
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceParallel kinetic resolution of aziridines via chiral phosphoric acid-catalyzed apparent hydrolytic ring-opening†
Juan
Liu
,
Yi-Ying
Du
,
Yu-Shi
He
,
Yan
Liang
,
Shang-Zhong
Liu
,
Yi-Yi
Li
and
Yi-Ming
Cao
 *
*
College of Science & China Key Laboratory of National Forestry and Grassland Administration on Pest Chemical Control, China Agricultural University, Beijing 100193, China. E-mail: caoym@cau.edu.cn
First published on 12th October 2023
Abstract
We report a chiral phosphoric acid catalyzed apparent hydrolytic ring-opening reaction of racemic aziridines in a regiodivergent parallel kinetic resolution manner. Harnessing the acyloxy-assisted strategy, the highly stereocontrolled nucleophilic ring-opening of aziridines with water is achieved. Different kinds of aziridines are applicable in the process, giving a variety of enantioenriched aromatic or aliphatic amino alcohols with up to 99% yields and up to >99.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 enantiomeric ratio. Preliminary mechanistic study as well as product elaborations were inducted as well.
:0.5 enantiomeric ratio. Preliminary mechanistic study as well as product elaborations were inducted as well.
Introduction
Enantiopure β-amino alcohols are frequently encountered in natural products and synthetic molecules1 which display diverse bioactivities and excellent potential for pharmaceutical use (Scheme 1a).2 In addition, these units are frequently used as chiral auxiliaries and ligands in asymmetric synthesis.3 Due to the significant importance of such essential structural motifs, considerable attention has been paid to their enantioselective construction,4 and a variety of methods have been reported.5 The catalytic asymmetric hydrolytic ring-opening of aziridines6 provides an inherently efficient and atom-economical approach for the construction of chiral β-amino alcohols. Nonetheless, water is a rather inert nucleophile that is difficult to activate and control,7 rendering the method a daunting challenge. In 2014, Feng and co-workers realized a chiral magnesium(II)-catalyzed direct asymmetric ring-opening of meso-aziridine with H2O as the nucleophile (Scheme 1b),7i and this is to the best of knowledge, the only work on non-enzymatic catalytic enantioselective hydrolytic ring-opening of aziridines reported so far.8 In organocatalysis, List and co-workers reported a chiral phosphoric acid catalyzed aziridine ring-opening reaction using carboxylic acid as the nucleophile,9 which can be considered as an indirect version of asymmetric hydrolysis. In this context, the development of direct asymmetric hydrolytic ring-opening of aziridines (Scheme 1c), especially in organocatalysis, is highly demanded.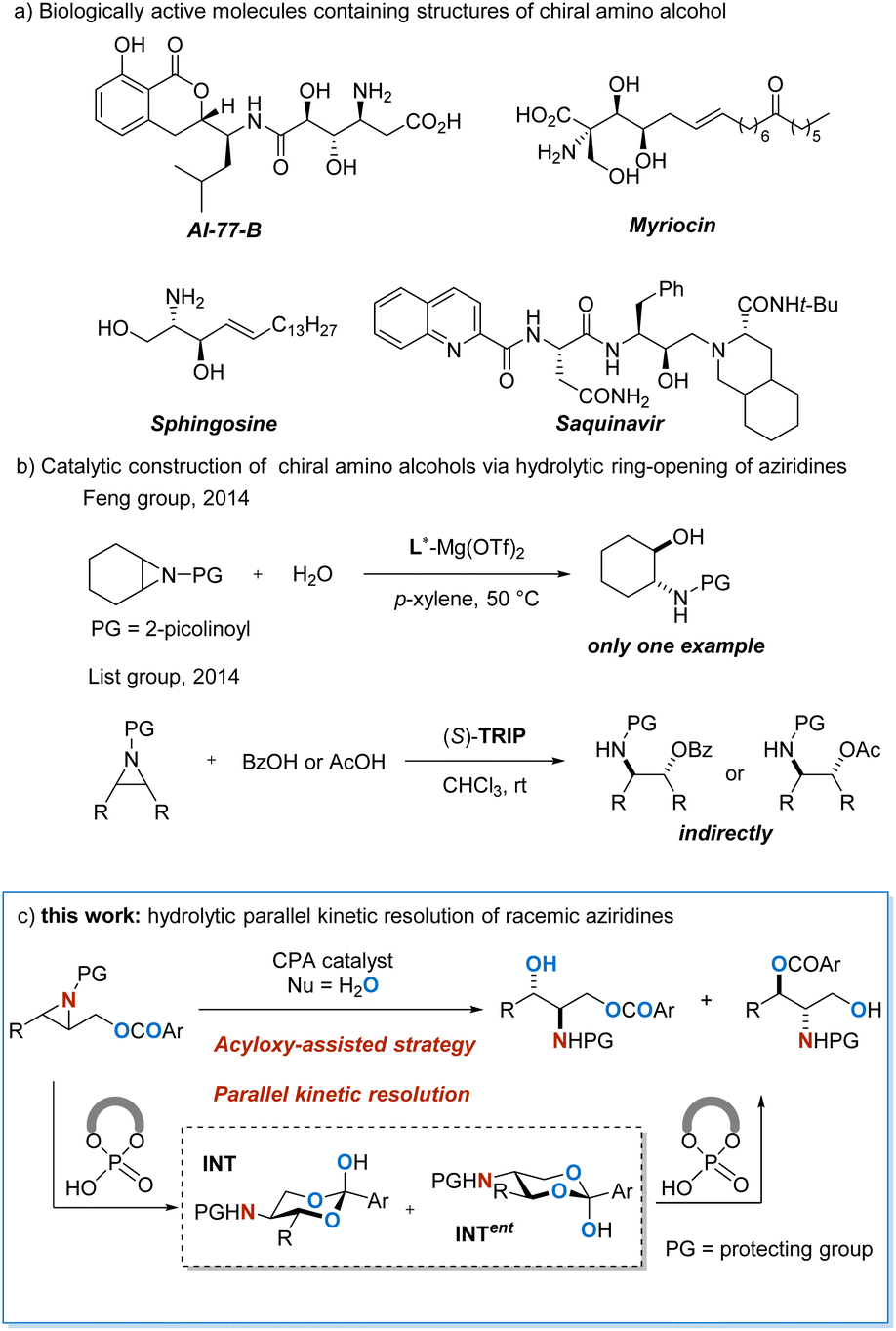 | ||
| Scheme 1 Enantioselective construction of amino alcohols via enantioselective hydrolytic ring-opening of aziridines. | ||
Recently, Christmann et al. reported a unique catalytic asymmetric halohydroxylation of unactivated alkenes using water as the nucleophile.10 In their strategy, an intramolecular assisting group is designed to activate H2O as a nucleophile by forming a temporally covalent interaction. We wondered whether a similar idea would facilitate the hydrolytic ring-opening of aziridines. To our delight, in the preliminary experiment, racemic aziridine (±)-1a′ successfully transformed into the corresponding products 2a′ and 3a′ in the presence of 4 equivalents of water and 10 mol% of chiral phosphoric acid C1 (ref. 11a) as the catalyst. The relationship between yields and ee is indicative of a parallel kinetic resolution process. When the acyl group was sterically substituted (3,5-di-tert-Bu), the enantioselectivity of the reaction was significantly improved, given the approach highly promising to be further optimized (Scheme 2).
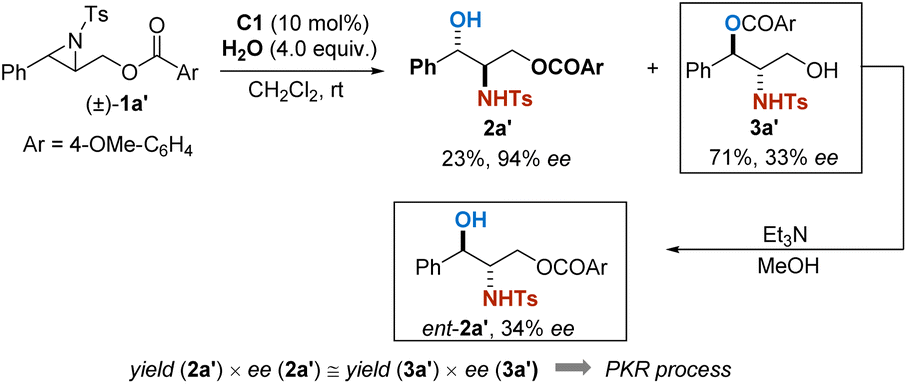 | ||
| Scheme 2 Preliminary study on the hydrolytic parallel kinetic resolution. | ||
Parallel kinetic resolution (PKR) is a unique type of resolution,12,13 in which the conversions of both enantiomers of a racemate proceed at similar rates, giving two separable chiral compounds as the products. This relatively uncommon class of reaction has considerable potential compared to traditional kinetic resolution (KR).14 However, it is a complicated task for one catalyst to accurately discriminate the enantiomers in a racemic mixture, yielding the two products in different directions according to their configurations. Therefore, in sharp contrast to the traditional resolution, much less successful examples have been reported on PKR.15 Most of the studies developed so far in this field involve metal catalysis,16 whilst examples of catalytic PKRs in enantioselective organocatalysis are, however, very limited.10,17
Chiral phosphoric acids (CPAs),14k,18 as an important class of organocatalysts, have been widely studied in asymmetric catalysis for their broad spectrum and efficient catalytic characteristics. Though, reports on CPA-catalyzed PKR are scarce. In 2010, List and co-workers reported CPA-catalyzed kinetic resolution of homoaldols via intramolecular transacetalization,19 wherein partial PKR was observed when the substrate was linear aliphatic substituted homoaldol; the group later reported a stereodivergent PKR in racemic diol acetalization20 and epoxide ring-opening21 with CPA as a catalyst. In 2011, Ding and co-workers demonstrated the CPA catalyzed Baeyer–Villiger oxidation of racemic cyclobutanones to form regioisomeric lactones in optically enriched forms.22 In 2019, Zheng and co-workers developed an efficient PKR of acyclic aliphatic syn-1,3-diol derived acetals in the presence of CPA.17e Very recently, Terada and co-workers reported a unique dynamic parallel kinetic resolution of the α-ferrocenyl cation initiated by the CPA catalyst.17f Although handful of examples were reported as mentioned, such a challenging field is still in its infancy, and new strategies as well as systematic development are highly desirable. Herein, we are glad to disclose a CPA-catalyzed asymmetric hydrolytic ring-opening of racemic aziridines via regiodivergent PKR, providing a new approach for the synthesis of chiral β-amino alcohols.
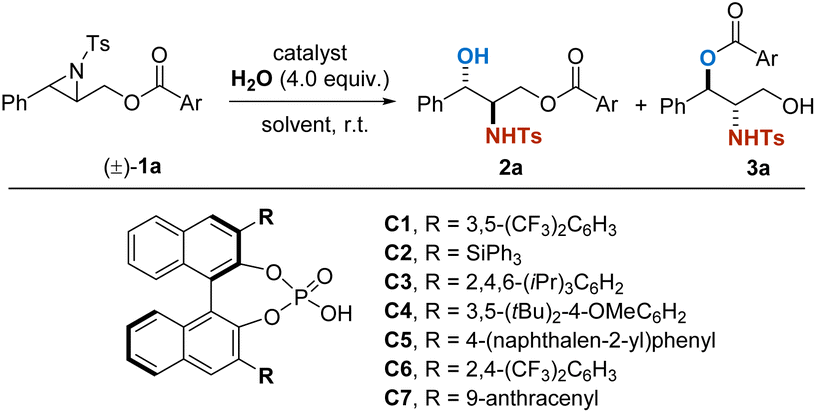
We commenced the investigation by using racemic aziridine 1a as a model substrate (Table 1). A variety of BINOL-derived CPA catalysts were evaluated (entries 2–7), and C7 (ref. 11b) was found to be the superior one, giving 2a with 99:1 enantiomeric ratio (er) and 3a with >99.5:0.5 er (entry 6). Notably, decreasing the catalyst loading from 10 mol% to 5 mol% resulted in almost the same outcomes in an acceptable reaction time (14 h, entry 7). The solvents were further screened and it was found that CH2Cl2 remains to be the optimal one. Ultimately, catalyst C7 in 5 mol% loading, together with CH2Cl2 as the solvent showed the best performance, giving 2a and 3a in excellent yields and enantioselectivities.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Solv. | Cat. (mol%) | Ratio 3/2 | er 2 | er 3 | Time (h) |
| a Unless otherwise noted, reactions were performed on a 0.05 mmol scale in 1.0 mL solvent at rt; >95% conversion was achieved after the indicated time; ratios were determined by 1H NMR analysis of the crude mixture; ers were determined by HPLC; Ar = 3,5-(t-Bu)2-4-MeO-C6H2. b The reactions were sluggish and substrates were not fully consumed. | |||||||
| 1 | C1 | CH2Cl2 | 10 | 1.12 | 98:2 |
94:6 |
2 |
| 2 | C2 | CH2Cl2 | 10 | 1.11 | 90:10 |
87.5:12.5 |
48 |
| 3 | C3 | CH2Cl2 | 10 | 1.07 | 99:1 |
96:4 |
14 |
| 4 | C4 | CH2Cl2 | 10 | 0.98 | 72:28 |
72.5:27.5 |
4 |
| 5 | C5 | CH2Cl2 | 10 | 1.09 | 97:3 |
95:5 |
2 |
| 6 | C7 | CH2Cl2 | 10 | 1.04 | 99:1 |
>99.5:0.5 |
3 |
| 7 | C7 | CH2Cl2 | 5 | 1.03 |
99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
>99.5:0.5
|
14 |
| 8 | C7 | Toluene | 5 | 0.94 | 97:3 |
98.5:1.5 |
16 |
| 9b | C7 | Et2O | 5 | 1.01 | 95:5 |
99:1 |
>72 |
| 10b | C7 | EtOAc | 5 | 1.02 | 95:5 |
97:3 |
>72 |
With the optimized conditions in hand, the substrate scope of this parallel kinetic resolution was assessed. Different racemic aryl substituted aziridines were evaluated (Table 2). Aziridines bearing both electron-withdrawing or -donating groups in para or meta positions on the phenyl ring proceeded smoothly and the corresponding products 2 and 3 were obtained in excellent yields and enantioselectivities (entries 2–11). Ortho-position-substituted racemic starting materials were also well tolerated under optimal conditions, providing excellent PKR performances, albeit a slower reaction rate was observed possibly due to steric hindrance (entries 12–14). The electronic properties of substituents on the phenyl rings of aziridines have a great impact on their reaction activities, for the electron-deficient substituents require a prolonged reaction time. It is worth noting that a sterically hindered 1-naphthyl derivative was feasible, giving the corresponding 2p in 45% yield with 97:3 er and 3p in 46% yield with 93.5:6.5 er (entry 16). Indene-derived aziridine 1s was also compatible, although with lower enantioselectivity (90:10 er, entry 19). Alcoholysis of 2a produced its amino alcohol derivative, of which the absolute configuration was unambiguously determined by X-ray crystallography (see Scheme 4a and ESI†).23
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R | 2 | 3 | Time (h) | ||||
| Yield (%) | er | Yield (%) | er | |||||
| a Unless otherwise noted, reactions were performed on a 0.1 mmol scale in 2 mL CH2Cl2 at rt for the indicated time; yields are of isolated products; ers were determined by HPLC; Ar = 3,5-(t-Bu)2-4-MeO-C6H2. b N-Mesyl aziridine was used as a substrate. c C6 was used as the catalyst. d ers are determined after derivatization. e Yields and ers are determined after derivatization. f 10 mol% catalyst was used. g C3 was used as the catalyst. | ||||||||
| 1 | C6H5 | 2a | 48 | 99:1 |
3a | 50 | >99.5:0.5 |
14 |
| 2 | 4-F-C6H4 | 2b | 49 | 99.5:0.5 |
3b | 50 | >99.5:0.5 |
14 |
| 3 | 4-Cl-C6H4 | 2c | 43 | 99:1 |
3c | 53 | 96:4 |
14 |
| 4 | 4-Br-C6H4 | 2d | 46 | 98:2 |
3d | 48 | 95:5 |
46 |
| 5b | 4-CF3-C6H4 | 2e | 46 | 98:2 |
3e | 51 | 97:3 |
48 |
| 6c | 4-Me-C6H4 | 2f | 43 | 98:2 |
3f | 48 | 90.5:9.5 |
5 |
| 7 | 3-F-C6H4 | 2g | 49 | 97.5:2.5 |
3g | 45 | >99.5:0.5 |
48 |
| 8 | 3-Cl-C6H4 | 2h | 47 | 97.5:2.5 |
3h | 48 | >99.5:0.5 |
60 |
| 9 | 3-Br-C6H4 | 2i | 46 | 98:2 |
3i | 48 | >99.5:0.5 |
72 |
| 10 | 3-MeO-C6H4 | 2j | 46 | 97.5:2.5 |
3j | 45 | 98:2 |
12 |
| 11 | 3-Me-C6H4 | 2k | 46 | 99:1 |
3k | 45 | 99.5:0.5 |
12 |
| 12 | 2-F-C6H4 | 2l | 48 | 99:1 |
3l | 48 | 99:1 |
72 |
| 13b | 2-Cl-C6H4 | 2m | 46 | 98.5:1.5 |
3m | 50 | 97.5:2.5 |
46 |
| 14b | 2-Br-C6H4 | 2n | 45 | 99.5:0.5 |
3n | 52 | 95.5:4.5 |
70 |
| 15b | 2-Me-C6H4 | 2o | 46 | 99.5:0.5 |
3o | 47 | 99:1 |
5 |
| 16c | 1-Naphthyl | 2p | 45 | 97:3 |
3p | 46 | 93.5:6.5 |
14 |
| 17f | 2-Naphthyl | 2q | 46 | 90:10 |
3q | 44 | 91:9 |
12 |
| 18g | 4-Phenyl | 2r | 44 | 99:1 |
3r | 46 | 98:2 |
12 |
| 19 |
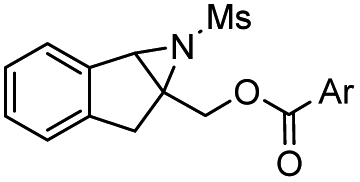
|
2s | 48 | 90:10 |
3s | 47 | 90:10 |
16 |
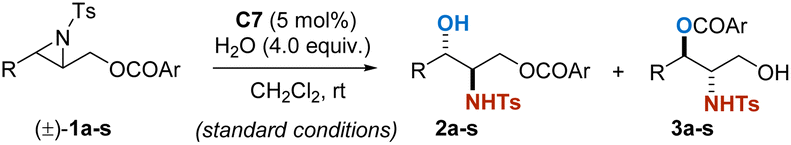
Encouraged by the satisfactory results obtained, we further broaden the substrate scope for PKR to alkyl-substituted aziridines, for their products, aliphatic amino alcohols, were found to be more common units in bioactive molecules. However, as the aliphatic aziridines are not electronically biased as aromatic ones, such substrates are relatively inert and hard to be activated for the transformation. As expected, aliphatic aziridine rac-1t as a substrate under the standard conditions resulted in a sluggish reaction. Therefore, several kinds of catalysts that are more electron-deficient were further screened (see the ESI†).24 Gratifyingly, C6 (R = 2,4-(CF3)2C6H3)24c as a catalyst was found to be satisfactory, resulting in 2t and 3t in excellent yield and high enantioselectivity (see the ESI†). Various kinds of alkyl-substituted aziridines reacted smoothly under the modified catalytic system, and a range of aliphatic chiral amino alcohols were synthesized (Table 3). Et, n-Pr, i-Pr and Bn substituted aziridines were well tolerated, providing the secondary alcohol products 2t–2w with 90:10 er to 95:5 er and the primary alcohol products 3t–3w with 90:10 er to 93:7 er (entries 1–4). Aziridines with terminal hydroxyl groups protected by silyl, benzyl and acyl-groups were amendable as well, and corresponding products were obtained in excellent yield and good to excellent enantioselectivity (entries 5–7). In addition, aziridine with an azide substitution was also applicable, which provided a convenient site for further derivatization (entry 8). It is noteworthy that the hydrolyses are all highly regioselective, although there is no electron effect provided by the aromatic ring as before in the examples in Table 2.
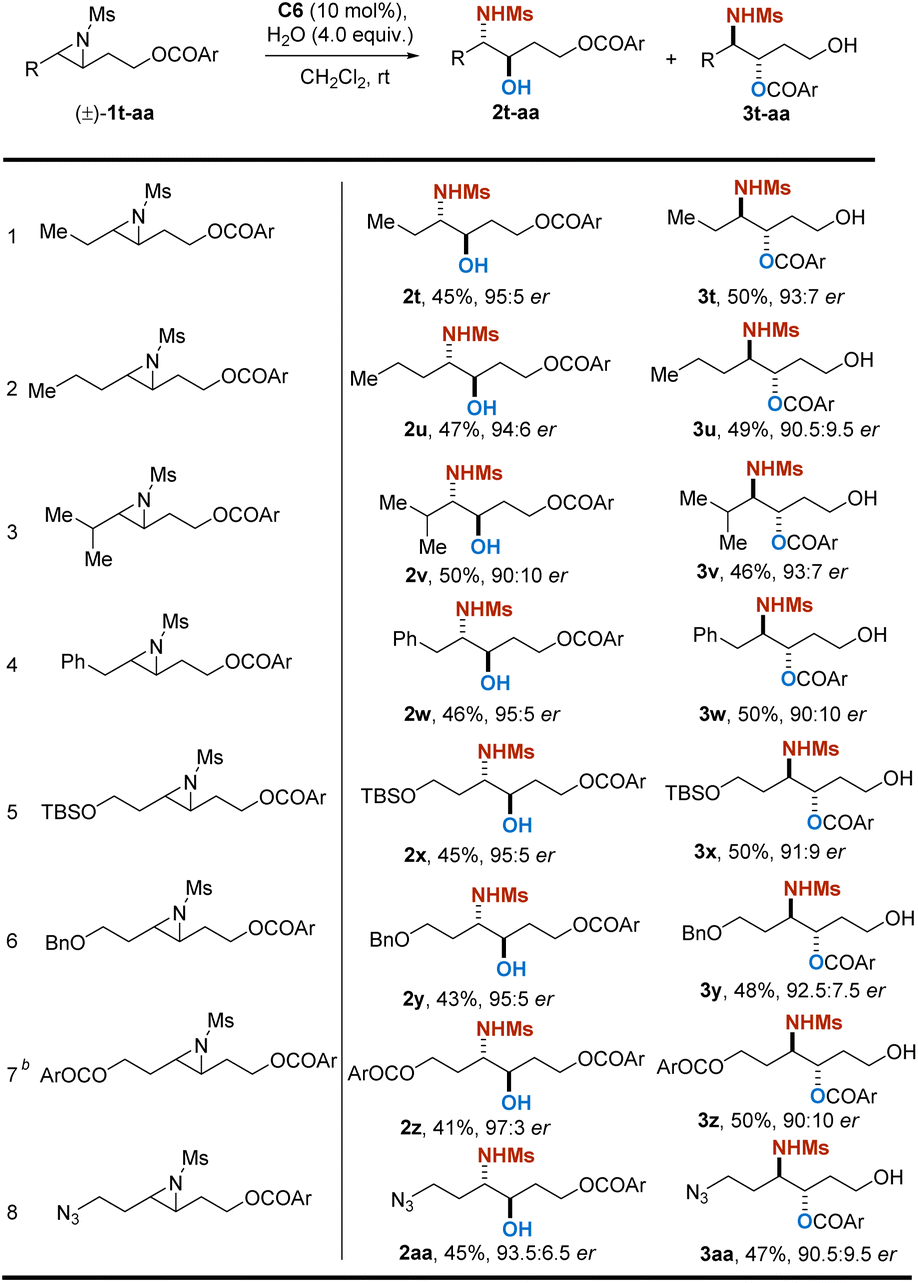
To further understand the mechanism of this developed reaction, control experiments were conducted (Scheme 3). Reaction employing Bn- instead of acyl-protected 2,3-aziridinyl alcohol was performed under the standard conditions, and no reaction took place, implying that the presence of an acyloxy moiety as an assisting group is crucial for the reaction to occur (Scheme 3a). When racemic constitutional isomers rac-2a or rac-3a were subjected to the standard conditions, no resolution was observed, which rules out the mechanisms involving interconversion between products (Scheme 3b). An isotopic labeling experiment was performed by using H218O instead of H2O (Scheme 3c), and the transformation resulted in an installation of both isomers with the 18O atom exclusively on the carbonyl group (see the ESI†). The outcome demonstrated that the addition of water proceeds via the assistance of acyloxyl but not a direct nucleophilic addition. Furthermore, enantiopure aziridine substrate 1o was subjected to the standard conditions. As anticipated, the reaction with (R)-C6 exclusively generated 2o, while the catalyst (S)-C6 with opposite configuration selectively afforded the corresponding product 3o (Scheme 3d). The performance of aziridine rac-1ae with two identical methyl substituents was evaluated, and this resulted in products 2ae and 3ae in almost racemic form. In contrast, when we subjected the disubstituted aziridine rac-1af with different substituents (methyl and n-propyl groups) to the reaction, the products 2af and 3af were both obtained with significantly increased er values (Scheme 3e). These results suggested that the steric difference at the carbon in the small ring distal to the acyloxy group is essential for the catalyst to discriminate substrates.
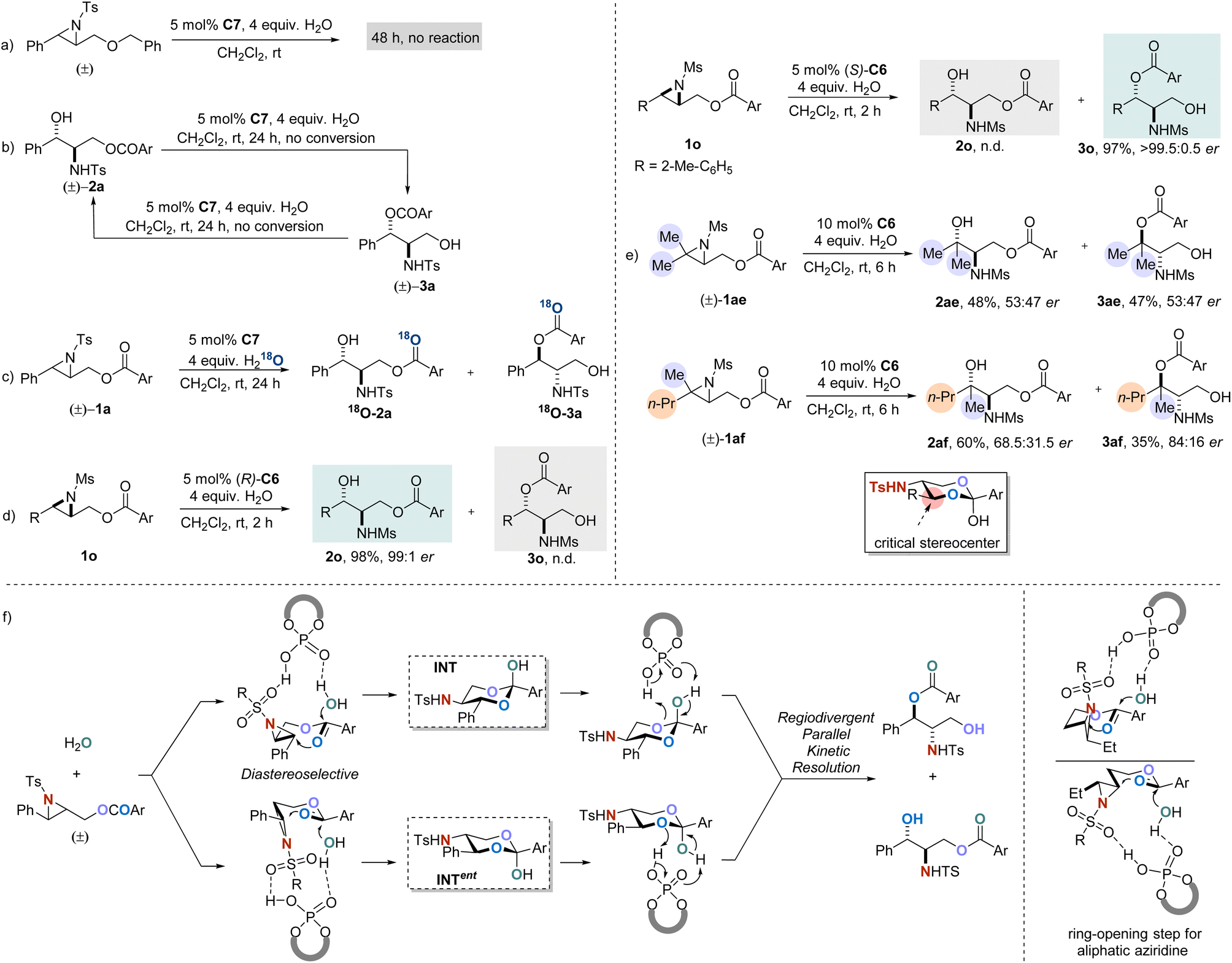 | ||
| Scheme 3 Preliminary study of the mechanism: (a) reaction of substrate without assisting group. (b) Products interconversion experiment. (c) Isotopic labeling experiment. (d) Reactions with enantiopure substrate. (e) Steric effect studies. (f) Proposed mechanism. | ||
On the basis of these results from the control experiments, a plausible reaction mechanism for the parallel kinetic resolution process is proposed (Scheme 3f). First, aziridine and H2O are both activated by CPA, and the two ends are connected by a double nucleophilic attack sequence throughout the carbonyl group in the substrate. During the course, the critical intermediates, racemic cyclic hemi-orthoesters (INT and ent-INT) containing three stereocenters are formed with excellent diastereoselectivity. Afterwards, the resulting two enantiomers of hemi-orthoester are regiodivergently protonated by CPA, leading to the corresponding final products.
Furthermore, transformations of the obtained products from this approach were presented for the elaboration of its synthetic utility (Scheme 4). Alcoholysis of isomer 3a readily provided 1,3-dihydroxy-2-amino compound 4a. Selective tosylation of 4a followed by treatment with Et3N regenerated a terminal aziridine 6a in 86% yield and 97.5:2.5 er. Reduction of 6a with LiAlH4 successfully afforded β-N-tosylaminoalcohol 7a in 95% yield and 99:1 er. In addition, treatment of 6a with t-BuOK gave aza-Payne rearrangement product 8a. The γ-N-tosylated amino alcohol 9a could be obtained by further transformation of 8a with LiAlH4 in 92% yield with maintained enantiopurity (Scheme 4a). Compound 9a is of interest as an intermediate in the synthesis of fluoxetine, which is a drug for treating depression and other disorders.25 Through the azide–alkyne cycloaddition, 2aa was transformed into a triazole (4aa) with almost complete retention of the stereochemical integrity (Scheme 4b).
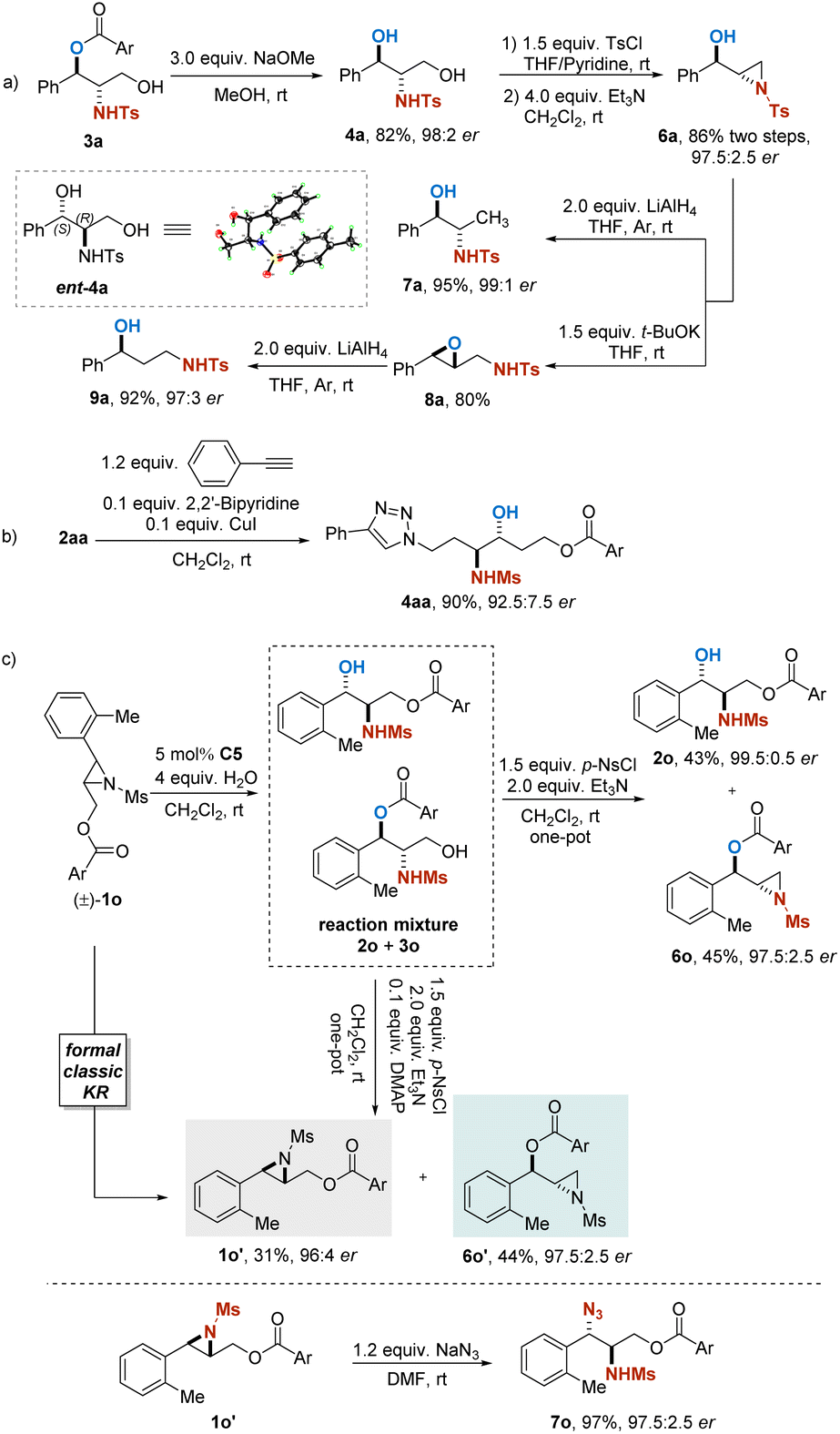 | ||
| Scheme 4 Transformations of the product: (a) derivatizations of 3a. (b) Azide–alkyne reaction of 2aa. (c) Reactions of 1o. | ||
As shown in Scheme 4c, treatment of the reaction mixture containing products 2o and 3o after the PKR with p-NsCl and Et3N resulted in a selective transformation, in which the primary alcohol 3o could be readily converted into terminal aziridine 6o with high efficiency, while the secondary alcohol 2o remained. However, when DMAP was additionally employed, the secondary alcohol 2o could be transformed into enantioenriched aziridine 1o′ as well. The obtained internal aziridine appeared to be one of the enantiomers of the starting materials in the PKR reaction, and this one-pot procedure resulted in a formal traditional KR of racemic aziridine 1o. Enantioenriched aziridines provided by the transformations mentioned above are synthetically useful for their propensity to undergo ring-opening reactions to give a series of functionalized molecules. For instance, by treatment of NaN3, chiral azido amide 7o could be regioselectively obtained in 97% yield with 97.5:2.5 er.
Conclusions
In summary, we reported an intriguing CPA-catalyzed asymmetric hydrolytic ring-opening of racemic aziridines via regiodivergent PKR. A wide array of aromatic and aliphatic aziridines were well compatible, providing amino alcohol derivatives in generally excellent yield and enantioselectivities. Preliminary mechanistic studies indicate that such a PKR process may have proceeded via the formation of cyclic hemi-orthoester intermediates and a subsequent regiodivergent resolution. In addition, facile and versatile transformations of the enantioenriched products demonstrated the utilities of the method in asymmetric synthesis. Further study on the mechanism as well as the applications of this methodology are underway.Data availability
All experimental data and detailed procedures are available in the ESI.†Author contributions
J. L. performed the main part of the experiments, and prepared the ESI† and manuscript. Y.-Y. D. participated in the synthesis of substrates, compound characterization, and ESI† preparation. Y.-S. H. and Y. L. participated in the synthesis of substrates and discussion. S.-Z. L. and Y.-Y. L. participated in the discussion and helped with the optimization of the manuscript. Y.-M. C. conceived and directed the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the National Key R&D Program (2022YFD1700403) and the 2115 Talent Development Program of China Agricultural University.Notes and references
- (a) I. Gallou and C. H. Senanayake, Chem. Rev., 2006, 106, 2843 CrossRef CAS PubMed; (b) P. Zhang, D. J. Dairaghi, J. C. Jaen and J. P. Powers, in Annual Reports in Medicinal Chemistry, ed. M. C. Desai, Academic Press, New York, 2013, vol. 48, p. 133 Search PubMed.
- (a) H. Umezawa, T. Aoyagi, H. Suda, M. Hamada and T. Takeuchi, J. Antibiot., 1976, 29, 97 CrossRef CAS PubMed; (b) J. Kobayashi, J.-F. Cheng, M. Ishibashi, M. R. Wälchli, S. Yamamura and Y. Ohizumi, J. Chem. Soc., Perkin Trans. 1, 1991, 1135 RSC; (c) Y. A. Hannun and C. M. Linardic, Biochim. Biophys. Acta, Biomembr., 1993, 1154, 223 CrossRef CAS PubMed; (d) Y. Ohta and I. Shinkai, Bioorg. Med. Chem., 1997, 5, 465 CrossRef CAS PubMed.
- (a) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS PubMed; (b) J. L. Vicario, D. Badia, L. Carrillo, E. Reyes and J. Etxebarria, Curr. Org. Chem., 2005, 9, 219 CrossRef CAS; (c) G. Della Sala, A. Russo and A. Lattanzi, Curr. Org. Chem., 2011, 15, 2147 CrossRef CAS; (d) U. V. S. Reddy, M. Chennapuram, C. Seki, E. Kwon, Y. Okuyama and H. Nakano, Eur. J. Org Chem., 2016, 2016, 4124 CrossRef.
- For selected reviews, see: (a) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS; (b) F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367 CrossRef CAS PubMed; (c) O. N. Burchak and S. Py, Tetrahedron, 2009, 65, 7333 CrossRef CAS; (d) T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem. – Eur. J., 2011, 17, 58 CrossRef CAS PubMed; (e) C. Weng, H. Zhang, X. Xiong, X. Lu and Y. Zhou, Asian J. Chem., 2014, 26, 3761 CrossRef CAS; (f) H. Sasai, in Comprehensive Organic Synthesis II, ed. P. Knochel, and G. A. Molander, Elsevier, Amsterdam, 2014, vol. 2, p. 543 Search PubMed.
- For selected examples, see: (a) S. Matsunaga, T. Yoshida, H. Morimoto, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 8777 CrossRef CAS PubMed; (b) B. M. Trost, J. Jaratjaroonphong and V. Reutrakul, J. Am. Chem. Soc., 2006, 128, 2778 CrossRef CAS PubMed; (c) J.-H. Xie, S. Liu, W.-L. Kong, W.-J. Bai, X.-C. Wang, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 4222 CrossRef CAS PubMed; (d) K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2012, 134, 12370 CrossRef CAS PubMed; (e) H.-C. Shen, Y.-F. Wu, Y. Zhang, L.-F. Fan, Z.-Y. Han and L.-Z. Gong, Angew. Chem., Int. Ed., 2018, 57, 2372 CrossRef CAS PubMed; (f) M. Tang, H. Gu, S. He, S. Rajkumar and X. Yang, Angew. Chem., Int. Ed., 2021, 60, 21334 CrossRef CAS PubMed.
- For selected reviews on ring-opening reaction of racemic aziridines: (a) X. E. Hu, Tetrahedron, 2004, 60, 2701 CrossRef CAS; (b) S. Stanković, M. D'Hooghe, S. Catak, H. Eum, M. Waroquier, V. V. Speybroeck, N. D. Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643 RSC; (c) R. Akhtar, S. A. R. Naqvi, A. F. Zahoor and S. Saleem, Mol. Diversity, 2018, 22, 447 CrossRef CAS PubMed; (d) Z. Chai, Synthesis, 2020, 52, 1738 CrossRef CAS . For selected reviews on ring-opening reaction of meso-aziridines:; (e) M. Pineschi, Eur. J. Org Chem., 2006, 2006, 4979 CrossRef; (f) C. Schneider, Angew. Chem., Int. Ed., 2009, 48, 2082 CrossRef CAS PubMed; (g) P.-A. Wang, Beilstein J. Org. Chem., 2013, 9, 1677 CrossRef PubMed; (h) P.-J. Yang and Z. Chai, Org. Biomol. Chem., 2023, 21, 465 RSC.
- For selected examples using water as a nucleophile: (a) M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936 CrossRef CAS PubMed; (b) J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 2687 CrossRef CAS PubMed; (c) S.-F. Zhu, C. Chen, Y. Cai and Q.-L. Zhou, Angew. Chem., Int. Ed., 2008, 47, 932 CrossRef CAS PubMed; (d) S.-F. Zhu, Y. Cai, H.-X. Mao, J.-H. Xie and Q.-L. Zhou, Nat. Chem., 2010, 2, 546 CrossRef CAS PubMed; (e) G. Dong, P. Teo, Z. K. Wickens and R. H. Grubbs, Science, 2011, 333, 1609 CrossRef CAS PubMed; (f) M. Gärtner, S. Mader, K. Seehafer and G. Helmchen, J. Am. Chem. Soc., 2011, 133, 2072 CrossRef PubMed; (g) N. Kanbayashi and K. Onitsuka, Angew. Chem., Int. Ed., 2011, 50, 5197 CrossRef CAS PubMed; (h) D. D. Ford, L. P. C. Nielsen, S. J. Zuend, C. B. Musgrave and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 15595 CrossRef CAS PubMed; (i) J. Li, Y. Liao, Y. Zhang, X. Liu, L. Lin and X. Feng, Chem. Commun., 2014, 50, 6672 RSC; (j) Q.-K. Kang, L. Wang, Q.-J. Liu, J.-F. Li and Y. Tang, J. Am. Chem. Soc., 2015, 137, 14594 CrossRef CAS PubMed; (k) X. Zhang, J. Li, H. Tian and Y. Shi, Chem. – Eur. J., 2015, 21, 11658 CrossRef CAS PubMed; (l) W. Guo, L. Martínez-Rodríguez, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 11037 CrossRef CAS PubMed; (m) W. Li, P. Zhou, G. Li, L. Lin and X. Feng, Adv. Synth. Catal., 2020, 362, 1982 CrossRef CAS; (n) Z. Xi, X.-J. Liu, Z. Guo, Z. Gao, Z.-X. Yu and H. Gao, Nat. Synth., 2023, 2, 778 CrossRef; (o) J. Li, Z. Wang, X. Wang and Y. Shi, Tetrahedron Chem, 2023, 6, 100039 CrossRef.
- For enzyme-catalyzed reactions, see: (a) T. Watabe and S. Suzuki, Biochem. Biophys. Res. Commun., 1972, 46, 1120 CrossRef CAS PubMed; (b) G. M. Lacourciere, V. N. Vakharia, C. P. Tan, D. I. Morris, G. H. Edwards, M. Moos and R. N. Armstrong, Biochemistry, 1993, 32, 2610 CrossRef CAS PubMed.
- M. R. Monaco, B. Poladura, M. D. de Los Bernardos, M. Leutzsch, R. Goddard and B. List, Angew. Chem., Int. Ed., 2014, 53, 7063 CrossRef CAS PubMed.
- Y.-M. Cao, D. Lentz and M. Christmann, J. Am. Chem. Soc., 2018, 140, 10677 CrossRef CAS PubMed.
- (a) T. Akiyama, H. Morita, J. Itoh and K. Fuchibe, Org. Lett., 2005, 7, 2583 CrossRef CAS PubMed; (b) D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2005, 127, 9360 CrossRef CAS PubMed.
- (a) E. Vedejs and X. Chen, J. Am. Chem. Soc., 1997, 119, 2584 CrossRef CAS; (b) H. B. Kagan, Tetrahedron, 2001, 57, 2449 CrossRef CAS.
- After the concept of PKR was introduced, it is also used to describe the process in which only one catalyst is involved. Such a reaction is similar to the concept of divergent reaction on a racemic mixture (divergent RRM). In this article, PKR and divergent RRM are equal.
- For selected reviews of KR, see: (a) H. B. Kagan and J. C. Fiaud, in Topics in Stereochemistry, ed. E. L. Eliel and S. H. Wilen, Wiley-VCH, Weinheim, 1988, vol. 18, p. 249 Search PubMed; (b) G. R. Cook, Curr. Org. Chem., 2000, 4, 869 CrossRef CAS; (c) J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5 CrossRef CAS; (d) D. E. J. E. Robinson and S. D. Bull, Tetrahedron: Asymmetry, 2003, 14, 1407 CrossRef CAS; (e) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS PubMed; (f) C. E. Müller and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 6012 CrossRef PubMed; (g) H. Pellissier, Adv. Synth. Catal., 2011, 353, 1613 CrossRef CAS; (h) G. Ma and M. P. Sibi, Chem. – Eur. J., 2015, 21, 11644 CrossRef CAS PubMed; (i) R. Gurubrahamam, Y.-S. Cheng, W.-Y. Huang and K. Chen, ChemCatChem, 2016, 8, 86 CrossRef CAS; (j) I. Kreituss and J. W. Bode, Acc. Chem. Res., 2016, 49, 2807 CrossRef CAS PubMed; (k) K. S. Petersen, Asian J. Org. Chem., 2016, 5, 308 CrossRef CAS PubMed; (l) W. Liu and X. Yang, Asian J. Org. Chem., 2021, 10, 692 CrossRef CAS.
- For selected reviews of PKR, see: (a) H. B. Kagan, Tetrahedron, 2001, 57, 2449 CrossRef CAS; (b) J. R. Dehli and V. Gotor, Chem. Soc. Rev., 2002, 31, 365 RSC; (c) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS PubMed; (d) R. R. Kumar and H. B. Kagan, Adv. Synth. Catal., 2010, 352, 231 CrossRef CAS; (e) L. C. Miller and R. Sarpong, Chem. Soc. Rev., 2011, 40, 4550 RSC; (f) N. Funken, Y.-Q. Zhang and A. Gansäuer, Chem. – Eur. J., 2017, 23, 19 CrossRef CAS PubMed.
- For selected related examples in metal catalysis, see: (a) F. Bertozzi, P. Crotti, F. Macchia, M. Pineschi and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 930 CrossRef CAS; (b) K. Tanaka and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 8078 CrossRef CAS PubMed; (c) K. Tanaka, Y. Hagiwara and M. Hirano, Angew. Chem., Int. Ed., 2006, 45, 2734 CrossRef CAS PubMed; (d) A. Gansäuer, C.-A. Fan, F. Keller and J. Keil, J. Am. Chem. Soc., 2007, 129, 3484 CrossRef PubMed; (e) C. K. Jana and A. Studer, Angew. Chem., Int. Ed., 2007, 46, 6542 CrossRef CAS PubMed; (f) D. Minato, Y. Nagasue, Y. Demizu and O. Onomura, Angew. Chem., Int. Ed., 2008, 47, 9458 CrossRef CAS PubMed; (g) R. Webster, C. Böing and M. Lautens, J. Am. Chem. Soc., 2009, 131, 444 CrossRef CAS PubMed; (h) B. Wu, J. R. Parquette and T. V. RajanBabu, Science, 2009, 326, 1662 CrossRef CAS PubMed; (i) A. Gansäuer, L. Shi and M. Otte, J. Am. Chem. Soc., 2010, 132, 11858 CrossRef PubMed; (j) D. Katayev, M. Nakanishi, T. Bürgi and E. P. Kündig, Chem. Sci., 2012, 3, 1422 RSC; (k) M. Mulzer, W. C. Ellis, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 1928 RSC; (l) B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Chem. Sci., 2014, 5, 1102 RSC; (m) Y. Xu, K. Kaneko, M. Kanai, M. Shibasaki and S. Matsunaga, J. Am. Chem. Soc., 2014, 136, 9190 CrossRef CAS PubMed; (n) D. Grosheva and N. Cramer, ACS Catal., 2017, 7, 7417 CrossRef CAS; (o) Y. Yuan, Z.-J. Zheng, L. Li, X.-F. Bai, Z. Xu, Y.-M. Cui, J. Cao, K.-F. Yang and L.-W. Xu, Adv. Synth. Catal., 2018, 360, 3002 CrossRef CAS; (p) X. Wang, Y. Luo, J. Li, C. Wang, Q. Liu, Y. He, S. Luo and Q. Zhu, ACS Catal., 2022, 12, 14918 CrossRef CAS.
- For selected related examples in organocatalysis, see: (a) J. M. Rodrigo, Y. Zhao, A. H. Hoveyda and M. L. Snapper, Org. Lett., 2011, 13, 3778 CrossRef CAS PubMed; (b) A. D. Worthy, X. Sun and K. L. Tan, J. Am. Chem. Soc., 2012, 134, 7321 CrossRef CAS PubMed; (c) F. Romanov-Michailidis, M. Pupier, L. Guénée and A. Alexakis, Chem. Commun., 2014, 50, 13461 RSC; (d) C. R. Shugrue, A. L. Featherston, R. M. Lackner, A. Lin and S. J. Miller, J. Org. Chem., 2018, 83, 4491 CrossRef CAS PubMed; (e) H. Yang and W.-H. Zheng, Org. Lett., 2019, 21, 5197 CrossRef CAS PubMed; (f) Y. Toda, T. Korenaga, R. Obayashi, J. Kikuchi and M. Terada, Chem. Sci., 2021, 12, 10306 RSC; (g) K. Yamashita, R. Hirokawa, M. Ichikawa, T. Hisanaga, Y. Nagao, R. Takita, K. Watanabe, Y. Kawato and Y. Hamashima, J. Am. Chem. Soc., 2022, 144, 3913 CrossRef CAS PubMed; (h) A. Adili, J.-P. Webster, C. Zhao, S. C. Mallojjala, M. A. Romero-Reyes, I. Ghiviriga, K. A. Abboud, M. J. Vetticatt and D. Seidel, ACS Catal., 2023, 13, 2240 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (b) M. Terada, Chem. Commun., 2008, 4097 RSC; (c) D. Kampen, C. M. Reisinger and B. List, in Asymmetric Organocatalysis, ed. B. List, Topics in Current Chemistry, Springer, Berlin, 2010, vol. 291, p. 201 Search PubMed; (d) M. Rueping, R. M. Koenigs and I. Atodiresei, Chem. – Eur. J., 2010, 16, 9350 CrossRef CAS PubMed; (e) M. Terada, Synthesis, 2010, 2010, 1929 CrossRef; (f) M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539 RSC; (g) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (h) R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142 RSC; (i) B. P. Neupane and G. K. Friestad, Curr. Org. Chem., 2022, 26, 991 CrossRef CAS.
- I. Čorić, S. Müller and B. List, J. Am. Chem. Soc., 2010, 132, 17370 CrossRef PubMed.
- J. H. Kim, I. Čorić, C. Palumbo and B. List, J. Am. Chem. Soc., 2015, 137, 1778 CrossRef CAS PubMed.
- M. R. Monaco, D. Fazzi, N. Tsuji, M. Leutzsch, S. Liao, W. Thiel and B. List, J. Am. Chem. Soc., 2016, 138, 14740 CrossRef CAS PubMed.
- S. Xu, Z. Wang, X. Zhang and K. Ding, Eur. J. Org Chem., 2011, 2011, 110 CrossRef.
- Deposition Number 2244430 (for ent-4a) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- (a) N. Momiyama, H. Okamoto, J. Kikuchi, T. Korenaga and M. Terada, ACS Catal., 2016, 6, 1198 CrossRef CAS; (b) N. Momiyama, H. Nishimoto and M. Terada, Org. Lett., 2011, 13, 2126 CrossRef CAS PubMed; (c) T. Sakamoto, J. Itoh, K. Mori and T. Akiyama, Org. Biomol. Chem., 2010, 8, 5448 RSC.
- X.-M. Kan, J. Zhu, P.-H. Li, Z.-C. Wu and P.-J. Yang, Tetrahedron, 2023, 132, 133263 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectral data, NMR-data, and HPLC-data. CCDC 2244430. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03899h |
| This journal is © The Royal Society of Chemistry 2023 |