DOI:
10.1039/D3SC03686C
(Edge Article)
Chem. Sci., 2023,
14, 12091-12097
Organocatalytic atroposelective synthesis of axially chiral N,N′-pyrrolylindoles via de novo indole formation†
Received
17th July 2023
, Accepted 4th October 2023
First published on 11th October 2023
Introduction
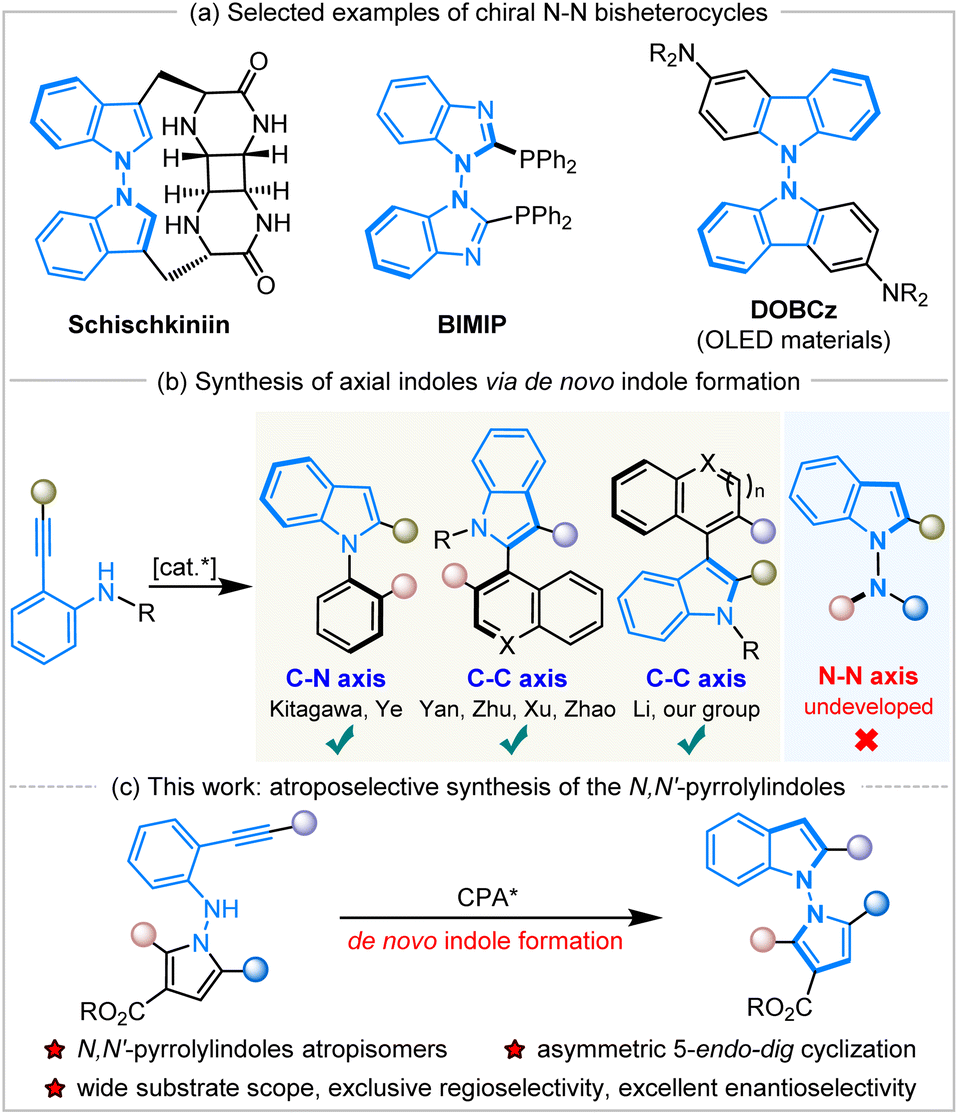
N–N atropisomers are widely present as core structures in a variety of important chiral molecules, such as natural products, chiral ligands and functional materials (Scheme 1a).1 In sharp contrast to the well-developed syntheses of C–C and C–N axially chiral compounds,2 the construction of N–N atropisomers has remained in an underdeveloped state in the decades since their discovery in 1931.3 The asymmetric catalytic synthesis of N–N axially chiral compounds was considered a challenging issue until 2021, when Li,4a Lu and Houk,4b and Liu's5a groups developed various synthetic methods to access these fascinating molecules promoted by chiral transition metals or organocatalysis.
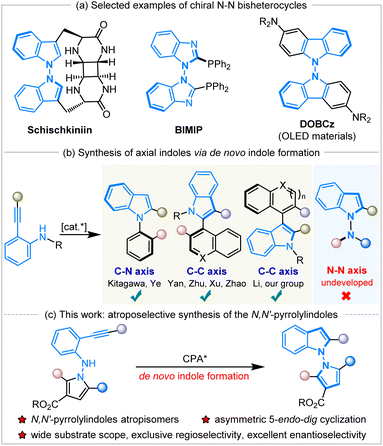 |
| | Scheme 1 Selected examples of N–N atropisomers and asymmetric de novo construction of indole atropisomers. | |
Subsequently, a great deal of attention has been paid to the development of efficient synthetic strategies and some progress has been achieved, mainly involving direct N–H functionalization,4 the functionalization of prochiral or racemic bis-heteroaryl rings,5de novo ring formation via a Paal–Knorr reaction,6 or dual-ring formation by cyclization and oxidation.7 In addition, the catalytic atroposelective synthesis of N–N axially chiral molecules via de novo indole formation has emerged as an attractive route.8 The 5-endo-dig cyclization of o-alkynylanilines is a versatile method for the de novo generation of indole derivatives, and a variety of chiral indole atropisomers9 have been well established through this catalytic enantioselective cyclization (Scheme 1b). To our knowledge, the documented protocols rely heavily on asymmetric transition metal catalysis, and there are only two examples using organocatalysis for the construction of axially chiral indoles via the 5-endo-dig cyclization of o-alkynylanilines. In 2019, Yan and workers developed chiral quinine-derived thiourea-catalyzed asymmetric cyclization of o-alkynylanilines to give chiral naphthyl-C2-indoles with a C–C axis via vinylidene orthoquinone methide intermediates with excellent results.9c Meanwhile, Ye and coworkers realized chiral CPA-catalyzed atroposelective 5-endo-dig cyclization of ynamides to access chiral N-arylindoles with a C–N axis in high yields with excellent enantioselectivities.9d However, the approach to accessing axially chiral N–N bis-heteroaryl atropisomers through the de novo formation of chiral indole rings by the organocatalytic annulation of o-alkynylanilines remains undeveloped and unprecedented. During the preparation of this manuscript, Sparr and coworkers reported an asymmetric Pd-catalyzed 5-endo-hydroaminocyclization for the synthesis of N–N atropisomeric bisindoles and indolyl-carbazoles with moderate results.9j
Herein, we have designed and synthesized a new type of molecule, N-pyrrole o-alkynylanilines, as the substrates, and applied them in Brønsted-acid CPA-catalyzed atroposelective 5-endo-dig cyclization to achieve the de novo formation of axially chiral N,N′-pyrrolylindoles (Scheme 1c). It should be noted that this is the first organocatalytic annulation of o-alkynylanilines to prepare N–N axially chiral compounds in a practical and atom-economic manner. This fascinating protocol also faces a lot of challenges, including: (1) the development of new and efficient synthetic strategies to access N–N axially chiral molecules with excellent enantioselective control, (2) the design and synthesis of new kind of substrates and chiral N–N bis-heteroaryl pyrrolylindoles atropisomers, (3) the discovery and application of these highly-valuable molecules in the field of synthetic and medicinal chemistry.
Results and discussion
Optimization of reaction conditions
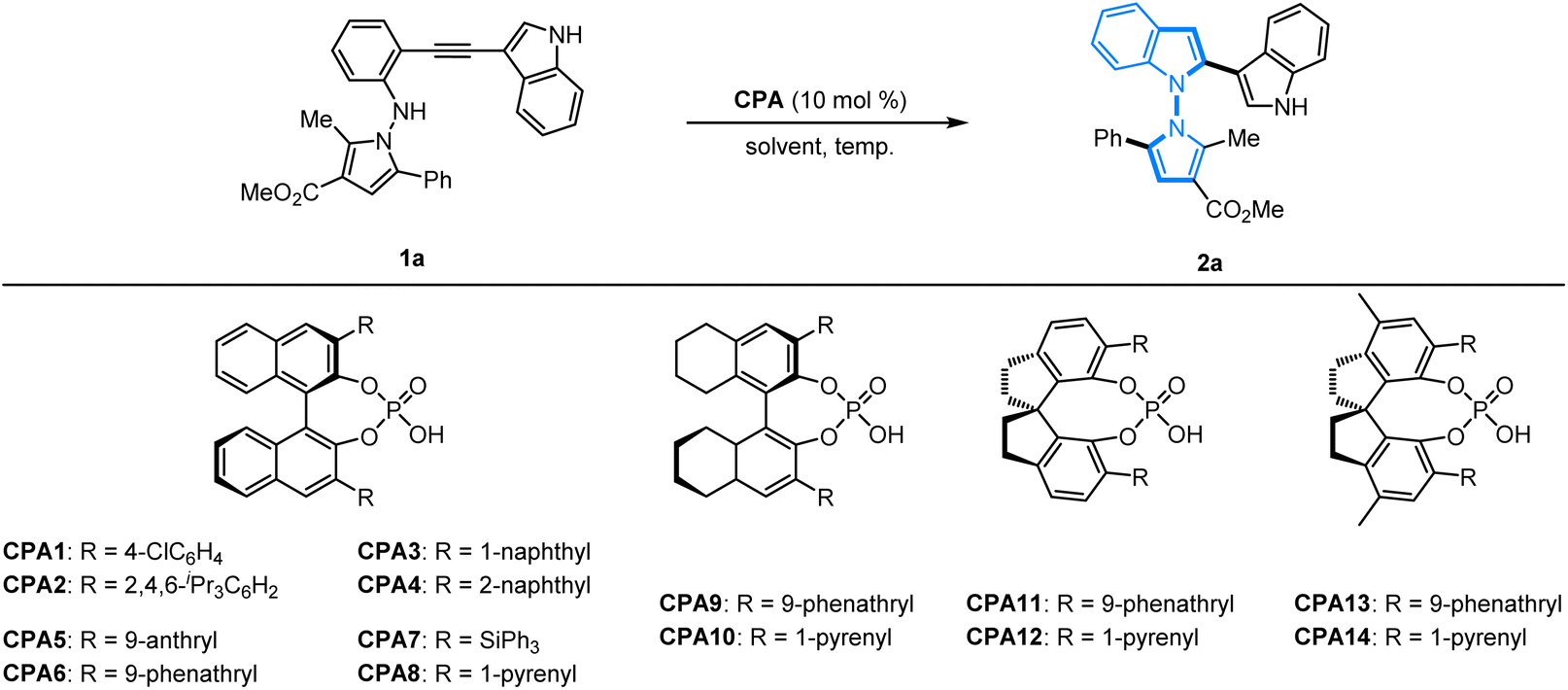
To verify this hypothesis, an initial investigation of the asymmetric 5-endo-dig-cyclization was begun with N-pyrrolo-acetylaniline 1a as the model substrate promoted by chiral phosphoric acids. A series of BINOL-based chiral phosphoric acids CPA1–CPA8 as the catalysts were firstly applied to promote this cyclization in CHCl3. Except for CPA3 with a 1-naphthyl group, the formation of the expected 2a with an N–N axis could be obtained in high yields but with poor enantioselectivity (Table 1, entries 1–8). H8-BINOL-based chiral phosphoric acids CPA9–CPA10 were also employed but, unfortunately, no further improvement was obtained (Table 1, entries 9 and 10). We found that SPINOL-derived CPA11–CPA14 could promote smooth cyclization, and the desired products could be obtained with better results (90–96% yields, 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :26–89:11 er, Table 1, entries 11–14). Pleasingly, SPINOL-derived CPA12 containing a 1-pyrenyl substituted group furnished the best results in terms of reactivity and enantioselectivity (93% yield, 89:11 er, Table 1, entry 12). A further solvent survey in the presence of CPA12 revealed that toluene is the most favorable choice, and the enantioselectivity of product 2a was greatly improved to 91:9 er (Table 1, entry 19). When the reaction temperature was gradually decreased to −10 °C, excellent enantioselectivity was obtained (90% yield, 95:5 er, Table 1, entry 22). It was found that the ee value was maintained without improvement at −40 °C, but lower reactivity was observed with only 50% yield (Table 1, entry 23). Accordingly, it was identified that the optimized protocol for the 5-endo-dig-cyclization of N-pyrrolo-acetylanilines should be conducted under conditions with SPINOL-derived CPA12 as the privileged catalyst in toluene at −10 °C.
:26–89:11 er, Table 1, entries 11–14). Pleasingly, SPINOL-derived CPA12 containing a 1-pyrenyl substituted group furnished the best results in terms of reactivity and enantioselectivity (93% yield, 89:11 er, Table 1, entry 12). A further solvent survey in the presence of CPA12 revealed that toluene is the most favorable choice, and the enantioselectivity of product 2a was greatly improved to 91:9 er (Table 1, entry 19). When the reaction temperature was gradually decreased to −10 °C, excellent enantioselectivity was obtained (90% yield, 95:5 er, Table 1, entry 22). It was found that the ee value was maintained without improvement at −40 °C, but lower reactivity was observed with only 50% yield (Table 1, entry 23). Accordingly, it was identified that the optimized protocol for the 5-endo-dig-cyclization of N-pyrrolo-acetylanilines should be conducted under conditions with SPINOL-derived CPA12 as the privileged catalyst in toluene at −10 °C.
Table 1 Optimization of reaction conditionsa
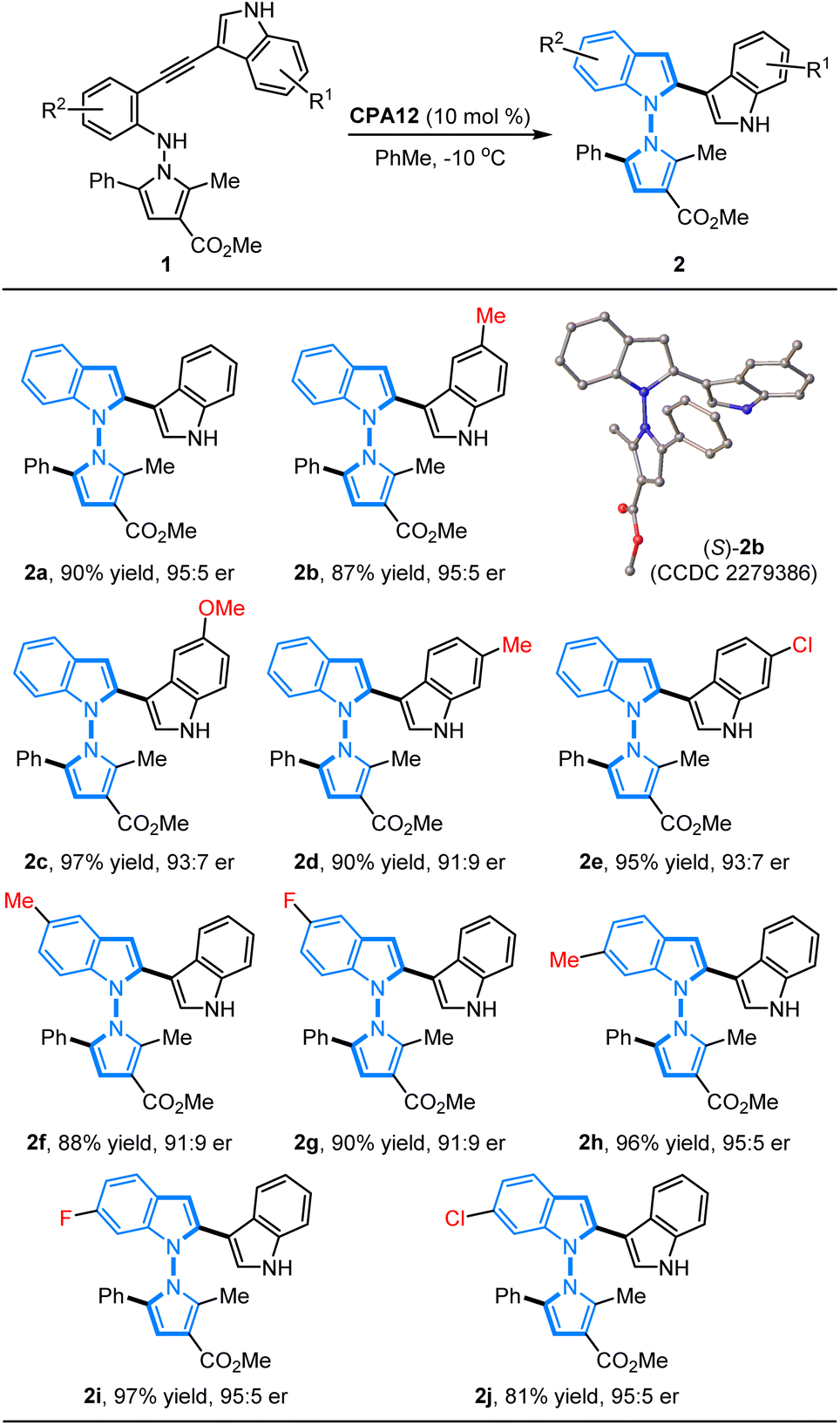
Substrate scope study
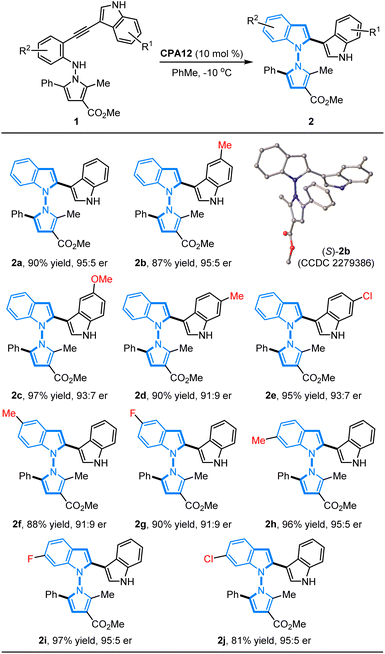
With the established optimal conditions in hand, we focused on an evaluation of the substrate generality of N-pyrrolo-acetylanilines for this CPA12-catalyzed 5-endo-dig-cyclization. As summarized in Table 2, a wide range of N-pyrrolo-acetylanilines with different substituted groups on the phenyl ring and indole ring were applied to prepare chiral pyrrolyindoles containing an N–N axis. It was found that the electronic properties and positions of the substituted groups have a negligible impact on the reactivity and enantioselectivity. The N-pyrrolo-acetylanilines bearing electron-rich (1b–1d) or electron-deficient (1e) groups on the indole ring were examined first, and the expected annulation products (2b–2e) were furnished in 87–97% yields with 91:9–95:5 er. In addition, the N-pyrrolo-acetylanilines (1f–1j) containing different substituted groups on the phenyl ring of the aniline motifs could be well accommodated to give the desired products (2f–2j) in good to high yields with excellent enantioselectivities (81–97% yields, 91:9–95:5 er). The absolute configuration of cycloadduct 2b was unambiguously determined as S by X-ray diffraction analysis (CCDC 2279386).10
Table 2 Substrate scope of N-pyrrolo-acetylanilinesa
|
All reactions were carried out with 0.2 mmol 1 in 2 mL of toluene catalyzed by CPA12 (10 mol%) at −10 °C. The yield is isolated yield. The er value was determined by chiral HPLC analysis.
|
|
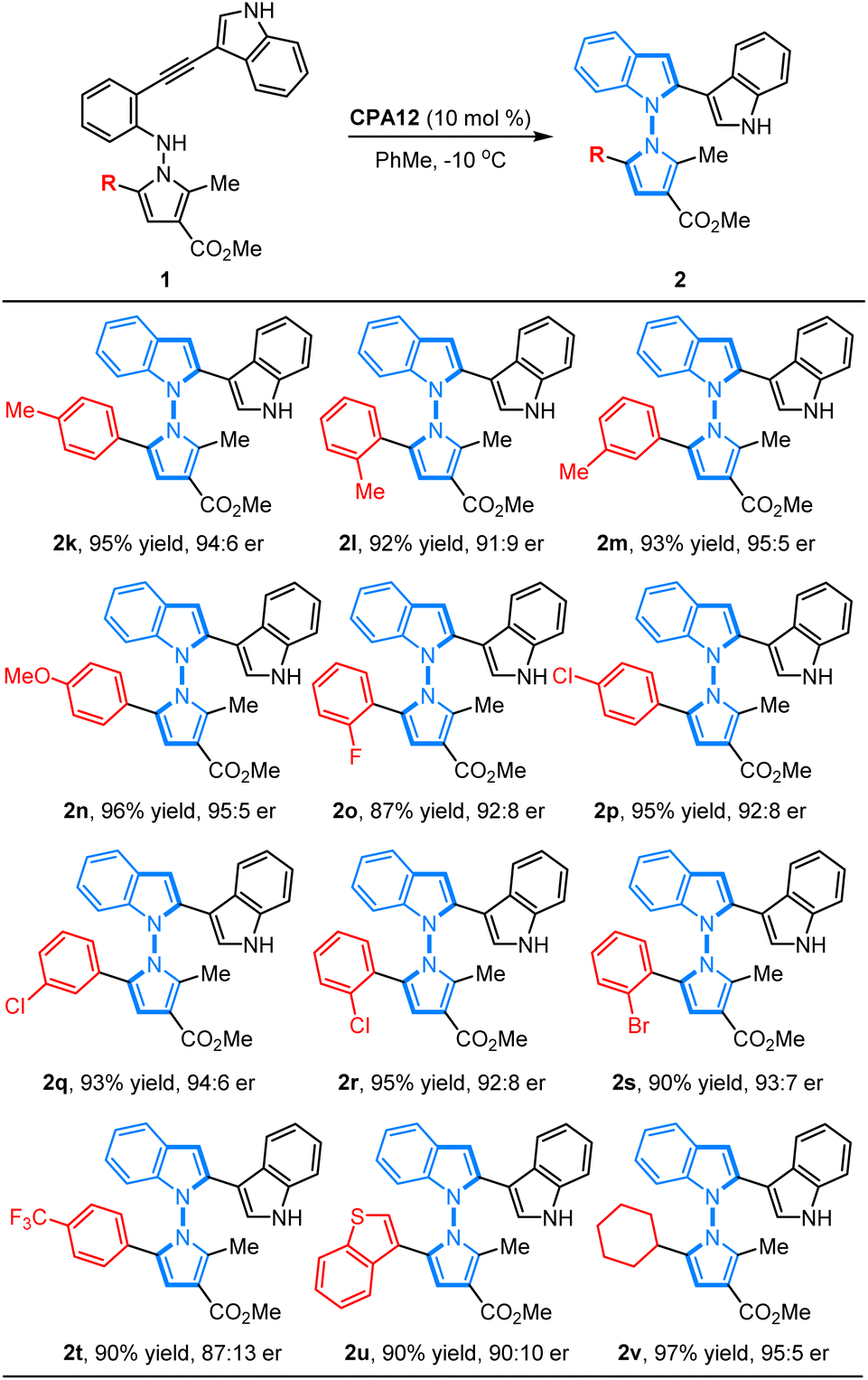
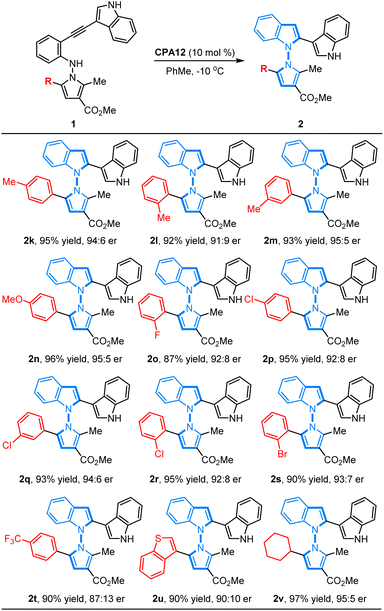
On the other hand, we also investigated the feasibility of the CPA12-catalyzed 5-endo-dig-cyclization of N-pyrrolo-acetylanilines with different substituted groups on the phenyl ring in the pyrrole motif. It was found that a wide range of N-pyrrolo-acetylanilines (1k–1t) bearing electron-rich groups or electron-deficient groups on the phenyl ring worked as good reaction partners, and this cyclization proceeded smoothly, resulting in the corresponding chiral products (2k–2t) in good to high yields with excellent enantioselectivities (87–96% yields, 87:13–95:5 er). It is noteworthy that the position of the substituted group did not affect the reactivity or enantioselectivity. Additionally, the heteroaryl-fused substrate 1u was well tolerated to deliver the corresponding product 2u in 90% yield with 90:10 er. Remarkably, the alkyl-substituted substrate 1v also worked well, leading to the expected product 2v in high yield with good enantioselectivity (97% yield, 95:5 er) (Table 3).
Table 3 Substrate scope of N-pyrrolo-acetylanilinesa
|
All reactions were carried out with 0.2 mmol 1 in 2 mL of toluene catalyzed by CPA12 (10 mol%) at −10 °C. The yield is isolated yield. The er value was determined by chiral HPLC analysis.
|
|
Scale-up experiments and synthetic applications
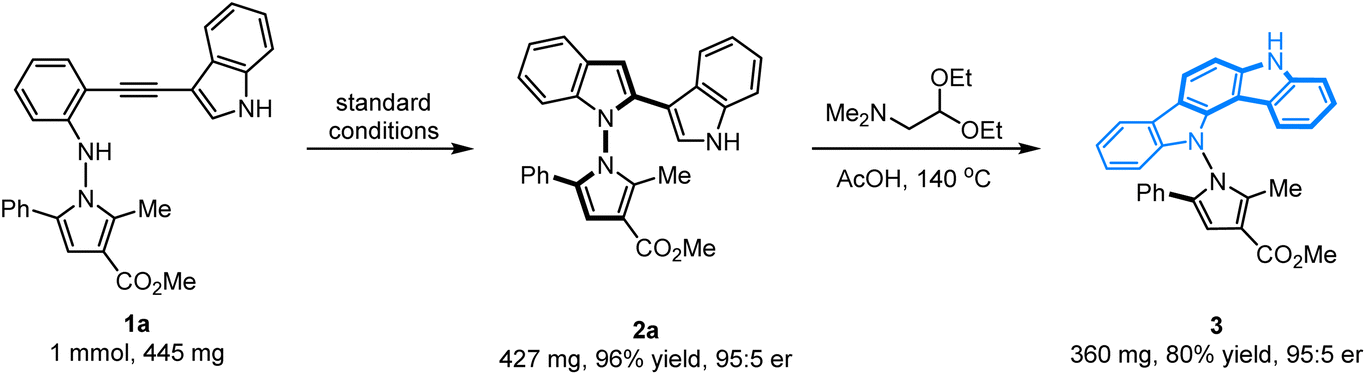
The configurational stability of the novel axially chiral N–N biheteroaryl pyrrolylindoles was investigated, and no erosion of enantiopurity was observed when the product 2a was heated in toluene at 130 °C for 48 h. Therefore, the configurational stability of these compounds could certainly show good tolerance for their further application in organic synthesis. To further highlight the application of this protocol for the synthesis of novel axially chiral biheteroaryl pyrrolylindoles, as shown in Scheme 2, the scale-up annulation reaction with 1 mmol substrate 1a was carried out under standard reaction conditions, and product 2a was easily accessible with high yield and maintained good enantioselectivity (96% yield, 95:5 er). In addition, compound 2a could undergo a condensation–cyclization reaction with 2,2-diethoxy-N,N-dimethylethan-1-amine to furnish product 3 containing an indolo[3,2-a]carbazole skeleton in 80% yield without erosion of the er value, which forms the key core structure in natural indolo[3,2-a]carbazole alkaloids from deep-water sponges of the genus Asteropus.11
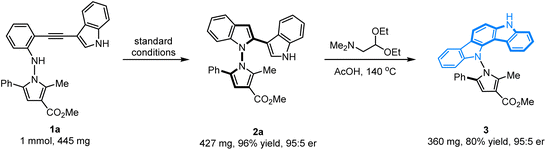 |
| | Scheme 2 Scale-up experiment and synthetic transformation. | |
Investigation of biological activities
In view of N–N atropisomers being core units in some natural products and bioactive molecules, we are interested in the potential biological activities of these new axially chiral N,N′-pyrrolylindoles. Consequently, the preliminary cytotoxic effects of randomly selected N,N′-pyrrolylindole products (2a, ent-2a, 2b, 2o, 2i and 2k) and transformation products (3 and ent-3) with an indolo[3,2-a]carbazole skeleton were then examined against some kinds of cancer cell lines, such as HL-60 cancer cells, A549 lung carcinoma cells, SMMC-7721 hepatoma cells, MDA-MB-231 breast adenocarcinoma cells, and SW480 colon cancer cells by MTS assay (MTS, 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxy-phenyl)-2-(4-sulfopheny)-2H-tetrazolium). As shown in Fig. 1a, preliminary evaluation of the results showed that these structurally-important molecules generally showed a high cell inhibitory rate against these cancer cells at a concentration of 40 μM. Then, we turned our attention to testing the corresponding cytotoxicity with half maximal inhibitory concentration (IC50) values of these three molecules in the low micromolar range, with the widely used anticancer drug cisplatin used as a comparison group (Fig. 1b). It is worth noting that compounds 2a and 2i exhibited impressive cytotoxicity against these five kinds of cancer cells with low IC50 values, which were generally better than the drug cisplatin. Compound 3 also showed favorable anticancer activities against A549 lung carcinoma cells, SMMC-7721 hepatoma cells and MDA-MB-231 breast adenocarcinoma cells. The distinctive preliminary results of these high-value axially chiral N,N′-pyrrolylindoles could to some extent provide potentiality in the drug discovery and development process.
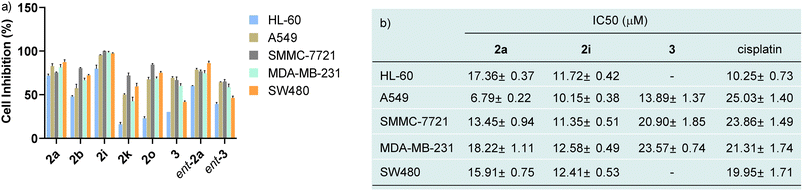 |
| | Fig. 1 (a) 40 μM of eight randomly selected chiral N,N′-pyrrolylindoles were incubated individually with HL-60 cancer cells, A549 lung carcinoma cells, SMMC-7721 hepatoma cells, MDA-MB-231 breast adenocarcinoma cells, and SW480 colon cancer cells. (b) The cell viability after incubation with different concentrations of three chiral N,N′-pyrrolylindoles and their IC50 values were determined in comparison to cisplatin. | |
Mechanistic studies
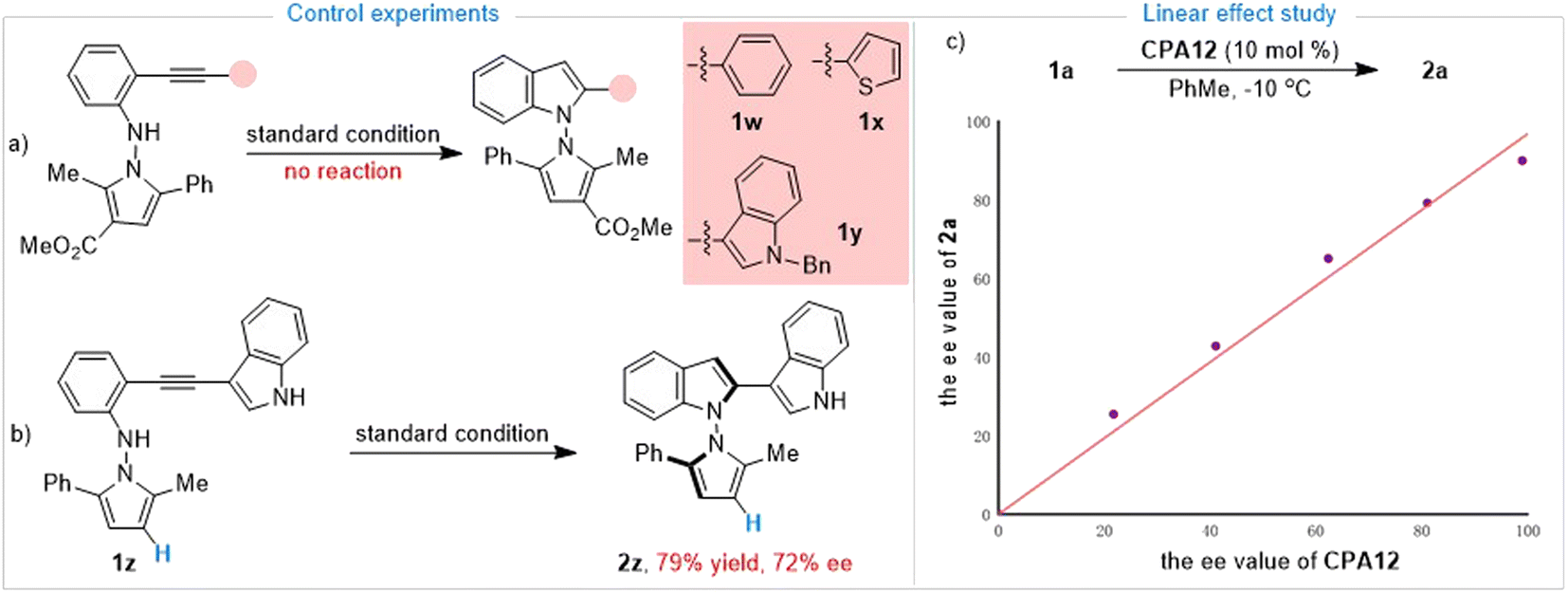
Having established the synthesis of axially chiral N,N′-pyrrolylindoles based on o-alkynylanilines via de novo indole formation, we were interested in the reaction mechanism. To explore the mechanistic insights for this protocol, as depicted in Scheme 3a, further investigations and control experiments were then carried out. Substrates 1w and 1x without an indole group and the N–Bn protected substrate 1y did not work under standard reaction conditions, leading to there being no desired product, which indicated that the indole group must be involved in the reaction pathway. In addition, substrate 1z in the absence of an ester group was also utilized to give the desired product 2z in 79% yield with 72% ee, which demonstrated that the ester group may play an important role in determining stereoselective control (Scheme 3b). Then, we paid attention to an investigation of the nonlinear effect between the enantioselectivity of 2a and the ee value of the CPA12 catalyst. As shown in Scheme 3c, linear correlation was observed, indicating that one active catalyst species was involved in the stereodetermination process.
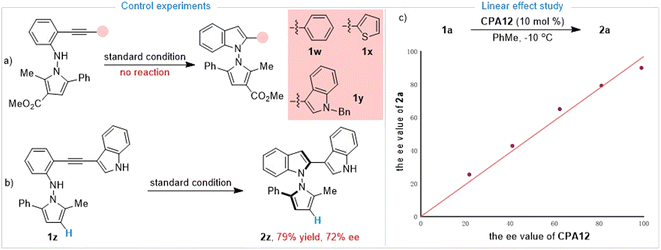 |
| | Scheme 3 Control experiments and linear effect study. | |
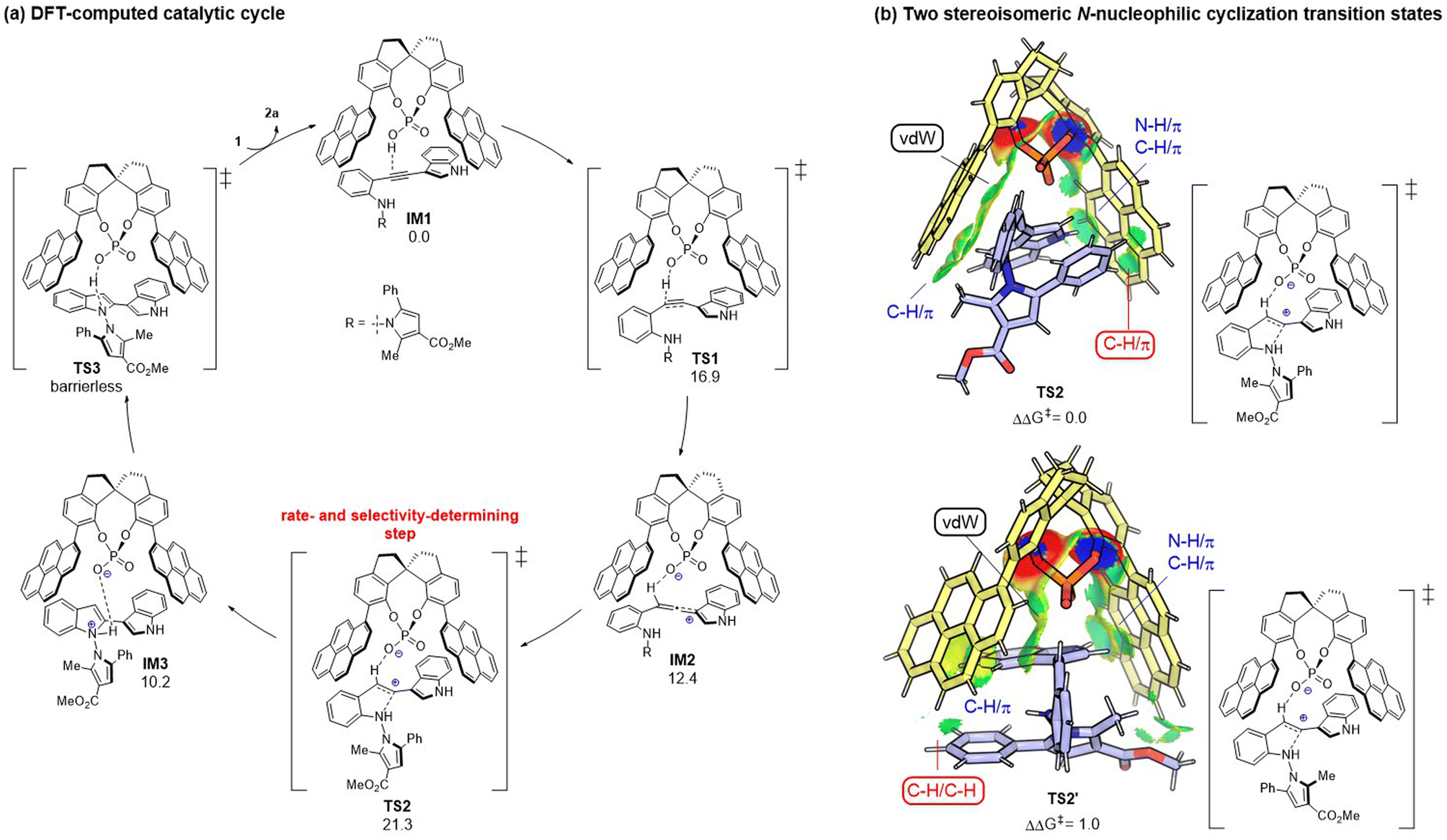
To gain mechanistic insights into the CPA-dependent stereoselectivity, density functional theory (DFT) calculations were performed with the Gaussian 09 package.12 As shown in Fig. 2a, the reaction starts with the hydrophosphoryloxylation of the alkyne moiety in the CPA-substrate adduct IM1viaTS1 with an energy barrier of 16.9 kcal mol−1, leading to allene-type intermediate IM2.13 Subsequently, the nucleophilic cyclization of the N atom in the aniline moiety on the central carbon of the allene moiety occurs viaTS2 (ΔG‡ = 21.3 kcal mol−1), forming protonated indole complex IM3. Finally, the deprotonation viaTS3 delivers product 2a and regenerates IM1 through product/substrate exchange. Herein, the transition state of proton transfer TS3 could not be located, which may be because proton transfer from ammonium to the phosphate anion could be regarded as an extremely facile process (see ESI† for more details). Overall, N-nucleophilic cyclization viaTS2 is the rate- and stereo-determining step that controls the axial chirality of the formed product. TS2 (leading to the major product) is found to be lower than TS2′ (leading to the minor product) by 1.0 kcal mol−1, whose computed result agrees with the experimental observations. Noncovalent interactions (NCIs) using IGMH analysis have been utilized to compare the two competing transition states (TS2vs.TS2′).14 As shown in Fig. 2b, the main difference between the two transition states is that TS2 has favorable C–H⋯π dispersion interactions between the phenyl group of the pyrrole moiety and the pyrenyl substituent in CPA; while only weak C–H/C–H ligand–substrate interactions can be detected in TS2′ of the analogous parts. Accordingly, the ligand–substrate dispersion interactions serve as the key factor controlling the axial chirality of the N,N′-pyrrolylindole products.
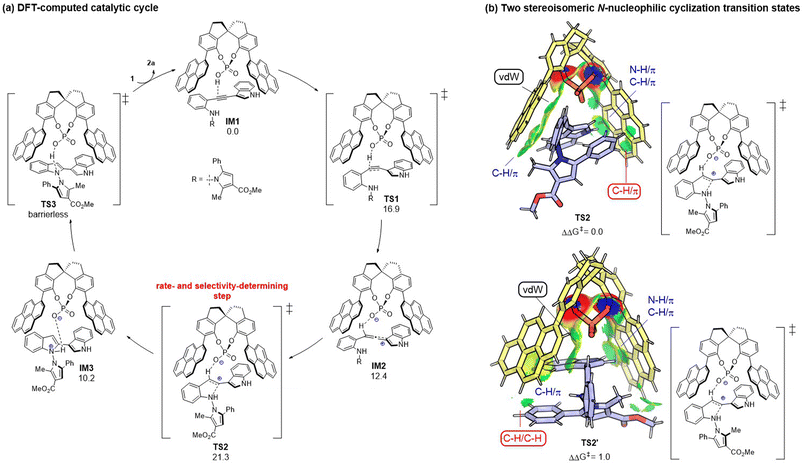 |
| | Fig. 2 (a) DFT-computed catalytic cycle with free energy given in kcal mol−1. (b) Origin of the stereoselectivity between TS2 and TS2′. Comparison of key NCIs by IGMH analysis. | |
Conclusions
In summary, we developed the first organocatalytic atroposelective synthesis of axially chiral N,N′-pyrrolylindoles based on o-alkynylanilines through CPA-catalyzed asymmetric 5-endo-dig cyclization via de novo indole formation. This elegant protocol provided a facile synthetic strategy, and a broad range of intriguing N,N′-pyrrolylindoles were obtained in high yields with exclusive regioselectivity and excellent atroposelectivities (up to 99% yield, >20:1 rr, 95:5 er). The scale-up synthesis and the synthetic transformation for the construction of an indolo[3,2-a]carbazole skeleton as a key core structure in natural product asteropusazoles exhibited potential utilization. Preliminary studies of biological activity suggested that these structurally important N,N′-pyrrolylindoles had a low IC50 value, which could offer an encouraging opportunity for drug discovery. DFT mechanistic explorations indicated that the stereochemistry was regulated by the CPA-promoted N-nucleophilic cyclization step, wherein the favored transition state is stabilized by more ligand–substrate C–H⋯π dispersion forces.
Data availability
All experimental procedures, characterisation data, mechanistic investigations, NMR spectra and HPLC spectra can be found in the ESI.†
Author contributions
C. J. W. conceptualized the project. C. J. W. and X. Q. D. supervised the investigation. C. S. W., Q. X., and H. R. Y. performed the research. Y. D. directed the DFT calculation, and H. X. performed the DFT calculation research. C. J. W., X. Q. D. and Y. D. co-wrote the paper. All authors analyzed the data, discussed the results, and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by NSFC (22071186, 22071187, 22073067, 22101216, 22271226, and 22371216), National Youth Talent Support Program, Hubei Province NSF (2020CFA036 and 2021CFA069), and Fundamental Research Funds for the Central Universities (2042022kf1180). The authors thank Dr Ran Zhang from the Core Facility of Wuhan University for his generous support in the X-ray structures analysis.
Notes and references
-
(a) P. Antognazza, T. Benincori, S. Mazzoli, F. Sannicolo and T. Pilati, Phosphorus Sulfur, 1999, 144, 405–408 CrossRef;
(b) M. Shoeb, S. Celik, M. Jaspars, Y. Kumarasamy, S. M. MacManus, L. Nahar, P. K. Thoo-Lin and S. D. Sarker, Tetrahedron, 2005, 61, 9001–9006 CrossRef CAS;
(c) X.-Y. Liu, Y.-L. Zhang, X. Fei, L.-S. Liao and J. Fan, Chem. - Eur. J., 2019, 25, 4501–4508 CrossRef CAS PubMed.
-
(a) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC;
(b) B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981–2988 CrossRef CAS;
(c) T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem.–Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed;
(d) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed;
(e) X.-F. Bai, Y.-M. Cui, J. Cao and L.-W. Xu, Acc. Chem. Res., 2022, 55, 2545–2561 CrossRef CAS PubMed;
(f) J. K. Cheng, S.-H. Xiang and B. Tan, Acc. Chem. Res., 2022, 55, 2920–2937 CrossRef CAS PubMed;
(g) P. Rodríguez-Salamanca, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem.–Eur. J., 2022, 28, e202104442 CrossRef PubMed;
(h) J. S. Sweet and P. C. Knipe, Synthesis, 2022, 54, 2119–2132 CrossRef CAS;
(i) Y.-J. Wu, G. Liao and B.-F. Shi, Green Synth. Catal., 2022, 3, 117–136 CrossRef;
(j) H.-H. Zhang and F. Shi, Acc. Chem. Res., 2022, 55, 2562–2580 CrossRef CAS PubMed;
(k) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed;
(l) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed;
(m) G. Ma and M. P. Sibi, Chem.−Eur. J., 2015, 21, 11644–11657 CrossRef CAS PubMed;
(n) D. Bonne and J. Rodriguez, Chem. Commun., 2017, 53, 12385–12393 RSC;
(o) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed;
(p) S. Zhang, G. Liao and B. Shi, Chinese J. Org. Chem., 2019, 39, 1522–1528 CrossRef CAS;
(q) Z. Li and S. Yu, Sci. Sin.: Chim., 2020, 50, 509–525 Search PubMed;
(r) J. A. Carmona, C. Rodríguez-Franco, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem. Soc. Rev., 2021, 50, 2968–2983 RSC;
(s) B.-C. Da, S.-H. Xiang, S. Li and B. Tan, Chin. J. Chem., 2021, 39, 1787–1796 CrossRef CAS;
(t) C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed;
(u) G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1855–1893 CrossRef CAS.
- C. Chang and R. Adams, J. Am. Chem. Soc., 1931, 53, 2353–2357 CrossRef CAS.
-
(a) W. Lin, Q. Zhao, Y. Li, M. Pan, C. Yang, G. H. Yang and X. Li, Chem. Sci., 2021, 13, 141–148 RSC;
(b) G.-J. Mei, J. J. Wong, W. Zheng, A. A. Nangia, K. N. Houk and Y. Lu, Chem, 2021, 7, 2743–2757 CrossRef CAS;
(c) M. Pan, Y. B. Shao, Q. Zhao and X. Li, Org. Lett., 2022, 24, 374–378 CrossRef CAS PubMed;
(d) C. Portolani, G. Centonze, S. Luciani, A. Pellegrini, P. Righi, A. Mazzanti, A. Ciogli, A. Sorato and G. Bencivenni, Angew. Chem., Int. Ed., 2022, 61, e202209895 CrossRef CAS PubMed.
-
(a) X.-M. Wang, P. Zhang, Q. Xu, C.-Q. Guo, D.-B. Zhang, C.-J. Lu and R.-R. Liu, J. Am. Chem. Soc., 2021, 143, 15005–15010 CrossRef CAS PubMed;
(b) Q. Xu, H. Zhang, F.-B. Ge, X.-M. Wang, P. Zhang, C.-J. Lu and R.-R. Liu, Org. Lett., 2022, 24, 3138–3143 CrossRef CAS PubMed;
(c) W. Yao, C.-J. Lu, L.-W. Zhan, Y. Wu, J. Feng and R.-R. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218871 CrossRef CAS PubMed;
(d) S.-Y. Yin, Q. Zhou, C.-X. Liu, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2023, 62, e202305067 CrossRef CAS PubMed.
-
(a) K.-W. Chen, Z.-H. Chen, S. Yang, S.-F. Wu, Y.-C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202116829 CrossRef CAS PubMed;
(b) Y. Gao, L. Y. Wang, T. Zhang, B. M. Yang and Y. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202200371 CrossRef CAS PubMed.
- L.-Y. Pu, Y.-J. Zhang, W. Liu and F. Teng, Chem. Commun., 2022, 58, 13131–13134 RSC.
-
(a) P. Zhang, Q. Xu, X.-M. Wang, J. Feng, C.-J. Lu, Y. Li and R.-R. Liu, Angew. Chem., Int. Ed., 2022, 61, e202212101 CrossRef CAS PubMed;
(b) Z.-H. Chen, T.-Z. Li, N.-Y. Wang, X.-F. Ma, S.-F. Ni, Y.-C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202300419 CrossRef CAS PubMed.
-
(a) N. Ototake, Y. Morimoto, A. Mokuya, H. Fukaya, Y. Shida and O. Kitagawa, Chem. - Eur. J., 2010, 16, 6752–6755 CrossRef CAS PubMed;
(b) Y. Morimoto, S. Shimizu, A. Mokuya, N. Ototake, A. Saito and O. Kitagawa, Tetrahedron, 2016, 72, 5221–5229 CrossRef CAS;
(c) L. Peng, K. Li, C. Xie, S. Li, D. Xu, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2019, 58, 17199–17204 CrossRef CAS PubMed;
(d) Z.-S. Wang, L.-J. Zhu, C.-T. Li, B.-Y. Liu, X. Hong and L.-W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202201436 CrossRef CAS PubMed;
(e) M. Tian, D. Bai, G. Zheng, J. Chang and X. Li, J. Am. Chem. Soc., 2019, 141, 9527–9532 CrossRef CAS PubMed;
(f) Y.-P. He, H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2020, 59, 2105–2109 CrossRef CAS PubMed;
(g) X. Li, L. Zhao, Z. Qi and X. Li, Org. Lett., 2021, 23, 5901–5905 CrossRef CAS PubMed;
(h) C.-S. Wang, L. Wei, C. Fu, X.-H. Wang and C.-J. Wang, Org. Lett., 2021, 23, 7401–7406 CrossRef CAS PubMed;
(i) W.-C. Yang, X.-B. Chen, K.-L. Song, B. Wu, W.-E. Gan, Z.-J. Zheng, J. Cao and L.-W. Xu, Org. Lett., 2021, 23, 1309–1314 CrossRef CAS PubMed;
(j) V. Hutskalova and C. Sparr, Synthesis, 2023, 55, 1770–1782 CrossRef CAS;
(k) L. Yu, J. Liu, S. Xiang, T. Lu, P. Ma and Q. Zhao, Org. Lett., 2023, 25, 522–527 CrossRef CAS PubMed.
- CCDC 2279386 ((S)-2b) contains the supplementary crystallographic data for this paper.
-
(a) F. Russell, D. Harmody, P. J. McCarthy, S. A. Pomponi and A. E. Wright, J. Nat. Prod., 2013, 76, 1989–1992 CrossRef CAS PubMed;
(b) X. Zheng, L. Lv, S. Lu, W. Wang and Z. Li, Org. Lett., 2014, 16, 5156–5159 CrossRef CAS PubMed.
- DFT studies were conducted at the M06-2X/def2-TZVP(SMD)//M06-2X/6-31G** level. See ESI† for computational details.
- Y.-B. Wang, P. Yu, Z.-P. Zhou, J. Zhang, J. Wang, S.-H. Luo, Q.-S. Gu, K. N. Houk and B. Tan, Nat. Catal., 2019, 2, 504–513 CrossRef CAS.
-
(a) S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed;
(b) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2279386. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03686c |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c,
Xiu-Qin
Dong
*c,
Xiu-Qin
Dong