Investigators Program for funding. Additionally, B. P. P. thanks NSF for support in the form of a predoctoral fellowship. The authors thank Dr David VanderVelde for NMR assistance and maintenance of the Caltech NMR facility, Dr Michael Takase and the Caltech XRD facility for XRD assistance, and Dr Mona Shahgholi for mass spectrometry assistance. The authors thank the Beckman Institute for their support of the Caltech XRD facility as well as the Dow Next Generation Instrument Grant.Notes and references
- For reviews of the polycyclic norcembranoids see: (a) Y. Li and G. Pattenden, Nat. Prod. Rep., 2011, 28, 429–440 RSC; (b) Y. Li and G. Pattenden, Nat. Prod. Rep., 2011, 28, 1269–1310 RSC; (c) R. A. Craig II and B. M. Stoltz, Chem. Rev., 2017, 117, 7878–7909 CrossRef PubMed . At the commencement of this study, there were nine known members of this natural product family. For more recently isolated and structurally characterized furanobutenolide-derived norcembranoids see: ; (d) J. Liu, J. Huang, T. Li, H. Ouyang, W.-H. Lin, X.-J. Yan, X. Yan and S. He, J. Org. Chem., 2022, 87, 9806–9814 CrossRef CAS PubMed; (e) Y. Du, L. Yao, X. Li and Y. Guo, Chin. Chem. Lett., 2023, 34, 107512 CrossRef CAS.
- For synthetic efforts toward ineleganolide see: (a) F. Tang and K. D. Moeller, Tetrahedron, 2009, 65, 10863–10875 CrossRef CAS; (b) K. D. Moeller, Synlett, 2009, 2009, 1208–1218 CrossRef; (c) F. Tang and K. D. Moeller, J. Am. Chem. Soc., 2007, 129, 12414–12415 CrossRef CAS PubMed; (d) E. J. Horn and C. D. Vanderwal, J. Org. Chem., 2016, 81, 1819–1838 CrossRef CAS PubMed; (e) R. A. Craig II, J. L. Roizen, R. C. Smith, A. C. Jones, S. C. Virgil and B. M. Stoltz, Chem. Sci., 2017, 8, 507–514 RSC.
- For synthetic efforts toward yonarolide see: Y. Ueda, H. Abe, K. Iguchi and H. Ito, Tetrahedron Lett., 2011, 52, 3379–3381 CrossRef CAS.
- Y. Li and G. Pattenden, Tetrahedron, 2011, 67, 10045–10052 CrossRef CAS.
- (a) M. Deng, X. Zhang, Z. Li, H. Chen, S. Zang and G. Liang, Org. Lett., 2019, 21, 1493–1496 CrossRef CAS PubMed; (b) N. J. Truax, S. Ayinde, K. Van, J. O. Liu and D. Romo, Org. Lett., 2019, 21, 7394–7399 CrossRef CAS PubMed.
- R. L. Beingessner, J. A. Farand and L. Barriault, J. Org. Chem., 2010, 75, 6337–6346 CrossRef CAS PubMed.
- G. Mehta and R. S. Kumaran, Tetrahedron Lett., 2001, 42, 8097–8100 CrossRef CAS.
- N. J. Hafeman, S. A. Loskot, C. E. Reimann, B. P. Pritchett, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2020, 142, 8585–8590 CrossRef CAS PubMed.
- Z. Meng and A. Fürstner, J. Am. Chem. Soc., 2022, 144, 1528–1533 CrossRef CAS PubMed.
- J. P. Tuccinardi and J. L. Wood, J. Am. Chem. Soc., 2022, 144, 20539–20547 CrossRef CAS PubMed.
- N. J. Truax, S. Ayinde, J. O. Liu and D. Romo, J. Am. Chem. Soc., 2022, 144, 18575–18585 CrossRef CAS PubMed.
- N. J. Hafeman, M. Chan, T. J. Fulton, E. J. Alexy, S. A. Loskot, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2022, 144, 20232–20236 CrossRef CAS PubMed.
- For a review of ring-closing metathesis in the context of macrocyclization see: A. Gradillas and J. Pérez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086–6101 CrossRef CAS PubMed.
- R. A. Craig II, J. L. Roizen, R. C. Smith, A. C. Jones and B. M. Stoltz, Org. Lett., 2012, 14, 5716–5719 CrossRef PubMed.
- C.-Y. Duh, S.-K. Wang, M.-C. Chia and M. Y. Chiang, Tetrahedron Lett., 1999, 40, 6033–6035 CrossRef CAS.
- For the isolation of scabrolide a see: (a) J. Sheu, A. F. Ahmed, R. Shiue, C. Dai and Y. Kuo, J. Nat. Prod., 2002, 65, 1904–1908 CrossRef CAS PubMed; (b) For the isolation of yonarolide see: ; (c) K. Iguchi, K. Kajiyama and Y. Yamada, Tetrahedron Lett., 1995, 36, 8807–8808 CrossRef CAS.
- In their report of the isolation of sinulochmodin C, the authors determine the absolute stereochemistry of two macrocyclic norcembranoids coisolated and report the absolute stereochemistry of sinulochmodin C, and the other polycyclic furanobutenolide-derived norcembranoids by analogy; see: (a) Y.-J. Tseng, A. F. Ahmend, C.-F. Dai, M. Y. Chiang and J.-H. Sheu, Org. Lett., 2005, 7, 3813–3816 CrossRef CAS PubMed; (b) The absolute stereochemistry of ineleganolide was very recently determined unambiguously by X-ray diffraction, and, in the same study, the absolute stereochemistry of scabrolide A was determined by ECD methods; see: W.-X. Cui, M. Yang, H. Li, S.-W. Li, L.-G. Yao, G. Li, W. Tang, C.-H. Wang, L.-F. Liang and Y.-W. Guo, Bioorg. Chem., 2020, 94, 103350 CrossRef CAS PubMed; (c) Furthermore, this research serves as confirmation of the absolute stereochemistry of scabrolide A..
- M. A. González, S. Ghosh, F. Rivas, D. Fischer and E. A. Theodorakis, Tetrahedron Lett., 2004, 45, 5039–5041 CrossRef.
- (a) S. Ohira, Synth. Commun., 1989, 19, 561–564 CrossRef CAS; (b) S. Müller, B. Liepold, G. J. Roth and H. J. Bestmann, Synlett, 1996, 521–522 CrossRef.
- J. Inanaga, K. Hirata, S. Hiroko, K. Tsutomu and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- (a) Z. G. Brill, H. K. Grover and T. J. Maimone, Science, 2016, 352, 1078–1082 CrossRef CAS PubMed; (b) D. Q. Thach, Z. G. Brill, H. K. Grover, K. V. Esguerra, J. K. Thompson and T. J. Maimone, Angew. Chem., Int. Ed., 2020, 59, 1532–1536 CrossRef CAS PubMed.
- Y. Izawa, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2004, 77, 2033–2045 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- M. Breunig, P. Yuan and T. Gaich, Angew. Chem., Int. Ed., 2020, 59, 5521–5525 CrossRef CAS PubMed.
- A similar epoxidation/Meinwald rearrangement strategy was attempted by our laboratory on the ineleganolide scaffold but also failed, requiring a reductive opening/oxidation sequence to prepare the analogous ketone. See ref. 2e for details.
- (a) T. V. RajanBabu, W. A. Nugent and M. S. Beattie, J. Am. Chem. Soc., 1990, 112, 6408–6409 CrossRef CAS; (b) T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997 CrossRef CAS.
- W. P. Griffith, S. V. Ley, G. P. Whitcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 21, 1625–1627 RSC.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- (a) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29–31 RSC; (b) I. Fleming and P. E. J. Sanderson, Tetrahedron Lett., 1987, 28, 4229–4232 CrossRef CAS.
- For reviews of [2 + 2] photocycloadditions see: (a) M. T. Crimmins, Chem. Rev., 1988, 88, 1453–1473 CrossRef CAS; (b) D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 2020, 1310–1326 CrossRef CAS; (c) N. Hoffman, Chem. Rev., 2008, 108, 1052–1103 CrossRef PubMed; (d) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed.
- For reviews of cyclobutane fragmentation strategies in synthesis see: (a) W. Oppolzer, Acc. Chem. Res., 1982, 15, 135–141 CrossRef CAS; (b) J. D. Winkler, C. M. Bowen and F. Liotta, Chem. Rev., 1995, 95, 2003–2020 CrossRef CAS.
- E. Piers, T. Wong and K. A. Ellis, Can. J. Chem., 1992, 70, 2058–2064 CrossRef CAS.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644–17655 CrossRef CAS PubMed.
- M. T. Crimmins and T. L. Reinhold, Enone Olefin [2+2] Photochemical Cycloadditions, in Organic Reactions, 1993, vol 44 Search PubMed; L. A. Paquette, in Organic Reactions, Wiley: Hoboken, NJ, 1993, pp. 297–599 Search PubMed.
- For a computational investigation of this mechanism, see: T. Zhang, A. Q. Cusumano, N. J. Hafeman, S. A. Loskot, C. E. Reimann, S. C. Virgil, W. A. Goddard III and B. M. Stoltz, J. Org. Chem., 2022, 87, 14115–14124 CrossRef CAS PubMed.
- R. Srinivasan and K. H. Carlough, J. Am. Chem. Soc., 1967, 89, 4932–4936 CrossRef CAS.
- For an example of this type of diastereoselectivity, see: (a) M. C. Pirrung, J. Am. Chem. Soc., 1979, 101, 7130–7131 CrossRef CAS; (b) M. C. Pirrung, J. Am. Chem. Soc., 1981, 103, 82–87 CrossRef CAS.
- (a) H. C. Kolb and S. V. Ley, Tetrahedron Lett., 1991, 32, 6187–6190 CrossRef CAS; (b) H. C. Kolb, S. V. Ley, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1992, 2735–2760 RSC.
- F.-Q. Huang, J. Xie, J.-G. Sun, Y.-W. Wang, X. Dong, L.-W. Qi and B. Zhang, Org. Lett., 2016, 18, 684–687 CrossRef CAS PubMed.
- H. Weinstabl, T. Gaich and J. Mulzer, Org. Lett., 2012, 14, 2834–2837 CrossRef CAS PubMed.
- E. J. Corey and P. L. A. Fuchs, Tetrahedron Lett., 1972, 13, 3769–3772 CrossRef.
- A. Gansäuer, H. Bluhm and M. Pierobon, J. Am. Chem. Soc., 1998, 120, 12849–12859 CrossRef.
- Similar preference for the formation of fused [4-4] systems (as opposed to bridging “crossed adducts” resulting from initial 1,5-cyclization) has been observed in intramolecular [2 + 2] reactions, particularly in cases in which the internal position of the reacting olefin is substituted. For examples see: (a) S. Wolff and W. C. Agosta, J. Org. Chem., 1981, 46, 4821–4825 CrossRef CAS; (b) S. Wolff and W. C. Agosta, J. Chem. Soc., Chem. Commun., 1981, 118–120 RSC; (c) S. Wolff and W. C. Agosta, J. Am. Chem. Soc., 1983, 105, 1292–1299 CrossRef CAS.
- (a) P. A. Greico, S. Gilman and M. Nishizawa, J. Org. Chem., 1976, 41, 1485–1486 CrossRef; (b) K. B. Sharpless and M. W. Young, J. Org. Chem., 1975, 40, 947–949 CrossRef CAS.
- (a) Y. Ge and S. Isoe, Chem. Lett., 1992, 139–140 CrossRef CAS; (b) S. D. Holmbo and S. V. Pronin, J. Am. Chem. Soc., 2018, 140, 5065–5068 CrossRef CAS PubMed.
- N. S. Simpkins and J. K. Cha, t-Butyl Hypochlorite, in Encyclopedia of Reagents for Organic Synthesis. Wiley, 2001 Search PubMed.
- K. Takasu, S. Nagao and M. Ihara, Tetrahedron Lett., 2005, 46, 1005–1008 CrossRef CAS.
- B. Gross, S.-J. Han, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2023 DOI:10.1021/jacs.3c02142.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1987989, 1987997, 1987999, 1988002, 2239956, 2239957, 2239959, 2239963 and 2239966. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00651d |
| ‡ NJH and SAL contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |