
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe structural and functional impacts of rationally designed cyclic peptides on self-assembly-mediated functionality
Taichi
Kurita
 a and
Keiji
Numata
*abc
a and
Keiji
Numata
*abc
aDepartment of Material Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: keiji.numata@riken.jp
bBiomacromolecules Research Team, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
cInstitute for Advanced Biosciences, Keio University, Nipponkoku 403-1, Daihouji, Tsuruoka, Yamagata 997-0017, Japan
First published on 18th November 2024
Abstract
Compared with their linear counterparts, cyclic peptides, characterized by their unique topologies, offer superior stability and enhanced functionality. In this review article, the rational design of cyclic peptide primary structures and their significant influence on self-assembly processes and functional capabilities are comprehensively reviewed. We emphasize how strategically modifying amino acid sequences and ring sizes critically dictate the formation and properties of peptide nanotubes (PNTs) and complex assemblies, such as rotaxanes. Adjusting the number of amino acid residues and side chains allows researchers to tailor the diameter, surface properties, and functions of PNTs precisely. In addition, we discuss the complex host–guest chemistry of cyclic peptides and their ability to form rotaxanes, highlighting their potential in the development of mechanically interlocked structures with novel functionalities. Moreover, the critical role of computational methods for accurately predicting the solution structures of cyclic peptides is also highlighted, as it enables the design of novel peptides with tailored properties for a range of applications. These insights set the stage for groundbreaking advances in nanotechnology, drug delivery, and materials science, driven by the strategic design of cyclic peptide primary structures.
 Taichi Kurita | Taichi Kurita received his BSc in 2020 (from Prof. Shunsaku Kimura's group) and MSc in 2022 from Kyoto University. Since 2022, he has been a PhD candidate under the supervision of Prof. Keiji Numata in the Department of Material Chemistry, Kyoto University. He is currently a research fellow of the Japan Society for the Promotion of Science (Research Fellowship for Young Scientists). His research interest is the development of polypeptide-based tough materials using rationally designed cyclic peptides. |
 Keiji Numata | Keiji Numata is currently a Full Professor at the Department of Material Chemistry, Kyoto University; Team Leader at the Biomacromolecules Research Team, RIKEN Center for Sustainable Resource Science; and Project Professor at the Institute for Advanced Biosciences, Keio University, Japan. He is also the Research Director for JST-ERATO Numata Organellar Reaction Cluster Project, Research Director for JST-COI-NEXT, and Research Director for the MEXT Program: Data Creation and Utilization-Type Material Research and Development Project. He investigates the biosynthesis and material design of structural proteins and polypeptides. |
1. Introduction
Peptides have revolutionized the biomedical and materials science fields, offering critical benefits such as biodegradability, biocompatibility, and minimal environmental toxicity.1–5 Among these, cyclic peptides stand out, drawing significant attention for their unique cyclic topology, which imparts stability superior to that of their linear counterparts.6–8 Such enhanced stability is critical in pharmaceutical contexts for improving membrane permeability and, consequently, the efficacy of drug delivery systems.9–13 In the field of materials chemistry, this distinctive cyclic topology facilitates self-assembly into complex structures with advanced functionalities that are unachievable by linear or monocyclic peptides,14,15 and these self-assembly processes dramatically increase the inherent advantages offered by cyclic peptide topology.In this review article, we highlight in-depth how strategic modifications in the design of cyclic peptide primary structures determine their assembly behavior and functional outcomes in material chemistry. We investigate peptide nanotube (PNT) formation through hydrogen bonding and complex structures arising from host–guest chemistry, revealing their potential for novel applications. We illustrate these concepts with practical examples, demonstrating the ingenuity underlying the rational design of cyclic peptides. Furthermore, we discuss breakthroughs in structural prediction tools that significantly expand our ability to customize cyclic peptides for precise assembly and applications. Such tools are instrumental in offering predictions that could revolutionize the design of cyclic peptides for breakthrough applications.
A thorough understanding of these principles sets the stage for the development of new cyclic peptide-based materials, increasing their potential for use in sophisticated and targeted functionalities for a variety of applications.
2. Tubular assembly of cyclic peptides
In nature, tubular structures, often composed of proteins, are essential for a variety of biological functions.16 For example, viruses, such as tobacco mosaic virus, use tubular-shaped structures to infect their hosts and store genetic material.17 Inspired by these biological activities, engineered nanotubes have potential application prospects in nanomaterials, such as antibiotics, drug delivery, and organic electronics. Recently, several groups have reported the synthesis of molecular nanotubes by forming covalent or noncovalent bonds with biologically relevant compounds. The Harada group developed tubular polymers by crosslinking α-cyclodextrins threaded through polyethylene glycol (PEG) chains (Fig. 1a).18 Shimizu et al. designed glycolipid-like molecules that self-assembled into lipid nanotubes with inner diameters, lengths, and wall thicknesses that could be precisely controlled (Fig. 1b).19,20 Another example involves the self-assembly of Phe–Phe dipeptides, which is the core sequence of amyloid proteins. These dipeptides form nanotubes or micron-sized tubes with 10 Å hydrophilic channels upon dilution or heating (Fig. 1c).21 In addition, the amphiphilic block polypeptides have been shown to form nanotubes with a adjustable diameter of approximately 80 nm in Tris-HCl buffer after heat treatment (Fig. 1d).22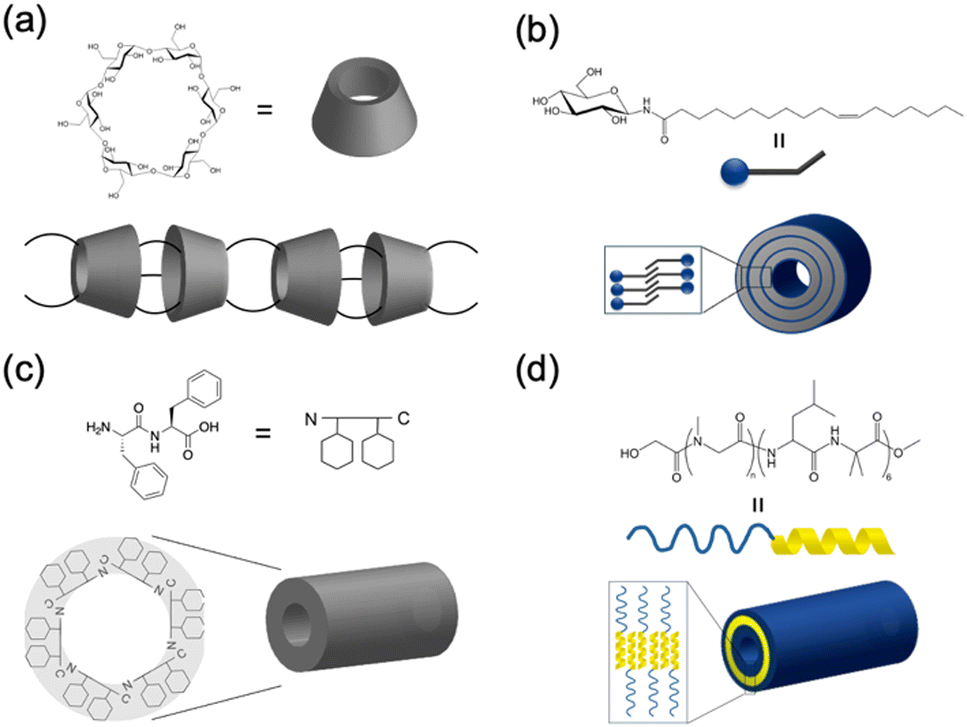 | ||
| Fig. 1 Schematic illustration of various biomolecular nanotubes: (a) cyclodextrin nanotubes, (b) lipid nanotubes, (c) hydrophilic PNTs, and (d) amphiphilic PNTs. | ||
Among the various methods for constructing tubular assemblies, self-assembling PNTs, which are stacked cyclic peptides stabilized by hydrogen-bonding interactions, are of particular interest because their structural and functional properties can be easily tuned.14,15,23 Self-assembling PNTs offer two advantages. First, the nanotube diameter can be easily controlled by changing the number of amino acid residues in each cyclic peptide. Second, by varying the amino acid side chains, the outer surface properties of the nanotubes can be easily modified. Thus, nanotubes tailored to specific applications can be prepared by properly designing the constituent cyclic peptides and optimizing the self-assembly conditions. The drawback of constructing PNTs from cyclic peptides lies in the limitations of cyclic peptide synthesis. Two main strategies have been developed for the synthesis of cyclic peptides: cyclization on resin and cyclization in solution. Cyclization on resin is efficient but requires specific side chain groups and several orthogonal protecting groups, whereas cyclization in solution offers more availability in peptide sequences but is more time-consuming due to slower cyclization carried out in dilute solutions and complicated purification steps. Nonetheless, the development of various peptide-ligation technologies over the years has provided valuable tools to overcome these limitations, enabling effective head-to-tail cyclic peptide synthesis.24 To date, several types of cyclic peptides have been stacked on PNTs and further classified primarily on the basis of their composition as cyclic peptides, including cyclic α-alt(D,L)-peptides (Fig. 2a),25 cyclic β-peptides (Fig. 2b),26 and cyclic α,γ-peptides (Fig. 2c).27 Although these peptides differ in their primary structure, they all have their peptide backbone carbonyl and amide protons oriented perpendicular to the plane of the ring. The formation of PNTs is driven by the ability of adjacent peptide subunits to form β-sheet-like hydrogen bonds across both sides of the ring structure, which is crucial for stabilizing their tubular structures.
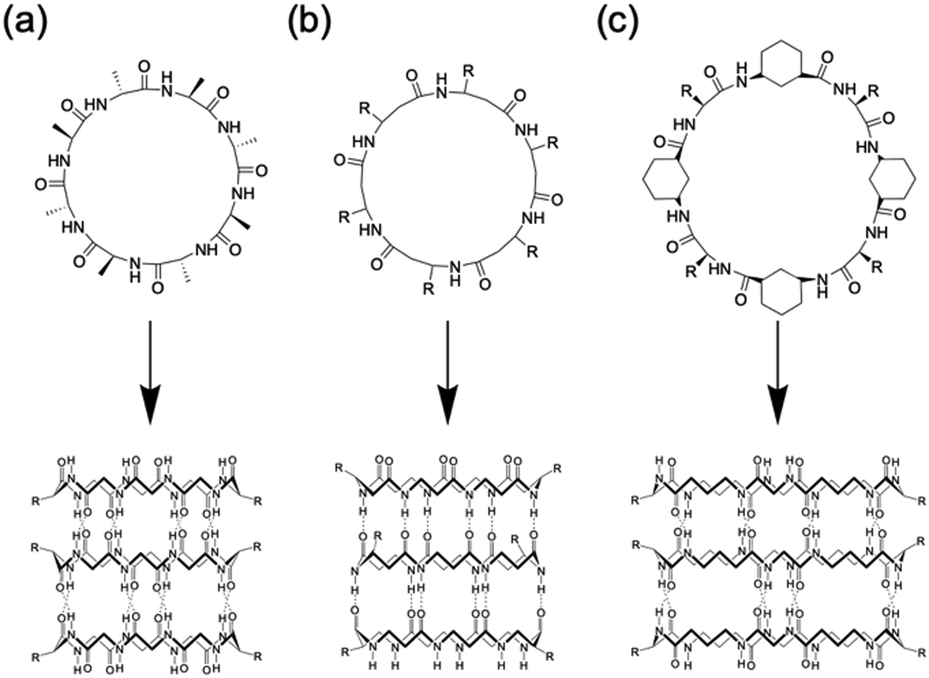 | ||
| Fig. 2 Classes of cyclic peptides that assemble into PNTs by forming hydrogen bonds: (a) cyclic α-alt(D,L)-peptides, (b) cyclic β-peptides, and (c) cyclic α,γ-peptides. | ||
2.1. Tubular assemblies of cyclic α-alt(D,L)-peptides
In 1993, Ghadiri et al. prepared the first well-characterized PNT using an octapeptide, cyclo[(L-Gln-D-Ala-L-Glu-D-Ala)2].25 This peptide sequence was chosen to increase solubility in basic aqueous solutions and prevent premature subunit association via coulombic repulsion. By controlling the acidification of the alkaline peptide solution, rod-shaped microcrystalline aggregates were formed. These structures were subsequently comprehensively characterized via transmission electron microscopy (TEM), electron diffraction, Fourier transform infrared (FT-IR) spectroscopy, and molecular modeling.25 The resulting peptide assemblies featured ordered hollow tubular structures with internal diameters of 7.5 Å and 4.73 Å between the ring-shaped subunits. The cyclic peptide units were stacked in an antiparallel orientation facilitated by backbone–backbone intermolecular hydrogen bonds. Building upon this foundation, Lambert et al. employed a similar pH-controlled self-assembly strategy to construct nanotubular microcrystals of a cyclo(D-Phe-L-Asp-D-Phe-L-Asn-D-Phe-L-Asp-D-Phe-L-Asn) that included two aspartic acid units, further advancing the field of PNT design.28Given the versatility of solid- and liquid-phase synthesis methods for producing cyclic peptides with arbitrary sequences, examining the effect of peptide sequence variations on PNTs formation is essential. In natural proteins, minor modifications to the amino acid sequence can drastically alter the biological functions of the resulting protein. Similarly, unique amino acid side chains are anticipated to modulate noncovalent interactions among cyclic peptides, including hydrogen bonding, electrostatic, van der Waals, and hydrophobic interactions. This variety of interactions between various D- and L-amino acid residues could potentially lead to the creation of PNTs with diverse structures and functionalities determined by the primary structures of the corresponding cyclic peptides.
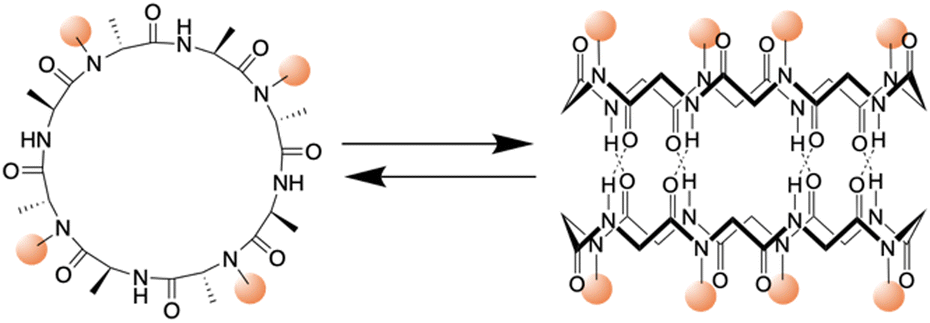 | ||
| Fig. 3 Schematic illustration of the chemical structure of a cyclic peptide with N-methylated amino acids and the cylindrical dimeric ensemble. | ||
Kobayashi and colleagues examined β-sheet structure formation during the self-assembly of cyclic peptides into PNTs (Fig. 4).33 Cyclic α-alt(D,L)-peptides can stack into two different β-sheet configurations: parallel and antiparallel. They directly evaluated the thermodynamic preference for antiparallel versus parallel β-sheet formation using two enantiomeric forms of the same peptide sequence, cyclo(L-Phe-D-MeN-Ala)4 and cyclo(D-Phe-L-MeN-Ala)4. In their study, a racemic mixture with an equal amount of each peptide yielded an equilibrium mixture of parallel and antiparallel ensembles distinguishable by 1H NMR spectroscopy. Moreover, the free energy of stabilization was determined for both β-sheet arrangements, and it was revealed that the antiparallel β-sheet structure was thermodynamically more stable than the parallel arrangement by 0.8 kcal mol−1.
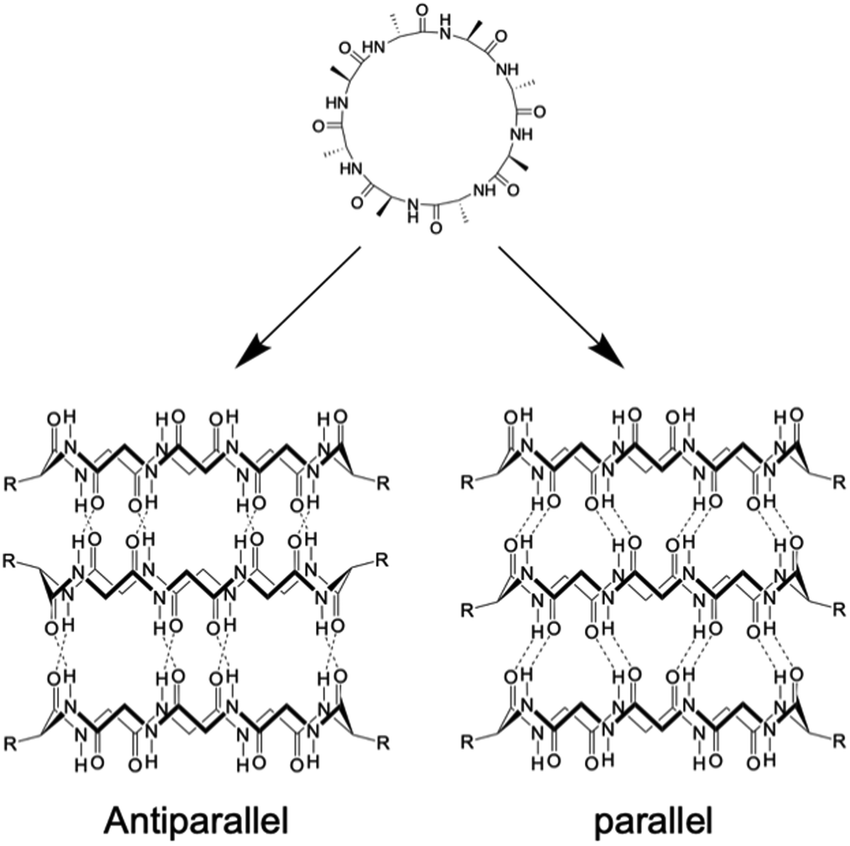 | ||
| Fig. 4 Schematic illustration of the formation of antiparallel and parallel β-sheet structures from cyclic α-alt(D,L) peptides. | ||
Building upon these molecular insights, Silk et al. studied the crystallization and structures of two specifically designed cyclic peptides, cyclo(L-Asp-D-Leu-L-Lys-D-Leu)2 and cyclo(L-Asp-D-Ala-L-Lys-D-Ala)2, to understand how side chain variations affect the stacking arrangements of PNTs (Fig. 5).34 For cyclo(L-Asp-D-Leu-L-Lys-D-Leu)2, which features larger Leu side chains, the experimental results confirmed the hypothesis that larger side chains facilitate antiparallel stacking. The crystal structure of this cyclic peptide with bulky side chains revealed the formation of continuous β-sheet-like nanotubes with extensive hydrogen bonding interactions between two neighboring peptides. In these structures, the cyclic peptides were aligned in an antiparallel fashion, supporting the idea that larger side chains enhance intermolecular interactions, which ultimately leads to the cyclic peptide adopting a more stable antiparallel orientation. Conversely, crystallographic analysis revealed that cyclo(L-Asp-D-Ala-L-Lys-D-Ala)2, which contains shorter side chains like Ala, adopted a parallel stacking arrangement. The shorter side chains facilitated closer packing of the nanotubes, which was hypothesized to be conducive for parallel β-sheet formation. These findings underscore the critical roles of side chain size and composition in determining the stacking manner of the cyclic peptides. The contrasting stacking behaviors observed for these two peptides, driven by differences in side-chain characteristics, illustrate a fundamental aspect of peptide design for engineering materials with specific structural and functional properties.
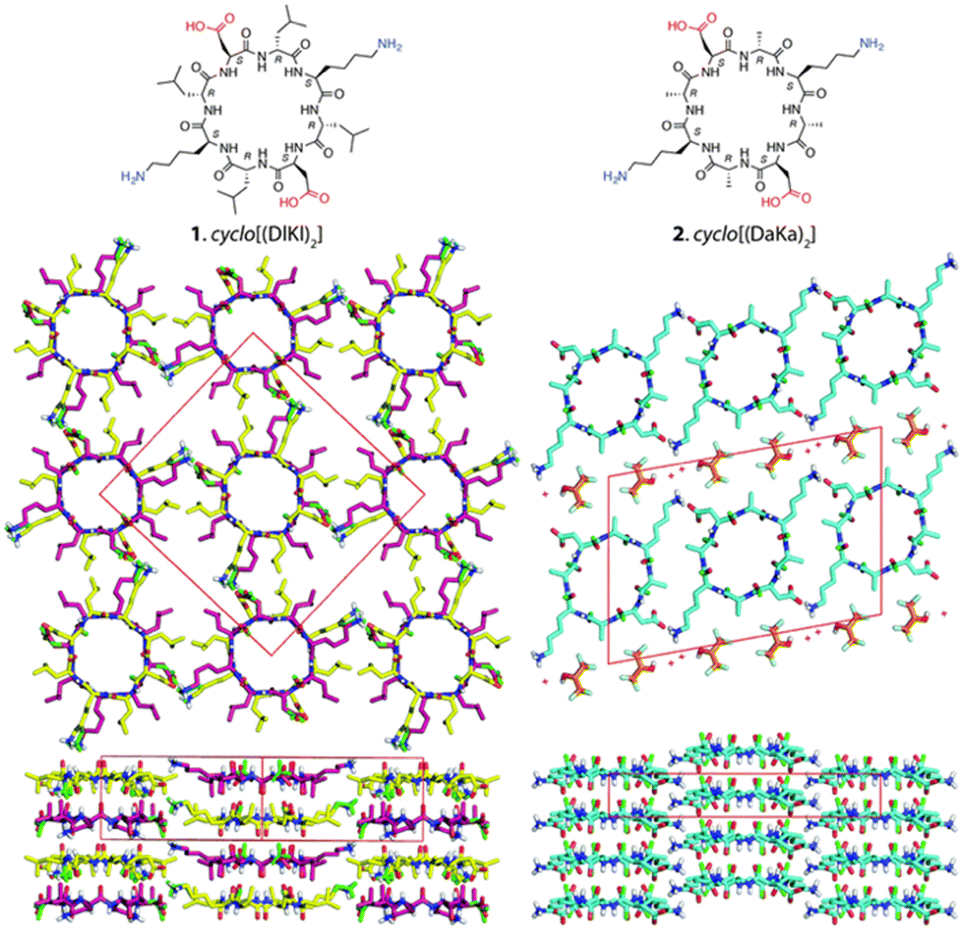 | ||
| Fig. 5 Chemical structures of cyclo(L-Asp-D-Leu-L-Lys-D-Leu)2 and cyclo(L-Asp-D-Ala-L-Lys-D-Ala)2 and crystal structures of their PNTs. Reproduced with permission from ref. 34 Copyright 2017, The Royal Society of Chemistry. | ||
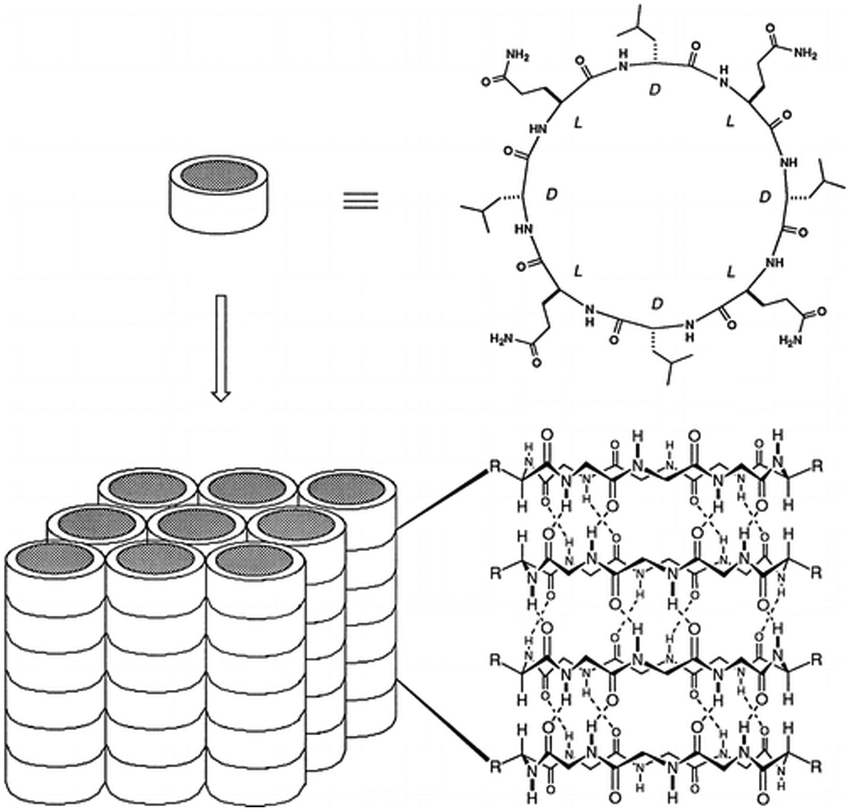 | ||
| Fig. 6 Schematic illustration of the formation of ordered parallel arrays of solid-state nanotubes from cyclic peptide structures with alternating D- and L-amino acids. The arrangement is dependent on the peptide sequence and conditions used. Reproduced with permission from ref. 35 Copyright 1996, American Chemical Society. | ||
Recent advances in supramolecular chemistry have demonstrated that rational design of the primary structure of cyclic peptides can lead to the generation of highly ordered and functional self-assembled structures. In particular, the works of the Montenegro group exemplify how strategic molecular design can facilitate the formation of complex supramolecular architectures from simple peptide units.36,37 In 2019, Insua et al. explored cyclic peptides that feature specific amino acid sequences with alternating D and L chirality, cyclo(D-Leu-L-Trp-D-Leu-L-His-D-Glu-L-Gln-D-His-L-Glu) (Fig. 7a).36 This design is crucial for guiding peptides to first assemble into one-dimensional nanotubes and subsequently into two-dimensional nanosheets. These peptides were composed of tailored hydrophobic and hydrophilic domains. The hydrophobic domain, which typically contained amino acids such as Leu and Trp, formed the internal hydrophobic core of nanotubes, which is essential for their structural stability. The hydrophilic domain, comprising charged and polar amino acids, such as Glu and His, facilitates external interactions, enabling the nanotubes to remain soluble and interact to form larger structures. These cyclic peptides adopt a β-sheet secondary structure that aids in the formation of cylindrical nanotubes, which are crucial for subsequent lateral packing into stable 2D nanosheets. Expanding on this foundation, Insua and Montenegro recently designed cyclo(L-Trp-D-Leu-L-Glu-D-His-L-Leu-D-Trp-L-Leu-D-Glu-L-His-D-Leu) (Fig. 7b).37 The introduced Trp residues acted as hydrophobic supramolecular hinges, enhancing control over their self-assembly process. These hinges allow the peptides to fold and organize more effectively and reach a critical polymerization threshold where lateral stacking becomes favorable, thus forming robust 2D networks. The precise placement of hydrophobic and hydrophilic residues, along with the structural flexibility provided by Trp hinges, allows for the controlled, dynamic assembly of peptides into monolayers that can adapt to environmental changes. Together, these studies underscore the importance of amino acid sequence and chirality when designing cyclic peptides that self-assemble into higher-order structures. By manipulating these parameters, researchers can create peptides that self-assemble into predetermined geometries and exhibit certain functional properties, such as responsiveness to external stimuli. This innovative approach holds great promise for the development of new materials with applications in biosensing, molecular transport, and responsive surfaces, reflecting a profound understanding of how molecular design can influence the macroscopic material properties.
 | ||
| Fig. 7 (a) Proposed model for the sequential 1D-to-2D self-assembly of cyclo(D-Leu-L-Trp-D-Leu-L-His-D-Glu-L-Gln-D-His-L-Glu). Reproduced with permission from ref. 36 Copyright 2020, American Chemical Society. (b) Rational design and structural scheme of self-assembling 2D nanotubular monolayers of cyclo(L-Trp-D-Leu-L-Glu-D-His-L-Leu-D-Trp-L-Leu-D-Glu--L-His-D-Leu). Reproduced with permission from ref. 37 Copyright 2023, The Royal Society of Chemistry. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds oriented perpendicular to the ring. As a result, they form PNTs via stacking of cyclic peptides through hydrogen bonds. As long as they consist of an even number of alternating D- and L-amino acid residues, a wide range of peptide sequences is compatible. As shown in Table 1, the stacking mode and lateral aggregation of PNTs varied depending on these amino acid sequences. While assemblies of 1D fibers and 2D sheets have been reported so far, the rational design of cyclic peptides is expected to provide access to a wide variety of other well-ordered self-assembled structures.
O bonds oriented perpendicular to the ring. As a result, they form PNTs via stacking of cyclic peptides through hydrogen bonds. As long as they consist of an even number of alternating D- and L-amino acid residues, a wide range of peptide sequences is compatible. As shown in Table 1, the stacking mode and lateral aggregation of PNTs varied depending on these amino acid sequences. While assemblies of 1D fibers and 2D sheets have been reported so far, the rational design of cyclic peptides is expected to provide access to a wide variety of other well-ordered self-assembled structures.
| Section | Peptide sequence | Characteristic feature | Key sequences | Ref. |
|---|---|---|---|---|
| 2.1. | Cyclo(L-Gln-D-Ala-L-Glu-D-Ala)2 | First example of PNT formation | Alternating sequences of D- and L-amino acids | 25 |
| Cyclo(D-Phe-L-Asp-D-Phe-L-Asn-D-Phe-L-Asp-D-Phe-L-Asn) | Acidification triggered PNT formation | Asp (pH-response) | 28 | |
| 2.1.1. | Cyclo(D-Leu-L-MeN-Leu)3 | Dimmer formation (Ka = 80 M−1 in CDCl3) | N-Methylation of Leu | 29 and 30 |
| Cyclo(L-Phe-D-MeN-Ala)4 | Dimmer formation (Ka = 2540 M−1 in CDCl3) | Number of amino acid residues in the cyclic peptide (8-mer had higher Ka than 6-mer) | 31 | |
| Cyclo(L-Phe-D-MeN-Ala)4 | Measuring the relative stability of parallel vs. antiparallel β-sheets | Replace chirality of the constituent amino acids | 33 | |
| Cyclo(D-Phe-L-MeN-Ala)4 | ||||
| Cyclo(L-Asp-D-Leu-L-Lys-D-Leu)2 | Antiparallel stacking arrangements | Side chain interactions between adjacent nanotubes | 34 | |
| Cyclo(L-Asp-D-Ala-L-Lys-D-Ala)2 | Parallel stacking arrangements | |||
| 2.1.2. | Cyclo(L-Gln-D-Ala)4 | Hydrophobicity of cyclic peptides (The intertubular hydrophobic interactions affected orienting nanotubes.) | 35 | |
| Cyclo(L-Gln-D-Val)4 | Less ordered lateral packing | |||
| Cyclo(L-Gln-D-Leu)4 | Tighter lateral packing of nanotubes than cyclo(L-Gln-D-Ala)4 | |||
| Cyclo(L-Gln-D-Phe)4 | Tighter lateral packing of nanotubes than cyclo(L-Gln-D-Ala)4 | |||
| Cyclo(D-Leu-L-Trp-D-Leu-L-His-D-Glu-L-Gln-D-His-L-Glu) | 2D nanosheets | Amphiphilic sequence | 36 | |
| Leu zippers and Trp stacks | ||||
| Cyclo(L-Trp-D-Leu-L-Glu-D-His-L-Leu-D-Trp-L-Leu-D-Glu-L-His-D-Leu) | 2D nanotubular monolayers | Amphiphilic sequence | 37 | |
| The positioning of two opposite hydrophobic ‘tryptophan hinges’ |
2.2. Tubular assemblies of cyclic β-peptides
As significant structural counterparts to cyclic α-alt(D,L)-peptides, cyclic β-peptides, which are derived from β-amino acids, show a pronounced ability to form stable PNTs. β-Peptides are notable for their structural integrity, strong resistance to enzymatic degradation and advantageous conformational properties, which may provide benefits compared with α-peptides.38 In 1997, Seebach et al. were the first to synthesize and characterize cyclic β-peptides.39 They designed and synthesized three stereoisomeric cyclic β-tetrapeptides composed of 3-amino butanoic acids, specifically cyclo(β3-HAla)4. Studies on cyclo(β3-HAla)4 confirmed that these peptide structures maintain a flat conformation with each subunit interconnected through four hydrogen bonds. Moreover, the rings stack efficiently due to the spatial configuration and hydrogen bonding capabilities of β-amino acids, resulting in tightly bound nanotubes. This precise molecular arrangement led to the formation of a structure with an inner pore of approximately 2.6 Å in diameter.The most distinctive feature of PNTs composed of β-amino acids is their ability to generate a significant macrodipole moment along their longitudinal axes. This is a result of the uniform orientation of all the amide bonds in the cyclic skeleton facing the same direction. Macrodipoles have been shown to accelerate electron transfer reactions along their orientation.40–42 Driven by the hypothesis that macrodipoles resulting from the stacking of cyclic β-peptides might enhance their performance as artificial transmembrane ion channels, Ghadiri et al. investigated the ion transport capabilities of cyclic β-peptides.43 They used three homochiral cyclic β-tetrapeptides, cyclo(β3-HTrp)4, cyclo(β3-HTrp-β-HLeu)2, and cyclo(β3-HLeu)4, to explore functionalities beyond the C2 symmetrical conformation reported by Seebach, which features a collapsed central hole in the peptide ring. Instead, they suggested that these cyclic β-tetrapeptides assume a C4 symmetrical conformation that maintains an internal diameter of 2.6–2.7 Å within a lipid membrane environment. Notably, cyclo(β3-HTrp)4 achieved an ion transport rate of 1.9 × 107 K+ ions per second, which is on par with that of the previously studied cyclic α-alt(D,L)-peptide cyclo[(L-Trp-D-Leu)3-L-Gln-D-Leu]. This performance is particularly important, given the much smaller internal diameter of the nanotube compared with its cyclic α-alt(D,L)-peptide counterpart, which typically measures 7–8 Å. This finding highlights the potential role of the macrodipole in influencing ion transport efficiency. However, a direct comparison between cyclic β-peptides and cyclic α-alt(D,L)-peptides with similar chemical structures and internal diameters is essential to draw a definitive conclusion.
In addition to their advantages in ion transport, the significant macroscopic dipole moments of PNTs indicate their potential applications in organic electronic nanomaterials with pyroelectric, piezoelectric, and semiconducting properties. However, two substantial challenges must be addressed to effectively utilize PNTs as valuable nanomaterials. The first challenge involved modifying the PNT surface to align the functional groups in a consistent pattern. The second challenge is achieving precise structural control when constructing the PNT bundles, as cyclic β-peptide PNTs tend to form bundles with a wide size distribution; this phenomenon is driven by strong dipole–dipole interactions due to the significant dipole moments along their axes. Here, we highlight the strategic approach applied by the Kimura group in the design of cyclic peptides to overcome these challenges.
To achieve oriented stacking of cyclic peptides, Tabata et al. designed a new cyclic peptide containing a sequence of ethylenediamine (ED) and succinic acid (SA), cyclo(β-Ala(nap)-ED-SA) (C3NES) (Fig. 8a and b).49,50 This sequence differs from that of its analog, cyclo(β-Ala(nap)-β-Ala-β-Ala) (C3NAA), in the orientation of the connecting amide bond. The former sequence enabled the formation of optimal intermolecular hydrogen bonding interactions along the PNT, but only when the cyclic peptides were stacked such that the ED-SA units were aligned on the same side of the nanotube (Fig. 8c). Such alignment results in the side chains being located on the same side of the PNT (Fig. 8d and e). Ohmura et al. designed cyclic peptides incorporating sequences of ED and SA, which were functionalized with the donor tetrathiafulvalene (TTF) and acceptor molecules (chloranil), specifically cyclo(β-Lys(TTF)-ED-SA) and cyclo(β-Asp(chloranil)-ED-SA), from which PNTs were constructed (Fig. 9).51 This design facilitated the linear alignment of TTF and chloranil along the nanotube axis and led to the generation of molecular dipole switching materials. This approach allows dipole switching without structural modifications, thereby ensuring minimal energy dissipation.
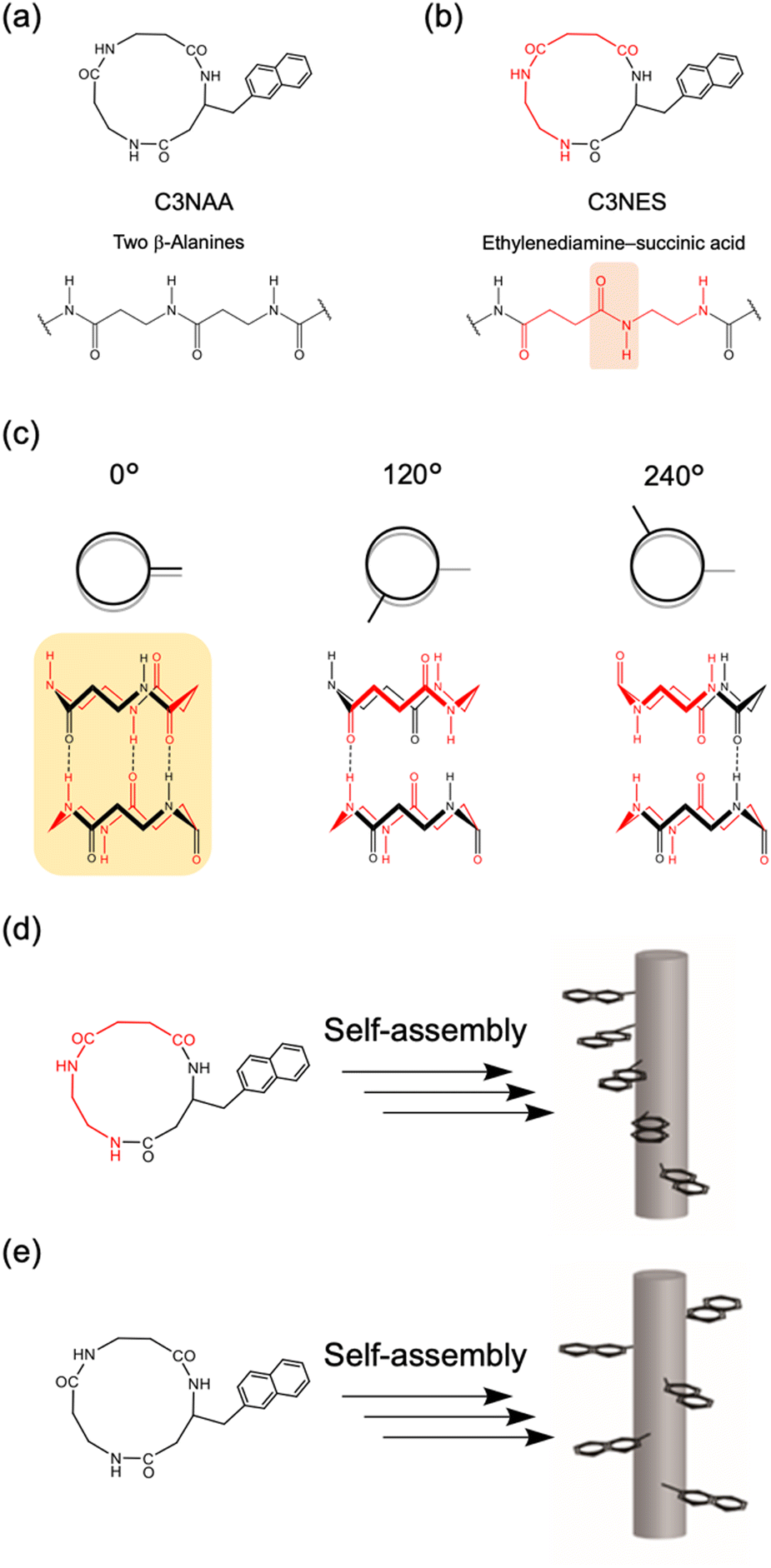 | ||
| Fig. 8 Molecular structures of cyclic tri-β-peptides of (a) C3NAA and (b) C3NES. (c) Structures of C3NES dimers, which have a sequence with initial rotations of 0°, 120°, and 240°. Schematic of PNTs formed from cyclic β-peptides with (d) or without (e) a sequence of ethylenediamine-succinic acid. | ||
 | ||
| Fig. 9 Chemical structures of cyclic peptides and schematic of the switch pairing of the charge–transfer complexes along the PNT accompanying the change in direction of the dipoles. Reproduced with permission from ref. 51 Copyright 2020, Wiley-VCH. | ||
Cyclic β-peptide-based PNTs exhibit a piezoelectric response due to the tilting of the amide bonds under an applied electric field (Fig. 10). In the emerging era of the internet of things (IoT), where sensors are essential for health monitoring, piezoelectric materials such as PNTs are prized for their ability to convert mechanical motion into electrical energy, which is ideal for miniaturized sensors and power sources.52,53 Recent advancements have focused on downsizing organic piezoelectric nanogenerators to sizes smaller than 1 μm.54–56 These devices minimally affect human activities and adapt well to the curved and corrugated surfaces of human organs. Leveraging their self-assembling and biocompatible properties, cyclic β-peptide-based PNTs have shown great potential in the development of piezoelectric devices ranging in size from several to tens of micrometers.
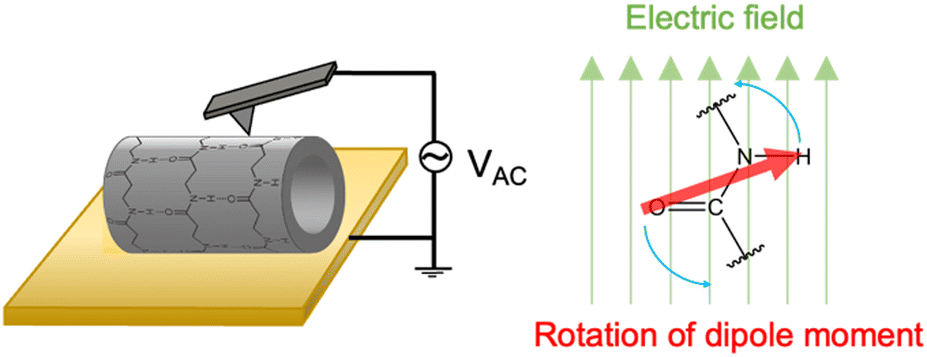 | ||
| Fig. 10 Schematic illustration of the mechanism of the piezoelectric properties of PNTs on a gold substrate. | ||
To assess the effect of side chain orientation on the piezoelectric properties of PNTs, two types of PNTs were evaluated: those composed of C3NAA with randomly oriented side chains and those composed of C3NES with aligned side chains.57 The C3NAA nanotube bundles demonstrated a high converse piezoelectric coefficient, 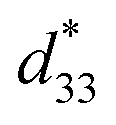 , of approximately 9 pm V−1. In contrast, the C3NES nanotube bundles exhibited a limited response to the applied electric field, with a
, of approximately 9 pm V−1. In contrast, the C3NES nanotube bundles exhibited a limited response to the applied electric field, with a 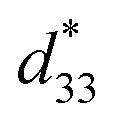 value of less than 1 pm V−1. This variation in the piezoelectric coefficient can be attributed to the extent of the non-centrosymmetric arrangement of peptide dipoles. In the case of the C3NAA nanotubes, all peptide bonds were unidirectionally aligned, leading to a highly noncentrosymmetric dipole distribution within the PNT. Conversely, in the C3NES nanotubes, two consecutive hydrogen bonding networks adopted an antiparallel orientation, disrupting the noncentrosymmetry and reducing the piezoelectric coefficient. These findings align with observations from other cyclic β-peptide-based nanotubes, where reduced symmetry typically enhances the piezoelectric properties.58
value of less than 1 pm V−1. This variation in the piezoelectric coefficient can be attributed to the extent of the non-centrosymmetric arrangement of peptide dipoles. In the case of the C3NAA nanotubes, all peptide bonds were unidirectionally aligned, leading to a highly noncentrosymmetric dipole distribution within the PNT. Conversely, in the C3NES nanotubes, two consecutive hydrogen bonding networks adopted an antiparallel orientation, disrupting the noncentrosymmetry and reducing the piezoelectric coefficient. These findings align with observations from other cyclic β-peptide-based nanotubes, where reduced symmetry typically enhances the piezoelectric properties.58
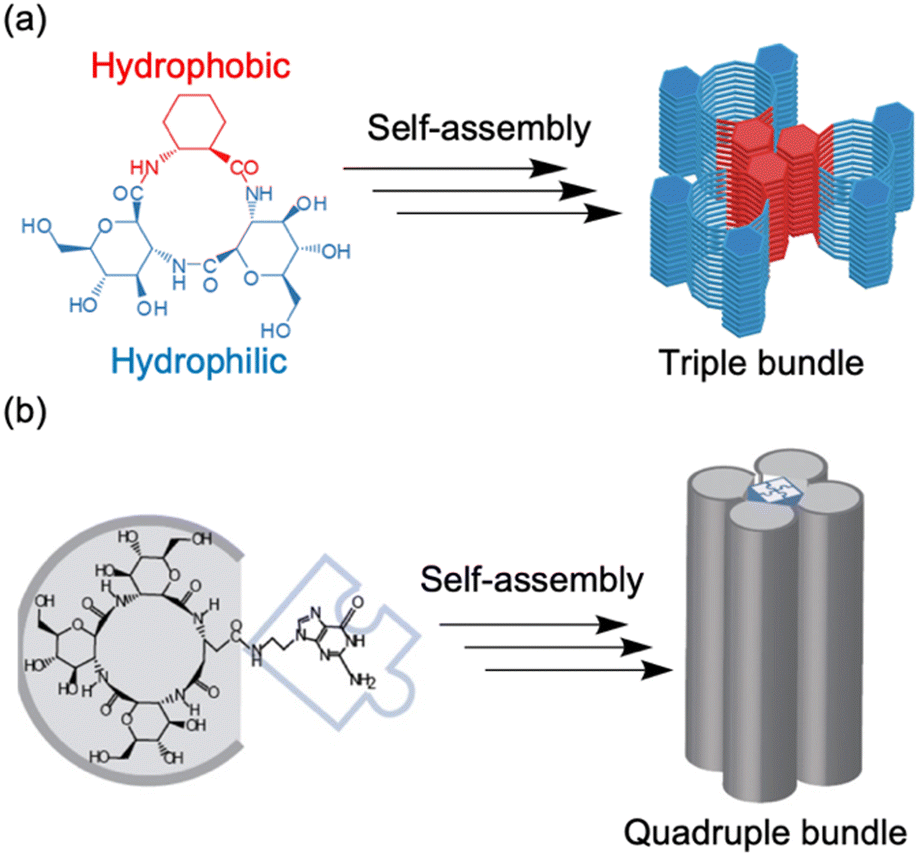 | ||
| Fig. 11 (a) Schematic illustration of the association of three amphiphilic cyclic tri-β-peptides. The hydrophobic cyclohexyl side chains were aligned linearly to make the surface hydrophobic. The other surface is hydrophilic, which leads to PNT association. (b) Schematic of self-assembly processes, from a cyclic tetra-β-peptide with a guanine moiety to a quadruple PNT bundle. Reproduced with permission from ref. 60 Copyright 2013, Wiley-VCH. | ||
Recently, the introduction of charged side-chain moieties to cyclic β-peptides was found to lead to the formation of single PNTs in aqueous solutions, preventing bundle formation by electrostatic repulsion.61 To prevent ionic functional groups from affecting the piezoelectric properties of PNTs, the authors focused on a helical peptide, which has a dipole moment from the C-terminus to the N-terminus, generating an electrostatic potential field.62 The authors designed CP3A8E, a cyclic β-peptide with a helical peptide attached to its side chain, to lead the formation of a single PNT (Fig. 12).63 A single PNT was constructed from CP3A8E by preventing close contact between the PNTs owing to the electrostatic repulsion and steric hindrance provided by the helical peptides. This study highlights the role of the primary structure of a cyclic peptide in controlling the hierarchical structure of the resulting PNT. Additionally, after this single PNT was constructed, we were able to compare its piezoelectric properties with those of PNT bundles. Piezoresponse force microscopy (PFM) measurements revealed that the  of the single PNTs was 1.39 ± 0.12 pm V−1. This value closely matches the
of the single PNTs was 1.39 ± 0.12 pm V−1. This value closely matches the 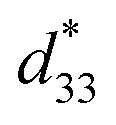 value of 1.34 ± 0.16 pm V−1 measured for bundled PNTs.58 These results indicate that the piezoelectric properties of PNTs are inherently linked to the deformation of their cyclic skeletons rather than their aggregate structure. In other words, the piezoelectric properties of PNTs remain unchanged when their size is reduced to a single unit. Given that a single PNT with a size of 4 nm represents the smallest known biomaterial-based piezoelectric material, PNTs composed of cyclic peptides are promising candidates for use as energy harvesters in biomedical applications, particularly in previously inaccessible microenvironments.
value of 1.34 ± 0.16 pm V−1 measured for bundled PNTs.58 These results indicate that the piezoelectric properties of PNTs are inherently linked to the deformation of their cyclic skeletons rather than their aggregate structure. In other words, the piezoelectric properties of PNTs remain unchanged when their size is reduced to a single unit. Given that a single PNT with a size of 4 nm represents the smallest known biomaterial-based piezoelectric material, PNTs composed of cyclic peptides are promising candidates for use as energy harvesters in biomedical applications, particularly in previously inaccessible microenvironments.
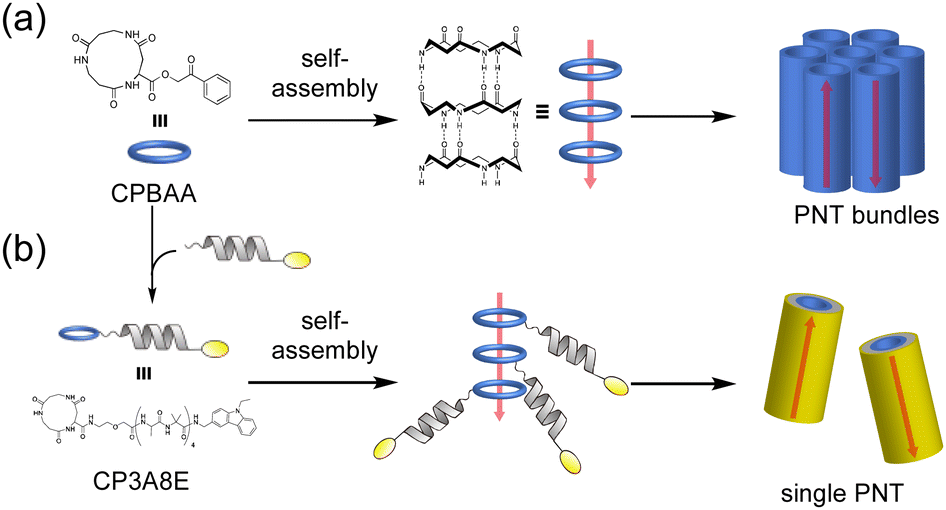 | ||
| Fig. 12 (a) Schematic illustration of PNT bundle formation with a usual cyclic β-peptide. (b) Schematic of single PNT formation from a cyclic β-peptide with a helical peptide. Each red arrow indicates the dipole moment of the PNT. Reproduced with permission from ref. 63 Copyright 2021, American Chemical Society. | ||
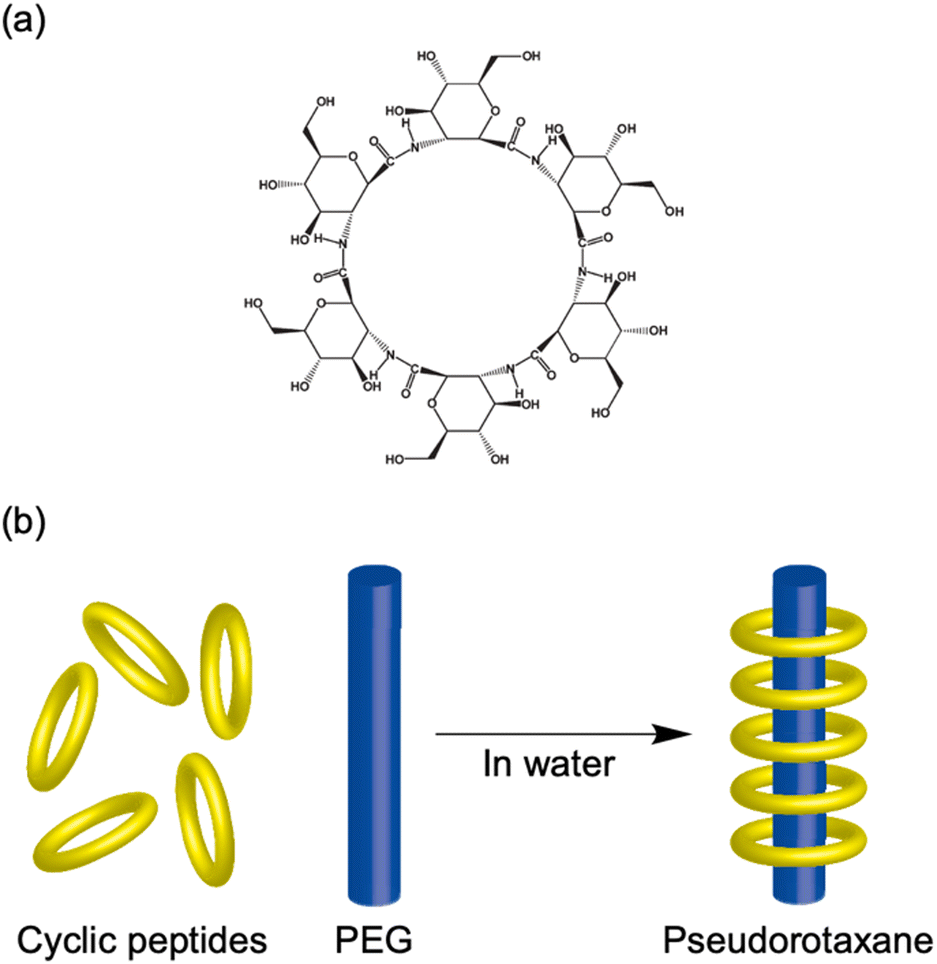 | ||
| Fig. 13 (a) Chemical structures of cyclic hexamers of β-glucosamino acid. (b) Schematic illustration of polypseudorotaxane formation from cyclic hexamers of β-glucosamino acid and poly(ethylene glycol). | ||
| Section | Peptide sequence | Characteristic feature | Key sequences | Ref. |
|---|---|---|---|---|
| 2.1. | Cyclo(β3-HAla)4. | First example of PNT from cyclic β-peptides | β-Pepitde | 39 |
| 2.1.1. | Cyclo(β3-HTrp)4 | Working as transmembrane ion channels | Cyclic β3-peptides adopted flat-ring C4 symmetrical conformation with an internal diameter of 2.6–2.7 Å, large enough to accommodate water and small ions. | 43 |
| Cyclo(β3-HTrp-β-HLeu)2, | Working as transmembrane ion channels | |||
| Cyclo(β3-HLeu)4 | Not working as transmembrane ion channels | β3-HTrp residues allowed PNTs incorporation into lipid membranes. | ||
| 2.2.1. | Cyclo(β-Ala(nap)-ED-SA) (C3NES) | The side chains located on the same side of the PNTs. | ED-SA sequences | 49 and 50 |
| Cyclo(β-Ala(nap)-β-Ala-β-Ala) (C3NAA) | The side chains were randomly distributed along the PNTs. | |||
| Cyclo(β-Lys(TTF)-ED-SA) and cyclo(β-Asp(chloranil)-ED-SA) | Formation of charge–transfer complexes | Linear alignment of donor and acceptor molecules induced by ED-SA sequences | 51 | |
| 2.2.2. | Cyclo[(GA)2ACHC] | Triple bundle formation of the PNTs | Amphiphilic cyclic β-peptide | 59 |
| Cyclo[(GA)3-Glu(G)] | Quadruple bundle formation of the PNTs | Introducing a guanine group into a cyclic peptide | 60 | |
| CP3A8E | Formation of single PNTs | Introducing a helical peptide into a cyclic peptide | 63 | |
| 2.2.3 | Cyclo(GA)6 | Pseudorotaxane formation with PEG | Cyclic hexa-β-peptide of GA with large inner pore of 5.1 Å and water solubility | 65 |
2.3. Tubular assemblies of cyclic α,γ-peptides
Recent advancements in peptide-based materials have led to significant interest in the development of PNTs featuring alternating α- and γ-amino acids. Granja et al. pioneered the use of cyclic γ-amino acids, such as cis-3-aminocycloalkane carboxylic acid (γ-Aca), to create novel self-assembling cyclic peptides.27 These cyclic γ residues confer unique structural and functional properties to PNTs, enabling their diverse applications in biological systems and materials science. The incorporation of cyclic γ-amino acids contributes to the rigid and flat conformation of the peptide backbone, which is essential for stacking and nanotube formation. This structural orientation also endows the internal cavity with novel features, such as the creation of a hydrophobic region within the otherwise hydrophilic lumen. This effect was achieved by strategically positioning a cycloalkane methylene group from γ-Aca into the lumen of the nanotube. In contrast to α- or β-peptide-based nanotubes, which have fixed cavities, γ-cyclic PNTs permit functional modifications at specific sites within the nanotubes, thus offering enhanced control over their internal properties. PNTs are typically composed of an even number of α- and cyclic γ-amino acids arranged in a planar ring-like conformation, which facilitates selective intermolecular interactions. This configuration promotes antiparallel stacking, driven by specific hydrogen bonding patterns between the amide groups of the α and γ residues, potentially resulting in more stable nanotube structures. Alternatively, parallel arrangements facilitated by interactions between the α-face and γ-face are also possible, which contributes to the stability of these assemblies. The ability to fine-tune the peptide rings and hydrophobicity of the internal cavity through simple chemical modifications enhances the utility of these assemblies, positioning them as versatile candidates for diverse material science and biotechnological applications. | ||
| Fig. 14 Heterodimeric supramolecular systems. The α,γ-Ach and α,γ-Acp cyclic peptide combination was used to obtain new heterodimeric structures. | ||
| Section | Peptide sequence | Characteristic feature | Key sequences | Ref. |
|---|---|---|---|---|
| 2.3.1. | Cyclo(γ-Aca-D-MeN-Ala)3 | First example of PNT from cyclic α,γ-peptides with γ–γ hydrogen bonding interactions | Alternating sequences of α- and γ-amino acids | 39 |
| Cyclo(D-Phe-MeN-γ-Aca)3 | First example of PNT from cyclic α,γ-peptides with α–α hydrogen bonding interactions | |||
| Cyclo(D-Leu-(1R,3S)-MeN-γ-Acp)3 | Heterodimer formation | Combination of Acp- and Ach-based cyclic α,γ-peptides | 67 | |
| Cyclo(D-Phe-(1R,3S)-MeN-γ-Ach)3 | ||||
| 2.3.2. | Cyclo(D-Leu-MeN-γ-Acp-D-Leu-γ-Ahf)2 | PNTs with hydrophilic cavity | γ-Ahf | 69 and 70 |
3. Supramolecular chemistry of cyclic peptides
3.1. Cation binding ability of cyclic peptides
The three-dimensional structures of naturally occurring cyclic peptides are closely related to their biological activities.73–76 Cyclization constrains the molecule overall, preconfiguring it for receptor binding and reducing the conformational entropy loss by this preconfiguration. Additionally, this reduction in entropy loss aids in complex formation. For example, the cyclodecapeptide antibiotic antamanide forms complexes with alkali metal cations, particularly potassium ions.77 Similarly, the cyclodepsipeptide valinomycin transports alkali metal cations across biological membranes after complex formation.78–81 These cation interactions have a significant influence on biological activity, promoting the synthesis of various synthetic peptides inspired by their natural counterparts. Researchers have evaluated the complex-forming abilities of these synthetic peptides and determined their conformations. Although researchers have studied the structures and affinities of individual cyclic peptides, not all combinations can be synthesized, leaving design guidelines imperfectly established. Pro plays a crucial role in the activity of these peptides.82–88 Pro is found in many naturally occurring peptide antibiotics and peptide antitoxins.89,90 Thus, it is beneficial to consider the structural implications of including Pro in cyclic peptides, because its pyrrolidine ring promotes the formation of a bend in the peptide chain, and this bending can restrict or halt the coordination of adjacent amide nitrogen donors to the metal ions. The rigidity imposed by the N–Cα bond in proline residues limits the formation of β-turns and restricts the available conformations. Therefore, Pro is frequently used in the design of synthetic cyclic peptides.3.2. Rotaxanes and catenanes from cyclic peptides
Complex formation significantly affects biological functions, a topic that is further detailed in other reviews.91 We believe that utilizing the receptor properties of cyclic peptides enables the formation of supramolecular assemblies with interlocked structures, such as rotaxanes. This is evident from the crucial role of crown ethers, which can also coordinate cations, in supramolecular chemistry.92 In nature, peptide [2]rotaxane systems exhibit specific bioactivities. Notably, peptide-based rotaxane microcin J25, one of the few natural molecules that inhibits RNA polymerase, is an ideal target for antimicrobial therapy.93–95 By synthesizing analogs that mimic these properties, we can potentially obtain new bioactive compounds with considerable utility. Furthermore, lasso peptides possess unique properties that make them suitable for various applications in medicinal chemistry and can serve as robust scaffolds. In addition, rotaxane crosslinked polymers are extremely tough owing to their freely movable crosslinking points, which equalize the tension in the polymer chain upon tensile deformation.96,97 Rotaxanes synthesized from peptides can serve as crosslinkers, increasing the toughness of the network polymers while exhibiting biodegradability and compatibility with polypeptides and proteins. Therefore, interlocking peptide structures are expected to have various applications. However, the methods by which interlocked structures, such as rotaxanes and catenanes, can be synthesized from cyclic peptides remain limited owing to specific technical and molecular constraints.Aucagne et al. used cyclo(L-Pro-L-Gly)4and5, which reported to form a γ-turn structure with its carbonyl oxygens pointing toward the center of the ring, demonstrating high affinity constants for metal ions and cations in acetonitrile.88 They reported the formation of [2]rotaxanes with yields of 56–63% via the construction of cationic diol threads and utilization of bulky acid chlorides to “stopper” the reactions with these macrocycles via ethane-1,2-diammonium and butane-1,4-diammonium templates (Fig. 15).98
 | ||
| Fig. 15 Synthetic scheme of cyclic peptide-based [2]rotaxanes from cyclo(PG)4 and diammonium threads. Reproduced with permission from ref. 98 Copyright 2006, American Chemical Society. | ||
All peptide-based rotaxanes have not been reported because peptides lack binding motifs with sufficient strength for stable rotaxane formation.99 Specific cyclic peptides can form assemblies through hydrogen bonding interactions between amide bonds. However, these bonds are not strong enough to allow axial molecules to penetrate the cyclic peptides.
The authors found that cyclo(L-Pro-L-Gly)4 forms complexes with primary amines via hydrogen bonding. Bearing this in mind, we successfully synthesized all peptide-based rotaxanes from cyclic peptides with a γ-turn structure, electrophiles, and nucleophiles via the active-template method100 (Fig. 16).101 This method facilitates the synthesis of rotaxanes through an axis-forming reaction inside a cyclic molecule. The successful synthesis of the peptide rotaxane was confirmed by high-performance liquid chromatography (HPLC) and electrospray ionization mass spectrometry (ESI-MS) in a yield of 1.2%. Combining experimental results with molecular dynamics (MD) simulation data, we determined that cyclic peptides with an inward-directed carbonyl oxygen and a spacious internal cavity are preferable for rotaxane synthesis.
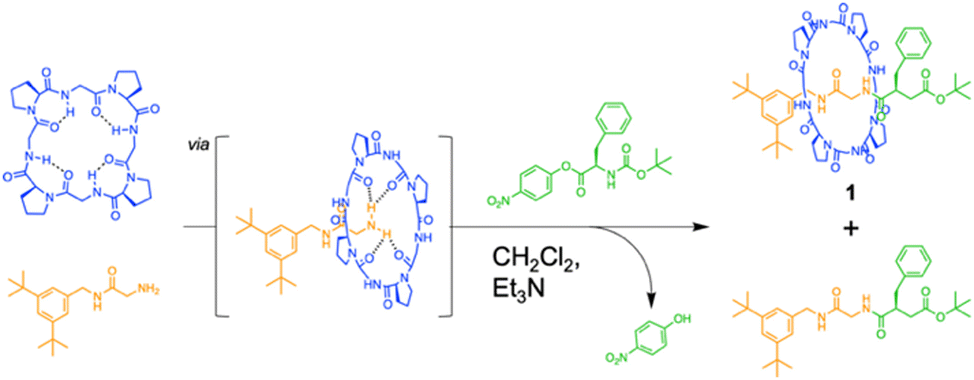 | ||
| Fig. 16 Synthesis of an all peptide-based [2]rotaxane by the cyclic peptide-accelerated N-acylation of primary amines. Reproduced with permission from ref. 101 Copyright 2024, American Chemical Society. | ||
Recent advances in peptide-based catenane formation have been reported using dynamic combinatorial libraries. These supramolecular systems are generated from reversible binding units that self-assemble around a specific receptor target, acetylcholine. Specifically, chiral dipeptides with a masked aldehyde functionality and hydrazide moiety (e.g., Pro–Phe) reversibly bind via hydrazone bonds. Surprisingly, among the potential diastereomers, only [2]catenane, consisting of two interlocked macrocyclic trimers, was formed.102 The acetylcholine trimethylammonium group was identified as the major template recognition site.
The relatively limited number of peptide-based rotaxanes and catenanes available clearly indicates ample room for further experimental and theoretical investigations in these intriguing fields. Peptide science is classically an interdisciplinary field that is expected to contribute significantly to bridging and strengthening the connection between synthetic organic chemistry, biology, and nanoscience. Within this framework, the unique modular nature of peptides and vast chemical diversity of natural and unnatural amino acids offer substantial advantages for the development and application of peptide-based rotaxanes and catenanes. Mechanically interlocked peptides and proteins exhibit unique properties that are not observed in conventional proteins, including thermal, mechanical, and chemical stability; proteolytic resistance; and dynamic features such as sliding and switching. A synthetic approach to generating interlocking structures opens new avenues for the creation of peptide- and protein-based materials with unprecedented functions.
4. Solution structure prediction of cyclic peptides via computational methods
As mentioned above, a wide variety of assemblies have been constructed via rational design of cyclic peptide primary structures, and various functions have been achieved. If we can predict the solution structures of cyclic peptides and the types of complexes that they form with various molecules, we can more easily and rationally design novel cyclic peptides on the basis of their predicted solution structures. These cyclic peptides are likely to form novel assemblies and perform innovative functions.Cyclic peptides tend to adopt multiple conformations in solution, which complicates the generation of structural models. This difficulty arises because experimental measurements, such as NMR and circular dichroism (CD) spectroscopy, often yield time- and ensemble-averaged data. Therefore, computational methods play a crucial role in studying the conformational ensembles of cyclic peptides and could provide insights that are challenging to obtain experimentally.
MD simulations are powerful tools for understanding the properties of peptides and proteins. However, conventional MD simulations often struggle to provide complete structural ensembles of cyclic peptides because of the large free energy barriers between conformations that are caused by ring strain. To overcome these barriers, several enhanced sampling methods have been developed, including replica exchange molecular dynamics (REMD),103 bias-exchange metadynamics (BE-META),104 multivariate molecular dynamics (McMD),105 accelerated molecular dynamics (aMD),106 and complementary coordinate molecular dynamics (CoCo-MD).107 Please refer to this review article for a discussion on the enhanced techniques used for sampling cyclic peptide conformations and the recent applications of MD simulations used with these methods to understand or design cyclic peptides.108 Owing to the development of these enhanced sampling techniques, it is now possible to study dozens of cyclic peptide sequences that target essential transitional motions within a reasonable timeframe. In addition to explaining known experimental results, MD simulations have recently played a leading role in making predictions, which have been subsequently verified experimentally.109–111 Lin et al. identified that conformational switches in cyclic peptides require coherent changes in coupled dihedral angles-specifically, either the ϕ and ψ angles of the same residue (ϕi and ψi) or the ψ angle of a residue and the ϕ angle of the next residue (ψi and ϕi+1).112 By targeting these coupled dihedrals using two-dimensional collective variables with BE-META simulations, they significantly reduced convergence times compared to standard methods, enhancing the efficiency of sampling conformational space. Expanding on this method, they systematically investigated more than 70 cyclic pentapeptides, aiming to understand how amino acid sequences control their structural ensembles, and developed a scoring function based on these MD simulation data to predict structural preferences, estimating that cyclo(L-Gly-L-Asn-L-Ser-L-Arg-L-Val) adopt a well-defined βII′ + αR conformation (a type-II′ β-turn with a tight αR turn opposing it). Further MD simulations confirmed this prediction, revealing 67% prevalence of the desired conformation. This result was further supported by NMR experiments.109 Extending the application of this predictive BE-META simulation, they explored the comprehensive structural ensembles of two families of cyclic hexapeptides: cyclo(L-Glyn-L-Ala{6−n}) and cyclo(L-Glyn-L-Val{6−n}). These two families contain a total of 27 cyclic peptides, taking into account sequence permutations and symmetry. Among these cyclic peptides, cyclo(L-Val-L-Val-L-Gly-L-Gly-L-Val-L-Gly) was uniquely predicted to adopt a single, highly populated conformation characterized by two βII turns at specific residues.113 They synthesized cyclo(L-Val-L-Val-L-Gly-L-Gly-L-Val-L-Gly) and confirmed the MD predictions using NMR spectroscopy, observing the anticipated well-defined structure. Additional simulations and experiments underscored the critical role of the Val residue at position 1 in stabilizing this conformation, emphasizing the significance of specific amino acid positions for structures of cyclic peptides.110 MD simulations also play a predictive role in designing new cyclic peptides as potential inhibitors of protein–protein interactions. The optimized BE-META sampling protocol accurately predicted the preorganized structure of a series of peptides, each with individual amino acid substitutions in a Kelch-like ECH-associated protein-1 (Keap1)-binding sequence derived from nuclear factor-erythroid 2 p45-related factor 2 (Nrf2). The experimental binding affinity results confirmed the binding affinity prediction using a simulated conformational ensemble. The culmination of their sequence optimization effort, cyclo(ortho-L-Cys-L-Asn-L-Pro-L-Glu-L-Thr-D-Ala-L-Glu-L-Cys), competes with the highest reported binding affinity for Keap1 to date (4.7 nM, compared to 2.8 nM for a disulfide-linked cyclic peptide of similar size114), indicating that MD simulations can design substitutions that promoted the desired conformations more effectively.111
Recent advancements in machine learning (ML) technologies, particularly the development of AlphaFold2 and RoseTTAFold, have demonstrated impressive capabilities for predicting protein structures.115–120 A notable extension of this technology is AfCycDesign, which is based on AlphaFold and has shown promising results in predicting the structures of cyclic peptide monomers, underscoring the adaptability of these models to various molecular configurations.121 However, cyclic peptides, typically comprising 5 to 15 amino acids, rarely form stable secondary structures or distinct folds, which presents significant challenges for these ML models in terms of accurately predicting the peptide structure in solution. Moreover, advanced ML models, which were developed with extensive protein-specific structural and evolutionary data, are not directly applicable to cyclic peptides. Cyclic peptides often lack detailed structural data owing to limited nuclear Overhauser effect (NOE) signal availability and are commonly produced through nonribosomal synthesis. This scarcity in data complicates the further enhancement of these models for such applications. Furthermore, because the training data for these models are derived primarily from proteins, predicting the structures of peptides that incorporate unnatural amino acids or exist in nonaqueous environments remains difficult. The pioneering work of the Lin group on structural ensembles achieved by MD and ML (StrEAMM) marked a significant advancement in the computational prediction of cyclic peptide structures.122 This innovative method combines MD simulations with ML techniques to predict the structural ensembles of cyclic peptides effectively. By performing MD simulations on 705 variants, they compiled a diverse dataset to train linear regression models. This approach enables the rapid prediction of structural ensembles of cyclic pentapeptides, giving results in less than one second per sequence across an extensive sequence space of approximately 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential pentapeptides (Fig. 17). To extend this approach, StrEAMM has been adapted for cyclic hexapeptides. They incorporated advanced ML techniques, including convolutional and graph neural networks, to capture nonlinear interactions, and these techniques significantly outperformed traditional models. These enhanced StrEAMM models demonstrated remarkable prediction accuracies, with R2 values reaching 0.97 for pentapeptides and 0.91 for hexapeptides.123
000 potential pentapeptides (Fig. 17). To extend this approach, StrEAMM has been adapted for cyclic hexapeptides. They incorporated advanced ML techniques, including convolutional and graph neural networks, to capture nonlinear interactions, and these techniques significantly outperformed traditional models. These enhanced StrEAMM models demonstrated remarkable prediction accuracies, with R2 values reaching 0.97 for pentapeptides and 0.91 for hexapeptides.123
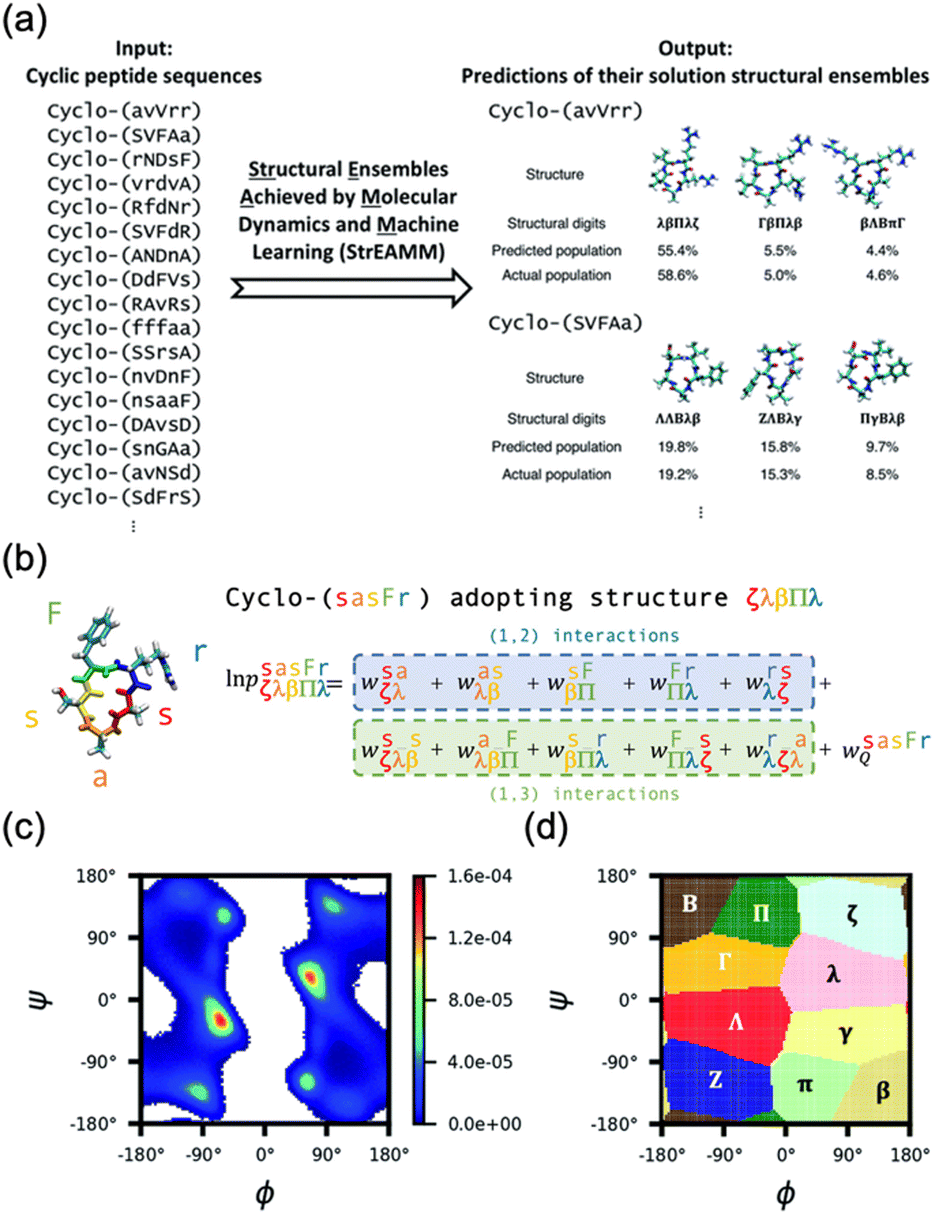 | ||
Fig. 17 Schematic illustration of StrEAMM. (a) A StrEAMM model leverages MD simulation results as the training dataset to rapidly predict the structural ensembles of cyclic peptides with both well-structured and non-well-structured sequences. (b) the StrEAMM linear regression models to predict the population for a sequence cyclo(X1X2X3X4X5) adopting a specific structure S1S2S3S4S5. The natural logarithm of the population of cyclo(X1X2X3X4X5) is the sum of the interaction weights for each (1,2) and (1,3) neighbor present. Xi is an amino acid; Si is a structural digit that represents a region of (ϕ, ψ) space (described in (c) and (d)); 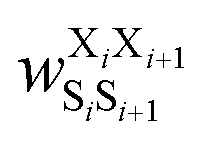 is the weight for residues XiXi+1 adopting structure SiSi+1; is the weight for residues XiXi+1 adopting structure SiSi+1; 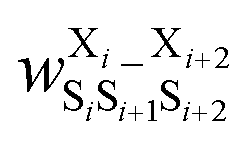 is the weight for residues Xi and Xi+2 adopting substructure SiSi+1Si+2; and is the weight for residues Xi and Xi+2 adopting substructure SiSi+1Si+2; and 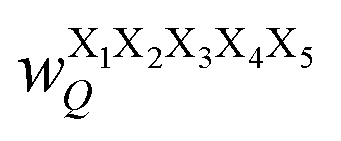 is related to the partition function, Q, for sequence X1X2X3X4X5 and ensures that all the structures’ populations sum to 1 for a given sequence. (c) The (ϕ, ψ) population density of cyclo(L-Gly)5. (d) The Ramachandran plot divided into 10 regions: L, l, G, g, B, b, P, p, Z, and z. For structural description based on the (ϕ, ψ) population density of cyclo(L-Gly)5. Reproduced with permission from ref. 122 and 123 Copyright 2021, The Royal Society of Chemistry and 2023, American Chemical Society. is related to the partition function, Q, for sequence X1X2X3X4X5 and ensures that all the structures’ populations sum to 1 for a given sequence. (c) The (ϕ, ψ) population density of cyclo(L-Gly)5. (d) The Ramachandran plot divided into 10 regions: L, l, G, g, B, b, P, p, Z, and z. For structural description based on the (ϕ, ψ) population density of cyclo(L-Gly)5. Reproduced with permission from ref. 122 and 123 Copyright 2021, The Royal Society of Chemistry and 2023, American Chemical Society. | ||
5. Conclusion and future outlook
Our comprehensive review underscores the transformative role of the primary structures of cyclic peptides, highlighting their profound impact on self-assembly processes and functionality (Fig. 18). This insight propels our ability to manipulate these structures, paving the way for the development of sophisticated biomaterials and groundbreaking nanotechnology. Strategically modifying peptide sequences, particularly the ring sizes and side chains, is crucial for directing the assembly behaviors of PNTs and enabling precise control over their structural and functional properties. Such modifications both shape the hierarchical structure and increase the functionality of PNTs, thereby broadening their applications from organic electronics to innovative solutions for ion channels, antibacterial materials, and targeted delivery systems. The synthesis of rotaxanes using cyclic peptides represents a thrilling advancement, opening new frontiers in molecular architecture. Rationally designed cyclic peptides can interact with other molecules and forge pathways to develop mechanically interlocked molecular architectures. These dynamic structures have immense potential, revolutionizing everything from drug delivery systems to the development of molecular machines and materials with unprecedented mechanical properties. | ||
| Fig. 18 Representative design protocols for rationally designed cyclic peptides, (a) cyclic β-peptides with helical peptides that form single PNTs and (b) cyclic peptides that form rotaxanes. | ||
The integration of computational tools and advanced simulation methods is expected to lead to significant breakthroughs in cyclic peptide research, enhancing both their understanding and application. These technologies not only increase our precision in predicting peptide behaviors but also innovate the way in which we design complex peptide structures. With cutting-edge sampling techniques, MD simulations will be crucial in unraveling the dynamic behaviors of cyclic peptides, fostering the development of new materials with enhanced properties. To fully exploit the potential of cyclic peptides, cross-disciplinary collaboration that merges insights from synthetic chemistry, biology, and materials science into a cohesive research effort will be essential. Ultimately, the success of these endeavors rests on the sophisticated design of the primary structures of cyclic peptides, underscoring the pivotal role of molecular architecture in unlocking their full functional potential.
Author contributions
T. K. and K. N. contributed to the writing of this article.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This research was financially supported by JST ERATO grant no. JPMJER1602, Japan (K. N.); Grant-in-Aid for Transformative Research Areas (B) grant no. JP20H05735 (K. N.); the MEXT Data Creation and Utilization-type MaTerial R&D project (K. N.); JST CREST grant no. JPMJCR2091 (K. N.) and JSPS Research Fellowships for Young Scientists (grant no. 23KJ1250 (K. T.)).Notes and references
- B. J. Pepe-Mooney and R. Fairman, Curr. Opin. Struct. Biol., 2009, 19, 483–494 CrossRef CAS PubMed.
- M. J. Harrington and P. Fratzl, Prog. Mater. Sci., 2021, 120, 1–40 CrossRef.
- B. E. Ramakers, J. C. van Hest and D. W. Lowik, Chem. Soc. Rev., 2014, 43, 2743–2756 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS.
- G. Abbenante, D. R. March, D. A. Bergman, P. A. Hunt, B. Garnham, R. J. Dancer, J. L. Martin and D. P. Fairlie, J. Am. Chem. Soc., 1995, 117, 10220–10226 CrossRef CAS.
- H. L. Sham, C. A. Rempel, H. Stein and J. Cohen, J. Chem. Soc., Chem. Commun., 1990, 666–667 RSC.
- R. M. Schultz, J. P. Huff, P. Anagnostaras, U. Olsher and E. R. Blout, Int. J. Pept. Protein Res., 1982, 19, 454–469 CrossRef CAS PubMed.
- J. Witek, S. Wang, B. Schroeder, R. Lingwood, A. Dounas, H. J. Roth, M. Fouche, M. Blatter, O. Lemke, B. Keller and S. Riniker, J. Chem. Inf. Model., 2019, 59, 294–308 CrossRef CAS.
- S. Ono, M. R. Naylor, C. E. Townsend, C. Okumura, O. Okada and R. S. Lokey, J. Chem. Inf. Model., 2019, 59, 2952–2963 CrossRef CAS PubMed.
- J. Witek, M. Muhlbauer, B. G. Keller, M. Blatter, A. Meissner, T. Wagner and S. Riniker, ChemPhysChem, 2017, 18, 3309–3314 CrossRef CAS.
- J. Witek, B. G. Keller, M. Blatter, A. Meissner, T. Wagner and S. Riniker, J. Chem. Inf. Model., 2016, 56, 1547–1562 CrossRef CAS PubMed.
- B. Over, P. Matsson, C. Tyrchan, P. Artursson, B. C. Doak, M. A. Foley, C. Hilgendorf, S. E. Johnston, M. D. T. Lee, R. J. Lewis, P. McCarren, G. Muncipinto, U. Norinder, M. W. Perry, J. R. Duvall and J. Kihlberg, Nat. Chem. Biol., 2016, 12, 1065–1074 CrossRef CAS PubMed.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed.
- R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448–1456 RSC.
- B. J. Pieters, M. B. van Eldijk, R. J. Nolte and J. Mecinovic, Chem. Soc. Rev., 2016, 45, 24–39 RSC.
- J. M. Alonso, M. L. Gorzny and A. M. Bittner, Trends Biotechnol., 2013, 31, 530–538 CrossRef CAS PubMed.
- A. Harada, J. Li and M. Kamachi, Nature, 1993, 364, 516–518 CrossRef CAS.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- T. Shimizu, H. Minamikawa, M. Kogiso, M. Aoyagi, N. Kameta, W. Ding and M. Masuda, Polym. J., 2014, 46, 831–858 CrossRef CAS.
- C. H. Gorbitz, Chem. – Eur. J., 2001, 7, 5153–5159 CrossRef CAS.
- T. Kanzaki, Y. Horikawa, A. Makino, J. Sugiyama and S. Kimura, Macromol. Biosci., 2008, 8, 1026–1033 CrossRef CAS.
- W. H. Hsieh and J. Liaw, J. Food Drug Anal., 2019, 27, 32–47 CrossRef CAS.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS.
- D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913–941 CrossRef CAS.
- M. Amorin, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2003, 125, 2844–2845 CrossRef CAS.
- M. E. Polaskova, J. N. Lambert and N. J. Ede, Aust. J. Chem., 1998, 51, 535 CrossRef CAS.
- M. Saviano, L. Zaccaro, A. Lombardi, C. Pedone, B. Blasio, X. Sun and G. P. Lorenzi, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 18, 27–36 CrossRef CAS.
- X. Sun and G. P. Lorenzi, Helv. Chim. Acta, 1994, 77, 1520–1526 CrossRef CAS.
- M. R. Ghadiri, K. Kobayashi, J. R. Granja, R. K. Chadha and D. E. McRee, Angew. Chem., Int. Ed. Engl., 1995, 34, 93–95 CrossRef CAS.
- T. D. Clark, J. M. Buriak, K. Kobayashi, M. P. Isler, D. E. McRee and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 8949–8962 CrossRef CAS.
- K. Kobayashi, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed. Engl., 1995, 34, 95–98 CrossRef CAS.
- M. R. Silk, J. Newman, J. C. Ratcliffe, J. F. White, T. Caradoc-Davies, J. R. Price, S. Perrier, P. E. Thompson and D. K. Chalmers, Chem. Commun., 2017, 53, 6613–6616 RSC.
- J. D. Hartgerink, J. R. Granja, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1996, 118, 43–50 CrossRef CAS.
- I. Insua and J. Montenegro, J. Am. Chem. Soc., 2020, 142, 300–307 CrossRef CAS.
- I. Insua, A. Cardellini, S. Diaz, J. Bergueiro, R. Capelli, G. M. Pavan and J. Montenegro, Chem. Sci., 2023, 14, 14074–14081 RSC.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- D. Seebach, J. L. Matthews, A. Meden, T. Wessels, C. Baerlocher and L. B. McCusker, Helv. Chim. Acta, 2004, 80, 173–182 CrossRef.
- H. Uji, T. Morita and S. Kimura, Phys. Chem. Chem. Phys., 2013, 15, 757–760 RSC.
- S. Yasutomi, T. Morita, Y. Imanishi and S. Kimura, Science, 2004, 304, 1944–1947 CrossRef CAS PubMed.
- T. Morita, S. Kimura, S. Kobayashi and Y. Imanishi, J. Am. Chem. Soc., 2000, 122, 2850–2859 CrossRef CAS.
- T. D. Clark, L. K. Buehler and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 651–656 CrossRef CAS.
- W. S. Horne, N. Ashkenasy and M. R. Ghadiri, Chem. – Eur. J., 2005, 11, 1137–1144 CrossRef CAS PubMed.
- N. Ashkenasy, W. S. Horne and M. R. Ghadiri, Small, 2006, 2, 99–102 CrossRef CAS PubMed.
- H. Uji, H. Kim, T. Imai, S. Mitani, J. Sugiyama and S. Kimura, Biopolymers, 2016, 106, 275–282 CrossRef CAS.
- R. J. Brea, M. E. Vazquez, M. Mosquera, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2007, 129, 1653–1657 CrossRef CAS.
- C. Reiriz, R. J. Brea, R. Arranz, J. L. Carrascosa, A. Garibotti, B. Manning, J. M. Valpuesta, R. Eritja, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2009, 131, 11335–11337 CrossRef CAS PubMed.
- Y. Kamano, Y. Tabata, H. Uji and S. Kimura, RSC Adv., 2019, 9, 3618–3624 RSC.
- Y. Tabata, S. Mitani and S. Kimura, J. Pept. Sci., 2016, 22, 391–396 CrossRef CAS PubMed.
- H. Ohmura, Y. Tabata, S. Kimura and H. Uji, Pept. Sci., 2020, 113, 1–8 Search PubMed.
- L. Atzori, A. Iera and G. Morabito, Comput. Networks, 2010, 54, 2787–2805 CrossRef.
- R. Bogue, Sens. Rev., 2014, 34, 137–142 CrossRef.
- Y. Sun, K. Y. Zeng and T. Li, Sci. China: Phys., Mech. Astron., 2020, 63, 278701 CAS.
- H. Yuan, P. Han, K. Tao, S. Liu, E. Gazit and R. Yang, Research, 2019, 2019, 1–13 CrossRef.
- B. Y. Lee, J. Zhang, C. Zueger, W. J. Chung, S. Y. Yoo, E. Wang, J. Meyer, R. Ramesh and S. W. Lee, Nat. Nanotechnol., 2012, 7, 351–356 CrossRef CAS.
- Y. Tabata, S. Mitani, H. Uji, T. Imai and S. Kimura, Polym. J., 2019, 51, 601–609 CrossRef CAS.
- Y. Tabata, Y. Kamano, H. Uji, T. Imai and S. Kimura, Chem. Lett., 2019, 48, 322–324 CrossRef CAS.
- Y. Ishihara and S. Kimura, J. Pept. Sci., 2010, 16, 110–114 CrossRef CAS.
- Y. Ishihara and S. Kimura, Biopolymers, 2013, 100, 141–147 CrossRef CAS.
- Y. Tabata, Y. Kamano, S. Kimura and H. Uji, RSC Adv., 2020, 10, 3588–3592 RSC.
- W. G. Hol, P. T. van Duijnen and H. J. Berendsen, Nature, 1978, 273, 443–446 CrossRef CAS PubMed.
- T. Kurita, T. Terabayashi, S. Kimura, K. Numata and H. Uji, Biomacromolecules, 2021, 22, 2815–2821 CrossRef CAS.
- F. Fujimura, T. Hirata, T. Morita, S. Kimura, Y. Horikawa and J. Sugiyama, Biomacromolecules, 2006, 7, 2394–2400 CrossRef CAS PubMed.
- T. Hirata, F. Fujimura and S. Kimura, Chem. Commun., 2007, 1023–1025 RSC.
- F. Fujimura, Y. Horikawa, T. Morita, J. Sugiyama and S. Kimura, Biomacromolecules, 2007, 8, 611–616 CrossRef CAS.
- R. J. Brea, M. Amorin, L. Castedo and J. R. Granja, Angew. Chem., Int. Ed., 2005, 44, 5710–5713 CrossRef CAS PubMed.
- R. J. Brea, L. Castedo, J. R. Granja, M. Á. Herranz, L. Sánchez, N. Martín, W. Seitz and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5291–5294 CrossRef CAS PubMed.
- M. Calvelo, A. Lamas, A. Guerra, M. Amorin, R. Garcia-Fandino and J. R. Granja, Chem. – Eur. J., 2020, 26, 5846–5858 CrossRef CAS PubMed.
- N. Rodríguez-Vázquez, M. Amorín, I. Alfonso and J. R. Granja, Angew. Chem., Int. Ed., 2016, 55, 4504–4508 CrossRef.
- N. Rodriguez-Vazquez, R. Garcia-Fandino, M. Amorin and J. R. Granja, Chem. Sci., 2016, 7, 183–187 RSC.
- H. L. Ozores, M. Amorin and J. R. Granja, J. Am. Chem. Soc., 2017, 139, 776–784 CrossRef CAS.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC.
- N. Lawrence, G. J. Philippe, P. J. Harvey, N. D. Condon, A. H. Benfield, O. Cheneval, D. J. Craik and S. Troeira Henriques, RSC. Chem. Biol., 2020, 1, 405–420 RSC.
- X. Luan, Y. Wu, Y. W. Shen, H. Zhang, Y. D. Zhou, H. Z. Chen, D. G. Nagle and W. D. Zhang, Nat. Prod. Rep., 2021, 38, 7–17 RSC.
- H. Zhang and S. Chen, RSC. Chem. Biol., 2022, 3, 18–31 RSC.
- V. T. Ivanov, A. I. Miroshnikov, N. D. Abdullaev, L. B. Senyavina, S. F. Arkhipova, N. N. Uvarova, K. K. Khalilulina, V. F. Bystrov and Y. A. Ovchinnikov, Biochem. Biophys. Res. Commun., 1971, 42, 654–663 CrossRef CAS PubMed.
- R. P. Daniele and S. K. Holian, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3599–3602 CrossRef CAS.
- L. Becucci, M. R. Moncelli, R. Naumann and R. Guidelli, J. Am. Chem. Soc., 2005, 127, 13316–13323 CrossRef CAS PubMed.
- E. E. Ross, B. Hoag, I. Joslin and T. Johnston, Langmuir, 2019, 35, 9410–9421 CrossRef CAS.
- J. R. Aviles-Moreno, F. Gamez, G. Berden, J. Martens, J. Oomens and B. Martinez-Haya, Phys. Chem. Chem. Phys., 2020, 22, 19725–19734 RSC.
- K. D. Kopple, A. Go, T. J. Schamper and C. S. Wilcox, J. Am. Chem. Soc., 1973, 95, 6090–6096 CrossRef CAS PubMed.
- K. D. Kopple, T. J. Schamper and A. Go, J. Am. Chem. Soc., 1974, 96, 2597–2605 CrossRef CAS PubMed.
- L. M. Gierasch, C. M. Deber, V. Madison, C. H. Niu and E. R. Blout, Biochemistry, 1981, 20, 4730–4738 CrossRef CAS PubMed.
- D. S. Eggleston, P. W. Baures, C. E. Peishoff and K. D. Kopple, J. Am. Chem. Soc., 1991, 113, 4410–4416 CrossRef CAS.
- V. Madison, M. Atreyi, C. M. Deber and E. R. Blout, J. Am. Chem. Soc., 1974, 96, 6725–6734 CrossRef CAS PubMed.
- M. Hollosi and T. Wieland, Int. J. Pept. Protein Res., 1977, 10, 329–341 CrossRef CAS.
- V. Madison, C. M. Deber and E. R. Blout, J. Am. Chem. Soc., 1977, 99, 4788–4798 CrossRef CAS PubMed.
- I. L. Karle, J. Karle, T. Wieland, W. Burgermeister, H. Faulstich and B. Witkop, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 1836–1840 CrossRef CAS PubMed.
- J. D. Gregory and L. C. Craig, J. Biol. Chem., 1948, 172, 839 Search PubMed.
- L. R. Gahan and R. M. Cusack, Polyhedron, 2018, 153, 1–23 CrossRef CAS.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- M. J. Bayro, J. Mukhopadhyay, G. V. Swapna, J. Y. Huang, L. C. Ma, E. Sineva, P. E. Dawson, G. T. Montelione and R. H. Ebright, J. Am. Chem. Soc., 2003, 125, 12382–12383 CrossRef CAS PubMed.
- O. Pavlova, J. Mukhopadhyay, E. Sineva, R. H. Ebright and K. Severinov, J. Biol. Chem., 2008, 283, 25589–25595 CrossRef CAS.
- M. O. Maksimov, S. J. Pan and A. James Link, Nat. Prod. Rep., 2012, 29, 996–1006 RSC.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- J. Sawada, D. Aoki, S. Uchida, H. Otsuka and T. Takata, ACS Macro Lett., 2015, 4, 598–601 CrossRef CAS PubMed.
- V. Aucagne, D. A. Leigh, J. S. Lock and A. R. Thomson, J. Am. Chem. Soc., 2006, 128, 1784–1785 Search PubMed.
- A. Moretto, M. Crisma, F. Formaggio and C. Toniolo, Biomol. Concepts, 2012, 3, 183–192 Search PubMed.
- S. D. P. Fielden, D. A. Leigh, C. T. McTernan, B. Perez-Saavedra and I. J. Vitorica-Yrezabal, J. Am. Chem. Soc., 2018, 140, 6049–6052 CrossRef CAS.
- T. Kurita, M. Higashi, J. Gimenez-Dejoz, S. Fujita, H. Uji, H. Sato and K. Numata, Biomacromolecules, 2024, 25, 3661–3670 CrossRef CAS.
- R. T. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. Sanders, Science, 2005, 308, 667–669 Search PubMed.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 Search PubMed.
- S. Piana and A. Laio, J. Phys. Chem. B, 2007, 111, 4553–4559 CrossRef CAS.
- N. Nakajima, H. Nakamura and A. Kidera, J. Phys. Chem. B, 1997, 101, 817–824 CrossRef CAS.
- D. Hamelberg, J. Mongan and J. A. McCammon, J. Chem. Phys., 2004, 120, 11919–11929 CrossRef CAS PubMed.
- A. Shkurti, I. D. Styliari, V. Balasubramanian, I. Bethune, C. Pedebos, S. Jha and C. A. Laughton, J. Chem. Theory Comput., 2019, 15, 2587–2596 CrossRef CAS.
- J. Damjanovic, J. Miao, H. Huang and Y. S. Lin, Chem. Rev., 2021, 121, 2292–2324 CrossRef CAS PubMed.
- D. P. Slough, S. M. McHugh, A. E. Cummings, P. Dai, B. L. Pentelute, J. A. Kritzer and Y. S. Lin, J. Phys. Chem. B, 2018, 122, 3908–3919 CrossRef CAS PubMed.
- A. E. Cummings, J. Miao, D. P. Slough, S. M. McHugh, J. A. Kritzer and Y. S. Lin, Biophys. J., 2019, 116, 433–444 CrossRef CAS PubMed.
- F. Fonseca Lopez, J. Miao, J. Damjanovic, L. Bischof, M. B. Braun, Y. Ling, M. D. Hartmann, Y. S. Lin and J. A. Kritzer, J. Chem. Inf. Model., 2023, 63, 6925–6937 CrossRef CAS PubMed.
- S. M. McHugh, J. R. Rogers, H. T. Yu and Y. S. Lin, J. Chem. Theory Comput., 2016, 12, 2480–2488 CrossRef CAS PubMed.
- S. M. McHugh, H. Yu, D. P. Slough and Y. S. Lin, Phys. Chem. Chem. Phys., 2017, 19, 3315–3324 RSC.
- M. C. Lu, Z. Y. Chen, Y. L. Wang, Y. L. Jiang, Z. W. Yuan, Q. D. You and Z. Y. Jiang, RSC Adv., 2015, 5, 85983–85987 Search PubMed.
- P. Bryant, G. Pozzati and A. Elofsson, Nat. Commun., 2022, 13, 1265 CrossRef CAS PubMed.
- P. Bryant, G. Pozzati, W. Zhu, A. Shenoy, P. Kundrotas and A. Elofsson, Nat. Commun., 2022, 13, 6028 Search PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C. C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Zemgulyte, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Zidek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 Search PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millan, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS.
- R. Krishna, J. Wang, W. Ahern, P. Sturmfels, P. Venkatesh, I. Kalvet, G. R. Lee, F. S. Morey-Burrows, I. Anishchenko, I. R. Humphreys, R. McHugh, D. Vafeados, X. Li, G. A. Sutherland, A. Hitchcock, C. N. Hunter, A. Kang, E. Brackenbrough, A. K. Bera, M. Baek, F. DiMaio and D. Baker, Science, 2024, 384, eadl2528 Search PubMed.
- S. A. Rettie, K. V. Campbell, A. K. Bera, A. Kang, S. Kozlov, J. De La Cruz, V. Adebomi, G. Zhou, F. DiMaio, S. Ovchinnikov and G. Bhardwaj, bioRxiv, 2023, preprint DOI:10.1101/2023.02.25.529956.
- J. Miao, M. L. Descoteaux and Y. S. Lin, Chem. Sci., 2021, 12, 14927–14936 Search PubMed.
- T. Hui, M. L. Descoteaux, J. Miao and Y. S. Lin, J. Chem. Theory Comput., 2023, 19, 4757–4769 Search PubMed.
| This journal is © the Owner Societies 2024 |