
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetallated dihydropyridinates: prospects in hydride transfer and (electro)catalysis
Leo W. T.
Parsons
and
Louise A.
Berben
 *
*
Department of Chemistry, University of California, Davis CA, 95616, USA. E-mail: laberben@ucdavis.edu
First published on 17th July 2023
Abstract
Hydride transfer (HT) is a fundamental step in a wide range of reaction pathways, including those mediated by dihydropyridinates (DHP−s). Coordination of ions directly to the pyridine ring or functional groups stemming therefrom, provides a powerful approach for influencing the electronic structure and in turn HT chemistry. Much of the work in this area is inspired by the chemistry of bioinorganic systems including NADH. Coordination of metal ions to pyridines lowers the electron density in the pyridine ring and lowers the reduction potential: lower-energy reactions and enhanced selectivity are two outcomes from these modifications. Herein, we discuss approaches for the preparation of DHP–metal complexes and selected examples of their reactivity. We suggest further areas in which these metallated DHP−s could be developed and applied in synthesis and catalysis.
 Leo W. T. Parsons | Leo Parsons was born in Seattle Washington, USA. He received a Bachelor of Science degree in Chemistry from Cal Poly, San Luis Obispo in 2017. He is currently a PhD candidate at the University of California Davis under the supervision of Professor Louise Berben, studying metal effects on electron and proton transfer to non-innocent ligand complexes. |
 Louise A. Berben | Louise Berben was born in Sydney, Australia. She received a Bachelor of Science degree with 1st class honours from the University of New South Wales in 2000, and in 2005 was awarded a PhD from the University of California Berkeley. After post-doctoral work at California Institute of Technology and Massachusetts Institute of Technology, Louise joined the faculty at the University of California Davis in 2009. Her research program focuses primarily on synthetic and physical inorganic chemistry with applications in electrocatalytic reduction of small molecules, solar fuels chemistry, and expanding the scope of Group 13 chemistry with redox-active ligand complexes. Her work has been recognized with the Alfred P. Sloan Foundation Fellowship, an NSF CAREER award, and with the Chemical Communications Emerging Investigator Lectureship. She is currently Director for the University of California Research Center on Direct Conversion of Captured CO2 into Chemicals and Fuels. |
1. Introduction
Electron transfer (ET) and proton transfer (PT) reactions are fundamental processes in chemistry. Indeed, most chemical transformations of small molecules and functional groups familiar to the organic chemist, e.g. aldol reactions, esterifications, epoxidations, reductive amination, nitrile hydrolysis to name a few, involve one or both of ET and PT and often in combination with other bond-making or -breaking events.1 For transformations limited to ET and PT only, these events commonly involve one electron and one proton (an H-atom transfer, H˙) or two electrons and a proton (a hydride transfer, H−). Hydrogenation reactions for reduction of ketones, imides or alkenes, and dehydrogenative oxidation reactions involve a total of two ET and two PT events equivalent to a molecule of H2; in most instances, this occurs with one proton (H+) and one hydride (H−) equivalent.2,3 Multiple site proton coupled electron transfer (MS-PCET) is not covered in this perspective.4Hydride transfer (HT) reagents in organic synthesis and catalysis will be discussed in this Perspective article with a focus on HT reactions mediated by functionalised dihydropyridinate (DHP−) molecules. The IUPAC definitions for dihydropyridine (DHP) and for DHP− are given in Scheme 1 and will be used throughout the remainder of this article. Following the Section 1: introduction, we discuss approaches to the synthesis of metallated DHP−s in Section 2; and then selected reaction chemistry of metallated DHP−s is addressed in Section 3. Open challenges and opportunities are mentioned throughout the text and discussed as an overview in Section 4.
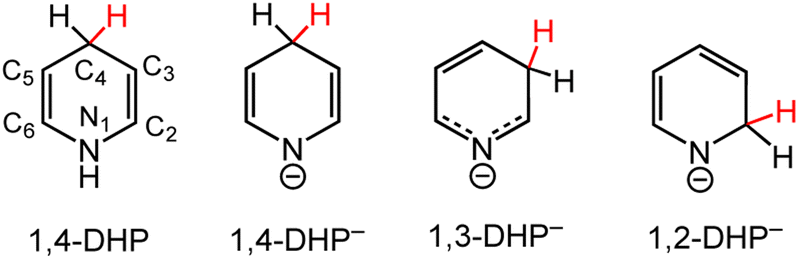 | ||
| Scheme 1 Dihydropyridine (DHP) and dihydropyridinate (DHP−) regio-isomers are numbered according to the carbon atoms in the pyridyl ring. | ||
HT is a mechanism for many chemical transformations that afford X′–H bonds, where X′ could be C, N, or another atom. DHP−s are already ubiquitous in organic synthesis.5–7 We demonstrate in this Perspective that their synthetic tunability and redox activity should enable a far greater scope for applications of DHP−s than is currently known for organic synthesis and catalysis.8–17 Advances in reaction selectivity and energy efficiency are required to use DHP−s in electrosynthesis, including the lowering of redox potentials for turnover of catalytic cycles. Theoretically, the formation of DHP−s in a catalytic cycle can be readily achieved via two ET (ET1 and ET2) and one PT (PT1) step following the HT; and those ET steps can be driven electrochemically (Scheme 2). However, it is well-known that organohydride catalysts require ∼1 V overpotentials to promote the turnover (or second) ET2 step in the cycle, and therefore, strategies for tuning the properties of DHP−s via the incorporation of functional groups and particularly by incorporation of metal ions are needed.18,19 Metal ions lower the reduction potential for ET but their effect on hydricity is largely unexplored although empirical plots of hydricity and E° suggest they are linearly correlated.20,21
 | ||
| Scheme 2 Possible catalytic cycle to enable electrochemical regeneration of DHP− for hydride transfer (HT) reactions. ET = electron transfer; PT = proton transfer. In some cases, the ordering of the ET2 and PT1 elementary steps is reversed. | ||
1.1 Bioinspired DHP− chemistry
The chemistry of DHP−s cannot be discussed without acknowledging the significant chemistry performed by organohydride-containing biochemical cofactors, which have long served as inspiration for the development of synthetic systems. More knowledge of organohydride-containing cofactors is likewise needed to better understand their function in biological systems. Many enzymatic processes transfer hydride equivalents, and those transformations are most often facilitated by cofactors that contain the DHP− functional group, including nicotinamide adenine dinucleotide (NAD)22,23 and the nickel pincer nucleotide (NPN) found in lactate racemase (LarA) (Chart 1).24,25 There are a handful of published structures in which the carbamoyl group (R–CONH2) of NAD+ is bound to a metal ion in a protein structure; example enzymes where this structure is found include isopropylmalate dehydrogenase,26 and pyrrolysine synthase.27 In isopropylmalate dehydrogenase, an activating potassium ion (K+) coordinates to the carbamoyl nitrogen and a glutamate side chain. This structural positioning and activating effect of K+ on the NAD+ ring helps HT to the NAD+ ring during substrate oxidation.28 In pyrrolysine synthase, a magnesium ion (Mg2+) is coordinated to the carbamoyl oxygen of NAD+. The NPN cofactor promotes a more limited set of chemistries relative to NAD+. As the name lactate racemase suggests, LarA mediates the racemisation of lactate, and that process proceeds following hydride abstraction from lactate by the NPN cofactor, to permit a C–C bond rotation before HT from NPN furnishes the racemised lactate molecule.29 There are also cofactors that facilitate HT in biology via O–H and N–H bond chemistries; they are outside the scope of this Perspective and so those cofactors will not be discussed.25,30–33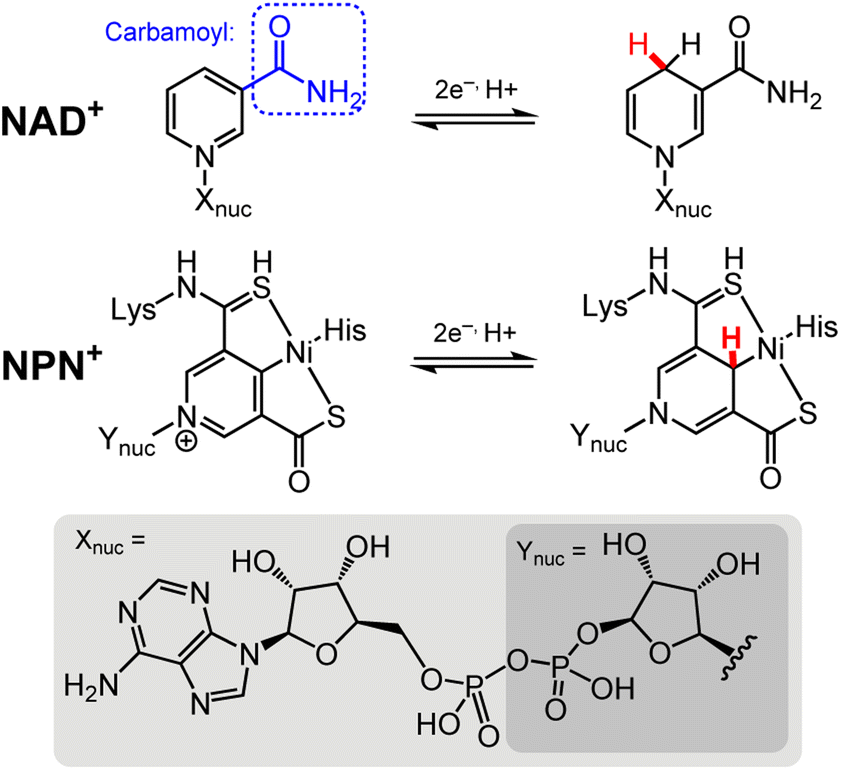 | ||
| Chart 1 Structures of the NAD+ and NPN+ cofactors in their oxidised and reduced forms.22,24, Lys = lysine; His = histidine; Xnuc = adenine dinucleotide (bottom, light grey shaded rectangle); Ynuc = mononucleotide (bottom, dark grey shaded rectangle). The site for HT chemistry is indicated by a red H atom. | ||
This Perspective will primarily focus on the structure–reactivity relationships that govern control of HT chemistry (and the formation of hydride reagents) since it is the nuances of the chemical structure that guide selectivity in chemical reactions. Opportunities for functionalisation of DHP−s are numerous and synthetically very accessible; they include N-coordination of metal ions, especially redox-inactive Lewis-acidic metal ions, and N-protonation. This viewpoint is provided through the lens of inorganic and coordination chemistry since metal-ion effects on DHP− chemistry are significant for accessing lower reduction potentials. Our focus will be on chemistry reported within the last 10 years; however, older examples may be cited to give context or for completeness in some areas. We make no attempt to comprehensively review the literature on DHP− chemistry and instead highlight only examples related to tuning the structures and function of DHP− reagents to lower their redox potentials and access greater utility and selectivity in their chemistry. In Section 2, we discuss a few advances in the synthesis of metal-containing DHP− reagents with a focus on achieving predictability in the regiochemistry of DHP− formation from the pyridyl precursor. In Section 3, we discuss the reaction chemistry of (DHP−)-based metal–ligand coordination complexes that have been employed in stoichiometric and catalytic HT chemistry. Throughout this Perspective article, the focus of our discussion will be on (1,4-DHP−)-containing reagents, which are structurally far more diverse than the model and native cofactors such as NPN and NAD.
2. Synthesis of metallated DHP− complexes (M-DHP−s)
Elimination of unwanted side reactions in stoichiometrically, catalytically, and electrocatalytically driven HT reactions is an important ongoing effort,34–36 with one approach being the development of milder conditions to achieve better regio- and chemo-selectivity as well as stereoselectivity.37–39 In electrosynthesis applications, milder reaction conditions, especially through lowered redox potentials, would also correlate with a more energy-efficient process. In this section, we begin with a brief overview of common organohydride reagents and then explore the potential to incorporate metal ions into DHP and DHP− structures; the resulting metallo-ligands may facilitate HT reaction chemistry. These synthetic approaches are relevant because HT reaction chemistry may be more widely employed if tuneable and regio-pure DHP− structures were more synthetically accessible. We will provide selected example syntheses of coordination complexes where the pyridyl N or C atom are the donor ligands. We will not discuss the preparation of functionalised DHP− ligand structures.40,41In coordination chemistry, a ligand is a group or molecule that can form a dative bond with a metal ion. Based on the structure of the DHP− core, which is common to (DHP−)-containing ligands, there are a few possible binding modes for metal ions to DHP−, and they may modulate the physical properties and reaction chemistry of DHP−. In general, synthetic DHP− ligands with variable N-coordination are either bi- or tridentate because the multi-dentate ligands form more stable metal complexes than a monodentate ligand due to the chelate effect.42 The most common structure in the coordination chemistry of DHP− comprises metal ions ligated by the pyridyl N or C atom (Chart 2 left). Functional groups on the DHP− core can also potentially be ligands for the metal ion (Chart 2 right); this outer sphere approach can position substrates near the DHP− hydride via docking to the metal and tune regioselectivity and reactivity as will be discussed in Section 3.4.
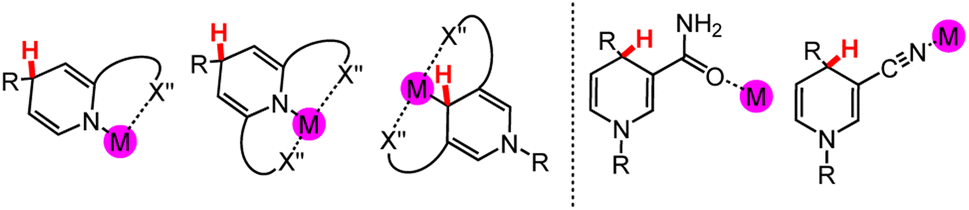 | ||
| Chart 2 (Left) Possible N- and C-coordinated DHP− ligands with typical examples of C- and N-alkylation patterns that support the coordination modes. (Right) Examples of functional groups on DHP− ligand cores, including carbamoyl and nitrile40 groups, that can bind metal ions or protons. M = a metal atom; X′′ = coordinating heteroatom. Again, the site for HT chemistry is indicated by a red H atom. | ||
2.1 Background on organohydride reagents
Here we provide a brief survey of organohydride reagents since their chemistry can inform the design of next-generation metal-containing DHP− reagents. Most commonly, the active C–H hydride in an organohydride reagent is found on a C atom of a N-containing heterocycle (Chart 3). The hydride-donor ability, often discussed in terms of the free energy for loss of hydride ion (hydricity, ΔGH–), of DHP− reagents spans roughly 50–100 kcal mol−1 in MeCN.18 NADH is often credited as inspiration for synthetic organohydrides that incorporate the pyridine ring; classic examples of this model NADH reagent class are 1-benzylnicotinamide (BNAH)43 and the Hantzsch ester (HEH)44 (Chart 3), which have enabled a great deal of HT reactivity and continue to be used in catalytic HT reactions.45 New organohydrides have also been developed from HEH and BNAH by functionalisation of those scaffolds: examples of this strategy include N- or C-functionalisation of pyridines, as illustrated by the structures of 3,5-bis(2,6-dimethylphenyl)-1-mesityl-1,2-dihydropyridine (BDMH)46 and 2,6-dimesityl-1-methyl-4-phenyl-1,4-dihydropyridine (DMPH) (Chart 3).35 Both BDMH and DMPH contain bulky mesityl substituents (Mes), which likely prevent association of molecules in solution, increasing their stability toward H2 evolution or other decomposition pathways. The mesityl substitution also changes the hydricity of the reagents, so that they are stronger hydride donors relative to BNAH. Carbamoyl is more electron-withdrawing compared to mesityl, so carbamoyl-containing BNAH has lower hydride-donor ability and more positive hydricity, ΔGH–, than Mes-containing BDMH and DMPH. Another class of organohydride based on the DHP− structure are fused ring systems, such as acridine (Acr)47 and phenanthridine (phen).48,49 The fused structures enable greater electron delocalisation across more atoms, so that the reduction potential for those structures is lowered, as is the hydride-donor strength relative to BNAH. In rough terms, fused-ring DHP− donors are roughly 5–20 kcal mol−1 weaker hydride donors than BNAH.18,50 Functionalisation of fused ring scaffolds has also been pursued to afford asymmetric phenanthridine organohydrides, ferrocene-based FENAM51,52 and [2.2]paracyclophane-based CYNAM53 (Chart 3 bottom); both FENAM and CYNAM enable access to stereoselective reduction products.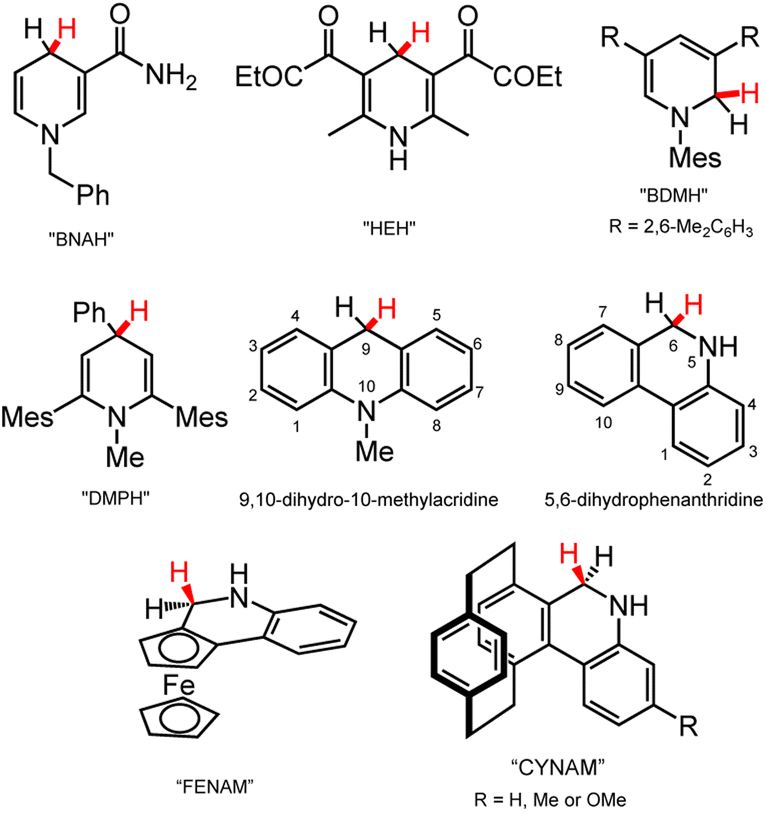 | ||
| Chart 3 Selected organohydride reagents,35,43–48 including NADH-inspired examples (BNAH, HEH), functionalised pyridines (BDMH, DMPH), and fused ring systems (acridine (Acr), phenanthridine (phen), FENAM, CYNAM). The site for HT chemistry is depicted with a red H atom. Mes = mesityl group. | ||
2.2 Preparation of M-DHP−s via reaction of pyridyl ligands with metal alkyls
A common synthetic route to DHP− coordination complexes is by reaction of the pyridyl form of the ligand with a metal–alkyl reagent, and this approach most often results in alkylation of the pyridine ring, which is activated by the N-coordination of the metal ion (Scheme 3 top).54 To indicate that the ligand has been reduced to the dihydropyridinate form, the negative charge of DHP−, and DHP− containing ligands, are explicitly shown; however, the positive charge of the M ion is understood and sometimes omitted. Groups 1 and 2 alkyl reagents have been explored most extensively and are very nucleophilic.55,56 The drawback of this approach for synthesising DHP−s is that it is not always selective for a product that is alkylated at the C2, C3 or C4 pyridyl positions. Work by Campora and coworkers has provided a host of examples with chelating ligands that have pyridyl at their core, and their reactions with various metal–alkyl reagents.39,57 Just one example is highlighted here; it is representative of the competition between thermodynamic and kinetic product outcomes and the challenges in achieving selectivity. Reaction of a diiminepyridine (I2P) with dibenzylmagnesium (Bn2Mg) results in isomers derived from unselective pyridine-ring substitution.58 At low temperature, C2 alkylation is favoured (−60 to 0 °C), while above 5 °C, a mixture of C3 and C4 alkylation products is present, but they ultimately converge to the C4 alkylation product. These results suggest the relative thermodynamic preference of the alkylation product is of the order: C2 < C3 < C4. The kinetic preference for 1,2- vs. 1,4-addition product has also been noted in other reactions producing M-DHP− complexes, such as the catalysed hydroboration of pyridines.59 Another common challenge in achieving DHP− synthesis with metal alkyls is avoiding the deprotonation of acidic groups contained in functionalised pyridines. This is illustrated by reactions of 2-methyl- or 4-methyl pyridine with alkyl lithium reagents, RLi,60 where dearomatization produces the enamide rather than alkylation of the pyridine ring (Scheme 3 bottom). Although undesirable for M-DHP− formation, this aromatization-dearomatization reactivity has been well documented in other reaction pathways.61–64 Similarly, other basic groups such amines should be avoided in the ligand structure due to similar protonation/deprotonation reactivity.65 | ||
| Scheme 3 (Top) The general reaction pathway for alkylation to make either 1,2-DHP−, 1,3-DHP− or 1,4-DHP− metal complexes, including the relative stability of the alkylation products. M = metal centre; Rn = alkyl group. (Bottom) Deprotonation of acidic functional groups is a possible side reaction with metal–alkyl reagents;60 avoiding this side reaction is a common challenge in the synthesis of M-DHP− compounds. | ||
Recent efforts have been made to enhance the selectivity of metal–alkyl reagents using chelating ligands as co-reagents as well as metathesis reactions to substitute the metal ion. Ongoing efforts in synthetic chemistry seek to improve the selectivity of metal–alkyl reagents in the functionalisation of aryl and heterocyclic compounds by the addition of chelating co-reagents.66–68 For example, using stoichiometric pyridine and tert-butyl lithium, tBuLi, followed by the addition of the chelating ligand Me6TREN, [tris(N,N-dimethyl-2-aminoethyl)amine], in hexane solution, the first report on the solid-state structure of a monomeric Li-DHP− was presented.69 Reactivity of pyridine with other butyl lithium reagents, n-, iso-, sec-butyl lithium (nBuLi, iBuLi, sBuLi), as well as with cheaper ligands PMDETA (N,N,N′,N′′,N′′ = pentamethyldiethylenetriamine), Me4AEE (bis-[2-(N,N-dimethylaminoethyl)]ether) and TMEDA (N,N,N′,N′-tetramethylethylenediamine), was later explored (Scheme 4).70 In each case, 1,2-DHP− was obtained and bulky alkyl substituents increased the solubility of the M-DHP− in non-polar solvents. Metathesis reactions with 1-lithio-2-tBu-1,2-DHP− (Li-2t-DHP−) and sodium or potassium tert-butoxide (tBuONa or tBuOK) resulted in 1-sodium-2-tBu-1,2-DHP− (Na-2t-DHP−) or 1-potassium-2-tBu-1,2-DHP− (K-2t-DHP−) complexes, respectively (Scheme 4, M-2t-DHP− pathway).71 The reactivity of these complexes including the influence of the metal ion will be discussed in greater detail in Section 3.
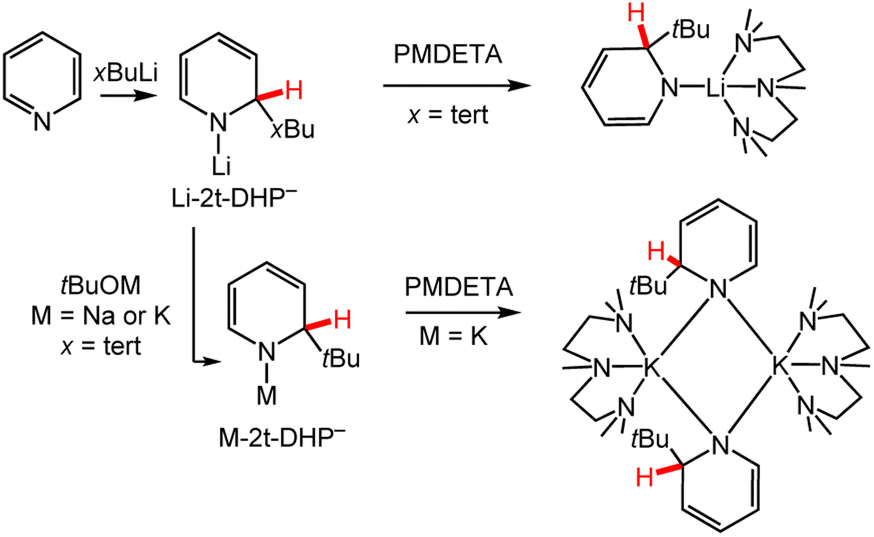 | ||
| Scheme 4 Use of chelating ligands to direct selective alkylation of pyridine for M-DHP− complex synthesis. Note that the Li-2t-DHP− and M-2t-DHP− products are formally neutral, with the “–” indicating the pyridinate form of the ligand. PMDETA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine; x = branching of butyl group, n-, iso-, sec-, and tert-.60,69–71 | ||
Given these factors, not all DHPs will be suitable for M-DHP− formation by metal alkyls. DHPs with acidic functional groups will have competing deprotonation pathways. However, with expanding efforts on selective metal–alkyl reagents and metathesis pathways, metal alkylation can serve as a good route to increase solubility of the M-DHP− and allow post-synthetic modifications by exchange of the metal ion.
2.3 Hydride transfer (HT) to pyridyl ligand–metal complexes
Regioselective HT to pyridines has been approached via two primary routes: the metal-catalysed hydroboration or hydrosilylation of pyridines and the direct reaction of pyridines with metal–hydride reagents. Hydroboration and hydrosilylation and metal-free approaches will not be discussed in detail since they produce N-borylated and N-silylated DHP−s and since boron and silicon are not metals, B-DHP−s and Si-DHP−s fall outside the scope of this Perspective.59,72–83 An exception are select N-borylated compounds whose metalloid character is informative of metal ion effects on DHP−s as discussed in Section 3.2.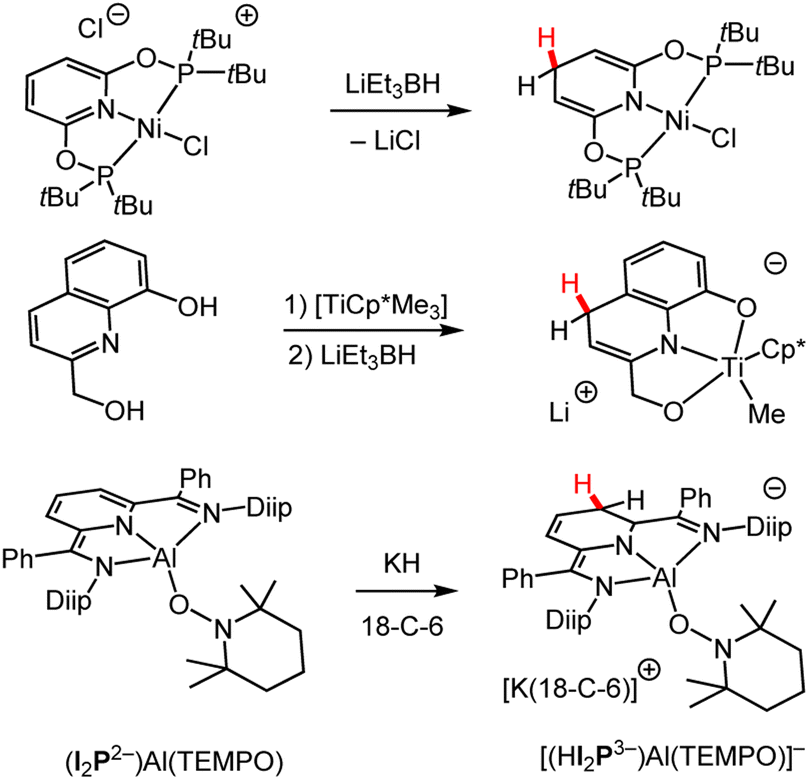 | ||
| Scheme 5 Examples of syntheses of M-DHP− complexes by the addition of various metal hydrides.84,85,89 Cp* = pentamethylcyclopentadienyl; Diip = 2,6-diisopropyphenyl; 18-C-6 = 18-crown-6 ether; I2P = diiminepyridyl; TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl. | ||
In some cases, the resonance pattern of the pyridine within a larger conjugated structure can be used to direct regioselective hydride formation. As an example, using the direct HT approach, selectivity for the 1,3-DHP− isomer of a tridentate diiminepyridine (I2P) ligand was demonstrated (Scheme 5 bottom).89 The identity of the metal in a metal hydride can also influence the regioselectivity of hydride addition. Comparison of Mg to calcium (Ca) reagents indicates that the larger ionic size of Ca favoured the formation of the M–(1,2-DHP−) complex while the smaller Mg ion gave both 1,2- and 1,4-DHP− products.90 This was explained in the primary literature where DFT calculations suggest high energy barriers required for Ca to form 1,4-DHP− products.
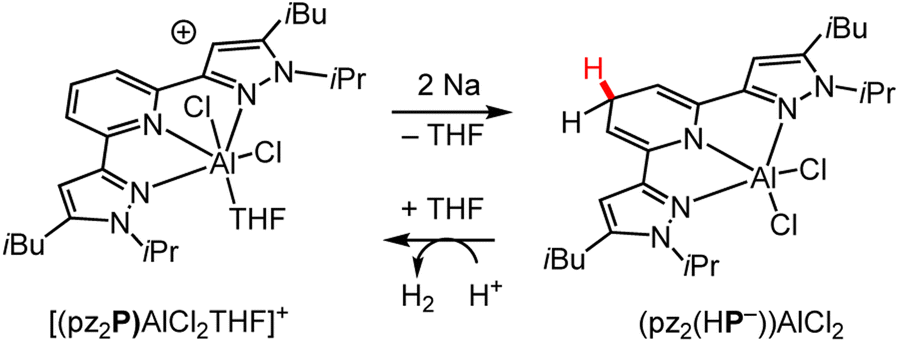 | ||
| Scheme 6 Synthesis of pz2(HP−)AlCl2,91 where pz is the substituted pyrazolyl shown and P is the pyridyl ligand backbone. HP− denotes the dihydropyridinate form, which is analogous to DHP−. Protons are believed to be scavenged from ligand equivalents during reduction by Na. | ||
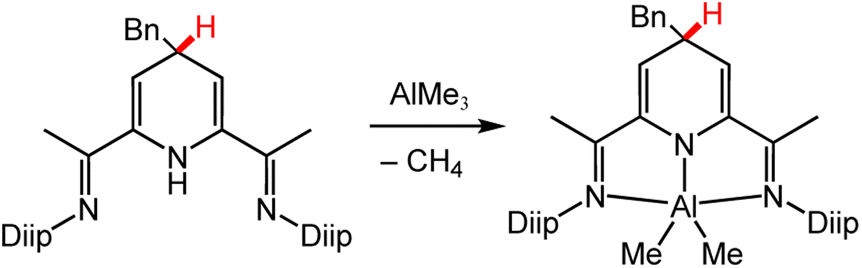 | ||
| Scheme 7 Deprotonation of DHP via metal–alkyl reaction to form an Al-DHP− complex.94 Diip = 2,6-diisopropyphenyl; Bn = benzyl group. | ||
2.4 Synthesis of C4-coordinated M-DHP−s
Metallation of the C4 pyridyl carbon in a chelating ligand has been achieved using nickel (Ni) and palladium (Pd) chemistry; both of these metals have accessible two-electron chemistry that facilitates oxidative addition of the C4–H pyridyl position. Both Ni and Pd complexes of bis(2-(diisopropylphosphaneyl)phenyl)pyridine (PCP) were synthesised by Agapie and coworkers, and bond metrics from single-crystal X-ray diffraction (XRD) reveal shortening of the C2–C3 and C5–C6 bonds in the pyridyl, indicative of DHP− formation (Scheme 8A and 8B).95 Inspired by the NCN cofactor in LarA, the Hu group has synthesised pincer ligands that are metallated at the C4 position by Ni.96,97 Metallation of the bis(diethylcarbamothioyl) methylpyridinium (SPS) ligand was achieved with bis(cyclooctadiene) nickel, Ni(COD)2 (Scheme 8C). Oxidation of benzyl alcohol or benzyl-α,α,-D2-alcohol to benzaldehyde or benzaldehyde-α-D1 respectively by the (SPS)NiCl complex confirmed that the pyridinate form of the (SPS)NiCl (analogous to DHP−, Scheme 8C, third complex) was accessible, as evidenced by H or D incorporation to the C4 position of the SPS ligand, even though it could not be isolated. Metallation of the C4 position can directly tune the electronics of the C4 atom by the nature of bonding between metal and C4, where strongly π-donating metals may increase the electron density relative to strongly π-accepting metals, offering another coordination mode for tuning DHP− electronics.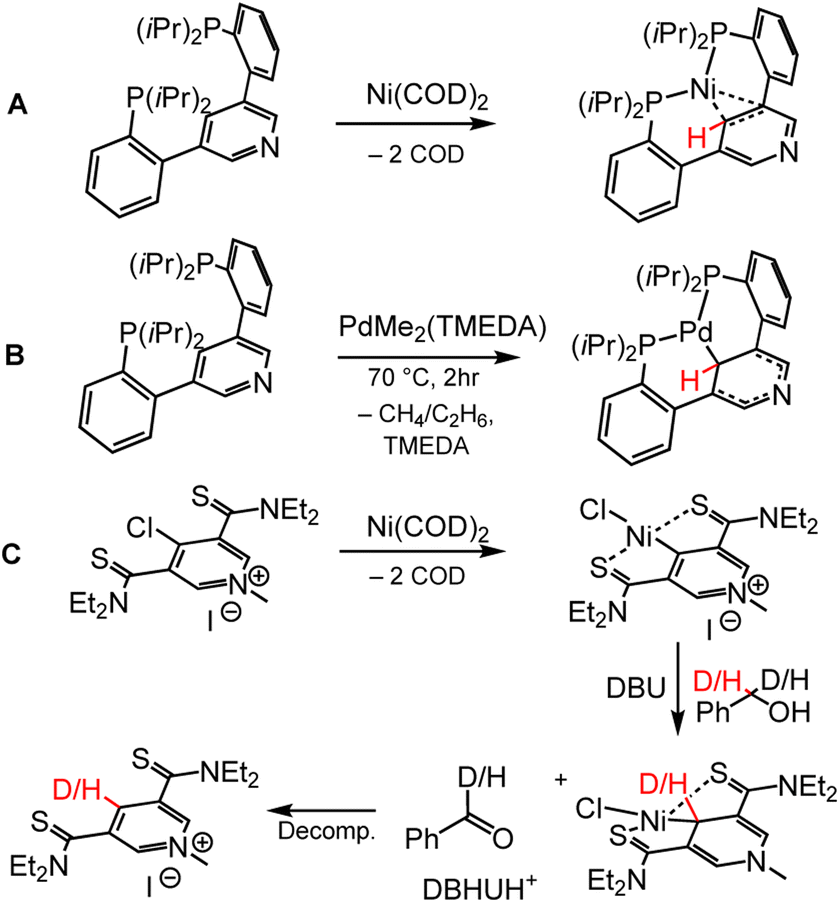 | ||
| Scheme 8 Metallation of the C4 position for the pincer ligands (A and B) PCP, bis(2-(diisopropyl-phosphane-yl)-phenyl)-pyridine, and (C) SPS, bis(diethyl-carbamo-thioyl) methyl-pyridinium.95–97 COD = cyclooctadiene; TMEDA = N,N,N′,N′-tetramethylethylenediamine; DBU = 1,8-diazabicyclo(5.4.0)undec-7-ene. Location of HT is shown with red H or D atoms. Isotope experiments show C4 of SPS ligand as hydride acceptor from benzyl alcohol, supporting proposed DHP− intermediate. | ||
3. Reactivity of M-DHP−s
In this section, we will outline and discuss possible roles for metal ions in tuning HT that is mediated by (DHP−)-based ligands. The potential for influences of metal ions on the properties and reactivity of DHP− reagents can in some cases be rationalised and predicted by structure–function relationships. Other effects of metal ions on DHP− chemistry arise from case-specific substrate interactions, or with ligand functional groups in the secondary coordination sphere. We will discuss both types of metal-ion effects.As a starting point, we will consider the electronic effects and reactivity consequences of pyridyl N-coordination to a metal ion. Electron donation from the pyridyl N atom to a cationic metal ion should result in lowered electron density on the pyridyl ring, lowering the reduction potential for the pyridyl ring; it will also raise the free energy for loss of hydride from the corresponding M-DHP−, which means that the ligated DHP− is a weaker hydride donor relative to free DHP−. In general, weaker hydride donors have the advantage of greater chemo- and regio-selectivity providing they have sufficient driving force for the reaction. Moreover, the hydricity of the DHP− should be tuneable by variation of the metal ion identity or oxidation state. In the context of a catalytic cycle for DHP− regeneration and HT to substrate, the lowered reduction potential of the bound DHP− will enable a lower applied reduction potential for catalytic turnover (Scheme 2). As an example from our research group, the reduction potential for I2P ligands is about −2.1 V vs. SCE in THF whereas the I2P−/2− couple for the Al-coordinated ligands in (I2P−)AlCl2 lies at −0.90 V vs. SCE.98,99 The advantages of a lower applied reduction potential include less chances for unwanted background reactions and a lower overall use of energy in the reaction. Tuneable hydride-donor abilities for the DHP−s could be very helpful in designing chemo- and regio-selective reactions (Scheme 9).18
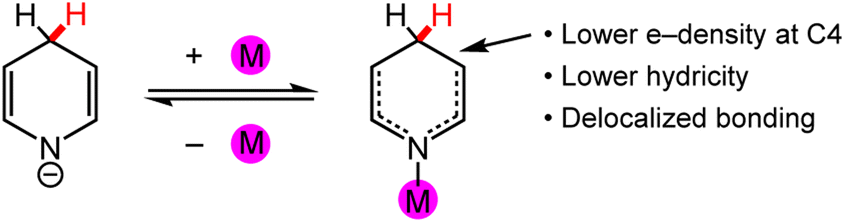 | ||
| Scheme 9 Common changes to DHP− upon coordination of a metal atom, M, at the pyridyl N atom. | ||
3.1 Influence of protons on DHP− reaction chemistry
Known Bronsted-acid effects on DHP− reaction chemistry can be used to illustrate or predict possible metal-ion effects on DHP− chemistry, since protons and Lewis-acidic metal ions both bring the influence of a cationic group on DHP− properties. The interest in Bronsted-acid effects on DHP− stems largely from systems with chiral anions, which therefore direct stereoselectivity in reduction reactions of organic molecules. However, the influence of the (non-chiral) proton is also hugely important. Insights from the extensive literature using Bronsted-acid/base activation of DHP− can be used to think about how the driving force for HT chemistry from DHP− is modulated by the cation. As an illustrative example, the reaction between HEH and BNA+ (Chart 3) has a free energy of ΔrG = + 2.5 kcal mol−1, and no reaction occurs in MeCN solution. In the presence of the strong base DBU (1,8-diazabicyclo(5.4.0)undec-7-ene), complete HT from HEH to BNA+ occurs rapidly with a reported free energy change of ΔrG = −16.6 kcal mol−1 (Scheme 10).100 Isotope labelling studies suggested a pre-equilibrium deprotonation of HEH precedes a rate-limiting HT step. More generally, these results demonstrate the significant influence that coordination at the pyridyl ring N atom has on the kinetics and thermodynamics of HT from a DHP− reagent.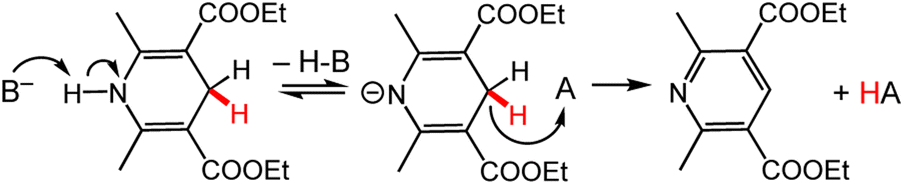 | ||
| Scheme 10 (Top) The proposed mechanism for reaction of HEH with a hydride acceptor (A) in the presence of a base (B−).100 For the other example in the text, the acceptor A would be BNA+; in both cases, the base B− is DBU. | ||
3.2 Reactivity of s- and p-block M-DHP−s
In this section, we discuss the reaction chemistry for known N-metallated DHP− ligands with metal ions drawn from the s- and p-block elements. These examples include stoichiometric HT reactions and electrocatalytic reactions where DHP− regeneration is mediated via successive ET–ET–PT reaction steps. | ||
| Scheme 11 Synthesis of (HAcr−)XLA, where XLA = BCl2+; XLA may also be other groups as indicated in the main text. (Left) Resonance forms of [(Acr)BCl2]+ and (right) (HAcr−){BCl2+}, the HT product, which is stabilised by the NAcr–B multiple bonding.102 | ||
A handful of examples exist where a catalytic HT reaction is enabled by regeneration of a DHP− electrochemically; these have mostly been studied within the past decade and involve organic DHP−s.18,19,103 The applied potentials needed to drive these catalytic reactions vary greatly depending on the solvent and choice of organohydride. Representative example ranges are 0.3 to −1.3 V vs. SCE in DMSO19,104 and −0.6 to −1.15 in MeCN.105,106 Metal-ion effects on electrocatalytic HT have been reported, and we highlight just one study here: the interconversion between [(pz2P)AlCl2THF]+ and (pz2(HP−))AlCl2 that was described in Section 2.3.91 The tridentate pyridyl-centred ligand, denoted as pz2P, is coordinated to an Al(III) centre. Electrochemical regeneration of (pz2(HP−))AlCl2 was demonstrated following its reaction with protons to liberate H2 at −1.2 V vs. SCE (Scheme 6). No evidence for dimerisation via pyridyl C4–C4 coupling of the proposed radical intermediate (pz2P˙)Al(THF)Cl2 formed following ET analogous to ET1 in Scheme 2, was reported in this work. This is consistent with the effects of the metal ion as discussed earlier in Section 3; i.e., if N-coordination of a metal ion results in lowered electron density on the pyridyl ring and especially at C4, then with metals such as Al, the C4–C4 coupling pathways should be disfavoured, which represents another advantage of N-metallation for organohydride reagents.
3.3 Reactivity of d-block M-DHP−s
Here we explore selected reactions from the past decade where hydride is transferred from a M-DHP− to a non-pyridine acceptor and consider the insights we can obtain from this reaction chemistry. Typical stoichiometric HTs from d-block M-DHP− complexes involve moderate to strong hydride acceptors with hydride-donor abilities in the range of 65 to ∼110 kcal mol−1 (the hydricity of methane was approximated from the trend in ΔGH– from substitution of phenyl groups on methane. ΔGH– increases in the order H–C(Ph)3 < H–CH(Ph)2 < H–CH2(Ph)).18 As examples, the DHP− form of zinc–I2P complexes reacts with B(C6F5)3,86 the cobalt pincer bis(diisopropylphenyl) imidazole pyridinate ligand complex, (HCNC−)CoN2, reacts with trityl chloride to yield triphenyl methane,109 and a rhenium-bipyridine-based DHP− reacts with methyl triflate to afford methane (Scheme 12).110 Hydrogenation of alkenes with (CNC)CoCl was achieved under H2, but no mechanistic studies were available to elucidate the hydride source; it was possible for both a Co–H or M-DHP− species to transfer hydride to the alkene.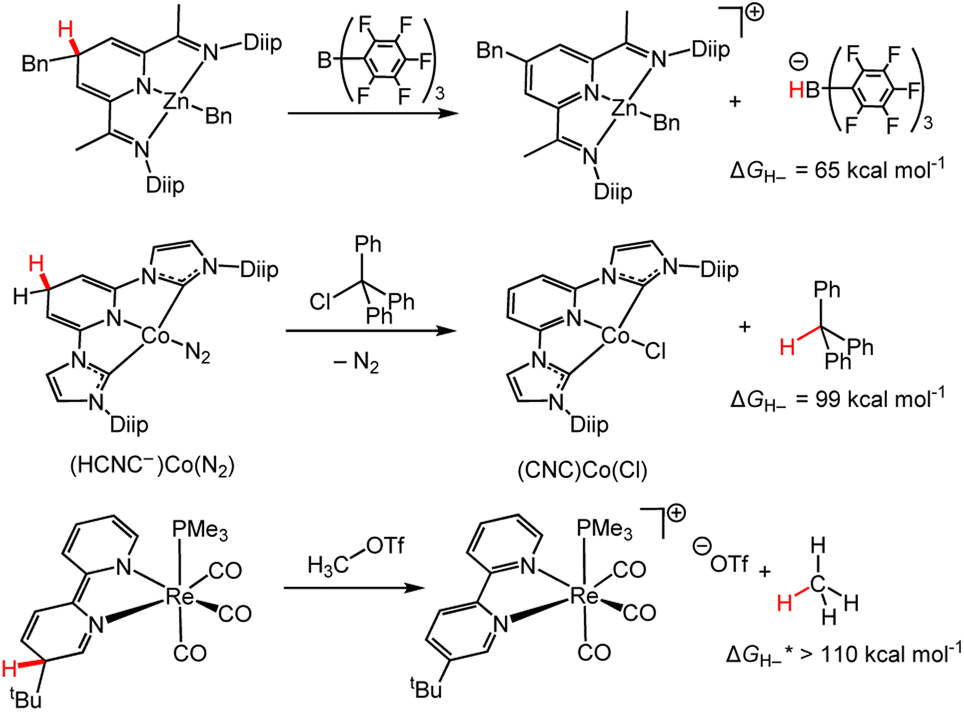 | ||
| Scheme 12 HTs from M-DHP− complexes. ΔGH– are the reported for the hydride acceptors.18,86,109,110 Bn = benzyl; Diip = 2,6-diisopropyphenyl; CNC = tridentate pincer ligand coordinating at 2 C and 1 N, as shown; OTf = triflate. | ||
As with (CNC)CoCl, ambiguity in the reactive site for HT—metal hydride (M–H) vs. M-DHP−—has been illustrated in other catalytic systems. Terpyridine DHP−s studied by Dub and coworkers are active catalysts for the hydroboration of ketones,111 reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC and CN bonds,112 and hydroboration of alkynes to vinyl borates.113 In each system, the active catalytic species bears both a M–H and a DHP− ligand. Furthermore, it is unclear from reactivity studies of bis((dialkylphosphaneyl)oxy)pyridine Ni complexes, (PONOP)NiCl+ (Scheme 13),114 whether a M-DHP− hydride or M–H hydride is involved in the HT. In reactions of (PONOP)NiCl+ and (Me4iPrPNP)NiBr+,87 where PONOP and Me4iPrPNP are both planar tridentate ligands (Scheme 13), stronger hydride-donating reagents tend to favour DHP− formation, while weaker hydride donors tend to favor M–H formation after multiple equivalents are added. In the case of (Me4iPrPNP)CoIICl2 (ref. 115) or (iPrPNP)CoIICl2,116 addition of one equivalent of sodium triethylborohydride, NaEt3BH, gives (Me4iPrPNP)CoICl or (iPrPNP)CoICl (Scheme 13). For (Me4tBuPNP)CoIICl2 various hydride donors do not afford isolable M-DHP− or metal–hydride products, and this may reflect low selectivity or another result (Scheme 13).117 All of the work in the foregoing paragraph is consistent with a trend where square planar d8 transition metal complexes favour DHP− formation when strong hydride donors are used.
O, CC and CN bonds,112 and hydroboration of alkynes to vinyl borates.113 In each system, the active catalytic species bears both a M–H and a DHP− ligand. Furthermore, it is unclear from reactivity studies of bis((dialkylphosphaneyl)oxy)pyridine Ni complexes, (PONOP)NiCl+ (Scheme 13),114 whether a M-DHP− hydride or M–H hydride is involved in the HT. In reactions of (PONOP)NiCl+ and (Me4iPrPNP)NiBr+,87 where PONOP and Me4iPrPNP are both planar tridentate ligands (Scheme 13), stronger hydride-donating reagents tend to favour DHP− formation, while weaker hydride donors tend to favor M–H formation after multiple equivalents are added. In the case of (Me4iPrPNP)CoIICl2 (ref. 115) or (iPrPNP)CoIICl2,116 addition of one equivalent of sodium triethylborohydride, NaEt3BH, gives (Me4iPrPNP)CoICl or (iPrPNP)CoICl (Scheme 13). For (Me4tBuPNP)CoIICl2 various hydride donors do not afford isolable M-DHP− or metal–hydride products, and this may reflect low selectivity or another result (Scheme 13).117 All of the work in the foregoing paragraph is consistent with a trend where square planar d8 transition metal complexes favour DHP− formation when strong hydride donors are used.
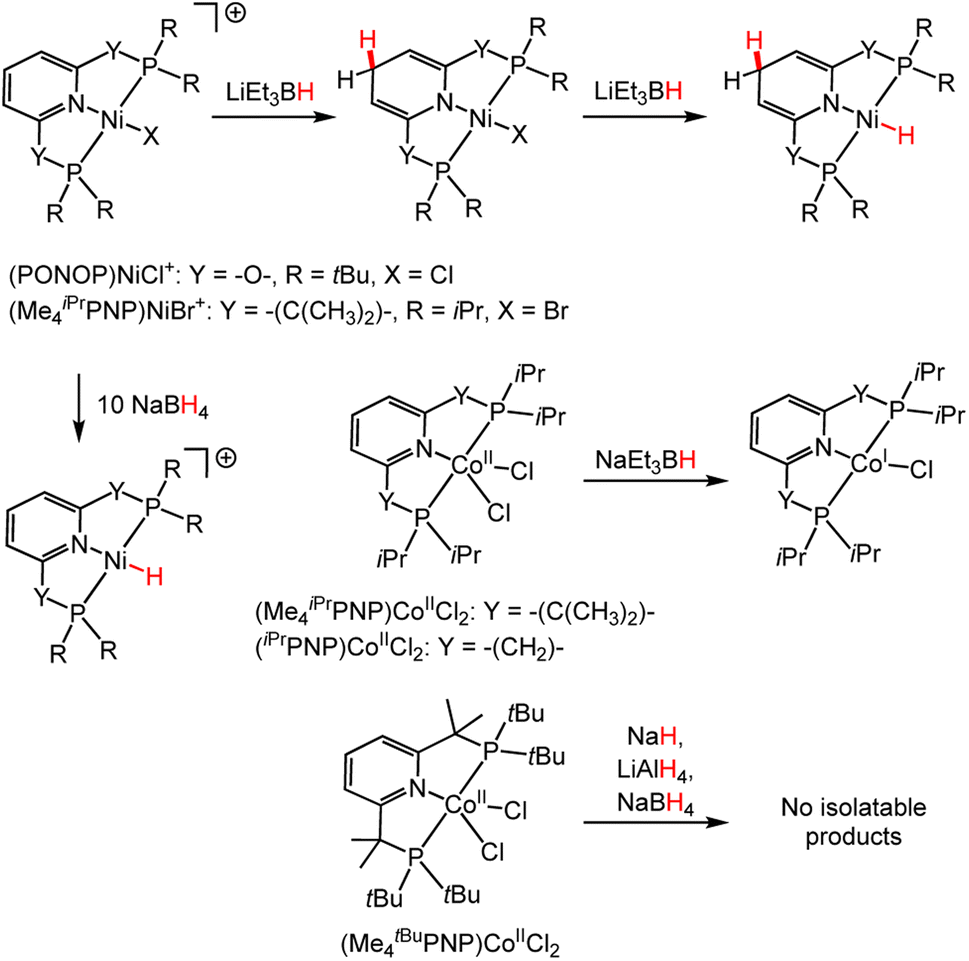 | ||
| Scheme 13 Reactions highlighting the influence of the ligands and metal ion on the HT selectivity: Ni complexes, (PONOP)NiCl+,114 and (Me4iPrPNP)NiBr+,87 and Co complexes (Me4iPrPNP)CoIICl2,115 (iPrPNP)CoIICl2 (ref. 116) and (Me4tBuPNP)CoIICl2.117 | ||
Effects of bimetallic activation on (DHP−)-containing ligands has also been studied but with only a few examples. A pincer ligand, 3,5-bis(2-phosphinophenyl)-pyridine, coordinates via two phosphorus (P) donors and the pyridyl C atom to a Ni or Pd centre; the pyridyl N atom can coordinate to a variety of Lewis acids, including metals, and alkyl substituents, which we denote as E.95 We abbreviate the complexes as [Ni], [Ni]–B(C6F5)3, [Ni]–Me and [Ni]–H, when E = lone pair, B(C6F5)3, Me+ and H+ (Scheme 14). Shifts in the 1H NMR spectra for the pyridyl C4 and C2/6 suggest stronger Ni-py π-bonding when E = Me+ than for E = B(C6F5)3 or E = lone pair. This is consistent with Me+ being more electron-withdrawing than the B(C6F5)3 group. CO was used to probe the electronic effects of the pyridine/metal centre, and wavenumber values for ν(CO) involving the Ni centre are 1930, 1976, 1966 and 1975 cm−1 for [Ni], [Ni]–B(C6F5)3, [Ni]–Me and [Ni]–H, respectively (Scheme 14, top). These data suggest that the electron-withdrawing substituents (E) can influence the electron density at the C4 carbon. Furthermore B(C6F5)3, Me+ and H+ all have a similar effect on electronic structure. Catalytic alkylation of pyridine was reported using AlR3 as Lewis acid to activate the pyridine ring;118 in this process, E = AlR3. Proposed mechanisms for the pyridine alkylation reaction have included Ni(0) insertion into the py C4 bond or a Ni-supported, direct HT from the py C4 to the alkene (Scheme 14, bottom).119 Recent computational and experimental work support the latter pathway. These results suggest that bimetallic pyridine systems as shown in Scheme 14, can support new HT reaction pathways, which incorporate reaction processes supported by transition metals.
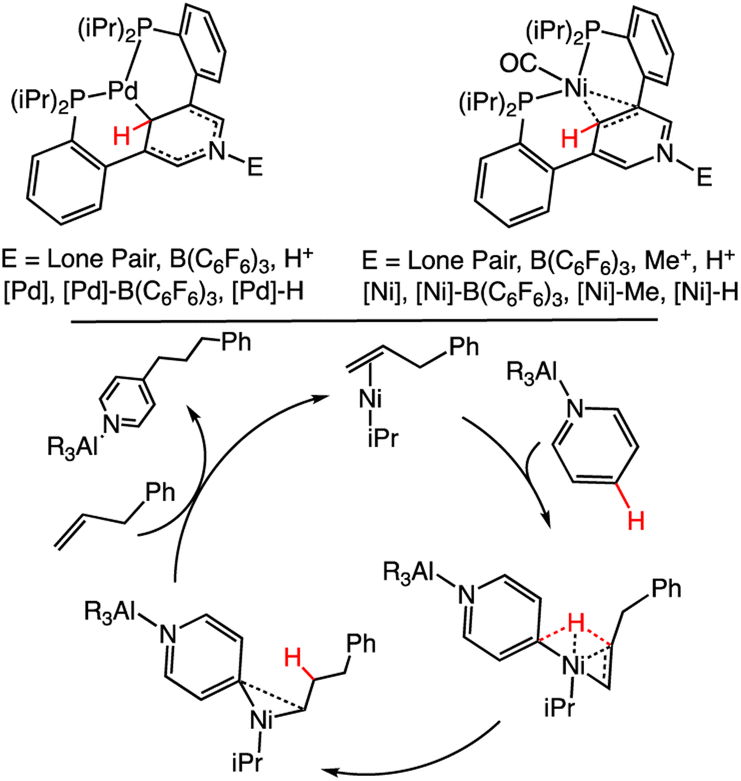 | ||
| Scheme 14 (Top): Structures of [Pd]–E and [Ni]–E.95 For the CO vibrational studies to probe the electronic effects on the [Ni]–E complex, the CO was bound directly to the Ni as shown. (Bottom): Proposed mechanism for C4 alkylation, which is believed to pass through a ligand-to-ligand HT rather than an oxidative insertion step.118 | ||
3.4 Secondary coordination sphere (SCS) effects on HT chemistry
In coordination chemistry, synthetic pyridyl ligands incorporating a carbamoyl functional group or other C3 substituent, have been studied as models for NADH since coordination of the carbamoyl group influences the HT reactivity of nicotinamide derivatives in biological systems.26,28 An illustrative synthetic example is HT from BNAH to B(Et)NA+, which does not spontaneously occur in solution.120 However, when a solution of BNA+ and [Ru(tpy)(bpy)H]+ was added to a solution of B(Et)NA+ and Et3N, HT was observed from the BNAH–Ru complex, [(BNAH)Ru(tpy)(bpy)]2+, to B(Et)NA+ (Scheme 15, top; tpy = terpyridine, bpy = bipyridine, BNAH (see Chart 3)). It is believed that coordination of the carbamoyl group of the in situ-formed BNAH–Ru complex increases the acidity of the –NH2 protons so that deprotonation by triethylamine occurs. This in turn enhances the hydride-donor ability of coordinated BNAH relative to free BNAH. It is also known that this reaction selectively produces the 1,4-DHP− regio-isomer of B(Et)NAH and no formation of the 1,2- or 1,6-DHP− regio-isomers was observed.121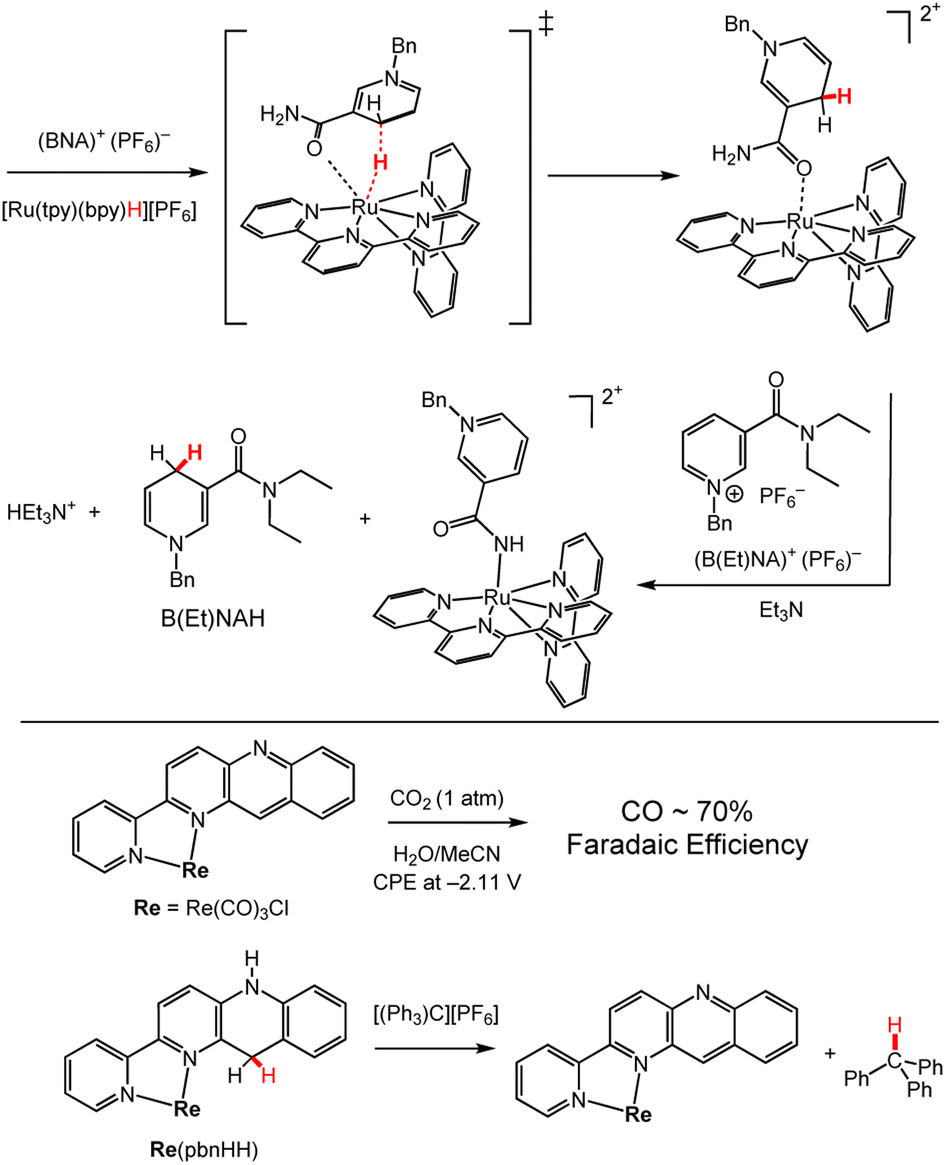 | ||
| Scheme 15 Examples of SCS effects on HT chemistry. (Top) Reaction between BNA+ and [Ru(tpy)(bpy)H]+ to form the [(BNAH)Ru(tpy)(bpy)]2+ complex, and its HT reaction with B(Et)NA+.119,120 Bn = benzyl; tpy = terpyridine; bpy = bipyridine. (Bottom) Reactions of Re(pbn) and Re(pbnHH).121 pbn = 2-(2-pyridyl)benzo[b]-1,5-naphthyridine; pbnHH denotes the ligand in its DHP− form. | ||
Other ligand modifications have shown promise for expanding HT reactivity by taking advantage of metal coordination to substrate. We highlight modification of Acr to 2-(2-pyridyl)benzo[b]-1,5-naphthyridine (pbn) providing a bidentate ligand where the metal centre is positioned in close proximity to the DHP/DHP− functional group.122 This positioning proves advantageous for HT reactions where the substrate can bind to a metal centre, rhenium (Re) in this case, and accept a hydride from pbn. This reactivity has been demonstrated for CO2, which had a binding constant of 40 M−1 to Re(pbn); controlled potential electrolysis experiments (CPE) at −2.11 V vs. Fc+/Fc, furnish CO with 70% faradaic efficiency, where Fc = ferrocene. More work has been done by the group of Tanaka with Rh(pbn) complexes, which can also facilitate catalytic HT to CO2 under electrocatalytic conditions.123 Hydricity of the of Re(pbnHH) was bracketed by reaction with trityl cation thermodynamic cycles, giving ΔGH– = ∼84 to ∼99 kcal mol−1. Analogous 9,10-dihydro-10-methylacridine is a stronger hydride donor with ΔGH– = ∼70 kcal mol−1 (Chart 3). The electron-withdrawing effects of the nitrogen and the coordination of the metal centre in Ru(pbnHH) likely led to the more positive hydricity values and lower hydride-donor ability.
We have provided two examples of SCS effects: first, metals coordination can modify the electronic properties of DHP functional groups; and second, molecular design allows for substrate coordination near the dihydroxy functional group of DHP−. Future systems can employ SCS effects to tune the electronics of the DHP− and or substrate.
4. Conclusions and outlook
We have described the influence of the metal ions on hydride formation and HT from M-DHP−s. Broadly speaking, metals lower the electron density on the pyridine ring, which in turn, lowers the reduction potential for electrochemical hydride formation and raises the free energy loss for HT of the M-DHP− relative to the non-metallated DHP− species. In Section 2, we described multiple avenues for synthesis and isolation of M-DHP−s. Alkylation with metal–alkyl reagents can provide access to M-DHP−s in a single step, provided the ligand does not have acidic protons. HT from a hydride donor can selectively provide the metallated 1,2-, 1,3- or 1,4-DHP− species through judicious choice of catalyst, hydride donor or ligand design. Additionally, deprotonation of a DHP to form a M-DHP− uses established deprotonation pathways to form anionic coordination compounds. By having multiple routes to M-DHP− formation, access to a developing library of these complexes becomes easier and will propel future studies, expanding HT chemistry in organic synthesis.Proof-of-principle reactions where HT is achieved from M-DHP− complexes were described in Section 3. A more accurate report for hydride-donating ability of M-DHP−s is warranted along with greater exploration of substrate scope. Based on prior work, it is likely that M-DHP−s can carry out the HT chemistry currently performed by organohydrides in synthetically relevant transformations; and this may lower the reduction potentials and make catalytic cycles such as those for electrocatalysis attainable (Scheme 2). The chemistry of M-DHP−s that are metallated at the C4 pyridyl position is underexplored at this time.
Current challenges in understanding reaction pathways of M-DHP−s often stem from the presence of a metal–hydride species and M-DHP− complex. This creates ambiguity in identifying the active HT species in solution, and therefore, difficulties in redesigning next-generation HT reagents. Post-synthetic modifications also offer an attractive way to further tune HT chemistry. For example, metal coordination to the pyridyl N of main-group-coordinated pbn systems tunes the metal centre. The resulting bimetallic systems can marry established transition-metal chemistry with metal-ion influences on DHP chemistry. Less electron-rich rings are more prone to metallation, and this metallation then can bring HT pathways facilitated by through-bond polarisation effects on the substrate or by the positioning of reactive groups.
Taken together, the history of metallated DHP− reagents offer a broad scope for further development of reaction chemistry particularly in the context of current interest in electrochemically driven organic transformations. Along with lowered energy demands, lower reduction potentials for ET provide benefits such as chemo- and regio-selectivity and the suppression of background H2 evolution. To expand the scope of M-DHP− chemistry, chemists can capitalise on the wealth of existing pyridine-centred organic scaffolds and ligands and develop metallation routes to produce tuneable HT reaction chemistry.
Author contributions
All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This manuscript is based on work supported by the National Science Foundation with award CHE-2054529.Notes and references
- S. Simic, E. Zukic, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Shortening synthetic routes to small molecule active pharmaceutical ingredients employing biocatalytic methods, Chem. Rev., 2022, 122, 1052–1126 CrossRef CAS PubMed.
- J. Wen, F. Wang and X. Zhang, Asymmetric hydrogenation catalyzed by first-row transition metal complexes, Chem. Soc. Rev., 2021, 50, 3211–3237 RSC.
- Z. An and J. Li, Recent advances in the catalytic transfer hydrogenation of furfural to furfuryl alcohol over heterogeneous catalysts, Green Chem., 2022, 24, 1780–1808 RSC.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, A Continuum of Proton-Coupled Electron Transfer Reactivity, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS.
- E. Carosati, P. Ioan, M. Micucci, F. Broccatelli, G. Cruciani, B. S. Zhorov, A. Chiarini and R. Budriesi, 1,4-Dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 2): action in other targets and antitargets, Curr. Med. Chem., 2012, 19, 4306–4323 CrossRef CAS.
- M. Rueping, J. Dufor and F. R. Schopke, Advances in Catalytic Metal-Free Reductions: From Bio-Inspired Concepts to Applications in the Organocatalytic Synthesis of Pharmaceuticals and Natural Products, Green Chem., 2011, 13, 1084–1105 RSC.
- A. M. F. Phillips and A. J. L. Pomberio, Recent Advances in Organocatalytic Enantioselective Transfer Hydrogenation, Org. Biomol. Chem., 2017, 15, 2307–2340 RSC.
- Z.-H. Zhu, Y.-X. Ding and Y.-G. Zhou, Transfer-catalyst-free biomimetic asymmetric reduction of 3-sulfonyl coumarins with a regenerable NAD(P)H model, Chem. Commun., 2022, 58, 3973–3976 RSC.
- G. Hamasaka, H. Tsuji, M. Ehara and Y. Uozumi, Mechanistic insight into the catalytic hydrogenation of nonactivated aldehydes with a Hantzsch ester in the presence of a series of organoboranes: NMR and DFT studies, RSC Adv., 2019, 9, 10201–10210 RSC.
- S. G. Ouellet, A. M. Walju and D. W. C. MacMillan, Enantioselective organocatalytic transfer hydrogenation reactions using hantzsch esters, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed.
- A. Erkkila, I. Majander and P. M. Pihko, Iminium catalysis, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed.
- J. W. Yang, M. T. Hechavarria Fonseca, N. Vignola and B. List, Metal-free, organocatalytic asymmetric transfer hydrogenation of α,β-unsaturated aldehydes, Angew. Chem., Int. Ed., 2005, 44, 108–110 CrossRef CAS PubMed.
- K. Akagawa, H. Akabane, S. Sakamoto and K. Kudo, Asymmetric transfer hydrogenation in aqueous media catalyzed by resin-supported peptide having a polyleucine tether, Tetrahedron: Asymmetry, 2009, 20, 461–466 CrossRef CAS.
- L. Bernardi and M. Fochi, A general catalytic enantioselective transfer hydrogenation reaction of β,β-disubstituted nitroalkenes promoted by a simple organocatalyst, Molecules, 2016, 21, 1000 CrossRef PubMed.
- A. M. F. Phillips and A. J. L. Pombeiro, Applications of hantzsch esters in organocatalytic enantioselective synthesis, Catalysis, 2023, 13, 419 Search PubMed.
- N. J. A. Martin, L. Ozores and B. List, Organocatalytic asymmetric transfer hydrogenation of nitroolefins, J. Am. Chem. Soc., 2007, 129, 8976–8977 CrossRef CAS PubMed.
- A. Midya, L. D. Khalse and P. Ghorai, Chiral amine catalyzed reductive aldol/reductive michael addition cascade towards enantioselective synthesis of benzannulated diquinanes, Eur. J. Org. Chem., 2023, 26, e20220140 CrossRef.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Thermodynamic and kinetic hydricities of metal-free hydrides, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Importance of proton-coupled electron transfer in cathodic regeneration of organic hydrides, Chem. Commun., 2019, 55, 5583–5586 RSC.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, Hydricity of tansition-metal hydrides: thermodynamic considerations for CO2 reduction, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Thermodynamic and kinetic hydricities of metal-free hydrides, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC.
- K. Kim and B. V. Plapp, Substitutions of Amino Acid Residues in the Substrate Binding Site of Horse Liver Alcohol Dehydrogenase Have Small Effects on the Structures but Significantly Affect Catalysis of Hydrogen Transfer, Biochemistry, 2020, 59, 862–879 CrossRef CAS.
- F. Kudo, T. Tsunoada, K. Yamaguchi, A. Miyanga and T. Eguchi, Stereochemistry in the Reaction of the myo-Inositol Phosphate Synthase Ortholog Ari2 during Aristeromycin Biosynthesis, Biochemistry, 2019, 58, 5112–5116 CrossRef CAS PubMed.
- B. Desguin, T. Zhange, P. Soumillion, P. Hols, J. Hu and R. P Hausinger, A Tethered Niacin-Derived Pincer Complex with a Nickel-Carbon Bond in Lactate Racemase, Science, 2015, 349, 66–69 CrossRef CAS PubMed.
- J. L. Nevarez, A. Turmo, J. Hu and R. P. Hausinger, Biological Pincer Complexes, ChemCatChem, 2020, 12, 4242–4254 CrossRef CAS.
- E. Graczer, A. Pallo, J. Olah, T. Szimler, P. V. Konarev, D. I. Severgun, A. Merli, P. Zavodsky, M. S. Weiss and M. Vas, Glutamate 270 Plays an Essential Role in K+-Activation and Domain Closure of Thermus Thermophilus Isopropylmalate Dehydrogenase, FEBS Lett., 2015, 589, 240–245 CrossRef CAS PubMed.
- F. Quitterer, P. Beck, A. Bacher and M. Groll, Structure and Reaction Mechanism of Pyrrolysine Synthase, Angew. Chem., Int. Ed., 2013, 52, 7033–7037 CrossRef CAS.
- A. Pallo, J. Olah, E. Graczer, A. Merli, P. Zavodsky, M. S. Weiss and M. Vas, Structural and Energetic Basis of Isopropylmalate Dehydrogenase Enzyme Catalysis, FEBS J., 2014, 281, 5063–5076 CrossRef CAS PubMed.
- S. Chatterjee, S. Gatreddi, S. Gupta, J. L. Nevarez, J. A. Rankin, A. Turmo, J. Hu and R. P. Hausinger, Unveiling the Mechanisms and Biosynthesis of a Novel Nickel-Pincer Enzyme, Biochem. Soc. Trans., 2022, 50, 1187–1196 CrossRef CAS PubMed.
- P. Marcheroux, B. Kappes and S. E. Ealick, Flavogenomics – A Genomic and Structural View of Flavin-Dependent Proteins, FEBS J., 2011, 278, 2625–2634 CrossRef PubMed.
- G. Gadda, Hydride Transfer Made Easy in the Reaction of Alcohol Oxidation Catalyzed by Flavin-Dependent Oxidases, Biochemistry, 2008, 47, 13745–13753 CrossRef CAS.
- S. Itoh, H. Kawakami and S. Fukuzumi, Electrochemical Behavior and Characterization of Semiquinone Radical Anion Species of Coenzyme PQQ in Aprotic Organic Media, J. Am. Chem. Soc., 1998, 120, 7271–7277 CrossRef CAS.
- A. McSkimming, T. Cheisson, P. J. Carroll and E. J. Schelter, Functional Synthetic Model for the Lanthanide-Dependent Quinoid Alcohol Dehydrogenase Active Site, J. Am. Chem. Soc., 2018, 140, 1223–1226 CrossRef CAS PubMed.
- A. Anne, J. Moiroux and J.-M. Saveant, Formal Hydride Transfer from NADH Analouges 1-Benzyl-4-tert-butyl-1,4 dihydronicotinamide as a Mechanistic Probe, J. Am. Chem. Soc., 1993, 115, 10224–10230 CrossRef CAS.
- Y. Hiaro, H. Eto, M. Teraoka and T. Kubo, A Strong Hydride Donating, Acid Stable and Reusable 1,4-Dihydropyridine for Selective Aldimine and Aldehyde Reductions, Org. Biomol. Chem., 2022, 20, 1671–1679 RSC.
- G. Bochao, W. Meng, X. Feng and H. Du, Regenerable Dihydrophenanthridine via Borane-Catalyzed Hydrogenation for the Asymmetric Transfer Hydrogenation of Benzoxazinones, Org. Lett., 2022, 24, 3955–3959 CrossRef PubMed.
- Z.-B. Zhao, J. Wang, Z.-H. Zhu, M.-W. Chen and Y.-G. Zhou, Enantioselective Synthesis of 2-Functionalized Tetrahydroquinolines through Biomimetic Reduction, Org. Lett., 2021, 23, 9112–9117 CrossRef CAS PubMed.
- C.-B. Bai, N.-X. Wang, Y. Xing and X.-W. Lan, Progress on Chiral NAD(P)H Model Compounds, Synlett, 2017, 28, 402–414 CAS.
- A. N. Kim and B. M. Stoltz, Recent Advances in Homogeneous Catalysts for the Asymmetric Hydrogenation of Heteroarenes, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS.
- F. Zhang, J. Jia, S. Dong, W. Wang and C.-H. Tung, Hydride Transfer from Iron(II) Hydride Compounds to NAD(P)+ Analouges, Organometallics, 2016, 35, 1151–1159 CrossRef CAS.
- A. Parthiban and P. Makam, 1,4-Dihydropyridine: Synthetic Advances, Medicinal and Insecticidal Properties, RSC Adv., 2022, 12, 29253–29290 RSC.
- C. E. Housecroft and A. G. Sharpe, in Inorganic Chemistry, Pearson, 4th edn, 2012, ch. 7, pp. 233 Search PubMed.
- D. Mauzerall and F. H. Westheimer, 1-Benzyldihydronicotinamide – A model for reduced DPN, J. Am. Chem. Soc., 1955, 8, 2261–2264 CrossRef.
- A. Hantzsch, Condensationsprodukte aus Aldehydammoniak und ketonartigen Verbindungen, Ber. Dtsch. Chem. Ges., 1881, 14, 1637–1638 CrossRef.
- C. Zheng and S.-L. You, Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- J. Auth, J. Padevet, P. Mauleon and A. Pfaltz, Pyridylidene-Mediated Dihydrogen Activation Coupled with Catalytic Imine Reduction, Angew. Chem., Int. Ed., 2015, 54, 9542–9545 CrossRef CAS PubMed.
- I.-S. H. Lee, H. J. Kil and Y. R. Ji, Reactivities of Acridine Compounds in Hydride Transfer Reactions, J. Phys. Org. Chem., 2007, 20, 484–490 CrossRef CAS.
- P. K. Giesbrecht, D. B. Nemez and D. E. Herbert, Electrochemical Hydrogenation of a Benzannulated Pyridine to a Dihydropyridine in Acidic Solution, Chem. Commun., 2018, 54, 338–341 RSC.
- Q.-A. Chen, K. Gao, Y. Duan, Z.-S. Ye, L. Shi, Y. Yang and Y.-G. Zhou, Dihydrophenanthridine: A New and Easily Regenerable NAD(P)H Model for Biomimetic Asymmetric Hydrogenation, J. Am. Chem. Soc., 2012, 134, 2442–2448 CrossRef CAS PubMed.
- W. W. Ellis, J. W. Raebiger, C. J. Curtis, J. W. Bruno and D. L. Dubois, Hydricities of BzNADH, C5H5Mo(PMe3)(CO)2H, and C5Me5Mo(PMe3)(CO)2H in Acetonitrile, J. Am. Chem. Soc., 2004, 126, 2378–2734 CrossRef.
- J. Wang, Z.-H. Zhu, M.-W. Chen, Q.-A. Chen and Y.-G. Zhou, Catalytic Biomimetic Asymmetric Reduction of Alkenes and Imines Enabled by Chiral and Regenerable NAD(P)H Models, Angew. Chem., Int. Ed., 2019, 58, 1813–1817 CrossRef CAS PubMed.
- J. Wang, Z.-B. Zhao, Y. Zhao, G. Luo, Z.-H. Zhu, Y. Luo and Y.-G. Zhou, Chiral and Regenerable NAD(P)H Models Enabled Biomimetic Asymmetric Reduction: Design, Synthesis, Scope, and Mechanistic Studies, J. Org. Chem., 2020, 85, 2355–2369 CrossRef CAS PubMed.
- Z.-H. Zhu, Y.-X. Ding, B. Wu and Y.-G. Zhou, Biomimetic Asymmetric Reduction of Tetrasubstituted Olefin 2,3-Disubstituted Inden-1-ones with Chiral and Regenerable NAD(P)H Model CYNAM, Org. Lett., 2021, 23, 7166–7170 CrossRef CAS.
- R. Arévalo, M. Espinal-Viguri, M. A. Huertos, J. Pérez and L. Riera, Dearomatization of transition metal-coordinated N-heterocyclic ligands and related chemistry, Adv. Organomet. Chem., 2016, 65, 47–114 CrossRef.
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 1st edn, 2009 Search PubMed.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley and Sons, 5th edn, 2009 Search PubMed.
- J. J. Sandoval, P. Palma, E. Alvarez, A. Rodriguez-Delgado and J. Campora, Dibenzyl and Diallyl 2,6-Bisiminopyridinezinc(II) Complexes: Selective Alkyl Migration to the Pyridine Ring Leads to Remarkably Stable Dihydropyridinates, Chem. Commun., 2013, 49, 6791–6793 RSC.
- J. J. Sandoval, P. Palma, E. Alvarez, J. Campora and A. Rodríguez-Delgado, Mechanism of Alkyl Migration in Diorganomagnesium 2,6-Bis(imino)pyridine Complexes: Formation of Grignard-Type Complexes with Square-Planar Mg(II) Centers, Organometallics, 2016, 35, 3197–3204 CrossRef CAS.
- M. Arrowsmith and M. S. Hill, Magnesium-Catalyzed Hydroboration of Pyridines, Organometallics, 2011, 30, 5556–5559 CrossRef CAS.
- A. R. Kennedy, R. E. Mulvey, R. I. Urgqhart and S. D. Robertson, Lithium, Sodium and Potassium Picolyl Complexes: Syntheses, Structures and Bonding, Dalton Trans., 2014, 43, 14265–14274 RSC.
- C. Gunanathan and D. Milstein, Metal-ligand Cooperation by Aromatization-Dearomatization: A New Paradigm in Bond Activation and “Green” Catalysis, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- T. Zell and D. Milstein, Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal-Ligand Cooperation by Aromatization/Dearomatization, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- C. D. Cates, T. W. Meyers and L. A. Berben, (IP)2GaIII Complexes Facilitate Net Two-Electron Redox Transformations (IP = α-Iminopyridine), Inorg. Chem., 2012, 51, 11891–11897 CrossRef CAS PubMed.
- T. Meyers and L. A. Berben, Redox Active Aluminum(III) Complexes Convert CO2 into MgCO3 or CaCO3 in a Synthetic Cycle Using Mg or Ca Metal, Chem. Commun., 2013, 49, 4175–4177 RSC.
- T. M. Meyers and L. A. Berben, Aluminum-Amido-Mediated Heterolytic Addition of Water Affords and Alumoxane, Organometallics, 2013, 32, 6647–6649 CrossRef.
- R. E. Mulvey, Avant-Garde Metalating Agents: Structural Basis of Alkali-Metal-Mediated Metalation, Acc. Chem. Res., 2009, 42, 743–755 CrossRef CAS.
- J. Garcia-Alvarez, E. Hevia and V. Capriati, Reactivity of Polar Organometallic Compounds in Unconventional Reaction Media: Challenges and Opportunities, Eur. J. Org. Chem., 2015, 31, 6779–6799 CrossRef.
- M. Uzelac and E. Hevia, Polar Organometallic Strategies for Regioselective C–H Metallation of N-Heterocyclic Carbenes, Chem. Commun., 2018, 54, 2455–2462 RSC.
- S. D. Robertson, A. R. Kennedy, J. J. Liggat and R. E. Mulvey, Facile Synthesis of a Genuinely Alkane-Soluble but Isolable Lithium Hydride Transfer Reagent, Chem. Commun., 2015, 51, 5452–5455 RSC.
- D. R. Armstrong, C. M. M. Harris, A. R. Kennedy, J. J. Liggat, R. McLellan, R. E. Mulvey, M. D. T. Urquhart and S. D. Robertson, Developing Lithium Chemistry of 1,2-Dihydropyridines: From Kinetic Intermediates to Isolable Characterized Compounds, Chem. – Eur. J., 2015, 21, 14410–14420 CrossRef CAS PubMed.
- S. A. Orr, A. R. Kennedy, J. J. Liggat, R. McLellan, R. E. Mulvey and S. D. Robertson, Accessible heavier s-block dihydropyridines: structural elucidation and reactivity of isolable molecular hydride sources, Dalton Trans., 2016, 45, 6234–6240 RSC.
- L. Hao, J. F. Harrod, A.-M. Lebuis, R. Shu, E. Samuel and H.-G. Woo, Homogenous catalytic hydrosilylation of pyridines, Angew. Chem., Int. Ed., 1998, 37, 3126–3129 CrossRef CAS.
- F. J. Harrod, R. Shu, H.-G. Woo and E. Samuel, Titanocene(III) Catalyzed Homogeneous Hydrosilation Hydrogenation of Pyridines, Can. J. Chem., 2001, 79, 1075–1085 CrossRef.
- D. V. Gutsulyak, A. van der Est and G. I. Nikonov, Facile Catalytic Hydrosilylation of Pyridines, Angew. Chem., Int. Ed., 2011, 50, 1384–1387 CrossRef CAS PubMed.
- M. S. Hill, D. J. MacDougall, G. Kociok-Kohn, M. F. Mahon and C. Weetman, Kinetically Directed Reactivity of Magnesium Dihydropyridides with Organoisocyanates, Organometallics, 2015, 34, 2509–2599 Search PubMed.
- A. Kaithal, B. Chatterjee and C. Gunanathan, Ruthenium-Catalyzed Regioselective 1,4-hydroboration of Pyridines, Org. Lett., 2016, 18, 3402–3405 CrossRef CAS PubMed.
- J. L. Lortie, T. Dudding, B. M. Gabidullin and G. I. Nikonov, Zinc-Catalyzed Hydrosilylation and Hydroboration of N-Heterocycles, ACS Catal., 2017, 7, 8454–8459 CrossRef CAS.
- J. Liu, J.-Y. Chen, M. Jia, B. Ming, J. Jia, R.-Z. Liao, C.-H. Tung and W. Wang, Ni−O Cooperation versus Nickel(II) Hydride in Catalytic Hydroboration of N-Heteroarenes, ACS Catal., 2019, 9, 3849–3857 CrossRef CAS.
- S. Bähr and M. Oestreich, A Neutral RuII Hydride Complex for the Regio- and Chemoselective Reduction of N-Silylpyridinium Ions, Chem. – Eur. J., 2018, 24, 5613–5622 CrossRef.
- S. Park, Recent Advances in Catalytic Dearomative Hydroboration of N-heteroarenes, ChemCatChem, 2020, 12, 3170–3185 CrossRef CAS.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- X. Fan, J. Zheng, Z. H. Li and H. Wang, Organoborane Catalyzed Regioselective 1,4-Hydroboration of Pyridines, J. Am. Chem. Soc., 2015, 137, 4916–4919 CrossRef CAS.
- E. N. Keyzer, S. S. Kang, S. Hanf and D. S. Wright, Regioselective 1,4-Hydroboration of Pyridines Catalyzed by an Acid-Initiated Boronium Cation, Chem. Commun., 2017, 53, 9434–9437 RSC.
- S. Kundu, W. W. Brennessel and W. D. Jones, Synthesis and Reactivity of New Ni, Pd, and Pt 2,6-Bis(di-tert-butylphosphinito)pyridine Pincer Complexes, Inorg. Chem., 2011, 50, 9443–9453 CrossRef CAS PubMed.
- R. Fandos, A. Rodriguez-Delgado, A. Rodriguez, I. Romero, J. A. Organero and E. Alvarez, Pathways to Metal-Ligand Cooperation in Quinoline-Based Titanium(IV) Pincers: Nonelectrophilic N-methylation, Deprotonation, and Dihydropyridine Formation, Organometallics, 2021, 40, 1838–1847 CrossRef CAS.
- J. J. Sondoval, E. Alvarez, P. Palma, A. Rodriguez-Delgado and J. Campora, Neutral Bis(imino)-1,4-dihydropyridinate and Cationic Bis(imino)pyridine σ-Alkylzinc(II) Complexes as Hydride Exchange Systems: Classic Organometallic Chemistry Meets Ligand-Centered, Biomimetic Reactivity, Organometallics, 2018, 37, 1734–1744 CrossRef.
- S. Lapointe, E. Khaskin, R. R. Fayzullin and J. R. Khusnutdinova, Nickel(II) Complexes with Electron-Rich, Sterically Hindered PNP Pincer Ligands Enable Uncommon Modes of Ligand Dearomatization, Organometallics, 2019, 38, 4433–4447 CrossRef CAS.
- T. Mehdoui, J.-C. Berthet, P. Thuery, L. Salmon, E. Riviere and M. Ephritikhine, Lanthanide(III)/Actinide(III) Differentiation in the Cerium and Uranium Complexes [M(C5Me5)2(L)]0,+ (L=2,2’-Bipyridine, 2,2’:6’,2’’-Terpyridine): Structural, Magnetic, and Reactivity Studies, Chem. – Eur. J., 2005, 11, 6994–7006 CrossRef CAS.
- T. J. Sherbow, L. W. T. Parsons, N. A. Phan, J. C. Fettinger and L. A. Berben, Ligand Conjugation Directs the Formation of a 1,3-Dihydropyridinate Regioisomer, Inorg. Chem., 2020, 59, 17614–17619 CrossRef CAS PubMed.
- J. Intemann, H. Bauer, J. Pahl, L. Maron and S. Harder, Calcium Hydride Catalyzed Highly 1,2-Selective Pyridine Hydrosilylation, Chem. – Eur. J., 2015, 21, 11452–11461 CrossRef CAS.
- T. J. Sherbow, J. C. Fettinger and L. A. Berben, Control of Ligand pKa Values Tunes the Electrocatalytic Dihydrogen Evolution Mechanism in a Redox-Active Aluminum(III) Complex, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed.
- P. Daw, A. Kumar, D. Oren, N. A. Espinosa-Jalapa, D. Srimani, Y. Diskin-Posner, G. Letius, L. J. W. Shimon, R. Carmieli, Y. Ben-David and D. Milstein, Redox Noninnocent Nature of Acridine-Based Pincer Complexes of 3d Metals and C−C Bond Formation, Organometallics, 2020, 39, 279–285 CrossRef CAS.
- G. Li, M. Lamberti, D. Pappalardo and C. Pellecchia, Random Copolymerization of ε-Caprolactone and Lactides Promoted by Pyrrolylpyridylamido Aluminum Complexes, Macromolecules, 2012, 45, 8614–8620 CrossRef CAS.
- M. Gallardo-Villagrán, F. Vidal, P. Palma, E. Álvarez, E. Y.-X. Chen, J. Cámpora and A. Rodríguez-Delgado, Aluminium(III) Dialkyl 2,6-Bisimino-4R-Dihydropyridinates(−1): Selective Synthesis, Structure and Controlled Dimerization, Dalton Trans., 2019, 48, 9104–9116 RSC.
- K. T. Horak, D. G. VanderVelde and T. Agapie, Tuning of Metal Complex Electronics and Reactivity by Remote Lewis Acid Binding to π-Coordinated Pyridine Diphosphine Ligands, Organometallics, 2015, 34, 4753–4765 CrossRef CAS.
- R. Shi, M. D. Wodrich, H.-J. Pan, F. F. Tirani and X. Hu, Functional Models of the Nickel Pincer Nucleotide Cofactor of Lactate Racemase, Angew. Chem., Int. Ed., 2019, 58, 16869–16872 CrossRef CAS PubMed.
- T. Xu, M. D. Wodrich, R. Scopelliti, C. Corminbeuf and X. Hu, Nickel Pincer Model of the Active Site of Lactate Racemase Involves Ligand Participation in Hydride Transfer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1241–1245 Search PubMed.
- E. J. Thompson and L. A. Berben, Electrocatalytic Hydrogen Production by an Aluminum(III) Complex: Ligand-Based Proton and Electron Transfer, Angew. Chem., Int. Ed., 2015, 54, 11642–11646 CrossRef CAS.
- T. W. Myers, T. J. Sherbow, J. C. Fettinger and L. A. Berben, Synthesis and Characterization of Bis(imino)pyridine Complexes of Divalent Mg and Zn, Dalton Trans., 2016, 45, 5989–5998 RSC.
- Z. Li, J.-D. Yang and J.-P. Cheng, Thermodynamic and Kinetic Studies of Hydride Transfer from Hantzsch Ester Under the Promotion of Organic Bases, Org. Chem. Front., 2021, 8, 876–882 RSC.
- S. Coffie, J. M. Hogg, L. Cailler, A. Ferrer-Ugalde, R. W. Murphy, J. D. Holbrey, F. Coleman and M. Swadzba-Kwasny, Lewis Superacidic Ionic Liquids with Tricoordinate Borenium Cations, Angew. Chem., Int. Ed., 2015, 127, 15183–15186 CrossRef.
- J. E. Radcliffe, J. J. Dunsford, J. Cid, V. Fasano and M. J. Ingleson, N-Heterocycle-Ligated Borocations as Highly Tunable Carbon Lewis Acids, Organometallics, 2017, 36, 4952–4960 CrossRef CAS PubMed.
- E. J. Lawrence, E. R. Clark, L. D. Curless, J. M. Courtney, R. J. Blagg, M. J. Ingleson and G. G. Wildgoose, Metal-free electrocatalytic hydrogen oxidation using frustrated Lewis pairs and carbon-based Lewis acids, Chem. Sci., 2016, 7, 2537–2543 RSC.
- S. Ilic, U. P. Kadel, Y. Basdogan, J. A. Keith and K. D. Glusac, Thermodynamic Hydricities of Biomimetic Organic Hydride Donors, J. Am. Chem. Soc., 2018, 140, 4569–4579 CrossRef CAS PubMed.
- P. Hapiot, J. Moiroux and J.-M. Savent, Electrochemistry of NADH/NAD+ Analogues. A Detailed Mechanistic Kinetic and Thermodynamic Analysis of the 10-Methylacridan/10-Methylacridimium Couple in Acetonitrile, J. Am. Chem. Soc., 1990, 112, 1337–1343 CrossRef CAS.
- A. Anne, P. Hapiot, J. Moiroux and J.-M. Savient, Electrochemistry of Synthetic Analogues of NAD Dimers, J. Electroanal. Chem., 1992, 331, 959–970 CrossRef CAS.
- R. McLellan, A. R. Kennedy, S. A. Orr, S. D. Robertson and R. E. Mulvey, Lithium Dihydropyridine Dehydrogenation Catalysis: A Group 1 Approach to the Cyclization of Diamine Boranes, Angew. Chem., Int. Ed., 2017, 56, 1036–1041 CrossRef CAS PubMed.
- R. McLellan, A. R. Kennedy, R. E. Mulvey, S. A. Orr and S. D. Robertson, 1-Alkali-metal-2-alkyl-1,2-dihydropyridines: Soluble Hydride Surrogates for Catalytic Dehydrogenative Coupling and Hydroboration Applications, Chem. – Eur. J., 2017, 23, 16853–16861 CrossRef CAS PubMed.
- R. P. Yu, J. M. Darmon, C. Milsmann, G. W. Margulieux, S. C. E. Stieber, D. DeBeer and P. J. Chirik, Catalytic Hydrogenation Activity and Electronic Structure Determination of Bis(arylimidazol-2-ylidene)pyridine Cobalt Alkyl and Hydride Complexes, J. Am. Chem. Soc., 2013, 135, 13168–13184 CrossRef CAS PubMed.
- R. Arevalo, R. López, L. R. Falvello, L. Riera and J. Perez, Building C(sp3) Molecular Complexity on 2,2’-Bipyridine and 1,10-Phenanthroline in Rhenium Tricarbonyl Complexes, Chem. – Eur. J., 2021, 27, 379–389 CrossRef CAS PubMed.
- G. Zhang, J. Wu, H. Zeng, M. C. Neary, M. Devany, S. Zheng and P. A. Dub, Dearomatization and Functionalization of Terpyridine Ligands Leading to Unprecedented Zwitterionic Meisenheimer Aluminum Complexes and Their Use in Catalytic Hydroboration, ACS Catal., 2019, 9, 874–884 CrossRef CAS.
- G. Zhang, J. Wu, S. Zheng, M. C. Neary, J. Mao, M. Flores, R. J. Trovitch and P. A. Dub, Redox-Noninnocent Ligand-Supported Vanadium Catalysts for the Chemoselective Reduction of C=X (X = O, N) Functionalities, J. Am. Chem. Soc., 2019, 141, 15230–15239 CrossRef CAS PubMed.
- G. Zhang, H. Zeng, S. Zheng, M. C. Neary and P. A. Dub, Vanadium-Catalyzed Stereo- and Regioselective Hydroboration of Alkynes to Vinyl Boronates, ACS Catal., 2022, 12, 5425–5429 CrossRef CAS.
- S. Kundu, W. W. Brennessel and W. D. Jones, Making M–CN bonds from M–Cl in (PONOP)M and (dippe)Ni Systems (M = Ni, Pd, and Pt) using t-BuNC, Inorg. Chim. Acta, 2011, 379, 109–114 CrossRef CAS.
- B. Lee, T. P. Pabst and P. J. Chirik, Effect of Pincer Methylation on the Selectivity and Activity in (PNP)Cobalt-Catalyzed C(sp2)−H Borylation, Organometallics, 2021, 40, 3766–3774 CrossRef CAS PubMed.
- J. V. Obligacion, S. P. Semproni, I. Pappas and P. J. Chirik, Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst Design, J. Am. Chem. Soc., 2016, 138, 10645–10653 CrossRef CAS PubMed.
- S. Lapointe, D. K. Pandey, J. M. Gallagher, J. Osborne, R. R. Fayzullin, E. Khaskin and J. R. Khusnutdinova, Cobalt Complexes of Bulky PNP Ligand: H2 Activation and Catalytic Two-Electron Reactivity in Hydrogenation of Alkenes and Alkynes, Organometallics, 2021, 40, 3617–3626 CrossRef CAS.
- Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, Selective C-4 Alkylation of Pyridine by Nickel/Lewis Acid Catalysis, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS PubMed.
- P. Naweephattana, B. Sawatlon and P. Surawatanawong, Insights into the Regioselectivity of Hydroheteroarylation of Allylbenzene with Pyridine Catalyzed by Ni/AlMe3 with N-Heterocyclic Carbene: The Concerted Hydrogen Transfer Mechanism, J. Org. Chem., 2020, 85, 11340–11349 CrossRef CAS.
- A. Kobayashi, H. Konno, K. Sakamoto, A. Sekine, Y. Ohashi, M. Iida and O. Ishitani, Transition Metal Complexes Coordinated by an NAD(P)H Model Compound and their Enhanced Hydride-Donating Abilities in the Presence of a Base, Chem. – Eur. J., 2005, 11, 4219–4226 CrossRef CAS PubMed.
- K. Koga, Y. Matsubara, T. Kosaka, K. Koike, T. Morimoto and O. Ishitani, Hydride Reduction of NAD(P)+ Model Compounds with a Ru(II)−Hydrido Complex, Organometallics, 2015, 34, 5530–5539 CrossRef CAS.
- Y. Matsubara, S. E. Hightower, J. Chen, D. C. Grills, D. E. Polyansky, J. T. Muckerman, K. Tanaka and E. Fujita, Reactivity of a fac-ReCl(α-Diimine)(CO)3 Complex with an NAD+ Model Ligand Toward CO2 Reduction, Chem. Commun., 2014, 50, 728–730 RSC.
- D. Ghosh, K. Kobayashi, T. Kajiwara, S. Kitagawa and K. Tanaka, Catalytic Hydride Transfer to CO2 using Ru-NAD-Type Complexes under Electrochemical Conditions, Inorg. Chem., 2017, 56, 11066–11073 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |