
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies in photochemical alcohol oxidation on noble-metal free nanomaterials as heterogeneous catalysts
Siqi
Li
,
Katherine H.
Morrissey
and
Bart M.
Bartlett
 *
*
Department of Chemistry, University of Michigan, 930 North University Avenue, Ann Arbor, Michigan, USA. E-mail: bartmb@umich.edu
First published on 16th August 2024
Abstract
Using light absorbing semiconductors to harvest solar energy has long been considered as a promising approach for driving chemical reactions. Photochemical oxidation of alcohols catalyzed by noble-metal free nanomaterials is particularly attractive due to the bio-derivable nature of the substrate and the wide availability of such catalysts. Here, a number of strategies that could facilitate the progress are discussed, where it is concluded that the photochemical alcohol oxidation process can benefit from both mechanistic optimization of the electron-transfer steps and structural engineering of the light-absorbing nanomaterials.
1. Introduction
Sunlight is the most abundant energy source within human grasp as fossil fuels reach their final century of projected storage.1,2 The direct conversion of sunlight to value-added products is ideal due to the renewable nature of solar energy. Among the approaches to harvesting solar irradiation, direct photochemical conversion chemistry stands out due to its simple device setup. Theoretically, solar-driven reactions catalysed by semiconductor photocatalysts (either as suspended particles or deposited as electrodes) can achieve both oxidation and reduction of selected redox couples present in the solution. Absorption of a photon with energy (hν) greater than the bandgap energy (Eg) of the semiconductor excites an electron from the valence band to the conduction band. The positively charged hole left behind in the valence band can oxidize a redox couple in solution with a less positive potential. Here, the oxidizing hole abstracts an electron from the target substrate, forming an oxidized substrate radical intermediate. Subsequently, this intermediate converts to product in a series of chemical steps. Accordingly, this process is hampered by both thermodynamic and kinetic barriers, which then requires the holes to be at slightly more positive potential to drive the forward reaction. Similarly, the electrons in the conduction band perform reduction on a redox couple with a less negative potential. In short, Eg determines the wavelength of incident light that can be absorbed, while the band edge positions of the semiconductor determine what redox reactions may occur thermodynamically. Note that for the semiconductor material to act as a catalyst, both redox half reactions must occur simultaneously and at same rate in order to maintain electroneutrality.Semiconductor photocatalysis research has traditionally focused on artificial photosynthesis (water oxidation and CO2 reduction) or water splitting (oxygen- and hydrogen-evolution reactions).3,4 However, recently it is recognized that alcohol oxidation is another promising half-reaction to couple with hydrogen evolution.5 In fact, alcohol oxidation may serve as a better redox partner for the hydrogen-evolution reaction due to faster kinetics than the oxygen evolution reaction. Moreover, industrially relevant commodity chemicals can be obtained through the 2-electron oxidation of primary alcohols to aldehydes or 4-electron oxidation to carboxylic acids. A number of alcohols are derivable from biomass feedstocks originated in agriculture practices, including 5-hydroxymethylfurfural, ethanol, 1-butanol, glycerol and others.6 Rather than using petroleum-based precursors, megatons of biomass can be obtained worldwide as building blocks for chemical synthesis. Among the options to obtain bio-derivable alcohols, biorefinery of ethanol has been industrialized with an annual production of >1015 gallons.7 Compared to the water oxidation reaction, the alcohol oxidation reaction (AOR) requires more precise mechanistic control to direct the selectivity towards a specific oxidation product due to multiple possible products. This presents another crucial challenge for implementing AOR catalysts. To use solar irradiation effectively, a considerable amount of exposed area is desired. This requires a large amount of material, as one would expect similarly to the silicon solar cell panels or other photovoltaic devices. This overarching condition determines that for large scale industrial chemical production, the price and abundance of the photocatalyst must be a critical factor. As such, materials with non-noble metal or metal-free elemental composition would be more practical and favourable. Thus, this review focuses on strategies that could be carried out to enable and optimize facile AORs, specifically on noble-metal-free semiconductor photocatalysts.
2. Noble-metal-free semiconductor nanoscale photocatalysts
The practise of semiconductor based photocatalysis on alcohol oxidation can be tracked back to last century, where anatase TiO2 and ZnO white pigments are applied as heterogeneous catalysts for oxidation reactions.8 These materials possess excellent chemical stability and simple preparation approaches, however their light absorbing capacity remains a bottleneck issue. The optical bandgaps of anatase TiO2 and wurtzite ZnO are 3.0 eV and 3.2 eV respectively, meaning that only UV photons may initiate carrier separation, resulting in poor use of the solar spectrum.9Fig. 1 shows the electronic structures of materials highlighted in this review. The dashed lines show the potentials for the water-splitting half reactions, as hydrogen evolution remains a highly sought criterion for solar energy conversion.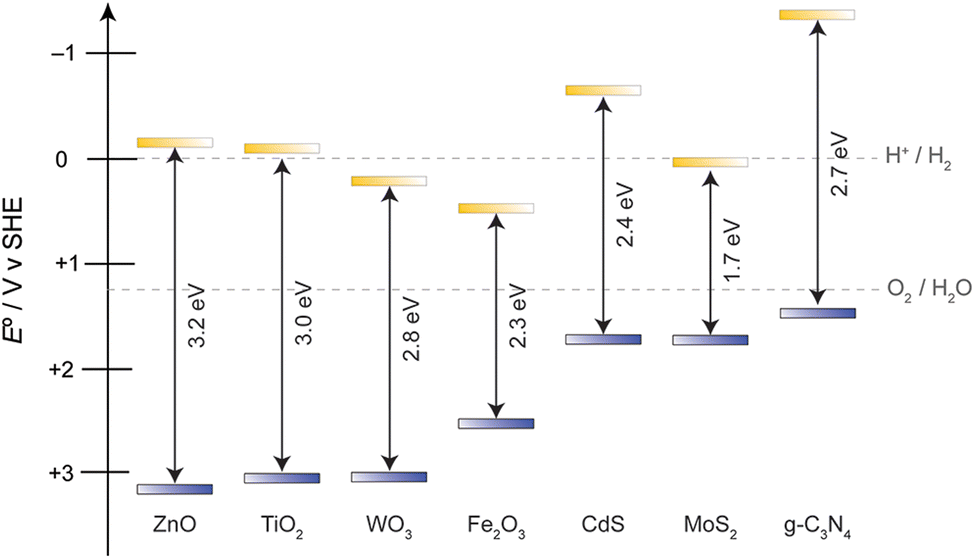 | ||
| Fig. 1 Band edge positions of noble-metal free photocatalysts highlighted in this review. | ||
Apart from oxides, metal chalcogenides (sulfides and selenides) are also heavily studied. In particular, CdS and MoS2 have shown promise as photocatalysts for the AOR.10,11 For these materials, the valence band is primarily composed of S(3p) or Se(4p) atomic orbitals, resulting in less positive valence band edges (following periodic trends in electronegativity) and thus smaller Eg compared to their oxide counterparts.12 This electronic structure makes metal chalcogenides attractive due to their more controllable oxidation capability as well as having better spectral overlap in the visible region. While the electronic structure is appealing, the chalcogenides do suffer from poor chemical stability in strong acids or highly oxidizing environments. Thus, when using chalcogenide AOR photocatalysts, the reaction conditions must be considered. The two common corrosion processes can proceed either through the presence of a strong Brønsted acid, which results in of H2S formation, or through the oxidation of S2− to SO42−, which is thermodynamically more stable when there is oxygen present (the SO42−/S2− couple is +0.3 V vs. SHE, much less positive than that of the 1.23 V O2/H2O redox couple),13 and has been confirmed by XPS analysis of metal sulfides after photocatalytic reactions.14 Consequently, when sulfide materials are employed in alcohol oxidation reactions, a sacrificial oxidant is usually desirable to avoid intermediates formed from the competing oxygen-reduction reaction.
Carbon based metal-free heterogeneous catalysts have attracted attention recent years because they possess excellent chemical stability, control of structure and composition, and have low environmental impact/toxicity compared to metal chalcogenides.15 Graphitic carbon nitride (g-C3N4) is a material having a planar structure in which sp2 hybridized C and N atoms form conjugated aromatic rings. Two major constructive units are usually observed for the overall bulk material, in s-triazine (one 6-membered ring) and tri-s-triazine (three 6-membered rings) with nitrogen atoms serving as the bridging anchor.16 This material stands out due to its desirable band positions, appropriate band gap for visible light absorption, facile synthesis, high chemical stability, and versatile use, which make it a key catalyst for targeting photochemical heterogeneous alcohol oxidation. Not only does its relatively negative valence band edge give rise to a milder oxidant (avoiding forming ˙OH, which is a very indiscriminate oxidant) for more controllable oxidation reactions, it also has a very negative conduction band edge, allowing more possible reduction reactions to couple with the oxidation. Moreover, bulk g-C3N4 is commonly prepared by a thermal polycondensation reaction, specifically through calcining C- and N-containing organic molecules.17 Consequently, it is malleable and can be developed into other morphologies and easily forms heterojunctions that provide better charge separation or absorption features when combined with other semiconductor photocatalysts.18
3. Direct and indirect mechanisms for photocatalytic alcohol oxidation
Heterogeneous oxidation of alcohols, or any substrates of interest, on semiconductors requires that the alcohol diffuses towards the photocatalyst and adsorbs onto the surface, and that valence-band hole transfer from the excited state of the photocatalyst occurs before recombination or other loss mechanisms. During the time that the adsorption persists, at least two direct oxidation mechanisms are supported. The first involves radical formation on the α carbon following the adsorption of benzyl alcohol on CdS and electron transfer. Subsequent O–H bond cleavage forms the aldehyde product.19 A second identified mechanism shows dissociative adsorption of the alcohol on TiO2, where it adsorbs as RO− and H+ separately, before the α-H cleavage proceeds. This mechanism is more prevalent for alcohols with more acidic protons on the hydroxyl groups, resulting in molecular hydrogen as the reduction product.20Direct alcohol oxidation by both mechanisms mentioned above has seen success on multiple materials in glycerol photooxidation on Au or bismuth oxyhalides,21–23 as well as ethanol on g-C3N4 or benzyl alcohols on Au–TiO2.24,25 However, many current examples require noble-metal based catalysts for plasmonic enhancement or for specific adsorption toward the alcohol substrate. Or they require the alcohol substrate to be easily oxidized to impart selectivity. For example, acetaldehyde, the two-electron oxidation product from ethanol, is easier to oxidize thermodynamically than ethanol itself. In cases like this, selectivity for aldehydes over acids or other further oxidized products can be a challenge.
The reaction yield and selectivity of such adsorption-based reactions are dictated by carrier recombination/trapping, adsorption/desorption kinetics, and parasitic reactions. Unwanted electron–hole recombination could proceed on trap states originating from bulk lattice defects, dopants, and surface defects. Side reactions are a leading cause of the chemical instability of the semiconductor itself during photocatalysis due to highly reactive charge carriers reacting with the solvent or oxygen at the semiconductor interface.26,27 Finally, competing chemical reactions with intermediates can proceed with the photogenerated charge carriers and form byproducts, leading to lower yield/rate of the targeted product.
As described earlier, semiconductor photocatalysts usually have fixed band positions that determines the thermodynamic potential of the electrons and holes. In aqueous conditions and/or aerobic conditions, forming active oxygen species as intermediates is likely and has been confirmed to contribute to alcohol oxidation as chemical oxidants alongside the photogenerated holes.28Fig. 2 shows the relevant standard reduction potentials of methanol oxidation with all the possible active oxygen species formed when a TiO2 photocatalyst is excited by incident light in water. While all these redox reactions are thermodynamically viable, kinetics determine the rate at which any particular reaction proceeds. Understanding the reactivity of these active oxygen species is crucial for achieving selective reactions. For example, isopropanol oxidation on anatase TiO2 is accompanied by O2 reduction to superoxide (O2−˙). This active oxygen radical intermediate ruins the selectivity.29 In another report using TiO2 as photocatalyst for aqueous ethanol oxidation, a mixture ranging from acetaldehyde, acetic acid, CO2 and even C–C bond cleavage products (formic acid, formates) are found.30 This serves as an example regarding the uncontrollable reactions that are possible because of the intrinsic band positions and their corresponding reaction intermediates. Thus, it is beneficial to maintain a sufficient quantity of adsorbed at the surface to ensure that photogenerated carriers are consumed fast enough to avoid parasitic side reactions.
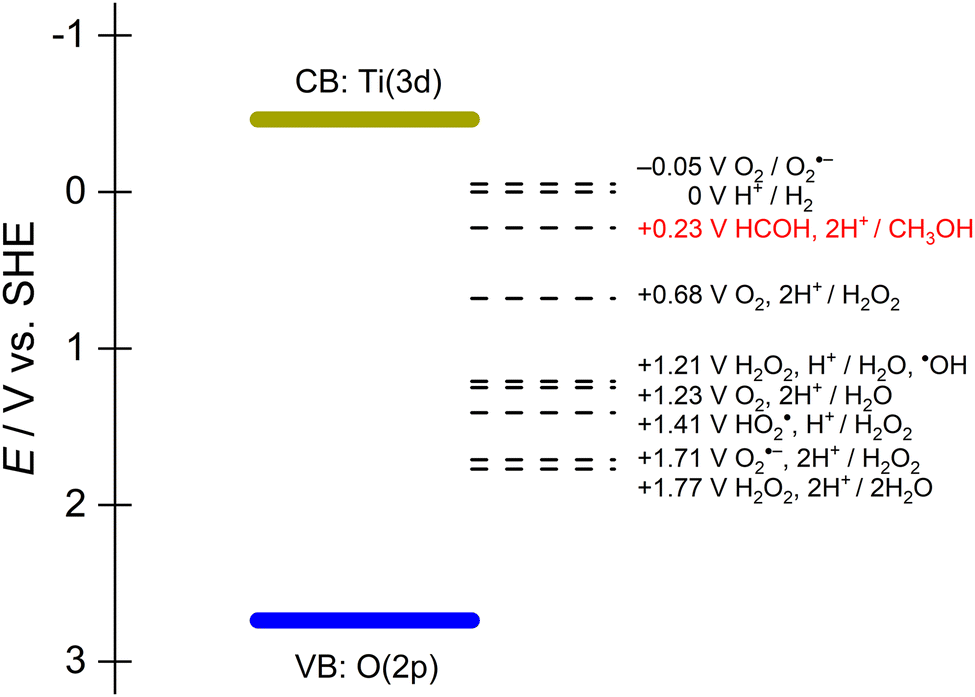 | ||
| Fig. 2 Standard potentials for methaol/formaldehyde, hydrogen evolution, and redox couples involving active oxygen species in water superimposed on the band edges of TiO2. | ||
One approach to avoid forming such high-energy intermediates is to use redox mediators to carry out the AOR indirectly and homogeneously. Here, the alcohol substrate does not adsorb to catalyst. Instead, redox mediator oxidation dominates the surface chemistry due to its fast kinetics and/or higher concentrations. Subsequently, the oxidized form of the mediator oxidizes the alcohol in solution, regenerating the reduced form of the mediator. Additionally, mediators alleviate the possible passivation of photocatalysts by bypassing reaction intermediates of the otherwise direct oxidation. The criteria for choosing redox mediators for alcohol oxidation are: (1) the mediator provides decent selectivity towards the desired product;31 (2) the mediator is inexpensive and readily available; and (3) most importantly, the mediator possesses fast kinetics for its oxidation on the photocatalyst, as well as fast kinetics for oxidizing the alcohol.32
With those considerations in mind, several categories of mediators have been employed towards alcohol oxidation reactions on semiconductor photocatalysts. Aside from molecular mediators which mostly contain noble-metal complexes, we focus on other homogeneous mediators that are either organic redox couples or anionic species as they exhibit better stability and less toxicity.
TEMPO-mediated photocatalytic alcohol oxidation on semiconductors
2,2,6,6-Tetramethylpiperidine N-oxide (TEMPO) stands as one of the most widely explored redox mediators for alcohol oxidation. TEMPO is a stable radical under ambient conditions, allowing it to be commercially stored and purchased. A scheme of redox conversions among TEMPO-related species is illustrated in Fig. 3. TEMPO can be oxidized to its oxoammonium form, TEMPO+, which is a strong oxidant that may extract 2 electrons and 1 H atom from the substrate to form a reduced hydroxide species, TEMPOH.33 Importantly, TEMPO possesses a stable backbone structure, reducing the parasitic reactions that could result from the unwanted functionalization of other functional groups. Cyclic voltammetry reveals that the standard reduction potential of the TEMPO+/TEMPOH redox couple is +0.88 V vs. SHE,34 conveniently positioned at less positive potentials than the valence band position of most semiconductor photocatalysts. Therefore, it can readily extract holes from the surface of semiconductors in a thermodynamically favored step. Potentiometry measurements also reveal that the TEMPO redox couple is nearly electrochemically reversible, indicating fast charge-transfer kinetics for regeneration from the oxidized oxoammonium form that reacts with alcohol.34 A well-recognized mechanism of TEMPO-mediated alcohol oxidation is presented in Fig. 4.35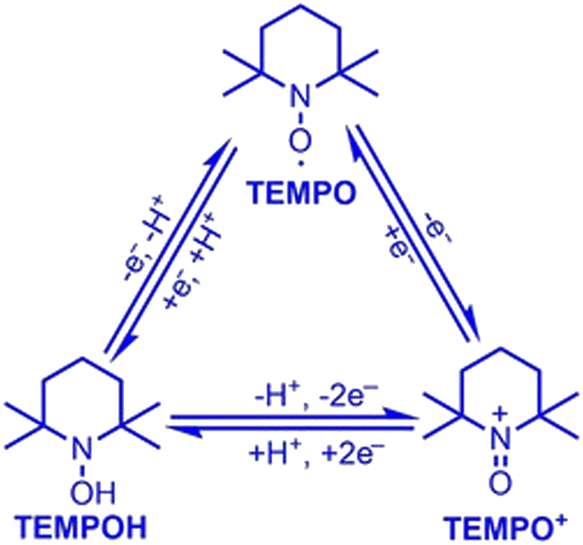 | ||
| Fig. 3 Electron and proton transfer of TEMPO–TEMPO+–TEMPOH conversion. Copyright: X. Lang and J. Zhao, Chem. – Asian J., 2018, 13, 599–613. | ||
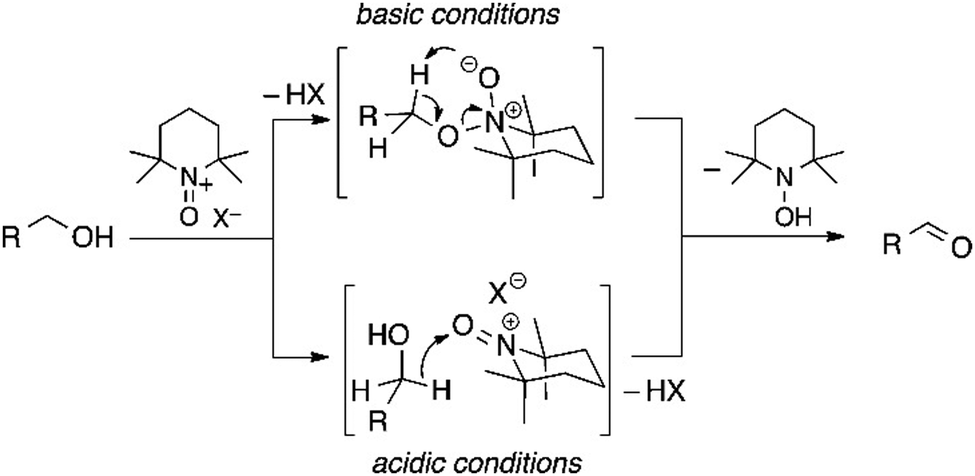 | ||
| Fig. 4 Projected alcohol oxidation driven by TEMPO+/TEMPOH couple in different conditions towards aldehyde. Copyright: M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757. | ||
This TEMPO-mediated mechanism has seen multiple applications when coupled with inorganic nanomaterials. A dye sensitized ZnO system for benzyl alcohol oxidation to benzaldehyde was developed with a reported 82% conversion with the presence of TEMPO. Notably, the conversion is only 12% without TEMPO, showcasing the significant improvement in reaction rate with the presence of mediator.36 A similar result is observed for eosin Y anchored anatase TiO2, where incorporating 2% TEMPO in the reaction mixture raised the overall conversion by ∼25%.37 A further study regarding both TEMPO and its derivatives with the assistance of EPR spectroscopy corroborates the mechanism illustrated in Fig. 5,38 where both high oxidation yield and selectivity towards aldehyde were observed for 15 aromatic alcohols.
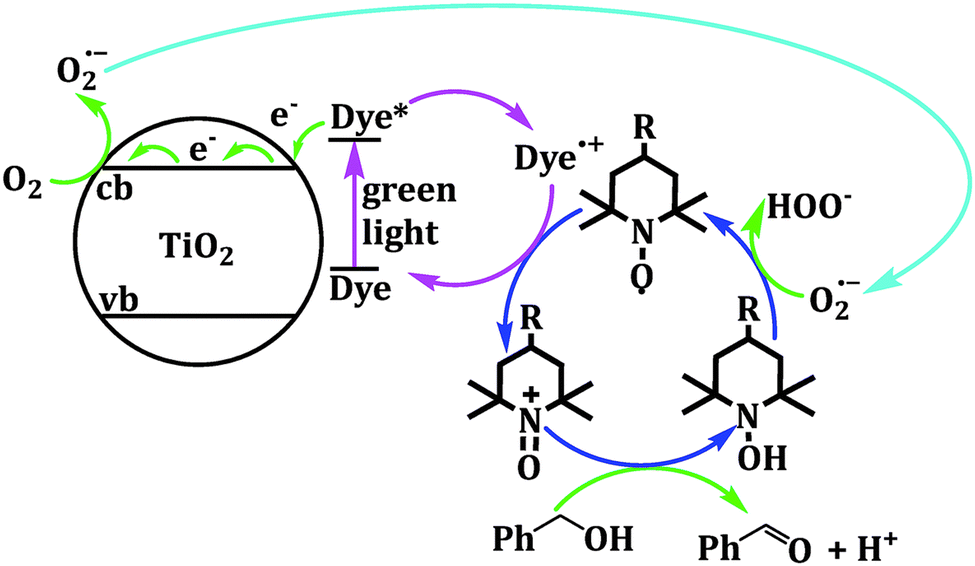 | ||
| Fig. 5 Proposed mechanism of TEMPO-derivative driven benzyl alcohol oxidation on dye-sensitized TiO2. Copyright: N. Wang, J. L. Shi, H. Hao, H. Yuan and X. Lang, Sustainable Energy Fuels, 2019, 3, 1701–1712. | ||
Anion mediated photocatalytic alcohol oxidation on semiconductors
While TEMPO and its derivatives have been significantly studied on both photochemical and photoelectrochemical alcohol oxidation, it is still a relatively expensive organic molecule with rather low oxidizing power. Thus, researchers have been searching for simpler alternatives that also provide comparable oxidizing capabilities. Naturally, inorganic anions dissolved in solution as electrolytes are considered, as they may be readily oxidized by valence-band holes in semiconductor materials and usually are present in large concentration. Radicals are usually formed when the otherwise stable anions are oxidized, subsequently working as very strong oxidants that can perform hydrogen-atom abstraction needed for alcohol oxidation.39 Considering the abundance of these anions and their significantly lower price, anion-mediated photocatalysis is a promising future direction for the field.Nitrate (NO3−) is one of the possible anions that can be used for alcohol oxidation due to its potential in forming nitrate radical (NO3˙), which is heavily involved in atmospheric chemistry.40 Work from our group shows that benzyl alcohol or 5-hydroxymethylfurfural (5-HMF) can be oxidized by CdS nanowires through the NO3−/NO3˙ mediated couple. A significant increase in the yield of benzaldehyde or 2,5-diformylfuran is observed when a metal nitrate salt is dissolved in acetonitrile solvent compared to the unmediated reaction. This marks the effectiveness of the oxidized nitrate radical as the homogeneous oxidant, illustrated in Fig. 6. A nitrate radical trap experiment carried out with tetramethylethylene supports NO3˙ formation during photocatalysis. H/D isotopic labeling of the benzylic position in the substrate alcohol showed that α-H abstraction is not rate determining, and that reduced oxygen species are not part of the mechanism. Curiously, Bu4NNO3 is not a good mediator in this case; a metal cation is required for the nitrate anion to function as a mediator effectively. Consequently, we proposed the mechanism, also included in Fig. 6.41
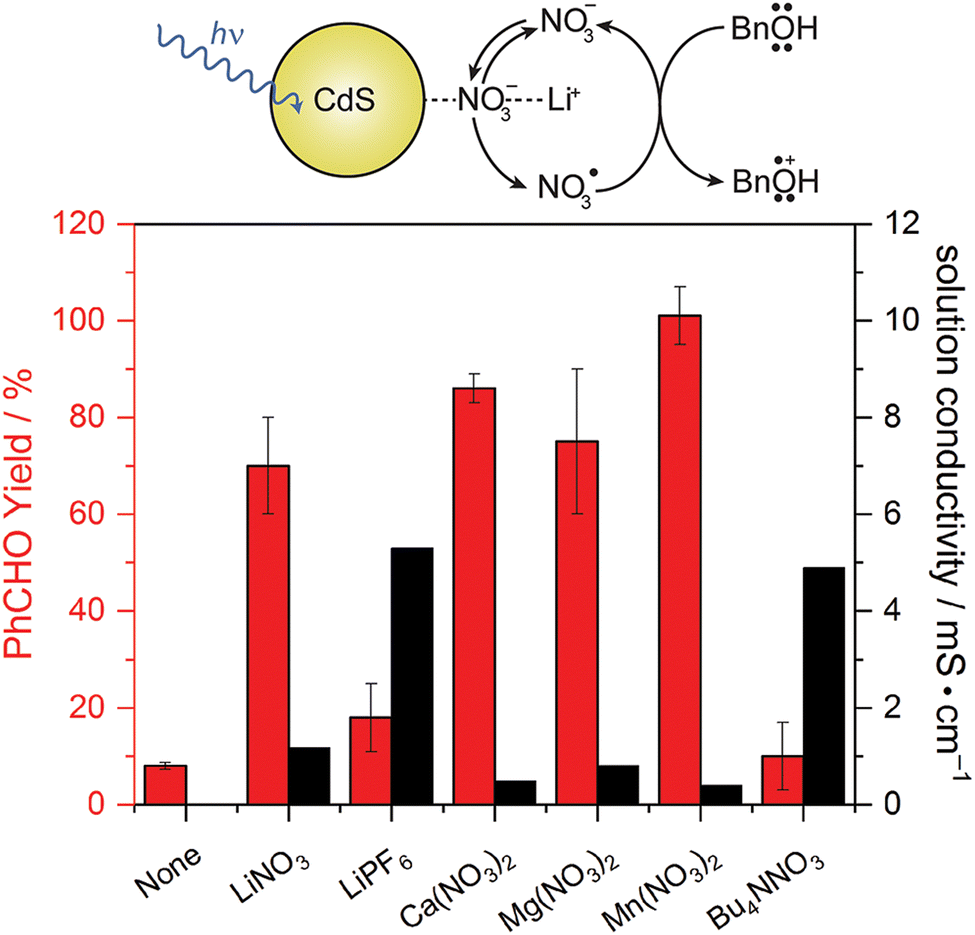 | ||
| Fig. 6 (top) Proposed mechanism of nitrate mediated benzyl alcohol oxidation with LiNO3 as a mediator. (down) Yield of benzaldehyde after 48 h irradiation with CdS and 50 mM mediator listed. Error bars represent deviations among 3 trials. Copyright: J. L. DiMeglio, A. G. Breuhaus-Alvarez, S. Li and B. M. Bartlett, ACS Catal., 2019, 9, 5732–5741. | ||
Similarily, other inorganic mediators have been employed in alcohol oxidation reactions. SO4˙−/SO42− is another redox couple of interest, where the electrochemical conversion between the redox couple is observed on boron-doped diamond electrodes42 and that the mediated approach where sulfate radical provides a 10–15 fold increase in rate compared with inert electrolytes on organic contaminant degradation.43 Switching gears towards photocatalysis, researchers probed the photocatalytic generation of sulfate radicals by P25-TiO2 or bismuth(III) phosphate (BPO) nanorods by oxizing 2,4-dichlorophenol (2,4-DCP). With the presence of sulfate, BPO exhibited faster oxidation that is attributed to the formation of sulfate radicals. The generation of radicals are assessed by radical trap experiments, where t-BuOH and MeOH are used as radical traps. Although these small alcohols are not considered as the authors’ main target, they do lower the yield of the phenol oxidation by consuming the radicals, highlighting the potential of using sulfate as a mediator for alcohol oxidation with a greater range of substrates. Fig. 7 shows the observed rate difference with the varied conditions, focusing on the presence of sulfate anion or radical scavengers. The decrease in rate on P25-TiO2 with added sulfate is attributed to the less positive VB position of TiO2 being unable to oxidize the sulfate effectively, thus adsorption of the SO42− blocks the ˙OH radical formation that could also oxidize the target alcohol. The surface of BPO has a weaker bond with the generated SO4˙− radical, allowing it to diffuse away and react with solution-phase substrates more easily.44 It is also noteworthy that the photochemical reaction is carried out in aqueous conditions, where the aforementioned NO3˙ radicals are less mobile and harder to solvate than in acetonitrile.45
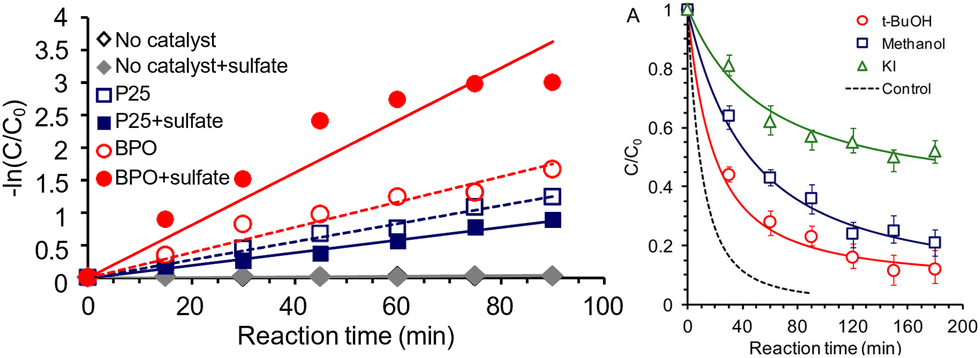 | ||
| Fig. 7 Rate comparison of 2,4-DCP oxidation under varied conditions. Left: With or without sulfate on BPO/P25-TiO2; Right: with or without radical scavengers. Copyright: G. Liu, S. You, Y. Tan and N. Ren, Environ. Sci. Technol., 2017, 51, 2339-2346. | ||
These examples emphasize the importance of carefully screening the semiconductor materials, the mediator, and the solvent when designing anion-mediated photochemical alcohol oxidation reactions, where band gap/band positions, surface adsorption features, standard redox potentials, solvent compatibility, strength of electrolyte, and solubility must be factored in.
Chloride ion is distinguished by its potential use in both electrochemical and photochemical ethanol oxidation to form 1,1-diethoxyethane,46 acetaldehyde/acetic acid,47 and ethylene oxide.48 In these cases, chloride also serves as mediator by being oxidized on the catalyst and carrying out homogeneous chemical reactions. If a heterogeneous photocatalyst can selectively oxidize chloride ion, the oxidized chlorine-containing species could work as a strong oxidant capable of carrying out alcohol oxidation homogeneously. Additionally, we believe the anion mediated strategy could circumvent the drawbacks encountered in non-aqueous electrocatalytic alcohol oxidation, which is the primary approach for ambient condition alcohol oxidation reactions, namely the low solution ionic conductivity and high viscoity that can limit the current. Overcoming these limitations becomes crucial as the substrate alcohol becomes bulkier in size and less polar.
4. Surface and morphology engineering of nanoscale AOR photocatalysts
Various approaches of material synthesis can result in products with vastly differentiated morphologies, surface compositions, or defects despite the same nominal elemental stoichiometry. For semiconductor photocatalysts, these intrinsic differences give rise to differing optical properties, carrier diffusion properties, and specified surface active sites that dictate the reaction rate and selectivity of alcohol oxidation reactions.Morphology control of crystalline semiconductor fabrication
Tuning reaction conditions, including controlling the heating profile and stoichiometry of directing agents in a thermal non-hydrolytic aminolysis reaction, gives shape control of ZnO nanostructures; nanoplates, nanobullets, nanorods, and nanocones can be selectively prepared.49 The morphologies of the materials are shown by TEM imaging in Fig. 8. When tested as photocatalyst for benzyl alcohol oxidation, the varying shapes exhibited different conversion rate in a follow up study, with nanocones showing the fastest rate, illustrated in Fig. 9.50 | ||
| Fig. 8 TEM images of the ZnO materials prepared by the reported method with different morphologies. (a) nanocones; (b) nanorods; (c) nanoplates. Credit: H. Weerathunga, C. Tang, A. J. Brock, S. Sarina, T. Wang, Q. Liu, H. Y. Zhu, A. Du and E. R. Waclawik, J. Colloid Interface Sci., 2022, 606, 588–599. | ||
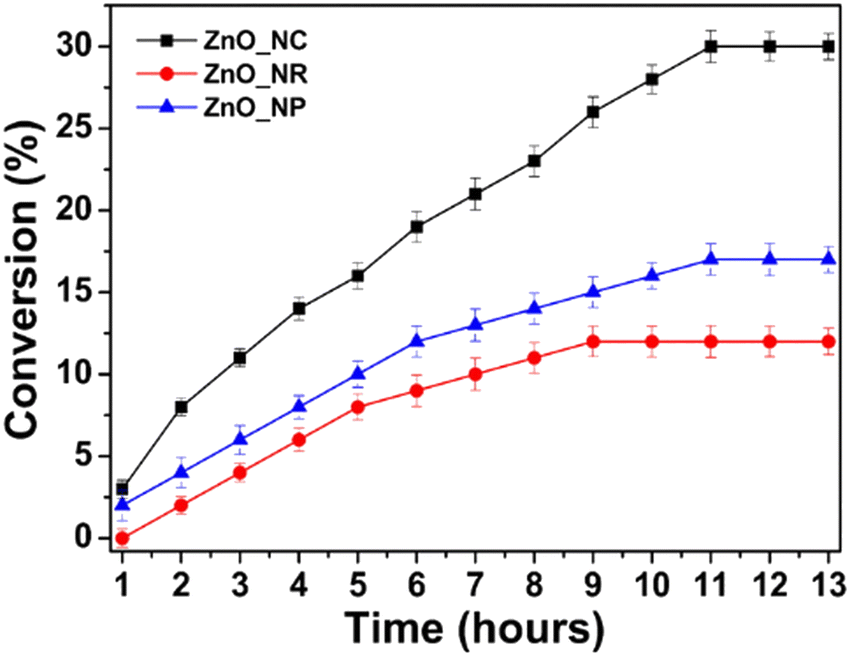 | ||
| Fig. 9 Percentage conversion of benzyl alcohol oxidation reaction over time powered by UV light under aerobic conditions with different shaped ZnO nanomaterials. Credit: H. Weerathunga, C. Tang, A. J. Brock, S. Sarina, T. Wang, Q. Liu, H. Y. Zhu, A. Du and E. R. Waclawik, J. Colloid Interface Sci., 2022, 606, 588–599. | ||
Among the morphologies, the critical difference that determines the rate mismatch is proposed to be the varied content of exposure of undercoordinated surface oxygen atoms. In the {10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1} facet of the hexagonal ZnO structure, such surface defective O atoms are most likely to be present. This facet is also thought to be most exposed in the nanocones, and the authors show that these defects provide a readily available site for benzyl alcohol to adsorb, as can be seen in Fig. 10. FTIR spectroscopy shows a 12 cm−1 shift in the O–H stretch when the alcohol adsorbs to these sites. This experimental observation is supported by DFT calculations as well, where the most negative adsorption energy is estimated for O-atom defect site, indicating it is the most thermodynamically favoured site when alcohol is to adsorb. Moreover, the DFT calculations reveal the electron distribution near the surface, where most of the electrons accumulate near Zn atoms and deplete near the O atoms. This distribution favours alcohol oxidation through the donation of electrons from the adsorbed alcohol to the oxygen-based valence band of the semiconductor. In conclusion, through precise control of the synthetic method, ZnO nanomaterials of a certain structure are obtained. Due to their difference in morphology, a desirable crystal facet is more exposed in the nanocones, on which the adsorption of benzyl alcohol is believed to be the strongest, leading to facile electron transfer and the fastest reaction rate of the studied nanostructures.
1} facet of the hexagonal ZnO structure, such surface defective O atoms are most likely to be present. This facet is also thought to be most exposed in the nanocones, and the authors show that these defects provide a readily available site for benzyl alcohol to adsorb, as can be seen in Fig. 10. FTIR spectroscopy shows a 12 cm−1 shift in the O–H stretch when the alcohol adsorbs to these sites. This experimental observation is supported by DFT calculations as well, where the most negative adsorption energy is estimated for O-atom defect site, indicating it is the most thermodynamically favoured site when alcohol is to adsorb. Moreover, the DFT calculations reveal the electron distribution near the surface, where most of the electrons accumulate near Zn atoms and deplete near the O atoms. This distribution favours alcohol oxidation through the donation of electrons from the adsorbed alcohol to the oxygen-based valence band of the semiconductor. In conclusion, through precise control of the synthetic method, ZnO nanomaterials of a certain structure are obtained. Due to their difference in morphology, a desirable crystal facet is more exposed in the nanocones, on which the adsorption of benzyl alcohol is believed to be the strongest, leading to facile electron transfer and the fastest reaction rate of the studied nanostructures.
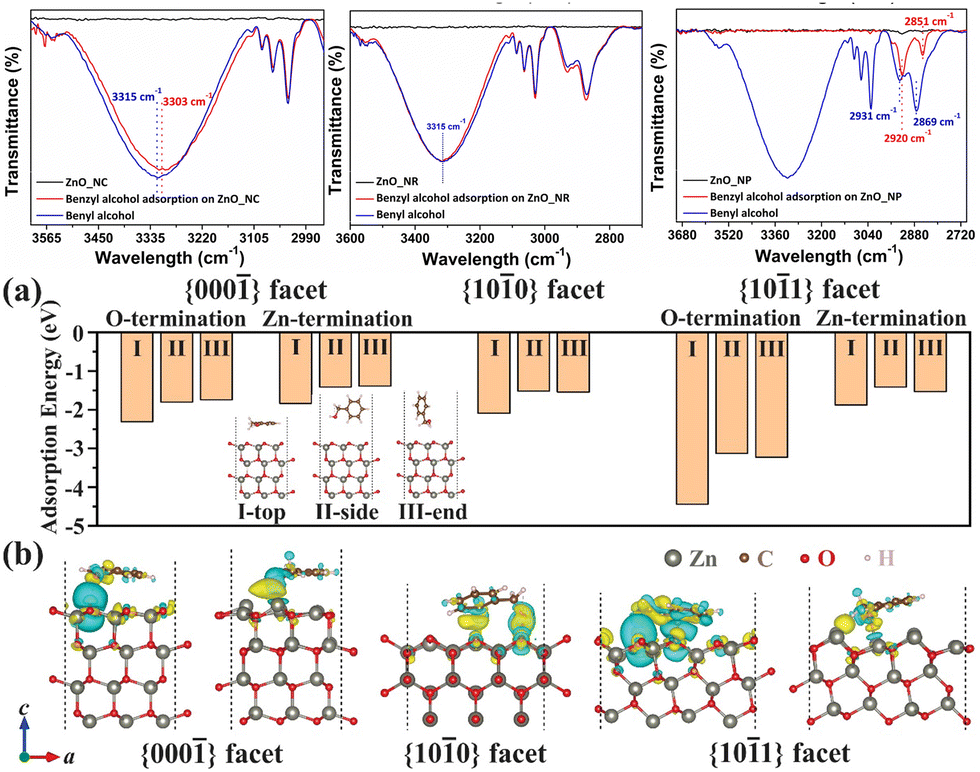 | ||
| Fig. 10 IR absorption of different ZnO nanomaterial before and after BnOH absorption. The range is focused on the O–H stretch. (a) Calculated absorption energy of benzyl alcohol in different modes on different facets of ZnO hexagons. (b) Electron density prediction of varied facets of ZnO with absorbed benzyl alcohol. Credit: H. Weerathunga, C. Tang, A. J. Brock, S. Sarina, T. Wang, Q. Liu, H. Y. Zhu, A. Du and E. R. Waclawik, J. Colloid Interface Sci., 2022, 606, 588–599. | ||
Surface area optimization during material synthesis
Another controllable morphological feature of semiconductor photocatalysts is the specific surface area. In most cases, a smaller particle size will lead to a higher surface area when normalized by mass, which is desirable for having more contact sites between the photocatalyst and substrate,51 better dispersibility in the reaction mixture,52 and longer charge-carrier lifetime by shortening the carrier diffusion.53 Thus, alternative synthesis methods have been introduced to reduce the particle size of desired catalyst. While hydrothermal/solvothermal synthesis remains one of the commonly used methods in nanoscale metal chalcogenides synthesis,54–56 in recent years, microwave-assisted synthesis has emerged as a more advanced approach. In traditional hydrothermal synthesis, the high-temperature, pressurized reaction vessel is usually filled with precursor solution and then placed in a convection oven. During a typical synthesis, the entire oven volume is heated to the desired temperature, of which the reaction solution is a miniscule part. The heating efficiency is therefore very low, and the ramping profile is not well controlled due to the mismatch between the temperature measured at the thermocouple and the temperature in the reaction vessel. Consequently, the heating rate of a conventional lab oven is usually limited to no more than 10 °C min−1 due to limitations of heating component.57 On the other hand, microwave heating can directly heat up the solvent within the reaction vessel. Polar solvents, depending on their dielectric constants and the dissolved ions in solution, can absorb microwave radiation and convert its energy into heat by exciting rotational modes of the molecules/ions which induces rapid collisions with other molecules/ions.58 Thus, as is shown in Fig. 11, the solvent molecules in the precursor heat up rapidly, thus heating the solute/reactant and resulting in swift bond activation and fast nucleation at multiple sites. | ||
| Fig. 11 Scheme of microwave assisted solvothermal reaction and conventional solvothermal reaction. Credit: N. Devi, S. Sahoo, R. Kumar and R. K. Singh, Nanoscale, 2021, 13, 11679–11711. | ||
Recently, with a modified microwave assisted solvothermal synthesis, our group has prepared WO3 nanoplatelets with a WCl6 precursor for the purpose of benzyl alcohol oxidation.59 Benzyl alcohol itself is used as the solvent for the precursor, while being the oxygen source. GC-MS analysis reveals the formation of benzyl chloride and dibenzyl ether as the byproducts, supporting the well-studied halide- and ether-elimination mechanisms of solvothermal synthesis.60 This preparation method turns out to be very desirable; crystalline WO3 nanoplatelets are observed without further annealing, indicated by the XRD pattern and TEM image in Fig. 12.
 | ||
| Fig. 12 (Left) X-ray diffraction data and (centre) TEM imaging of the obtained microwave synthesized WO3; (Right) concentration of benzaldehyde, oxidation product of benzyl alcohol, over time during WO3 photocatalysis. Black squares: microwave synthesized nanoplatelets; Red circles: commercial material. Credit: K. D. McDonald and B. M. Bartlett, RSC Adv., 2019, 9, 28688–28694. | ||
More importantly, these WO3 nanoplatelets show a high BET surface area of 37 m2 g−1, an order of magnitude higher than that of the commercially available material (2.6 m2 g−1). Using ∼460 nm blue LED irradiation under O2 atmosphere, oxidation of benzyl alcohol and 5-hydroxymethylfurfural are carried out both with the nanoplatelets and the commercial WO3. An obvious difference in the rate of benzyl alcohol oxidation between the two materials are observed, as can be seen from the kinetic data in Fig. 12. We attribute the difference in rate to the significant difference in surface area. Additionally, with XPS analysis, we observe surface tungsten reduction (W6+ to W5+) after illumination, which is attributed to trapped conduction-band electrons that do not reduce O2. This electron trapping is also supported by EPR spectroscopy and substrate-free control reactions, where no oxidation nor reduction reaction occurs. While WO3 is very chemically stable in acidic and non-aqueous conditions, this work highlights the need to couple the alcohol oxidation reaction with a facile reduction reaction to avoid the surface reduction of W6+.
Surface re-engineering to optimize performance of present material
Similar observations are also confirmed for ZnO, in which researchers synthesized 1D-nanorods from commercial ZnO and compared the materials against also commercially available ZnO.61 The ZnO 1D-nanorods are prepared by grinding bulk ZnO followed by a solvothermal synthesis with aqueous H2O2 as the solvent. This treatment drastically altered the morphology, as is shown in Fig. 13. When tested for photo-catalytic benzyl alcohol oxidation, the 1D nanorods showed faster conversion rate and much better selectivity towards benzaldehyde, outcompeting the bulk commercial ZnO particles. In this case, the authors assessed and compared the two catalysts in various directions. It is understood through gas adsorption experiments that the 1D-nanorods have larger specific surface area compared to the bulk material. Apart from this difference, two more important factors also contribute to the enhanced reaction rate. The nanorods showed a preference for benzyl alcohol adsorption compared to benzaldehyde adsorption, resulting in facile desorption of the aldehyde product, and thus avoiding overoxidation. This explains the higher selectivity of the aldehyde on the nanorods, compared to bulk ZnO where barely any adsorption preference is observed, as shown in Fig. 14. Similar arguments have also been made for TiO2 photocatalysts.62,63 In addition, the carrier lifetime of both materials was assessed by photoluminescence spectroscopy. A much smaller PL intensity is observed for the 1D-nanorods. This result indicates a longer carrier lifetime that agrees with other reports regarding 1D materials compared to bulk materials.64 | ||
| Fig. 13 (Top) SEM imaging of (a) 1D-nanorod ZnO and (b) bulk ZnO. (Bottom) TEM imaging of the obtained nanorods. Credit: Z. R. Tang, X. Yin, Y. Zhang and Y. J. Xu, RSC Adv., 2013, 3, 5956–5965. | ||
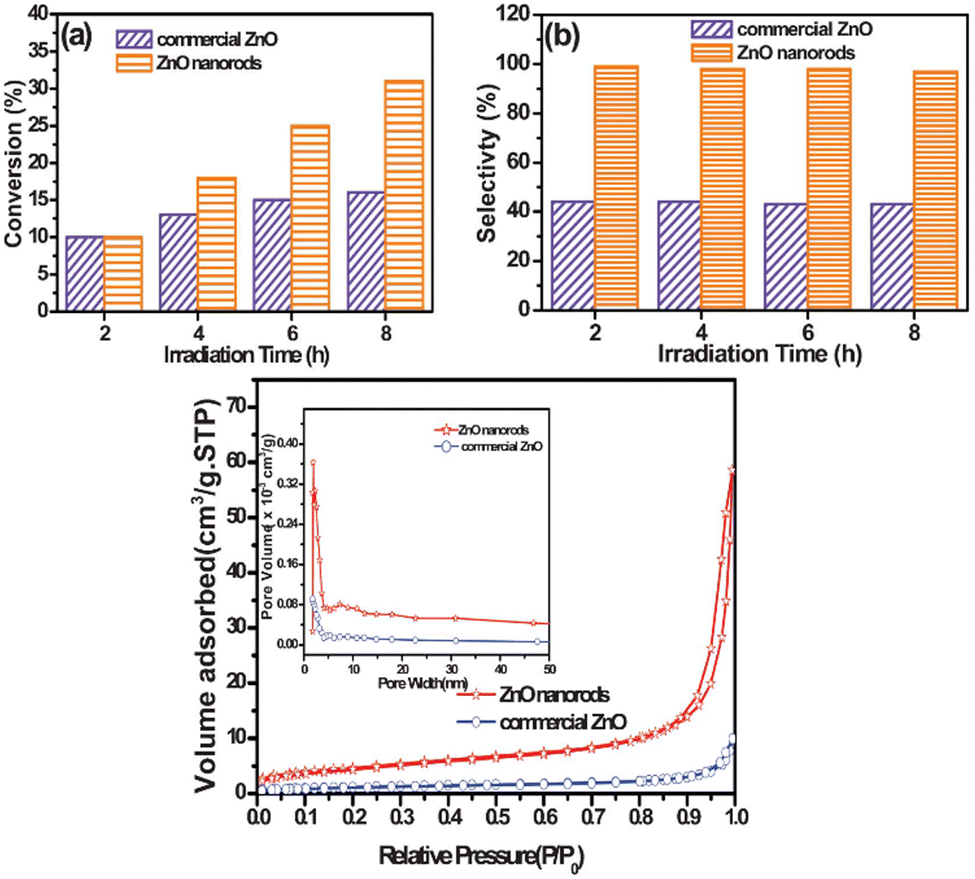 | ||
| Fig. 14 (Top) Yield and selectivity of benzaldehyde from benzyl alcohol oxidation driven by ZnO over time, where the performance of 1D-nanorod and commercial ZnO is compared. (Bottom) Gas adsorption measurements of the materials. Credit: Z. R. Tang, X. Yin, Y. Zhang and Y. J. Xu, RSC Adv., 2013, 3, 5956–5965. | ||
Together, even with the same catalyst–substrate combination, the rate and selectivity of alcohol oxidation reactions can be significantly optimized. By altering the preparation protocols, it is possible to achieve higher surface area and controllable shapes of nanostructures, which could be crucial in controlling the exposure of active crystal facets or sites. Moreover, engineering the surface of readily available materials may modify the adsorption/desorption profiles at the surface, optimizing the selectivity of desired oxidation product. As an improvement of direct alcohol oxidation, both rate and selectivity can be adjusted by the engineering of the photocatalyst. Importantly, in cases where indirect alcohol oxidation is not viable, tuning the adsorption/desorption of certain functional groups can also dictate the selectivity of reactions.
5. Advanced techniques for investigating mechanisms and characterizing materials
Conventional characterization methods focus mostly on ex situ discovery of materials’ properties. The experiments usually are carried out on the semiconductor material before and after photocatalysis to examine changes in morphology, crystallinity, and bonding environment. In this case, information regarding reactivity and stability can be gained, yet insight into what happens during the reaction is limited. It is desirable to know dynamics during the reactions, including photocatalyst synthesis. Thus, using in situ characterization techniques can provide better direct evidence in understanding mechanism as well as insight in materials development. We describe 3 powerful techniques in the following paragraphs: pXRD, FT-IR, and XAS, although we note that not all of these examples have been applied to the photocatalytic AOR, they represent a great opportunity for new research in the field – both catalyst synthesis/modification and mechanistic analysis.In our previous sections, we highlighted that the crystallinity of the synthesized material matters towards photocatalytic activity and selectivity. In situ powder X-ray diffraction (pXRD) carried out during synthesis can inform and control the crystal structure of the photocatalyst. In synthesizing WO3 nanomaterials, Kang et al. used in situ pXRD to determine the most calcination conditions for fine-tuning the hexagonal-monoclinic phase transition. The XRD measurement provided clear information on what temperature the phase transition starts, as well as the ratio of the phases. The corresponding data is shown in Fig. 15. The authors used this insight to synthesize a series of WO3 material with hexagonal phase content ranging from 0–100%. The BET surface areas of the two phases are similar, but WO3 having 88.1% hexagonal phase shows the fastest rate for photocatalysis (RhB dye degradation) by forming the most efficient heterojunction between the phases.65 Although using in situ XRD on photocatalyst powders during photocatalysis can be challenging, we believe this technique can be significantly helpful in controlled material synthesis.
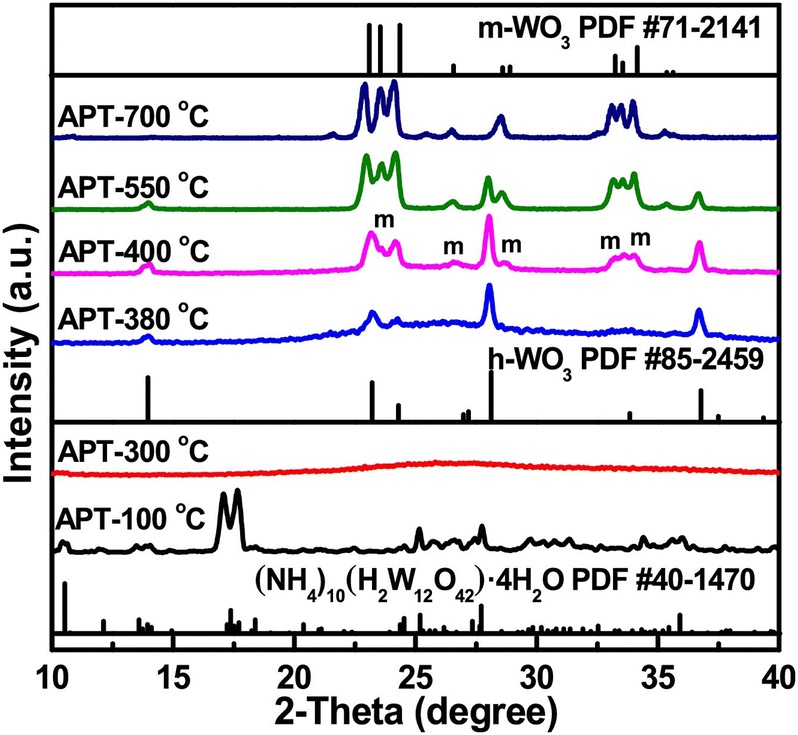 | ||
| Fig. 15 In situ XRD data during the calcination process of WO3. Credit: M. Kang, J. Liang, F. Wang, X. Chen, Y. Lu and J. Zhang, Mater. Res. Bull., 2020, 121, 110614. | ||
In situ FT-IR spectroscopy is a powerful tool in identifying reaction intermediates as well as probing the adsorption/desorption behavior at the photocatalyst surface. Previous research with this technique has been heavily applied to electrochemical mechanistic studies, where ethanol oxidation reaction on Pd is deciphered by exposing the intermediates. Adsorbed species including –OCO (acetate), –CO (acetyl) as well as acetic acid molecules were directly observed by the in situ measurement.66 With careful flow-cell device construction, Haselmann et al. designed an attenuated total reflection (ATR)-FTIR system in which the growth of Pt–TiO2 as well as methanol oxidation reaction are monitored in situ.67 They were able to track the progression of FT-IR signal at the surface upon irradiation of the TiO2 with varied Pt loading, shown in Fig. 16. The time-resolved data provide significant insight into the reaction mechanism. For low Pt loadings, methyl formate is observed as a bimolecular intermediate. At higher Pt loading, formaldehyde is predominantly observed as the intermediate for the direct oxidation towards CO2.
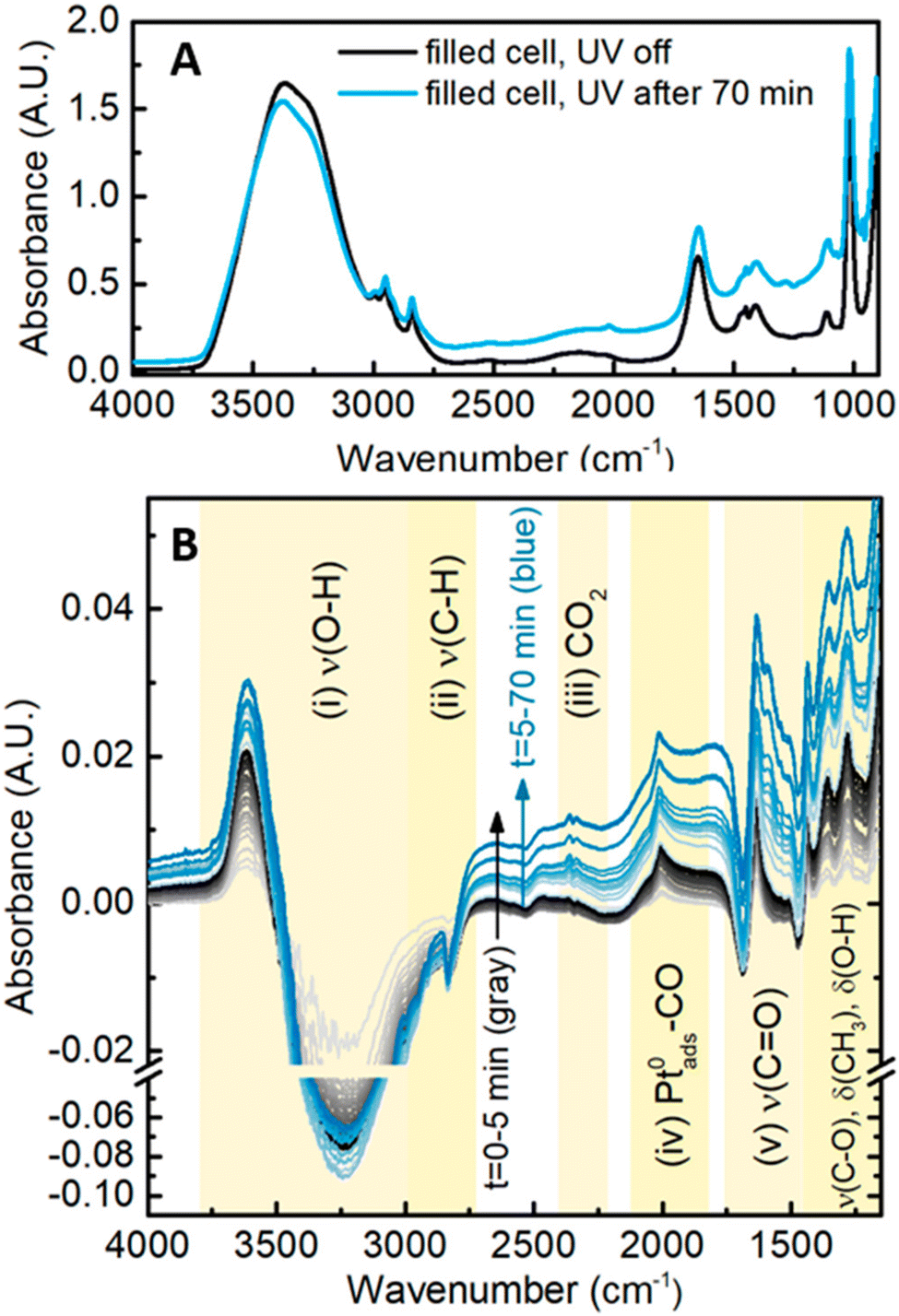 | ||
| Fig. 16 IR spectra during UV illumination with the Pt–TiO2 material. (A) Spectra before and after illumination; (B) difference spectra with a time interval of 10 s. Credit: G. M. Haselmann, B. Baumgartner, J. Wang, K. Wieland, T. Gupta, C. Herzig, A. Limbeck, B. Lindl and D. Eder, ACS Catal., 2020, 10(5), 2964–2977. | ||
Apart from tracking surface adsorbed species, another commonly used in situ technique is X-ray absorption spectroscopy (XAS), a strong tool in studying both the electronic state and bonding environment of the photocatalyst by X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), respectively. Liu et al. studied a Ru–RuOx/C3N4 heterojunction interface with both techniques. Here, in situ XANES was employed at high temperature, tracking the oxidation state of Ru atoms during catalysis. Similarly, EXAFS confirmed the increase of Ru–O coordination features, indicating that a Ru–RuOx interface is formed, as is shown in Fig. 17. This example shows the potential in tracking the structural change of a material during a photocatalytic reaction.68 Additionally, Hou et al. used both XAS techniques to assess WO3 photocatalyst changes during N2 reduction. The authors observed XANES edge shifting to lower energy, and it is thus understood that the W6+ undergoes reduction during illumination, agreeing with our group's report; additionally, EXAFS indicates that the coordination number on W for the W–O bond drops gradually throughout the reaction.69
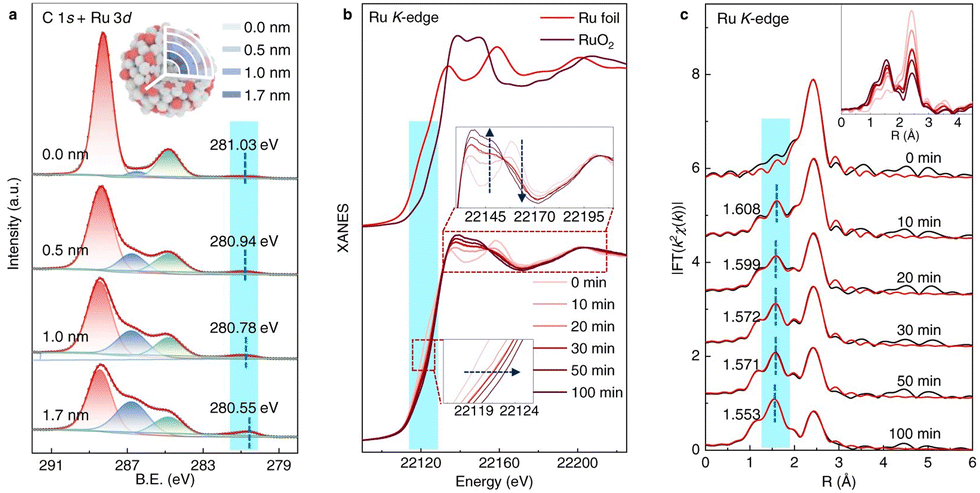 | ||
| Fig. 17 (a) XPS data at varied depths of the material. (b) In situ XANES spectra recorded at Ru K-edge during heating process. (c) In situ EXAFS spectra recorded during heating process. Inset: Overlay of EXAFS region among the different time points. Credit: D. Liu, T. Ding, L. Wang, H. Zhang, L. Xu, B. Pang, X. Liu, H. Wang, J. Wang, K. Wu and T. Yao, Nat. Commun., 2023, 14(1), 1720. | ||
Finally, many researchers have turned to theory and simulation for guidance in understanding the mechanisms involved in photochemical AORs.70–73 This approach has proven to be powerful, especially in developing a predictive adsorption/desorption model based on binding energies, the estimation of surface active sites, and even crystal structure alternations during photocatalysis. Theory and simulation circumvent the technical limitations for some characterization techniques and often help corroborate the experimental results, which in turn validate the theory/simulations.74 For instance, Sun et al. used DFT calculations to study the influence of introducing S-defects in In2S3 photocatalysts.75 By modelling In2S3 slabs both with and without S-vacancies, the authors were able to calculate the DOS for both materials, as well as the simulate oxygen gas behaviour adjacent to the surface. From these results, the authors found an obvious increase in DOS near the valence-band maximum, indicating swift electron excitation upon irradiation and suppressed recombination. Moreover, O2 gas has a −0.31 eV adsorption energy on the S-vacant slab, indicating that O2 is more likely to adsorb and react to form a reduced oxygen species that can serve as a strong oxidant towards alcohols. One such result is shown in Fig. 18.
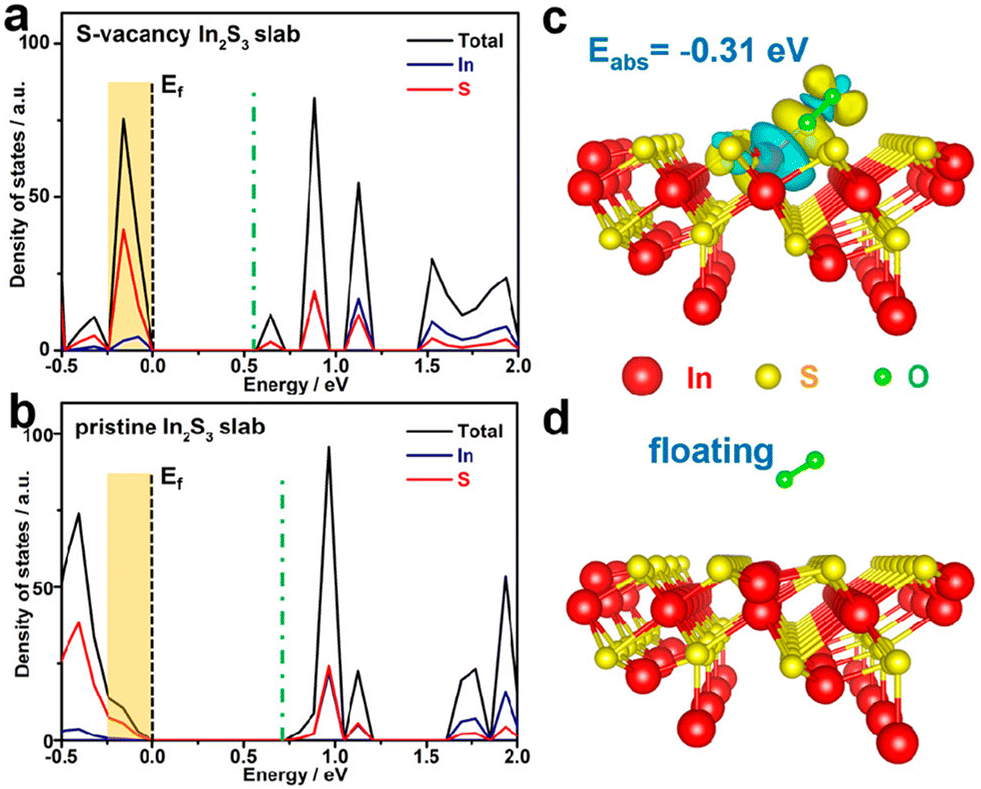 | ||
| Fig. 18 (a) and (b) DFT calculated DOS for In2S3 slabs with and without S-vacancy. (c) and (d) Illustration of simulated O2 interaction with both slabs. Credits: X. Sun, L. Sun, X. Zhang, J. Xie, S. Jin, H. Wang, X. Zheng, X. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141(9), 3797–3801. | ||
This understanding provided guidance both to material synthesis and design. With the synthesized S-vacant In2S3 photocatalyst, the authors could corroborate the calculation results with experiments, where much higher conversion towards aldehyde products were observed for 10 benzylic alcohol substrates. This example shows the power of combining advanced in situ measurements with simulations for discovering new ways to control key factors to direct reactivity, including appropriate band positions, sufficiently strong adsorption of the alcohol substrate, efficient separation and reaction of charge carriers, and finally product selectivity, which are critical for the materials synthesized to be used as an ideal alcohol oxidation photocatalyst.76–78
Conclusions and outlook
Nanoparticle-driven photochemical alcohol oxidation reactions represent a promising way to convert alcohols to value-added products using solar energy. While the conversion is usually thermodynamically feasible, slow kinetics and energy losses within the nanomaterials limit the rate and selectivity of the oxidation reactions. Multiple strategies can be employed to optimize these reactions, including using a redox mediator to alter the reaction pathways, by bridging the heterogeneous and homogeneous process, designing nanomaterial syntheses that focus on active facets, reducing the particle size of to achieve higher surface area, and re-engineering the catalyst to optimize chemical and optical properties. Despite the encouraging examples discussed, there remain several bottlenecks in photochemical alcohol oxidation that provide new research opportunities. There are still no materials that meets the three requirements having efficient solar absorption across the entire visible spectrum (CdS, ZnInS2, etc), excellent chemical stability during photocatalysis, (TiO2, ZnO), and being low cost for industrial use. It is thus necessary to combine two or more of the strategies discussed Moreover, a focus on the indirect oxidation mechanisms helps to circumvent side reactions controlled by adsorption/desorption phenomena, and tuning the indirect path can allow greater selectivity. Additionally, contributions from advanced in situ technologies as well as powerful simulations can strongly contribute to understanding reaction mechanism and provide accurate directions for experimental advancement, resulting in the newer generation photocatalysts. The strategies presented here have great potential for making photochemical alcohol oxidation practical, and a welcome addition to the larger landscape of problems for solar energy conversion.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CdS and WO3 research from our group highlighted in this article is supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award DE-SC-006587 (Catalysis Science).References
- Resource: https://www.bp.com/en/global/corporate/energy-economics/statistical-review-of-world-energy.html.
- J. Tollefson, Nature, 2018, 556, 422–425 CrossRef CAS PubMed.
- Y. Hermans, A. Klein, H. P. Sarker, M. N. Huda, H. Junge, T. Toupance and W. Jaegermann, Adv. Funct. Mater., 2020, 30, 1910432 CrossRef CAS.
- S.-J. Woo, S. Choi, S.-Y. Kim, P. S. Kim, J. H. Jo, C. H. Kim, H.-J. Son, C. Pac and S. O. Kang, ACS Catal., 2019, 9, 2580–2593 CrossRef CAS.
- X. Liu, X. Duan, W. Wei, S. Wang and B. J. Ni, Green Chem., 2019, 21, 4266–4289 RSC.
- R. K. Prasad, S. Chatterjee, P. B. Mazumder, S. K. Gupta, S. Sharma, M. G. Vairale, S. Datta, S. K. Dwivedi and D. K. Gupta, Chemosphere, 2019, 231, 588–606 CrossRef CAS.
- J. Zhang, E. Yoo, B. H. Davison, D. Liu, J. A. Schaidle, L. Tao and Z. Li, Green Chem., 2021, 23, 9534–9548 RSC.
- G. Irick Jr, J. Appl. Polym. Sci., 1972, 16, 2387–2395 CrossRef.
- S. N. Frank and A. J. Bard, J. Phys. Chem., 1977, 81, 1484–1488 CrossRef CAS.
- Z. Chai, T. T. Zeng, Q. Li, L. Q. Lu, W. J. Xiao and D. Xu, J. Am. Chem. Soc., 2016, 138, 10128–10131 CrossRef CAS.
- Y. Hu, X. Yu, Q. Liu, L. Wang and H. Tang, Carbon, 2022, 188, 70–80 CrossRef CAS.
- X. Wu, S. Xie, H. Zhang, Q. Zhang, B. F. Sels and Y. Wang, Adv. Mater., 2021, 33, 2007129 CrossRef CAS PubMed.
- D. O. Hall and G. Grassi, Photoconversion Processes for Energy and Chemicals, Taylor & Francis, 2005 Search PubMed.
- J. L. DiMeglio and B. M. Bartlett, Chem. Mater., 2017, 29, 7579–7586 CrossRef CAS.
- L. Liu, Y. P. Zhu, M. Su and Z. Y. Yuan, ChemCatChem, 2015, 7, 2765–2787 CrossRef CAS.
- J. Cui, X. Lu, M. Guo, M. Zhang, L. Sun, J. Xiong, R. Zhang, X. Li, Y. Qiao, D. Li, M. Guo and Z. Yu, Catal. Sci. Technol., 2023, 13, 940–957 RSC.
- Z. Jiang, X. Zhang, H. S. Chen, X. Hu and P. Yang, ChemCatChem, 2019, 11, 4558–4567 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- X. Xiang, B. Zhu, J. Zhang, C. Jiang, T. Chen, H. Yu, J. Yu and L. Wang, Appl. Catal., B, 2023, 324, 122301 CrossRef CAS.
- M. Eder, M. Tschurl and U. Heiz, J. Phys. Chem. Lett., 2023, 14, 6193–6201 CrossRef CAS.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48(5), 1403–1412 CrossRef CAS.
- L. Wen, X. Zhang and F. F. Abdi, Mater. Today Energy, 2024, 101648 CrossRef CAS.
- P. Limpachanangkul, P. Nimmmanterdwong, L. Liu, M. Hunsom, P. Piumsomboon and B. Chalermsinsuwan, Sci. Rep., 2023, 13(1), 14936 CrossRef CAS.
- V. Shvalagin, S. Kuchmiy, M. Skoryk, M. Bondarenko and O. Khyzhun, Mater. Sci. Eng., B, 2021, 271, 115304 CrossRef CAS.
- A. Kumar, P. Choudhary and V. Krishnan, Appl. Surf. Sci., 2022, 578, 151953 CrossRef CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568 RSC.
- S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4(7), 456–475 CrossRef CAS.
- V. Augugliaro, M. Bellardita, V. Loddo, G. Palmisano, L. Palmisano and S. Yurdakal, J. Photochem. Photobiol., C, 2012, 13, 224–245 CrossRef CAS.
- H. Goto, Y. Hanada, T. Ohno and M. Matsumura, J. Catal., 2004, 225(1), 223–229 CrossRef CAS.
- F. Guzman, Z. Yu and S. S. C. Chuang, Chem. Ind., 2009, 123, 463–472 CAS.
- A. J. Bosco, S. Lawrence, C. Christopher, S. Radhakrishnan, A. A. Joseph Rosario, S. Raja and D. Vasudevan, J. Phys. Org. Chem., 2015, 28, 591–595 CrossRef CAS.
- B. Ganem, J. Org. Chem., 1975, 40, 1998–2000 CrossRef CAS.
- X. Lang and J. Zhao, Chem. – Asian J., 2018, 13, 599–613 CrossRef CAS.
- T. Suga, Y. J. Pu, K. Oyaizu and H. Nishide, Bull. Chem. Soc. Jpn., 2004, 77, 2203–2204 CrossRef CAS.
- M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757 CrossRef CAS PubMed.
- V. Jeena and R. S. Robinson, Chem. Commun., 2012, 48, 299–301 RSC.
- Y. Zhang, Z. Wang and X. Lang, Catal. Sci. Technol., 2017, 7, 4955–4963 RSC.
- N. Wang, J. L. Shi, H. Hao, H. Yuan and X. Lang, Sustainable Energy Fuels, 2019, 3, 1701–1712 RSC.
- C. B. Bai, N. X. Wang, X. W. Lan, Y. J. Wang, Y. Xing, J. L. Wen, X. W. Gao and W. Zhang, Sci. Rep., 2016, 6, 20163 CrossRef.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, A. M. Laura, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St. Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed.
- J. L. DiMeglio, A. G. Breuhaus-Alvarez, S. Li and B. M. Bartlett, ACS Catal., 2019, 9, 5732–5741 CrossRef CAS.
- J. Davis, J. C. Baygents and J. Farrell, Electrochim. Acta, 2014, 150, 68–74 CrossRef CAS.
- A. Farhat, J. Keller, S. Tait and J. Radjenovic, Environ. Sci. Technol., 2015, 49, 14326–14333 CrossRef CAS PubMed.
- G. Liu, S. You, Y. Tan and N. Ren, Environ. Sci. Technol., 2017, 51, 2339–2346 CrossRef CAS.
- S. P. Mezyk, T. D. Cullen, K. A. Rickman and B. J. Mincher, Int. J. Chem. Kinet., 2017, 49, 635–642 CrossRef CAS.
- S. Li and B. M. Bartlett, J. Am. Chem. Soc., 2021, 143, 15907–15911 CrossRef CAS PubMed.
- A. G. Breuhaus-Alvarez, S. Li, N. Z. Hardin and B. M. Bartlett, J. Phys. Chem. C, 2021, 125, 26307–26312 CrossRef CAS.
- C. Lucky, T. Wang and M. Schreier, ACS Energy Lett., 2022, 7, 1316–1321 CrossRef CAS.
- J. M. De Corrado, J. F. Fernando, M. P. Shortell, B. L. Poad, S. J. Blanksby and E. R. Waclawik, ACS Appl. Nano Mater., 2019, 2, 7856–7869 CrossRef CAS.
- H. Weerathunga, C. Tang, A. J. Brock, S. Sarina, T. Wang, Q. Liu, H. Y. Zhu, A. Du and E. R. Waclawik, J. Colloid Interface Sci., 2022, 606, 588–599 CrossRef CAS PubMed.
- H. Cheng, J. Wang, Y. Zhao and X. Han, RSC Adv., 2014, 4, 47031–47038 RSC.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- F. Chen, T. Ma, T. Zhang, Y. Zhang and H. Huang, Adv. Mater., 2021, 33, 2005256 CrossRef CAS PubMed.
- J. Yu and X. Yu, Environ. Sci. Technol., 2008, 42, 4902–4907 CrossRef CAS PubMed.
- W. Shi, S. Song and H. Zhang, Chem. Soc. Rev., 2013, 42, 5714–5743 RSC.
- J. Kundu, S. Khilari and D. Pradhan, ACS Appl. Mater. Interfaces, 2017, 9, 9669–9680 CrossRef CAS PubMed.
- N. Devi, S. Sahoo, R. Kumar and R. K. Singh, Nanoscale, 2021, 13, 11679–11711 RSC.
- C. O. Kappe, A. Stadler and D. Dallinger, Microwaves in Organic and Medicinal Chemistry, 2nd edition, Wiley-VCH, Weinheim, 2012 Search PubMed.
- K. D. McDonald and B. M. Bartlett, RSC Adv., 2019, 9, 28688–28694 RSC.
- S. Hilaire, M. J. Süess, N. Kränzlin, K. Bieńkowski, R. Solarska, J. Augustyński and M. Niederberger, J. Mater. Chem. A, 2014, 2, 20530–20537 RSC.
- Z. R. Tang, X. Yin, Y. Zhang and Y. J. Xu, RSC Adv., 2013, 3, 5956–5965 RSC.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279 CrossRef CAS.
- R. H. Li, H. Kobayashi, J. F. Guo and J. Fan, J. Phys. Chem. C, 2011, 115, 23408 CrossRef CAS.
- T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2009, 131, 8485 CrossRef CAS PubMed.
- M. Kang, J. Liang, F. Wang, X. Chen, Y. Lu and J. Zhang, Mater. Res. Bull., 2020, 121, 110614 CrossRef CAS.
- Y. Y. Yang, J. Ren, Q. X. Li, Z. Y. Zhou, S. G. Sun and W. B. Cai, ACS Catal., 2014, 4(3), 798–803 CrossRef CAS.
- G. M. Haselmann, B. Baumgartner, J. Wang, K. Wieland, T. Gupta, C. Herzig, A. Limbeck, B. Lindl and D. Eder, ACS Catal., 2020, 10(5), 2964–2977 CrossRef CAS.
- D. Liu, T. Ding, L. Wang, H. Zhang, L. Xu, B. Pang, X. Liu, H. Wang, J. Wang, K. Wu and T. Yao, Nat. Commun., 2023, 14(1), 1720 CrossRef CAS PubMed.
- T. Hou, Y. Xiao, P. Cui, Y. Huang, X. Tan, X. Zheng and Y. Zou, Adv. Energy Mater., 2019, 9(43), 1902319 CrossRef CAS.
- L. Zhao, B. Zhang, X. Xiao, F. L. Gu and R. Q. Zhang, J. Mol. Catal. A: Chem., 2016, 420, 82–87 CrossRef CAS.
- H. Kobayashi and S. Higashimoto, Appl. Catal., B, 2015, 170, 135–143 CrossRef.
- E. Safaei, S. Mohebbi and M. Irani, J. Sol–Gel Sci. Technol., 2018, 87, 170–182 CrossRef CAS.
- J. Goclon and K. Winkler, Mol. Catal., 2020, 482, 110747 CrossRef CAS.
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11(8), 2003216 CrossRef CAS.
- X. Sun, L. Sun, X. Zhang, J. Xie, S. Jin, H. Wang, X. Zheng, X. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141(9), 3797–3801 CrossRef CAS PubMed.
- X. Yang, S. Zhang, P. Li, S. Gao and R. Cao, J. Mater. Chem. A, 2020, 8, 20897–20924 RSC.
- L. Yu, Y. Lin and D. Li, Appl. Catal., B, 2017, 216, 88–94 CrossRef CAS.
- G. Qian, P. Jiang, Y. Shen, Y. Leng, P. T. Wai, K. Zhang and A. Haryono, Nanotechnology, 2021, 32(23), 235602 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |