
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Brønsted acid-mediated thiazole synthesis from sulfoxonium ylides†
Joe L.
Smy
 ,
Roxanne
Ifill
and
Storm
Hassell-Hart
*
,
Roxanne
Ifill
and
Storm
Hassell-Hart
*
Department of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK. E-mail: s.hassell-art@sussex.ac.uk
First published on 1st October 2024
Abstract
A Brønsted acid-mediated insertion of thioureas/thioamides into sulfoxonium ylides to synthesise 40 thiazoles (34–95% yields) under mild, metal-free conditions is described. This process is scalable, substrate-tolerant (including both α-substituted and heterocyclic ylides/groups) and was successfully applied to the late-stage functionalisation of the complex chemical probe molecule (+)-JQ1.
Thiazoles are an important class of biologically active scaffold.1–5 They are highly versatile motifs forming the core of many pharmaceuticals6–8 and have recently been identified as a potential rigid exit vector in the growing field of VHL-based PROTACS.9 Historically, these motifs were prepared via the Hantzsch thiazole synthesis, a condensation reaction between an α-halo ketone and thiourea.10 Despite this method's prevalence, it suffers from significant limitations, most notably associated with the stability, toxicity and availability of the α-halo ketone precursor.11,12 To circumvent the need for α-halo carbonyls, novel synthetic strategies have been developed to replace these groups, including the use of diazo compounds,13,14 copper-catalysed condensations of oximes with anhydrides and KSCN,15 base-mediated cyclisations of substituted isocyanides with α-oxodithioesters,16 and more recently the use of sulfoxonium ylides.11,17,18
Since their discovery in the 1930s,19 sulfoxonium ylides have proven to be highly versatile intermediates in organic synthesis. Traditionally, these reagents are utilised in the construction of small ring systems, such as cyclopropanes, aziridines and epoxides.20,21 In 1993, pioneering work in the Baldwin group demonstrated sulfoxonium ylides potential as metal carbenoid precursors, enabling their formal insertion into N–H bonds.22 Since this report, sulfoxonium ylides have seen increased use as replacements for problematic diazo compounds, owing to their inherent stability, ease of synthesis and amenability to industrial-scale use.23–25 However, despite these recent advances, sulfoxonium ylide chemistry remains under-explored.
Previously, both our and the Wu group, reported novel routes to thiazoles via iridium or rhodium-catalysed carbenoid insertion of sulfoxonium ylides with thioureas (Fig. 1A).11,18 In both cases, the major limitations were the use of high loadings of expensive/precious metal catalysts and elevated reaction temperatures. To improve sustainability, reduce costs, and increase the substrate scope, the development of a mild metal-free synthesis of thiazoles from sulfoxonium ylides is highly desirable.26
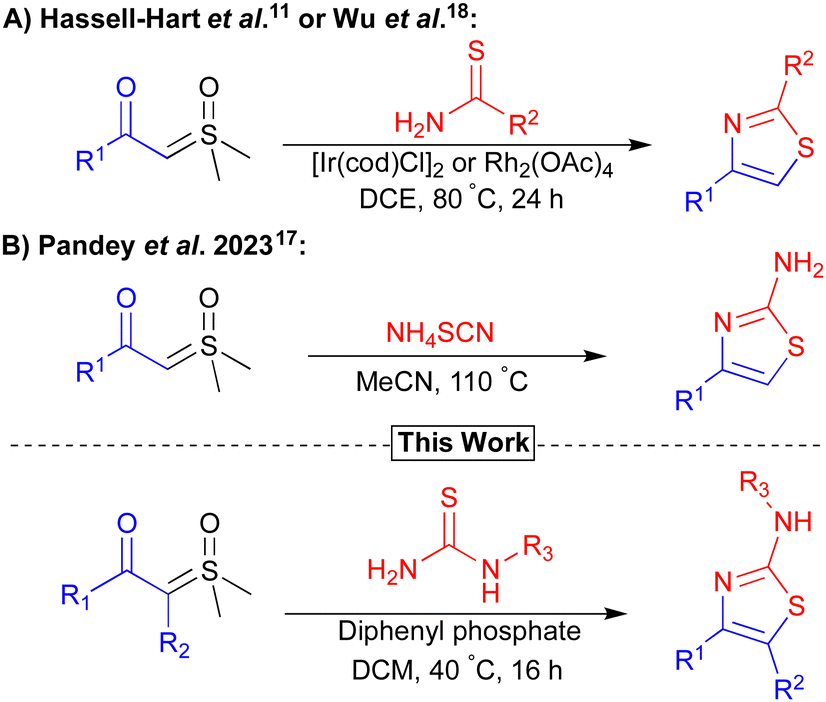 | ||
| Fig. 1 Previous selected strategies (A and B) to access thiazole motifs from sulfoxonium ylides and our work. | ||
Brønsted acids have been reported as alternatives to transition metal catalysts for the activation of sulfoxonium ylides. The Nugent and Burtoloso groups demonstrated this reactivity for the acid-mediated insertions of chloride and thiols respectively.12,27 More recently, Pandey et al. published the synthesis of α-thiocyanoketones from the reaction between ammonium thiocyanate and sulfoxonium ylides.17 In this study, Pandey et al. also prepared a small subset of 2-aminothiazoles from the reaction of amines with a thiocyanoketone intermediate under metal-free conditions, albeit using high temperatures (110 °C) with limited amine/ylide scope (Fig. 1B). We envisaged that an acid-mediated ylide activation, followed by the insertion/cyclisation of a thiourea/thioamide nucleophile, would provide a mild method to access a diverse set of thiazole products. Reported herein is a convenient, widely tolerant, scalable, method that achieves this goal (Fig. 1).
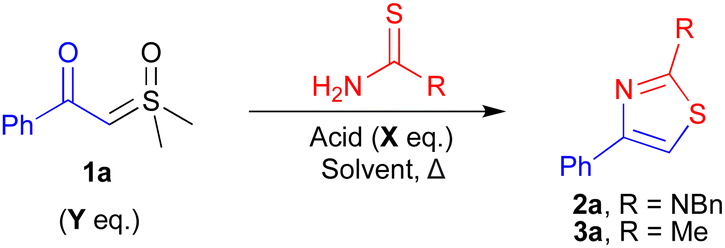
We began our investigations by exploring the reactions of the ylide 1a with the model thioamide/thiourea substrates thioacetamide and N-benzylthiourea (Table 1). Initially, we screened the previously reported conditions for the acid-mediated insertion of chloride (entry 1)27 or thiols (entry 2)12 in our model reaction with thioacetamide. Both reactions were moderately successful, giving the desired thiazole 3a in 14% and 33% yields respectively. Now confident in the viability of our system, we carried out a full solvent, acid, and temperature screen (also see ESI†). From our initial solvent/temperature screening, we identified that dichloromethane (DCM) at 40 °C gave the best conversion to thiazole 3a with diphenyl phosphate (DPP) as the acid (59%, entry 3). No starting ylide conversion was observed in the absence of DPP (entry 4). This suggests the reaction occurs via previously proposed ylide protonation and subsequent nucleophilic attack (see ESI†).12 Surprisingly, increasing the reaction temperature in DCE reduced conversion to 3a (42%) (entry 5). Next, we evaluated the performance of different acids (entries 6–8) (also see ESI†). We found that replacing DPP with TFA gave a similar yield of 64% (entry 9) compared to 59% (entry 3). Further increases in conversion were observed upon diluting the reaction mixture (1.0 M to 0.2 M) and increasing the equivalents of acid (from 0.2 eq.) (entries 10–12). Pleasingly, the more benign DPP gave improved conversions relative to TFA, with 0.5 eq. being the optimal loading (entry 12). Next, we explored the suitability of these conditions for the synthesis of 2-aminothiazole 2a using N-benzylthiourea (entries 13 and 14). Good transferability of the reaction conditions was observed, although, one equivalent of acid was required to give higher conversions (79%) to the thiazole 2a (entry 14). Finally, we found that using a slight excess of the ylide led to higher conversions, affording the thiazoles 3a and 2a in excellent 82% and 88% yields respectively (entries 15 and 16).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Acid | Acid (X eq.) | Ylide (Y eq.) | R | NMR conversiona (%) |
| a Calculated relative to an internal 1,3,5-trimethoxybenzene standard. Reaction conditions: b THF (1.0 M), 70 °C, thioacetamide (1.5 eq.), 24 h. c Solvent (1.0 M), 40 °C, thioacetamide (1.5 eq.), 16 h. d Solvent (1.0 M), 80 °C, thioacetamide (1.5 eq.), 16 h. e DCM (0.2 M), 40 °C, nucleophile (1.5 eq.), 16 h. f DCM (0.2 M), 40 °C, nucleophile (1.0 eq.), 16 h. | ||||||
| 1b | THF | MsOH | 0.2 | 1.0 | Me | 14 |
| 2c | MeCN | DPP | 0.2 | 1.0 | Me | 33 |
| 3c | DCM | DPP | 0.2 | 1.0 | Me | 59 |
| 4c | DCM | DPP | 0.0 | 1.0 | Me | 0 |
| 5d | DCE | DPP | 0.2 | 1.0 | Me | 42 |
| 6c | DCM | MsOH | 0.2 | 1.0 | Me | 45 |
| 7c | DCM | HNO3 | 0.2 | 1.0 | Me | 47 |
| 8c | DCM | AcOH | 0.2 | 1.0 | Me | 6 |
| 9c | DCM | TFA | 0.2 | 1.0 | Me | 64 |
| 10e | DCM | TFA | 1.0 | 1.0 | Me | 69 |
| 11e | DCM | DPP | 1.0 | 1.0 | Me | 70 |
| 12e | DCM | DPP | 0.5 | 1.0 | Me | 77 |
| 13e | DCM | DPP | 0.5 | 1.0 | NBn | 73 |
| 14e | DCM | DPP | 1.0 | 1.0 | NBn | 79 |
| 15 | DCM | DPP | 0.5 | 1.25 | Me | 82 |
| 16 | DCM | DPP | 1.0 | 1.25 | NBn | 88 |
With optimised reaction conditions in hand, we first investigated the scope of thioureas for this reaction (Scheme 1A). The reaction conditions tolerated a broad range of functional groups, enabling access to thiazoles containing a range of aromatic 2b, 2d, 2e (73–82%), alkene 2g (75%), free amine 2m (81% vs. 72% on gram-scale), phenolic 2c (49%), alkyl 2a, 2j, 2l (68–78%) and heteroaromatic 2h, 2k (76–95%) substituents in good to excellent yields. Of note, we also synthesised the anti-inflammatory drug fanetizole (2f) in a high 84% yield, comparable to our previously reported iridium route (83%).11
 | ||
| Scheme 1 Synthesis of thiazoles from the acid-mediated cyclisation of thioureas and thioamides with sulfoxonium ylides. | ||
Pleasingly, we found the use of selenourea under our optimised conditions was well tolerated, giving selenazole 2n in 65% yield.
To further explore the scope of our reaction, we next investigated changing the ylide precursor (Scheme 1A). Aromatic ylides bearing both electron-donating 2o, 2t, 2p, 2v (4-Me, 2-OMe, 4-OMe, 1,3-benzodioxole) and electron-withdrawing 2q-2s (4-Cl, 4-NO2, 4-CF3) groups were well tolerated under the reaction conditions. The thiazole scaffolds containing the ‘Cl’ and ‘NO2’ handles offer the potential for further elaboration.11 Furthermore, alkyl ylides were also tolerated delivering methyl 2u (61%), cyclopropyl 2a′ (69%) and cyclobutyl 2b′ (61%) thiazoles. Notably, styrene-derived thiazole 2y was obtained in a high 83% yield exclusively as the more thermodynamically stable E-isomer, as determined by 1H NMR spectroscopy (3JHH = 15.6 Hz). Moreover, excellent compatibility was observed for the thiophene-derived ylide, giving 2x in 78% yield. Surprisingly, the presence of basic centres in both the ylide (2w, 2z) and thiourea (2i) were tolerated, albeit requiring elevated temperatures. We propose this is due to the formation of nitrogen salts which are solubilised at 80 °C. In the case of piperidine (2z) an extra equivalent of acid was required due to the increased basicity. Finally, we are pleased to report the first examples of the synthesis of trisubstituted thiazoles 2c′–2f′ directly from α-substituted sulfoxonium ylides. Despite concerns about the steric environment, the reactions proceeded smoothly in 39-74% yield under the standard reaction conditions.
Following the successful exploration of synthesising 2-aminothiazole derivatives, we revisited the insertion of thioamide nucleophiles (Scheme 1B). Outside of the model substrate 3a, we found that significant increases in yield were observed upon raising the reaction temperature to 80 °C (see 3b and 3c), likely due to their reduced nucleophilicity relative to thioureas. Despite this, the insertion of thioamides afforded thiazoles in moderate yields ranging from 36–69% (3a–3f). In addition to the formation of the desired thiazole, we surprisingly observed the formation of methylthioether side products (3a′–3f′).
Thiomethylated thiazole 3a′ was isolated in 10% yield, with the site of SMe incorporation confirmed by comparison with spectroscopic data in the literature.28 In all thioamide cases, significant thiomethylation was observed, ranging between 7–14% by LCMS. This could account for the lower yields of the thioamide-derived thiazoles relative to the thiourea examples.
Finally, we attempted the late-stage introduction of a thiazole to (rac)-ibuprofen and the BET bromodomain inhibitor (+)-JQ1 (Scheme 1C).29 Gratifyingly, under our optimised conditions, we obtained the free 2-aminothiazoles derivatives of ibuprofen (2g′, 54%) and (+)-JQ1 (4b, 54%) without racemisation of the stereogenic centre. This was confirmed via1H NMR spectroscopy utilising Pirkle's alcohol as a chiral solvating agent, in comparison to the racemate (see ESI†). The introduction of the NH2-thiazole is of interest in medicinal chemistry, for example providing a rigid handle/linker for the synthesis of PROTACs.30,31
In summary, we have developed a scalable, mild, metal-free method to access diverse thiazole scaffolds from the insertion of thioamide/thioureas into sulfoxonium ylide precursors. This methodology proved particularly effective for obtaining 2-aminothiazoles and was successfully applied to the functionalisation of the highly complex probe molecule (+)-JQ1. Whilst exploring the insertion of thioamides, we were surprised to observe the formation of thiomethylated side products. Detailed mechanistic investigations probing SMe incorporation are currently ongoing in our lab and may enable novel access to thiomethylated derivatives, which could serve as trisubstituted cores for further elaboration.
We would also like to acknowledge and thank Prof. John Spencer and Dr Barnaby W. Greenland for their input and guidance, as well as Dr Graham Marsh of Tocris Biotechne for providing the racemic sample of JQ1. We would finally also like to thank the University of Sussex for funding this project.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M. F. Arshad, A. Alam, A. A. Alshammari, M. B. Alhazza, I. M. Alzimam, M. A. Alam, G. Mustafa, M. S. Ansari, A. M. Alotaibi, A. A. Alotaibi, S. Kumar, S. M. B. Asdaq, M. Imran, P. K. Deb, K. N. Venugopala and S. Jomah, Molecules, 2022, 27, 3994 CrossRef PubMed.
- S. Johannsen, R. M. Gierse, A. Krüger, R. L. Edwards, V. Nanna, A. Fontana, D. Zhu, T. Masini, L. P. de Carvalho, M. Poizat, B. Kieftenbelt, D. M. Hodge, S. Alvarez, D. Bunt, A. Lacour, A. Shams, K. A. Meissner, E. E. de Souza, M. Dröge, B. van Vliet, J. den Hartog, M. C. Hutter, J. Held, A. R. Odom John, C. Wrenger and A. K. H. Hirsch, ACS Infect. Dis., 2024, 10, 1000–1022 CrossRef CAS PubMed.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed, M. H. Abdellattif, M. S. Nafie and M. K. Hassan, Front. Chem., 2021, 9 DOI:10.3389/fchem.2021.694870.
- A. Singh, D. Malhotra, K. Singh, R. Chadha and P. M. S. Bedi, J. Mol. Struct., 2022, 1266, 133479 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. Steinberg, Clin. Ther., 2007, 29, 2289–2308 CrossRef CAS.
- G. G. Zhanel, A. R. Golden, S. Zelenitsky, K. Wiebe, C. K. Lawrence, H. J. Adam, T. Idowu, R. Domalaon, F. Schweizer, M. A. Zhanel, P. R. S. Lagacé-Wiens, A. J. Walkty, A. Noreddin, J. P. Lynch and J. A. Karlowsky, Drugs, 2019, 79, 271–289 CrossRef CAS.
- A. P. Lea and D. Faulds, Drugs, 1996, 52, 541–546 CrossRef CAS PubMed.
- J. Krieger, F. J. Sorrell, A. A. Wegener, B. Leuthner, F. Machrouhi-Porcher, M. Hecht, E. M. Leibrock, J. E. Müller, J. Eisert, I. V. Hartung and S. Schlesiger, ChemMedChem, 2023, 18, e202200615 CrossRef CAS.
- A. Hantzsch and J. H. Weber, Ber. Dtsch. Chem. Ges., 1887, 20, 3118–3132 CrossRef.
- S. Hassell-Hart, E. Speranzini, S. Srikwanjai, E. Hossack, S. M. Roe, D. Fearon, D. Akinbosede, S. Hare and J. Spencer, Org. Lett., 2022, 24, 7924–7927 CrossRef CAS PubMed.
- R. M. P. Dias and A. C. B. Burtoloso, Org. Lett., 2016, 18, 3034–3037 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, Y. G. Rao and A. V. Narsaiah, Tetrahedron Lett., 2008, 49, 2381–2383 CrossRef CAS.
- M. Luo, L. Li, S. Chen, Q. Yan, J. Lv, J. Zeng, H. Wang, S. Gu and F. Chen, J. Org. Chem., 2024, 89, 5038–5048 CrossRef CAS.
- X. Tang, J. Yang, Z. Zhu, M. Zheng, W. Wu and H. Jiang, J. Org. Chem., 2016, 81, 11461–11466 CrossRef CAS.
- K. R. Kiran, T. R. Swaroop, N. Rajeev, S. M. Anil, K. S. Rangappa and M. P. Sadashiva, Synthesis, 2020, 1444–1450 CAS.
- A. Sharma, A. K. Gola and S. K. Pandey, Chem. Commun., 2023, 59, 10247–10250 RSC.
- Y. Chen, S. Lv, R. Lai, Y. Xu, X. Huang, J. Li, G. Lv and Y. Wu, Chin. Chem. Lett., 2021, 32, 2555–2558 CrossRef CAS.
- C. K. Ingold and J. A. Jessop, J. Chem. Soc., 1930, 713–718 RSC.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353–1364 CrossRef CAS.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 867–868 CrossRef CAS.
- J. E. Baldwin, R. M. Adlington, C. R. Godfrey, D. W. Gollins and J. G. Vaughan, J. Chem. Soc., Chem. Commun., 1993, 1434–1435 RSC.
- P. B. Momo, A. N. Leveille, E. H. E. Farrar, M. N. Grayson, A. E. Mattson and A. C. B. Burtoloso, Angew. Chem., Int. Ed., 2020, 59, 15554–15559 CrossRef CAS.
- I. K. Mangion, I. K. Nwamba, M. Shevlin and M. A. Huffman, Org. Lett., 2009, 11, 3566–3569 CrossRef CAS.
- C. Molinaro, P. G. Bulger, E. E. Lee, B. Kosjek, S. Lau, D. Gauvreau, M. E. Howard, D. J. Wallace and P. D. O’Shea, J. Org. Chem., 2012, 77, 2299–2309 CrossRef.
- J. R. Ludwig and C. S. Schindler, Chemistry, 2017, 2, 313–316 CrossRef.
- D. Wang, M. D. Schwinden, L. Radesca, B. Patel, D. Kronenthal, M. H. Huang and W. A. Nugent, J. Org. Chem., 2004, 69, 1629–1633 CrossRef PubMed.
- M. Hori, K. Nogi, A. Nagaki and H. Yorimitsu, Asian J. Org. Chem., 2019, 8, 1084–1087 CrossRef.
- G. Jiang, W. Deng, Y. Liu and C. Wang, Mol. Med. Rep., 2020, 21, 1021–1034 Search PubMed.
- N. A. Zografou-Barredo, A. J. Hallatt, J. Goujon-Ricci and C. Cano, Bioorg. Med. Chem., 2023, 88–89, 117334 CrossRef PubMed.
- R. I. Troup, C. Fallan and M. G. J. Baud, Explor. Target. Antitumor. Ther., 2020, 1, 273–312 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03905j |
| This journal is © The Royal Society of Chemistry 2024 |