
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Secondary structure changes as the potential H2 sensing mechanism of group D [FeFe]-hydrogenases†
Ivan
Voloshyn
 a,
Conrad
Schumann
b,
Princess R.
Cabotaje
b,
Afridi
Zamader‡
b,
Henrik
Land
b and
Moritz
Senger
*ab
a,
Conrad
Schumann
b,
Princess R.
Cabotaje
b,
Afridi
Zamader‡
b,
Henrik
Land
b and
Moritz
Senger
*ab
aDepartment of Chemistry – BMC, Biochemistry, Uppsala University, 75120 Uppsala, Sweden. E-mail: moritz.senger@kemi.uu.se
bDepartment of Chemistry – Ångström Laboratory, Molecular Biomimetics, Uppsala University, 75120 Uppsala, Sweden
First published on 5th September 2024
Abstract
[FeFe]-hydrogenases function as both H2 catalysts and sensors. While catalysis is well investigated, details regarding the H2 sensing mechanism are limited. Here, we relate protein structure changes to H2 sensing, similar to light-driven bio-sensors. Our results highlight how identical cofactors incorporated in alternative protein scaffolds serve different functions in nature.
In nature, similar enzymes can serve multiple functions. A prime example of this versatility are the metalloenzyme [FeFe]-hydrogenases, which are mainly known for their high catalytic hydrogen (H2) turnover rates, making them an attractive target in the field of sustainable fuels research.1 Besides catalysis, certain [FeFe]-hydrogenases are proposed to have a H2 sensory function with the potential to regulate cellular metabolism.2–4 The involvement of [FeFe]-hydrogenases in the H2 metabolism of microbes shines new light on their role in medicine and health research.5,6 [FeFe]-hydrogenases share the same cofactor, known as the H–cluster and are composed of a diiron site linked to a [4Fe–4S] cluster via the sulphur atom of a cysteine residue. The two irons of the diiron site are ligated by a carbon monoxide (CO) and cyanide (CN−) ligand each, and share a bridging CO molecule and an azadithiolate (ADT) bridge (Fig. 1A). [FeFe]-hydrogenases can be grouped into different phylogenetic groups (group A–G).7 These groups exhibit variations in the second coordination sphere of the cofactor and overall protein architecture.1 The most extensively studied group A [FeFe]-hydrogenases are highly active, while groups C and D are proposed to serve a sensory function.1,8 High turnover rates in group A have been associated with a sulphur-rich second coordination sphere of the diiron site and an alternative configuration of their unique proton transfer pathway (PTP) upon reduction.9,10 In line with observations for the sensory [FeFe]-hydrogenases of group C, the second coordination sphere of the H–cluster in group D representative [FeFe]-hydrogenase from Thermoanaerobacter mathranii, TamHydS, lacks these sulphur-rich amino acids characteristic of group A [FeFe]-hydrogenases.3,4,9,11 The low turnover rates of characterized representatives from groups C and D and the relative stability of the reduced potential signalling state, Hred, upon H2 exposure in TamHydS further implies a role in signalling rather than catalysis.3,12 Moreover, we recently identified a novel PTP in group D of which the operational mechanism remains to be elucidated. (Fig. 1A).9 Notably, the outer coordination sphere for TamHydS features a C-terminal extension which harbours an additional [4Fe–4S] cluster motif (Fig. 1B). The group C sensory [FeFe]-hydrogenase from Thermotoga maritima, TamHydS, features the same C-terminal FeS–cluster domain that is followed by a Per–Arnt–Sim (PAS) domain, which in combination with a Ser/Thr protein phosphatase might regulate downstream group A catalytic [FeFe]-hydrogenases.2,4 However, in group D, the absence of this PAS domain necessitates an alternative sensing cascade, leading to their classification as putatively sensory [FeFe]-hydrogenases. The mechanism by which H2 sensing is facilitated in group D enzymes remains an open question.
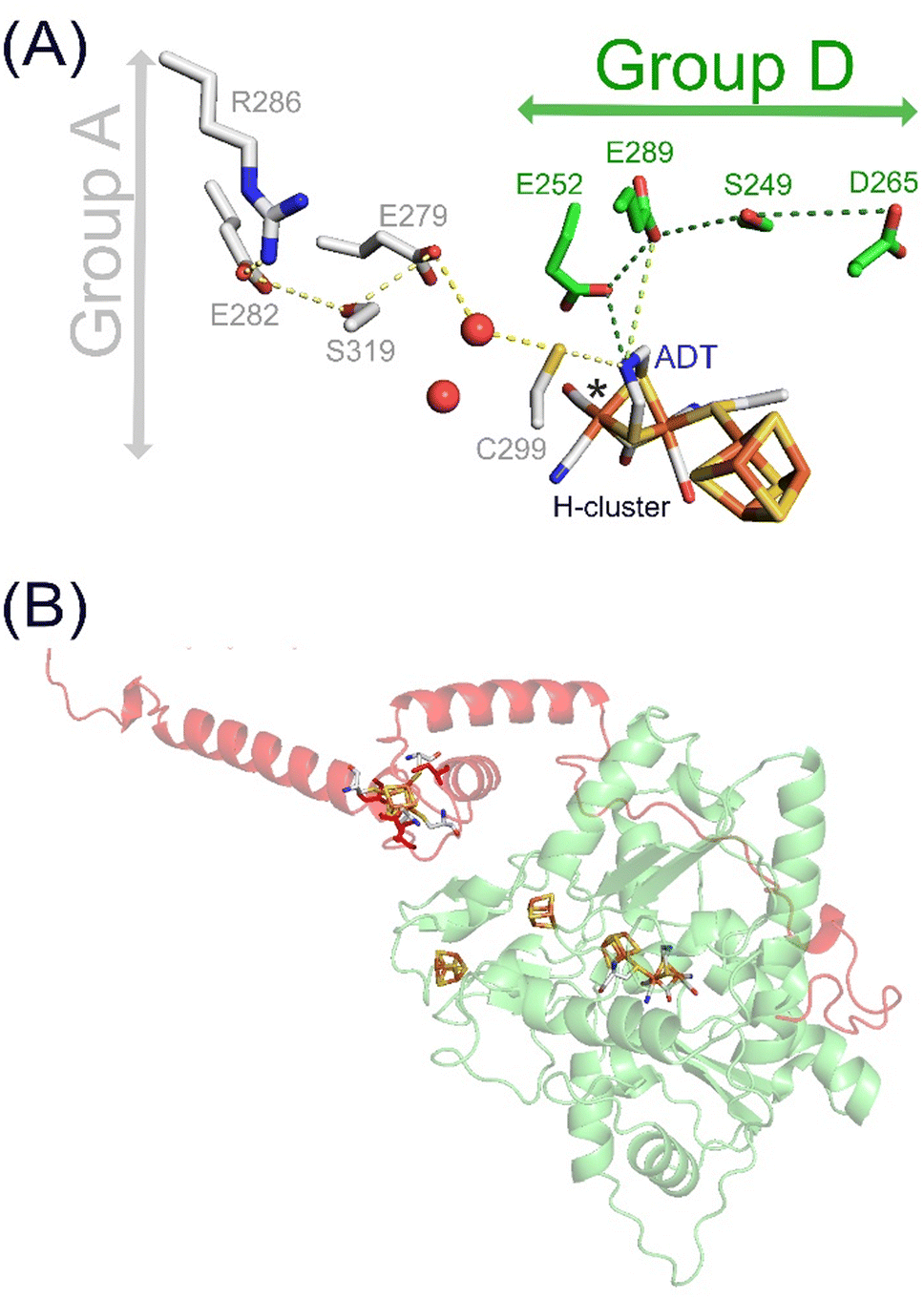 | ||
| Fig. 1 H–cluster, PTPs and AlphaFold model of TamHydS. (A) The H–cluster and its connection to PTPs in group A and D [FeFe]-hydrogenases. Hydrogen bonding networks constitute the PTPs that are composed of amino acids and water molecules (red spheres) conserved within either group A (grey sticks) or group D9 (green sticks). (B) The YASARA-generated homology model (green) of TamHydS3 was generated on the basis of CpI's crystal structure (PDB ID 4XDC)13 and the C-terminal domain was generated by AlphaFold (red). The [4Fe–4S] clusters and the H–cluster are shown as sticks (C: grey, Fe: orange, S: yellow, N: blue, O: red) RMSD values indicate closer alignment of the homology model (1.541 Å) than the AlphaFold model (3.692 Å) with CpI. The fourth [4Fe–4S] cluster in the C-terminal domain is visually represented by manually integrating a [4Fe–4S] cluster from the CpI structure into the TamHydS AlphaFold model, binding to cysteines C379, C382, C387, and C404 (red sticks), via PyMOL. | ||
Here, we investigate the putative sensory function of group D [FeFe]-hydrogenase, TamHydS. Our genomic analysis supports a putative signalling function, particularly when compared to sensory group C enzymes. We demonstrate the enrichment of the potential signalling state, Hred, in two ways, via photoreduction or exposure to H2. In contrast to the catalytic group A [FeFe]-hydrogenases, we do not observe a rearrangement of the H-bonding network of the PTP when Hred is populated. Instead, we detect a secondary structural rearrangement using Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy. We propose that this secondary structure change is involved in the signalling mechanism of TamHydS. In a broader sense, our results shine light on how nature evolved to facilitate different functions via alternative protein scaffolds harbouring the identical cofactors.
To substantiate the putative H2 sensing role, we analysed the genome of T. mathranii surrounding TamHydS (Fig. 2). We identified two operons located in direct vicinity harbouring genes for TamHydS and a group A [FeFe]-hydrogenase respectively. In Operon 1 upstream to the TamHydS gene, a Ser/Thr protein kinase is encoded that is proposed to be involved in signal transduction.2 Further downstream, we identified a sequence that could be assigned to either a phosphotransferase, a polymerase and histidinol phosphatase (PHP), or a phosphoesterase PHP domain protein. Any of these could assume the putative signal transfer role of the Ser/Thr protein phosphatase found in the genomic context of group C [FeFe]-hydrogenases. However, in group C the phosphatase is proposed to be regulated by the PAS domain, which is absent in group D. Instead, the T. mathranii genome encodes a DRTGG protein immediately following the TamHydS gene. This DRTGG protein has been implicated in the regulation of pyrophosphatase in Clostridium perfringens and Desulfitobacterium hafniense.14,15 In Operon 2, we found a group A [FeFe]-hydrogenase with a NADH dehydrogenase (ubiquinone), a histidine kinase and a NADH dehydrogenase (quinone) found upstream. This resembles the group A [FeFe]-hydrogenases commonly encoded downstream in the genome of organisms containing group C sensory [FeFe]-hydrogenases.1,2,4,11 Collectively, these findings strongly suggest an involvement of TamHydS in H2 sensing and signal transduction.
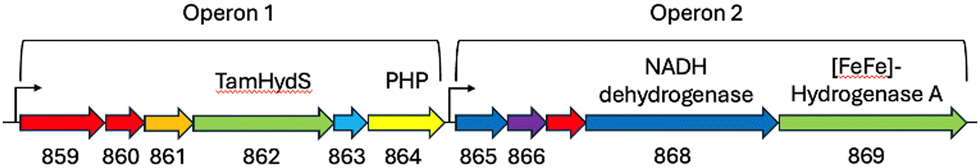 | ||
| Fig. 2 Genome region surrounding TamHydS. Both TamHydS and a group A [FeFe]-hydrogenase are present (light green) in neighbouring operons. directly upstream of TamHydS a Ser/Thr protein kinase is found (861, orange), downstream, a DRTGG protein (863, light blue), followed by PHP domain protein (864, yellow) are found. In Operon 2 a NADH dehydrogenase (ubiquinone) and NADH dehydrogenase (quinone) are marked as blue, 865 and 868 respectively. A His kinase (866, purple) can be found downstream of the group A [FeFe]-hydrogenase. Conserved hypothetical proteins present in the region are marked as red arrows. Promoters are labelled as black arrows. | ||
We investigated the potential H2 sensing mechanism of TamHydS at a molecular level via ATR-FTIR difference spectroscopy. A solution of purified TamHydS enzyme was deposited on the surface of the ATR crystal, dried and rehydrated under humidified N2 gas as reported previously.16 We populated the potential signalling state, Hred, in two ways (i) via H2 uptake and (ii) via photo-reduction (Fig. 3 inset). Upon exposure to H2 gas, we observed the formation of the diiron site reduced state and the potential signalling state, Hred, to the expense of the oxidised resting state, Hox. The corresponding difference spectrum (Fig. 3 red) showed positive bands corresponding to Hred (2063, 2032, 1922, 1896, 1802 cm−1) and negative bands associated with Hox (2082, 2074, 1970, 1948, 1787 cm−1), respectively. Populating Hredvia our previously developed photo-reduction protocol10 resulted in a difference spectrum nearly identical to that obtained via H2 exposure (Fig. 3 black spectrum). The band positions of Hred enriched via the photo-reduction approach are slightly shifted (ca. 1 cm−1) mainly to lower wavenumbers, compared to the Hred signature enriched via H2 exposure, most likely due to additional reduction of F-clusters.17,18 Furthermore, we detected a μCO band (1801–1803 cm−1) associated with Hred for both reduction methods. We were able to monitor the co-population of all bands assigned to a single redox state over the course of the photo-reduction experiment (Table S1 and Fig. S1, ESI†). Beyond the fingerprint region of the H–cluster ligand bands (2120–1780 cm−1), we detected difference features in the region of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations (1750–1600 cm−1), which will be discussed in detail in the next section.
O vibrations (1750–1600 cm−1), which will be discussed in detail in the next section.
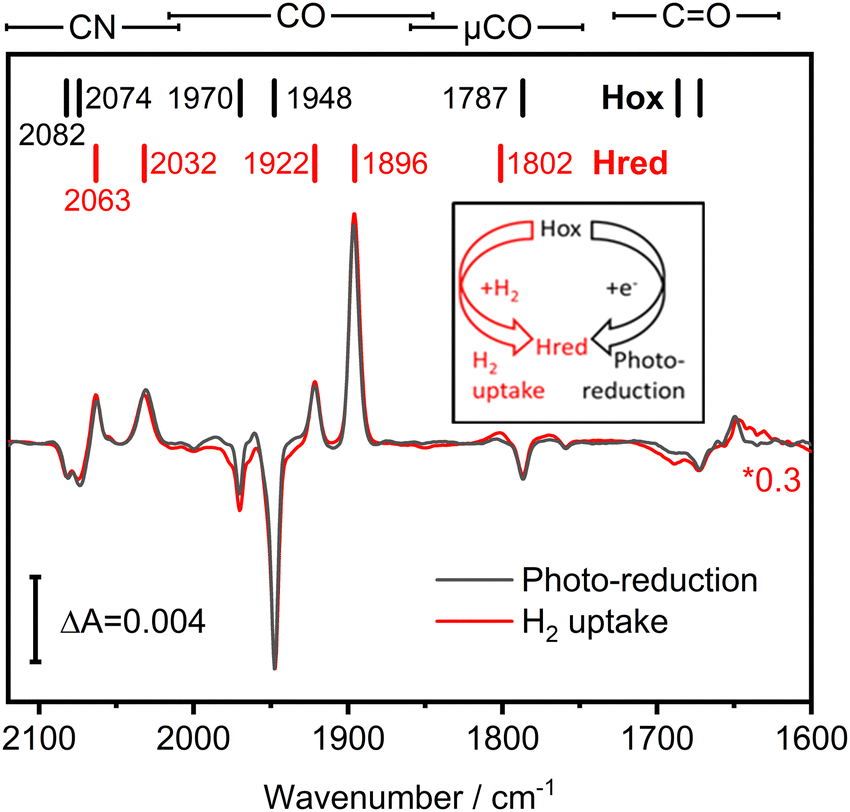 | ||
| Fig. 3 FTIR difference spectra showing the population of the signalling state, Hred, via H2 uptake and via photo-reduction. We observed population of the reduced state Hred (positive peaks: 2063, 2032, 1922, 1896, 1802 cm−1) and a de-population of Hox (negative peaks: 2082, 2074, 1970, 1948, 1787 cm−1) upon exposure of TamHydS to either H2 (red spectrum) or illumination of TamHydS in the presence of eosin Y and EDTA as sacrificial electron donor (black spectrum, photo-reduction). The respective vibrational regions are indicated above the graph. Band positions are indicated by coloured bars. The H2 uptake spectrum is scaled by 0.3 for better comparability. (inset) Scheme illustrating the two ways to populate Hred starting from Hox either via H2 uptake or photo-reduction. | ||
In Fig. 4, the difference spectra in the CO region of the Hox to Hred transition induced via photo-reduction for group D [FeFe]-hydrogenase TamHydS (black) and group A representative CrHydA1 (blue, data from ref10) are overlaid. These spectra are scaled based on the negative band for Hox to facilitate comparison (Fig. S2, ESI†). The CO region can be separated into the CO vibrations of carboxylic acid residues found between 1750–1690 cm−1 and of CO vibrations from the peptide back bone found at 1690–1600 cm−1, which is denoted as amide I vibration (Fig. 4).19,20 In CrHydA1, the difference features related to carboxylic acid residues between 1750–1690 cm−1 with its most prominent peaks at 1715 and 1700 cm−1 report a rearrangement of the H-bonding network in their unique PTP.10,21TamHydS is missing the carboxylic acids assigned to the 1700/1715 cm−1 feature in group A. Furthermore, no similar difference features were detected from 1750–1690 cm−1, indicating no changes in the H-bonding network of the novel PTP upon reduction. Instead, we observed difference features in the amide I CO region (1690–1600 cm−1). The main difference features are negative peaks at 1687 and 1672 cm−1, as well as a positive peak at 1649 cm−1, which all dominate the H2 induced difference spectrum as well (Fig. 4 and Fig. S2, ESI†). The 1687/1672/1649 cm−1 difference signals correlate with the CO and CN− bands associated with Hox and Hred, respectively (Fig. S1, ESI†). Note that in the HydA1 difference spectrum, no changes of comparable intensity could be detected in the amide I CO region.
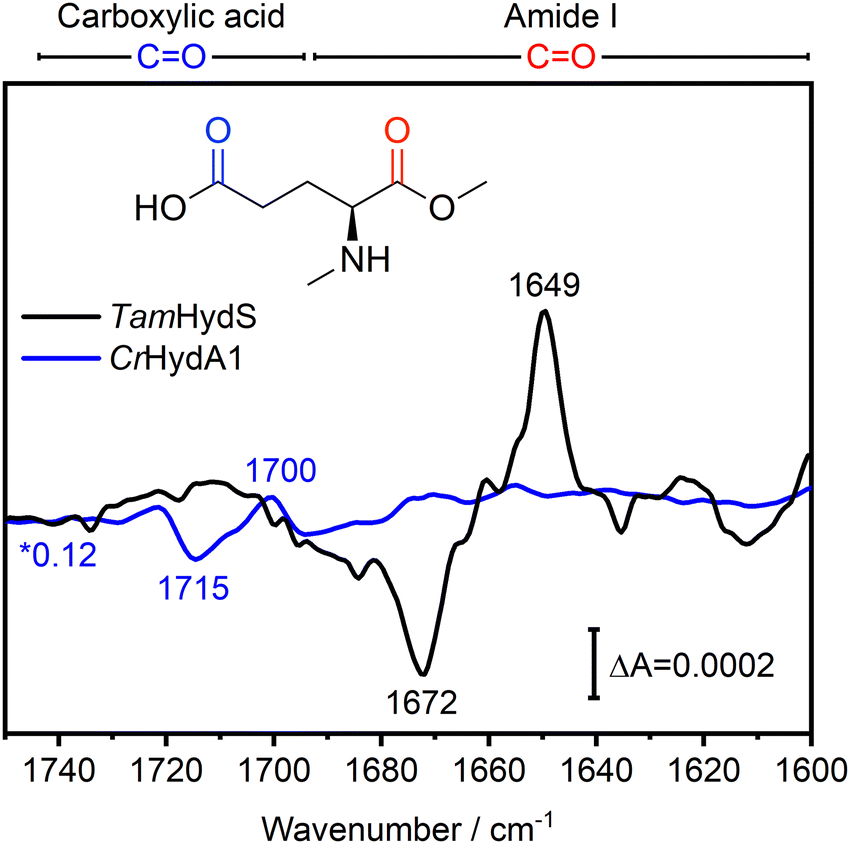 | ||
| Fig. 4 FTIR difference spectra of the CO region indicate secondary structure changes associated with the Hox to Hred transition in TamHydS. In the Hred–Hox difference spectrum of TamHydS induced via photo-reduction (black) we observed a large difference feature in the amide I CO region with a positive peak at 1649 cm−1 and negative peaks at 1687/1672 cm−1 which we assign to a change in secondary structure. In the difference spectrum of the same transition in group A [FeFe]-hydrogenase HydA1 (blue spectrum) no changes in the amide I region were detected. Instead, in CrHydA1 a difference feature in the carboxylic acid CO region (1715 and 1700 cm−1) indicate a rearrangement of the hydrogen bonding network of the PTP. In the same region no difference features indicative of a similar rearrangement of the PTP can be detected in TamHydS. The respective vibrational regions are indicated above the graph. The HydA1 spectrum is scaled by a factor of 0.12 to match the intensity of the negative Hox band to allow for comparison (Fig. S2, ESI†). CrHydA1 data modified from ref. 10. | ||
When considering the alternative PTP of TamHydS, it would be tempting to assign the difference bands at 1671 cm−1 and 1648 cm−1 to changes of the carboxylic acid residue(s) (E252 and E289) that are close to the H–cluster, in analogy to group A [FeFe]-hydrogenases (compare Fig. 1). However, these difference bands are two to three times more intense and are found at lower wavenumbers when compared to group A [FeFe]-hydrogenase. Importantly, the CO vibrations of carboxylic acid residues (1750–1690 cm−1) in proteins are normally not found at these low wavenumbers (1672 and 1649 cm−1).19,20
In HydA1, we could assign the low wavenumber band at 1681 cm−1 to the deprotonation of an arginine. However, no arginine is found in the putative PTP of TamHydS.9 Upon H/D exchange, the negative band at 1672 cm−1 shifts by 2 cm−1 and the positive band at 1649 cm−1 by 4 cm−1 to lower wavenumbers (Fig. S3, ESI†). These minor shifts argue against arginine or localised water molecules as the origin of the difference feature. The exclusion of these possibilities supports our assignment of the observed bands to changes in amide I vibrations induced by secondary structure changes. Similar amide I band positions were assigned to secondary structure changes upon ferredoxin binding in group A [FeFe]-hydrogenase recently.22
Several characteristics of TamHydS indicate its role as a H2 sensor. Once populated, the potential signalling state, Hred, exhibits high stabilitiy.3,12 Furthermore, the low turnover rates disfavour a catalytic purpose, similar to what has been observed for group C sensory [FeFe]-hydrogenases.3,12 The alternative function of TamHydS is additionally supported by its different PTP mechanism when compared to group A representative HydA1. In catalytically highly active group A [FeFe]-hydrogenases, the alternative configuration of the PTP upon reduction was proposed as one of the factors enabling fast catalysis.10,21 In contrast in TamHydS, we detected no such alternative configuration of the PTP that would favour the faster depopulation of the potential signalling state.
Instead, we detected secondary structure changes that we propose to be involved in a potential sensing mechanism. In other biological sensors, large secondary structure changes are well established to facilitate signalling (e.g., the light-sensitive phytochrome proteins).23 In H2 sensing group C [FeFe]-hydrogenases, a PAS domain protein is the most likely signal receptor that triggers a subsequent signal cascade.2 In TamHydS, this PAS domain is absent. However, we identified key enzymes in the genome region that could constitute an alternative signalling cascade. The secondary structure change detected in this study provides the first insight on how H2 sensing in [FeFe]-hydrogenases is facilitated at a molecular level. Understanding and being able to modulate the H2 metabolism in microbes, in particular in antibiotic resistant pathogens, holds promising potential for future treatment of diseases. From a more fundamental research perspective our study gives insight on how nature tunes the function of enzymes harbouring identical cofactors.
M. S. designed experiments, performed the FTIR spectroscopy, analysed data and wrote the first draft of the manuscript. I. V. performed genomic analysis. A. Z. performed the synthesis work. C. S., P. R. C. and H. L. isolated the proteins. P. R. C. performed structural alignment. All authors were involved in the analysis and revision of the manuscript and have given approval to the final version of the manuscript.
The authors thank Leopold Fichet for contributions to the FTIR spectroscopy experiments. The work presented in this article is supported by the Novo Nordisk Foundation (NNF23OC0085682 to M. S.) and by the Swedish Research Council (2023-04593 to M. S. and I. V.). The Olle Engkvist stiftelse (grant no 220-0226 to M. S.) is gratefully acknowledged for funding.
Data availability
The methods and data supporting the findings of this study are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS.
- Y. Zheng, J. Kahnt, I. H. Kwon, R. I. Mackie and R. K. Thauer, J. Bacteriol., 2014, 196, 3840–3852 CrossRef PubMed.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Nemeth, N. Polidori, L. S. Meszaros, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 12789–12801 RSC.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rudiger, W. Lubitz and H. Ogata, J. Am. Chem. Soc., 2018, 140, 1057–1068 CrossRef CAS PubMed.
- S. L. Benoit, R. J. Maier, R. G. Sawers and C. Greening, Microbiol. Mol. Biol. Rev., 2020, 84 DOI:10.1128/mmbr.00092-00019.
- F. Carbonero, A. C. Benefiel and H. R. Gaskins, Nat. Rev. Gastroenterol. Hepatol., 2012, 9, 504–518 CrossRef CAS PubMed.
- C. Greening, P. R. Cabotaje, L. E. Valentin Alvarado, P. M. Leung, H. Land, T. Rodrigues-Oliveira, R. I. Ponce-Toledo, M. Senger, M. A. Klamke, M. Milton, R. Lappan, S. Mullen, J. West-Roberts, J. Mao, J. Song, M. Schoelmerich, C. W. Stairs, C. Schleper, R. Grinter, A. Spang, J. F. Banfield and G. Berggren, Cell, 2024, 187, 3357–3372 CrossRef CAS PubMed.
- S. Morra, Front. Microbiol., 2022, 13, 853626 CrossRef PubMed.
- P. R. Cabotaje, K. Walter, A. Zamader, P. Huang, F. Ho, H. Land, M. Senger and G. Berggren, ACS Catal., 2023, 13, 10435–10446 CrossRef CAS PubMed.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knor, U. P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS PubMed.
- N. Chongdar, P. Rodriguez-Macia, E. J. Reijerse, W. Lubitz, H. Ogata and J. A. Birrell, Chem. Sci., 2023, 14, 3682–3692 RSC.
- H. Land, P. Ceccaldi, L. S. Meszaros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2019, 10, 9941–9948 RSC.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, Chem. Sci., 2016, 7, 959–968 RSC.
- H. Tuominen, A. Salminen, E. Oksanen, J. Jamsen, O. Heikkila, L. Lehtio, N. N. Magretova, A. Goldman, A. A. Baykov and R. Lahti, J. Mol. Biol., 2010, 398, 400–413 CrossRef CAS PubMed.
- V. A. Anashkin, A. Salminen, V. N. Orlov, R. Lahti and A. A. Baykov, Arch. Biochem. Biophys., 2020, 692, 108537 CrossRef CAS PubMed.
- M. Senger, T. Kernmayr, M. Lorenzi, H. J. Redman and G. Berggren, Chem Commun., 2022, 58, 7184–7187 RSC.
- P. Rodríguez-Maciá, N. Breuer, S. DeBeer and J. A. Birrell, ACS Catal., 2020, 10, 13084–13095 CrossRef.
- P. Rodriguez-Macia, K. Pawlak, O. Rudiger, E. J. Reijerse, W. Lubitz and J. A. Birrell, J. Am. Chem. Soc., 2017, 139, 15122–15134 CrossRef CAS PubMed.
- V. A. Lorenz-Fonfria, Chem. Rev., 2020, 120, 3466–3576 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- J. Duan, A. Hemschemeier, D. J. Burr, S. T. Stripp, E. Hofmann and T. Happe, Angew. Chem., Int. Ed., 2023, 62, e202216903 CrossRef CAS PubMed.
- S. Şahin, J. Brazard, T. B. M. Adachi, R. D. Milton and S. T. Stripp, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-pnm11.
- H. Takala, A. Björling, O. Berntsson, H. Lehtivuori, S. Niebling, M. Hoernke, I. Kosheleva, R. Henning, A. Menzel, J. A. Ihalainen and S. Westenhoff, Nature, 2014, 509, 245–248 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, infrared band patterns of different redox states, FTIR difference spectra of the Hox to Hred transition and band kinetics, H/D exchange data, fit parameters, annotated genome region of T. mathranii. See DOI: https://doi.org/10.1039/d4cc03098b |
| ‡ Current address: Laboratoire d’Electrochimie Moléculaire (LEM), Université Paris Cité, CNRS, F-75006, Paris, France. |
| This journal is © The Royal Society of Chemistry 2024 |