
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic dextrin nanoparticles for effective intracellular delivery of cytochrome C in cancer therapy†
Ankita
Sarkar
,
Sanchita
Sarkhel
,
Deepali
Bisht
and
Amit
Jaiswal
 *
*
School of Biosciences and Bioengineering, Indian Institute of Technology Mandi, Kamand, Mandi, 175075 Himachal Pradesh, India. E-mail: j.amit@iitmandi.ac.in
First published on 24th November 2023
Abstract
Intracellular protein delivery shows promise as a selective and specific approach to cancer therapy. However, a major challenge is posed by delivering proteins into the target cells. Despite the development of nanoparticle (NP)-based approaches, a versatile and biocompatible delivery system that can deliver active therapeutic cargo into the cytosol while escaping endosome degradation remains elusive. In order to overcome these challenges, a polymeric nanocarrier was prepared using cationic dextrin (CD), a biocompatible and biodegradable polymer, to encapsulate and deliver cytochrome C (Cyt C), a therapeutic protein. The challenge of endosomal escape of the nanoparticles was addressed by co-delivering the synthesized NP construct with chloroquine, which enhances the endosomal escape of the therapeutic protein. No toxicity was observed for both CD NPs and chloroquine at the concentration tested in this study. Spectroscopic investigations confirmed that the delivered protein, Cyt C, was structurally and functionally active. Additionally, the delivered Cyt C was able to induce apoptosis by causing depolarization of the mitochondrial membrane in HeLa cells, as evidenced by flow cytometry and microscopic observations. Our findings demonstrate that an engineered delivery system using CD NPs is a promising platform in nanomedicine for protein delivery applications.
1. Introduction
In the last few decades, cancer has become one of the major reasons for world-wide mortality and it also reduces quality and well-being of life, which leads to an early death. Conventional cancer treatment mainly includes chemotherapy and radiation therapy, which collectively cause severe toxicity to normal healthy cells as well as undesirable side effects with multi-organ damage.1,2 For several years, researchers have been engaged in developing suitable and less toxic mediated therapies as an alternative to conventional chemotherapy and radiation therapy to combat the associated risks to healthy cells.3–5 The development of nanotechnology has led to the emergence of various anticancer treatment methods,6–9 among which therapeutic protein delivery has been deemed of utmost importance in recent times. This is because, unlike chemotherapy where the anticancer drugs are not specific to the cancer tissue and damage healthy cells, and drug resistance develops upon repeated administration,10 a therapeutic protein offers increased specificity, selectivity, and reduced toxicity towards healthy cells.11 Similarly, unlike radiation therapy, protein-based therapies do not alter the gene sequence or normal functioning of a cell.12 Moreover, when compared to genetic drugs, proteins directly act on targets and using cells’ own metabolism, it further follows the triggered signalling pathway without causing any permanent gene mutation.13 However, proteins are generally membrane-impermeable due to their macromolecular structure14 so they are hindered in reaching the cytosol of a cell or in some cases the absence of a specific cell-membrane receptor restricts proteins from entry to a cell.15 Proteins are also highly susceptible to enzymatic degradation or proteolysis upon cellular entry.16,17 Moreover, proteins cannot be administered orally due to their instability in the digestive system.15 Therefore, delivery of proteins to cells remains a challenge.Nanotechnology has revolutionized medical fields, allowing for targeted cancer therapy through the use of nano-sized vehicles that deliver therapeutic proteins.18,19 This approach also utilizes the enhanced permeation and retention (EPR) effect to achieve target specificity.20 However, a major challenge associated with nanocarrier systems is achieving a high protein loading efficiency with simultaneous release of protein at the target site in its active form.21 Many researchers have devised methods for effective cargo protein encapsulation employing various kinds of nanocarriers, including liposomes,22 synthetic polymers,23 natural polymers, polymeric micelles14 and mesoporous silica nanoparticles, in order to best achieve this aim.1,24–26 It is anticipated that among these, NPs generated from naturally occurring polysaccharides will find a way towards clinical usage in the treatment of cancer due to their outstanding biocompatibility and low immunogenicity.27,28 In particular, the use of carbohydrate-based polymeric NPs to deliver proteins is a very promising strategy due to their advantages in terms of (i) biocompatibility, (ii) biodegradability, (iii) ease of synthesis, (iv) protection of the encapsulated cargo protein from proteolysis or enzymatic degradation, and (v) controlled and sustained release of the therapeutic cargo.29
Taking into account the aforementioned factors, we developed a protein delivery nanocarrier system utilizing dextrin, a naturally occurring low-molecular-weight polysaccharide derived from the partial hydrolysis of starch. Dextrin is an FDA-approved polymer categorized as generally regarded as safe (GRAS), and it is both affordable and easily obtainable.30 It has been widely used in numerous applications in food, textiles, adhesives etc., and clinically it is routinely used in peritoneal dialysis solution.27 Our study highlights the potential of using cationic dextrin nanoparticles (CD NPs) for protein delivery in cancer therapy. We previously synthesized CD NPs for chemotherapeutic drug delivery,31 and in this study we demonstrated their ability to encapsulate and deliver the crucial mitochondrial protein, cytochrome C, to cancer cells. With its pivotal role in inducing apoptosis, the successful delivery of cytochrome C using CD NPs holds great promise for targeted and effective cancer treatment.32 It facilitates the electron transport chain in the mitochondria and has been identified as the initiator of the apoptotic signalling pathway.24 Only in response to an internal or external signal (such as DNA damage), Cyt C can leave the inner mitochondria,21 and it eventually triggers the caspase cascade, which in turn initiates the downstream apoptotic signalling pathway.24 However, there are still some drawbacks to this method. A few cytosolic proteins that control the outer mitochondrial membrane can occasionally become mutated and dysfunctional, which prevents the release of Cyt C into the cytosol.32 In addition, many cancer cells attempt to avoid the self-death process by suppressing the release of cytochrome C.15 Therefore, by directly delivering the protein into the cytoplasm from an external source, the problem of Cyt C inhibition or suppression will be resolved, and there will be rapid onset of the downstream apoptotic pathway.33
We used CD NPs to deliver Cyt C and induce apoptotic cell death in cancer cells (Scheme 1). CD NPs were synthesized using ionic gelation and Cyt C was encapsulated within the polymer matrices (Scheme 1a). However, endocytosis is the main method of cellular entrance, and endosome maturation occurs when they fuse with lysosomes, which can hinder the effectiveness of protein delivery.34 To address this obstacle, we delivered chloroquine (CQ) along with the protein-loaded CD NPs to enhance endosome escape. The co-delivery of CQ with Cyt C-loaded CD NPs allowed the efficient release of the therapeutic protein into the cytosol and induced apoptosis, leading to cancer cell death (Scheme 1b).
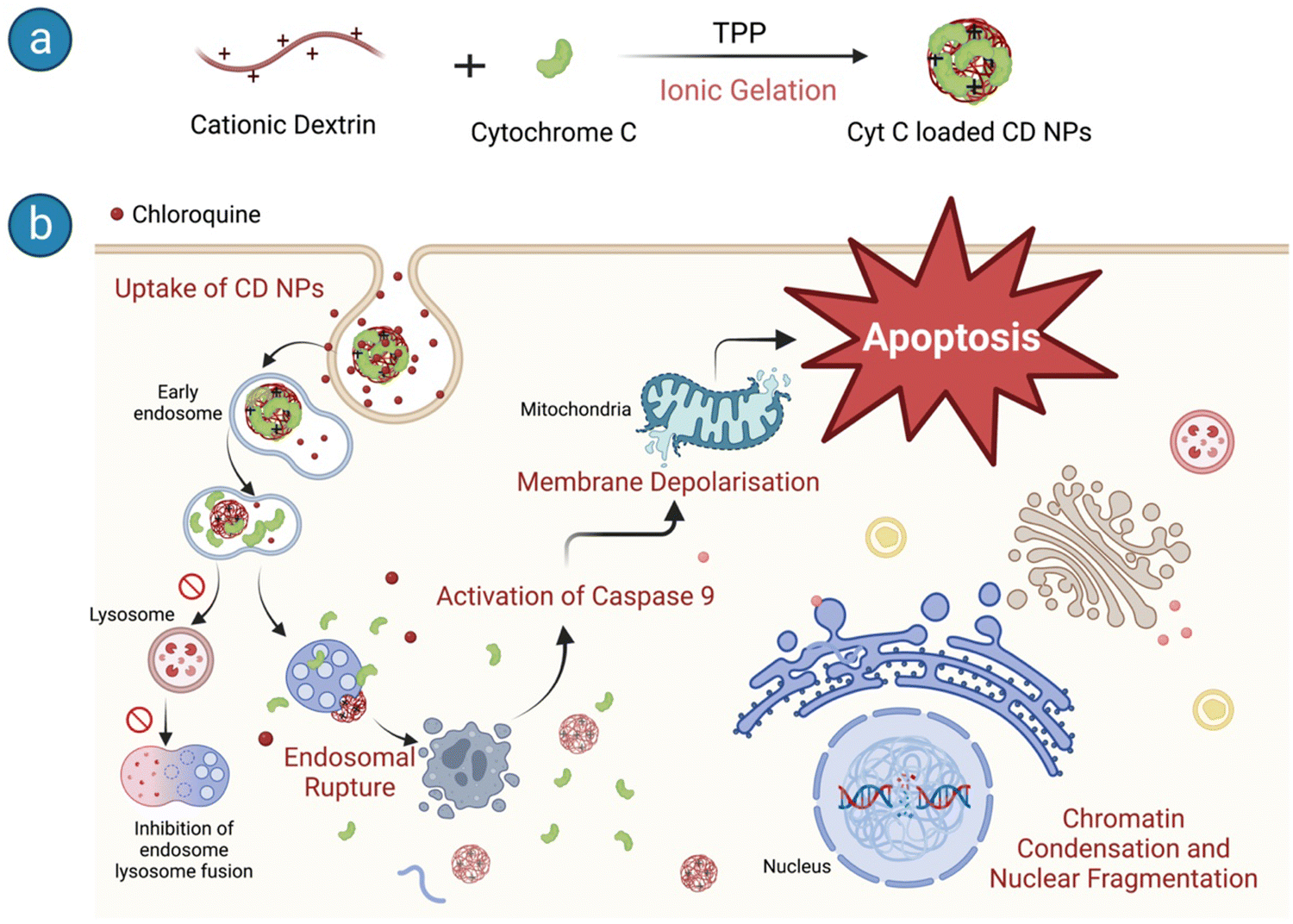 | ||
| Scheme 1 Schematic representation of (a) Cyt C-loaded CD NP synthesis and (b) the downstream pathways of apoptosis in HeLa cells upon entry. | ||
2. Materials and methods
2.1. Materials
Dextrin from maize starch, glycidyltrimethylammonium chloride (GTMAC), sodium tripolyphosphate (TPP), sodium hydroxide (NaOH), cytochrome C from equine heart, β-mercaptoethanol, cell proliferation assay kit II (XTT), and fluoroshield with DAPI mounting media were purchased from Sigma-Aldrich. Annexin V/PI staining kit was purchased from BD. JC-1 dye was purchased from Thermo Fisher Scientific. Primary monoclonal antibodies (rabbit mAb) for western blot were purchased from CST (cell signalling technologies). Horseradish-conjugated secondary antibody (anti-rabbit origin) was purchased from Merck. ABTS substrate was purchased from SRL. Ethanol (EtOH) was purchased from Merck, Germany. DMEM culture medium, fetal bovine serum (FBS), 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA), penicillin–streptomycin (Pen-Strep), and sodium pyruvate were obtained from Thermo Fisher Scientific (Gibco). Ultrapure water (resistivity = 18.2 MΩ cm) was used in all experiments.2.1. Synthesis and characterization of Cyt C-encapsulated CD NPs
Dextrin was initially cationized by incorporating quaternary ammonium groups onto the polymeric backbone using GTMAC following a process reported earlier.31,35,36 Cyt C was suspended in an alkaline buffer of pH 11, higher than its pI (10.8), which made the suspended protein negatively charged. The protein suspension (500 μg) was allowed to stir along with the polymer cationic dextrin (CD) at a concentration of 0.5 mg mL−1 for 30 min to interact electrostatically with each other. This step was followed by drop-wise addition of anionic sodium tripolyphosphate (TPP) at a concentration of 2 mg mL−1 to the aqueous mixture under constant stirring. The resulting Cyt C-loaded CD NPs were recovered by centrifugation with an equal amount of acetone added to the solution to remove excess unbound protein from the NPs in the supernatant. The % loaded amount of Cyt C was calculated with the standard curve using the absorbance at 409 nm through UV-vis spectroscopy with the following equation: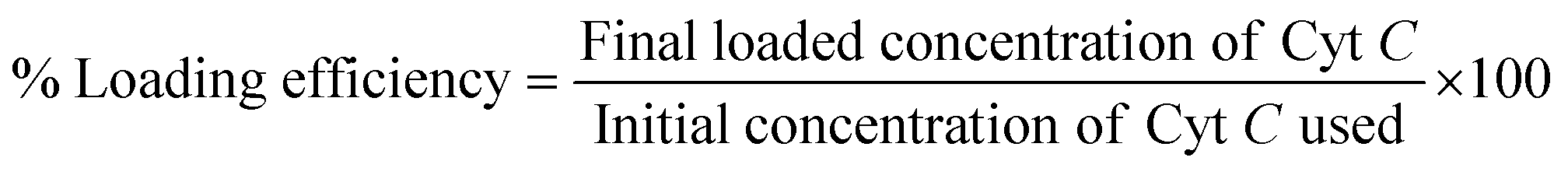
To visualize the prepared nanoparticles under SEM, equal volumes of prepared CD-Cyt C NPs (500 μL) and acetone (500 μL) were collected in a microcentrifuge tube and centrifuged at 4000 rpm for 15 min at 4 °C. Following centrifugation, the supernatant was decanted, and the tube was air-dried for some time to remove the residual acetone. Later, 500 μL of DI water was added to resuspend the pellet. The 100-fold diluted sample was drop-cast onto piranha-cleaned silicon wafers and air-dried overnight. The next day, samples were visualized and images were obtained under a scanning electron microscope (Nova Nano SEM450, FEI).
2.2. pH-dependent release of Cyt C from CD NPs
The release of Cyt C from CD NPs was carried out through a dialysis technique using a 100 kD MWCO dialysis membrane. Membranes containing the loaded particles were suspended in physiological and acidic buffers (pH 7.4 and 5.5, respectively) and incubated at room temperature for a designated period of time. At each time point, 1 mL of the dialysing buffer was collected and analyzed by UV-vis spectrometer to measure the concentration of released Cyt C (λmax = 409 nm). Subsequently, equal volumes of the respective buffers were replaced each time.2.3. Structural and functional stability of Cyt C before and after release from the nanoparticle
2.4. Cell culture
HeLa cells and A549 cells were maintained in a 25 mm tissue culture flask in DMEM media supplemented with 10% FBS, 1% sodium pyruvate, 1% L-glutamine, 1% Pen-Strep, and 1% non-essential amino acids in a humidified incubator with 5% CO2 at 37 °C.2.5. FITC conjugation and cellular uptake of CD NPs
The conjugation of CD polymer with FITC was performed as reported by Sarkar et al.31 Briefly, 1 mg of FITC was mixed with 1 mg of CD polymer in a total volume of 5 ml and stirred for 6 h in dark, followed by a dialysis process to remove excess FITC from the solution. Finally, the aqueous solution was lyophilized and then used for the cellular internalization experiment.For the investigation of the cellular internalization process, HeLa cells were cultured on a coverslip with a cell density of 2 × 104 cells. The next day, the cells were incubated with 5 μg ml−1 of FITC-tagged CD NPs. Another coverslip with untreated cells was kept as control. Cells were washed thrice with PBS after incubation and then fixed with 4% formaldehyde for 10 min. The fixed cells were again washed with PBS and mounted on a glass slide with fluoroshield + DAPI-stained mounting media. The cellular internalization and distribution of FITC-labelled CD NPs in HeLa cells was visualized using a fluorescence microscope.
2.6. In vitro cell viability assay
The viability of HeLa cells and A549 cells after treatment with the NP construct was determined using an XTT-based cell viability assay according to the manufacturers’ protocol. Cells (104 cells per well) were seeded in a 96-well plate as described above. The next day, cells were incubated with 1 and 1.5 mg mL−1 of Cyt C-loaded CD NPs, respectively, with and without co-treatment of chloroquine (0.1 mM) in serum-free DMEM media. 5 h after treatment, the cells were washed and the media was replaced with serum-supplemented DMEM media and incubated for a further 19 h. Afterwards, the cells were washed with PBS and XTT reagent mixture was added to each well followed by 3 h of incubation at 37 °C. Absorbance was measured using a plate reader at 490 nm and the absorbance at 690 nm was used to subtract the background. The experiment was performed in triplicate, and the % cell viability was calculated as follows: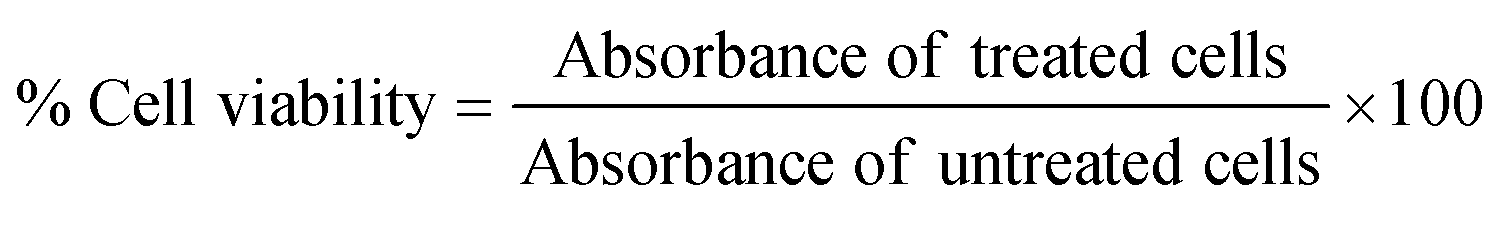
2.7. Studies to evaluate induction of apoptosis
3. Results and discussion
3.1. Synthesis and characterization of Cyt C-encapsulated CD NPs
Cationic dextrin nanoparticles were prepared following a protocol of ionic gelation using quaternary dextrin and TPP, as previously reported by our group but with modifications.31 The process involves complexation of positively charged quaternarnized dextrin with negatively charged TPP molecules, leading to the formation of spherical nanoparticles. The therapeutic protein, Cyt C, was loaded on the CP NPs during the synthesis of the CD NPs. However, the synthesis was carried out at pH 11 instead of physiological pH. This change in pH was done to enable the encapsulation of the protein within the CD NPs. The protein Cyt C has a pI of 10.8 and possesses a net positive charge at physiological pH due to the presence of lysine residues.15,32,38 As a result, the positively charged Cyt C could not interact efficiently with the quaternized dextrin due to similar charges at physiological pH and was not encapsulated. Hence, the protein Cyt C was suspended in a buffer of pH 11, above the isoelectric point of Cyt C, rendering a net negative charge to Cyt C. The surface charges of Cyt C and CD polymer were quantified at buffers of pH 7.4 and pH 11 using a zeta sizer and the results are shown in Fig. 1(a). Cyt C possess a net positive charge at pH 7.4; however, it has a net negative charge at pH 11. At pH 11, there was a slight decrease in the zeta potential of CD from pH 7.4, but it still possesses a net positive charge. Thus, the synthesis of the CD NPs was carried out at pH 11 where Cyt C was added, which can now interact with the oppositely charged CD and get encapsulated in the NPs during the process of ionic gelation using TPP as the anionic crosslinker. The prepared CD NPs were then characterized using SEM and AFM and the corresponding images are shown in Fig. 1(b) and (c), respectively. Uniformly distributed NPs were observed in both the SEM and AFM images with an average size of 100 ± 30 nm. NTA analysis of the prepared CD NPs demonstrated a hydrodynamic diameter of 98 nm (mode) with a PDI of 0.19 (Fig. S1, ESI†). The zeta potential of the Cyt C-loaded CD NPs was determined to be +6.3 mV. | ||
| Fig. 1 Characterization of the Cyt C-loaded CD NPs. (a) Zeta potential measurements of cytochrome C and CD polymer at pH 7.4 and pH 11. (b) SEM and (c) AFM images of Cyt C-loaded CD NPs. | ||
3.2. Cytochrome C loading and release from CD NPs
Cyt C has a characteristic absorption at 409 nm which allows for the quantification of the protein in a sample (Fig. 2(a)). A standard plot for Cyt C was prepared by taking the absorption of varying concentration of the protein Cyt C. The CD NPs do not have any significant absorbance at 409 nm (Fig. 2(a)) and the amount of Cyt C loaded into the nanoparticles was determined based on the absorbance of the Cyt C-loaded CD NPs. The loading efficiency was calculated using a standard curve (Fig. S2, ESI†) and was found to be ∼20%.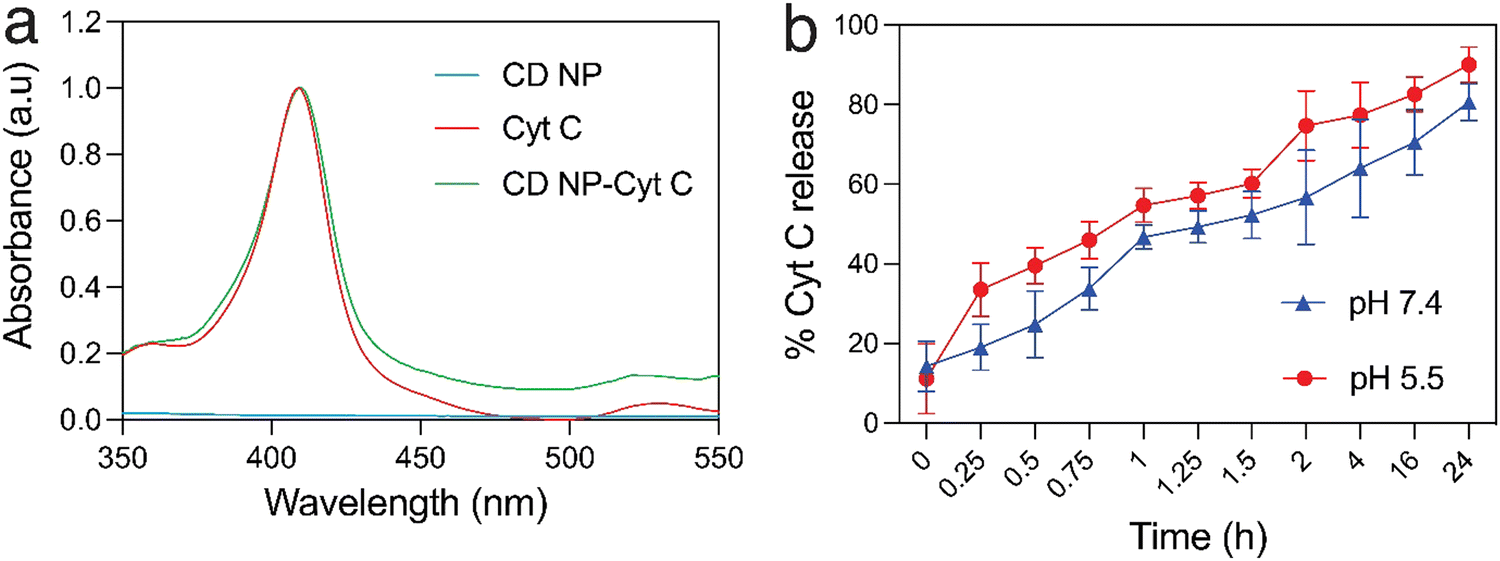 | ||
| Fig. 2 Evaluating the loading and release of cytochrome C in CD NPs: (a) UV-visible absorption spectra of Cyt C, CD NPs and Cyt C-loaded CD NPs. (b) pH-dependent Cyt C release from CD NPs at physiological pH (7.4) and acidic pH (5.5). | ||
Consequently, we investigated the release of Cyt C from the CD NPs through a dynamic dialysis technique using a dialysis membrane (MWCO: 100 kD). The membrane containing Cyt C-encapsulated CD NPs were suspended in buffers at two different pH, 7.4 and 5.5, which mimic the physiological pH and the acidic endosome pH, respectively.39 The amount of Cyt C released in the aqueous media was estimated by monitoring the absorbance of Cyt C at 409 nm through UV-vis spectroscopy (Fig. 2(b)). At both pH, there was an increase in the cumulative release of the drug with time. However, at acidic pH 5.5, the release of Cyt C was more with ∼40% of Cyt C released within 30 min, whereas only ∼25% was released within 30 min at pH of 7.4. Almost 90% of the protein was released within 24 h, at pH 5.5, whereas ∼75% of Cyt C was released at pH 7.4 in 24 h, which is a lot less than that released at the acidic pH. Since the encapsulation of Cyt C was carried out at a higher pH in order to achieve better encapsulation due to the charge reversal of Cyt C above its pI, when the NPs encounter a buffer with lower pH than its pI, the surface charge of the Cyt C changes, which leads to destabilization in the electrostatic interaction with the cationic dextrin polymer, leading to its release from the CD NPs into the aqueous medium. This justifies the higher release of Cyt C at pH 5.5 as compared to pH 7.4.
3.3. Structural and functional stability of cytochrome C before and after release
The structural and functional stability of the released Cyt C was investigated using circular dichroism and enzyme kinetics study. Fig. 3(a) shows the CD spectra of free Cyt C and that released from the CD NPs. The near-UV CD spectrum (190–320 nm) provides information on the tertiary structure of the protein. The spectrum of Cyt C in this range is mainly contributed by four Phe residues, four Tyr residues, one Trp residue, and two thioether bonds. There are two minima at 210 and 223 nm in the near-UV CD spectrum of native Cyt C, which correspond to Trp-59.1 The Cyt C released at pH 5.5 after 24 h also exhibited two minima at 210 and 223 nm, which clearly demonstrated that the structure of the Cyt C remained unchanged. This confirms the structural stability of the released Cyt C, which did not undergo any conformational change during the loading and the release process.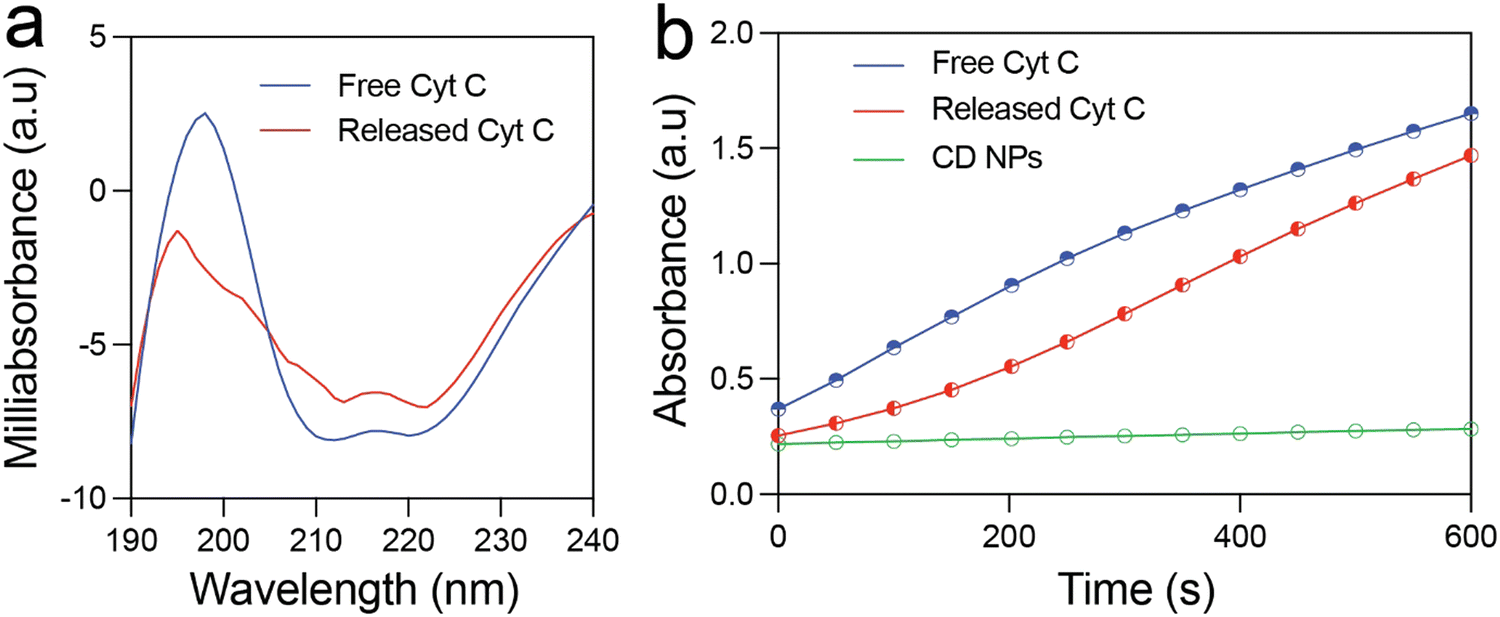 | ||
| Fig. 3 Characterization of the protein released from CD NPs. (a) Near-UV CD spectra of Cyt C released from CD NPs and free Cyt C at various pH conditions. (b) Enzyme kinetics study of native and released Cyt C for the oxidation of ABTS substrate. | ||
Further, it is prerequisite for a protein to remain functionally active after encapsulation and release from NPs for therapeutic applications. In order to investigate the functional activity of the released Cyt C, we estimated its enzymatic activity by means of ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) oxidation assay and compared it with that of free Cyt C. Fig. 3(b) illustrates the formation of oxidised ABTS with respect to time monitored using UV-vis spectroscopy. Free Cyt C clearly shows an increase in the product formation as indicated by the rise in absorbance at 430 nm. The released Cyt C also demonstrated a clear increase in absorbance at 430 nm with respect to time, clearly demonstrating its activity in the oxidation of ABTS. The CD NPs did not show any absorbance rise, demonstrating that they do not possess any enzymatic activity. These results clearly validated the functional activity of the released Cyt C.
3.4. Cellular internalization of FITC-conjugated CD NPs
Being positively charged, CD NPs are expected to accelerate the process of cellular entry. In the present study, the cellular uptake of CD NPs was evaluated using fluorescence microscopy. To execute the internalization process, CD NPs were conjugated with a fluorescent dye, FITC, to visualize the uptake of the NPs. HeLa cells were treated with 5 μg mL−1 of FITC-conjugated CD NPs for 6 h. Following incubation, the cells were stained with DAPI and observed under a fluorescence microscope. The control untreated cells showed no green fluorescence; however, blue DAPI-stained nuclei were clearly observed (Fig. 4). Conversely, the FITC-labelled CD NP-treated cells showed the presence of green fluorescence around the blue DAPI-stained nuclei, thus demonstrating the successful internalization of FITC-labelled CD NPs inside the cells.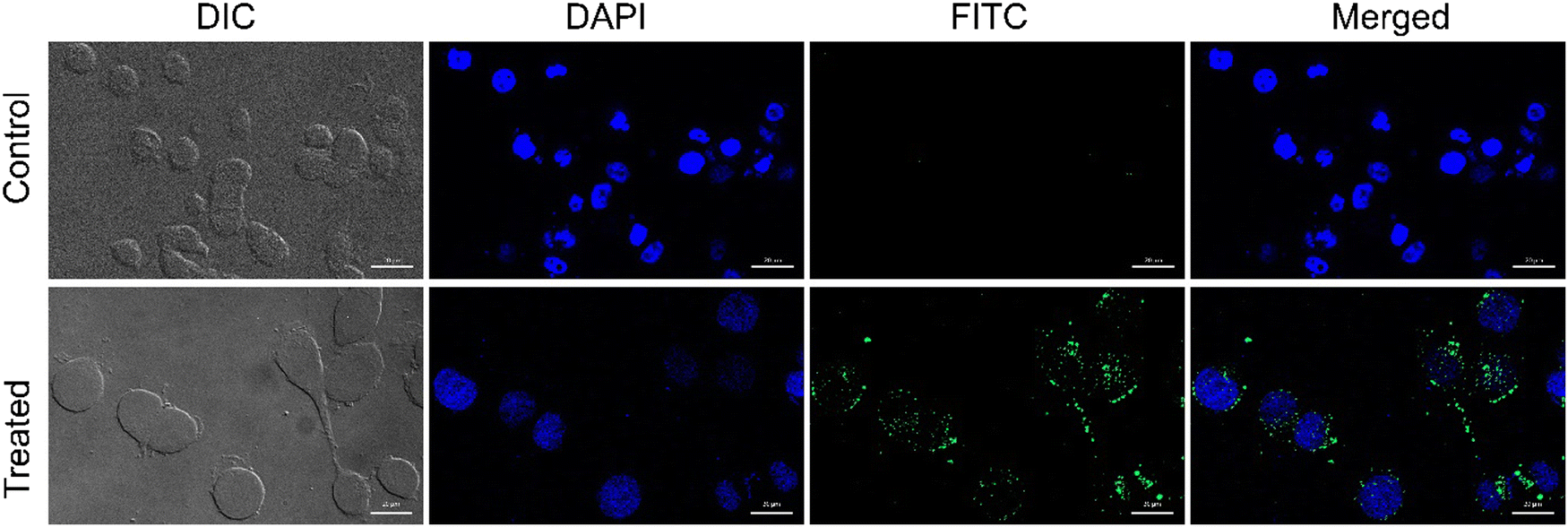 | ||
| Fig. 4 Determination of in vitro cellular internalization of FITC-conjugated CD NPs (5 μg ml−1) in HeLa cells at 6 h. Visualized under fluorescence microscope. The upper panel is untreated control cells and the lower panel shows cells 6 h post-treatment, where blue fluorescence corresponds to DAPI-stained nuclei and green fluorescence corresponds to FITC-conjugated CD NPs. Images were captured at 60× objective. Scale bar: 20 μm. | ||
3.5. In vitro cell viability assay
After confirming the functional and structural stability of the released Cyt C, we attempted to assess the effect of Cyt C in inducing cell death upon its delivery using CD NPs post-CQ treatment using XTT-based cell viability assay. Firstly, HeLa cells were treated with varying concentrations of only Cyt C (10–250 μg mL−1) for 24 h to understand if free Cyt C can be taken up by cells and induce cell death. The cell viability assay showed that there was no significant cell death (Fig. 5(a)) and the overall viability remained above 98%. This suggested that Cyt C was unable to show any toxic effect, which can be attributed to the impermeability cell membrane to the protein due to its macromolecular form. The cells treated with Cyt C-loaded CD NPs also did not show any significant toxicity when treated at concentrations of 1 and 1.5 mg mL−1 for 24 h. This was due to the endosomal entrapment of the protein Cyt C, which was not released into the cell cytoplasm and thus could not induce apoptosis. The Cyt C entrapped within the endosome is subsequently degraded upon maturation of the endosome to an endolysosome. In order to let the CD NPs-Cyt C release into the cell cytosol, the drug CQ was employed, which is known to act as an endosomal disruptor. Though CQ is a well-known antimalarial drug that has been in use for a long time, recently it has been the prime focus for increasing the process of ‘endosomal escape’ in cancer therapy.34 Chloroquine, being a very small molecule, can easily enter into cells and its basic nature also helps them to enter inside the endosome.40 Upon entering the acidic endosome, it will itself get protonated and sequester protons. H+ ions are thus recruited within the endosome through an ATPase proton channel situated at the periphery of the endosomal membrane. As a result, chloride ions will also accompany and accumulate within endosomes and together will lead to increased osmotic pressure. Subsequently, diffusion of water into the endosome will occur, which will lead to swelling and rupture of the endosomal membrane, followed by release of the entrapped materials into the cytosol. This whole process is termed the ‘proton sponge effect’.41,42 Hence, we investigated the viability of cells by co-delivering CQ (0.1 mM) along with Cyt C-encapsulated CD NPs to HeLa cells. A drastic reduction in the percentage of viable cells was observed for both the treated concentrations with 46% cell death at 1 mg mL−1 and 88% at 1.5 mg mL−1 which is evident from Fig. 5(b). Interestingly, cells treated with only CQ (0.1 mM) exhibited no cytotoxicity. Additionally, we investigated the viability of cells treated with Cyt C + CQ (0.1 mM) at various Cyt C concentrations, the range of which is equivalent to the loaded concentration within the CD NPs. 24 h post-treatment, cell viability remained above 94% (Fig. S3, ESI†), indicating that Cyt C + CQ was not able to cause cell death. Therefore, it can be established from the results that the CD NPs as a delivery vehicle aided Cyt C in cellular uptake, whereas CQ on the other hand enabled the Cyt C-CD NP construct to escape from the endosome into the cytosol without causing any cytotoxicity. Overall, the cell death upon treatment with CD NPs-Cyt C and CQ can be attributed to the action of released Cyt C.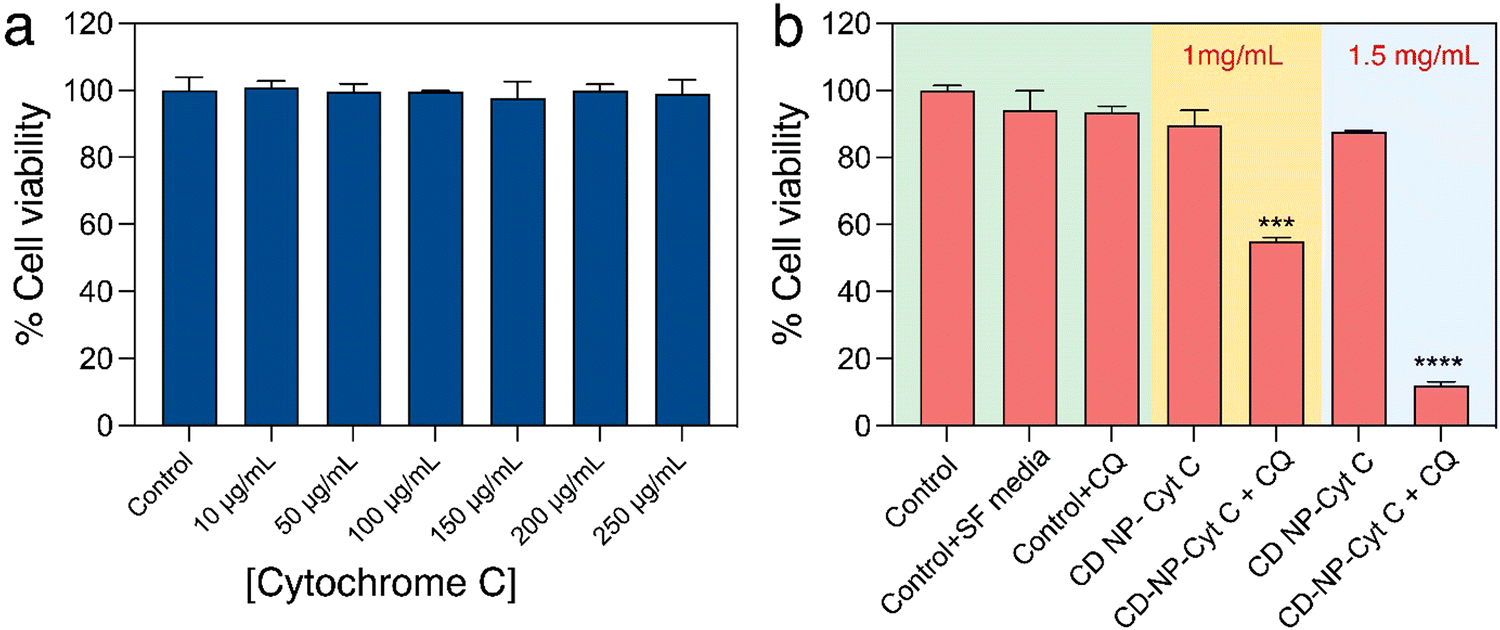 | ||
| Fig. 5 In vitro cell viability assay of HeLa cells treated with different concentration of (a) Cyt C and (b) Cyt C-loaded CD NPs with and without chloroquine. Data presented as mean ± SD (**p < 0.01, ***p < 0.001, and ****p < 0.0001). | ||
Besides HeLa cells, cell viability assay was also performed in A549 cells (lung carcinoma epithelial cells) to validate the strategy of cellular toxicity using CD NPs-Cyt C with and without the use of CQ. The results shown in Fig. S4 (ESI†) indicate ∼30% cell death after treating with 1 mg mL−1 of Cyt C-encapsulated CD NPs + CQ and ∼60% cell death at 1.5 mg mL−1 treated concentration, thus exhibiting the generality of the strategy where the cell death action is due to the released Cyt C along with CQ as an endosomal escape agent.
3.6. Studies on induction of apoptosis
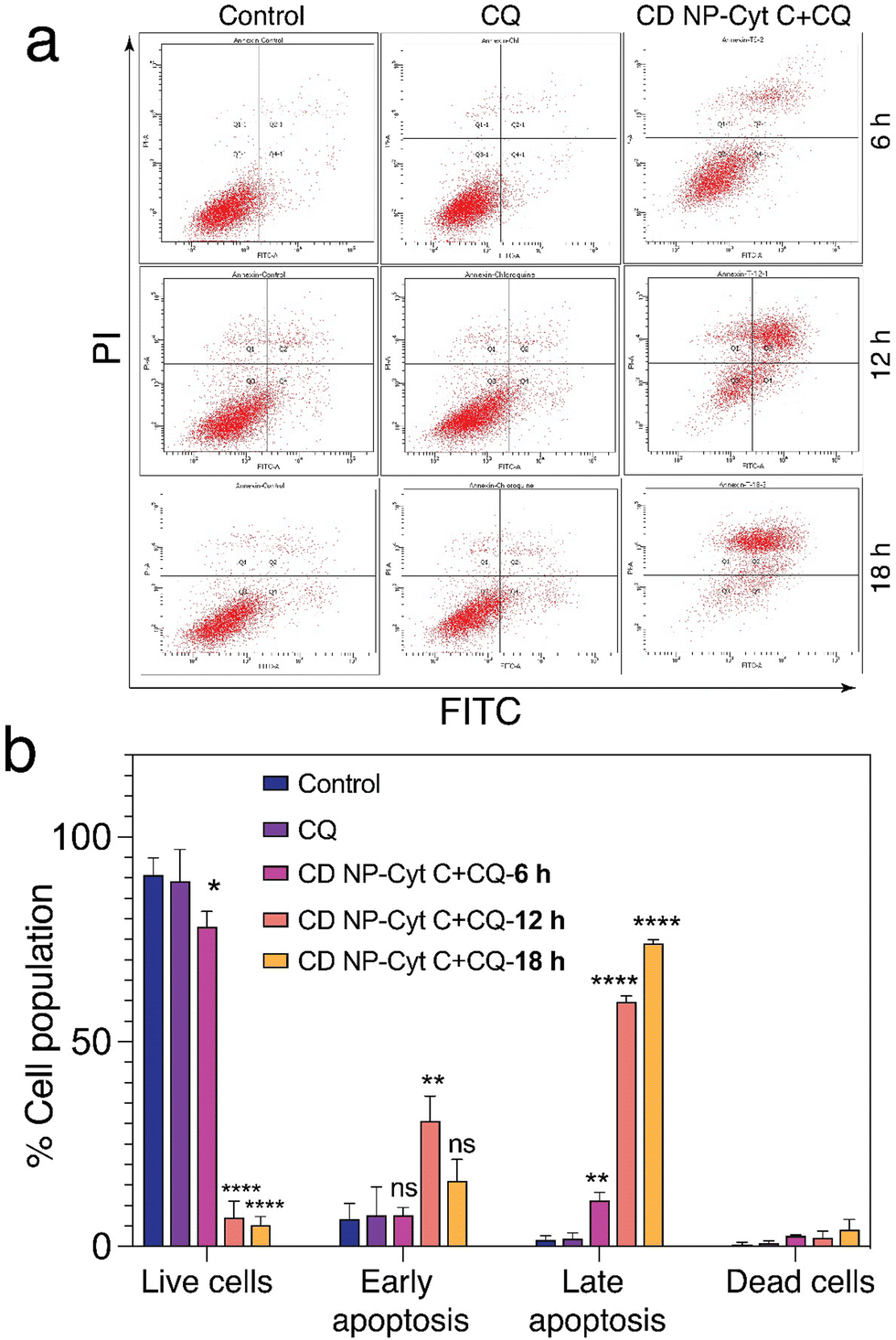 | ||
| Fig. 6 Evaluation of apoptotic events in HeLa cells. (a) Flow cytometric analysis of HeLa cells treated with 1.5 mg mL−1 Cyt C-loaded CD NPs in combination with chloroquine (0.1 mM) using annexin V/PI staining. (b) Graphical representation of the flow cytometry data. Data presented as mean ± SD (**p < 0.01, ***p < 0.001, and ****p < 0.0001). | ||
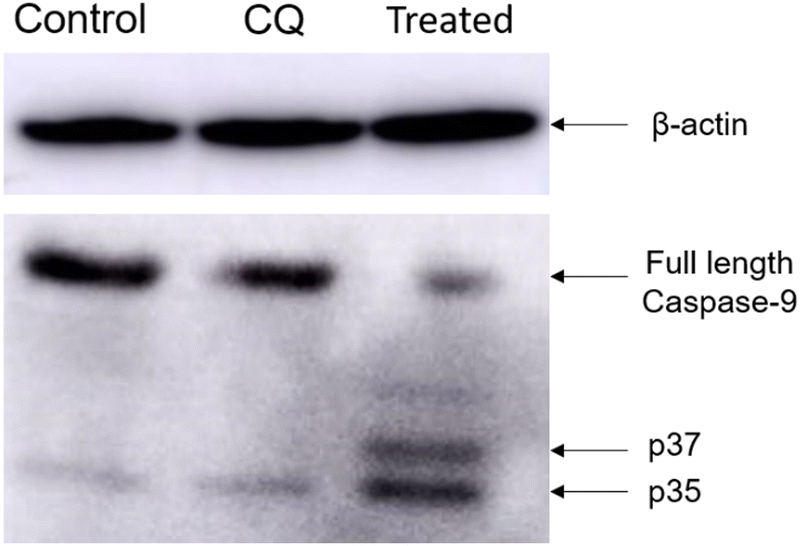 | ||
| Fig. 7 Evaluation of caspase-9 activation upon treatment with Cyt C-CD NPs in HeLa cells through western blot analysis. From left to right: control (without any treatment), CQ-treated, and treated with 1.5 mg mL−1 Cyt C-loaded CD NPs in combination with chloroquine (0.1 mM). | ||
Next, we examined the mitochondrial membrane potential (ΔΨM) at various time intervals post-treatment with CD NPs-Cyt C + CQ using the dye JC-1. JC-1 is a cationic lipophilic dye that emits green fluorescence in its monomeric form. JC-1 can selectively enter intact mitochondria and accumulate at a high concentration, leading to the formation of a reversible complex known as J-aggregates. In contrast with JC-1 monomeric form, J-aggregates exhibit red fluorescence. When there is a decrease in ΔΨM, the formation of J-aggregates will decrease and most of the dye will remain in its monomeric form, emitting green fluorescence. Thus, based on the permeation of the JC1 dye and its red/green fluorescence, the state of the mitochondrial membrane potential can be assessed. In our study, the untreated and CQ-treated HeLa cells exhibited more than 90% red fluorescence attributed to J-aggregates and a healthy condition of the cells as observed through flow cytometry analysis (Fig. 8(a) and (b)), whereas cells treated with Cyt C-encapsulated CD NPs in combination with CQ exhibited a decrease in the red fluorescence with time. The red fluorescence decreased from 80% to 70% to 50% after 6 h, 12 h and 18 h of treatment, respectively. Thus, a time-dependent decrease in percentage of red fluorescence and corresponding increase in green fluorescence suggested a time-dependent depolarization of ΔΨM. Overall, this confirms mitochondrial membrane depolarization when Cyt C is administered externally, leading to apoptosis.
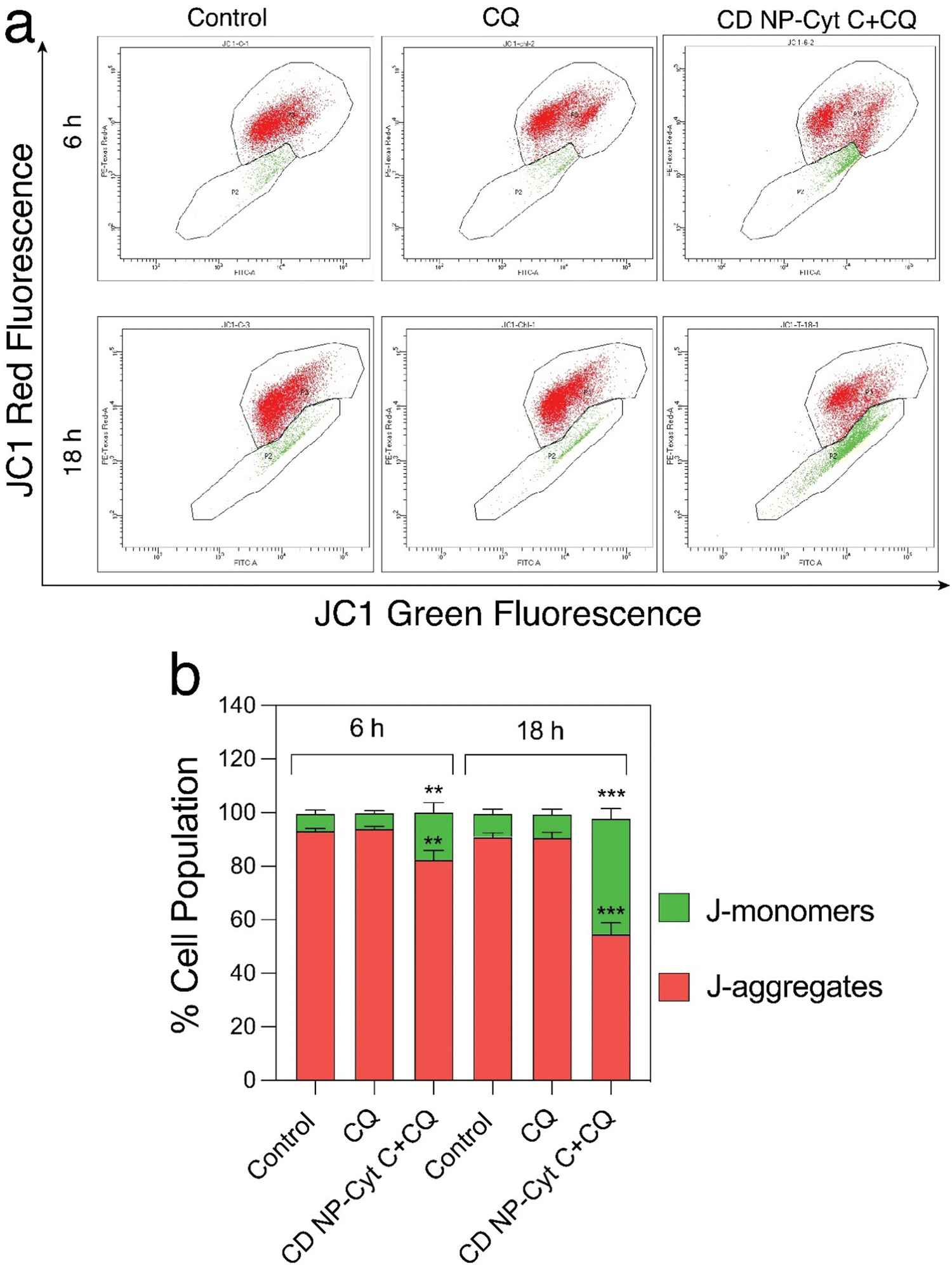 | ||
| Fig. 8 Mitochondrial depolarization assay. (a) Flow cytometric analysis of mitochondrial state of HeLa cells treated with 1.5 mg mL−1 Cyt C-loaded CD NPs in combination with chloroquine (0.1 mM) using JC-1 dye. The green colour represents the J-monomers and the red colour represents the J-aggregates. (b) Graphical representation of the flowcytometric data. Data presented as mean ± SD (**p < 0.01, ***p < 0.001, and ****p < 0.0001). | ||
Fluorescence microscopy for the same was performed and the extent of red and green fluorescence after CD NPs-Cyt C + CQ treatment was observed. The colocalised image of the control untreated cells, as depicted in Fig. 9, displays a yellowish colour that corresponds to the presence of both JC-1 monomers and a high concentration of J-aggregates, whereas in treated cells, a decrease in red fluorescence was observed with more green fluorescence, which corroborates the loss of ΔΨM. The overall findings suggest that depolarization of the mitochondrial membrane occurred upon treatment.
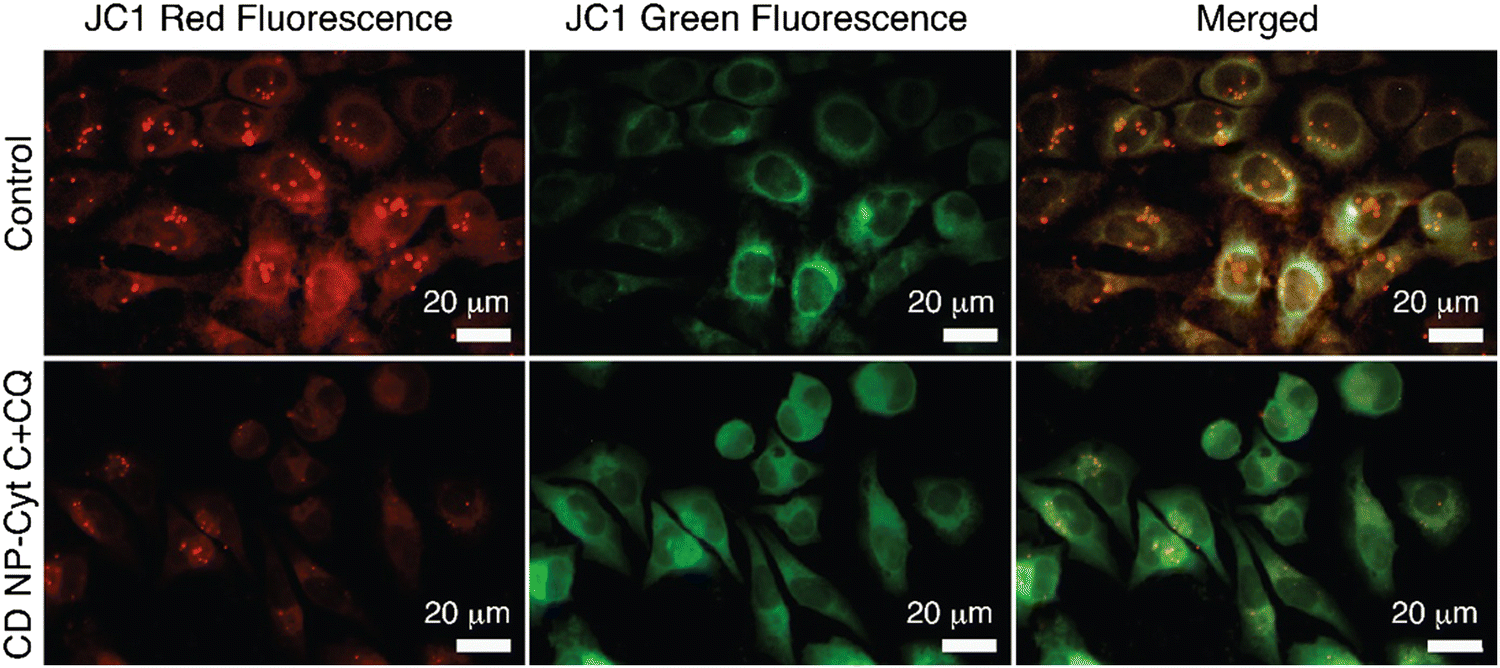 | ||
| Fig. 9 Confocal microscopic images for the visualization of mitochondrial membrane depolarization in HeLa cells after treatment with CD NPs-Cyt C + CQ using the dye JC-1. The top panel represents the control HeLa cells and the bottom panel represents the HeLa cells treated with 1.5 mg mL−1 Cyt C-loaded CD NPs in combination with chloroquine (0.1 mM). | ||
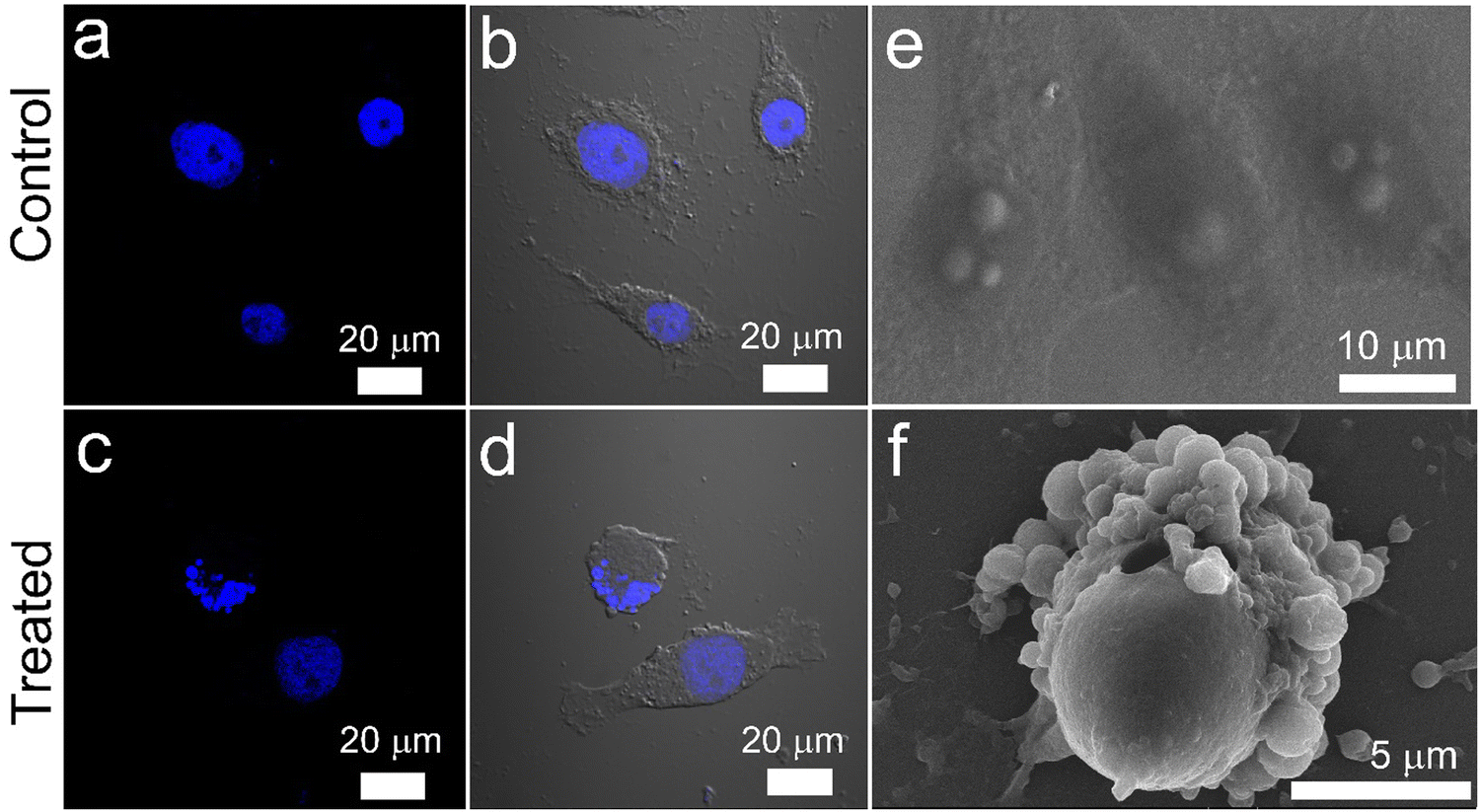 | ||
| Fig. 10 Nuclear fragmentation and membrane blebbing analysis. Confocal microscopic images showing nuclear fragmentation of HeLa cells stained with DAPI: (a) and (b) Untreated control cells and (c) and (d) cells treated with 1.5 mg mL−1 Cyt C-loaded CD NPs in combination with chloroquine (0.1 mM). Visualization of plasma membrane blebbing of HeLa cells showing apoptosis using SEM: (e) untreated control cells and (f) cells treated with 1.5 mg mL−1 Cyt C-loaded CD NPs in combination with chloroquine (0.1 mM). | ||
4. Conclusion
Ensuring compatibility between the engineered delivery system and the therapeutic cargo without affecting its activity and stability is a significant challenge in protein delivery. In this study, we developed a biocompatible and biodegradable NP construct using material obtained from natural sources. The CD NPs were synthesized using ionic gelation and encapsulated with the therapeutic protein Cyt C. Co-delivery of CQ, a drug that enhances endosomal release through the proton sponge effect, was used to overcome the challenge of inefficient release of the therapeutic cargo from endosomes. The bioactivity of the intracellular released Cyt C was maintained even in acidic pH. Our experimental findings suggest that externally administered Cyt C rapidly activates the caspase-9 protein and helps in the formation of pores in the mitochondria, leading to a decrease in mitochondrial membrane potential and ultimately inducing apoptosis. This approach offers a simple strategy for developing a delivery system for efficient encapsulation and direct delivery of therapeutic proteins to overcome the challenges posed by resistant cancer cells that inhibit the release of Cyt C upon stimulus.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the BioX centre and the Advanced Materials Research Centre (AMRC), Indian Institute of Technology Mandi for research and infrastructure facility. AS and SS acknowledge MoE and PMRF, respectively, for providing doctoral fellowships. The scheme and TOC images were prepared using Biorender.com and the authors acknowledge it.References
- J. Méndez, M. Morales Cruz, Y. Delgado, C. M. Figueroa, E. A. Orellano and M. Morales, et al., Delivery of chemically glycosylated cytochrome c immobilized in mesoporous silica nanoparticles induces apoptosis in HeLa cancer cells, Mol. Pharmaceutics, 2014, 11(1), 102–111 CrossRef PubMed.
- Radiation therapy side effects. NIH, Available from: https://www.cancer.gov/about-cancer/treatment/types/radiation-therapy/side-effects.
- G. Muthiah and A. Jaiswal, Can the Union of Prodrug Therapy and Nanomedicine Lead to Better Cancer Management?, Adv. NanoBiomed Res., 2022, 2(1), 2100074 CrossRef CAS.
- S. N. Bhatia, X. Chen, M. A. Dobrovolskaia and T. Lammers, Cancer nanomedicine, Nat. Rev. Cancer, 2022, 22(10), 550–556 CrossRef CAS PubMed.
- M. J. Nirmala, U. Kizhuveetil, A. Johnson, G. Balaji, R. Nagarajan and V. Muthuvijayan, Cancer nanomedicine: a review of nano-therapeutics and challenges ahead, RSC Adv, 2023, 13(13), 8606–8629 RSC.
- R. Khandelia, A. Jaiswal, S. S. Ghosh and A. Chattopadhyay, Gold nanoparticle–protein agglomerates as versatile nanocarriers for drug delivery, Small, 2013, 9(20), 3494–3505 CrossRef CAS PubMed.
- R. Khandelia, A. Jaiswal, S. S. Ghosh and A. Chattopadhyay, Polymer coated gold nanoparticle–protein agglomerates as nanocarriers for hydrophobic drug delivery, J. Mater. Chem. B, 2014, 2(38), 6472–6477 RSC.
- S. Roy, A. Sarkar and A. Jaiswal, Poly (allylamine hydrochloride)-functionalized reduced graphene oxide for synergistic chemo-photothermal therapy, Nanomedicine, 2019, 14(3), 255–274 CrossRef CAS PubMed.
- P. Singh, P. Haloi, K. Singh, S. Roy, A. Sarkar and R. Choudhary, et al., Palladium Nanocapsules for Photothermal Therapy in the Near-Infrared II Biological Window, ACS Appl. Mater. Interfaces, 2023, 15(33), 39081–39098 CrossRef CAS PubMed.
- M. Majidinia, M. Mirza-Aghazadeh-Attari, M. Rahimi, A. Mihanfar, A. Karimian and A. Safa, et al., Overcoming multidrug resistance in cancer: Recent progress in nanotechnology and new horizons. IUBMB, Life, 2020, 72(5), 855–871 CAS.
- S. Li, J. Zhang, C. Deng, F. Meng, L. Yu and Z. Zhong, et al., Redox-sensitive and intrinsically fluorescent photoclick hyaluronic acid nanogels for traceable and targeted delivery of cytochrome c to breast tumor in mice, ACS Appl. Mater. Interfaces, 2016, 8(33), 21155–21162 CrossRef CAS PubMed.
- R. Baskar, K. A. Lee, R. Yeo and K.-W. Yeoh, Cancer and radiation therapy: current advances and future directions, Int. J. Med. Sci., 2012, 9(3), 193 CrossRef PubMed.
- C. Xu, C. Lei and C. Yu, Mesoporous silica nanoparticles for protein protection and delivery, Front. Chem., 2019, 7, 290 CrossRef CAS PubMed.
- X. S. Sun, M.-S. Jang, Y. Fu, J. H. Lee, D. S. Lee and Y. Li, et al., Intracellular delivery of cytochrome C using hypoxia-responsive polypeptide micelles for efficient cancer therapy, Mater. Sci. Eng., C, 2020, 114, 111069 CrossRef CAS PubMed.
- L. J. Delinois, O. De León-Vélez, A. Vázquez-Medina, A. Vélez-Cabrera, A. Marrero-Sánchez and C. Nieves-Escobar, et al., Cytochrome c: Using Biological Insight toward Engineering an Optimized Anticancer Biodrug, Inorganics, 2021, 9(11), 83 CrossRef CAS PubMed.
- H. Chang, J. Lv, X. Gao, X. Wang, H. Wang and H. Chen, et al., Rational design of a polymer with robust efficacy for intracellular protein and peptide delivery, Nano. Lett., 2017, 17(3), 1678–1684 CrossRef CAS PubMed.
- S. K. Kim, M. B. Foote and L. Huang, The targeted intracellular delivery of cytochrome C protein to tumors using lipid-apolipoprotein nanoparticles, Biomaterials, 2012, 33(15), 3959–3966 CrossRef CAS PubMed.
- S. Hong, D. W. Choi, H. N. Kim, C. G. Park, W. Lee and H. H. Park, Protein-based nanoparticles as drug delivery systems, Pharmaceutics, 2020, 12(7), 604 CrossRef CAS PubMed.
- F. Moncalvo, M. I. Martinez Espinoza and F. Cellesi, Nanosized delivery systems for therapeutic proteins: Clinically validated technologies and advanced development strategies, Front. Bioeng. Biotechnol., 2020, 8, 89 CrossRef PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2021, 20(2), 101–124 CrossRef CAS PubMed.
- M. Morales-Cruz, A. Cruz-Montañez, C. M. Figueroa, T. González-Robles, J. Davila and M. Inyushin, et al., Combining stimulus-triggered release and active targeting strategies improves cytotoxicity of cytochrome c nanoparticles in tumor cells, Mol. Pharmaceutics, 2016, 13(8), 2844–2854 CrossRef CAS PubMed.
- L. Liguori, B. Marques, A. Villegas-Mendez, R. Rothe and J.-L. Lenormand, Liposomes-mediated delivery of pro-apoptotic therapeutic membrane proteins, J. Controlled Release, 2008, 126(3), 217–227 CrossRef CAS PubMed.
- I. Dominguez-Martinez, F. Joaquin-Ovalle, Y. Ferrer-Acosta and K. H. Griebenow, Folate-decorated cross-linked cytochrome c nanoparticles for active targeting of non-small cell lung carcinoma (NSCLC), Pharmaceutics, 2022, 14(3), 490 CrossRef CAS PubMed.
- E. Choi, D.-K. Lim and S. Kim, Hydrolytic surface erosion of mesoporous silica nanoparticles for efficient intracellular delivery of cytochrome c, J. Colloid Interface Sci., 2020, 560, 416–425 CrossRef CAS PubMed.
- B. Zhang, Z. Luo, J. Liu, X. Ding, J. Li and K. Cai, Cytochrome c end-capped mesoporous silica nanoparticles as redox-responsive drug delivery vehicles for liver tumor-targeted triplex therapy in vitro and in vivo, J. Controlled Release, 2014, 192, 192–201 CrossRef CAS PubMed.
- C. Guo, Y. Zhang, Y. Li, L. Zhang, H. Jiang and J. Tao, et al., Gold nanoparticle-guarded large-pore mesoporous silica nanocomposites for delivery and controlled release of cytochrome c, J. Colloid Interface Sci., 2021, 589, 34–44 CrossRef CAS PubMed.
- D. Hreczuk-Hirst, D. Chicco, L. German and R. Duncan, Dextrins as potential carriers for drug targeting: tailored rates of dextrin degradation by introduction of pendant groups, Int. J. Pharm., 2001, 230(1–2), 57–66 CrossRef CAS PubMed.
- A. Jaiswal, A. Chattopadhyay and S. S. Ghosh, Functional chitosan nanocarriers for potential applications in gene therapy, Mater. Lett., 2012, 68, 261–264 CrossRef CAS.
- Y. Han, J. Zhou, Y. Hu, Z. Lin, Y. Ma and J. J. Richardson, et al., Polyphenol-based nanoparticles for intracellular protein delivery via competing supramolecular interactions, ACS Nano, 2020, 14(10), 12972–12981 CrossRef CAS PubMed.
- D. M. Silva, C. Nunes, I. Pereira, A. S. Moreira, M. R. M. Domingues and M. A. Coimbra, et al., Structural analysis of dextrins and characterization of dextrin-based biomedical hydrogels, Carbohydr. Polym., 2014, 114, 458–466 CrossRef CAS PubMed.
- A. Sarkar, S. Roy, P. Bhatia and A. Jaiswal, Quaternary ammonium substituted dextrin-based biocompatible cationic nanoparticles with ultrahigh pH stability for drug delivery, J. Appl. Polym. Sci, 2023, 140(11), e53626 CrossRef CAS.
- S. Santra, C. Kaittanis and J. M. Perez, Cytochrome C encapsulating theranostic nanoparticles: a novel bifunctional system for targeted delivery of therapeutic membrane-impermeable proteins to tumors and imaging of cancer therapy, Mol. Pharmaceutics, 2010, 7(4), 1209–1222 CrossRef CAS PubMed.
- M. Saxena, Y. Delgado, R. K. Sharma, S. Sharma, S. L. P. D. L. Guzmán and A. D. Tinoco, et al., Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate, PLoS One, 2018, 13(4), e0195542 CrossRef PubMed.
- M. Hajimolaali, H. Mohammadian, A. Torabi, A. Shirini, M. Khalife Shal and H. B. Nezhad, et al., Application of chloroquine as an endosomal escape enhancing agent: new frontiers for an old drug, Expert Opin. Drug Delivery, 2021, 18(7), 877–889 CrossRef CAS PubMed.
- S. Roy, M. Kumari, P. Haloi, S. Chawla, V. B. Konkimalla and A. Kumar, et al., Quaternary ammonium substituted pullulan accelerates wound healing and disinfects Staphylococcus aureus infected wounds in mouse through an atypical ‘non-pore forming’pathway of bacterial membrane disruption, Biomater. Sci., 2022, 10(2), 581–601 RSC.
- S. Roy, P. Haloi, R. Choudhary, S. Chawla, M. Kumari and V. B. Konkimalla, et al., Quaternary Pullulan-Functionalized 2D MoS2 Glycosheets: A Potent Bactericidal Nanoplatform for Efficient Wound Disinfection and Healing, ACS. Appl. Mater. Interfaces., 2023, 15(20), 24209–24227, DOI:10.1021/acsami.3c04390.
- I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins, J. Am. Chem. Soc., 2007, 129(28), 8845–8849 CrossRef CAS PubMed.
- A. Macone, S. Masciarelli, F. Palombarini, D. Quaglio, A. Boffi and M. C. Trabuco, et al., Ferritin nanovehicle for targeted delivery of cytochrome C to cancer cells, Sci. Rep., 2019, 9(1), 1–7 CrossRef CAS PubMed.
- A. Sarkar, S. Roy, P. Sanpui and A. Jaiswal, Plasmonic Gold Nanorattle Impregnated Chitosan Nanocarrier for Stimulus Responsive Theranostics, ACS Appl. Bio Mater., 2019, 2(11), 4812–4825 CrossRef CAS PubMed.
- M. A. A. Al-Bari, Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases, Pharmacol. Res. Perspect., 2017, 5(1), e00293 CrossRef PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, Endosomal escape pathways for delivery of biologicals, J. Controlled Release, 2011, 151(3), 220–228 CrossRef CAS PubMed.
- B. Zhang and S. Mallapragada, The mechanism of selective transfection mediated by pentablock copolymers; Part II: Nuclear entry and endosomal escape, Acta Biomater., 2011, 7(4), 1580–1587 CrossRef CAS PubMed.
- M. Van Engeland, L. J. Nieland, F. C. Ramaekers, B. Schutte and C. P. Reutelingsperger, Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure, Cytometry, 1998, 31(1), 1–9 CrossRef CAS PubMed.
- C. Riccardi and I. Nicoletti, Analysis of apoptosis by propidium iodide staining and flow cytometry, Nat. Protoc., 2006, 1(3), 1458–1461 CrossRef CAS PubMed.
- C. Garrido, L. Galluzzi, M. Brunet, P. Puig, C. Didelot and G. Kroemer, et al., Mechanisms of cytochrome c release from mitochondria, Cell Death Differ., 2006, 13(9), 1423–1433 CrossRef CAS PubMed.
- I. Marzo, C. Brenner, N. Zamzami, S. A. Susin, G. Beutner and D. Brdiczka, et al., The permeability transition pore complex: a target for apoptosis regulation by caspases and Bcl-2–related proteins, J. Exp. Med., 1998, 187(8), 1261–1271 CrossRef CAS PubMed.
- T. Xia, C. Jiang, L. Li, C. Wu, Q. Chen and S.-S. Liu, A study on permeability transition pore opening and cytochrome c release from mitochondria, induced by caspase-3 in vitro, FEBS Lett., 2002, 510(1–2), 62–66 CrossRef CAS PubMed.
- V. Barcelo-Bovea, I. Dominguez-Martinez, F. Joaquin-Ovalle, L. A. Amador, E. Castro-Rivera and K. Medina-Álvarez, et al., Optimization and characterization of protein nanoparticles for the targeted and smart delivery of cytochrome c to non-small cell lung carcinoma, Cancers, 2020, 12(5), 1215 CrossRef CAS PubMed.
- L. J. Pagliari, M. J. Pinkoski and D. R. Green, Apoptosis signaling: a means to an end, Handbook of Cell Signaling, Elsevier, 2010, pp. 2535–2543 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NTA analysis of CD NPs, standard curve plot of Cyt C and cell viability plot of Cyt C + chloroquine in HeLa cells and in vitro cell viability assay of A549 cells treated with different concentration of Cyt C-loaded CD NPs with and without chloroquine. See DOI: https://doi.org/10.1039/d3cb00090g |
| This journal is © The Royal Society of Chemistry 2024 |