
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vacuum ultraviolet photodissociation of sulfur dioxide and its implications for oxygen production in the early Earth's atmosphere†
Yao
Chang‡
 a,
Yanlin
Fu‡
a,
Zhichao
Chen‡
a,
Zijie
Luo
ab,
Yarui
Zhao
a,
Zhenxing
Li
a,
Weiqing
Zhang
a,
Guorong
Wu
a,
Bina
Fu
*ade,
Dong H.
Zhang
aef,
Michael N. R.
Ashfold
c,
Xueming
Yang
*aef and
Kaijun
Yuan
*ade
a,
Yanlin
Fu‡
a,
Zhichao
Chen‡
a,
Zijie
Luo
ab,
Yarui
Zhao
a,
Zhenxing
Li
a,
Weiqing
Zhang
a,
Guorong
Wu
a,
Bina
Fu
*ade,
Dong H.
Zhang
aef,
Michael N. R.
Ashfold
c,
Xueming
Yang
*aef and
Kaijun
Yuan
*ade
aState Key Laboratory of Molecular Reaction Dynamics, Dalian Coherent Light Source, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, 116023, China. E-mail: kjyuan@dicp.ac.cn; bina@dicp.ac.cn; xmyang@dicp.ac.cn
bMarine Engineering College, Dalian Maritime University, Liaoning 116026, China
cSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK
dUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
eHefei National Laboratory, Hefei 230088, China
fDepartment of Chemistry, Center for Advanced Light Source Research, College of Science, Southern University of Science and Technology, Shenzhen 518055, China
First published on 1st August 2023
Abstract
The emergence of molecular oxygen (O2) in the Earth's primitive atmosphere is an issue of major interest. Although the biological processes leading to its accumulation in the Earth's atmosphere are well understood, its abiotic source is still not fully established. Here, we report a new direct dissociation channel yielding S(1D) + O2(a1Δg/X3Σg−) products from vacuum ultraviolet (VUV) photodissociation of SO2 in the wavelength range between 120 and 160 nm. Experimental results show O2 production to be an important channel from SO2 VUV photodissociation, with a branching ratio of 30 ± 5% at the H Lyman-α wavelength (121.6 nm). The relatively large amounts of SO2 emitted from volcanic eruptions in the Earth's late Archaean eon imply that VUV photodissociation of SO2 could have provided a crucial additional source term in the O2 budget in the Earth's primitive atmosphere. The results could also have implications for abiotic oxygen formation on other planets with atmospheres rich in volcanically outgassed SO2.
Introduction
The provenance of oxygen (O2) remains a crucial topic in the history of the Earth's evolution. The Earth's present atmosphere is notable for its remarkably high concentration of O2 (∼21% by volume), whereas geological and geochemical constraints suggest that free oxygen was anything but plentiful during the first half of the Earth's 4.5 billion year history.1–3 A permanent rise to appreciable concentrations of O2 in the atmosphere, known as the “Great Oxidation Event (GOE)”,4–8 is estimated to have occurred ∼2.4 billion years ago (2.4 Ga). The GOE could have been a consequence of the emergence of oxygenic photosynthesis. Alternatively, O2 biogenesis may be much older; several pieces of evidence point to the first emergence of oxygenic photosynthesis long before the GOE (as early as ∼3.8 Ga).9–14 If so, the emergence of the GOE could have been a consequence of an abiotic shift in the balance of oxidants and reductants at the Earth's surface, i.e., at early times, the biogenically produced O2 was effectively consumed via reaction with reduced compounds, thereby suppressing O2 levels (<10−6 of the present atmospheric level (PAL))15–17 but, eventually, this source-sink balance shifted in favour of O2 accumulation. Recent trace metal studies, e.g. of molybdenum and rhenium enrichment in the crust,10,18 suggested “whiffs” of O2 in the late Archaean (2.5–2.7 Ga), i.e., intermittent periods before the GOE when the O2-sources overwhelmed the sinks. Such whiffs of oxygen have been suggested as a possible trigger for the GOE, but their duration, magnitude, and sources remain unknown.Apart from biogenic processes leading to O2 production, the widely accepted abiotic route to forming O2 is the three-body recombination reaction O + O + M → O2 + M, involving O atoms produced by photolysis of CO2 or other oxygen-containing molecules.19–22 Direct O2 production via vacuum ultraviolet (VUV) photodissociation of CO2 has also been identified, but deduced to be a very minor process compared with the indirect three-body recombination.23 Dissociative electron attachment to CO2 has also been shown to lead to direct O2 production.24 Haqq-Misra et al.25 have suggested another abiotic O2 production pathway, in which H2O2 was first produced from the by-products of H2O photolysis, and then converted to O2via disproportionation reactions. Modelled O2 concentrations up to 10−7 PAL were reported. These findings have all informed knowledge of the history of the Earth's atmosphere and our understanding of planetary atmospheres and of interstellar photochemical processes.
SO2 has also been considered as a possible source of oxygen. Astronomical observations have identified high SO2 concentrations in the atmosphere of terrestrial exoplanets like Venus26,27 and Io,28,29 which are largely attributable to outgassing from volcanoes. In the Earth's late Archaean, prior to the GOE, subaerial volcanic degassing became important, yielding gases much richer in sulfur and dominated by SO2.30,31 The photochemistry of volcanic SO2 has been studied extensively in recent decades and has commonly been linked to the origin of the sulfur mass independent fractionation (S-MIF) in ancient rock samples.32–34 SO2 should be the major component from volcano eruptions, but the role of SO2 photochemistry in the formation of molecular oxygen in the Earth's early atmosphere has hitherto been largely ignored.
SO2 has two strong absorption bands in the UV region (see Section S2 and Fig. S2 in the ESI†). The more intense band, assigned to excitation from the ground ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 1A1(11A′ in Cs) state to the
1A1(11A′ in Cs) state to the ![[C with combining tilde]](https://www.rsc.org/images/entities/char_0043_0303.gif) 1B2(21A′) state, spans the wavelength range 185 < λ < 235 nm.35 The other strong band at longer wavelengths, λ ∼ 240–350 nm, is associated with transition to the
1B2(21A′) state, spans the wavelength range 185 < λ < 235 nm.35 The other strong band at longer wavelengths, λ ∼ 240–350 nm, is associated with transition to the ![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) 1B1(21A′′) state. The 1B1(21A′′) state potential energy surface displays a conical intersection with that of the first excited singlet state (the Ã1A2(11A′′) state).33,36,37 Weak absorption at yet longer wavelengths is attributed to the spin-forbidden transition to the ã3B1 state. Photoexcitation of SO2 at wavelengths λ ≤ 219 nm results in predissociation, primarily to SO and O fragments, driven by non-adiabatic couplings to the lower-lying dissociative singlet and triplet states.38–41 As Fig. S2† shows, the SO2 absorption spectrum at shorter (VUV) wavelengths displays several intense diffuse absorption bands assigned to transitions to Rydberg states.
1B1(21A′′) state. The 1B1(21A′′) state potential energy surface displays a conical intersection with that of the first excited singlet state (the Ã1A2(11A′′) state).33,36,37 Weak absorption at yet longer wavelengths is attributed to the spin-forbidden transition to the ã3B1 state. Photoexcitation of SO2 at wavelengths λ ≤ 219 nm results in predissociation, primarily to SO and O fragments, driven by non-adiabatic couplings to the lower-lying dissociative singlet and triplet states.38–41 As Fig. S2† shows, the SO2 absorption spectrum at shorter (VUV) wavelengths displays several intense diffuse absorption bands assigned to transitions to Rydberg states.
Early investigations of the VUV photolysis of SO2 found indirect experimental evidence for the S + O2 channel through detection of OH fluorescence from SO2/H2 mixtures.42 More recently, Rosch et al.43 reported the first direct evidence for the S(3P) + O2 channel from SO2 photolysis at 193 nm but, in the absence of quantitative measurements, the assessment of the importance (or otherwise) of O2 production from VUV photodissociation of SO2 in the Earth's primitive atmosphere was not possible. The recent development of the intense VUV free electron laser (FEL) at the Dalian Coherent Light Source (DCLS) provides a unique tool for studying molecular photofragmentation dynamics across the entire VUV range.22,44,45 Here, we present careful experimental studies of the S(1D) + O2 product channel following SO2 photolysis at various wavelengths in the range 120 < λ < 160 nm using the VUV-pump and VUV-probe time-sliced velocity-map imaging (TS-VMI) technique, along with complementary electronic structure calculations. The quantitative assessment of this channel suggests that the VUV photochemistry of SO2 could have been an important additional source of O2 in the Earth's atmosphere in the late Archaean.
Results and discussion
The verification of the S(1D) + O2 product channel
In this study, the photodissociation dynamics of SO2 have been investigated using the recently constructed VUV pump and VUV probe TS-VMI apparatus, which is equipped with two independently tunable VUV laser radiation sources (see Section S1 and Fig. S1 in the ESI†). A pulsed supersonic molecular beam generated from a gas mixture of about 1% SO2 in Ar was irradiated with two counter-propagating VUV beams. The VUV FEL output was used to excite SO2 molecules to different Rydberg states at wavelengths in the range 120 < λ < 160 nm. The S(1D) photofragments were then resonantly ionized with λ = 130.092 nm photons, which were generated using a table-top VUV source and a difference frequency four-wave mixing (FWM) scheme, involving two 212.556 nm photons and one 580.654 nm photon that were overlapped in a Kr gas cell. Post-ionization, the S(1D) photoproducts were detected by the high resolution VMI detector.Fig. 1 shows time-sliced ion images of the S(1D) photofragments recorded following photolysis of SO2 at VUV wavelengths λ = 121.6, 133.1, 140.0 and 150.0 nm, respectively. Additional images, taken at λ = 125.1, 130.1, 144.1 and 154.1 nm, are displayed in Fig. S3 of the ESI.† The double headed arrow in Fig. 1 shows the direction of the polarization vector of the photolysis laser. Well-resolved, concentric rings with different intensities are clearly observable in the displayed images. These structures can be readily assigned to the population of different vibrational levels of the O2 co-product in its ground (X3Σg−) or first excited (a1Δg) electronic state arising via the photodissociation channel (1),
| SO2 + hv → S(1D) + O2. | (1) |
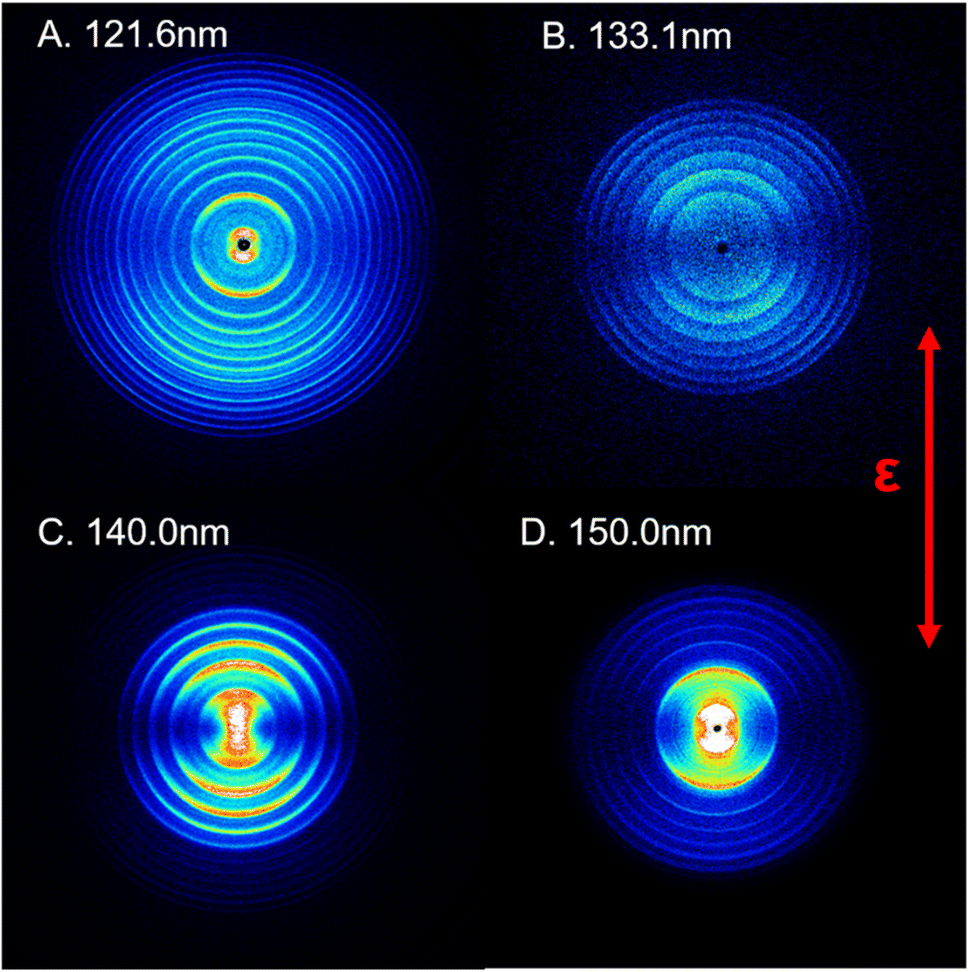 | ||
| Fig. 1 Time-sliced images of the S(1D) products from SO2 photodissociation. The images were recorded at photolysis wavelengths λ = (A) 121.6 nm, (B) 133.1 nm, (C) 140.0 nm and (D) 150.0 nm. The double headed red arrow indicates the polarization direction of the photodissociation laser, ε. The concentric ring features reflect the population of different vibrational levels of the coincident O2(X3Σg−/a1Δg, v) products. | ||
The triple dissociation channel yielding S(1D) + O + O products has a threshold energy of ∼12.3 eV46 and is thus not accessible in the present experiment. The use of an off-axis biconvex LiF lens as the exit window for the four-wave mixing cell ensured that the 212.556 nm and 580.654 nm laser beams were dispersed from the photodissociation/photoionization region, thereby eliminating the possibility of unintended secondary dissociation of any primary SO fragments (from the rival SO + O dissociation channel) by absorption of another UV or IR photon. In addition, both the VUV FEL beam and the 130.092 nm probe beam were kept defocused to minimize any two-photon excitation effects. These steps ensured that no processes other than channel (1) yielded S(1D) fragments under the prevailing experimental conditions.
The thermochemical threshold for process (1), i.e. D0[SO2(, v = 0) → S(1D) + O2(X, v = 0)], is ∼7.1 eV (corresponding to an excitation wavelength, λ ∼ 175 nm).46 Any energy provided by the VUV photon (Ehv) in excess of this threshold energy will be deposited into the fragment kinetic energy and/or into the internal energy (Eint) of the O2 products. The radii of the well resolved ring structures in the TS-VMI images can be used to determine the velocity distribution of the S(1D) products. These velocities can then be converted to a total kinetic energy release P(ET) spectrum of the S(1D) + O2 products using linear momentum conservation arguments. Fig. 2 and S4 (in the ESI†) display the P(ET) spectra of the S(1D) + O2 products obtained by integrating signals over all angles in the respective images. The internal energy distributions of the O2 co-products, Eint[O2] formed at each wavelength can then be obtained from the corresponding P(ET) spectrum using the law of energy conservation (eqn (2)).
| Ehv − D0 = Eint[O2] + ET[S(1D) + O2], | (2) |
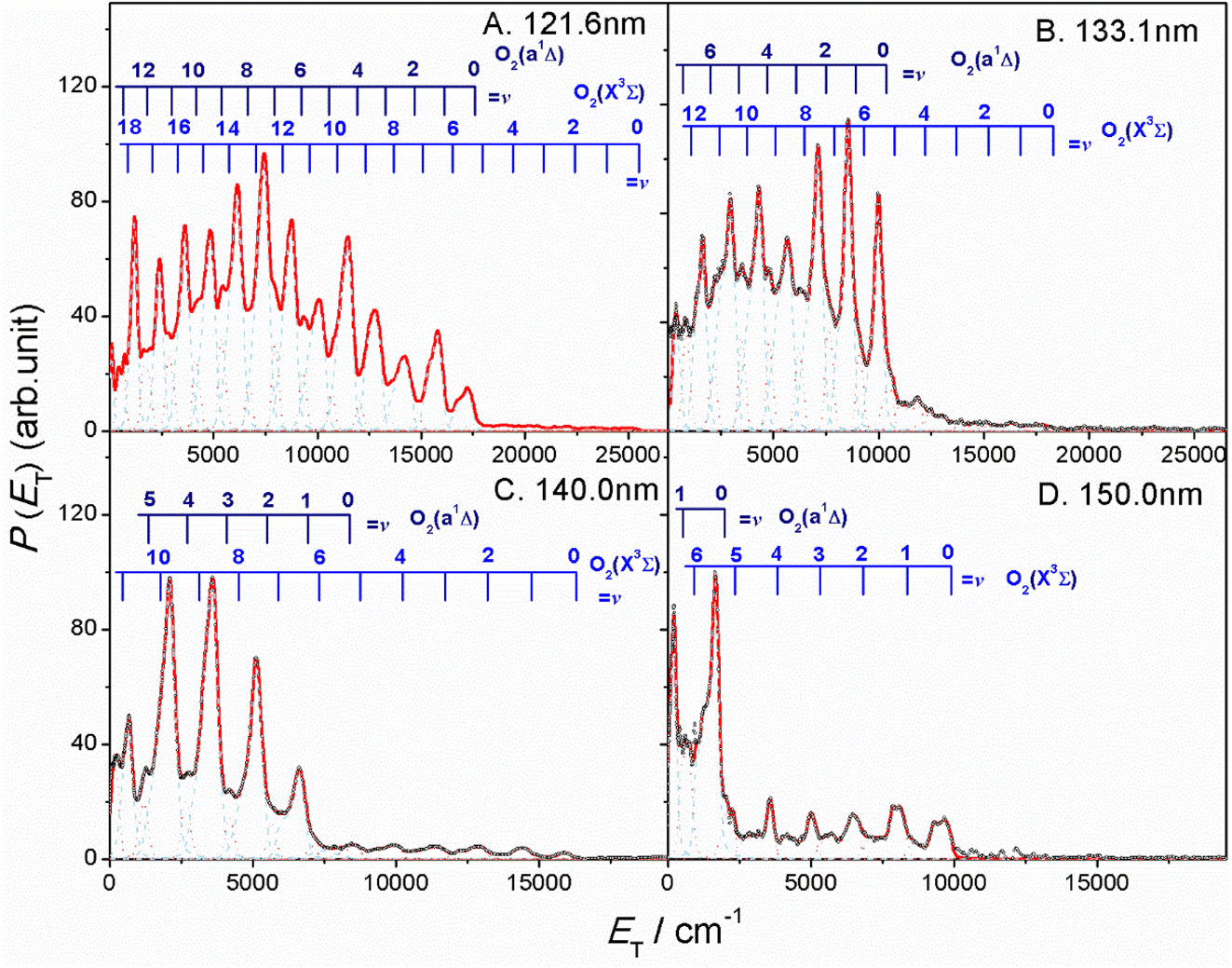 | ||
| Fig. 2 The product total kinetic energy (P(ET)) spectra for S(1D) + O2 products following photolysis at λ = (A) 121.6 nm, (B) 133.1 nm, (C) 140.0 nm and (D) 150.0 nm, derived from the images shown in Fig. 1, in red, along with the best-fit simulation of the spectra, in cyan dashed lines and orange dotted lines. The superposed combs indicate the ET values associated with the formation of the various vibrational levels of O2(X3Σg−/a1Δg, v). | ||
Each panel in Fig. 2 reveals two vibrational progressions for the O2 co-products, which can be assigned to the population of different vibrational levels of the ground (X3Σg−) and first excited electronic (a1Δg) state (the zero-point level of which lies 7882 cm−1 above that of the ground state46). The onset of the strong progression in each P(ET) spectrum accords well with the threshold of the S(1D) + O2(a1Δg) product channel, providing unambiguous evidence for the formation of molecular O2 in the VUV photodissociation of SO2. Fig. 3 shows the relative populations of the O2 products formed in the two electronic states following SO2 photolysis at each wavelength studied, obtained from simulation of the P(ET) spectra (Fig. 2 and S4†). Clearly, the S(1D) + O2(X3Σg−) channel is more important at the longest wavelengths investigated, but the S(1D) + O2(a1Δg) channel becomes increasingly dominant upon tuning to shorter wavelengths. The O2(a1Δg) and O2(X3Σg−) fragments formed at all but the very longest wavelengths both display inverted vibrational state population distributions, spanning a wide range of vibrational levels (Fig. S5 and S6 in the ESI†). The finite bandwidth of the FEL source precludes detailed discussion of the rotational energy disposal in the O2 products from the observed vibrational peak profiles.
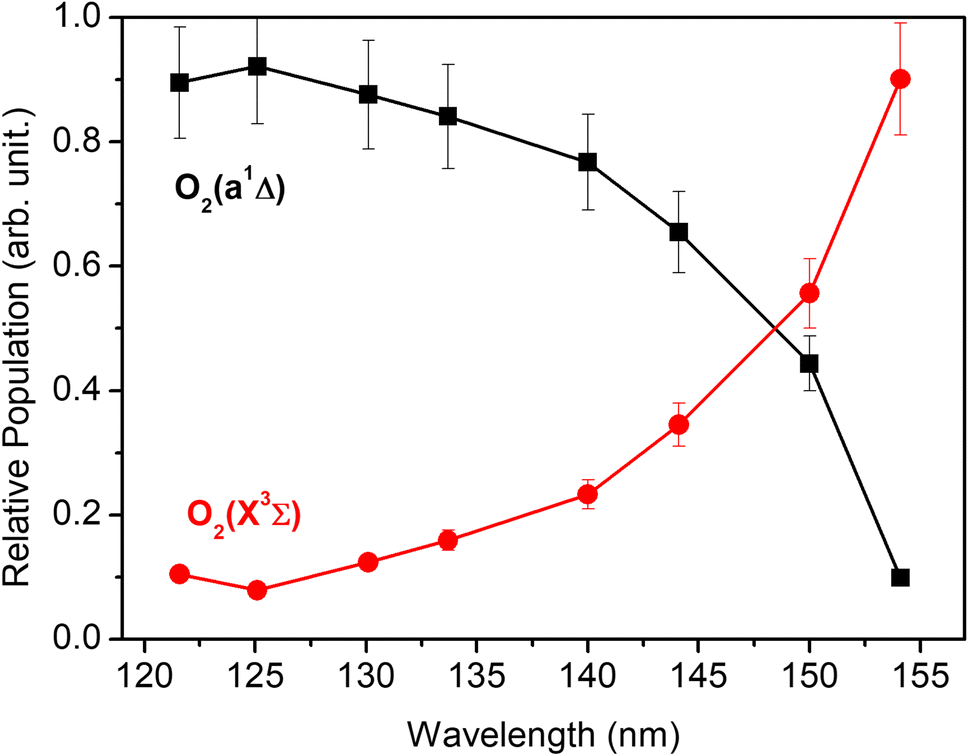 | ||
| Fig. 3 The relative population of the O2(a1Δg) and O2(X3Σg−) products as a function of photolysis wavelength. | ||
Potential energy surface (PES) calculations were also performed to gain insights into possible dissociation mechanisms for SO2 molecules excited to low lying Rydberg states. Fig. 4 depicts two possible dissociation pathways. The left half of Fig. 4 illustrates a triplet-state pathway, in which SO2 is initially excited to the 53A′′ Rydberg state, and then undergoes non-adiabatic coupling to the 43A′′ PES. The SO2 molecule evolves on the 43A′′ state PES, and one O-atom roams away from the SO partner and then returns to abstract the other O-atom. This mechanism involves an S–O–O intermediate and eventual dissociation to S(1D) + O2(X3Σg−) products on the 53A′′ PES. The right half of Fig. 4 illustrates a dissociation pathway via the singlet-state manifold. Initial photoexcitation in this case is to the 51A′′ state, which is followed by non-adiabatic coupling to the 31A′′ PES and dissociation to form S(1D) + O2(a1Δg) products. In this pathway, the interbond angle reduces, the two oxygen atoms approach towards a cyclic-SO2 intermediate and the O2 molecule is ejected. The identification of these pathways supports the experimental observation of O2(X3Σg−) or O2(a1Δg) products following VUV photodissociation of SO2. More details of the theoretical calculations are provided in Section S5 and Fig. S9 of the ESI.†
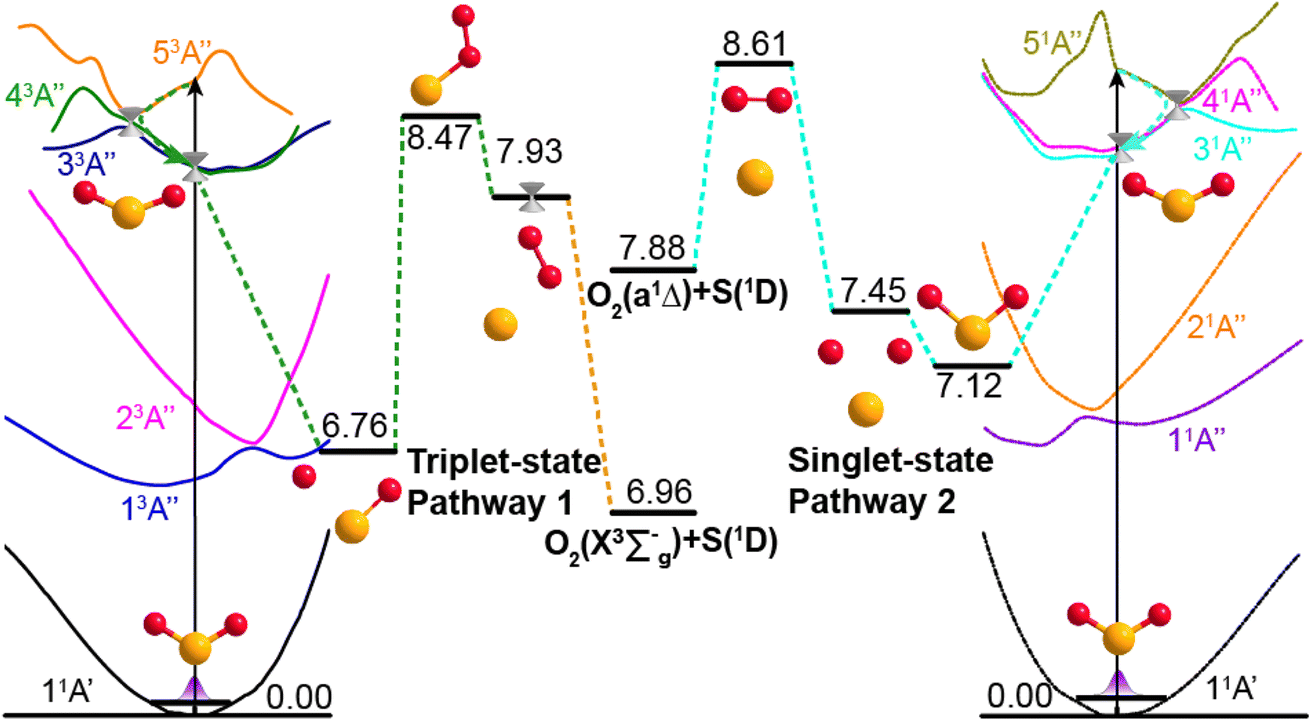 | ||
| Fig. 4 Dissociation pathways of SO2 leading to O2(X3Σg−) + S(1D) products via the triplet state manifold (left) and to O2(a1Δg) + S(1D) products via the singlet state manifold. The schematics of the simplified PECs for the ground and the first five excited states of 3A′′ symmetry (left) and the first five states of 1A′′ symmetry (right) support different paths leading to S + O2 product formation. Dashed arrows indicate possible non-adiabatic pathways and the geometries and energies (in eV) of key intermediate structures along the dissociation pathways are also indicated. The horizontal axis represents the reaction coordinate, and the vertical axis is the potential energy (in eV). | ||
The quantum yield of S(1D) + O2 products
To quantify the importance of O2 production from SO2 photolysis in the Earth's primitive atmosphere, we have sought to determine the branching ratios of all active dissociation channels. Since the H Lyman-α wavelength (121.6 nm) is most abundant in stellar VUV radiation and about 80% of all SO2 photodissociation events occur around the Lyman-α line,47 we have taken this as the representative wavelength to measure the branching ratios. Three fragment channels O(1D) + SO, O(1S) + SO and S(1D) + O2 formed by 121.6 nm photolysis have been detected using the VUV pump and VUV probe technique (Section S4 and Fig. S7 in the ESI†). Attempts to detect O(3P), S(3P) and S(1S) products under similar experimental conditions yielded no observable signals, suggesting that branching into each of the O(3P) + SO, S(3P) + O2 and S(1S) + O2 channels is negligible at this wavelength. By calibrating the detection efficiencies for the O(1D), O(1S) and S(1D) fragments (Section S4 and Fig. S8 in the ESI†), the branching ratio of the S(1D) + O2 product channel was determined to be ∼30 ± 5% at λ = 121.6 nm (as a fraction of all the detectable channels).Insights into the O2 budget in the ancient atmosphere
The present experimental results imply that O2 production is a significant process following VUV excitation of SO2. This finding could have profound implications for understanding the evolution of O2 in the Earth's primitive atmosphere. Current scenarios assume that biogenic O2 production started long before the GOE,10–14 but that any potential accumulation of O2 was offset by its consumption in reactions with reduced compounds emanating from the Earth's interior, i.e. O2 consumption balanced its production at these early times. This source-sink balance then shifted in favour of O2 accumulation, and finally triggered the GOE. This critical shift, relying on appreciable O2 production, is poorly understood. Geological studies suggest that the composition of the primitive atmosphere was probably determined by outgassing, through volcanism, and that the redox state of volcanic gases differs markedly between subaerial and submarine eruptions.30,31,48 Volcanic gases that erupted subaerially have generally equilibrated at high temperatures and low pressures with magmas close to the fayalite–magnetite–quartz buffer. Consequently, subaerial volcanism emitted SO2-dominated gas into the primitive atmosphere. Volcanic SO2 gas could reach the stratosphere during eruptions, whereupon photodissociation would contribute an additional O2 source. Such a scenario would introduce additional O2 sources that, temporarily, would exceed the available sinks.Here we attempt to estimate the total O2 production from the VUV photochemistry of volcanic SO2 in the late Archaean. We start by assuming that the average SO2 emission per year from volcanoes in the late Archaean was the same as the modern volcanic outgassing rate on Earth (∼2.3 × 1013 gram per year (g per year)).49 When the volcanism subsided, SO2 was rapidly removed from the atmosphere by continued photolysis, gas-phase reactions and rain-out. Given that O2 production from SO2 photochemistry has here been shown to be a prominent pathway, we attempt to quantify the possible contribution of SO2 by assuming that VUV photodissociation coverts 1% of the total emitted SO2 directly into O2. The accumulated amount of O2 in the atmosphere from VUV photodissociation of SO2 during the late Archaean eon (∼200 million years) can then be estimated as follows,
| WO2 = 2.3 × 1013 g per year × 200 × 106 year × 1% = 4.6 × 1019 g | (3) |
The total mass of O2 in the current Earth's atmosphere is ∼1.07 × 1021 g. Based on the above assumptions, the summed O2 from SO2 photolysis (eqn (3)) could be ∼4.3% of the present level of atmospheric O2 and this estimate should probably be viewed as a lower limit as it is likely that SO2 emissions and the UV flux were both higher in the late Archaean. Such arguments imply that O2 production from volcanic SO2 photochemistry could have provided substantial (and probably sporadic) contributions to the atmospheric O2 budget, given prevailing assumptions that most of the O2 sinks were already balanced by biogenic O2 production.10–14
We note that volcanic SO2 clouds and SO2-based aerosols typically only survive in the atmosphere for a short time (several weeks to a few years) before sinking, and that most incident VUV excitation of SO2 would likely occur in the stratosphere during the volcanic eruptions. (The incipient O3 column in the late Archean is transparent to VUV radiation and the CO2 column is also transparent to VUV photons around 121.6 nm.50) Within the average lifetime of SO2 in the atmosphere, the transient accumulation of O2 from SO2 photolysis can be estimated as ∼1018 to 1019 molecules per cm2 or ∼10−5 to 10−6 PAL. (This range was estimated assuming (i) a photon flux at λ ∼ 121.6 nm of ∼2 × 1012 photons per cm2 per s,47 (ii) that all VUV photons can be absorbed by SO2 clouds during the volcanic eruption and (iii) atmospheric lifetimes of SO2 between one month and one year). Such events could have led to short-lived “oxygen oases”, i.e., localized or regional areas with significantly elevated O2 during the volcanic eruption. The photochemical activity of SO2 gas at 2.5–2.7 Ga has been linked with the abrupt rise of S-MIF signatures in sedimentary rocks around this time.1,51,52 Spikes in Mo, Se, and Re concentrations at 2.5 to 2.66 Ga, and their isotopic excursions, have also recently been interpreted in terms of transient sources, or “whiffs”, of O2.10,18,53,54 The present findings are consistent with these scenarios. We propose that the VUV photochemistry of volcanic SO2 can lead to efficient production of molecular O2 and could have led to transient and localised accumulations of O2 in the atmosphere before the GOE. Such transient elevations of O2 from SO2 photochemistry merit further consideration as a possible trigger for the GOE.
Furthermore, the elemental sulfur produced via channel (1) from SO2 photochemistry might polymerize into S2, S3, S4, etc., and end up contributing to insoluble S8-containing aerosols,55 the recycling of which could be responsible for the S-MIF signature in sedimentary rocks. If so, the tectonic reorganization, the abrupt rise of S-MIF signatures in sedimentary rocks and the “whiffs” of O2 in the late Archaean might be far from coincidental. Tectonics control volcanic eruptions, and the photochemistry of volcanic SO2 contributes to S-MIF and boosts the O2 budget.
O2 production from SO2 photochemistry could also be relevant in the contemporary atmospheres of other planets. For example, a layer of volcanic SO2 has been observed in the Venusian atmosphere, spanning heights from 48–65 km above the surface and with a measured maximal abundance of ∼130 ppm.56 More recently, O2 molecules in the a1Δg state have also been detected (through the O2(a1Δg) dayglow) in the Venusian atmosphere.57 The present study provides unambiguous evidence for O2(a1Δg) formation via SO2 photolysis at wavelengths around the H Lyman-α transition. Clearly, this is an O2 production mechanism that merits further scrutiny and, if necessary, incorporation into photochemical models for all planets with rich volcanically outgassed SO2.
Data availability
The data supporting this study are available within the main text and the ESI.†Author contributions
K. Y. conceived the research. K. Y. and X. Y. designed the experiments and supervised the research. Y. C., Z. C., Z. L., Y. Z. and Z. L. performed the experiments. W. Q. Z., G. R. W., and X. M. Y. operated the FEL facility. Y. F., B. F., and D. Z. performed the theoretical calculations. Y. C., B. F., K. Y., M. N. R. A. and X. Y. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The experimental work was supported by the National Natural Science Foundation of China (Grant No. 22241304, 22225303, and 22203093), the National Natural Science Foundation of China (NSFC Center for Chemical Dynamics (Grant No. 22288201)), the Scientific Instrument Developing Project of the Chinese Academy of Sciences (Grant No. GJJSTD20220001), the Innovation Program for Quantum Science and Technology (2021ZD0303304), and the Innovation Fund Project of Dalian Institute of Chemical Physics (DICP I202112). The theoretical work was supported by the National Natural Science Foundation of China (Grant No. 22173099) and the Innovation Program for Quantum Science and Technology (2021ZD0303305). X. Yang also thanks the Guangdong Science and Technology Program (Grant No. 2019ZT08L455 and 2019JC01X091) and the Shenzhen Science and Technology Program (Grant No. ZDSYS20200421111001787). Y. Chang thanks the Special Research Assistant Funding Project of Chinese Academy of Sciences and the China Postdoctoral Science Foundation (Grant No. 2021M693118).References
- J. Farquhar, H. Bao and M. Thiemens, Science, 2000, 289, 756–758 CrossRef CAS PubMed.
- H. D. Holland, Geochim. Cosmochim. Acta, 2009, 73, 5241–5255 CrossRef CAS.
- D. E. Canfield, Annu. Rev. Earth Planet. Sci., 2005, 33, 1–36 CrossRef CAS.
- H. D. Holland, Geochim. Cosmochim. Acta, 2002, 66, 3811–3826 CrossRef CAS.
- H. D. Holland, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 903–915 CrossRef CAS.
- A. Bekker, H. D. Holland, P. L. Wang, D. Rumble 3rd, H. J. Stein, J. L. Hannah, L. L. Coetzee and N. J. Beukes, Nature, 2004, 427, 117–120 CrossRef CAS.
- G. Luo, S. Ono, N. J. Beukes, D. T. Wang, S. Xie and R. E. Summons, Sci. Adv., 2016, 2, e1600134 CrossRef PubMed.
- G. Luo, X. Zhu, S. Wang, S. Zhang and C. Jiao, Sci. China: Earth Sci., 2022, 65, 1646–1672 CrossRef CAS.
- J. J. Brocks, G. A. Logan, R. Buick and R. E. Summons, Science, 1999, 285, 1033–1036 CrossRef CAS PubMed.
- A. D. Anbar, Y. Duan, T. W. Lyons, G. L. Arnold, B. Kendall, R. A. Creaser, A. J. Kaufman, G. W. Gordon, C. Scott, J. Garvin and R. Buick, Science, 2007, 317, 1903–1906 CrossRef CAS PubMed.
- M. T. Rosing and R. Frei, Earth Planet. Sci. Lett., 2004, 217, 237–244 CrossRef CAS.
- K. L. French, C. Hallmann, J. M. Hope, R. Buick, J. J. Brocks and R. E. Summons, Mineral. Mag., 2013, 77, 1110 Search PubMed.
- B. Rasmussen, I. R. Fletcher, J. J. Brocks and M. R. Kilburn, Nature, 2008, 455, 1101–1104 CrossRef CAS PubMed.
- T. W. Lyons, C. T. Reinhard and N. J. Planavsky, Nature, 2014, 506, 307–315 CrossRef CAS.
- A. A. Pavlov and J. F. Kasting, Astrobiology, 2002, 2, 27–41 CrossRef CAS PubMed.
- K. Zahnle, M. Claire and D. Catling, Geobiology, 2006, 4, 271–283 CrossRef CAS.
- D. C. Catling and K. J. Zahnle, Sci. Adv., 2020, 6, eaax1420 CrossRef CAS PubMed.
- J. Meixnerova, J. D. Blum, M. W. Johnson, E. E. Stueken, M. A. Kipp, A. D. Anbar and R. Buick, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2107511118 CrossRef CAS PubMed.
- J. F. Kasting, S. C. Liu and T. M. Donahue, J. Geophys. Res.: Oceans, 1979, 84, 3097–3107 CrossRef CAS.
- Y. Chang, Y. Yu, F. An, Z. Luo, D. Quan, X. Zhang, X. Hu, Q. Li, J. Yang, Z. Chen, L. Che, W. Zhang, G. Wu, D. Xie, M. N. R. Ashfold, K. Yuan and X. Yang, Nat. Commun., 2021, 12, 2476 CrossRef CAS PubMed.
- J. F. Kasting and D. Catling, Annu. Rev. Astron. Astrophys., 2003, 41, 429–463 CrossRef CAS.
- Y. Chang, F. An, Z. Chen, Z. Luo, Y. Zhao, X. Hu, J. Yang, W. Zhang, G. Wu, D. Xie, K. Yuan and X. Yang, Nat. Commun., 2021, 12, 6303 CrossRef CAS PubMed.
- Z. Lu, Y. C. Chang, Q. Z. Yin, C. Y. Ng and W. M. Jackson, Science, 2014, 346, 61–64 CrossRef CAS PubMed.
- X. D. Wang, X. F. Gao, C. J. Xuan and S. X. Tian, Nat. Chem., 2016, 8, 258–263 CrossRef CAS PubMed.
- J. Haqq-Misra, J. F. Kasting and S. Lee, Astrobiology, 2011, 11, 293–302 CrossRef CAS PubMed.
- X. Zhang, M.-C. Liang, F. Montmessin, J.-L. Bertaux, C. Parkinson and Y. L. Yung, Nat. Geosci., 2010, 3, 834–837 CrossRef CAS.
- A. I. Stewart, D. E. Anderson, Jr., L. W. Esposito and C. A. Barth, Science, 1979, 203, 777–779 CrossRef CAS PubMed.
- C. T. Russell and M. G. Kivelson, Science, 2000, 287, 1998–1999 CrossRef CAS PubMed.
- R. Hu, S. Seager and W. Bains, Astrophys. J., 2013, 769, 6 CrossRef.
- F. Gaillard, B. Scaillet and N. T. Arndt, Nature, 2011, 478, 229–232 CrossRef CAS PubMed.
- L. R. Kump and M. E. Barley, Nature, 2007, 448, 1033–1036 CrossRef CAS PubMed.
- S. O. Danielache, C. Eskebjerg, M. S. Johnson, Y. Ueno and N. Yoshida, J. Geophys. Res., 2008, 113, D17314 CrossRef.
- C. Xie, X. Hu, L. Zhou, D. Xie and H. Guo, J. Chem. Phys., 2013, 139, 014305 CrossRef PubMed.
- S. Ono, Annu. Rev. Earth Planet. Sci., 2017, 45, 301–329 CrossRef CAS.
- C. Leveque, A. Komainda, R. Taieb and H. Koppel, J. Chem. Phys., 2013, 138, 044320 CrossRef.
- A. R. Hoy and J. C. D. Brand, Mol. Phys., 1978, 36, 1409–1420 CrossRef CAS.
- J. Heicklen, N. Kelly and K. Partymiller, Rev. Chem. Intermed., 1980, 3, 315–404 CrossRef CAS.
- H. Katagiri, T. Sako, A. Hishikawa, T. Yazaki, K. Onda, K. Yamanouchi and K. Yoshino, J. Mol. Struct., 1997, 413–414, 589–614 CrossRef CAS.
- P. C. Ray, M. F. Arendt and L. J. Butler, J. Chem. Phys., 1998, 109, 5221–5230 CrossRef CAS.
- A. Okazaki, T. Ebata and N. Mikami, J. Chem. Phys., 1997, 107, 8752–8758 CrossRef CAS.
- S. Becker, C. Braatz, J. Lindner and E. Tiemann, Chem. Phys., 1995, 196, 275–291 CrossRef CAS.
- C. Lalo and C. Vermeil, J. Photochem., 1975, 3, 441–454 CrossRef CAS.
- D. Rosch, R. Almeida, B. Sztaray and D. L. Osborn, J. Phys. Chem. A, 2022, 126, 1761–1774 CrossRef CAS PubMed.
- Y. Chang, Y. Yu, H. Wang, X. Hu, Q. Li, J. Yang, S. Su, Z. He, Z. Chen, L. Che, X. Wang, W. Zhang, G. Wu, D. Xie, M. N. R. Ashfold, K. Yuan and X. Yang, Nat. Commun., 2019, 10, 1250 CrossRef PubMed.
- J. Zhou, Y. Zhao, C. S. Hansen, J. Yang, Y. Chang, Y. Yu, G. Cheng, Z. Chen, Z. He, S. Yu, H. Ding, W. Zhang, G. Wu, D. Dai, C. M. Western, M. N. R. Ashfold, K. Yuan and X. Yang, Nat. Commun., 2020, 11, 1547 CrossRef CAS PubMed.
- On the basis of thermodynamic calculations with the data available from the thermochemical network (https://atct.anl.gov) and references therein.
- M. W. Claire, J. Sheets, M. Cohen, I. Ribas, V. S. Meadows and D. C. Catling, Astrophys. J., 2012, 757, 95 CrossRef.
- Z.-X. Anser Li and C.-T. Aeolus Lee, Earth Planet. Sci. Lett., 2004, 228, 483–493 CrossRef.
- S. A. Carn, V. E. Fioletov, C. A. McLinden, C. Li and N. A. Krotkov, Sci. Rep., 2017, 7, 44095 CrossRef CAS PubMed.
- D. L. Huestis and J. Berkowitz, Adv. Geosci., 2010, 25, 229–242 Search PubMed.
- J. Farquhar, M. Peters, D. T. Johnston, H. Strauss, A. Masterson, U. Wiechert and A. J. Kaufman, Nature, 2007, 449, 706–709 CrossRef CAS.
- D. E. Canfield, K. S. Habicht and B. Thamdrup, Science, 2000, 288, 658–661 CrossRef CAS PubMed.
- A. J. Kaufman, D. T. Johnston, J. Farquhar, A. L. Masterson, T. W. Lyons, S. Bates, A. D. Anbar, G. L. Arnold, J. Garvin and R. Buick, Science, 2007, 317, 1900–1903 CrossRef CAS.
- E. E. Stüeken, R. Buick and A. D. Anbar, Geology, 2015, 43, 259–262 CrossRef.
- D. Babikov, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3062–3067 CrossRef CAS.
- E. Marcq, B. Bézard, P. Drossart, G. Piccioni, J. M. Reess and F. Henry, J. Geophys. Res., 2008, 113, E00B07 CrossRef.
- J. C. Gérard, S. W. Bougher, M. A. López-Valverde, M. Pätzold, P. Drossart and G. Piccioni, Space Sci. Rev., 2017, 212, 1617–1683 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03328g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |