
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deep eutectic solvent assisted preparation of cellulose nanofibers and graphene composite films for supercapacitors†
Zhongzheng
Ma
a,
Yi
Duan
a,
Yongqi
Deng
 a,
Hongdong
Quan
b,
Xiuguo
Yang
b,
Hongyan
Li
b,
Luqian
Ye
b,
Bingxia
Xu
c and
Lifeng
Yan
*a
a,
Hongdong
Quan
b,
Xiuguo
Yang
b,
Hongyan
Li
b,
Luqian
Ye
b,
Bingxia
Xu
c and
Lifeng
Yan
*a
aDepartment of Chemical Physics, University of Science and Technology of China, Jinzai Road 96, Hefei, 230026, Anhui, China. E-mail: lfyan@ustc.edu.cn
bInner Mongolia Key Laboratory of Polyol Chemical New Material Enterprise, Chifeng Ruiyang Chemical Co. Ltd, Pingzhuang, Ruiyang, 024076, Inner Mongolia, China
cKey Laboratory of Anhui for Tobacco Chemistry, Hefei, 230088, Anhui, China
First published on 15th May 2023
Abstract
Flexible cellulose nanofiber-modified graphene composite has been prepared using rice husk and graphite as the feedstocks. First, cellulose nanofibers (CNFs) were obtained from designated rice husk by green choline hydroxide (ChOH) and L-necropine hydrochloride deep eutectic solvent (DES), which can efficiently promote the dispersion of graphene oxide and relatively reduced graphene oxide (rGO) to form rGO/CNF composite membranes. The composite film shows good bending and low resistance to charge transfer. Electrochemical testing studies reveal that the as-prepared rGO/CNF composite membranes can work as an efficient electrode for supercapacitors, and it shows a high area-specific capacitance (382 mF cm−2 when the current density is 1 mA cm−2) and good cycle stability (6% capacitance loss over 3000 cycles at 10 mA cm−2). The rGO/CNF composite has potential applications as electrodes for flexible supercapacitors.
Sustainability spotlightAlternative biomass feedstocks and green solvents for functional materials are basic elements of sustainability, while high-performance energy storage devices are emergency necessary. Here, the green solvent deep eutectic solvent was used to treat rice husk directly for cellulose nanofibers (CNF), and the flexible composite of CNF and graphene has been prepared. The composite film can be used as the flexible electrode for supercapacitors with high area-specific capacitance for sustainable energy. |
Introduction
With global energy scarcity, high-efficiency energy storage devices have attracted much attention, and the main energy storage devices entering commercialization today are lithium-ion batteries and supercapacitors.1–3 In the research on supercapacitors, electrode materials have been a hot research topic.4–8 Carbon materials are favored due to their high electrical conductivity, easy availability of raw materials, and low production costs, and commercial electrodes are still mainly made of carbon materials.9–11 Since 2004, when graphene was first introduced, it has received attention from the fields of materials, energy, and biomedicine.12–14 Graphene is a two-dimensional nanomaterial with advantages such as high specific surface area, excellent electrical conductivity, high mechanical properties, and good thermal conductivity, and is considered an excellent choice for electrodes in energy storage devices,15 and the energy storage field was also the first important area where graphene was commercialised.16A great deal of research into graphene-based electrodes has focused on how to increase the specific capacitance of the electrode by increasing the specific surface area, but this brings with it a reduction in the density and strength of the material. These materials often have a fluffy structure that makes them extremely weak and prone to collapse and deformation during use. In addition, although the material itself has little mass, the electrolyte enters the pores and fills them, decreasing energy storage capacity but increasing the mass of the device. These problems can hinder the application of graphene-based electrodes in portable electronic devices.17 In addition, graphene, regardless of the degree of oxidation, tends to aggregate together in solvents and substrates, to the detriment of the excellent properties of graphene itself. To make graphene uniformly dispersed and to prepare better graphene composites, researchers often ensure the dispersion of graphene through in situ polymerization, graphene modification, and the addition of dispersants.18–20
Recently, with the application of biomass in energy storage, biomass materials such as cellulose have been increasingly used in graphene-based composites.21–24 Cellulose, especially cellulose nanofibres, is mostly compounded with conductive materials to prepare flexible electrodes for energy storage devices such as supercapacitors.25–27 It has been demonstrated that the addition of cellulose can alleviate the agglomeration of graphene flakes and facilitate the dispersion of graphene.28 In addition, it has also been demonstrated that deep eutectic solvent (DES) can insert the lamellae of graphene due to its viscosity and strong hydrogen bonding, preventing the aggregation and settling of graphene,29–31 and the combination of DES and cellulose may further promote the dispersion of graphene nanosheets and avoid the formation of aggregates. Recently, Liu et al. prepared nitrogen-doped composite electrodes using DES of choline chloride and urea to composite cellulose nanofibres with reduced graphene in a simple hydrothermal environment.29 In this method, the aggregation of the lamellae was attenuated by the protonation of the graphene surface due to the pH adjustment of the system by DES. In addition, graphene also showed significant doping due to the presence of urea.29
In this work, in order to further exploit lignocellulose and explore the application of DES, we used the DES of choline hydroxide and L-necropine hydrochloride to swell the cellulose under the assistance of ultrasound, allowing partial cellulose strip into cellulose nanofibres and destroying the amorphous aggregates of cellulose. Then, doped graphene-cellulose composite films were prepared by mixing the suspension of cellulose nanofiber and the DES with graphene oxide by a simple hydrothermal method. The composite films showed good flexibility with reasonable doping rates of heteroatoms. The high area-specific capacitance demonstrated in the two-electrode test remains quite high after 3000 cycles of charge and discharge. We hope that such a strategy will lead to further applications of lignocellulose with DES in graphene-based composite electrodes (Scheme 1).
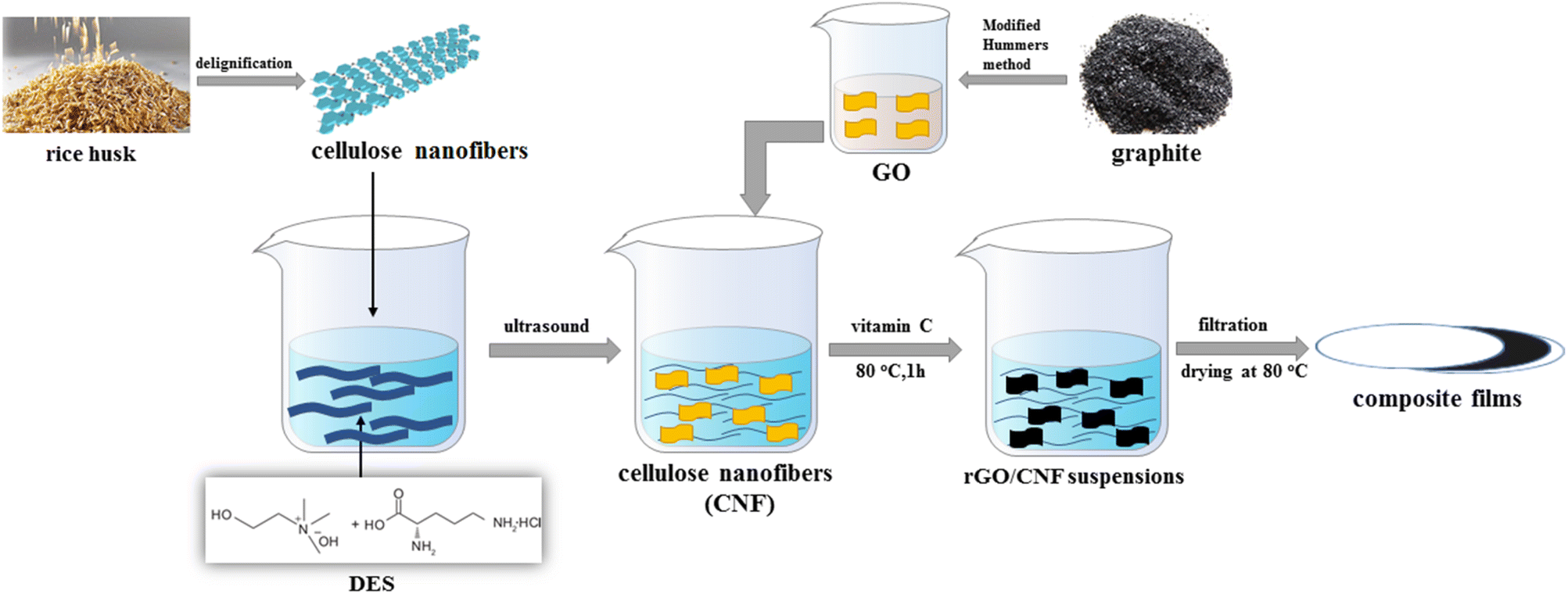 | ||
| Scheme 1 Schematic illustration of the preparation of cellulose nanofibers and reduced graphene oxide composite films. | ||
Results and discussion
To reduce the energy consumption in obtaining cellulose nanofibres and to produce cellulose that retains some of the larger fibers, we used a combination of DES pretreatment and powerful sonication to obtain a partial nano-fibrillated cellulose suspension. The DES selected for the study will neutralize the reaction during the mixing process, alleviating the strong alkalinity of ChOH, and both are green and non-polluting. Due to the strong hydrogen bonding interactions of DES, the bleached cellulose is dispersed in DES and the crystalline areas of the cellulose are gradually dissolved and the amorphous areas are gradually swollen, allowing the fibrous structure of the cellulose to be stripped, which greatly increases the accessibility of the ultrasound treatment. Under intense mechanical treatment, the hydrogen bonding between the amorphous large cellulose fibers is gradually disrupted and dispersed to form a network of cellulose nanofibres.32To further characterize the effect of the DES pretreatment process on the preparation of cellulose nanofibres, comparative experiments were done with 90 °C heat treatment for 6 h (DC-6), 90 °C heat treatment for 12 h (DC-12), and 400 W microwave treatment for 6 min (DC-MW). After the treatment, a gel-like suspension of cellulose nanofiber was obtained (Fig. S1†). To observe the morphology of the cellulose, we subjected the sonicated cellulose suspension for TEM characterization, as shown in Fig. 1. Cellulose in the picture shows a dendritic network structure, which can be seen in the field of view at 500 nm, where the cellulose is stripped from the end, showing fine branches of a few nanometres at the thinnest point and curved fibers of about 100 nm at the backbone. At a field of view of 100 nm, it can be observed that each fibril bundle exhibits streaks of fine lines, which also demonstrate the mechanism of fibril peeling and that these fibrils are in a network-like structure. It is worth noting that as the heat treatment time increases, there are more particles in the field of view, which could be crystals formed by DES precipitation after solvent drying or denaturing at high temperatures. Especially after the microwave treatment, cellulose is stripped more severely and more fine strips of fibers of a few nanometres appear, but with this comes the appearance of more crystalline particles of DES. After microwave treatment, the solution appears as a solid–liquid partition. The liquid part was a low-viscosity black liquid, which indicated that some of the cellulose was dissolved and pyrolyzed, and changed into small molecules or even carbon dots after the microwave treatment. Therefore, considering the protection of DES and the cellulose yield, we used the suspension of DC-6 for the subsequent experiments.
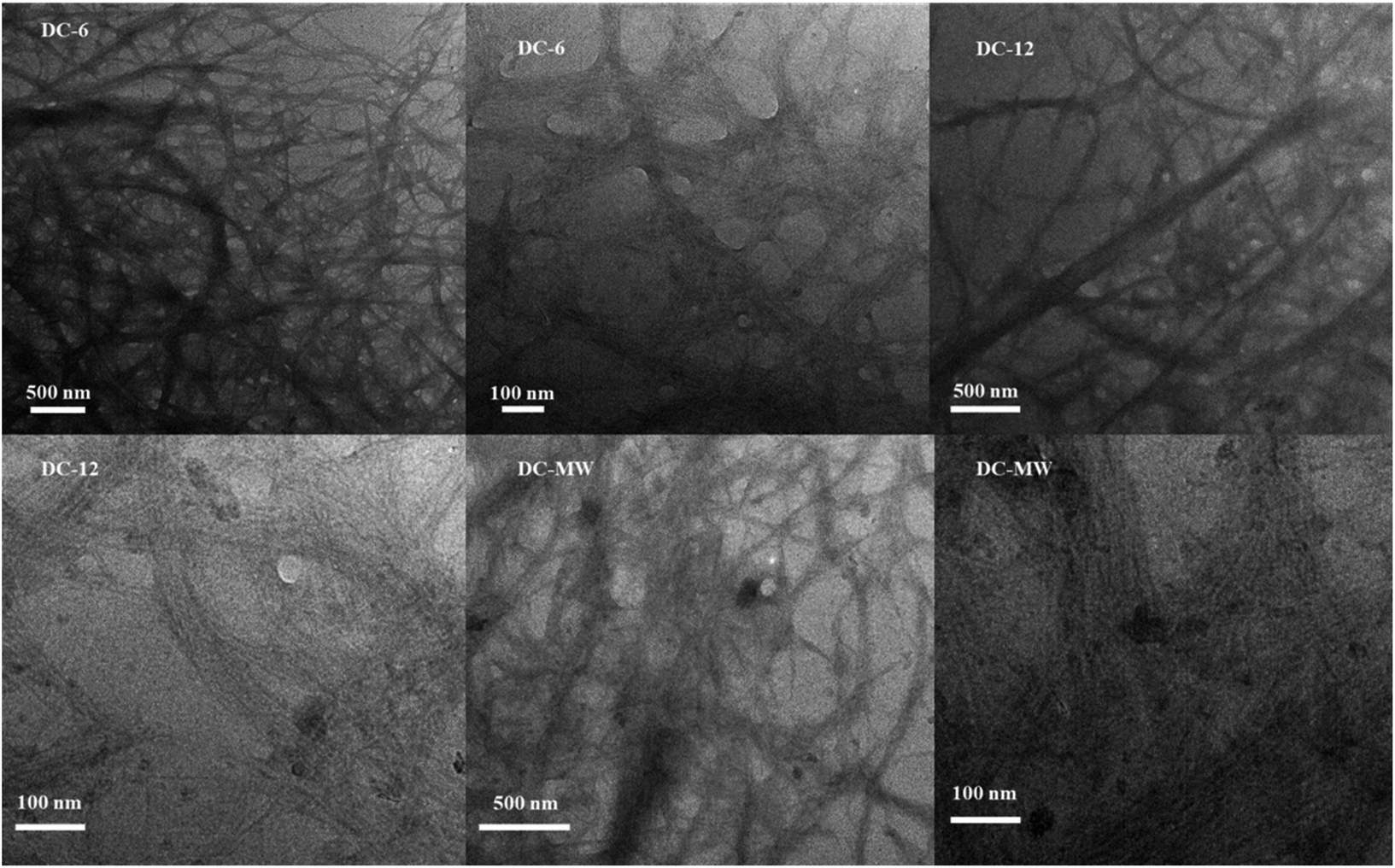 | ||
| Fig. 1 TEM images of cellulose nanofiber suspensions under different experimental conditions. | ||
According to previous studies, DES and cellulose can have an effect on the dispersion of reduced graphene. To demonstrate whether cellulose and DES used in this experiment have the same effect, we prepared reduced graphene suspensions under different conditions, as shown in Fig. 2a. Images from left to right are reduced graphene (rGO) suspension without DES and cellulose, reduced graphene (CrGO) suspension with cellulose suspension, reduced graphene (DrGO) suspension with 200 μL DES, and CG-2 (cellulose graphene) suspension. Suspensions in the graphs are all in a cooled and rested state for 24 h after the reduction was completed. It can be seen that agglomeration and settling of the reduced graphene were prevented due to the addition of DES and cellulose. This effect may arise from the dispersion of graphene by the hydrogen bonding interaction of cellulose and DES, while the principle of DES may also include a first-order increase in viscosity and protonation upon its incorporation. The flexibility of this composite film CGF-2 (cellulose graphene film) is visualized by simple bending and folding, as shown in Fig. 2b and c. This composite film can be easily bent and folded in pairs, which also illustrates its potential for use in flexible electrodes.
 | ||
| Fig. 2 (a) Reduced graphene suspension under different experimental conditions (b) CGF-2 bending photo (c) folding photo. | ||
To further observe the surface microstructure of the cellulose/rGO composite and the distribution of the components in the material, SEM tests were carried out, as shown in Fig. 3. Compared to other film production methods, the composite film obtained by this method has a smooth surface and is easy to bend, and the thickness of the composite film was measured to be 40–80 μm. The material prepared from pure rGO showed surface folds due to the disordered stacking of graphene, but with the addition of cellulose suspension, the surface graphene folds were significantly reduced, and the surface and sub-surface dendritic distribution of cellulose, which also surface cellulose and DES had a dispersing effect on the graphene flakes. In particular, in the picture of CGF-2, the distribution of multi-scale cellulose can be clearly observed and entangled with each other, and such a structure can lead to greater rigidity and flexibility. In CGF-3, due to the increase in DES, some of the crystalline particles of DES can also be observed after drying and losing water. Together, these samples demonstrate the dispersing effect of the addition of cellulose and DES on graphene. Fig. 3e and f show the SEM cross-section images of CGF-2 and CGF-3, respectively. Clearly, compact films were obtained with homogenous structures, indicating the good dispersion of the contents. In addition, the CGF-2 film was also analyzed by SEM at various enlarged scales, and clearly, as shown in Fig. S2,† the graphene nanosheets were distributed homogeneously in the composited film. Fig. S3† shows the SEM cross-section image of the CGF film at various resolutions, and clearly, the film is a multilayer structure, while the inner layer has similar structural properties, indicating that the surface and the bulk composition are homogeneous.
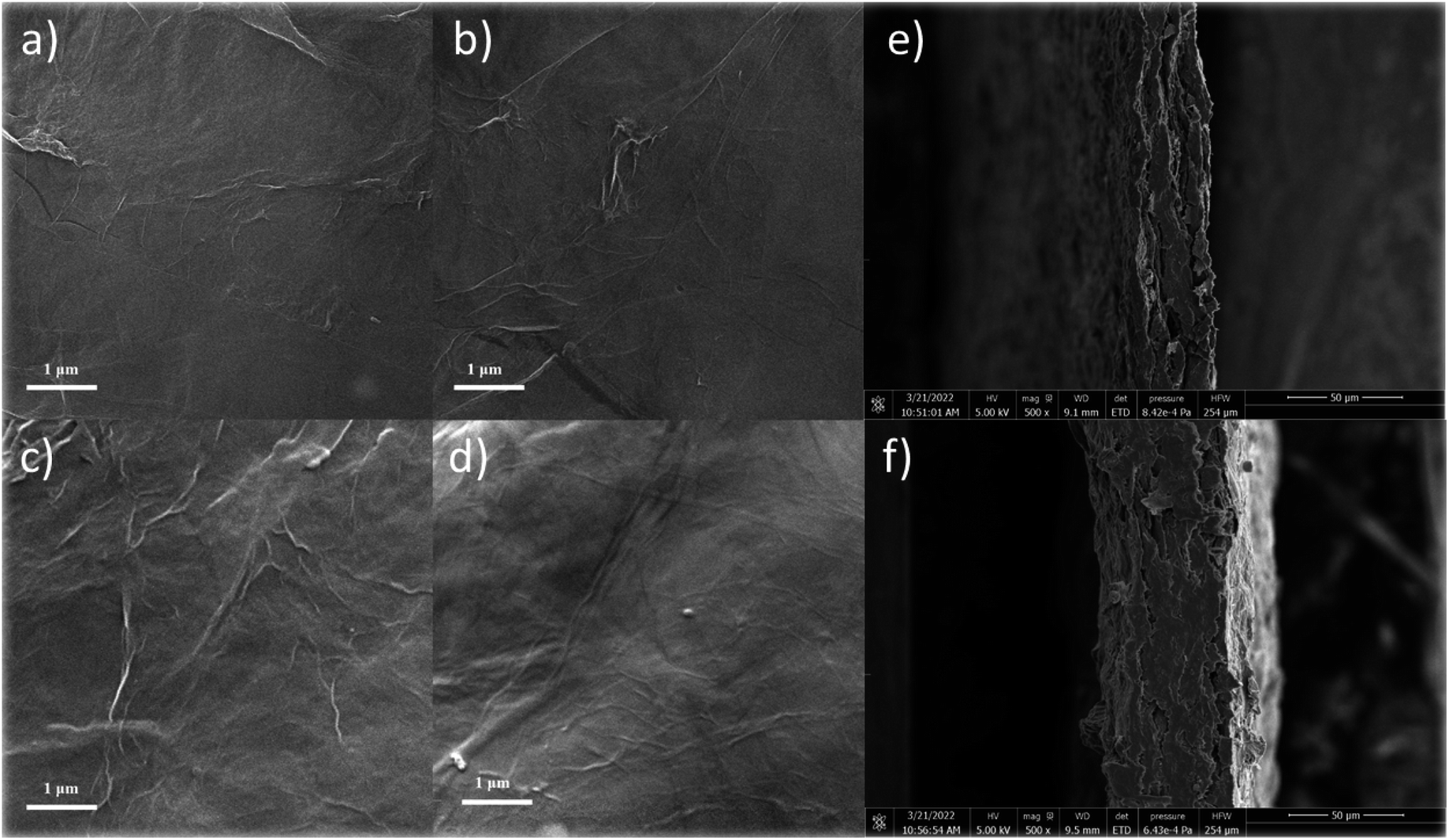 | ||
| Fig. 3 SEM images of (a) rGO, (b) CGF-1, (c) CGF-2, (d) CGF-3 films, and SEM cross-section images of CGF-2 (e) and CGF-3 (f). | ||
Fourier transform infrared spectroscopy (FTIR) has been used to characterize the reduction of graphene in the composite film and the formation and role of DES present in the system, as shown in Fig. 4. Curve a in the figure shows the FTIR spectrum of bleached cellulose, where the absorption peaks at 3420 cm−1 and 2924 cm−1 correspond to the cellulose –OH vibration and the –C–H stretching vibration in the glucose unit, respectively. The absorption peak around 2900 cm−1 was present in all samples and is the most common characteristic peak for organic compounds. The absorption peaks at 1650 cm−1, 1427 cm−1, and 1042 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, –OH bending vibrations, and C–O stretching vibrations, respectively, which are also characteristic of cellulose, demonstrating the good purity and undamaged structure of the bleached cellulose.33 To further distinguish the absorption peaks from DES in the composite film, we performed an infrared test on DES as well (curve b in Fig. 4). Due to the presence of large amounts of –NH2 and –OH in DES, a large and broad absorption peak appears near 3400 cm−1. The absorption peaks from 1645 cm−1, 1480 cm−1, 1400 cm−1, and 1086 cm−1 correspond to the bending vibration of –NH2, in-plane bending vibration of CH2, symmetric stretching vibration of the carboxylate, and the C–N stretching vibration, respectively, which correspond to the characteristic peaks of L-ornithine hydrochloride. In contrast, the presence of an absorption peak at 950 cm−1 attributed to C–C vibrations is evidence for the presence of Ch+. Similarly, the IR spectra of GO, rGO, and CGF-2 (curves c, d, and e in Fig. 4) show absorption peaks corresponding to –OH in the region above 3000 cm−1. While the peaks at 1737 cm−1, 1623 cm−1, 1404 cm−1, 1257 cm−1, and 1042 cm−1 correspond to CO stretching vibrations, –OH bending vibrations, and C–O stretching vibrations.18 Compared to GO, the peaks of rGO and CGF-2 corresponding to oxygen-containing functional groups weaken or even disappear, which also proves that L(+)-ascorbic acid can partially reduce graphene either in pure water or by mixing DES with cellulose. In particular, CGF-2 has a number of overlapping characteristic peaks due to the addition of DES and cellulose, bringing about a change in peak shape, which also demonstrates the homogeneous mixing of the composites. In addition, the appearance of characteristic peaks for –NO2 at 1545 cm−1 and 1380 cm−1 in CGF-2, the weakening of the peak intensity of the choline cation at 950 cm−1 and the weakening of the characteristic peak for L-ornithine hydrochloride are evidence of partial DES incorporation into graphene in the form of nitrogen doping.
O stretching vibrations, –OH bending vibrations, and C–O stretching vibrations, respectively, which are also characteristic of cellulose, demonstrating the good purity and undamaged structure of the bleached cellulose.33 To further distinguish the absorption peaks from DES in the composite film, we performed an infrared test on DES as well (curve b in Fig. 4). Due to the presence of large amounts of –NH2 and –OH in DES, a large and broad absorption peak appears near 3400 cm−1. The absorption peaks from 1645 cm−1, 1480 cm−1, 1400 cm−1, and 1086 cm−1 correspond to the bending vibration of –NH2, in-plane bending vibration of CH2, symmetric stretching vibration of the carboxylate, and the C–N stretching vibration, respectively, which correspond to the characteristic peaks of L-ornithine hydrochloride. In contrast, the presence of an absorption peak at 950 cm−1 attributed to C–C vibrations is evidence for the presence of Ch+. Similarly, the IR spectra of GO, rGO, and CGF-2 (curves c, d, and e in Fig. 4) show absorption peaks corresponding to –OH in the region above 3000 cm−1. While the peaks at 1737 cm−1, 1623 cm−1, 1404 cm−1, 1257 cm−1, and 1042 cm−1 correspond to CO stretching vibrations, –OH bending vibrations, and C–O stretching vibrations.18 Compared to GO, the peaks of rGO and CGF-2 corresponding to oxygen-containing functional groups weaken or even disappear, which also proves that L(+)-ascorbic acid can partially reduce graphene either in pure water or by mixing DES with cellulose. In particular, CGF-2 has a number of overlapping characteristic peaks due to the addition of DES and cellulose, bringing about a change in peak shape, which also demonstrates the homogeneous mixing of the composites. In addition, the appearance of characteristic peaks for –NO2 at 1545 cm−1 and 1380 cm−1 in CGF-2, the weakening of the peak intensity of the choline cation at 950 cm−1 and the weakening of the characteristic peak for L-ornithine hydrochloride are evidence of partial DES incorporation into graphene in the form of nitrogen doping.
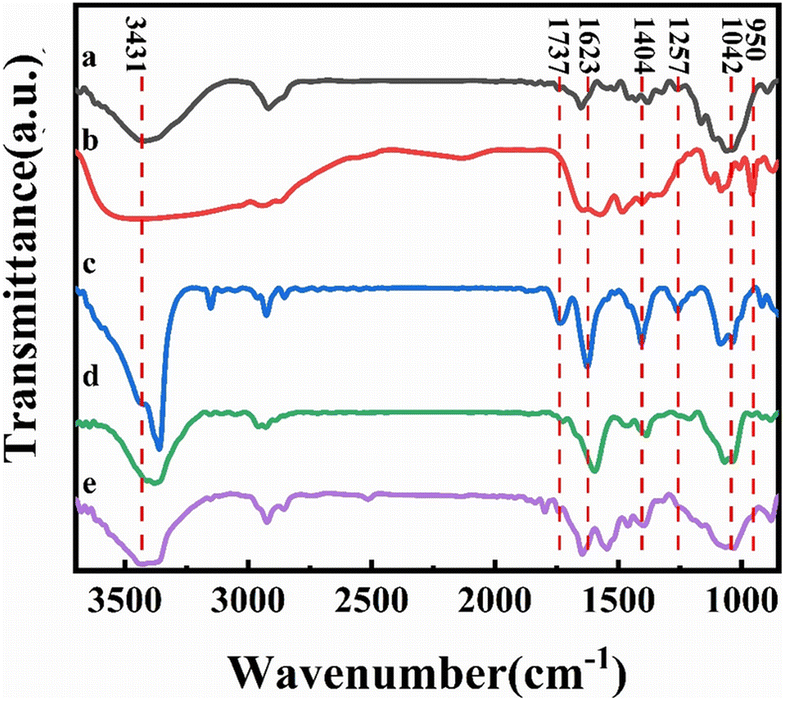 | ||
| Fig. 4 FTIR spectra (a) cellulose; (b) DES; (c) GO; (d) rGO; (e) CGF-2. | ||
Fig. 5a shows the XRD of the bleached cellulose, GO, and composite film samples, mainly to observe the reduction of graphene in the samples. The two diffraction peaks of bleached cellulose, located at 15.5° and 21.9°, respectively, correspond to the (110) and (200) faces of the cellulose I crystal. Both peaks can also be observed in the composite film, which indicates that after the DES pretreatment and sonication, the amount of regenerated cellulose is minimal, and no shift in crystalline type has occurred. Such a structure is also consistent with the mechanism of DES pretreatment being dominated by solubilization. The GO diffraction peak at 10.5° in the (001) plane is much less intense in the composite sample, and its peak decreases with the addition of DES and cellulose, indicating that the presence of DES and cellulose in the system not only does not affect the reduction of L(+)-ascorbic acid but also facilitates the reduction to some extent. The new appearance of a broad diffraction peak at 23°, the (002) plane, in the composite film samples is also evidence of a high level of reduction taking place. Comparing the composite film samples with each other, it can be observed that the intensity of the diffraction peak at 10.5° decreases with the addition of cellulose and DES, which indicates that the presence of DES and cellulose has a facilitating effect on the reduction process, which may also be related to the better dispersion giving greater accessibility to the L(+)-ascorbic acid.
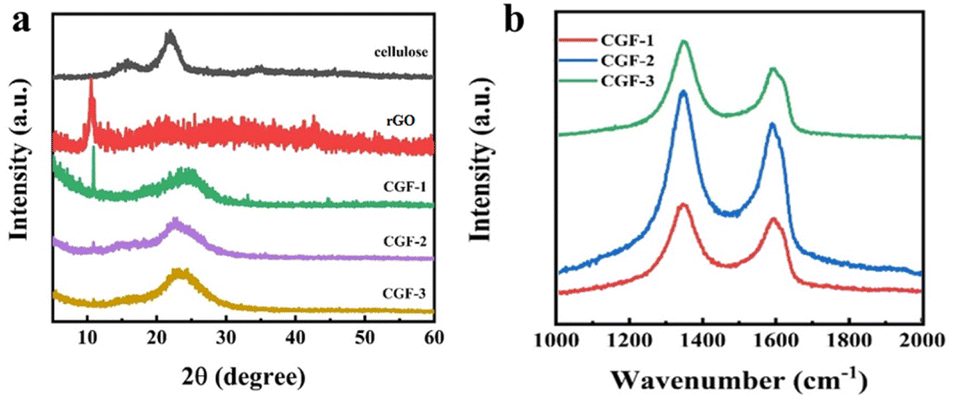 | ||
| Fig. 5 (a) XRD patterns of cellulose, rGO, and the composite films of CGF-1, CGF-2, and CGF-3; (b) Raman patterns of composite films. | ||
In addition, Raman spectroscopy was used to characterize the crystallinity of the sample, and the degree of graphitization corresponds to the electrical conductivity, which is also a fundamental property of the electrode material, as shown in Fig. 5b. The two peaks in the composite film sample are located at 1348 cm−1 and 1590 cm−1, corresponding to the D and G bands of the carbon material.34 The disordered structure (A1g vibrational mode) and defects in the carbon material started the production of the D-band, while the G-band came from the stretching motion of all sp2 atom pairs in the carbon ring or long chain (E2g vibrational mode). The presence of high-intensity G-bands in all samples indicates that the graphitization is incomplete and defects are present. The defects arise due to the entry of heteroatoms during the hydrothermal reduction process. To determine the degree of disorder during the charring process, we calculated the intensity ratios in the D and G bands. Ratios of ID/IG were 1.14 (CGF-1), 1.16 (CGF-2), and 1.10 (CGF-3), with little variation, compared to values generally around 1 for graphene oxide, demonstrating that the disorder of graphene increases considerably as the reduction proceeds. The difference in disorder between the composite film samples illustrates the effect of DES on the reduction process and suggests that the presence of a rich defect structure in the composite implies the successful entry of heteroatoms with a more rapid Faraday reaction, favoring the capacitive performance of the electrode.
The surface chemistry and state of the composite film samples were characterized by XPS (Fig. 6). The obvious peaks (Fig. 6a) are C (283.58 eV), N (399.65 eV), and O (532.65 eV), and the two peaks located at 101.65 eV and 152.65 eV correspond to Si. The elemental composition of the different samples proves that the presence of DES has a strong influence on the doping of N in graphene, as shown in Table 1. The elemental composition of the composite film samples, especially the N content, is greatly enhanced compared to rGO and is most likely related to the denaturation and redox process of DES during the hydrothermal reduction process. It is worth mentioning that after the aforementioned characterization, CGF-2 exhibited excellent reduction levels and graphitization with an elemental composition of C (78.2 at%), N (18.4 at%), and O (3.3 at%), so we selected CGF-2 for subsequent characterization. The N 1s spectrum (Fig. 6b) is divided into three peaks corresponding to pyrrole-N at 400.2 eV, graphite-N at 402.3 eV, and oxide-N at 408.1 eV, showing N doping at different positions in the reduced graphene backbone.35 The O 1s spectrum (Fig. 6c) is decomposed into three peaks located at 531.9, 532.8, and 533.6 eV, which can be attributed to CO, C–O, and O–CO, showing that the activation of graphene by L(+)-ascorbic acid reduction and doping with DES has changed the form of its O functional group present. CGF-2 is rich in pyrrole-N, a nitrogen that reacts with H+ in a redox reaction to provide pseudo-capacitance, which will give a boost to the specific capacity of the electrode.36 The N-doping mechanism may be the contribution of high-intensity compact ultrasound.37 The entry of graphite-N into the carbon skeleton will significantly enhance the electrical conductivity, which is also an extremely important factor for the electrode material. In addition, as a function of oxygen, phenolic oxygen may also undergo redox reactions to increase capacitance, but this results in a decrease in conductivity.35,38 However, CGF-2 has a moderate oxygen content and theoretically good electrochemical properties due to the abundant presence of N. In addition, the doping of O and N heteroatoms and related functional groups improves the wettability of the electrode, increasing the area that can be contacted by the electrolyte and promoting redox reactions. Wang et al. reported that the oxygen-containing functional groups could enhance the capacitance performance of TRGs by introducing abundant pseudocapacitance active sites through reversible Faradaic redox reactions.39 Chen et al. also found that the presence of oxygen functional groups is detrimental to the capacitive performance of porous carbon electrodes in organic electrolytes.40 Rohani et al. prepared a multilayer perceptron neural network (MLP-NN) with various N/O for co-doped AC electrode materials and found that the N/O ratio is the key parameter for performance.41
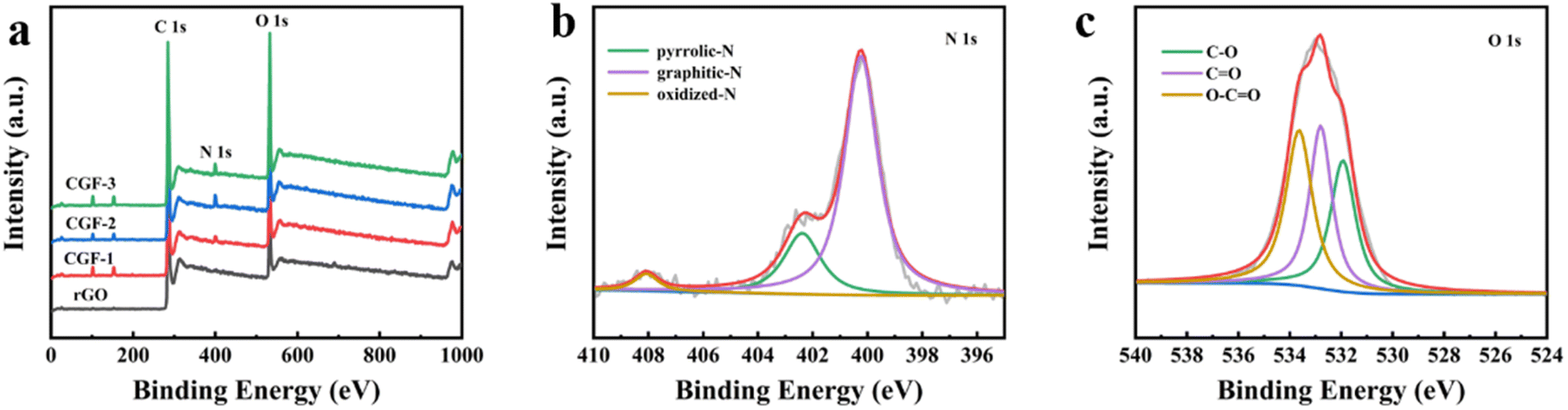 | ||
| Fig. 6 (a) XPS spectra of rGO and composite film samples; (b) N 1s spectra of CGF-2; (c) O 1s spectra of CGF-2. | ||
| Samples | C (at%) | N (at%) | O (at%) |
|---|---|---|---|
| rGO | 87.5 | 12.1 | 0.4 |
| CGF-1 | 80.6 | 17.4 | 2.0 |
| CGF-2 | 78.2 | 18.4 | 3.3 |
| CGF-3 | 78.1 | 19.6 | 2.3 |
Overall, CGF-2 has an excellent composite structure, good flexibility, a high degree of graphitization, and excellent heteroatom doping that give the material excellent potential as a high-performance flexible electrode material for supercapacitors and other energy conversion and storage devices. The present method provides a milder and environmentally friendly heteroatom doping scheme compared to pyrolytic doping, which is beneficial for the commercial application and sustainable development of cellulose/graphene flexible composite films.
In order to characterize the electrochemical properties of the different samples and to select the best samples for complete testing, cyclic voltammetry (CV) curves were therefore obtained with electrodes prepared from different samples at a scan rate of 5 mV s−1, as shown in Fig. 7a. In 1 M H2SO4, the CGF electrode produced a fast current response at the positive and negative electrodes at the initial stage of the scan and was fast and smooth, showing a quasi-rectangular CV curve, which can be inferred as a characteristic of the bilayer capacitance. The CGF-2 has a larger and better-shaped curve than the other two samples (with essentially equal charge and discharge times), which suggests a faster ion transfer rate and a larger specific capacity.42 GCD tests were carried out on the CGFs electrodes at a current density of 1 mA cm−2 (Fig. 7b) and the curves all showed a near isosceles triangular shape, consistent with the performance of a near ideal bilayer capacitor, exhibiting good ion transport within the composite. CGF-2 shows the largest specific capacitance and better bilayer capacitance performance in both CV and GCD curves, which is also the result of its doping effect and the coordinated effect of reduced graphene, making it the best choice of electrode material. To further characterize the electrochemical behavior of CGF-2 electrodes, we have performed CV and GCD tests on CGF-2 at different scanning speeds or current densities, as shown in Fig. 7c and d, both of which exhibit excellent capacitive properties. As the scanning speed increases, it shows a significantly higher current, the value of which is approximately proportional to the square root of the scanning rate, which also proves that its charge storage is perhaps a diffusion-controlled process, so the efficiency of the ion diffusion channel is particularly important, while the electrodes made by hot pressing have thinner and shorter channels. The graph gradually changes from rectangular to shuttle-shaped with an increasing scan rate, which is related to the polarization of the pseudocapacitive material. It is worth noting that the CGF-2 electrode shows excellent multiplicative performance, with the GCD curve maintaining a quasi-triangular shape even at a current density of 10 mA cm−2, suggesting that the low thickness and layered structure of the material itself reduce transport resistance during rapid charging and discharging and that the good doping results in a rapid redox response.
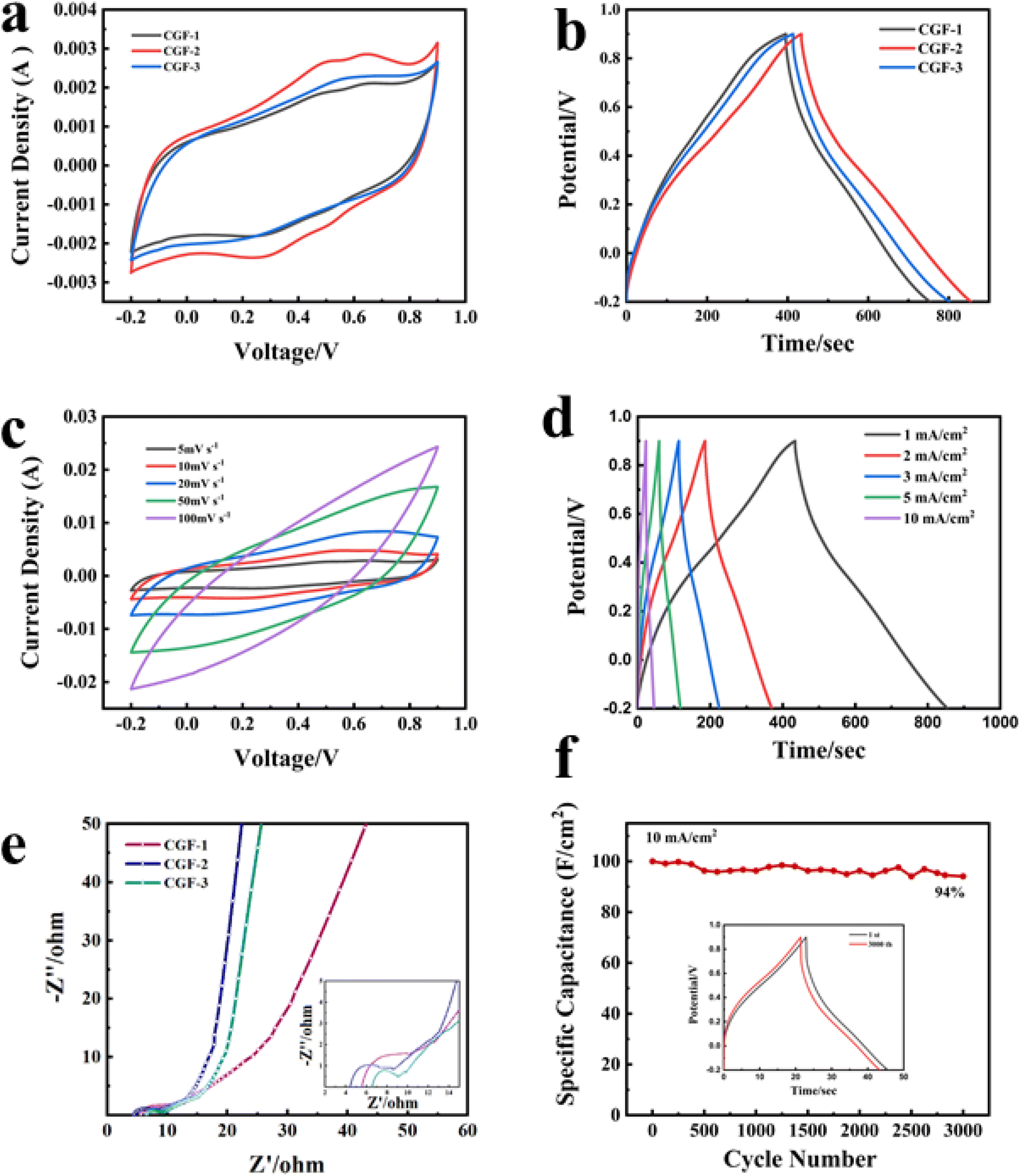 | ||
| Fig. 7 Three-electrode test (a) CV comparison graph of composite film sample at 5 mV s−1 sweep rate; (b) GCD comparison graph of composite film sample at the current density of 1 mA cm2; (c) CV graph of CGF-2 at different sweep rates; (d) GCD graph of CGF-2 at different current densities; (e) Nyquist plots of composite film sample (insert: Nyquist plots in the high-frequency region); (f) cycling stability of CGF-2 at the current density of 10 mA cm−2 and GCD comparison graph before and after scanning. | ||
Fig. 7e shows the AC impedance plots for the CGFs. All samples show typical curves with a semicircle due to the charge transfer resistance (RC) at the electrode/electrolyte interface in the mid to high-frequency region and a sloping straight line due to the diffusion resistance (RW) of the electrolyte ions towards the electrode in the low-frequency region. The intersection of the curves with the horizontal coordinates represents the intrinsic ohmic resistance (Rs), including the resistance between the electrode material, the electrolyte and the active material, and the collector fluid.43 The slope of the low-frequency region displayed fast ion diffusion, which was conducive to fast charge and discharge. As can be judged from the intercept position, CGF-2 has the smallest Rs (4.4 Ω) compared to CGF-1 (5.6 Ω) and CGF-3 (6.5 Ω). In terms of curve shape, CGF-2 exhibits a semicircle with the smallest radius, which means that it has the least resistance to charge transfer at the electrode/electrolyte interface, facilitating the rapid formation of the bilayer, and these indirectly give the reason for its extremely good multiplicative performance. In addition, CGF-2 exhibits a more vertical slope of the low-frequency region, which is due to its superior doping and reduction compared to other samples, making ion diffusion easier and indicating better capacitive performance. These results are consistent with those of CV and GDC. The specific capacitance of the electrodes reached 382 mF cm−2 at a current density of 1 mA cm−2 and remained at 200 mF cm−2 at a high current density of 10 mA cm−2. The high specific capacitance of CGF-2 should be attributed to the synergy between a reasonable degree of reduction and excellent nitrogen doping efficiency, providing a large number of ion-accessible active sites for interfacial charge accumulation and redox reactions. To further characterize the long-period durability of the electrode, we carried out charge/discharge cycling tests. The electrode still had a high capacitance retention of 94% after 3000 repeated cycles at a current density of 10 mA cm−2 (Fig. 7f), and the graphical change in GCD before and after the cycle was minimal, thus demonstrating the good long-term cycling stability of the CGF-2 electrode.
In the as-prepared composite material, the abundant functional groups on cellulose fibers can provide strong interactive sites to bind graphene nanosheets, well-distributed over the cellulose fibers with macroporous texture. It overcomes the low porosity of graphene papers, while the cellulose nanofibers in the as-prepared composite electrode can significantly absorb the electrolyte and act as electrolyte reservoirs to facilitate ion transport, resulting in good rate capability and long cyclic stability with high capacitance.
Experimental
Materials
Rice husk (80 mesh) was collected from Shandong, China. Choline hydroxide and ornithine hydrochloride were purchased from Shanghai Yuanye Bio-Technology Co., Ltd. Sodium hydroxide (NaOH), graphite (C), potassium persulphate (K2S2O8), phosphorus pentoxide (P2O5), concentrated sulphuric acid (H2SO4), potassium permanganate (KMnO4), hydrochloric acid (HCl), and L(+)-ascorbic acid (VC) were provided by Sinopharm Chemical Reagent Co., Ltd.Preparation of bleached cellulose
Mix 2 g of rice husk, 43 mL of deionized water, and 1.5 g of NaOH and add 7 mL of H2O2 dropwise to it. To prevent the liquid from expanding and leaking, use a condenser tube to reflux during the reaction and slowly ramp up to 90 °C. Wait for the bubbles to disappear from the system and filter out the solid. Repeat the experiment until the solid turns white.Preparation of a cellulose suspension containing some cellulose nanofibres
40% aqueous solution of ChOH (24.7 g) and L-ornithine hydrochloride (7.56 g) were mixed with 1 g of bleached cellulose and the mixture was sonicated at 800 W for 5 min. The homogenized mixture was heated at 90 °C for 6 h. Deionized water was added to the appealing mixture to a total of 200 mL, stirred for 10 min, and then placed in ultrasound (800 W) for 1 h to obtain the suspension.Preparation of graphene oxide
Graphene oxide aqueous phase dispersions were prepared by a modified Hummers' method.44 Briefly, graphite powder, 1 g each of potassium persulphate, and phosphorus pentoxide were mixed with 10 mL of concentrated sulphuric acid and reacted at 80 °C for 6 h. After cooling, in an ice bath, water is added and diluted. The rinsing with water was continued during the filtration process until the filtrate was neutral. The dried sample was reacted with 30 mL of sulphuric acid and 4 g of potassium permanganate under an ice bath and held at 38 °C for 2 h, followed by continuous addition of water and warming to 90 °C and taken out for cooling. To the mixture, 5 mL of H2O2 was added and finally, the precipitate was washed twice with HCl and collected by centrifugation. The precipitate was dried to give graphene oxide (GO).Preparation of cellulose/nitrogen-doped reduced graphene composite film
36 mg of graphene oxide was mixed with the sonicated cellulose suspension, 360 mg of vitamin C was added, stirred well, and heated in a water bath at 80 °C for 1 h. The resulting mixture was filtered using 0.22 μm of mixed cellulose to obtain a gel-like solid. The solid was held at 80 °C for 2 h between two Teflon sheets pressed using an iron block as a weight. The resulting composite membrane is a circle with a diameter of 4 cm. The resulting sample was named CGF (cellulose graphene film), while CGF-1, CGF-2, and CGF-3 are the samples according to the amount of cellulose suspension added (1 mL, 2 mL, and 3 mL), while graphene reduced by vitamin C in pure water without the addition of cellulose suspension was referred to as rGO.Material characterization
Fourier transform infrared (FTIR) spectra were used to characterize the degree of crosslinking of the gel in the range of 400–4000 cm−1. The surface morphology of the aerogel and hierarchical porous carbon was monitored by scanning electron microscopy (SEM) and transmission electron microscopy (TEM, JEM-2100F). The degree of graphitization and crystallinity of graded porous carbon could be monitored using Raman spectrometry (Renishaw, inVia, under excitation at 532 nm), and X-ray diffraction (XRD, PANalytical, X'Pert Pro, using Cu Kα radiation). The redox reaction was monitored by X-ray photoelectron spectroscopy (XPS, Kratos Axis supra+, using Al kα monochromatic X-ray source).Electrochemical measurements
To facilitate electrochemical testing and subsequent calculations, cellulose/nitrogen-doped reduced graphene composite films were cut into 1 cm × 1 cm square and used as working electrodes. The graphene loading of each electrode was approximately 3 mg. To characterize the electrochemical performance of the individual electrodes, a 1 M aqueous sulphuric acid solution was used as the electrolyte, Ag/AgCl as the reference electrode, and a platinum wire as the counter electrode. Three-electrode testing was carried out using a CHI660D electrochemical workstation. Cyclic voltammetry (CV), alternating current impedance (EIS), and constant current charge/discharge (GCD) were used to calculate the specific capacitance of the electrodes and to characterize their multiplicative properties.Conclusions
In summary, we used DES as a swelling agent to reduce the energy consumption for the preparation of cellulose nanofibres and further used it as a dispersant and doping precursor for reduced graphene to prepare cellulose/nitrogen-doped reduced graphene composite films by simple hydrothermal reduction, demonstrating the feasibility of preparing nitrogen-doped graphene composites at low temperature and low consumption. The resulting reduced graphene has an enhanced nitrogen content of 3 at% compared to reduced graphene in a pure water environment. The experimental results demonstrate that the electrodes exhibit excellent bendability due to the addition of DES and cellulose. The electrochemical results show that the electrochemical performance is significantly affected by DES. The cellulose/nitrogen-doped reduced graphene composite films exhibit significantly enhanced area-specific capacitance (382 mF cm−2 at a current density of 11 mA cm−2 in a three-electrode system) and high cycling stability (6% loss of capacitance for 3000 cycles at 10 mA cm−2), demonstrating the potential of graphene composite film materials for applications such as supercapacitors and sensors. This is a potential method for the preparation of flexible electrodes directly from lignocellulose and graphene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key R&D Program of China (No. 2020YFA0710700), the National Natural Science Foundation of China (No. 51873201), and the Fundamental Research Funds for the Central Universities (No. YD2060002015).Notes and references
- S. Shleev, E. González-Arribas and M. Falk, Curr. Opin. Electrochem., 2017, 5, 226–233 CrossRef CAS.
- J. Libich, J. Máca, J. Vondrák, O. Čech and M. Sedlaříková, J. Energy Storage, 2018, 17, 224–227 CrossRef.
- R. Reece, C. Lekakou and P. A. Smith, ACS Appl. Mater. Interfaces, 2020, 12, 25683–25692 CrossRef CAS PubMed.
- Q. Zhu, D. Zhao, M. Cheng, J. Zhou, K. A. Owusu, L. Mai and Y. Yu, Adv. Energy Mater., 2019, 9, 1901081 CrossRef.
- F. Xu, Z. Tang, S. Huang, L. Chen, Y. Liang, W. Mai, H. Zhong, R. Fu and D. Wu, Nat. Commun., 2015, 6, 7221 CrossRef PubMed.
- H. Feng, H. Hu, H. Dong, Y. Xiao, Y. Cai, B. Lei, Y. Liu and M. Zheng, J. Power Sources, 2016, 302, 164–173 CrossRef CAS.
- Y. Liu, Y. Shen, L. Sun, J. Li, C. Liu, W. Ren, F. Li, L. Gao, J. Chen, F. Liu, Y. Sun, N. Tang, H. M. Cheng and Y. Du, Nat. Commun., 2016, 7, 10921 CrossRef CAS PubMed.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- Q. Dou and H. S. Park, Energy Environ. Mater., 2020, 3, 286–305 CrossRef CAS.
- P. Ghimire, C. Gunathilake, N. P. Wickramaratne and M. Jaroniec, Carbon, 2017, 121, 408–417 CrossRef CAS.
- Q. Xu, X. Wang, J. Cheng, L. Zhang, F. He and H. Xie, RSC Adv., 2020, 10, 36504–36513 RSC.
- Q. Li, X. Guo, Y. Zhang, W. Zhang, C. Ge, L. Zhao, X. Wang, H. Zhang, J. Chen, Z. Wang and L. Sun, J. Mater. Sci. Technol., 2017, 33, 793–799 CrossRef CAS.
- W. Zhang, C. Xu, C. Ma, G. Li, Y. Wang, K. Zhang, F. Li, C. Liu, H. M. Cheng, Y. Du, N. Tang and W. Ren, Adv. Mater., 2017, 29, 2104952 Search PubMed.
- J. Zeng, C. Xu, T. Gao, X. Jiang and X. B. Wang, Carbon Energy, 2021, 193–224 CrossRef CAS.
- T.-R. Cui, D. Li, X.-R. Huang, A.-Z. Yan, Y. Dong, J.-D. Xu, Y.-Z. Guo, Y. Wang, Z.-K. Chen, W.-C. Shao, Z.-Y. Tang, H. Tian, Y. Yang and T.-L. Ren, Appl. Sci., 2022, 12, 4688 CrossRef.
- M. Horn, B. Gupta, J. MacLeod, J. Liu and N. Motta, Curr. Opin. Green Sustainable Chem., 2019, 17, 42–48 CrossRef.
- Z. Li, S. Gadipelli, H. Li, C. A. Howard, D. J. L. Brett, P. R. Shearing, Z. Guo, I. P. Parkin and F. Li, Nat. Energy, 2020, 5, 160–168 CrossRef CAS.
- W. Shang, J. Li, A. Rabiei Baboukani, Y. Wen, D. Kong, N. Peng and J. Jiang, Appl. Surf. Sci., 2020, 511, 145518 CrossRef CAS.
- J. Luo, L. Yang, D. Sun, Z. Gao, K. Jiao and J. Zhang, Small, 2020, 16, e2003426 CrossRef PubMed.
- L. Li, M. Zhou, L. Jin, Y. Mo, E. Xu, H. Chen, L. Liu, M. Wang, X. Chen and H. Zhu, Materials, 2020, 13, 4069 CrossRef CAS PubMed.
- D. K. Kulal, R. V. Khose, D. A. Pethsangave, P. H. Wadekar and S. Some, ChemistrySelect, 2019, 4, 4568–4574 CrossRef CAS.
- H. Xing, F. Zhang, Y. Lu, B. Zhai, S. Zhai, Q. An and C. Yu, RSC Adv., 2016, 6, 79366–79371 RSC.
- O. P. Troncoso and F. G. Torres, Int. J. Mol. Sci., 2020, 21, 6532 CrossRef CAS PubMed.
- Y. Chen, P. Pötschke, J. Pionteck, B. Voit and H. Qi, J. Mater. Chem. A, 2018, 6, 7777–7785 RSC.
- A. Kafy, A. Akther, L. Zhai, H. C. Kim and J. Kim, Synth. Met., 2017, 223, 94–100 CrossRef CAS.
- S. Tan, J. Li, L. Zhou, P. Chen, D. Xu and Z. Xu, J. Mater. Sci., 2018, 53, 11648–11658 CrossRef CAS.
- V. Kuzmenko, N. Wang, M. Haque, O. Naboka, M. Flygare, K. Svensson, P. Gatenholm, J. Liu and P. Enoksson, RSC Adv., 2017, 7, 45968–45977 RSC.
- X. Wang, K. Wan, P. Xie, Y. Miao and Z. Liu, Molecules, 2021, 26, 4891 CrossRef CAS PubMed.
- Q. Liu, W. Sun, T. Yuan, S. B. Liang, F. Peng and C. L. Yao, Carbohydr. Polym., 2021, 272, 118514 CrossRef CAS PubMed.
- M. Shakourian-Fard, S. M. Taimoory, H. R. Ghenaatian, G. Kamath and J. F. Trant, J. Mol. Liq., 2021, 327, 114850 CrossRef CAS.
- Y. Liu, X. Li, X. Zhou and Y. Cui, New J. Chem., 2021, 45, 15637–15646 RSC.
- W. Yu, C. Wang, Y. Yi, H. Wang, Y. Yang, L. Zeng and Z. Tan, Cellulose, 2020, 28, 175–188 CrossRef.
- J. Wang, Y. Wang, Z. Ma and L. Yan, Green Energy Environ., 2020, 5, 232–239 CrossRef.
- J. Chang, Z. Gao, X. Wang, D. Wu, F. Xu, X. Wang, Y. Guo and K. Jiang, Electrochim. Acta, 2015, 157, 290–298 CrossRef CAS.
- S. Ghosh, S. Barg, S. M. Jeong and K. Ostrikov, Adv. Energy Mater., 2020, 10, 2001239 CrossRef CAS.
- L. Chen, J. Deng, S. Hong and H. Lian, J. Sol-Gel Sci. Technol., 2018, 86, 795–806 CrossRef CAS.
- G. Chen, S. Tang, Y. Song, X. Meng, J. Yin, Y. Xia and Z. Liu, Chem. Eng. J., 2019, 361, 387–397 CrossRef CAS.
- L. Hu, J. Hou, Y. Ma, H. Li and T. Zhai, J. Mater. Chem. A, 2016, 4, 15006–15014 RSC.
- C.-M. Chen, Q. Zhang, M.-G. Yang, C.-H. Huang, Y.-G. Yang and M.-Z. Wang, Carbon, 2012, 50, 3572–3584 CrossRef CAS.
- S. Yuan, X. Huang, H. Wang, L. Xie, J. Cheng, Q. Kong, G. Sun and C.-M. Chen, J. Energy Chem., 2020, 51, 396–404 CrossRef.
- M. Rahimi, M. H. Abbaspour-Fard and A. Rohani, J. Power Sources, 2022, 521, 230968 CrossRef CAS.
- C. Ma, Z. Li, J. Li, Q. Fan, L. Wu, J. Shi and Y. Song, Appl. Surf. Sci., 2018, 456, 568–576 CrossRef CAS.
- H. Peng, J. Zhou, K. Sun, G. Ma, Z. Zhang, E. Feng and Z. Lei, ACS Sustain. Chem. Eng., 2017, 5, 5951–5963 CrossRef CAS.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2su00107a |
| This journal is © The Royal Society of Chemistry 2023 |