
Expedient (3+3)-annulation of carbonyl ylides with azaoxyallyl cations: formal access to oxa-benzo[c]azepin-3-ones†
Kshitiz
Verma
 ,
Hemanga
Bhattacharyya
,
Sharajit
Saha
and
Tharmalingam
Punniyamurthy
*
,
Hemanga
Bhattacharyya
,
Sharajit
Saha
and
Tharmalingam
Punniyamurthy
*
Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati-781039, India. E-mail: tpunni@iitg.ac.in
First published on 21st October 2024
Abstract
The cascade carbon–carbon and carbon–nitrogen bond formation between in situ generated carbonyl ylides and azaoxyallyl cations, facilitated by Rh-catalysis and a base, has been achieved to furnish oxa-benzo[c]azepin-3-ones. Substrate scope, functional group diversity, scale-up and post-synthetic utilities are the important practical features.
Benzo[c]azepin-3-ones are privileged structural frameworks due to their interesting biological and medicinal properties.1 For example, they are a key scaffold found in the galanthamine family and exhibit biological activities like vitronectin receptor antagonist,1a tyrosine kinase inhibitor1c and opioid receptor antagonist1d (Fig. 1). Considerable efforts have thus been made on the construction of these scaffolds utilizing intramolecular cyclization processes, radical cyclization,2a olefin-metathesis,2b Pictet–Spengler cyclization2c and Meyers lactamization2dvia multi-step synthesis of intricate substrate precursors.2e Similarly, oxa-polycyclic motifs are the structural constituent of numerous compounds that are of pharmaceutical interest.3 The development of effective synthetic strategies for the assembly of these structural frameworks would thus be valuable.
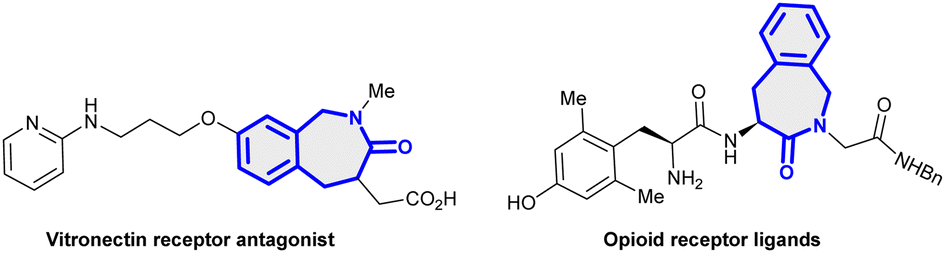 | ||
| Fig. 1 Biologically active benzo[c]azepin-3-one scaffolds. | ||
Diazocarbonyl compounds are versatile substrate precursors for constructing complex structural frameworks.4–6 In particular, α-diazoesters are the prominent precursors for the generation of carbonyl ylides that are three-atom synthons for the (3+2) and (4+3)-cycloadditions to afford oxa-bridged scaffolds, which are otherwise difficult to achieve in a single-step.7 However, the (3+3)-annulation of carbonyl ylides with suitable 1,3-dipolar coupling partners is scarce.8 More recently, Werz,8a Schneider8b and Gao8c groups have reported the (3+3)-annulation using the acid-catalyzed dipolarophiles with carbonyl ylides (Scheme 1a). Meanwhile, α-halohydroxamates have gained significant attention for constructing amide containing scaffolds due to their stability, reactivity and easy accessibility. In the presence of a base, they undergo dehydrohalogenation to generate in situ azaoxyally cations, which are reactive 1,3-dipolar synthons useful for forming biologically relevant scaffolds.9 Herein, we wish to report a cascade carbon–carbon and carbon–nitrogen bond formation of carbonyl ylides with azaoxyallyl cations that have been generated in situ using Rh-catalysis in the presence of a base to afford oxa-benzo[c]azepin-3-one frameworks at room temperature (Scheme 1b). The main challenge in the developed annulation is the simultaneous activation of both the synthons to facilitate the desired reaction while suppressing the side reactions. Additionally, constructing seven membered rings remains a challenging goal, possibly due to relative instability, and non-bonding interaction in the transition states.10 This method offers an effective synthetic route for the (3+3)-annulation of carbonyl ylides with azaoxyallyl cations with a broad substrate scope, scalability, and useful post-synthetic applications.
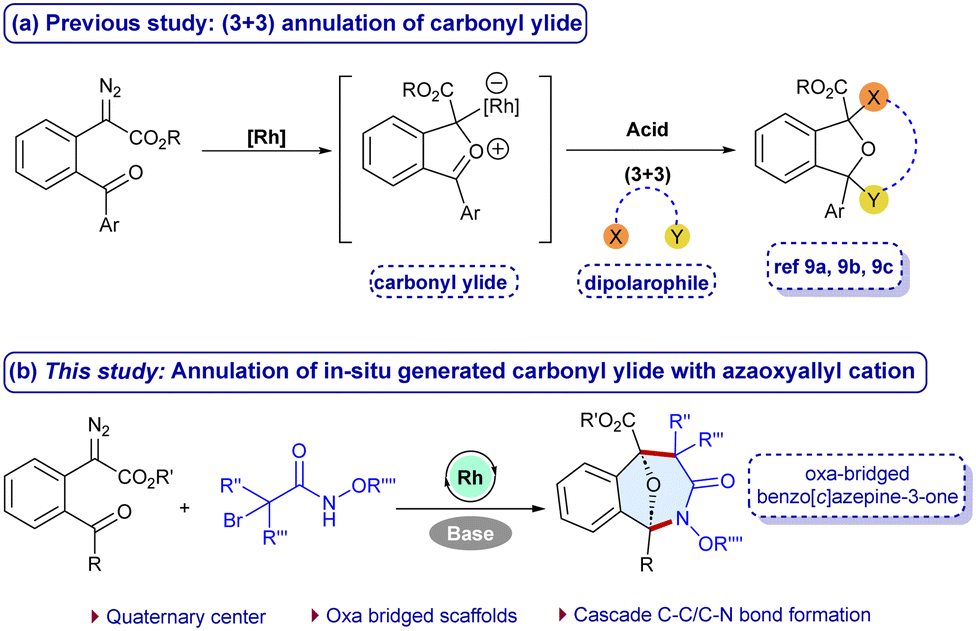 | ||
| Scheme 1 Approaches for (3+3)-annulation of carbonyl ylides. | ||
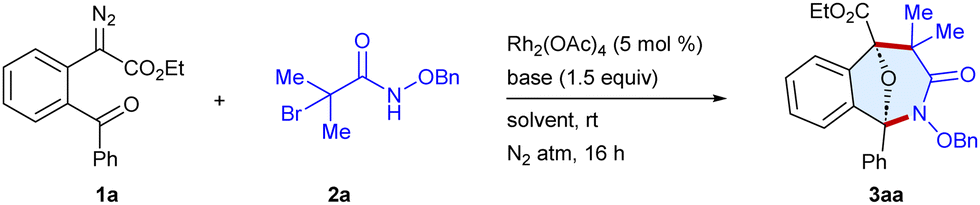
First, we commenced the optimization studies using ethyl 2-(2-benzoylphenyl)-2-diazoacetate 1a and N-(benzyloxy)-2-bromo-2-methylpropanamide 2a as the model substrates (Table 1 and Table S1, ESI†). To our delight, a reaction occurred to furnish the tricyclic scaffold 3aa in 43% yield when the substrates were stirred with Rh2(OAc)4 (5 mol%) and Cs2CO3 (1.5 equiv.) in CHCl3 under a N2 atmosphere for 16 h at room temperature (entry 1). In an array of bases screened viz. K2CO3, Cs2CO3, NaHCO3 and DBU, the former gave the best result (entries 1–4). The yield increased to 86% employing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CH2Cl2 and HFIP as a solvent due to enhanced stabilization of both the transient species via hydrogen bonding,9a,9i whereas using CH2Cl2 alone gave 73% yield (entry 5) and HFIP alone gave 68% yield (entry 8). In contrast, toluene and CH3CN were not effective and the formation of 3aa was not observed.
:1 mixture of CH2Cl2 and HFIP as a solvent due to enhanced stabilization of both the transient species via hydrogen bonding,9a,9i whereas using CH2Cl2 alone gave 73% yield (entry 5) and HFIP alone gave 68% yield (entry 8). In contrast, toluene and CH3CN were not effective and the formation of 3aa was not observed.
|
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.11 mmol), Rh2(OAc)4 (5 mol%), base (1.5 equiv.), solvent (2 mL), rt, N2 atm, 16 h. b Isolated yield. n.d. = not detected. HFIP = 1,1,1,3,3,3-hexafluoroisopropanol. | |||
| 1 | Cs2CO3 | CHCl3 | 43 |
| 2 | K2CO3 | CHCl3 | 65 |
| 3 | NaHCO3 | CHCl3 | Trace |
| 4 | DBU | CHCl3 | 58 |
| 5 | K2CO3 | CH2Cl2 | 73 |
| 6 | K2CO3 | Toluene | n.d. |
| 7 | K2CO3 | CH3CN | n.d. |
| 8 | K2CO3 | HFIP | 68 |
| 9 | K 2 CO 3 | CH 2 Cl 2 :HFIP | 86 |
Having the optimized conditions in hand, the scope of the procedure was examined with a series of α-diazo esters 1b–s utilizing N-(benzyloxy)-2-bromo-2-methylpropanamide 2a as a standard substrate (Scheme 2). The reaction of alkyl substituted diazo esters at the para-position of the phenyl rings such as methyl 1b, ethyl 1c and tert-butyl 1d gave the target products 3ba–da in 73–84% yields. In addition, halogen and electron-donating group-bearing diazo esters, fluoro 1e, bromo 1f and ethoxy 1g underwent reaction to afford the cyclic scaffolds 3ea–ga in 77–82% yields. Likewise, the diazo esters with electron withdrawing trifluoromethyl 1h, nitro 1i and carboxylate 1j groups at the para-position underwent annulation to produce 3ha–ja in 64–77% yields, whereas the substrates with fluoro 1k and methoxy 1l groups at the meta-position furnished 3ka and 3la in 82% and 78% yields, respectively. Similar results were observed with diazo esters bearing ortho-methyl 1m and trifluoromethyl 1n groups, furnishing 3ma and 3na in 75% and 66% yields, respectively. Intriguingly, disubstituted 1o–p and thiophene 1q tethered diazo esters were able to afford the cyclic scaffolds 3oa–qa in 63–81% yields. Moreover, the methyl substituted ester moiety of diazo ester 1r gave 3ra in 83% yield, whose structure was determined by single crystal X-ray analysis (CCDC= 2372696, see ESI†). Gratifyingly, changing the phenyl ring to tert-butyl diazo ester 1s successfully gave the target heterocycle 3sa in 74% yield.
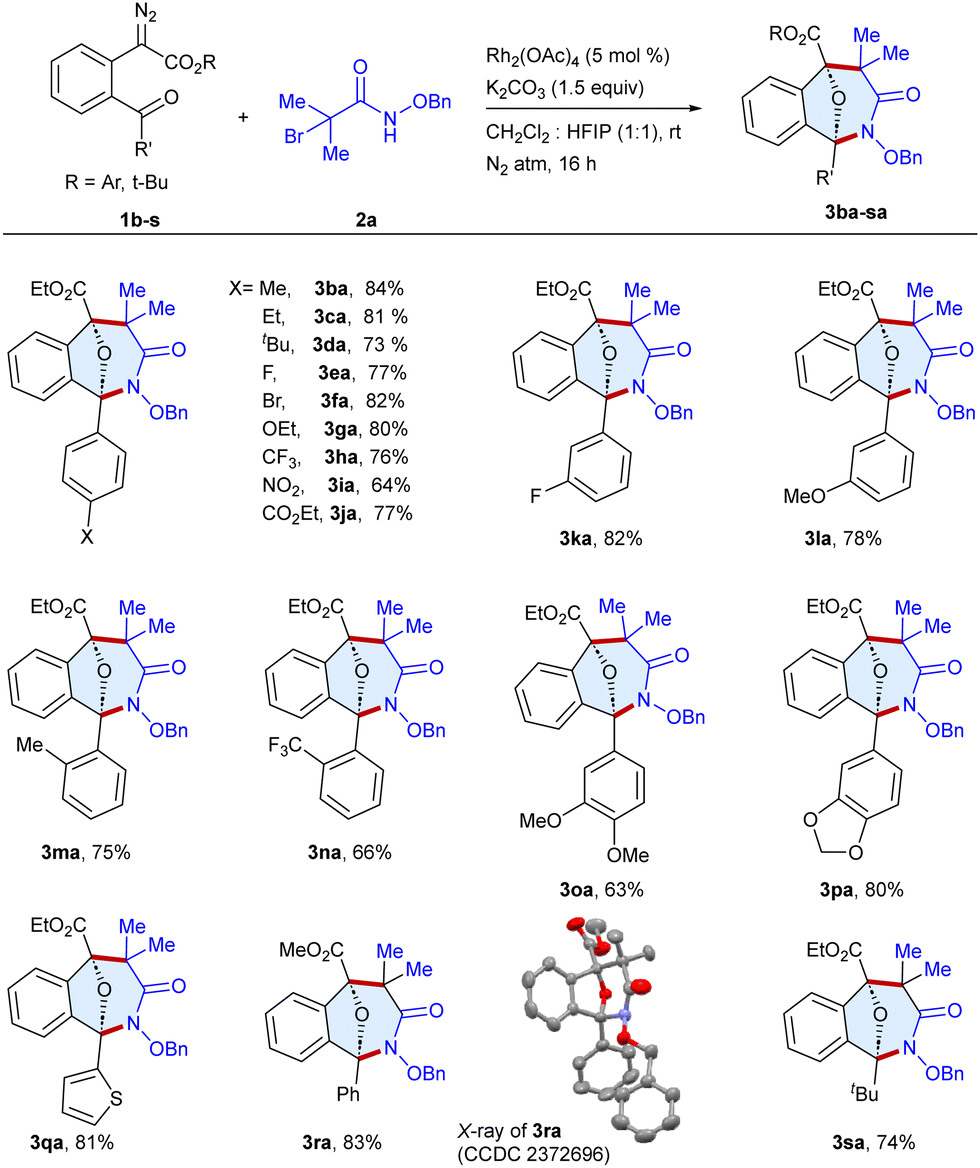 | ||
Scheme 2 Scope of α-diazo esters.a,b a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reaction conditions: 1b–s (0.1 mmol), 2a (0.11 mmol), Rh2(OAc)4 (5 mol %), K2CO3 (0.15 mmol), CH2Cl2:HFIP (1:1, 2 mL), rt, N2 atm, 16 h. bIsolated yield. Reaction conditions: 1b–s (0.1 mmol), 2a (0.11 mmol), Rh2(OAc)4 (5 mol %), K2CO3 (0.15 mmol), CH2Cl2:HFIP (1:1, 2 mL), rt, N2 atm, 16 h. bIsolated yield. | ||
The scope of the procedure was further examined for the annulation of a series of α-halohydroxamates 2b–j with ethyl 2-(2-benzoylphenyl)-2-diazoacetate 1a as a standard substrate (Scheme 3). The reaction of the substrates bearing methoxy 2b and ethoxy 2c protecting groups on the nitrogen atom of haloamide afforded 3ab and 3ac in 82% and 75% yields, respectively, whereas tert-butyl bearing 2d was an unsuccessful substrate, which might be due to steric hindrance. Furthermore, phenoxy 2e and allyloxy 2f bearing haloamides underwent reaction to furnish 3ae and 3af in 81% and 68% yields, respectively. Moreover, the reaction of the para-methyl benzyl 2g substituted haloamide afforded 3ag in 78% yield, whereas the reaction of α-bromoamide with a monomethyl group 2h yielded 3ah in a trace amount, which may be due to the formation of a less stable carbocation. Moreover, the substrate 2i bearing a diethyl group gave 3ai in 70% yield, whereas 2j with cyclohexyl yielded 3aj in a trace amount.
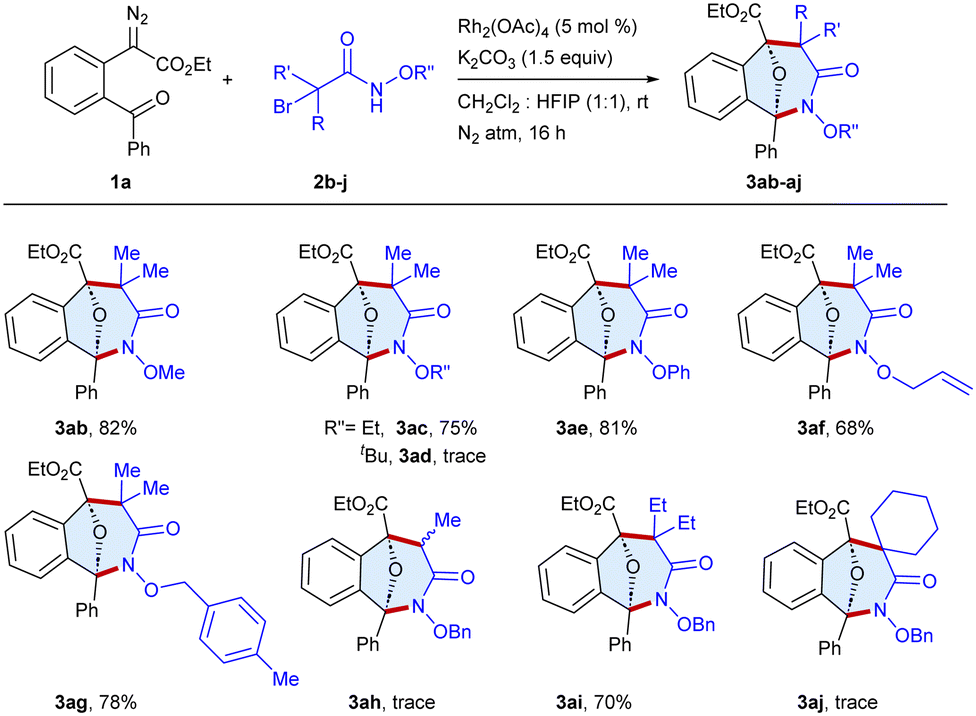 | ||
| Scheme 3 Scope of α-halohydroxamate.a,b aReaction conditions: 1a (0.1 mmol), 2b–j (0.11 mmol), Rh2(OAc)4 (5 mol %), K2CO3 (0.15 mmol) CH2Cl2:HFIP (1:1, 2 mL), rt, N2 atm, 16 h. bIsolated yield. | ||
To get insights into the reaction pathway, the radical scavenger experiments were carried out using 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT) and the formation of 3aa was observed in 75% and 72% yields, respectively (Scheme 4a), which indicates that the reaction may not involve a radical pathway. Furthermore, the reaction of ester 1a with N-(benzyloxy)-2-bromoacetamide 2k was unable to give 3ak, which indicates that the generation of a tertiary carbocation is essential for the reaction (Scheme 4b). In addition, the reaction of 1a with α-halohydroxamate 2a in the absence of K2CO3 was unable to yield 3aa; instead the dimer114 of the carbonyl ylide has been isolated in 58% yield (Scheme 4c), which suggests that the reaction involves the carbonyl ylide pathway. Moreover, the reaction of α-diazo ester 1a with α-halohydroxamate 2a in the absence of Rh2(OAc)4 failed to yield 3aa (Scheme 4d). Thus, the Rh-catalyst and base are crucial for generating the transient intermediates, carbonyl ylides and azaoxyallyl cations. In addition, the reaction of 1a and 2a was examined using (R)-BINAP and chiral phosphoric acid; however, 3aa was obtained in racemic form (ESI,† Table S3).
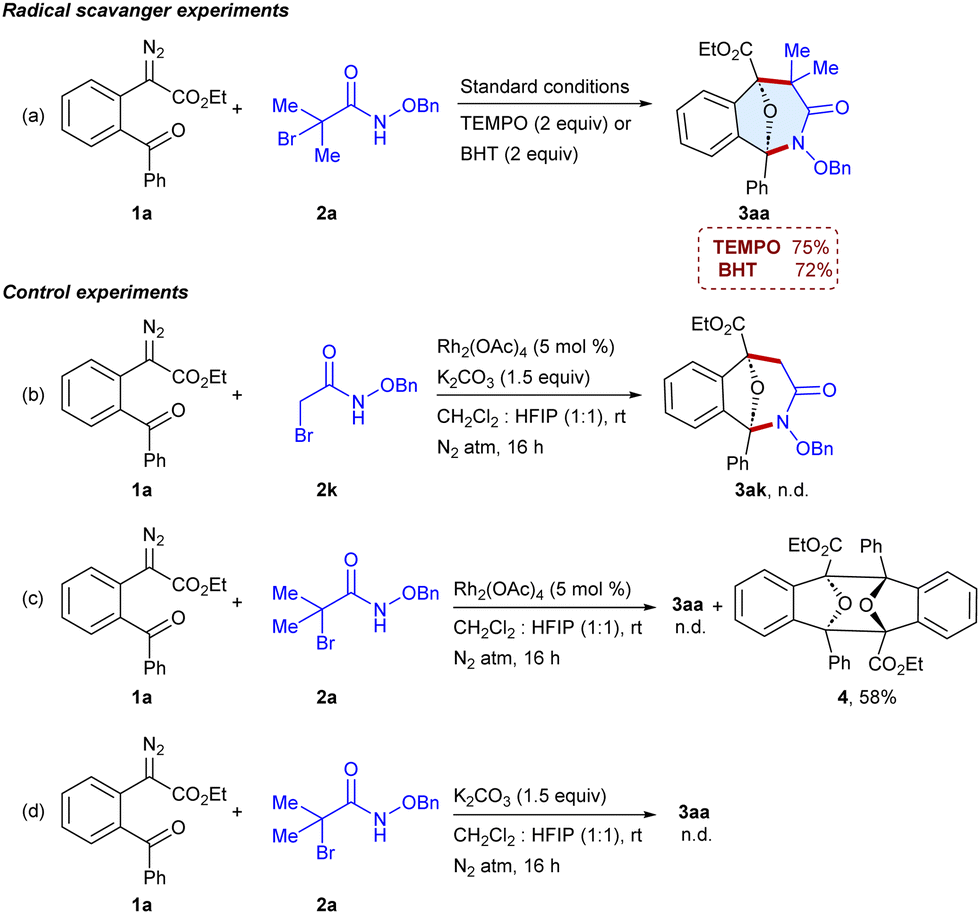 | ||
| Scheme 4 Preliminary mechanistic investigations. | ||
Based on the experimental results and literature precedents,7–9 a plausible reaction pathway is proposed (Scheme 5). Firstly, the Rh-catalyzed decomposition of α-diazoester 1a, followed by intramolecular capture of the rhodium carbene by the aryl keto group, leads to the formation of the carbonyl ylide A. At the same time, α-halohydroxamate 2a can undergo base-mediated dehydrohalogenation to produce azaoxyallyl cation B. Both the in situ generated A and B can undergo the (3+3)-annulation in a concerted fashion to give the target heterocycle 3aa (Path A). Alternatively, B can undergo a nucleophilic attack by A to form C that can lead to annulation in a step-wise fashion to produce 3aa (Path B).
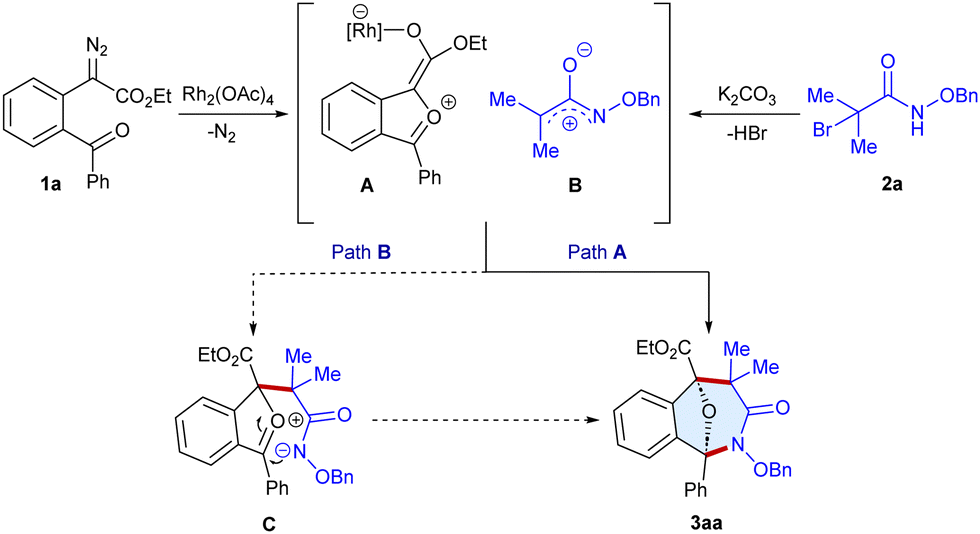 | ||
| Scheme 5 Plausible reaction pathway. | ||
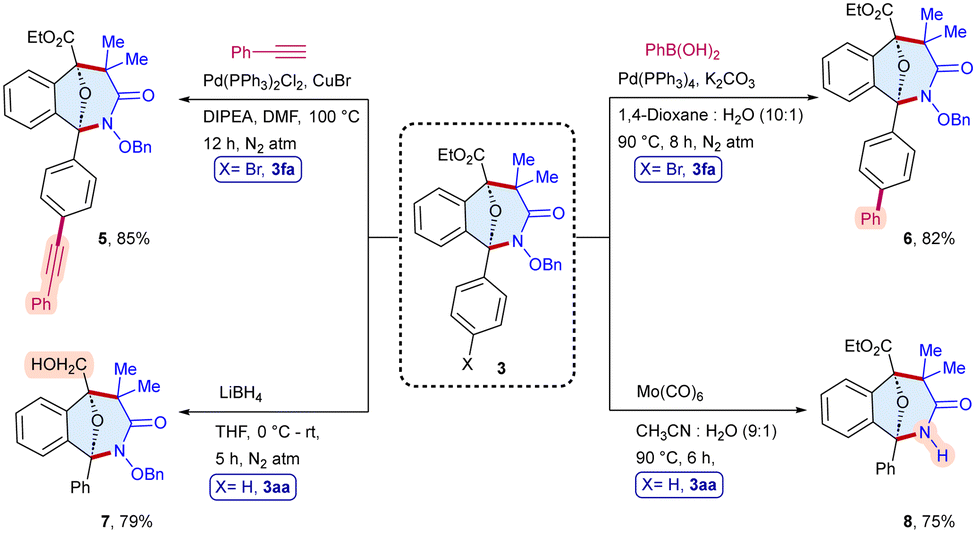
To demonstrate the practical applicability, we conducted a scale-up synthesis, the reaction of α-diazo ester 1a with α-halohydroxamate 2a as the representative examples, which afforded 3aa in 68% (312 mg) yield (Scheme 6). Moreover, the products were modified to produce diverse scaffolds (Scheme 7). The oxa-bridged benzo[c]azepin-3-one 3fa was coupled with phenylacetylene to give 5 in 85% yield, whereas Suzuki coupling with phenylboronic acid provided 6 in 82% yield. In addition, the reduction of the ester group of 3aa in the presence of LiBH4 furnished 7 in 79% yield. Likewise, the N–O bond cleavage of 3aa has been achieved to afford 8 in 75% yield.
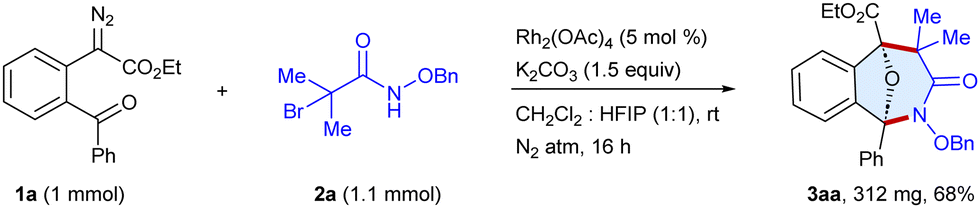 | ||
| Scheme 6 Scale-up synthesis. | ||
 | ||
| Scheme 7 Post synthetic transformations. | ||
In summary, we describe the reaction of the carbonyl ylides and azaoxyallyl cations using Rh-catalysis in the presence of K2CO3 to assemble diverse oxa-benzo[c]azepin-3-ones in a 1:1 mixture of CH2Cl2 and HFIP at room temperature. The substrate scope, functional group diversity, scale-up, selectivity and post-synthetic utilities are the essential practical features.
We thank SERB for the financial support (SCP/2022/000256 and CRG/2022/002778), Department of Chemistry, CIF and NECBH (BT/NER/143/SP44675/2023) for NMR, mass and X-ray analyses (DST-FIST) (SR/FST/CS-II/2017/23c). One of us (K. V) acknowledges the Ministry of Education for the Prime Minister's Research Fellowship (PMRF). T. P. thanks SERB for the J. C. Bose Fellowship (JCB/2022/000037).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) W. H. Miller, D. P. Alberts, P. K. Bhatnagar, W. E. Bondinell, J. F. Callahan, R. R. Calvo, R. D. Cousins, K. F. Erhard, D. A. Heerding, R. M. Keenan, C. Kwon, P. J. Manley, K. A. Newlander, S. T. Ross, J. M. Samanen, I. N. Uzinskas, J. W. Venslavsky, C. C. Yuan, R. C. Haltiwanger, M. Gowen, S. M. Hwang, I. E. James, M. W. Lark, D. J. Rieman, G. B. Stroup, L. M. Azzarano, K. L. Salyers, B. R. Smith, K. W. Ward, K. O. Johanson and W. F. Huffman, J. Med. Chem., 2000, 43, 22 CrossRef CAS PubMed; (b) S. Lilienfeld, CNS Drug Rev., 2002, 8, 159 CrossRef CAS PubMed; (c) P. Ruzza, A. Calderan, A. Donella-Deana, B. Biondi, L. Cesaro, A. Osler, S. Elardo, A. Guiotto, L. A. Pinna and G. Borin, Biopolymers., 2003, 71, 478 CrossRef CAS PubMed; (d) S. Ballet, D. Feytens, R. D. Wachter, M. D. Vlaeminck, E. D. Marczak, S. Salvadori, C. de Graaf, D. Rognan, L. Negri, R. Lattanzi, L. H. Lazarus, D. Tourwé and G. Balboni, Bioorg. Med. Chem. Lett., 2009, 19, 433 CrossRef CAS PubMed; (e) C. Betti, J. Starnowska, J. Mika, J. Dyniewicz, L. Frankiewicz, A. Novoa, M. Bochynska, A. Keresztes, P. Kosson, W. Makuch, J. Van Duppen, N. N. Chung, J. Vanden Broeck, A. W. Lipkowski, P. W. Schiller, F. Janssens, M. Ceusters, F. Sommen, T. Meert, B. Przewlocka, D. Tourwé and S. Ballet, ACS Med. Chem. Lett., 2015, 6, 1209 CrossRef CAS PubMed.
- (a) A. Kamimura, Y. Taguchi, Y. Omata and M. Hagihara, J. Org. Chem., 2003, 68, 4996 CrossRef CAS PubMed; (b) A. Couture, S. Lebrun, E. Deniau and P. Grandclaudon, Synthesis., 2011, 2011, 669 CrossRef; (c) M. So, T. Kotake, K. Matsuura, M. Inui and A. Kamimura, J. Org. Chem., 2012, 77, 4017 CrossRef CAS PubMed; (d) M. Jida, O. Van der Poorten, K. Guillemyn, Z. Urbanczyk-Lipkowska, D. Tourwé and S. Ballet, Org. Lett., 2015, 17, 4482 CrossRef CAS PubMed; (e) A. Petuškovs and K. Shubin, Chem. Heterocycl. Compd., 2016, 52, 530 CrossRef.
- (a) R. A. Shenvi, C. A. Guerrero, J. Shi, C.-C. Li and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7241 CrossRef CAS PubMed; (b) S. K. Sahu, P. K. Behera, P. Choudhury, M. Sethi, S. Jena and L. Rout, New J Chem., 2021, 45, 11018 RSC; (c) I. V. Kulakov, A. L. Stalinskaya, S. Y. Chikunov and Y. V. Gatilov, New J Chem., 2021, 45, 3559 RSC.
- For examples of total synthesis, see: (a) S. Nakamura, Y. Sugano, F. Kikuchi and S. Hashimoto, Angew. Chem., Int. Ed., 2006, 45, 6532 CrossRef CAS PubMed; (b) V. Navickas, D. B. Ushakov, M. E. Maier, M. Ströbele and H.-J. Meyer, Org. Lett., 2010, 12, 3418 CrossRef CAS PubMed; (c) J. P. Sorrentino and R. A. Altman, ACS Med. Chem. Lett., 2022, 13, 707 CrossRef CAS PubMed For examples of C–H activation, see: ; (d) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (e) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed; (f) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed; (g) S. Sharma, S. H. Han, S. Han, W. Ji, J. Oh, S.-Y. Lee, J. S. Oh, Y. H. Jung and I. S. Kim, Org. Lett., 2015, 17, 2852 CrossRef CAS PubMed; (h) J. Li, M. Tang, L. Zang, X. Zhang, Z. Zhang and L. Ackermann, Org. Lett., 2016, 18, 2742 CrossRef CAS PubMed.
- For examples of insertion and photo-redox catalysis, see: (a) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (c) F.-P. Wu, C. C. Chintawar, R. Lalisse, P. Mukherjee, S. Dutta, J. Tyler, C. G. Daniliuc, O. Gutierrez and F. Glorius, Nat. Catal., 2024, 7, 242 CrossRef CAS; (d) Z. Zhang and V. Gevorgyan, Chem. Rev., 2024, 124, 7214 CrossRef CAS PubMed; (e) S. Biswas and C. Bolm, Org. Lett., 2024, 26, 1511 CrossRef CAS PubMed.
- For examples of cycloaddition, see: (a) N. Shimada, M. Anada, S. Nakamura, H. Nambu, H. Tsutsui and S. Hashimoto, Org. Lett., 2008, 10, 3603 CrossRef CAS PubMed; (b) V. V. Pagar, A. M. Jadhav and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 20728 CrossRef CAS PubMed; (c) G. D. Titov, G. I. Antonychev, M. S. Novikov, A. F. Khlebnikov, E. V. Rogacheva, L. A. Kraeva and N. V. Rostovskii, Org. Lett., 2023, 25, 2707 CrossRef CAS PubMed.
- (a) H. Suga, K. Inoue, S. Inoue and A. Kakehi, J. Am. Chem. Soc., 2002, 124, 14836 CrossRef CAS PubMed; (b) H. Li, S. A. Bonderoff, B. Cheng and A. Padwa, J. Org. Chem., 2014, 79, 392 CrossRef CAS PubMed; (c) H. Lam, Z. Qureshi, M. Wegmann and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 16185 CrossRef CAS PubMed; (d) A. Suneja, H. J. Loui and C. Schneider, Angew. Chem., Int. Ed., 2020, 59, 5536 CrossRef CAS PubMed; (e) Q. Pang, J. Zhou, Y. Wu, W.-J. Zhou, W.-F. Zuo, G. Zhan and B. Han, Org. Lett., 2022, 24, 1362 CrossRef CAS PubMed; (f) P. Jia, Z. Lin, S. Yan, J. Liang, C. Luo, R. Lai, L. Hai, Z. Yang and Y. Wu, Org. Lett., 2023, 25, 5134 CrossRef CAS PubMed.
- (a) M. Petzold, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2019, 58, 6225 CrossRef CAS PubMed; (b) H. J. Loui, A. Suneja and C. Schneider, Org. Lett., 2021, 23, 2578 CrossRef CAS PubMed; (c) L. Tu, S. Li, L.-M. Gao, B.-W. Tang, Y.-S. Zheng and J.-K. Liu, J. Org. Chem., 2024, 89, 9031 CrossRef CAS PubMed.
- For examples, see: (a) C. S. Jeffrey, K. L. Barnes, J. A. Eickhoff and C. R. Carson, J. Am. Chem. Soc., 2011, 133, 7688 CrossRef CAS PubMed; (b) A. Acharya, D. Anumandla and C. S. Jeffrey, J. Am. Chem. Soc., 2015, 137, 14858 CrossRef CAS PubMed; (c) A. Acharya, K. Montes and C. S. Jeffrey, Org. Lett., 2016, 18, 6082 CrossRef CAS PubMed; (d) A. Acharya, J. A. Eickhoff, K. Chen, V. J. Catalano and C. S. Jeffrey, Org. Chem. Front., 2016, 3, 330 RSC; (e) B. Baldé, G. Force, L. Marin, R. Guillot, E. Schulz, V. Gandon and D. Lebœuf, Org. Lett., 2018, 20, 7405 CrossRef PubMed; (f) M. C. DiPoto and J. Wu, Org. Lett., 2018, 20, 499 CrossRef CAS PubMed; (g) A. El Bouakher, A. Martel and S. Comesse, Org. Biomol. Chem., 2019, 17, 8467 RSC; (h) P. Karjee, S. Mandal, B. Debnath, N. Namdev and T. Punniyamurthy, Chem. Commun., 2023, 59, 8270 RSC; (i) Y. Kim and S.-G. Kim, Molecules, 2024, 29, 1221 CrossRef CAS PubMed.
- For selected examples, see: (a) P. A. Wender, C. O. Husfeld, E. Langkopf, J. A. Love and N. Pleuss, Tetrahedron, 1998, 54, 7203 CrossRef CAS; (b) P. A. Wender, C. M. Barzilay and A. J. Dyckman, J. Am. Chem. Soc., 2001, 123, 179 CrossRef CAS PubMed; (c) G. Zuo and J. Louie, J. Am. Chem. Soc., 2005, 127, 5798 CrossRef CAS PubMed; (d) M.-B. Zhou, R.-J. Song, C.-Y. Wang and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 10805 CrossRef CAS PubMed; (e) X. Xu, P. Liu, X.-Z. Shu, W. Tang and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 9271 CrossRef CAS PubMed.
- R. N. Warrener, I. G. Pitt and R. A. Russell, J. Chem. Soc., Chem. Commun., 1982, 1195 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2372696. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04946b |
| This journal is © The Royal Society of Chemistry 2024 |