
Functional mimicry of sea urchin biomineralization proteins with CaCO3-binding peptides selected by phage display†
Elke
Völkle (nee Evgrafov)‡
a,
Fabian
Schulz
a,
Julia Maxi
Kanold
b,
Monika
Michaelis
 c,
Kerstin
Wissel
d,
Franz
Brümmer
b,
Anna S.
Schenk
e,
Sabine
Ludwigs
f,
Joachim
Bill
a and
Dirk
Rothenstein
*a
c,
Kerstin
Wissel
d,
Franz
Brümmer
b,
Anna S.
Schenk
e,
Sabine
Ludwigs
f,
Joachim
Bill
a and
Dirk
Rothenstein
*a
aDept. Bioinspired Materials, Institute for Materials Science, University of Stuttgart, Heisenbergstraße 3, 70569 Stuttgart, Germany. E-mail: dirk.rothenstein@imw.uni-stuttgart.de
bInstitute for Biomaterials and Biomolecular Systems & Scientific Diving Group (WiTUS), University of Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany
cBiomolecular and Materials Interface Research Group, Interdisciplinary Biomedical Research Centre, School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK
dDept. Chemical Materials Synthesis, Institute for Materials Science, University of Stuttgart, Heisenbergstraβe 3, 70569 Stuttgart, Germany
ePhysical Chemistry IV, Department of Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
fIPOC – Functional Polymers, Institute of Polymer Chemistry (IPOC), University of Stuttgart, Stuttgart 70569, Germany
First published on 10th October 2023
Abstract
The intricate process of biomineralization, e.g. in sea urchins, involves the precise interplay of highly regulated mineralization proteins and the spatiotemporal coordination achieved through compartmentalization. However, the investigation of biomineralization effector molecules, e.g. proteins, is challenging, due to their very low abundance. Therefore, we investigate the functional mimicry in the bioinspired precipitation of calcium carbonate (CaCO3) with artificial peptides selected from a peptide library by phage display based on peptide-binding to calcite and aragonite, respectively. The structure-directing effects of the identified peptides were compared to those of natural protein mixes isolated from skeletal (test) structures of two sea urchin species (Arbacia lixula and Paracentrotus lividus). The calcium carbonate samples deposited in the absence or presence of peptides were analyzed with a set of complementary techniques with regard to morphology, polymorph, and nanostructural motifs. Remarkably, some of the CaCO3-binding peptides induced morphological features in calcite that appeared similar to those obtained in the presence of the natural protein mixes. Many of the peptides identified as most effective in exerting a structure-directing effect on calcium carbonate crystallization were rich in basic amino acid residues. Hence, our in vitro mineralization study further highlights the important, but often neglected, role of positively charged soluble organic matrices associated with biological and bioinspired CaCO3 deposition.
Introduction
Proteins are fundamental components of life that perform a wide range of functions. One of their key roles is to regulate metabolic and cellular processes as enzymes, hormones, and antibodies through protein–protein interactions. In addition to these organic–organic interactions, proteins are also involved in the formation of biogenic minerals, referred to as biominerals, which characteristically represent organic–inorganic hybrid materials with complex hierarchical structures and remarkable properties.1 In biomineralization processes, the binding partners of the proteins are typically inorganic components including solvated ions, amorphous mineral species or the facets of crystals.2 Biominerals are found in a wide variety of organisms, ranging from unicellular microorganisms (e.g. coccolithophores3) to complex multicellular organisms, including humans.4 They play an important role in the formation of hard tissues such as vertebrate bones,5 teeth,6 and mollusk shells,7 thus providing structural support and protection to the organism. The formation of biominerals is a complex process that involves the molecular recognition of an inorganic target, such as a substrate surface, ion, or particle, by a protein. This molecular recognition is in many cases essential for the deposition, stabilization and assembly of inorganic mineral species and the transformation of precursor phases. As a tight interaction between water-soluble proteins and the (forming) mineral is often a prerequisite for a structure- and process-directing effect, it is not unusual for the organic matrix to become occluded into the final crystalline product,8,9 thus leading to the formation of organic–inorganic hybrid materials, whose mechanical properties are strongly affected by the incorporation of the soft organic component.10,11 By studying biomineralization processes at the molecular level and translating fundamental concepts of biological crystallization control (e.g. mineral deposition directed by soluble and insoluble organic matrices, compartmentalization, isotropic amorphous species) into synthetic model systems,12–15 scientists aim for a better understanding of how the underlying design principles can eventually be harnessed for biomedical applications, such as implants or bone substitutes.Calcium carbonate (CaCO3), in particular, is a ubiquitous inorganic component of many biominerals such as in sea urchin skeletons16 and mollusk shells.17 In a geological context, CaCO3 occurs in three anhydrous crystalline polymorphs-calcite, aragonite and vaterite – along with hydrated and amorphous modifications. The selectivity of the polymorph largely depends on environmental parameters (e.g. pressure, temperature), with calcite being the thermodynamically most stable crystalline phase under ambient conditions. Mineral deposition in the presence of biomineralization proteins, by contrast, can lead to the stabilization of polymorphs that would usually not form under the given environmental conditions or even induce the formation of stable or transient amorphous phases.18,19 The specific protein-inorganic interactions along with the physicochemical boundary conditions for mineral deposition often lead to a hierarchical organization spanning several length scales from the macroscopic level down to the nanoscale. In this way, biomineralization can also result in nanogranular crystalline structures with long-range texture, such that the protein-interconnected crystallites are in near-perfect crystallographic register and the superstructures show a quasi-single crystalline character in its entity. Biologically, such a highly ordered arrangement of the mineral building blocks, termed mesocrystal,20 has been observed, for example, in sea urchin test plates and spines,21 which show single-crystalline behavior in polarized light microscopy and XRD analysis.22
Sea urchins have long been used as model organism to study biomineralization processes. The biological deposition of mineral species is a spatiotemporally regulated process, i.e. sea urchins are capable of precipitating calcium carbonate phases with different structural features (e.g. spine, test, teeth) optimized for specific functions spatially separated from each other and at different stages of development (larvae, mature).23,24 The resulting mineralized structures consists of CaCO3 (>95 wt%), MgCO3 (5 wt%) and biomolecules (0.1 wt%), mainly proteins but also glycoproteins and polysaccharides.25 Although, more than 100 proteins are known to be associated with mineralized sea urchin tissues, their exact function in directing the crystallization process often remains obscure,26 mostly because the low concentration of the organic matrix in mineralized tissues makes it difficult to extract proteins in sufficient quantities for their characterization. However, some examples of proteins involved in biomineralization have indeed been isolated and the corresponding genes were identified by reverse genetics of CaCO3-mineralizing organisms, including sea urchin species.27,28 Moreover, it was shown that sea urchins from different orders, namely the black sea urchin (Arbacia lixula, Arbacioida) and the purple sea urchin (Paracentrotus lividus, Camarodonta), show similarities in their protein composition,29 thus pointing to a potential correlation between the amino acid sequence and the functionality in terms of interfering with calcium carbonate mineralization. Different protein families were shown to be associated with the biomineralization processes.26,29–32 While the majority of proteins that were identified in biogenic calcium carbonate, specifically mollusk shell proteins33–35 and sea urchin proteins,29 are negatively charged (i.e. rich in Asp and Glu residues), basic protein sequences (i.e. positively charged domains) have also been reported.30
The fundamental principles of biomineralization, such as confinement, crystallization pathways via amorphous precursors, and in particular, the control of mineralization by soluble and insoluble organic matrices, have been successfully transferred to synthetic systems. To investigate the effects of artificial soluble structure-directing additives,36in vitro model systems for the mineralization of CaCO3 have been designed. As the literature on biomineralization-associated proteins has long been dominated by reports on acidic protein domains, bioinspired synthetic approaches aiming to mimic structural features of biological crystallization often use negatively charged polyelectrolytes to control mineralization pathways and products.37 However, there are a growing number of publications discussing the effects of cationic (i.e. amine-functionalized) polymer additives and their role is increasingly being recognized.38–40 In bioinspired polymer-mediated mineralization, synthetic polyelectrolytes have been shown to exert multiple effects. Examples include the stabilization of transient amorphous precursors of hydrated colloids even in the presence of minute amounts of organic molecules,41 promotion of the formation and stabilization of an amorphous precursor phase by suppressing crystalline growth,42 altering the pH value of the reaction solution,41 or affecting the composition and water content of the mineral phase, resulting in a manifold of different crystal morphologies.41
While a large variety of soluble additives, have been investigated as structure-directing agents in calcium carbonate precipitation, including ions like Mg2+,43 small molecules such as individual amino acids,44 and polyelectrolytes,45 block copolymers,42 as well as acidic and basic (poly) amino acids,43 a specific molecular interaction between the additive and the inorganic phase is often not directly addressed experimentally. To gain access to a sufficient amount of biomolecules with a specific binding capacity to inorganic materials, so called inorganic-binding peptides, the phage display method has been successfully applied.46,47 Phage display uses a pseudo-randomized peptide library presented on the surface of M13 phages (bacterial viruses). Peptides are selected from this peptide library based on their ability to bind to a target material, where the interaction is based on the specific amino acid sequence of the peptide. Such inorganic-binding peptides have been shown to mineralize engineering materials such as zinc oxide, thereby controlling material properties.13,14 In a biological context, peptides that show a significant effect on the morphology or polymorphism of the synthetic organic/inorganic hybrid materials may be indicative of the sequence of natural mineralization proteins. In this way, a feedback loop between the biogenic minerals and the synthetic model systems is generated, which may help to identify previously unknown proteins or protein domains in biomineralization proteins.
In our study, we present a systematic investigation into the bioinspired precipitation of calcium carbonate using selected peptides obtained from a peptide library through phage display, based on their binding affinity to geological calcite and aragonite. To assess their structure-directing effects, we compared them with natural protein mixes extracted from the calcitic tests of two distinct sea urchin species, Arbacia lixula and Paracentrotus lividus, belonging to different orders. We applied the calcite- and aragonite-binding peptides, along with the biologically derived soluble proteins, as organic additives in the in vitro mineralization of CaCO3 and determined their influence on the (nano-)morphology and polymorphism of the resulting crystals with scanning electron microscopy (SEM), X-ray diffraction (XRD), and small-angle X-ray scattering (SAXS). The most effective peptides that led to calcite structures as seen for the natural biomineralization proteins, were basic peptides. This observation further emphasizes the importance of basic soluble organic molecules in biomineralization.
Materials and methods
Materials
Agar agar, bovine serum albumin (BSA), CaCl2·2H2O, glycine, isopropyl β-D-thiogalactoside (IPTG), (NH4)2CO3, NaCl, NaOCl, polyethylene glycol-8000, Tris–HCl, tryptone, yeast extract, 5-Bromo-4-chloro-3-indolyl-β-D-galactoside (XGal) were purchased from Roth (Germany) or Sigma–Aldrich (Germany) and were used as received. All solutions were prepared using ultrapure deionized water (ddH2O, 18.2 MΩ, Arium 611UF, Sartorius).Phage display
Aragonite- and calcite-binding peptides were isolated from a random 12-mer peptide library (New England Biolabs, Inc.) by the phage display technique. The peptide library is expressed as N-terminal fusion protein with the minor coat protein p3 of M13 phages. The peptides are separated by a short spacer sequence (GGGS) from the p3 coat protein. General phage methods were conducted according to the manufacturer's recommendations.For phage display experiments, geological aragonite (Alpha Aesar, Product no. 42523, LOT N04A029) and calcite (Alpha Aesar, Product no. 44520, LOT N04A030) substrates were applied as received from the manufacturer. Aragonite and calcite substrates of approximately 9 mm2 were applied for the biopanning procedure. The target substrates were washed twice in Tris buffered saline (TBS), pH 7.5 supplemented with 0.1% Tween 20. The random phage library (1.5 × 1011 phages) was incubated with the aragonite and calcite substrates, respectively, under light agitation in TBS, 0.1% Tween 20 for one hour at RT. Unbound phages were removed from the substrate by washing 10 times in TBS, 0.5% Tween 20. The remaining phages were eluted applying 1 mL Glycine Elution Buffer. The eluate was neutralized with 150 μL of 1 M Tris–HCl, pH 9.1. The eluted phages were amplified in E. coli ER2738 and purified by polyethylene glycol-8000/sodium chloride (PEG/NaCl) precipitation. In the following bio panning rounds at least 1.3 × 1010 phages of the restricted library were applied. To enrich the specificity of aragonite and calcite-binding sequences, in total 5 panning rounds were carried out. From each experiment, 50 randomly selected phage clones were analyzed by DNA sequencing.
Extraction of CaCO3-associated proteins from sea urchin skeletons
The extraction of CaCO3-associated protein was performed according to Kanold et al.25 Skeletons from the sea urchin strains Arbacia lixula and Paracentrotus lividus were mechanically cleaned. Adherent organic debris was removed by incubation under agitation in NaOCl (0.26% active chlorine) for 72 h. Afterwards, skeletons were extensively rinsed in ddH2O and dried at 37 °C and were pulverized in a mortar. For decalcification, 30 g of skeleton powder were suspended in 70 mL ddH2O and 400 mL of cold 10% acetic acid were drop-wise added with an electronic burette, flow rate of 100 μL every 5s, at 4 °C to the final pH 4 to 5. Non-solubilized material, acid insoluble matrix (AIM), was removed by filtering (pore size 200 μm) and centrifugation 30 min, 4500 rpm at 4 °C. The AIM pellet was washed in ddH2O and stored at −80 °C. The soluble fraction, acid soluble matrix (ASM), was filtered (pore size 0.45 μm) and concentrated with a centrifugal concentrator with 10 kDa molecular weight cut off size (MWCO) (Vivaspin 20, Sartorius, Göttingen, Germany) for 45 min at 4500 rpm and 4 °C. The concentrated ASM sample was dialyzed 2 kDa MWCO at 4 °C for 40 h and lyophilized at 1 mbar and −70 °C within three days. The extraction of acid-soluble matrix proteins (ASM) from sea urchin skeletons was conducted through four repetitions, yielding consistent results in terms of protein extraction efficiency. The protein extraction process was carried out using 15 to 30 grams of initial material. Specifically, from A. lixula, 1.4, 2.0, and 6.1 milligrams of ASM were successfully extracted, representing a range of 0.01 to 0.02 weight percent (wt%). In the case of P. lividus, 3.5 milligrams of ASM (0.01%) were obtained from 33 grams of starting material. These results align well with previous studies focused on the extraction of sea urchin proteins, both in terms of the total matrix protein content and the yield of ASM.16,25,29ASM and AIM protein fractions were analyzed by 1D SDS/PAGE (12% acrylamide). 10 mg AIM sample was mixed with 100 μL 2x Laemmli buffer and 100 μL ddH2O. The AIM-PAGE samples represented only a fraction of the AIM sample since some proteins were not solubilized in Laemmli buffer. ASM samples were resuspended in 2x Laemmli buffer:ddH2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to a final concentration of 2 mg mL−1. Gels were silver stained afterwards.
:1) to a final concentration of 2 mg mL−1. Gels were silver stained afterwards.
CaCO3-binding peptides
CaCO3-binding peptides for mineralization experiments were purchased from EMC microcollections GmbH (Germany) and used as received from the supplier. The purity of the peptides was assessed using high-performance liquid chromatography (HPLC) and was found to be greater than 80%.Secondary structure prediction of CaCO3-binding peptides
Starting from the amino acid sequence in fasta format three secondary structure prediction servers are used.48–51De novo secondary structure predictions are performed with the PEP-Fold server.52–54 PEP-Fold predicts first structural alphabet (SA) letters from the sequence using a hidden Markov model approach.55 These fragments are assembled by a greedy procedure driven by a modified optimized potential for efficient structure prediction (OPEP) coarse grained force field56 energy score followed by a clustering procedure.In vitro mineralization of CaCO3
CaCO3 was precipitated by slow diffusion of gaseous NH3 and CO2 formed by the decomposition of solid ammonium carbonate into a 10 mM CaCl2 solution according to a protocol described by Albeck et al.57 The aqueous CaCl2 stock solution was freshly prepared in ddH2O and sterile-filtered (pore size 0.2 μm) prior to use. The mineralization was performed in 16 well chamber slides in a total volume of 500 μL per well (Lab-Tek, Thermo Fisher Scientific, USA). To ensure gas exchange, a hot cannula was used to poke one hole per well into the lid of the chamber slide. Organic additives, ASM and CaCO3-binding peptides, were diluted to final concentrations of 500, 100, 30, 20, 10, 5, and 1 μg mL−1. Solutions with 2 mg mL−1 of the structure-directing organic matrix were additionally prepared for the CaCO3-binding peptides. The chamber slides were placed under vacuum in a sealed desiccator, containing a 5 cm Petri dish with solid ammonium carbonate, and the prepared crystallization solutions were incubated in the forming vapor phase at 4 °C for three days. Afterwards, the chamber slides were removed from the desiccator, the supernatant was decanted and the CaCO3 crystals were washed twice with ddH2O and dried at 37 °C. All mineralization samples were prepared in triplicate.Mineralization products were analyzed by scanning electron microscopy (SEM), X-ray diffraction (XRD), and small-angle X-ray scattering (SAXS).
CaCO3 sample characterization
All two-dimensional data sets were reduced to one-dimensional traces of the modulus of the scattering vector q (q = 4πsin(2θ/λ), where 2θ represents the scattering angle) vs. the intensity I(q) and corrected for sample transmission and background intensity (by subtraction of an empty tape pocket). Additionally, the Laue background (ILaue) was subtracted from the data based on a Porod fit (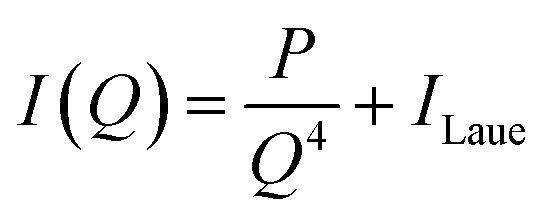 , P being the Porod constant) prior to T-parameter analysis as previously reported (
, P being the Porod constant) prior to T-parameter analysis as previously reported (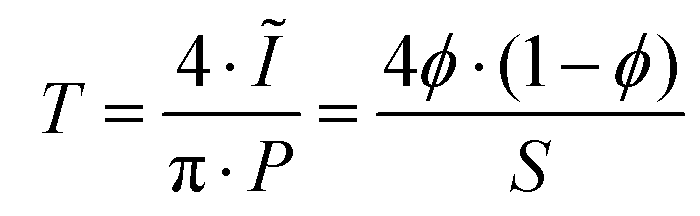 , where
, where 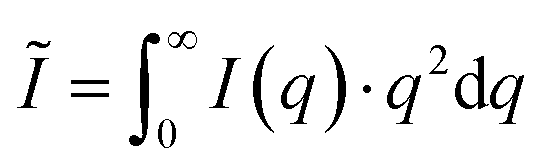 represents the integral intensity, and S and ϕ being the specific surface area and the volume fraction of the scattering objects).58–60 Due to the strong scattering contribution of the large external surfaces of the powder grains in the low q limit, data points with q < 0.3 nm−1 were approximated by a rectangle curve when calculating the integral intensity Ĩ. For dilute platelet-shaped heterogeneities within a sample, the T-parameter, which generally represents an average measure for the volume fraction per surface area, can be interpreted in terms of the thickness D of the discs with T = 2D.61
represents the integral intensity, and S and ϕ being the specific surface area and the volume fraction of the scattering objects).58–60 Due to the strong scattering contribution of the large external surfaces of the powder grains in the low q limit, data points with q < 0.3 nm−1 were approximated by a rectangle curve when calculating the integral intensity Ĩ. For dilute platelet-shaped heterogeneities within a sample, the T-parameter, which generally represents an average measure for the volume fraction per surface area, can be interpreted in terms of the thickness D of the discs with T = 2D.61
Results
Natural sea urchin proteins are often present in low abundance within the hard tissue, and their isolation can be challenging, potentially leading to loss or degradation. To circumvent these limitations, we turned to synthetic CaCO3 binding peptides that exhibit mineralization properties similar to those of natural sea urchin proteins. These peptides were carefully selected from a random peptide library based on their strong binding affinity to mature crystal material, effectively mimicking the biomineralization function observed in sea urchins. By characterizing and studying these synthetic peptides, we aim to gain insights into the nature and composition of the elusive natural biomineralization proteins.Phage display and selection of peptides
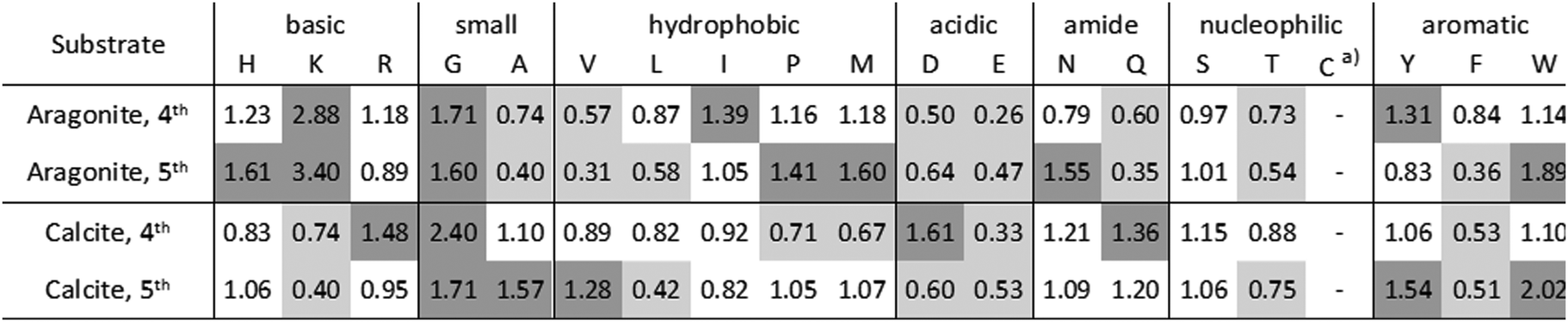
Peptides, binding to geological aragonite or calcite were selected from a peptide library by phage display. Phage clones, expressing individual peptides, were analyzed after the 4th and 5th biopanning round. In total, 33 aragonite-binding peptides and 39 calcite-binding peptides were identified from each 50 analyzed phage clones (Tables S1 and S2, ESI†).Five aragonite-binding peptide sequences were repeatedly isolated. The calculated isoelectric points (pI) of the isolated aragonite-binding peptides ranged from 4.47 to 12.3, with two accumulations at pH 8 (22.7% of the peptides) and pH 10 (29.5% of the peptides). The calcite-binding peptides were more diverse compared to the aragonite-binding peptides. The pI values of the calcite-binding peptides spanned a range from 3.81 to 11.12, with an accumulation at pH 7 (28.2% of the peptides). The amino acid residue frequencies of both, the aragonite- and the calcite-binding peptides, were referenced to the frequencies of the naïve peptide library (Table 1). The quotient of an amino acid frequency in the isolated peptide pool and in the initial peptide library is indicative of its enrichment (criterion: quotient > 1.25) or depletion (criterion: quotient < 0.75). Enriched amino acid residues may have a beneficial effect on the binding of peptides, while depleted amino acid residues might have an adverse effect on the interaction. Aragonite-binding peptides showed an enrichment of lysine (K) and to a lesser extend of histidine (H). The incorporation of lysine into CaCO3 had been shown previously in in vitro mineralization experiments, where only lysine was incorporated to detectable amounts, while histidine and arginine were below the detection limit.62 Furthermore, negatively charged amino acids were depleted in both, aragonite- and calcite-binding peptides. This is remarkable, since acidic proteins are known to control CaCO3 mineralization in biogenic systems,63 including marine organisms, like sponges64 and mollusks.34 Moreover, the majority of the proteins associated with the tests of A. lixula and P. lividus are also mainly acidic.16,25 We also identified some similarities between the selected peptides and natural biomineralization proteins. Glycine (G) and tyrosine (Y) were enriched in aragonite- and calcite-binding peptides. These amino acids are also present in increased amounts in the proteins16,25,29 of Pinctada fucata, the Akoya pearl oyster, a marine mollusk species.65,66
|
Calcium carbonate precipitation in the presence of biogenic protein mixtures and peptides
In a next step, the structure-directing effect of biomineralization-associated proteins isolated form sea urchin tests and CaCO3-binding peptides on the in vitro mineralization of CaCO3 were investigated and compared. In the mineralization experiments, only the designed peptides with demonstrated mineral interaction capabilities were included. The preselection process via phage display enabled to avoid the extensive screening of random peptides, ensuring that only effective peptides were utilized. The acid-soluble biomineralization proteins of sea urchin tests were characterized by 1D polyacrylamide gel electrophoresis (SDS-PAGE) and 2D PAGE.16,25,29 Briefly, the proteins accumulated in a range between 25 kDa and 40 kDa with distinct focal points at 40 kDa, 36 kDa, and 27 kDa. The isoelectric points of the majority of ASM from A. lixula were within the acidic to neutral range (pH 3–6.5), with only a very small fraction of low molecular weight ASM having a pI > pH 9.29 In case of P. lividus more proteins in the basic pH regime were identified.16 In the experimental set up, CaCO3 was mineralized in vitro from aqueous CaCl2 solutions where the precipitation was induced by the influx of atmospheric CO2 and NH3 through the slow decomposition of ammonium carbonate in an enclosed chamber.57 | ||
| Fig. 1 Effect of skeletal proteins from Arbacia lixula on the in vitro mineralization of CaCO3. Extracted CaCO3-associated proteins from A. lixula skeletons were applied in the bioinspired mineralization of CaCO3via a slow diffusion method. The mineralization was performed in the presence of protein concentrations ranging from 1 μg mL−1 to 500 μg mL−1. In protein-mediated precipitation, the deposited CaCO3 particles showed a complex morphology, whereas regular, rhombohedral particles were synthesized in the absence of proteins. Scale bars 10 μm. | ||
 | ||
| Fig. 2 Effect of skeletal proteins from Paracentrotus lividus on the in vitro mineralization of CaCO3. Extracted CaCO3-associated proteins from P. lividus skeletons were applied in the bioinspired mineralization of CaCO3via a slow diffusion method. The mineralization was performed in the presence of protein concentrations ranging from 1 μg mL−1 to 500 μg mL−1. In protein-mediated precipitation, the deposited CaCO3 particles showed a complex morphology, whereas regular, rhombohedral particles were synthesized in the absence of proteins. Scale bars 10 μm. | ||
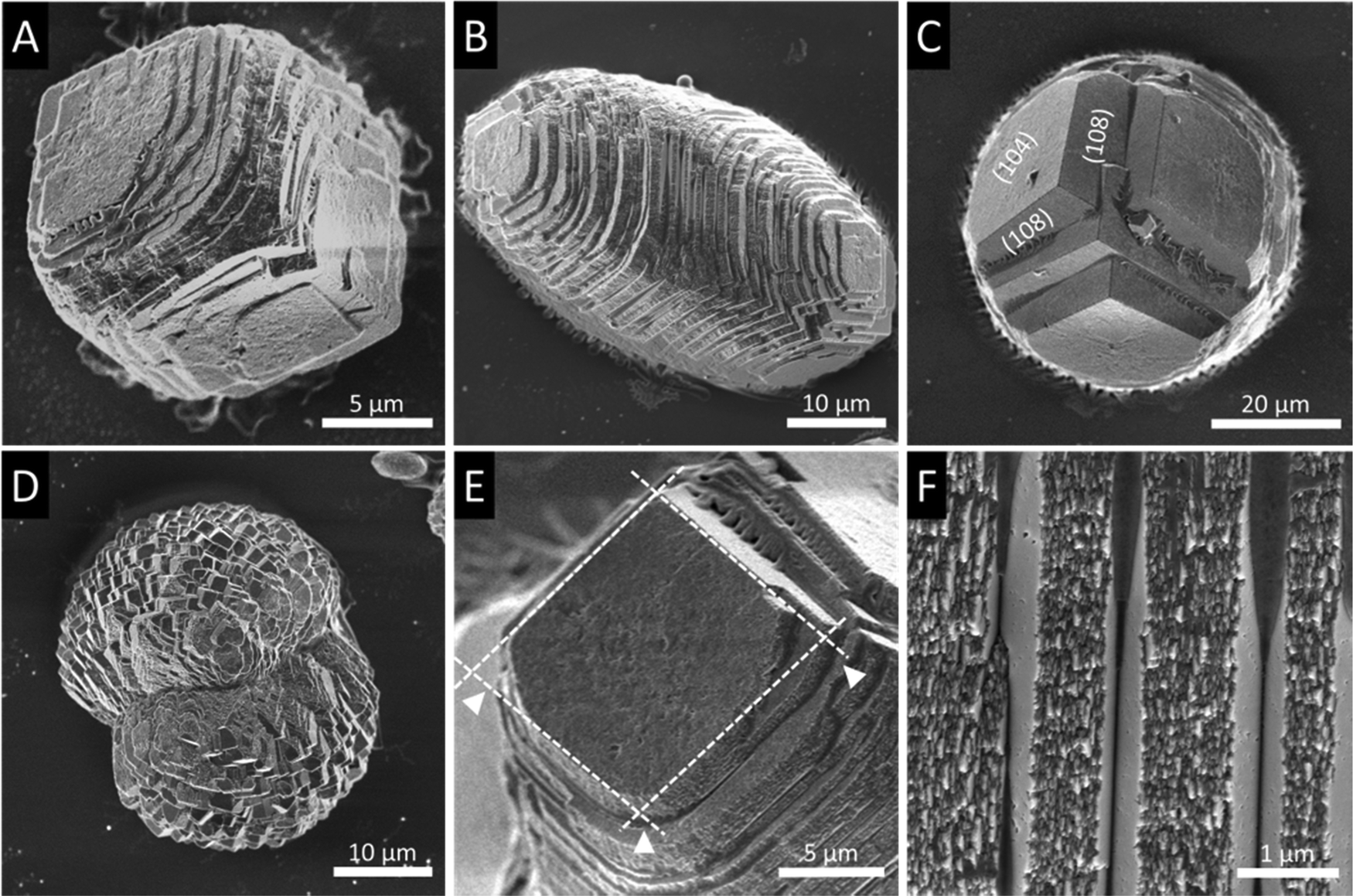 | ||
| Fig. 3 Details of the surface topography of CaCO3 particles obtained by in vitro mineralization in the presence of sea urchin protein mixtures. (A) The formation of pronounced steps blurs the sharp-edged rhombohedral particle morphology (probably initial stage). (B) Elongated, cigar-shaped particles are formed. (C) Top view of an elongated particle oriented along the principle axis. (D) Dumbbell-shaped particles with a pronounced constriction in the centre. (E) The (104) rhombic surfaces (white frame) usually have an intact obtuse angle, which is opposed by a corner that is not completely expressed or has been degraded. Likewise, the acute angles are not fully developed (arrowheads). (F) The particle surface appears to consist of co-aligned platelets with a nanogranular substructure. | ||
 | ||
| Fig. 4 Effect of peptide A-04-09 on the in vitro mineralization of CaCO3. The aragonite-binding peptide was applied in the bioinspired mineralization of CaCO3via a slow diffusion of NH3 and CO2 induced by the decomposition of ammonium carbonate into a CaCl2 solution. The mineralization was performed in the presence of protein concentrations ranging from 1 μg mL−1 to 2 mg mL−1. | ||
 | ||
| Fig. 5 Effect of peptide A-04-17 on the in vitro mineralization of CaCO3. The aragonite-binding peptide was applied in the bioinspired mineralization of CaCO3via a slow diffusion of NH3 and CO2 induced by the decomposition of ammonium carbonate into a CaCl2 solution. The mineralization was performed in the presence of protein concentrations ranging from 1 μg mL−1 to 2 mg mL−1. | ||
|
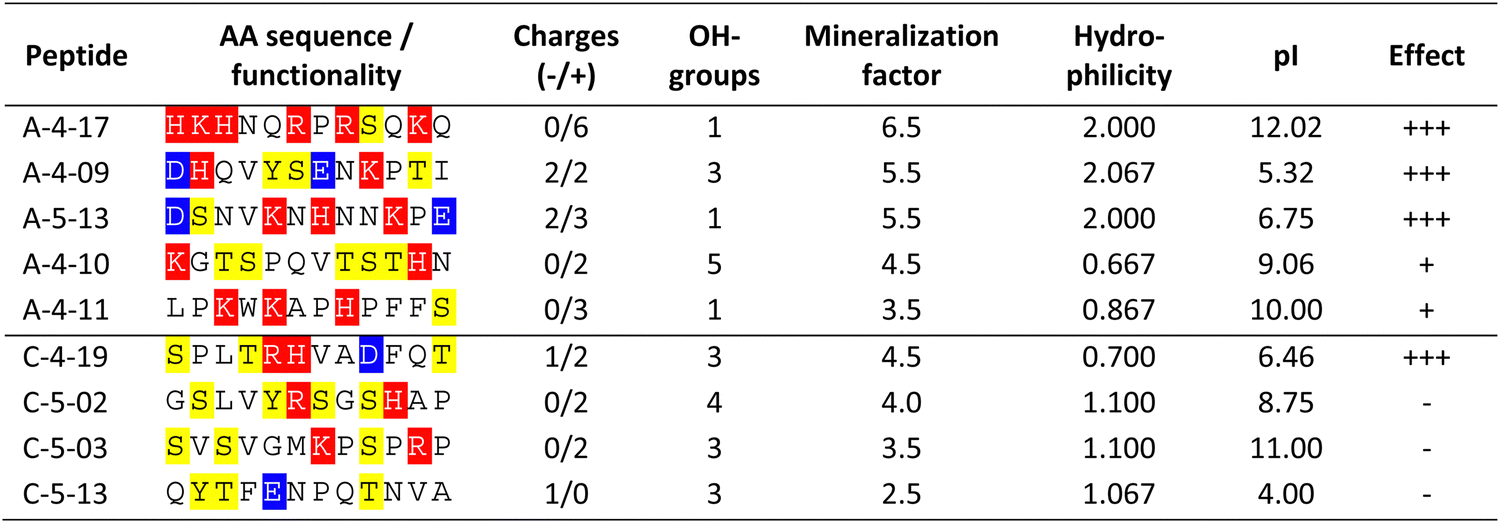
The conformation of the most effective peptides (A-4-17, A-4-09, A-5-13, and C-4-19; Table 2) was assessed using different secondary structure prediction servers (see Fig. S2, ESI†). The modern machine learning-based techniques NetSurfP 3.050 and PSSpred,51 applied here, as well as de novo predictions, offer significantly high accuracy and showed that all investigated peptides predominantly adopt an unordered conformation.
In addition, to the afore mentioned sea urchin protein concentrations, the peptides were applied at a concentration of 2 mg mL−1. All peptides had at least one charged amino acid side chain (Table 2). The aragonite-binding peptides applied in the mineralization experiments exhibit a higher proportion of charged amino acids compared to the calcite-binding peptides (Table 2), thus facilitating electrostatic interactions. In contrast, calcite-binding peptides contained a higher proportion of amino acids with hydroxyl groups (i.e. serine, threonine, tyrosine) that may promote the formation of hydrogen bonds.
In general, in the in vitro precipitation system, the calcite-binding peptides showed a minor impact on particle morphology compared to the aragonite-binding peptides. The aragonite-binding peptides A-04-09 (Fig. 4) and A-04-17 (Fig. 5) demonstrated the largest effect on particle morphology at almost all concentrations tested. Furthermore, four peptides showed a strong effect on the morphology of the CaCO3 particles at a peptide concentration of 2 mg mL−1 (Fig. 6 and Table 2). Among these most potent structure-directing additives, were three aragonite-binding peptides (A-4-09, A-5-13, and A-4-17) and one calcite-binding peptide (C-4-19). In the presence of these peptides, calcium carbonate particles with shapes derived from a rhombohedron with one elongated axis were formed, similar to the crystal morphologies obtained in in vitro mineralization experiments with sea urchin proteins. At low peptide concentrations (5 μg mL−1 to 500 μg mL−1) the two aragonite-binding peptides A-4-09 and A-4-17 still had a visible effect on the particle morphology (Fig. 4 and 5). Furthermore, the c-axis elongation was reduced at low peptide concentration compared to particles synthesized at high peptide concentrations, thus almost globular particles with a staircase-morphology were formed. The greatest influence on the particle morphology was achieved with peptides with a large number of charged amino acids and hydroxyl-functionalized amino acids (Fig. 6 and Table 2). The mineralization factor was calculated using the count of charged amino acids and hydroxyl groups in the amino acid residues. Each charged amino acid contributed 1 point, while each hydroxyl group contributed 0.5 points to the peptide's score. The cumulative points represented the mineralization factor, a parameter representing an estimate for the capacity of the peptides to interact with ionic species or mineral facets, thus reflecting the effectiveness of the peptides on in vitro mineralization. The higher the value, the stronger the impact on particle morphology. Only peptide C-4-19 deviated from this finding and showed a great influence on the morphology even at a lower mineralization factor. A correlation of pI and changes in morphology was not found, as reported for other mineralization proteins.69 As seen at the mineralization with sea urchin proteins, the surfaces of the particles sometimes showed voids and gaps that might be attributed to similar organic–inorganic interaction mechanisms. The peptides, however, usually required a higher concentration than the natural proteins to be effective. This difference might be in part due to the fact that the peptides were selected solely on the basis of their binding capacity to an inorganic substrate, while other potentially important interactions, e.g., with a liquid mineral precursor or cooperative effects between different proteins, cannot be considered due to the experimental design.
 | ||
| Fig. 6 Mineralization of CaCO3 in the presence of peptides selected by phage display. Four of the investigated peptides (A-4-09, A-5-13, A-4-17, and C-4-19) showed a significant impact on the particle morphology similar to the effects of sea urchin proteins (compare Fig. 3). Selected micrographs are recorded from reaction products obtained with 2 mg mL−1 peptide. | ||
The polymorphs of the crystalline CaCO3 precipitates were determined by XRD measurements (Fig. 7). Samples with low (10 μg mL−1) and high (500 μg mL−1) peptide concentrations in the mineralization solutions were examined. The XRD diffraction patterns of all the in vitro CaCO3 mineralization products showed calcite as the only polymorph regardless of the nature and concentration of the peptide additives. Although the peptides A-4-09 and A-4-17 were selected for their binding to aragonite, the polymorph of the synthesized particles was calcite. The observation that polymorph selectivity was independent of the concentration of the peptide, i.e. 10 μg mL−1vs. 500 μg mL−1, clearly demonstrates that the peptides had a big influence on the morphology but not on the crystallographic modification of the synthesized CaCO3. Since calcite was also precipitated in samples without peptide additives, we conclude that the polymorphism in this experimental setup is mainly driven by thermodynamic boundary conditions and not affected by the interaction with the peptides and proteins.
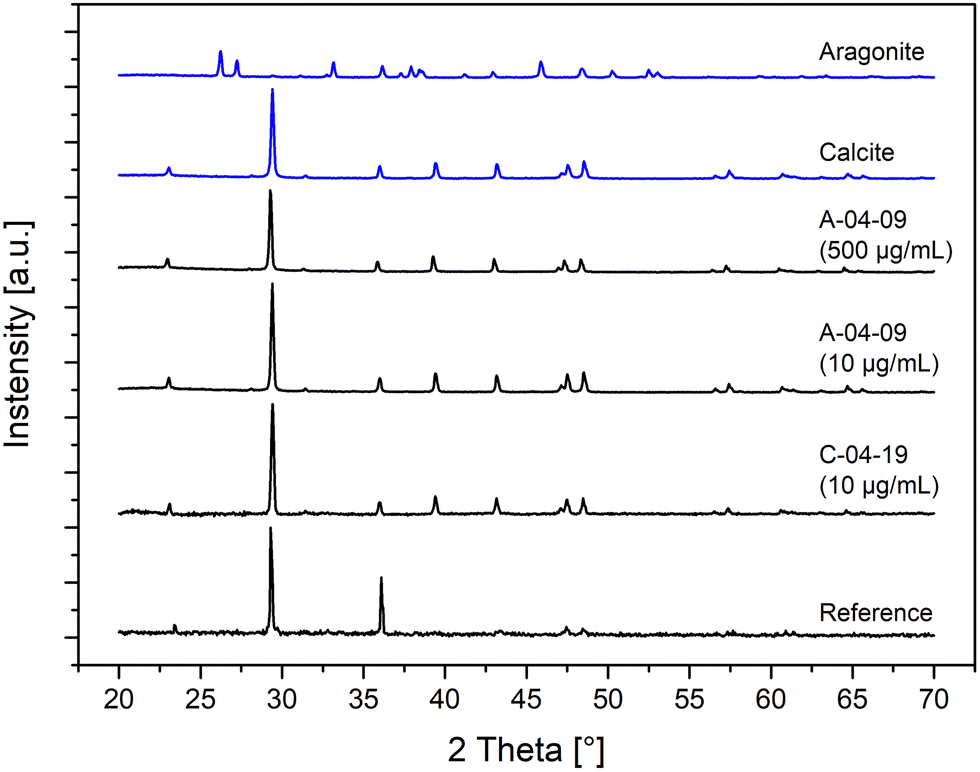 | ||
| Fig. 7 Polymorphism of in vitro mineralized and geological CaCO3. The polymorph of CaCO3 mineralized in the presence of aragonite (A)- and calcite (C)-binding peptides was determined by capillary powder X-ray diffraction (XRD). For comparison, the diffractogram of a reference sample of calcium carbonate, which was mineralized without peptide additives, is shown. The comparison with diffractograms of geological (blue lines) aragonite (Alpha Aesar, Product no. 42523, LOT N04A029) and calcite (Alpha Aesar, Product no. 44520, LOT N04A030) indicated that calcite was deposited, both in the absence and presence of peptides independent of the original binding target of the peptides. | ||
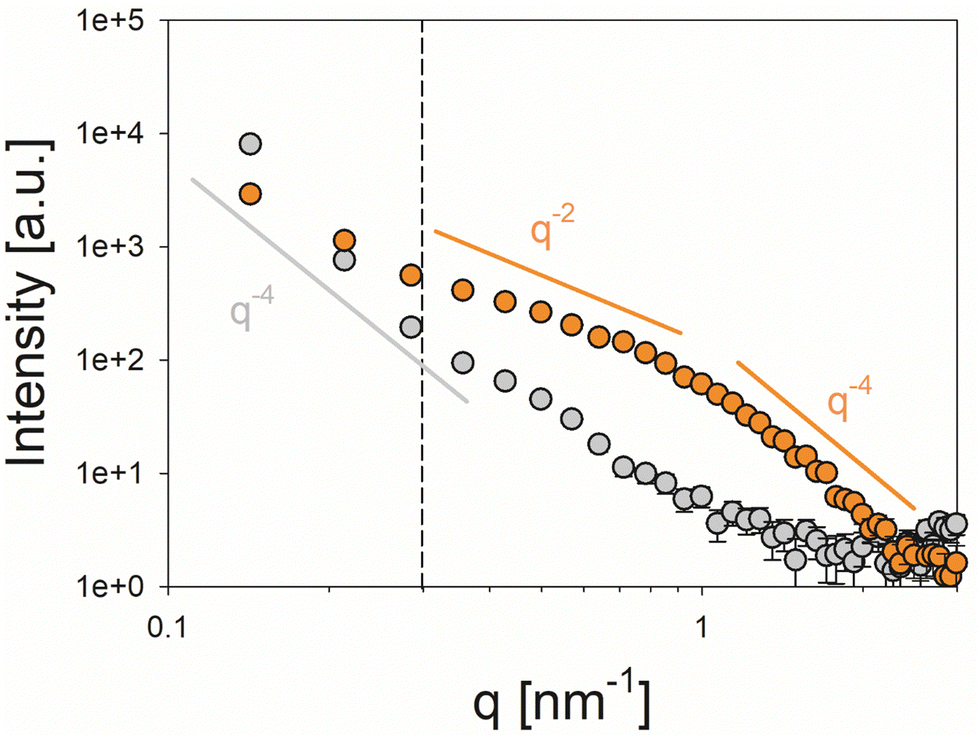 | ||
| Fig. 8 Orientation-averaged plots of the small-angle X-ray scattering (SAXS) intensity versus the modulus of the scattering vector q (log–log representation) recorded for powdered samples of control calcite (gray symbols, no additives) and calcite crystals prepared in the presence of peptide A-04-09 ([peptide] = 500 μg mL−1, orange symbols). While the control sample shows an intensity decay of roughly I(q) ∝ q−4 according to Porod's law over the entire q-range covered by the experiment, the scattering profile of the peptide-mediated crystals provides indications for structural complexity at the nanometer scale. The dashed line indicates the cutoff q = 0.3 nm−1 between the low q regime, where the scattering profile of the peptide-mediated crystals is dominated by their external facets and the intermediate q-range, where a curve shape roughly following an exponential decay of I(q) ∝ q−2 is observed. The latter indicates plate-like nanostructural motifs (presumably in the form of organic occlusions). | ||
In SAXS analysis, the control calcite sample exhibited a feature-less scattering profile following a Porod-like (I(q) ∝ q−4) behavior72 over the entire investigated q range, which corresponds to larger crystals with smooth micron-sized external facets and provides no indication of an internal nanostructure (Fig. 8, grey symbols). In the presence of the peptide, by contrast, the scattering curve of the mineralized structures showed a more complex profile suggesting a pronounced substructure at the nanoscale presumably originating from internal porosity/organic occlusions within the crystals (Fig. 8, orange symbols). Even though the signal in the low q-regime (q < 0.3 nm, vertical dashed line) appeared to be dominated by the external facets of the micron-scale powder grains, a bent curve shape which can be described by a I(q) ∝ q−2 power law characteristic of oblate scattering objects is seen at larger values of q. T-Parameter analysis revealed a mean characteristic size of T = 2.28 nm of the nanostructural motifs, which can be associated with a thickness of D = 1.14 nm in the case of dilute (non-interacting) platelet-shaped scattering objects (Fig. S6, Porod and Kratky plots for the determination of the Porod constant P and the integral intensity Ĩ, respectively, ESI†).
The nanosized granularity of the peptide-mediated crystals may reflect on a formation mechanism that proceeds via the assembly of amorphous calcium carbonate (ACC) particles, which only subsequently crystallize. The peptide additives can interact with the amorphous CaCO3 and become occluded during crystallization. Such a process has been demonstrated for the occlusion of anionic polymers into calcite along so-called transition bars during the crystallization of CaCO3 from ACC.73 Therefore, we assume that the peptides are incorporated in the CaCO3 crystals. This conclusion is further supported by the finding that such nanoscale structures are not detectable in the reference sample.
Discussion
Biomineralization proteins control the formation of inorganic materials at very low concentrations. In the natural system, the total organic content in tests of the sea urchin A. lixula is approximately 0.12% w/w,25 and the sea urchin proteins have been reported to intercalate into calcite crystals.70 A morphological effect at low protein doses was also shown in the in vitro mineralization of CaCO3 particles presented here. Interestingly, not only the proteins from sea urchin tests affected the morphology but, in addition, CaCO3-binding peptides selected by phage display. The effect of proteins on the mineralization is connected to their physicochemical properties, in particular to their net charge and hydrophilicity.74 Early mineralization processes in sea urchin larvae were reported to occur at neutral to light alkaline conditions,75 similar to the pH value of phage display selection. Therefore, no fundamental differences in the amino acid composition of the interacting proteins and peptides are expected as a function of pH. Differences between natural mineralization proteins and phage-display selected inorganic-binding peptides may account on other reasons. The lattice constants of geological calcite and the sea urchin test calcite are slightly different,76 which is due to integrated organic molecules and, more importantly, to the incorporation of Mg2+ ions into the biogenic CaCO3. These lattice differences may also contribute to variations in binding sequences. The strong influence of sea urchin matrix proteins on in vitro mineralization could be due to the glycosylation of these proteins. Up to 1.8% of the matrix proteins are glycosylated,25 and glycosylation may enhance the effect on mineralization, e.g. by increasing the hydrophilicity of proteins. The majority of sea urchin biomineralization proteins identified so far are acidic. However, basic mineralization proteins are also found in sea urchins associated with tests and spicules.16,25,29,77,78 A prominent example is the spicule matrix protein SM50 from the purple sea urchin Strongylocentrotuts purpuratus79 that stabilizes amorphous calcium carbonate.71 Interestingly, in vitro, the basic glycine rich region (GRR) domain (pI 10.48) of SM50 protein interferes with calcite formation by stabilization of prenucleation clusters,71 thus controlling the mineralization process. The exact mechanisms of crystallization are still under debate with two different theories are discussed, i.e. the classical theory of ion-by-ion crystallization and the non-classical crystallization via an amorphous precursor.80 Accordingly, the organic–inorganic interaction in biomineralization was reported to occur either with an amorphous precursor or with crystalline particles. The non-classical crystallization mechanism via an ACC precursor is supported by some findings. In natural sea urchin biomineralization, the formation of skeletons progresses through a transient phase of ACC.81,82In vitro, it was shown that protein domains interact with ACC and stabilize these structures.71 Inspired from such basic biomineralization proteins, the positively charged polymer poly(allylamine hydrochloride) (PAH) was shown to induce a liquid-like mineral precursor phase in the early stages of polymer-mediated in vitro crystallization of calcite, resulting in distinct particle morphologies.38 However, it has also been shown that negatively charged polymers, which are considered standard as structure-directing additives in mineralization processes, can sequester and interact with ions in order to initiate mineralization. Polystyrene sulfonate (PSS) was shown to complex Ca2+ to form the primary species for crystallization in solution.83 In addition, other negatively charged polymers like polyacrylic acid and polyaspartic acid were shown to interact with precursor species and step edges of CaCO3 crystals.74,84,85In the designed mineralization peptides, one of the prerequisites for effectively influencing the mineralization process was found to be the presence of charges within the peptide. In addition, polar hydroxyl groups also play a minor role in this context. The peptides are identified as predominantly adopting unordered conformations. The conformational instability was reported as a prevalent feature of bio-molecules involved in the recognition and binding to solid materials.86–89 Moreover, the inherent binding affinity of peptides is determined by the amino acid sequence and the secondary structure. It is assumed that conformationally labile peptides enhance their adaptability to interfacial features on surfaces, leading to the creation of accessible side chain regimes.86
The experimental approach for bioinspired calcium carbonate mineralization is significantly simplified compared to the biological situation. Many conditions cannot be exactly replicated in the experiment, e.g. due to the incomplete identification of all involved components, and the inability to reproduce the temporal and spatial distribution of precursor molecules. The proteins extracted from sea urchin tests strongly suggest a function of this organic matrix in the mineralization process. This is particularly evident in the impact of these proteins on the morphology of the in vitro mineralization product as compared to pure calcite. The structure-directing effects of CaCO3-binding peptides can be inferred in the context of the formation of structural motifs reminiscent of those observed in the presence of sea urchin proteins. However, since the peptide selection is based on the binding affinity of peptides to calcite or aragonite, the peptide selectivity has to be carefully interpreted. Consequently, a distinction should be made between the selectivity regarding peptide binding to target substrates and the effects of the additives on structural parameters e.g. the morphology in the bioinspired mineralization of CaCO3. In the experimental set up, calcite was formed as the only polymorph, which does not necessarily hint towards a poor peptide selectivity but may rather be explained by the reaction conditions in the artificial model system, e.g. the thermodynamic boundary conditions.
Biomolecules and also high concentrations of ions (Mg2+) can decelerate biomineralization in the beginning by inhibiting the transformation of ACC into the crystalline phase.71,90 This temporal delay may allow control over the mineralization process. Furthermore, it was suggested that the conformation of peptides may play a role in mineralization71 and that structurally disordered proteins may match unstructured ACC precursors.91,92 In the progress of biomineralization, the ACC is subsequently transformed into calcite,45 aragonite and vaterite,80 respectively. Contrasting the interaction of proteins and peptides with ACC, it has been reported that in vitro rhombohedral calcite crystals are formed first and grow to a size of about 100 to 500 nm and only then the interaction with bioorganic polymers changes their morphology.45 In the further course of in vitro mineralization, positively and negatively charged additives bind to different sites of the nascent rhombohedral calcite crystals with distinct effects on particle morphology. Rhombohedral calcite exposing (104) faces have acute and obtuse step edges that are possible sites of interaction. Mg2+ ions interact with growing calcite crystals preferentially at acute step edges,93 leading to the elongation along the c-axis.45 Negatively charged polymers like PSS interact with the obtuse step of calcite and with the (001) calcite surface which leads to the formation of truncated edges and the formation of polyhedral particles.45,83 Accordingly, negative and positive protein domains could simultaneously interact with calcite crystals, each in their own mode of action.
The morphological changes of calcite particles are correlated with the concentration of peptides, as shown for the most effective peptides A-04-09 and A-04-17 (Fig. 4 and 5). Such a correlation has also been demonstrated for the polymer PSS.45 Proteins as well as other soluble additives, e.g. block copolymers and polyelectrolytes, showed control over the polymorph as well as the morphology and the texture of the crystal.59 The concentration of mineralization peptides also influences the kinetics of the mineralization processes, where low peptide concentrations promote and high concentrations repress growth rate.74,84 The adsorption of proteins could accelerate the growth rate in biomineralization.74,94,95 It was speculated, that the interaction reduces the diffusion barrier by disturbing the layer of water molecules adsorbed on the surface in order to lower the energy barrier.74 Furthermore, a positive effect on the nucleation was also shown for synthetic macromolecules.83
Conclusion
Inorganic-binding peptides, which interact with calcite and aragonite, respectively, were selected by phage display. While most of the biomineralization proteins associated with sea urchin calcite up to now were found to be acidic in nature, many CaCO3-binding peptides identified in our study are enriched in basic amino acid residues. We showed that the peptides, obtained by phage display, were able to control the morphology of nascent CaCO3 crystallites in in vitro mineralization experiments. Moreover, the influences over crystallization exerted by CaCO3-binding peptides and natural biomineralization proteins from sea urchins lead to similar particle morphologies. In this in vitro mineralization approach, CaCO3-binding peptides with a significant number of positively charged amino acids effectively change particle morphologies towards more elongated shapes. However, the sea urchin proteins have a greater influence on mineralization, as they provoke pronounced morphological modulations even at low protein concentrations. Although some of the peptides were isolated on aragonite binding, the polymorph of the crystalline phase is not affected by the inorganic-binding peptides, as reported for some natural systems. Additional prerequisites present in the natural systems, e.g. compartmentalization, mineralization vesicles, and cooperative interaction of several proteins, might be necessary to control the polymorphism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Carl Zeiss Foundation and the University Stuttgart for the financial support of the Projekthaus NanoBioMater. The authors thank Prof. Joachim Spatz, MPI for Medical Research, for the accession to SEM and Dr Marc Widenmeyer for XRD measurements. A.S.S. acknowledges support from the Carl Zeiss Foundation in the framework of a postdoctoral fellowship at the University of Stuttgart, and from the Bavarian Academy of Sciences and Humanities (BAdW) via a Young Academy fellowship. The group of Prof. Frank Gießelmann, University of Stuttgart, is thanked for providing access to analytical infrastructure for small-angle X-ray scattering measurements.References
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed.
- S. E. Wolf, in Synthetic Inorganic Chemistry, ed. E. J. M. Hamilton, Elsevier, 2021, pp. 433–490 DOI:10.1016/B978-0-12-818429-5.00006-5.
- A. W. Skeffington and A. Scheffel, Curr. Opin. Biotechnol, 2018, 49, 57–63 CrossRef CAS PubMed.
- M. Ayoubi, A. F. van Tol, R. Weinkamer, P. Roschger, P. C. Brugger, A. Berzlanovich, L. Bertinetti, A. Roschger and P. Fratzl, Adv. Healthcare Mater., 2021, 10, 2100113 CrossRef CAS PubMed.
- P. Fratzl, H. S. Gupta, P. Roschger and K. Klaushofer, Nanotechnology, 2010, 345–360, DOI:10.1002/9783527628155.nanotech060.
- J.-B. Forien, C. Fleck, P. Cloetens, G. Duda, P. Fratzl, E. Zolotoyabko and P. Zaslansky, Nano Lett., 2015, 15, 3729–3734 CrossRef CAS PubMed.
- A. G. Checa, Front. Marine Sci., 2018, 5, 353 CrossRef.
- B. Bayerlein, L. Bertinetti, B. Bar-On, H. Blumtritt, P. Fratzl and I. Zlotnikov, Adv. Funct. Mater., 2016, 26, 3663–3669 CrossRef CAS.
- C. Gilow, E. Zolotoyabko, O. Paris, P. Fratzl and B. Aichmayer, Cryst. Growth Des., 2011, 11, 2054–2058 CrossRef CAS.
- M. E. Kunitake, L. M. Mangano, J. M. Peloquin, S. P. Baker and L. A. Estroff, Acta Biomater., 2013, 9, 5353–5359 CrossRef CAS PubMed.
- J. Ihli, A. S. Schenk, S. Rosenfeldt, K. Wakonig, M. Holler, G. Falini, L. Pasquini, E. Delacou, J. Buckman, T. S. Glen, T. Kress, E. H. R. Tsai, D. G. Reid, M. J. Duer, M. Cusack and F. Nudelman, Nat. Commun., 2021, 12, 5383 CrossRef CAS PubMed.
- Y.-Y. Kim, K. Ganesan, P. Yang, A. N. Kulak, S. Borukhin, S. Pechook, L. Ribeiro, R. Kröger, S. J. Eichhorn, S. P. Armes, B. Pokroy and F. C. Meldrum, Nat. Mater., 2011, 10, 890–896 CrossRef CAS PubMed.
- S. Kilper, S. J. Facey, Z. Burghard, B. Hauer, D. Rothenstein and J. Bill, Adv. Funct. Mater., 2018, 28, 1705842 CrossRef.
- S. Kilper, T. Jahnke, M. Aulich, Z. Burghard, D. Rothenstein and J. Bill, Adv. Mater., 2019, 31, 1805597 CrossRef PubMed.
- Z.-Z. Li, M. Li, Y.-X. Feng, Z. Zhang, Y.-E. Wen, Q.-Q. Huang, Z. Lu, H. Bai, H.-L. Wang, B.-B. Xu, H. Cölfen, P. Fratzl, S. Amini and Y. Jiang, Chem. Mater., 2021, 33, 9240–9251 CrossRef CAS.
- K. Karakostis, I. Zanella-Cléon, F. Immel, N. Guichard, P. Dru, T. Lepage, L. Plasseraud, V. Matranga and F. Marin, J. Proteomics, 2016, 136, 133–144 CrossRef CAS PubMed.
- I. M. Weiss, N. Tuross, L. Addadi and S. Weiner, J. Exp. Zool., 2002, 293, 478–491 CrossRef CAS PubMed.
- L. Kabalah-Amitai, B. Mayzel, P. Zaslansky, Y. Kauffmann, P. Clotens and B. Pokroy, J. Struct. Biol., 2013, 183, 191–198 CrossRef CAS PubMed.
- W. J. E. M. Habraken, A. Masic, L. Bertinetti, A. Al-Sawalmih, L. Glazer, S. Bentov, P. Fratzl, A. Sagi, B. Aichmayer and A. Berman, J. Struct. Biol., 2015, 189, 28–36 CrossRef CAS PubMed.
- E. V. Sturm and H. Cölfen, Chem. Soc. Rev., 2016, 45, 5821–5833 RSC.
- J. Seto, Y. Ma, S. A. Davis, F. Meldrum, A. Gourrier, Y.-Y. Kim, U. Schilde, M. Sztucki, M. Burghammer, S. Maltsev, C. Jäger and H. Cölfen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3699–3704 CrossRef CAS PubMed.
- A. Berman, L. Addadi and S. Weiner, Nature, 1988, 331, 546–548 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161–1164 CrossRef CAS PubMed.
- F. H. Wilt, Zool. Sci., 2002, 19(253–261), 259 Search PubMed.
- J. M. Kanold, N. Guichard, F. Immel, L. Plasseraud, M. Corneillat, G. Alcaraz, F. Brümmer and F. Marin, FEBS J., 2015, 282, 1891–1905 CrossRef CAS PubMed.
- K. Mann, A. J. Poustka and M. Mann, Proteome Sci., 2008, 6, 22 CrossRef PubMed.
- E. Sodergren, G. M. Weinstock, E. H. Davidson, R. A. Cameron, R. A. Gibbs, R. C. Angerer, L. M. Angerer, M. I. Arnone, D. R. Burgess, R. D. Burke, J. A. Coffman, M. Dean, M. R. Elphick, C. A. Ettensohn, K. R. Foltz, A. Hamdoun, R. O. Hynes, W. H. Klein, W. Marzluff, D. R. McClay, R. L. Morris, A. Mushegian, J. P. Rast, L. C. Smith, M. C. Thorndyke, V. D. Vacquier, G. M. Wessel, G. Wray, L. Zhang, C. G. Elsik, O. Ermolaeva, W. Hlavina, G. Hofmann, P. Kitts, M. J. Landrum, A. J. Mackey, D. Maglott, G. Panopoulou, A. J. Poustka, K. Pruitt, V. Sapojnikov, X. Song, A. Souvorov, V. Solovyev, Z. Wei, C. A. Whittaker, K. Worley, K. J. Durbin, Y. Shen, O. Fedrigo, D. Garfield, R. Haygood, A. Primus, R. Satija, T. Severson, M. L. Gonzalez-Garay, A. R. Jackson, A. Milosavljevic, M. Tong, C. E. Killian, B. T. Livingston, F. H. Wilt, N. Adams, R. Bellel, S. Carbonneau, R. Cheung, P. Cormier, B. Cosson, J. Croce, A. Fernandez-Guerra, A.-M. Genevière, M. Goel, H. Kelkar, J. Morales, O. Mulner-Lorillon, A. J. Robertson, J. V. Goldstone, B. Cole, D. Epel, B. Gold, M. E. Hahn, M. Howard-Ashby, M. Scally, J. J. Stegeman, E. L. Allgood, J. Cool, K. M. Judkins, S. S. McCafferty, A. M. Musante, R. A. Obar, A. P. Rawson, B. J. Rossetti, I. R. Gibbons, M. P. Hoffman, A. Leone, S. Istrail, S. C. Materna, M. P. Samanta, V. Stolc, W. Tongprasit, Q. Tu, K.-F. Bergeron, B. P. Brandhorst, J. Whittle, K. Berney, D. J. Bottjer, C. Calestani, K. Peterson, E. Chow, Q. A. Yuan, E. Elhaik, D. Graur, J. T. Reese, I. Bosdet, S. Heesun, M. A. Marra, J. Schein, M. K. Anderson, V. Brockton, K. M. Buckley, A. H. Cohen, S. D. Fugmann, T. Hibino, M. Loza-Coll, A. J. Majeske, C. Messier, S. V. Nair, Z. Pancer, D. P. Terwilliger, C. Agca, E. Arboleda, N. Chen, A. M. Churcher, F. HallboÌoÌk, G. W. Humphrey, M. M. Idris, T. Kiyama, S. Liang, D. Mellott, X. Mu, G. Murray, R. P. Olinski, F. Raible, M. Rowe, J. S. Taylor, K. Tessmar-Raible, D. Wang, K. H. Wilson, S. Yaguchi, T. Gaasterland, B. E. Galindo, H. J. Gunaratne, C. Juliano, M. Kinukawa, G. W. Moy, A. T. Neill, M. Nomura, M. Raisch, A. Reade, M. M. Roux, J. L. Song, Y.-H. Su, I. K. Townley, E. Voronina, J. L. Wong, G. Amore, M. Branno, E. R. Brown, V. Cavalieri, V. Duboc, L. Duloquin, C. Flytzanis, C. Gache, F. O. Lapraz, T. Lepage, A. Locascio, P. Martinez, G. Matassi, V. Matranga, R. Range, F. Rizzo, E. Rottinger, W. Beane, C. Bradham, C. Byrum, T. Glenn, S. Hussain, G. Manning, E. Miranda, R. Thomason, K. Walton, A. Wikramanayke, S.-Y. Wu, R. Xu, C. T. Brown, L. Chen, R. F. Gray, P. Y. Lee, J. Nam, P. Oliveri, J. Smith, D. Muzny, S. Bell, J. Chacko, A. Cree, S. Curry, C. Davis, H. Dinh, S. Dugan-Rocha, J. Fowler, R. Gill, C. Hamilton, J. Hernandez, S. Hines, J. Hume, L. Jackson, A. Jolivet, C. Kovar, S. Lee, L. Lewis, G. Miner, M. Morgan, L. V. Nazareth, G. Okwuonu, D. Parker, L.-L. Pu, R. Thorn and R. Wright, Science, 2006, 314, 941–952 CrossRef PubMed.
- G. Zhang, X. Fang, X. Guo, L. Li, R. Luo, F. Xu, P. Yang, L. Zhang, X. Wang, H. Qi, Z. Xiong, H. Que, Y. Xie, P. W. H. Holland, J. Paps, Y. Zhu, F. Wu, Y. Chen, J. Wang, C. Peng, J. Meng, L. Yang, J. Liu, B. Wen, N. Zhang, Z. Huang, Q. Zhu, Y. Feng, A. Mount, D. Hedgecock, Z. Xu, Y. Liu, T. Domazet-Lošo, Y. Du, X. Sun, S. Zhang, B. Liu, P. Cheng, X. Jiang, J. Li, D. Fan, W. Wang, W. Fu, T. Wang, B. Wang, J. Zhang, Z. Peng, Y. Li, N. Li, J. Wang, M. Chen, Y. He, F. Tan, X. Song, Q. Zheng, R. Huang, H. Yang, X. Du, L. Chen, M. Yang, P. M. Gaffney, S. Wang, L. Luo, Z. She, Y. Ming, W. Huang, S. Zhang, B. Huang, Y. Zhang, T. Qu, P. Ni, G. Miao, J. Wang, Q. Wang, C. E. W. Steinberg, H. Wang, N. Li, L. Qian, G. Zhang, Y. Li, H. Yang, X. Liu, J. Wang, Y. Yin and J. Wang, Nature, 2012, 490, 49–54 CrossRef CAS PubMed.
- J. M. Kanold, F. Immel, C. Broussard, N. Guichard, L. Plasseraud, M. Corneillat, G. Alcaraz, F. Brümmer and F. Marin, Comp. Biochem. Physiol., Part D: Genomics Proteomics, 2015, 13, 24–34 CAS.
- F. Marin, G. Luquet, B. Marie and D. Medakovic, Current Topics in Developmental Biology, Academic Press, 2007, vol. 80, pp. 209–276 Search PubMed.
- F. Marin, N. Roy and B. Marie, Front. Biosci., 2012, 4, 1099–1125 CrossRef PubMed.
- M. Suzuki, A. Iwashima, M. Kimura, T. Kogure and H. Nagasawa, Mar. Biotechnol., 2013, 15 Search PubMed.
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, Science, 2009, 325, 1388–1390 CrossRef CAS PubMed.
- Y. Politi, J. Mahamid, H. Goldberg, S. Weiner and L. Addadi, CrystEngComm, 2007, 9 Search PubMed.
- S. E. Wolf, I. Lieberwirth, F. Natalio, J.-F. Bardeau, N. Delorme, F. Emmerling, R. Barrea, M. Kappl and F. Marin, Faraday Discuss., 2012, 159, 433–448 RSC.
- L. A. Gower and D. A. Tirrell, J. Cryst. Growth, 1998, 191, 153–160 CrossRef CAS.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- B. Cantaert, Y.-Y. Kim, H. Ludwig, F. Nudelman, N. A. J. M. Sommerdijk and F. C. Meldrum, Adv. Funct. Mater., 2012, 22, 907–915 CrossRef CAS.
- A. S. Schenk, B. Cantaert, Y.-Y. Kim, Y. Li, E. S. Read, M. Semsarilar, S. P. Armes and F. C. Meldrum, Chem. Mater., 2014, 26, 2703–2711 CrossRef CAS.
- O. Nahi, A. N. Kulak, S. Zhang, X. He, Z. Aslam, M. A. Ilett, I. J. Ford, R. Darkins and F. C. Meldrum, Adv. Sci., 2023, 10, 2203759 CrossRef CAS PubMed.
- M. A. Bewernitz, D. Gebauer, J. Long, H. Cölfen and L. B. Gower, Faraday Discuss., 2012, 159, 291–312 RSC.
- Y.-Y. Kim, A. N. Kulak, Y. Li, T. Batten, M. Kuball, S. P. Armes and F. C. Meldrum, J. Mater. Chem., 2009, 19, 387–398 RSC.
- A. S. Schenk, H. Zope, Y.-Y. Kim, A. Kros, N. A. J. M. Sommerdijk and F. C. Meldrum, Faraday Discuss., 2012, 159, 327–344 RSC.
- S. Mijowska, I. Polishchuk, A. Lang, E. Seknazi, C. Dejoie, S. Fermani, G. Falini, N. Demitri, M. Polentarutti, A. Katsman and B. Pokroy, Chem. Mater., 2020, 32, 4205–4212 CrossRef CAS.
- Y. Y. Kim, C. L. Freeman, X. Q. Gong, M. A. Levenstein, Y. W. Wang, A. Kulak, C. Anduix-Canto, P. A. Lee, S. B. Li, L. Chen, H. K. Christenson and F. C. Meldrum, Angew. Chem., Int. Ed., 2017, 56, 11885–11890 CrossRef CAS PubMed.
- S. Kilper, T. Jahnke, K. Wiegers, V. Grohe, Z. Burghard, J. Bill and D. Rothenstein, Materials, 2019, 12, 904 CrossRef CAS PubMed.
- D. Rothenstein, B. Claasen, B. Omiecienski, P. Lammel and J. Bill, J. Am. Chem. Soc., 2012, 134, 12547–12556 CrossRef CAS PubMed.
- P. Y. Chou and G. D. Fasman, Biochemistry, 1974, 13, 211–222 CrossRef CAS PubMed.
- T. Ashok Kumar, Wide Spectrum, 2013, 1, 15–19 Search PubMed.
- M. H. Høie, E. N. Kiehl, B. Petersen, M. Nielsen, O. Winther, H. Nielsen, J. Hallgren and P. Marcatili, Nucleic Acids Res., 2022, 50, W510–W515 CrossRef PubMed.
- R. Yan, D. Xu, J. Yang, S. Walker and Y. Zhang, Sci. Rep., 2013, 3, 2619 CrossRef PubMed.
- Y. Shen, J. Maupetit, P. Derreumaux and P. Tufféry, J. Chem. Theory Comput., 2014, 10, 4745–4758 CrossRef CAS PubMed.
- P. Thévenet, Y. Shen, J. Maupetit, F. Guyon, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2012, 40, W288–W293 CrossRef PubMed.
- A. Lamiable, P. Thévenet, J. Rey, M. Vavrusa, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2016, 44, W449–W454 CrossRef CAS PubMed.
- A. C. Camproux, R. Gautier and P. Tufféry, J. Mol. Biol., 2004, 339, 591–605 CrossRef CAS PubMed.
- J. Maupetit, P. Tuffery and P. Derreumaux, Proteins: Struct., Funct., Bioinf., 2007, 69, 394–408 CrossRef CAS PubMed.
- S. Albeck, J. Aizenberg, L. Addadi and S. Weiner, J. Am. Chem. Soc., 1993, 115, 11691–11697 CrossRef CAS.
- P. Fratzl, S. Schreiber and K. Klaushofer, Connect. Tissue Res., 1996, 34, 247–254 CrossRef CAS PubMed.
- A. S. Schenk, I. Zlotnikov, B. Pokroy, N. Gierlinger, A. Masic, P. Zaslansky, A. N. Fitch, O. Paris, T. H. Metzger, H. Cölfen, P. Fratzl and B. Aichmayer, Adv. Funct. Mater., 2012, 22, 4668–4676 CrossRef CAS.
- Y.-Y. Kim, A. S. Schenk, J. Ihli, A. N. Kulak, N. B. J. Hetherington, C. C. Tang, W. W. Schmahl, E. Griesshaber, G. Hyett and F. C. Meldrum, Nat. Commun., 2014, 5, 4341 CrossRef CAS PubMed.
- P. Fratzl, J. Stat. Phys., 1994, 77, 125–143 CrossRef.
- S. Borukhin, L. Bloch, T. Radlauer, A. H. Hill, A. N. Fitch and B. Pokroy, Adv. Funct. Mater., 2012, 22, 4216–4224 CrossRef CAS.
- S. Weiner and L. Hood, Science, 1975, 190 Search PubMed.
- J. Aizenberg, M. Ilan, S. Weiner and L. Addadi, Connect. Tissue Res., 1996, 34, 255–261 CrossRef CAS PubMed.
- D. Tsukamoto, I. Sarashina and K. Endo, Biochem. Biophys. Res. Commun., 2004, 320, 1175–1180 CrossRef CAS PubMed.
- M. Suzuki, E. Murayama, H. Inoue, N. Ozaki, H. Tohse, T. Kogure and H. Nagasawa, Biochem. J., 2004, 382, 205–213 CrossRef CAS PubMed.
- K. Delak, J. Giocondi, C. Orme and J. S. Evans, Cryst. Growth Des., 2008, 8, 4481–4486 CrossRef CAS.
- N. E. Pingitore, S. B. Fretzdorff, B. P. Seitz, L. Y. Estrada, P. M. Borrego, G. M. Crawford and K. M. Love, J. Sediment. Res., 1993, 63, 641–645 CAS.
- A. Hernandez-Hernandez, A. B. Rodriguez-Navarro, J. Gomez-Morales, C. Jimenez-Lopez, Y. Nys and J. M. Garcia-Ruiz, Cryst. Growth Des., 2008, 8, 1495–1502 CrossRef CAS.
- A. Berman, L. Addadi, Å. Kvick, L. Leiserowitz, M. Nelson and S. Weiner, Science, 1990, 250, 664–667 CrossRef CAS PubMed.
- A. Rao, J. Seto, J. K. Berg, S. G. Kreft, M. Scheffner and H. Cölfen, J. Struct. Biol., 2013, 183, 205–215 CrossRef CAS PubMed.
- G. Porod, Kolloid-Z., 1951, 124, 83–114 CrossRef CAS.
- L. Dai, X. Cheng and L. B. Gower, Chem. Mater., 2008, 20, 6917–6928 CrossRef CAS.
- S. Elhadj, J. J. De Yoreo, J. R. Hoyer and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19237–19242 CrossRef CAS PubMed.
- M. Stumpp, M. Y. Hu, F. Melzner, M. A. Gutowska, N. Dorey, N. Himmerkus, W. C. Holtmann, S. T. Dupont, M. C. Thorndyke and M. Bleich, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18192–18197 CrossRef CAS PubMed.
- E. Zolotoyabko, E. N. Caspi, J. S. Fieramosca, R. B. Von Dreele, F. Marin, G. Mor, L. Addadi, S. Weiner and Y. Politi, Cryst. Growth Des., 2010, 10, 1207–1214 CrossRef CAS.
- B. T. Livingston, C. E. Killian, F. Wilt, A. Cameron, M. J. Landrum and O. Ermolaeva, Dev. Biol., 2006, 300 Search PubMed.
- M. R. Illies, M. T. Peeler, A. M. Dechtiaruk and C. A. Ettensohn, Dev. Genes Evol., 2002, 212, 419–431 CrossRef CAS PubMed.
- Y. Katohfukui, T. Noce, T. Ueda, Y. Fujiwara, N. Hashimoto, T. Higashinakagawa, C. E. Killian, B. T. Livingston, F. H. Wilt, S. C. Benson, H. M. Sucov and E. H. Davidson, Dev. Biol., 1991, 145, 201–202 CrossRef CAS PubMed.
- M. H. Nielsen, S. Aloni and J. J. De Yoreo, Science, 2014, 345, 1158–1162 CrossRef CAS PubMed.
- Y. Politi, Y. Levi-Kalisman, S. Raz, F. Wilt, L. Addadi, S. Weiner and I. Sagi, Adv. Funct. Mater., 2006, 16, 1289–1298 CrossRef CAS.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461–465 CAS.
- T. Wang, H. Cölfen and M. Antonietti, J. Am. Chem. Soc., 2005, 127, 3246–3247 CrossRef CAS PubMed.
- S. Elhadj, E. A. Salter, A. Wierzbicki, J. J. De Yoreo, N. Han and P. M. Dove, Cryst. Growth Des., 2006, 6, 197–201 CrossRef CAS.
- S. Sun, L.-B. Mao, Z. Lei, S.-H. Yu and H. Cölfen, Angew. Chem., Int. Ed., 2016, 55, 11765–11769 CrossRef CAS PubMed.
- M. Hnilova, E. E. Oren, U. O. S. Seker, B. R. Wilson, S. Collino, J. S. Evans, C. Tamerler and M. Sarikaya, Langmuir, 2008, 24, 12440–12445 CrossRef CAS PubMed.
- S. Collino and J. S. Evans, Biomacromolecules, 2008, 9, 1909–1918 CrossRef CAS PubMed.
- E. E. Oren, R. Notman, I. W. Kim, J. S. Evans, T. R. Walsh, R. Samudrala, C. Tamerler and M. Sarikaya, Langmuir, 2010, 26, 11003–11009 CrossRef CAS PubMed.
- J. M. Slocik, A. O. Govorov and R. R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS PubMed.
- J. Seto, Y. Ma, S. A. Davis, F. Meldrum, A. Gourrier, Y.-Y. Kim, U. Schilde, M. Sztucki, M. Burghammer, S. Maltsev, C. Jäger and H. Cölfen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3699–3704 CrossRef CAS PubMed.
- A. L. Boskey and E. Villarreal-Ramirez, Matrix Biol., 2016, 52–54, 43–59 CrossRef CAS PubMed.
- J. S. Evans, Bioinformatics, 2012, 28, 3182–3185 CrossRef CAS PubMed.
- J. Xu, J. H. Wang, M. Hong and H. H. Teng, Am. Mineral., 2016, 101, 1104–1112 CrossRef.
- G. Fu, S. R. Qiu, C. A. Orme, D. E. Morse and J. J. De Yoreo, Adv. Mater., 2005, 17, 2678–2683 CrossRef CAS.
- I. W. Kim, S. Collino, D. E. Morse and J. S. Evans, Cryst. Growth Des., 2006, 6, 1078–1082 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb01584j |
| ‡ Current affiliation Dept. Cellular Biophysics, Max Planck Institute for Medical Research, Heisenbergstraße 3, 70569 Stuttgart, Germany |
| This journal is © The Royal Society of Chemistry 2023 |