
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interstitial and substitutional doping of Mn2+ in 2D PEA2PbBr4 and BA2PbBr4 perovskites†
Udara M.
Kuruppu
 a,
Alvaro J.
Magdaleno
cd,
Anuraj S.
Kshirsagar
a,
Bruno
Donnadieu
a,
Ferry
Prins
*cd and
Mahesh K.
Gangishetty
*ab
a,
Alvaro J.
Magdaleno
cd,
Anuraj S.
Kshirsagar
a,
Bruno
Donnadieu
a,
Ferry
Prins
*cd and
Mahesh K.
Gangishetty
*ab
aDepartment of Chemistry, Mississippi State University, Mississippi State, Mississippi 39762, USA. E-mail: mg2234@msstate.edu
bDepartment of Physics and Astronomy, Mississippi State University, Mississippi State, Mississippi 39762, USA
cCondensed Matter Physics Center (IFIMAC), Autonomous University of Madrid, 28049, Madrid, Spain. E-mail: ferry.prins@uam.es
dDepartment of Condensed Matter Physics, Autonomous University of Madrid, 28049, Madrid, Spain
First published on 22nd November 2024
Abstract
Mn2+ doping imposes intriguing optoelectronic properties on lead-halide perovskites; however, its impact on their crystal structure remains unclear. This study investigates the consequences of interstitial and substitutional Mn2+ doping on the lattice-strain and interplanar spacings of 2D perovskites and correlates the structural changes to their optical properties.
Doping with a foreign metal ion is an attractive strategy to improve the properties of semiconductors such as lead-halide perovskites. Due to the flexible nature of perovskite structures, a wide range of metal ions have been explored. Among these, Mn2+ has been most extensively studied due to its bright characteristic emission and paramagnetic nature.1,2 Despite these extensive studies, the nature of Mn2+ doping such as interstitial or substitutional, and the role of host lattice on the doping is still ambiguous. There have been several contradicting results reported on the type of Mn2+ doping. Some reports showed lattice contraction due to substitutional Mn2+ doping, while others demonstrated lattice expansion owing to their interstitial nature. Generally, the lattice expansion and contraction can be identified by the shift in their p-XRD peaks. Recently, Torma et al. observed a shift in the XRD peak to lower angles and demonstrated interstitial Mn2+ doping in Cs2PbI2Cl2 using nano-XRD and nano-XRF measurements.3 On the contrary, Dutta et al. and Hou et al. observed a peak shift to the higher angles in XRD after doping with Mn2+ in 2D Cs2PbBr4 nanoplatelets and 3D CsPb(BrCl)3 perovskite nanocrystals.4,5 Interestingly, Nag et al. observed no shift in the XRD peak when Mn2+ (<1%) is doped in 2D BA2PbBr4 perovskite.6 Therefore, a comprehensive understanding of the nature of doping, its dependence on the host lattice structure, and its impact on the crystal structure, lattice strain, and optical properties of perovskites is required.
In this study, we utilize Mn2+ doping in two different 2D perovskite hosts: (i) phenylethylammonium lead bromide (PEA2PbBr4) and (ii) butylammonium lead bromide (BA2PbBr4), to investigate their structural changes and link these changes to their optical properties. These two perovskites are selected by considering the difference in their structural rigidity owing to the difference in their A-site ions, although both adopt similar layered 2D structures of the Ruddlesden–Popper (RP) type.7 Consequently, our study reveals that the nature of Mn2+ doping is significantly different in both PEA2PbBr4 and BA2PbBr4. We observed a striking difference in their XRD patterns after Mn2+ doping. PEA2PbBr4 showed lattice expansion perpendicular to the plane of the 2D inorganic framework (interstitial nature) after doping with Mn2+ whereas BA2PbBr4 showed a lattice contraction along the 2D plane (substitutional doping). Due to the manifestation of a soft lattice with a layered structure of 2D perovskites, the Mn2+ doping was feasible, and the changes in their crystal structure were easily noticeable after Mn2+ incorporation. Subsequently, this resulted in a dramatic difference in their emission; orange (Mn2+) emission in Mn2+:PEA2PbBr4 is more intense (∼10× higher) than that of Mn2+:BA2PbBr4. Upon tracking the exciton diffusion using transient photoluminescence microscopy (TPLM), we found that the exciton transport in PEA2PbBr4 is unaffected, whereas a noticeable difference in the exciton transport was observed in BA2PbBr4 with Mn2+ doping.
To commence our study, undoped and a series of Mn2+ doped 2D perovskites were synthesized using acid-initiated precipitation (detailed methodology is available in the ESI†). The ICP-MS analyses reveal that the amount of Mn2+ increases with an increase in the concentration of Mn2+ salts in the precursors (shown in Table S3, ESI†). However, the maximum feasible doping amount was 4.8% and 2.0% of Mn2+ (respective to the molar percentage of Pb2+) for PEA2PbBr4 and BA2PbBr4, respectively. Such low doping levels of Mn2+ are attributed to a difference in the ionic radii of Mn2+ (0.83 Å) compared to that of Pb2+ (1.19 Å).6,8 The crystal structures of PEA2PbBr4 and BA2PbBr4 were analyzed by scXRD and shown in Fig. 1a and b (see Tables S1 and S2 for refined data, ESI†). Both perovskites show 2D layered Ruddlesden–Popper (RP) type structures and exhibit a higher degree of preferential orientation along the c-axis. Further, their p-XRD patterns were recorded after doping with various amounts of Mn2+ (Fig. 1c and d), which was and used for strain analyses. From p-XRD, we found that both perovskites retain the RP structures even after doping with Mn2+.
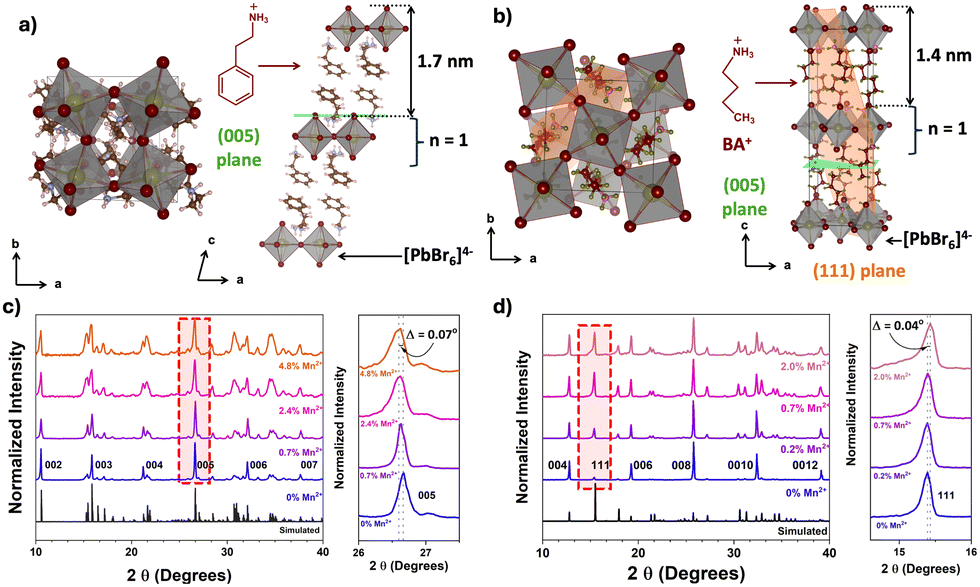 | ||
| Fig. 1 Crystal structures of (a) PEA2PbBr4 and (b) BA2PbBr4. The pXRD patterns of undoped and Mn2+ doped (c) PEA2PbBr4 and (d) BA2PbBr4. The 2D perovskite lattices of PEA2PbBr4 and BA2PbBr4 are composed of a single (n = 1) inorganic layer of [PbBr6]4− octahedra. The PEA+ and BA+ cations act as organic spacers between the octahedral layers of PEA2PbBr4 and BA2PbBr4, respectively. | ||
In the case of PEA2PbBr4, both undoped and Mn2+ doped perovskites show equally spaced and sharp diffraction peaks in p-XRD (Fig. 1c), denoted as (002), (003), (004), etc., and agree with the simulated pattern of a triclinic phase (lattice parameters a ≠ b ≠ c, α ≠ β ≠ γ) derived from scXRD. All these peaks arise from the same family of planes {00l} where (l = n), indicating the high degree of orientation with stacking layers. The periodic peaks due to these (00l) diffractions imply the spacing between the two layers, since these planes are perpendicular to the stacking axis (c-axis), see Fig. 1a. The periodicity obtained from the diffraction peaks was 1.648 ± 0.007 nm, indicating that each layer is ∼1.65 nm apart, as shown in Fig. 1a. After doping with Mn2+, no major changes in the p-XRD pattern were observed; however, a shift in some XRD peaks with slight broadening was observed. These peaks are at ∼26.6° and at ∼37.6°, which correspond to the (005) plane and the (007) plane, respectively, as shown in the magnified region of Fig. 1c and Fig. S1 (ESI†). The magnitude of the shift was Δ2θ005 = 0.07° and Δ2θ007 = 0.09°. Generally, given the difference in the ionic radii of Pb2+ (1.19 Å) and Mn2+ (0.83 Å), if Pb2+ is replaced by smaller Mn2+, a lattice contraction is expected with a shift in the XRD peaks to higher angular scale. On the contrary, in this case, the XRD peaks shifted to lower angles, indicating that there is a lattice expansion after doping with Mn2+ ions. This also implies that the Mn2+ ions are not replacing Pb2+. Since this shift is observed for (005) and (007) peaks, the lattice expansion occurs in an out-of-plane direction (perpendicular to (005) and (007) planes) along the c-axis, as shown in Fig. 1a. This is also evident from an increase in the spacing of (005) planes, as shown in Fig. 2a. Similar trends in p-XRD were observed by Torma et al.3 By using nano-XRD, they demonstrated that Mn2+ occupies the interstitial position.3
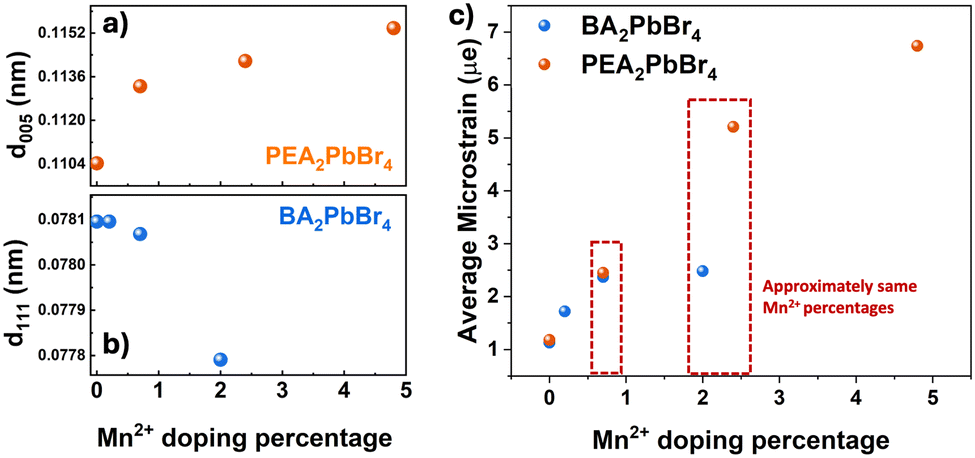 | ||
| Fig. 2 The change in d-spacing against Mn2+ concentration of (a) the (005) plane in PEA2PbBr4 (b) the (111) plane in BA2PbBr4 and (c) microstrain vs. Mn2+ doping percentage. | ||
In the case of 2D BA2PbBr4, the powder XRD patterns of both undoped and Mn2+ doped show equally spaced and sharp diffraction peaks indexed as (002), (004), (006), etc (Fig. 1d). This diffraction pattern aligns well with the simulated pattern obtained from the reference and scXRD data, confirming the orthorhombic Ruddlesden–Popper crystal structure (lattice parameters a ≠ b ≠ c, α = β = γ = 90°). All these peaks arise from the same family of planes {00l}, where (l = 2n) indicates highly oriented crystallites. The periodicity, obtained from {00l} family peaks, was found to be 1.366 ± 0.008 nm. Note that this interlayer spacing is smaller than the spacing in PEA2PbBr4 perovskite. Unlike PEA2PbBr4, neither a peak shift nor broadening of the (00l) planes was observed upon doping, indicating these planes are unaffected after the incorporation of Mn2+ in BA2PbBr4. Interestingly, in addition to the (00l) family peaks, another peak at ∼15.5°, corresponds to the (111) plane, was observed (Fig. 1d). This (111) plane cuts the organic and inorganic layers diagonally, as shown in Fig. 1b.9 Upon a close examination, a slight shift in the ∼15.5° peak to higher angles was observed (magnified region of Fig. 1d). This shift was more prominent at higher (>1%) Mn2+ doping, and almost negligible at lower doping levels. The magnitude of this shift was Δ2θ = 0.04°. The shift in this peak to higher angular scale implies there is a lattice contraction along the out-of-plane direction along the ab-plane (perpendicular to the (111) plane and the c-axis), as shown in Fig. 1b. This is further corroborated by the decrease in d-spacing of (111) planes, as shown in Fig. 2b. From the crystal structure in Fig. 1b, this ab plane represents the plane of the inorganic framework; the lattice contraction along this plane implies that there is shrinkage of the inorganic framework. This is possible only when the central atom, Pb2+, is replaced by an atom with a smaller ionic radius such as Mn2+. Therefore, from these analyses, it is evident that the Mn2+ in BA2PbBr4 is substituting Pb2+, whereas, in the case of PEA2PbBr4, the Mn2+ is occupying interstitial sites.6,10 This difference in occupancy, despite the similar layered RP type structure, could be attributed to either the smaller interlayer spacing in BA2PbBr4 compared to PEA2PbBr4 or a difference in the lattice rigidity owing to difference in their organic spacers (aromatic vs. aliphatic). Perhaps this substitutional nature of Mn2+ doping is limiting the amount of Mn2+ that can be incorporated into BA2PbBr4, since a maximum of only ∼2% doping was achievable synthetically.
Regardless of the crystal phases, type of spacers, and interstitial or substitutional doping, the incorporation of Mn2+ into the perovskite host can induce strain in the lattice. Generally, from the changes in the p-XRD peaks, one can estimate the strain in the lattice. A peak shift with a slight broadening in PEA2PbBr4 after Mn2+ doping may be an indication of a homogenous strain caused by the uniform expansion of the lattice. Whereas a moderate change in peak position in the case of BA2PbBr4, suggesting that the magnitude of the lattice strain is less in the case of BA2PbBr4.11 To quantitatively compare the microstrain induced by Mn2+ incorporation in these two host lattices, we employed the Williamson–Hall method (Fig. S2, ESI†).12 A Voigt function, which is convoluted Lorentzian and Gaussian functions, was used to fit the p-XRD peaks and derive the FWHM for the strain analyses. From Fig. 2c, a striking difference in the microstrain was observed at higher Mn2+ levels. At >2% of Mn2+ doping, the strain in the PEA2PbBr4 is approximately three times higher than that of BA2PbBr4, implying that the interstitial doping can induce more strain in the lattice than the substitutional doping.
Next, the impact of these discrepancies in the Mn2+ occupancy on the optical properties of perovskites was investigated. We recorded absorption, photoluminescence (PL), and exciton diffusion in PEA2PbBr4 and BA2PbBr4 perovskites with and without Mn2+ doping. As shown in Fig. S3 (ESI†), both pristine perovskites exhibit sharp excitonic features in the UV-vis absorption spectra with similar band-edge absorption at 2.89 eV (430 nm) for PEA2PbBr4 and at 2.85 eV (435 nm) for BA2PbBr4 (Fig. S4, ESI†). The absorbance remains unchanged upon Mn2+ doping of the pristine 2D perovskites. Supporting this, we observed no change in the excitation (PLE) spectra after Mn2+ doping (Fig. S6, ESI†). The PL spectra of both pristine 2D perovskites exhibit two emission features around 410 nm and 430 nm, similar to other studies.13 As these are 2D layered perovskites, the lower energy peak at 430 nm is due to the recombination of edge excitons, while the higher energy transition at 410 nm is designated to interior excitons.10,13 When Mn2+ is introduced into both BA2PbBr4 and PEA2PbBr4, a new broad, intense emission at 600 nm was observed. This 600 nm emission has been assigned to 4T1g → 6A1g d–d transition in Mn2+.6,10 These Mn states are typically sensitized from the host's excitation, and the emission originates from the excitonic energy transfer from the host to the dopant ion (Fig. S5, ESI†).6 Interestingly, the intensity of the Mn2+ peak (600 nm) with respect to the host peak (∼430 nm) is significantly different in Mn2+:BA2PbBr4 compared to Mn2+:PEA2PbBr4 (Fig. S7, ESI†); this is true even in the case of samples with the same amount of Mn2+ (Fig. S13, ESI†). For instance, the PL intensity ratio between dopant and host emission in ∼2% Mn2+-doped PEA2PbBr4 suggests an equal intensity dopant peak with the host. Whereas in BA2PbBr4, at the same Mn2+ loading, the intensity of the dopant peak was only ∼1/4th of the host peak (Fig. S7, ESI†). Consequently, under the 365 nm excitation, the color of Mn2+:PEA2PbBr4 crystals appeared significantly different from Mn2+:BA2PbBr4 (Fig. S9–S11, ESI†). The emission color tends to move towards orange (λEm = 600 nm) in Mn2+:PEA2PbBr4; however, a more prominent blue emission in Mn2+:BA2PbBr4 was observed. These deviations in the PL spectra of Mn2+:BA2PbBr4 and Mn2+:PEA2PbBr4 crystals show a clear distinction in the CIE 1931 (Commission Internationale de l’Eclairage) chromaticity diagram in Fig. S14 (ESI†).
Furthermore, the time-resolved photoluminescence (TRPL) was recorded at 430 nm and 435 nm for both undoped PEA2PbBr4 and BA2PbBr4 on the order of nanoseconds (Fig. 3c, and f). The average lifetimes decrease with an increase in Mn2+ levels, as shown in Tables S4 and S5 (ESI†). Under the same excitation fluence, the average lifetime of undoped PEA2PbBr4 (∼8.2 ns) is significantly higher than undoped BA2PbBr4 (∼3 ns). With a small amount of Mn2+ doping, there is a dramatic decrease in the lifetime of the PEA2PbBr4 when compared to BA2PbBr4, indicating an efficient host-dopant energy transfer in Mn2+:PEA2PbBr4. This also complements the intense Mn2+ PL in Mn2+:PEA2PbBr4 compared to Mn2+:BA2PbBr4. Subsequently, the PLQY of the host is decreased (Fig. 3c) and the Mn2+ is increased with an increase in the Mn2+ doping (Fig. 3f). Both the Mn2+ doped 2D perovskites showed a similar decay profile with a prolonged lifetime on the order of microseconds (Fig. S8, ESI†) regardless of the host structure due to the forbidden nature of Mn2+ d–d (4T1g → 6A1g) transitions.10 As expected, the PLQYMn of this Mn2+ emission at 600 nm increased with an increase in the Mn2+ doping in both PEA2PbBr4 and BA2PbBr4. However, this increase in PLQYMn is more prominent in PEA2PbBr4 compared to BA2PbBr4, reaching a maximum of 43.8% (Fig. 3f). From all these studies, it is evident that the host-to-dopant ion energy transfer is more efficient in the case of Mn2+:PEA2PbBr4 compared to Mn2+:BA2PbBr4. Such differences in optical properties hints towards the important contributions from the nature of doping. In addition, the difference in their crystal phases, and the lattice rigidity due to the difference in their organic spacers may play a critical role in the energy transfer. Regardless, introducing foreign Mn2+ ions into the lattice can further perturb the host structure, consequently, the dynamics of exciton transport and the host-to-dopant energy transfer can be affected. After photoexcitation, the excitons generated in the host perovskites diffuse within the inorganic framework before they get trapped in the Mn2+ states. Therefore, the nature of doping, the location of the dopant ions, and strain in the inorganic framework play a critical role in exciton diffusion and energy transport dynamics.14,15 The efficient host-dopant energy transfer despite the greater strain in Mn2+:PEA2PbBr4 perovskites could be due to either (i) the interstitial doping facilitating the smooth sailing of excitons without affecting the landscape of 2D inorganic framework or (ii) the energy transfer barrier for Mn2+ may be lower in Mn2+:PEA2PbBr4. The weak Mn2+ emission in the case of Mn2+:BA2PbBr4 perhaps due to the strain created within the inorganic framework due to the lattice contraction by substitutional doping. This strain may be affecting the exciton transport by creating non-radiative defects caused by an inhomogeneous landscape. Consequently, these excitons decay non-radiatively before reaching Mn2+ states. Also, the difference in contribution towards the non-radiative sites from rigid aromatic PEA cations and labile aliphatic BA cations cannot be ruled out.
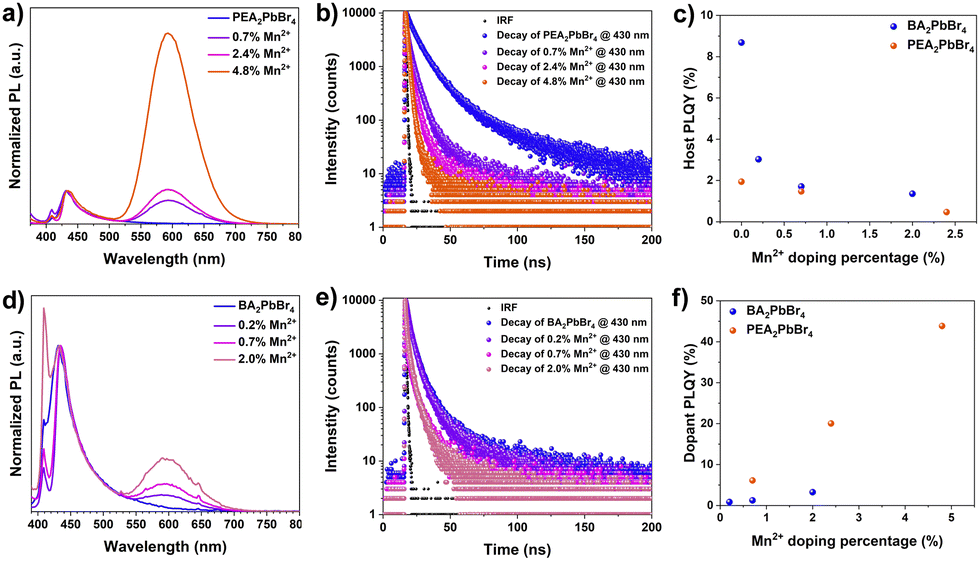 | ||
| Fig. 3 (a) PL spectra of Mn2+:PEA2PbBr4. (b) TRPL of Mn2+:PEA2PbBr4. (c) Host PLQY against Mn2+ percentage. (d) PL spectra of Mn2+:BA2PbBr4. (e) TRPL of Mn2+:BA2PbBr4. (f) Dopant PLQY against Mn2+ percentage. | ||
To unveil these dynamics and study the impact of the different doping nature of the two perovskites on exciton transport, we employ TPLM, which allows for direct visualization of in-plane exciton transport. For this, we specifically chose Mn2+-doped PEA2PbBr4 and BA2PbBr4 with comparable Mn2+ doping percentages. Fig. 4a shows the resulting diffusion maps of the undoped and Mn2+ doped materials. For both undoped perovskites (Fig. 4a, lower panels), fast spatial expansion of the initial exciton population is observed. In contrast, for doped perovskites, the initial fast diffusion is followed by a stagnation of the expansion, consistent with excitons reaching dopant sites and undergoing energy transfer. We can quantify the rate of expansion by fitting the variance σ of the population for each time slice and calculating the mean-square displacement (MSD(t) = σt2 − σ0−), as shown for the different doping levels in Fig. 4b and c for PEA2PbBr4 and BA2PbBr4, respectively. From the early time dynamics (t < 1 ns), we can extract the diffusivity of the exciton population in the lattice (MSD = 2Dt), as indicated by the dashed lines in Fig. 4b and c. For the undoped cases, we observe a higher diffusivity for PEA2PbBr4 (0.22 cm2 s−1) compared to BA2PbBr4 (0.13 cm2 s−1). This is consistent with earlier reports and can be explained by the difference in the softness of the inorganic lattice and a stronger exciton–phonon coupling in BA2PbBr4, which slows down transport.16 Upon doping, it is interesting to note that the diffusivity PEA2PbBr4 remains unaffected (Fig. 4b). In contrast, the expansion is notably slower for the highest (2%) doping concentration in BA2PbBr4, where the diffusivity drops to 0.08 cm2 s−1. Interestingly the same sample, BA2PbBr4 with 2% Mn2+ doping, showed more prominent peak shift in the p-XRD owing to the lattice distortions along the inorganic 2D framework, highlighting the important role of lattice distortions in the exciton diffusion. Further, the lower diffusivity in BA2PbBr4 can partially explain the lower PLQY of the Mn2+ doping in that system, with slower diffusion leading to less efficient energy transfer.
 | ||
| Fig. 4 (a) Diffusion maps of undoped and Mn2+ doped PEA2PbBr4 and BA2PbBr4. The extracted mean-square displacement as a function of time for (b) PEA2PbBr4 and (c) BA2PbBr4 with different doping levels. | ||
In conclusion, Mn2+ doping induces distinct structural changes by occupying different preferential sites in PEA2PbBr4 and BA2PbBr4 2D perovskite hosts lattice, interstitial in the former and substitutional in the latter. Interestingly, the resultant crystal shows intense Mn2+ emission, which is more efficient in PEA2PbBr4 compared to BA2PbBr4. The discrepancy in optical properties is most likely due to the nature of doping and its impact on the crystal lattice, including dopant-induced in-plane and out-of-plane lattice strain. Consequently, we observed a striking difference in the exciton diffusion. However, a few aspects like the interstitial site of Mn2+ in crystal, the local structure around Mn2+ at the interstitial site, and the role of organic spacers require further exploration.
This work was partially funded by the European Union (ERC, EnVision, project number 101125962). FP acknowledges support from the Spanish Ministry of Science and Innovation (CEX2023-001316-M). MG, AS, and UK acknowledge the support from DOE (DE-SC0024214).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M. D. Smith, et al. , Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS PubMed.
- Y. Li, et al. , ChemComm, 2022, 58, 941–944 RSC.
- A. J. Torma, et al. , ACS Nano, 2021, 15, 20550–20561 CrossRef CAS PubMed.
- S. K. Dutta, et al. , ACS Energy Lett., 2019, 4, 343–351 CrossRef CAS.
- S. Hou, et al. , Joule, 2018, 2, 2421–2433 CrossRef CAS.
- T. Sheikh and A. Nag, J. Phys. Chem. C, 2019, 123, 9420–9427 CrossRef CAS.
- F. C. Grozema, et al. , J. Phys. Chem. C, 2020, 124, 28201–28209 CrossRef PubMed.
- C. Zhou, et al. , Mater. Sci. Eng., R, 2019, 137, 38–65 CrossRef.
- S. A. Cuthriell, et al. , Adv. Mater., 2022, 34, 2202709 CrossRef CAS PubMed.
- A. Biswas, et al. , Chem. Mater., 2017, 29, 7816–7825 CrossRef CAS.
- A. Khorsand Zak, et al. , Solid State Sci., 2011, 13, 251–256 CrossRef CAS.
- R. Kamal Yadav and P. Chauhan, Indian J. Pure Appl. Phys., 2019, 57, 881–890 Search PubMed.
- T. Jin, et al. , Nat. Commun., 2023, 14, 1–9 Search PubMed.
- D. B. Straus and C. R. Kagan, J. Phys. Chem. Lett., 2018, 9, 1434–1447 CrossRef CAS PubMed.
- C. M. Mauck and W. A. Tisdale, Trends Chem., 2019, 1, 380–393 CrossRef CAS.
- M. Seitz, et al. , Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04074k |
| This journal is © The Royal Society of Chemistry 2024 |