
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and catalytic activity of heterobimetallic Au/M (M = RhIII, IrIII) complexes with ditopic mono- and triphosphane ligands†
Ivana
Predarska
 a,
Wieland
Körber
a,
Peter
Lönnecke
a,
Dmitri
Gelman
*b and
Evamarie
Hey-Hawkins
*a
a,
Wieland
Körber
a,
Peter
Lönnecke
a,
Dmitri
Gelman
*b and
Evamarie
Hey-Hawkins
*a
aLeipzig University, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bThe Hebrew University, Institute of Chemistry, Edmond Safra Campus, 9190401 Jerusalem, Israel. E-mail: dmitri.gelman@mail.huji.ac.il
First published on 26th June 2024
Abstract
A series of heterobimetallic complexes Au/M (M = RhIII, IrIII) were prepared on the basis of two ditopic ligands: a monophosphane ligand L1H and a triphosphane ligand L2H. The complexes were fully characterised, including single-crystal X-ray diffraction studies. Catalytic activity of cationic L1/AuI/IrIII and L2/AuI/IrIII bis(trifluoromethane)sulfonimide was analysed through their capacity to induce allenyl ether rearrangement and cycloisomerisation of N-propargyl benzamide. While cationic L1/AuI/IrIII showed some ability to induce allenyl ether rearrangement, no conversion was observed for cationic L2/AuI/IrIII. Similarly, N-propargyl benzamide could undergo cycloisomerisation in the presence of cationic L1/AuI/IrIII, whereas cationic L2/AuI/IrIII was again inactive. These findings highlight how crucial the surroundings of the metal centre are to the catalytic activity. Catalytic activity is only possible when Au has a free coordination site; the gold complex becomes inactive when the tridentate ligand is present.
Introduction
In the context of homogeneous catalysis using organometallic compounds, the focus has historically been on single-site catalysts to prevent the formation of multinuclear compounds.1 However, in biological systems, especially those involved in redox transformations of small molecules like water or carbon dioxide, multimetallic assemblies and their reactivity are crucial.2 Some biological systems rely on the synergistic activation of substrates by multiple metal centres.3 For instance, nickel-containing carbon monoxide dehydrogenase (Ni–CODH) utilises both nickel and iron centres to convert carbon dioxide to carbon monoxide and vice versa.4,5Comprehension of the interactions and catalytic characteristics of bi- and polymetallic centres within metalloenzymes has exerted a substantial influence on various scientific disciplines, including the realm of synthetic chemistry. Significant attention has been garnered in the field of coordination chemistry and catalysis, particularly regarding multimetallic complexes.2 These complexes offer a unique platform for expanding the repertoire of homogeneous catalysts. Heterobimetallic compounds, composed of two different metal ions incorporated into a singular ligand framework, have the potential to broaden the range of redox states and coordination possibilities for substrates due to intermetallic collaboration driven by electronic interactions or the concurrent activation of one or more substrates.6–8 The versatile nature of metal pairings in heterobimetallic complexes offers a possibility to streamline diverse catalytic processes into a unified one-pot tandem reaction.7–10 As a result, these complexes have been documented to exhibit superior catalytic activity and selectivity compared to their mono- and homobimetallic counterparts.9,10
Despite their distinctive catalytic characteristics, the synthesis of well-defined heterobimetallic complexes and their practical application in catalysis presents many challenges on account of the potential for non-specific chelation of metal ions by both binding sites or ion exchange in solution. To mitigate this, the construction of elaborate ligand scaffolds characterised by two distinct binding domains, with the capacity to discriminate between two distinct metal ions, is indispensable.11
One class of ligands that has received steadily growing interest in the context of homogeneous catalysis over the past decades is pincer ligands and their associated metal complexes.12,13 That is due to their robust tridentate coordination, effective metal–ligand cooperation, and high thermal and kinetic stability, thereby minimising metal leaching during the catalytic cycle. Additionally, fine-tuning of the electronic and steric properties of the metal centre can be afforded with these ligands.14–17 A plethora of different types of pincer ligands have been developed over the years. Their complexes have been used as sensors, crystalline switches, and, most frequently, as precatalysts for various reactions.14 Particularly relevant in catalysis are those containing phosphorus coordination sites owing to their ability to stabilise metal centres in high and low oxidation states.18,19
We have recently created a range of heterobimetallic complexes using a ditopic ligand setup (L2, Scheme 1) with a pincer PPP-type binding site and a phenylpyridine binding site. These include a Mo0/RhIII, Mo0/IrIII, Cr0/IrIII, W0/IrIII and a Co0/IrIII complex.20–22 Among the described compounds, the Mo0/IrIII complex exhibited the highest activity in the context of homogeneous carbon dioxide hydrogenation, resulting in the formation of formate salts.20,22 Herein, we are expanding this approach by introducing additional heterobimetallic Au/M complexes (M = RhIII, IrIII) built upon the same ligand system. Additionally, an analogous ligand with a monophosphane binding site was prepared (L1H, Scheme 1) and is hereby reported along with its corresponding Au/M complexes (M = RhIII, IrIII). Selected analogous complexes based on both ligands L1H and L2H were tested for their catalytic activity, namely, their capacity to induce allenyl ether rearrangement and cycloisomerisation of N-propagyl benzamide.
 | ||
| Scheme 1 Synthesis of heterometallic Au/M complexes (M = Rh, Ir; Cp* = C5Me5). L2H was described previously.20 | ||
Results and discussion
Synthesis and characterisation of ligand L1H
The heteroditopic ligand L1H was designed to afford two distinct coordination sites.24 The lithiation of 2-(4-bromophenyl)pyridine at −60 °C with nBuLi followed by the reaction with diisopropylchlorophosphane gave the monophosphane, which was protected in situ with sulfur, to yield L1H in 88% yield. Protection was required to avoid the coordination of phosphorus during the cyclometallation step. L1H was characterised by multinuclear NMR spectroscopy. Expectedly, for sulfurised tertiary phosphanes, the 31P{1H} NMR spectrum of L1H revealed a singlet at 66.4 ppm. The structure was further verified via high-resolution ESI(+) mass spectrometry and single crystal X-ray diffraction.Cyclometallation
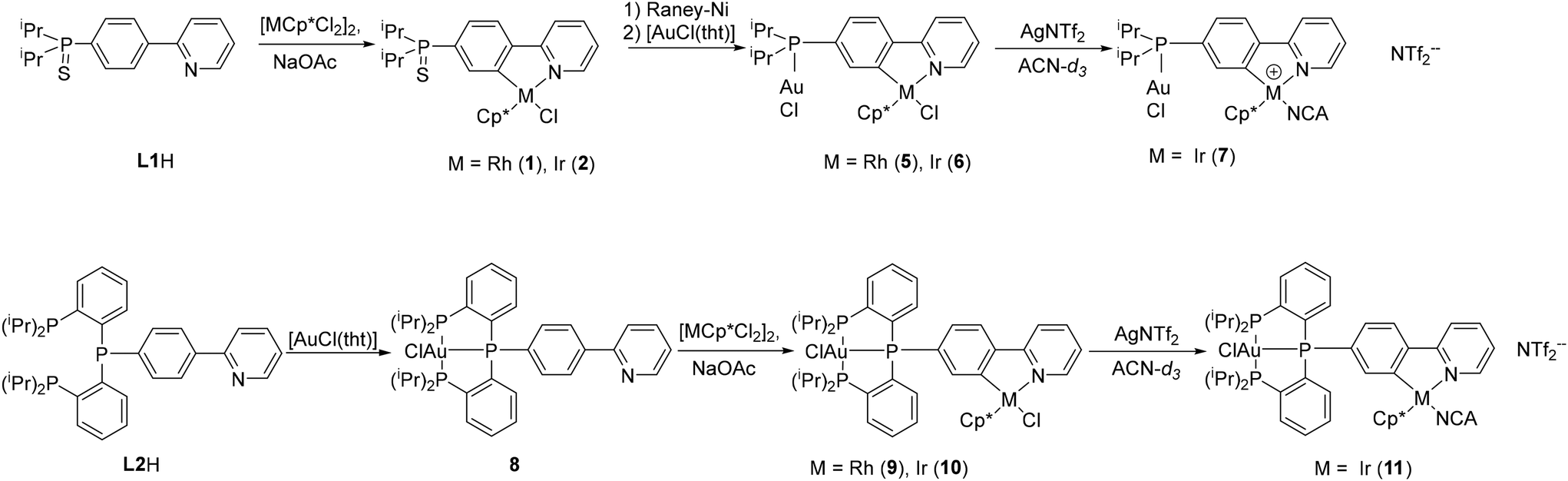
The cyclometallation step was performed according to the literature procedure.23 The phosphane sulfide L1H was reacted with dimeric [MCp*Cl2]2 (M = Rh, Ir) in the presence of excess NaOAc. The cyclorhodate complex 1 and cycloiridate complex 2 were isolated in 97% yield and fully characterised. In the 31P{1H} NMR spectra, singlets were observed at 67.0 ppm or 66.7 ppm for 1 or 2, respectively. In the 1H and 13C{1H} NMR spectra, cyclometallation leads to separate signals for each atom at the phenyl ring. Similarly, the methine protons are no longer equivalent in both complexes and split up into two sets of signals. Similar chemical shifts were found for most protons in the 1H NMR spectra of both complexes. The only exception are the methyl protons of the Cp* moiety with a singlet signal at 1.70 ppm for 2 and 1.66 ppm for 1. The 13C{1H} NMR spectra also show significant similarities, the difference being in the metal-bound carbon atoms: the cyclopentadienyl carbon atoms are observed at 96.3 ppm for 1, split into a doublet due to the coupling with Rh (1JCRh = 6.3 Hz). In comparison, a singlet at 89.1 ppm is observed for 2. The phenyl ring carbon atoms resonate at 163.0 ppm (d, 3JCP = 8.8 Hz) for 2 and a doublet of doublets at 178.1 ppm (dd, 1JCRh = 32.5, 3JCP = 8.4 Hz) is observed for 1. The comparable 2-phenylpyridine rhodium complex [RhCp*(ppy)Cl] showed a higher 1JCRh coupling constant of 86.8 Hz.30 A similar drop in the 1JCRh coupling constant was observed for the rhodacycle of 2-(4-fluorophenyl)pyridine (dd, 1JCRh = 33.4, 3JCF = 4.3 Hz).31Slow decomposition of the complexes was observed over time in CDCl3 (followed by 1H NMR spectroscopy). Single crystals suitable for X-ray diffraction analysis could be obtained by slow cooling of saturated solutions in MeOH (for 1) or acetonitrile (ACN) (for 2). The molecular structures are depicted in Fig. 1.
 | ||
| Fig. 1 Molecular structure and labelling scheme of complexes 1 (top), 2 (centre) and 3 (bottom). Hydrogen atoms are omitted for clarity and displacement ellipsoids are drawn at 50% probability level. | ||
Complex 1 crystallised in the monoclinic space group P21 with one molecule and one cocrystallised methanol molecule in the asymmetric unit. The chloride ligand forms a hydrogen bond with the hydroxy group (d(O1–Cl1) = 3.220(4) Å). Compound 2 crystallised in the orthorhombic space group Pna21 with one molecule in the asymmetric unit. Both structures show a piano stool coordination of the metal centre. The bond lengths and angles are very similar to the closely related 2-phenylpyridine complexes previously published (see Table S2, ESI†).30
Compared to the parent complexes, the M1–C9 bond (2.036(1) Å for 1 and 2.032(5) Å for 2) are shorter by 1–2 pm. Such subtle changes have been observed for complexes bearing other electron-withdrawing groups in para position (such as CN, CF3, F) and show the higher acidity of the ortho-H compared to 2-phenylpyridine.23,31–33 The M1–Cl1 bond is almost perpendicular to the C9–M1–N1 plane.
Desulfurisation
Several deprotection methods are known for phosphine sulfides.34–36 Reduction of phosphine sulfides in complexes 1 and 2 with strong hydrides was avoided because of possible decomposition of the metallacycle, which was shown by Norton et al.37 Addition of a strong nucleophilic phosphane, such as PEt3 to the cyclorhodate complex 1, did not result in the sulfur transfer, but over time led to complexation of RhIII. Using freshly activated RANEY® nickel (RANEY®-Ni) in THF at 50 °C gave the phosphane 3. The progress of the reaction was monitored by 31P{1H} NMR spectroscopy, and, if necessary, small amounts of activated RANEY®-Ni were added until conversion was completed. Due to the poor solubility of 1 and 2 in THF, the measurement time had to be increased to ensure all of the starting material was completely consumed. The free phosphane was directly used for further reactions after a rough yield determination to avoid side reactions, including oxidation or complexation.Complex 3 was isolated and characterised by NMR spectroscopy, HR-ESI(+)-MS as well as single crystal X-ray diffraction, while complex 4 was not isolated, but used directly for the formation of heterometallic complex 6. The 31P{1H} NMR spectra of both complexes display a singlet between 10 to 15 ppm depending on the solvent used and the coordinated metal. Compared to the sulfurised precursor, the 1H NMR signals in 3 are shifted highfield in good accordance with the lesser electron-withdrawing effect of the free phosphane. The most affected protons are those of the phenylene and isopropyl groups. The same effect was observed in the 13C{1H} NMR spectrum for the phenyl carbon atom bound to phosphorus (shifted from 146.5 to 144.2 ppm).
Under high-resolution ESI(+)-MS conditions, the main signal at 508.1646 m/z could be attributed to the desired complex 3 with a lost chloride ligand [M − Cl]+, typical for halide complexes.
Crystals of 3 (Fig. 1, bottom) were obtained by layering a THF solution with n-hexane. 3 crystallised in the monoclinic space group P21n with one molecule in the asymmetric unit. Desulfurisation had little impact on the Rh–C9 bond length. The Rh–Cl1 bond is 1 pm shorter, possibly due to the lack of the hydrogen bond of the solvent observed in 1. As in 1, the Rh1–Cl1 bond is perpendicular to the N1–Rh–C1 plane. The sum of bond angles in the rhodacycle did not change (539.7° for 3, 539.9° for 1 (ideal 540°)).
Gold complexation
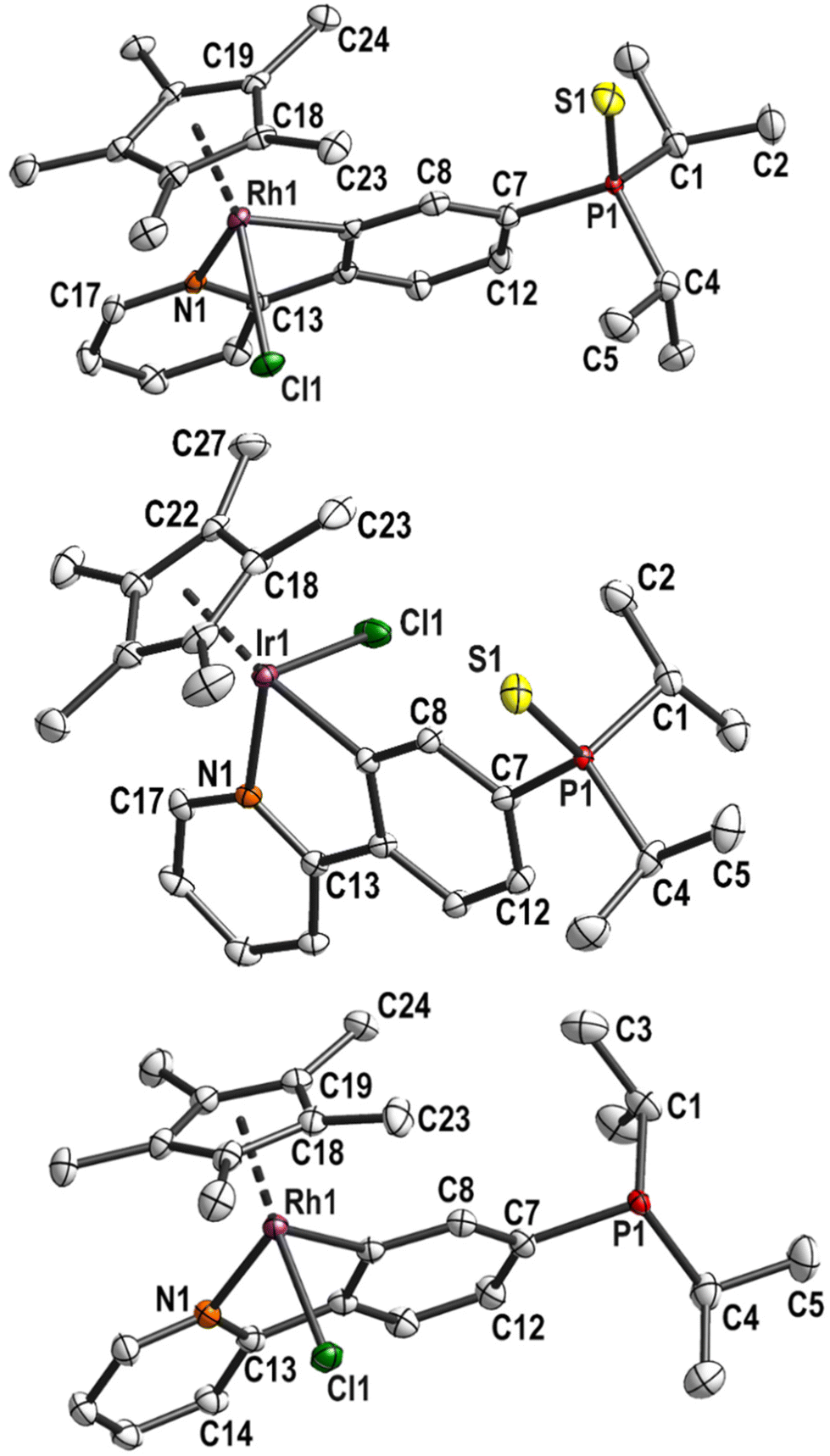
The cyclometalated phosphane complexes 3 and 4 were reacted with [AuCl(tht)] (tht = tetrahydrothiophene) to give the heterobimetallic complexes 5 (M = Rh) and 6 (M = Ir) quantitatively (Scheme 1). It is worth noting that our attempt to reverse the order of metalation reactions were proved inefficient in the past.20 The 31P{1H} NMR spectra of the reaction solutions indicated complete conversion by a downfield shift of the signal to 60.0 ppm (from 10 to 15 ppm in 3 and 4). The 1H and 13C{1H} NMR spectra show similar features as observed for the sulfurised precursors. In CDCl3, the 1H NMR spectra showed very similar shifts for most signals, with the only exception of the methyl protons of the Cp* moiety with a singlet at 1.56 ppm for 5 and 1.71 ppm for 6. For 5, the signals in the aromatic region between 7.81 and 7.64 ppm (dt and dd) are well separated, but overlap to a multiplet for 6. The 13C{1H} NMR spectra also show great similarities, the difference being in the metal-bound carbon atoms. The cyclopentadienyl carbon atoms are observed at 96.7 (d, 1JCRh = 6.3 Hz) for 5 and 89.0 ppm (s) for 6. The phenyl ring carbon atoms are observed as a doublet of doublets at 179.8 ppm (dd, 1JCRh = 33.0, 3JCP = 9.1 Hz) for 5, and as a doublet at 164.0 ppm (d, 3JCP = 9.7 Hz) for 6.Single crystals suitable for X-ray diffraction analysis of the heteronuclear complexes were obtained from a boiling saturated acetonitrile solution by stepwise cooling with a thermostat over several days. The use of a thermostat prevented the formation of powders. Attempts to crystallise 5 from a DCM solution with Et2O or MeOH lead to decomposition. The compounds are more stable in chlorinated solvents, but solvent removal was accompanied by partial decomposition. On the other hand, the compounds were found to be stable toward light and in an acetonitrile solution.
Both complexes crystallised isostructurally in the monoclinic space group P21 with one molecule in the asymmetric unit. The Au1–P1 bond lengths of 2.245(1) Å (5) and 2.239(2) Å (6) as well as the Au1–Cl1 bond lengths (2.301(2) Å (5); 2.300(2) Å (6)) are comparable to other phosphane gold(I) complexes such as [AuCl(PPh3)],38 [AuCl(PCy3)],39 [AuCl(PiPr3)]40 and [AuCl(PCy2Ph)].41 The gold atoms are in linear coordination with P1–Au1–Cl1 bond angles of 176.75° (5) and 176.7° (6) (Fig. 2).
 | ||
| Fig. 2 Molecular structure and labelling scheme of complexes 5 (left) and 6 (right). Hydrogen atoms are omitted for clarity and displacement ellipsoids are drawn at the 50% probability level. | ||
The bond lengths and angles around Rh and Ir are very similar to the precursor complexes, respectively. As there are no major deviations from the normal coordination environment, it can be assumed that both metal centres can work in tandem in catalysis. The P–Au–Cl unit in both complexes is oriented in the direction of the Cp* ligand with torsion angles Cp*(centroid)–M1⋯P1–Au1 around 15° (Table S3†). This is similar to the phosphane sulfide precursors and is probably the result of steric interaction between the isopropyl groups and the Cp* ligand. In the free phosphane rhodacycle complex 6, this torsion angle amounts to 60°, which minimises the steric interaction of the isopropyl groups with the aryl group. Since there are greater steric interactions in the gold(I) complexes, the P atom is slightly dislocated from the phenyl plane by 0.271(7) Å (5) and 0.291(9) Å (6), respectively.
The intramolecular distances between the metal atoms are 6.2471(6) Å (5) and 6.2344(4) Å (6). The chloride ligands are further apart (8.025(3) Å (5) and 8.047(3) Å (6)) since the metal–chloride vectors are staggered (Cl1–Au1⋯M1–Cl2 torsion angles of 110.34(6)° (5) and 111.32(6)° (6)). The most similar dinuclear AuI/IrIII or AuI/RhIII complexes in the literature were bis-carbene complexes by Hahn et al. and showed a larger distance between the metal atoms in the solid state (Table S4†).42
Anion exchange reactions
Cationic piano-stool type complexes were found to work better for most catalytic reactions in comparison to their neutral counterparts,43 due to the higher availability of the catalytically active species with a free active site for the reaction. Here, the ion exchange reaction of 6 was conducted by the addition of AgNTf2 and subsequent isolation by the precipitated AgCl salt via filtration, as this might have an impact on the catalytic performance.44The reaction was performed in CD3CN which acts as a weak ligand and can prevent rapid exchange reactions. Due to the poor solubility of 6 in acetonitrile, CD2Cl2 was added. With one equiv. AgNTf2, the reaction was incomplete, while two equiv. AgNTf2 led to the formation of the desired bis-cationic complex 7, which was investigated in situ by NMR spectroscopy and HRMS. No crystals could be obtained, and the stability upon solidification was questionable. HR-ESI(+)-MS showed the same signal as found for 6, suggesting the presence of 6 ([M − Cl]+) or the monocationic complex with one NTf2 anion ([M − NTf2]+) as an impurity. However, several other signals support the formation of the desired product, namely a signal corresponding to [M − NTf2]+ (m/z = 1075.1053, 37% rel. abundance), furthermore the acetonitrile adduct [M − (NTf2)2 + (NCCH3)2]2+ (m/z = 438.6191, 1% rel. abundance), as well as the “naked” dinuclear complex [M − (NTf2)2]2+ (m/z = 397.5958, <1% rel. abundance).
Synthesis and characterisation of complexes based on ligand L2H
The synthesis of the triphosphane ligand L2H was previously described by our group.20 To afford heterobimetallic complexes based on this ditopic ligand, the two distinctively different coordination environments available were approached with stepwise coordination of the two metals.Contrary to the monophosphane ligand L1H, in the case of L2H, at first, the phosphane site was employed in complexation using chlorido(tetrahydrothiophene)gold(I) ([AuCl(tht)]) as the gold(I) source. Complex 8 was obtained quantitatively and fully characterised with multinuclear NMR spectroscopy, ESI-MS, elemental analysis, and FT-IR. Compared to the complexes based on the monophosphane ligand L1H, the biggest difference is observed in the 31P NMR spectrum with doublet and triplet signals displayed with a P,P coupling constant of ca. 115 Hz. Complex 8 showed high stability in DCM over weeks and did not manifest sensitivity toward light.
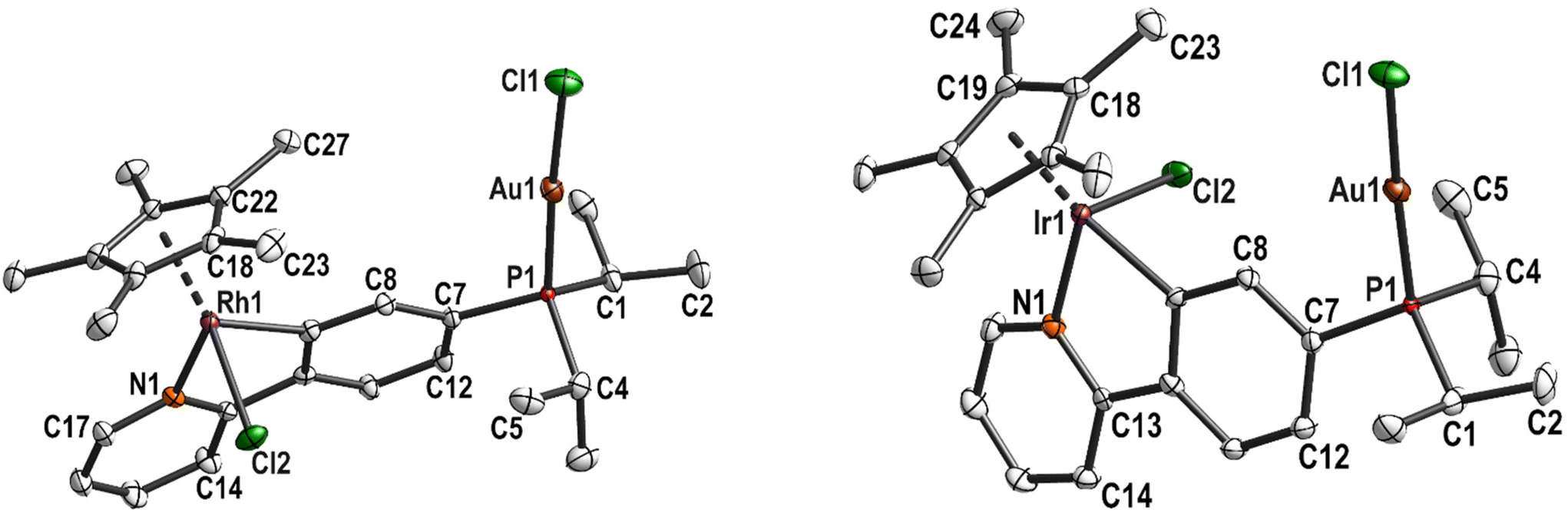
Crystals were obtained by layering a DCM solution of 8 with n-hexane. 8 crystallised in the monoclinic space group P21/c with two independent molecules in the asymmetric unit (Fig. 3). The complex molecules are most likely marginally disordered with a ratio of 0.9845(4)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.0155(4). This disorder is only detectable for the most electron-rich atoms Au1 and Au2. The bond lengths and angles involving Au1 or Au2 are comparable.
:0.0155(4). This disorder is only detectable for the most electron-rich atoms Au1 and Au2. The bond lengths and angles involving Au1 or Au2 are comparable.
 | ||
| Fig. 3 Molecular structure and labelling scheme of 8. Both independent molecules are shown. Hydrogen atoms are omitted for clarity and displacement ellipsoids are drawn at the 50% probability level. | ||
Complex 8 was subsequently involved in cyclometallation in the same way as described for the monophosphane ligand L1H, with [MCp*Cl2]2 (M = Rh or Ir, Cp* = C5Me5) and excess of NaOAc. The resulting complexes 9 (M = Rh) and 10 (M = Ir) were fully characterised, including single crystal structure determinations (Fig. 4). The gold–rhodium complex 9 crystallised in the monoclinic space group P21/c, the gold–iridium complex 10 in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Selected bond lengths and angles of 8, 9, and 10 are given in Table S5.† Further crystallographic details for these complexes are given in Table S1 (ESI†).
. Selected bond lengths and angles of 8, 9, and 10 are given in Table S5.† Further crystallographic details for these complexes are given in Table S1 (ESI†).
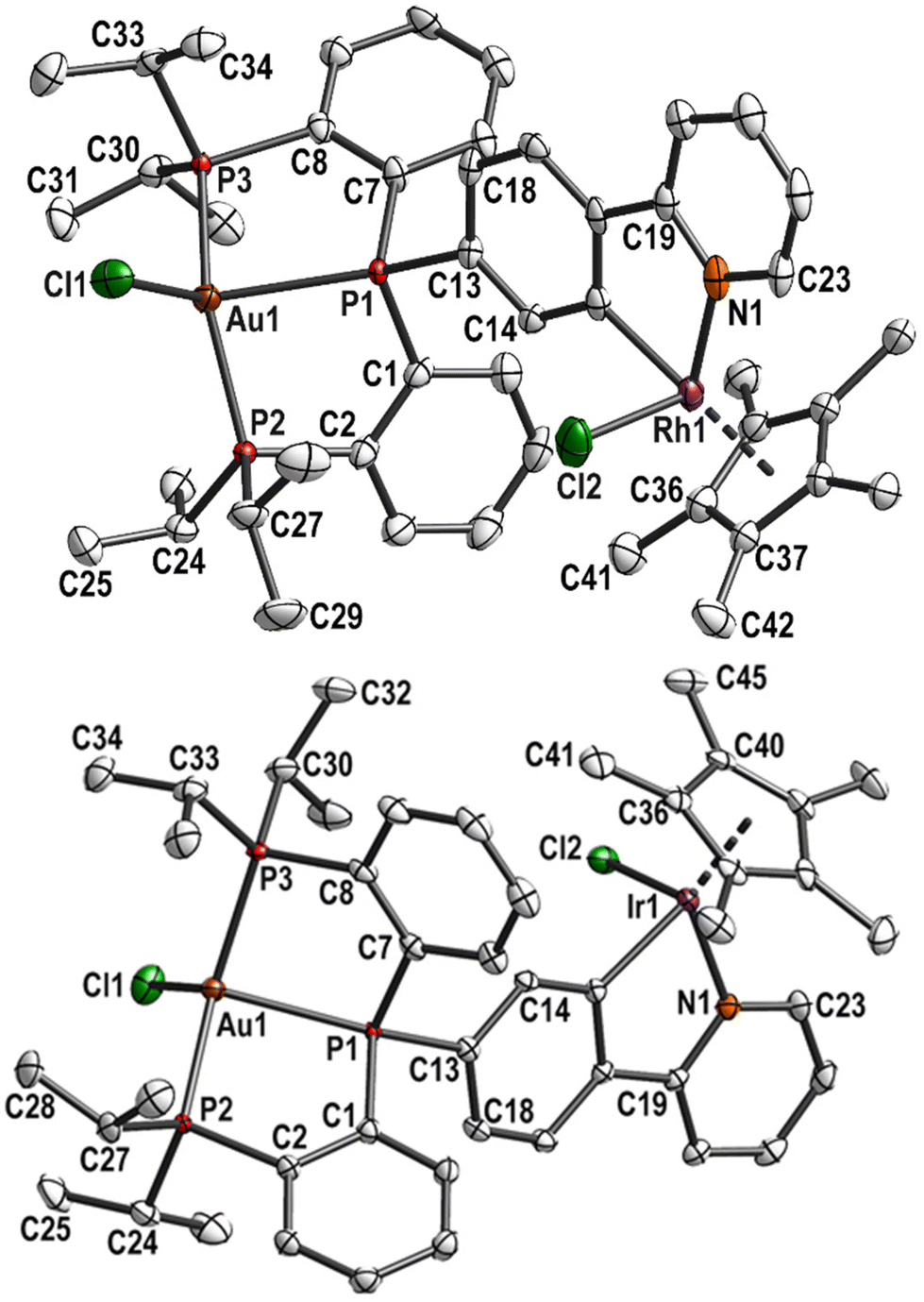 | ||
| Fig. 4 Molecular structure and labelling scheme of 9 (top) and 10 (bottom). Hydrogen atoms are omitted for clarity and displacement ellipsoids are drawn at the 50% probability level. | ||
Finally, to obtain a complex with a more easily accessible active site, an anion exchange reaction was performed for complex 10 with AgNTf2 in acetonitrile. The formation of 11 was confirmed in situ with multinuclear NMR spectroscopic analysis. The exchange of the Cl anion with NTf2 led to a downfield shift of the phosphorus signals in the 31P NMR spectrum and a smaller P,P coupling constant.
Catalytic activity
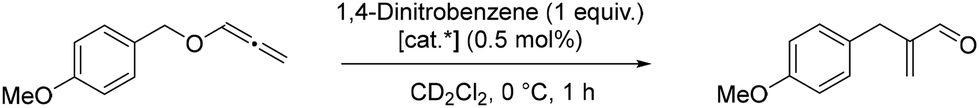 | ||
| Scheme 2 Allenyl ether rearrangement. [cat*] = complex 7 or 11. | ||
With 7 as catalyst, the consumption of the starting material increased by increasing the temperature from 0 °C to room temperature. However, the conversion was still incomplete after 14.5 h at room temperature. Additionally, besides the desired aldehyde, the formation of a side product was observed. Although we did not investigate the nature and origin of the side product, based on previous reports in the literature, this is presumably the hydrolysis product as hydrolysis is described as a side reaction observed for some combinations of ligands with weakly coordinating counterions. Hashmi et al.43 managed to avoid hydrolysis in this transformation by using the isolated catalyst [Au(PPh3)]NTf2 instead of a mixture of [AuCl(PPh3)] (2 mol%) and AgNTf2 (5 mol%), a combination first reported by Ramana et al.45 The same group proposed the mechanism for the gold(I) catalysed [1,3] O→C rearrangement of allenyl ethers.46 Thus, the reaction proceeds via the initial coordination of the oxygen of the allenyl ether to gold(I), leading to the elongation of the carbinol C–O bond. In case of sufficient electrophilicity of the oxygen substituent, the C–O bond is cleaved, leading to the formation of a contact-ion that subsequently undergoes the [1,3]-rearrangement.46
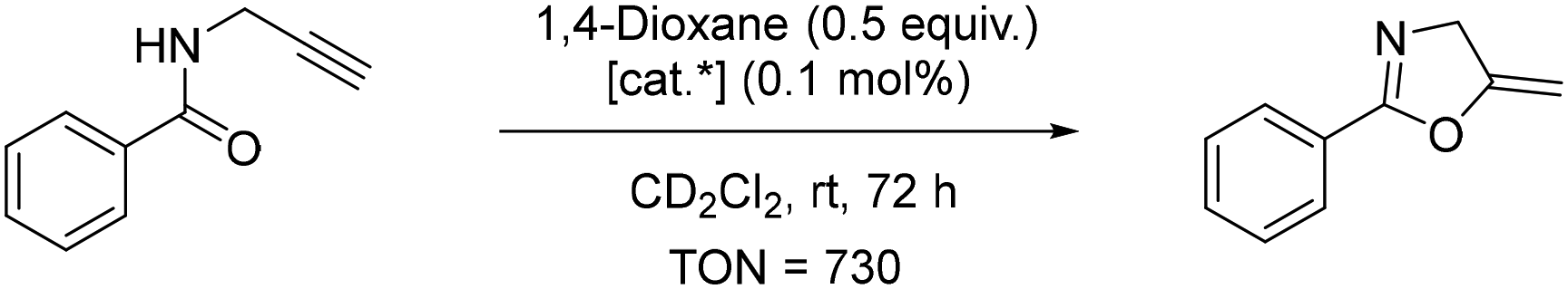 | ||
| Scheme 3 Cycloisomerisation of N-propargyl benzamide. [cat*] = complex 7 or 11. | ||
On the other hand, complex 7 as a catalyst led to the complete conversion of the starting material within 72 hours. The turnover number (TON) of 7 was calculated after 24 h to be 730, comparable to the complex [Au(PtBu3)]NTf2 with TON = 590.43 Other gold(I) catalysts have also demonstrated the capacity to induce cycloisomerisation. Zuccaccia et al.50 have even demonstrated that a range of NHC–Au–X [NHC = (1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene); X− = BF4−, OTf−, OTs−, TFA−] catalysts could induce the cycloisomerisation in a wide set of green solvents (cyclohexanone, isopropyl acetate, ethyl acetate, furfuryl alcohol, γ-valerolactone, propylene carbonate and propionic acid) in a comparable or better manner with respect to traditional volatile organic solvents. Furthermore, the catalyst activity is related to the basic strength of the anion, and the performances of the catalysts decrease gradually with increasing basicity and hydrogen-bond acceptor power of X−. Kinetic experiments and DFT calculations conducted indicate that both the characteristics of the solvent and counterion should be taken into account.50
After the successful cycloisomerisation induced by 7, 2 equiv. of HSiEt3 were added to the same NMR tube to test if the heterobimetallic complex 7 can facilitate tandem catalysis. However, no reaction was observed within 22 h. When an additional amount of 7 was added (0.9 mol%), a grey precipitate formed, indicating the decomposition of the catalyst.
Conclusions
Heterobimetallic complexes Au/M (M = RhIII, IrIII) were synthesised using two ditopic ligands, namely a monophosphane L1H and a triphosphane L2H, and fully characterised, including single-crystal X-ray diffraction analyses for most complexes. The catalytic activity of cationic 7 (L1/AuI/IrIII) and 11 (L2/AuI/IrIII) in allenyl ether rearrangement and cycloisomerisation of N-propargyl benzamide was studied. While 7 showed some ability to induce allenyl ether rearrangement, 11 did not facilitate any conversion. Likewise, N-propargyl benzamide was converted to oxazole in the presence of 7, but no reaction was observed when 11 was used as a catalyst. These results demonstrate the importance of the metal centre's coordination sphere in catalytic processes.20–22 Gold(I) is only catalytically active when coordinated by a monophosphane ligand, while the tridentate ligand led to loss of catalytic activity. Furthermore, while the gold(I) centre in cationic complex 7 was catalytically active, the second metal (IrIII) could not be involved in tandem catalysis under the conditions reported here. More importantly, we demonstrated the universality of the ligand framework for designing heterobimetallic catalysts for future catalytic applications.Experimental
Materials and methods
Reactions were performed using standard Schlenk line techniques. Reagents were purchased from chemical vendors and used without further purification unless noted otherwise. PCl3 was degassed, distilled, and stored under a nitrogen atmosphere over 4 Å molecular sieves prior to use. [RhCp*Cl2]2, [IrCp*Cl2]2 (Cp* = C5Me5),23 PiPr2Cl,24 and the triphosphane ligand bis[2-(diisopropylphosphanyl)phenyl]-4-(2-pyridyl)phenylphosphane (L2H)20 were synthesised according to literature procedures. All other compounds are commercially available. NMR spectra were recorded at 25 °C with a Bruker AVANCE DRX 400 spectrometer (1H NMR: 400.13 MHz, 13C NMR: 100.16 MHz, 31P NMR: 161.97 MHz). TMS was used as the internal standard for 1H and 13C NMR spectra; spectra of other nuclei were referenced to TMS. Mass spectra (HR-ESI-MS) were recorded on an FT-ICR-MS Bruker–Daltonics ESI mass spectrometer (APEX II, 7 T). Elemental analyses were carried out with a Heraeus VARIO EL oven. IR spectra were measured between 4000 and 400 cm−1 as KBr pellets on an FT-IR Spectrum 2000 spectrometer by PerkinElmer. All solvents were purified and degassed with an MBRAUN Solvent Purification System SPS-800. Dichloromethane (DCM) was stored under nitrogen over 4 Å molecular sieves. Tetrahydrofuran (THF) was distilled over K and benzophenone under nitrogen atmosphere and stored over 4 Å molecular sieves. Acetonitrile (ACN) was dried over P2O5 for 3 d, distilled over P2O5, and stored under N2 atmosphere and over 3 Å molecular sieves. All deuterated solvents (CDCl3, CD2Cl2, CD3CN) were dried over 3 Å molecular sieves, degassed, and stored under N2 atmosphere over 3 Å molecular sieves. Crystallographic data were collected on a Gemini diffractometer (Rigaku Oxford Diffraction) using Mo-Kα radiation and ω-scan rotation. Data reduction was performed with CrysAlisPro,25 including the program SCALE3 ABSPACK for an empirical absorption correction. With the exception of structures L1H, 1, and 3, an additional analytical numeric absorption correction was applied using a multifaceted crystal model based on expressions derived by Clark and Reid.26 All structures were solved by dual space methods with SHELXT27 and refined with SHELXL.28 Non-hydrogen atoms were refined with anisotropic displacement parameters. For L1H, hydrogen atoms were located with a difference-density Fourier map. For all other structures, hydrogen atoms were calculated on idealised positions using the riding model. Structure figures were generated with DIAMOND-4.29CCDC deposition numbers given in Table S1 in the ESI† contain the supplementary crystallographic data for this paper.
Synthesis of monophosphane sulfide ligand L1H
2-(4-Bromophenyl)pyridine (2.483 g, 10.6 mmol, 1.05 equiv.) was dissolved in THF (30 ml) and cooled to −60 °C. To the cooled solution, nBuLi in Et2O (8.0 ml, 11.1 mmol, 1.1 equiv.) was added dropwise, and the reaction mixture was stirred for one hour. Diisopropylchlorophosphane (1.54 g, 10.1 mmol, 1.0 equiv.) was dissolved in Et2O, cooled to −60 °C and added dropwise to the lithiated 2-phenylpyridine. The reaction mixture was left to warm up to room temperature overnight. Water (30 ml) was added to the solution, the layers were separated, and the organic layer was washed with water (2 × 10 ml) and brine (10 ml). The aqueous layers were extracted with DCM (3 × 10 ml), and the combined organic layers were dried over MgSO4. The solvent was removed, resulting in a yellow oil. The pure monophosphane ligand was obtained upon column chromatography with n-hexane containing a gradually increasing amount of ethyl acetate (5–10%). The monophosphane ligand (1.153 g, 4.25 mmol, 1 equiv.) was dissolved in THF (5 ml). S8 (0.136 g, 4.25 mmol, 1 equiv.) and a catalytic amount of NEt3 (1 drop) was added, and the reaction mixture was stirred at 50 °C for 24 h. Pure monophosphane sulfide L1H was obtained upon column chromatography with n-hexane containing a gradually decreasing amount of ethyl acetate (40–10%). Colourless single crystals (prisms) of L1H were obtained by slow solvent evaporation from a saturated DCM/Et2O solution. Yield: 1.13 g (88%).1H NMR (400.1 MHz, CDCl3): δ = 1.09 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 6 H, 2′′-H), 1.24 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 6 H, 4′′-H), 2.55 (heptd, 3JHH = 7.0 Hz, JHP = 1.3 Hz, 2 H, 1′′ and 3′′-H), 7.31 (tt, 3JHH = 8.0 Hz, 4JHH = 1.7 Hz, 1 H, 2′-H), 7.81 (m, 3JHH = 8.0 Hz, 3JHH = 5.0 Hz, 4JHH = 1.7 Hz, 2 H, 3′ and 4′-H), 7.97 (m, 3JHH = 8.0 Hz, 4JHH = 2.3 Hz, 2 H, 2-H), 8.11 (dd, 3JHH = 8.0 Hz, 4JHH = 2.3 Hz, 2 H, 3-H), 8.74 (dt, 3JHH = 5.0 Hz, 4JHH = 1.4 Hz, 1 H, 5′-H) ppm. 13C{1H} NMR (100.6 MHz, CDCl3): δ = 15.7 (s, C2′′), 16.5 (s, C4′′), 27.6 (d, 1JCP = 51 Hz, C1′′ and C3′′), 121.0 (s, 3′), 122.9 (s, C2′), 126.6 (d, 3JCP = 11 Hz, C3), 132.6 (d, 2JCP = 8.5 Hz, C2), 137.2 (s, C4′), 141.9 (s, C1), 149.6 (s, C5′), 156.1 (s, C1′) ppm. 31P{1H} NMR (161.97 MHz, CDCl3): δ = 66.4 ppm.
HR-MS (CH3OH): (C17H22NPS; [M + H]+, pos. ESI) calcd: 304.1283, found: 304.1281.
Synthesis of complexes 1 and 2
Monophosphane sulfide L1H (61 mg, 0.2 mmol, 1 equiv.) was dissolved in DCM or ACN (for reactions with M = Rh and Ir, respectively). [MCp*Cl2]2 (M = Rh, Ir; 0.1 mmol, 0.5 equiv.) and anhydrous NaOAc (49 mg, 0.6 mmol, 3 equiv.) were added, and the reaction mixture was stirred at room temperature or 50 °C (for reactions with M = Rh and Ir, respectively). Single crystals of 1 (orange plates) were obtained by slow cooling of a saturated solution in MeOH, while for 2, obtained as orange prisms, a saturated solution in ACN was used.1: Yield: 114.6 mg (97%).
1H NMR (400.1 MHz, CDCl3): δ = 1.12 (dd, 3JHP = 10 Hz, 3JHH = 7.0 Hz, 3 H, 2′′-H), 1.16 (dd, 3JHP = 10 Hz, 3JHH = 7.0 Hz, 3 H, 3′′-H), 1.21 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 3 H, 5′′-H), 1.31 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 3 H, 6′′-H), 1.66 (s, 15 H, Cp*–Me), 2.50 (heptd, 3JHH = 7.0 Hz, 2JHP = 1.3 Hz, 1 H, 1′′-H), 2.59 (heptd, 3JHH = 7.0 Hz, 2JHP = 1.3 Hz, 1 H, 4′′-H), 7.22 (m, 3JHH = 6.4 Hz, 4JHH = 1.6 Hz, 1 H, 4′-H), 7.56 (m, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 1 H, 6-H), 7.68 (dd, 3JHH = 8.0 Hz, 4JHH = 2.6 Hz, 1 H, 5-H), 7.77 (m, 3JHH = 8.0 Hz, 4JHH = 1.6 Hz, 1 H, 3′-H), 7.85 (dt, 3JHH = 8.0 Hz, 4JHH = 1.2 Hz, 1 H, 2′-H), 8.31 (dd, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 1 H, 2-H), 8.79 (dt, 3JHH = 6.4 Hz, 4JHH = 1.0 Hz, 1 H, 5′-H) ppm. 13C{1H} NMR (100.6 MHz, CDCl3): δ = 9.3 (s, Cp*–Me), 16.0 (s, C2′′ and C3′′), 16.8 (t, 2JCP = 2.6 Hz, C5′′ and C6′′), 27.7 (d, 1JCP = 51 Hz, C1′′), 28.2 (d, 1JCP = 51 Hz, C4′′), 96.3 (d, 1JCRh = 6.3 Hz, Cp*), 119.9 (s, C2′), 122.6 (s, C4′), 122.9 (s, C5), 126.6 (d, 2JCP = 9.3 Hz, C6), 128.4 (d, C4), 137.3 (s, C3′), 140.5 (d, 2JCP = 6.3 Hz, C2), 146.5 (d, 1JCP = 2.7 Hz, C1), 151.4 (s, C5′), 164.4 (s, C1′), 178.1 (dd, 1JCRh = 32.5 Hz, 3JCP = 8.4 Hz C3) ppm. 31P{1H} NMR (161.97 MHz, CDCl3): δ = 67.0 ppm.
Elemental analysis for C27H36ClNPRhS calcd: C 56.30, H 6.30, N 2.43, found: C 55.11, H 5.52, N 2.32.
2: Yield: 131.9 mg (97%).
1H NMR (400.1 MHz, CDCl3): δ = 1.12 (dd, 3JHP = 13 Hz, 3JHH = 7.0 Hz, 3 H, 2′′-H), 1.16 (dd, 3JHP = 13 Hz, 3JHH = 7.0 Hz, 3 H, 3′′-H), 1.21 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 3 H, 5′′-H), 1.30 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 3 H, 6′′-H), 1.70 (s, 15 H, Cp*–Me), 2.48 (heptd, 3JHH = 7.0 Hz, 1 H, 1′′-H), 2.59 (heptd, 3JHH = 7.0 Hz, 1 H, 4′′-H), 7.16 (m, 3JHH = 6.5 Hz, 4JHH = 1.4 Hz, 1 H, 4′-H), 7.52 (m, 3JHH = 10.0 Hz, 4JHH = 1.5 Hz, 1 H, 6-H), 7.73 (m, 3JHH = 10.0, 3JHH = 8.0 Hz, 4JHH = 2.6 Hz, 2 H, 5-H and 3′-H), 7.90 (d, 3JHH = 8.0 Hz, 1 H, 2′-H), 8.31 (dd, 3JHH 11.2 Hz, 4JHH = 1.5 Hz, 1 H, 2-H), 8.74 (dd, 3JHH = 6.5 Hz, 4JHH = 1.3 Hz, 1 H, 5′-H) ppm. 13C{1H} NMR (100.6 MHz, CDCl3): δ = 9.2 (s, Cp*–Me), 16.2 (s, C2′′ and C3′′), 17.0 (s, C5′′ and C6′′), 27.7 (d, 1JCP = 50.5 Hz, C1′′), 28.5 (d, 1JCP = 50.5 Hz, C4′′), 89.1 (s, Cp*), 119.9 (s, C2′), 123.2 (d, 3JCP = 11.7 Hz, C5), 123.5 (s, C4′), 125.9 (d, 2JCP = 9.4 Hz, C6), 128.8 (s, C6′), 129.5 (s, C3′), 137.4 (s, 2JCP = 6.3 Hz, C2), 139.8 (d, 1JCP = 7.8 Hz, C1), 147.3 (d, 4JCP = 2.7 Hz, C4), 151.7 (s, C5′), 163.0 (d, 3JCP = 8.8 Hz C3), 166.4 (s, C1′) ppm. 31P{1H} NMR (161.97 MHz, CDCl3): δ = 66.7 ppm.
HR-MS (CH3OH): (C27H36ClNPIrS; [M − Cl]+, pos. ESI) calcd: 630.1929, found: 630.1920.
Desulfurisation reaction: preparation of 3 and 4
Complex 1 (25 mg, 0.434 mmol) was dissolved in THF (4 ml). Freshly activated RANEY®-Ni was added, and the reaction mixture was heated to 50 °C. The progress of the reaction was monitored with 31P{1H} NMR, and if necessary, small amounts of activated RANEY®-Ni were added until conversion was complete.Yield: 145.3 mg (60%), orange prisms.
1H NMR (400.1 MHz, CDCl3): δ = 1.02 (dd, 3JHP = 10 Hz, 3JHH = 7.0 Hz, 3 H, 2′′-H), 1.03 (dd, 3JHP = 10 Hz, 3JHH = 7.0 Hz, 3 H, 3′′-H), 1.12 (dd, 3JHP = 15 Hz, 3JHH = 7.0 Hz, 3 H, 5′′-H), 1.18 (dd, 3JHP = 15 Hz, 3JHH = 7.0 Hz, 3 H, 6′′-H), 1.64 (s, 15 H, Cp*–Me), 2.14 (heptd, 3JHH = 7.0 Hz, JHP = 1.2 Hz, 1 H, 1′′-H), 2.23 (heptd, 3JHH = 7.0 Hz, JHP = 3.2 Hz, 1 H, 4′′-H), 7.14 (tt, 3JHH = 8.0 Hz, 4JHH = 1.4 Hz, 1 H, 4′-H), 7.19 (tt, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 1 H, 6-H), 7.57 (d, 3JHH = 8.0 Hz, 1 H, 5-H), 7.71 (td, 3JHH = 8.0 Hz, 4JHH = 1.6 Hz, 1 H, 3′-H), 7.78 (dt, 3JHH = 8.0 Hz, 4JHH = 1.2 Hz, 1 H, 2′-H), 7.94 (dd, 3JHH = 6.8 Hz, 4JHH = 1.4 Hz, 1 H, 2-H), 8.75 (dt, 3JHH = 5.5 Hz, 4JHH = 1.0 Hz, 1 H, 5′-H) ppm. 13C{1H} NMR (100.6 MHz, CDCl3): δ = 9.2 (s, Cp*–Me), 18.8 (d, 2JCP = 7.0 Hz, C2′′), 19.5 (d, 2JCP = 10.0 Hz, C3′′), 20.0 (d, 2JCP = 18.6 Hz, C5′′), 20.2 (d, 2JCP = 18.6 Hz, C6′′), 23.2 (d, 1JCP = 12 Hz, C1′′), 23.3 (d, 1JCP = 12 Hz, C4′′), 96.0 (d, 1JCRh = 6.3 Hz, Cp*), 119.3 (s, C2′), 122.1 (s, C4′), 122.5 (d, 3JCP = 8.0 Hz, C5), 129.2 (d, 2JCP = 20.0 Hz, C6), 137.0 (s, C3′), 137.2 (s, C4), 142.8 (d, 2JCP = 7.1 Hz, C2), 144.2 (s, C1), 151.2 (s, C5′), 165.2 (s, C1′), 177.7 (dd, 1JCRh = 32.2 Hz, 3JCP = 5.7 Hz, C3) ppm. 31P{1H} NMR (161.97 MHz, CDCl3): δ = 13.7 ppm.
HR-MS (CH3OH): (C27H36ClNPRh; pos. ESI) calcd for [M − Cl]+: 508.1635, found: 508.1646.
Complex 4 (0.434 mmol, dissolved in THF (4 ml)) was prepared in the same way as complex 3, but was directly used in the next step to give complex 6.
Synthesis of heterobimetallic complexes 5 and 6
Free phosphane compound 3 or 4 (120 mg or 135 mg, respectively, 0.21 mmol, 1 equiv.) was dissolved in DCM and reacted with [AuCl(tht)] (68 mg, 0.21 mmol, 1 equiv.) at room temperature for 15 min to afford precipitated heterobimetallic complexes 5 (M = Rh, orange prisms) and 6 (M = Ir, orange prisms) quantitatively.5: 1H NMR (400.1 MHz, CD2Cl2): δ = 1.06 (dd, 3JHP = 13 Hz, 3JHH = 7.0 Hz, 3 H, 2′′-H), 1.10 (dd, 3JHP = 13 Hz, 3JHH = 7.0 Hz, 3 H, 3′′-H), 1.20 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 3 H, 5′′-H), 1.25 (dd, 3JHP = 17 Hz, 3JHH = 7.0 Hz, 3 H, 6′′-H), 1.56 (s, 15 H, Cp*–Me), 2.49 (heptd, 3JHH = 7.0 Hz, 2JHP = 1.7 Hz, 2 H, 1′′ and 4′′-H), 7.21 (tt, 3JHH = 6.4 Hz, 4JHH = 1.7 Hz, 1 H, 4′-H), 7.28 (tt, 3JHH = 9.0 Hz, 4JHH = 1.7 Hz, 1 H, 6-H), 7.64 (dd, 3JHH = 8.0 Hz, 4JHH = 2.2 Hz, 1 H, 5-H), 7.75 (td, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 1 H, 3′-H), 7.81 (dt, 3JHH = 8.0 Hz, 4JHH = 1.2 Hz, 1 H, 2′-H), 8.14 (dd, 3JHH = 12.7 Hz, 4JHH = 1.6 Hz, 1 H, 2-H), 8.70 (dt, 3JHH = 5.5 Hz, 4JHH = 1.0 Hz, 1 H, 5′-H) ppm. 13C{1H} NMR (100.6 MHz, CD2Cl2): δ = 9.4 (s, Cp*–Me), 18.7 (d, 2JCP = 17.4 Hz, C2′′ and C3′′), 19.7 (d, 2JCP = 13.7 Hz, C5′′ and C6′′), 25.4 (d, 1JCP = 20.3 Hz, C1′′), 25.7 (d, 1JCP = 19.8 Hz, C4′′), 96.7 (d, 1JCRh = 6.3 Hz, Cp*), 120.3 (s, C2′), 123.2 (d, 3JCP = 10.4 Hz, C5), 123.7 (s, C4′), 126.5 (d, 1JCP = 49.0 Hz, C1), 128.3 (d, 2JCP = 10.3 Hz, C6), 137.9 (s, C3′), 144.9 (d, 2JCP = 12.9 Hz, C2), 147.9 (s, C4), 152.0 (s, C5′), 164.2 (s, C1′), 179.8 (dd, 1JCRh = 33.0 Hz, 3JCP = 9.1 Hz, C3) ppm. 31P{1H} NMR (161.97 MHz, CD2Cl2): δ = 60.5 ppm.
HR-MS (CH3OH): (C27H36AuCl2NPRh; [M − Cl]+, pos. ESI) calcd: 740.0989, found: 740.0924.
6: 1H NMR (400.1 MHz, CDCl3): δ = 1.13 (dd, 3JHP = 10.0 Hz, 3JHH = 7.0 Hz, 3 H, 2′′-H), 1.17 (dd, 3JHP = 10.0 Hz, 3JHH = 7.0 Hz, 3 H, 3′′-H), 1.27 (dd, 3JHP = 18.6 Hz, 3JHH = 7.0 Hz, 3 H, 5′′-H), 1.32 (dd, 3JHP = 18.6 Hz, 3JHH = 7.0 Hz, 3 H, 6′′-H), 1.71 (s, 15 H, Cp*–Me), 2.52 (heptd, 3JHH = 7.0 Hz, 2JHP = 1.7 Hz, 2 H, 1′′ and 4′′-H), 7.19 (tt, 3JHH = 6.4 Hz, 4JHH = 1.5 Hz, 1 H, 4′-H), 7.30 (tt, 3JHH = 9.0 Hz, 4JHH = 1.7 Hz, 1 H, 6-H), 7.74 (m, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 2 H, 5-H and 3′-H), 7.90 (d, 3JHH = 8.0 Hz, 1 H, 2′-H), 8.25 (dd, 3JHH = 13.0 Hz, 4JHH = 1.6 Hz, 1 H, 2-H), 8.76 (d, 3JHH = 5.5 Hz, 1 H, 5′-H) ppm. 13C{1H} NMR (100.6 MHz, CDCl3): δ = 9.0 (s, Cp*–Me), 18.4 (d, 2JCP = 4.8 Hz, C2′′ and C3′′), 19.4 (d, 2JCP = 4.4 Hz, C5′′ and C6′′), 24.9 (d, 1JCP = 34.6 Hz, C1′′), 25.3 (d, 1JCP = 34.2 Hz, C4′′), 89.0 (s, Cp*), 119.8 (s, C2′), 123.4 (d, 3JCP = 11.0 Hz, C5), 123.6 (s, C4′), 126.5 (d, 1JCP = 50.0 Hz, C3), 127.2 (d, 2JCP = 10.7 Hz, C6), 137.3 (s, C3′), 143.5 (d, 2JCP = 12.9 Hz, C2), 147.9 (s, C4), 151.6 (s, C5′), 164.0 (d, 3JCP = 9.7 Hz, C1), 165.9 (s, C1′) ppm. 31P{1H} NMR (161.97 MHz, CDCl3): δ = 60.0 ppm.
HR-MS (CH3OH): (C27H36AuCl2IrNP; [M − Cl]+, pos. ESI) calcd: 830.1556, found: 830.1559.
Elemental analysis for C27H36AuCl2IrNP calcd: C 37.46, H 4.19, N 1.62, found: C 37.78, H 3.84, N 1.58.
Anion exchange reaction: preparation of 7
Complex 6 (17.3 mg, 0.02 mmol, 1 equiv.) was dissolved in CD2Cl2 (1 ml) and CD3CN (1 ml). AgNTf2 (15.5 mg, 0.04 mmol, 2 equiv.) was added, and the reaction mixture was stirred at room temperature for 15 min. The precipitate (AgCl) was filtered off, and the filtrate was analysed in situ.1H NMR (400.1 MHz, CD3CN/CD2Cl2): δ = 1.12 (dd, 3JHP = 6.0 Hz, 3JHH = 7.0 Hz, 3 H, 2′′-H), 1.13 (dd, 3JHP = 6.0 Hz, 3JHH = 7.0 Hz, 3 H, 3′′-H), 1.28 (dd, 3JHP = 19.0 Hz, 3JHH = 7.0 Hz, 3 H, 5′′-H), 1.31 (dd, 3JHP = 19.0 Hz, 3JHH = 7.0 Hz, 3 H, 6′′-H), 1.72 (s, 15 H, Cp*–Me), 2.66 (heptd, 3JHH = 7.0 Hz, 2 H, 1′′ and 4′′-H), 7.43 (m, 3JHH = 5.7 Hz, 4JHH = 1.5 Hz, 1 H, 4′-H), 7.46 (m, 3JHH = 8.0 Hz, 4JHH = 1.6 Hz, 1 H, 4′-H), 7.90 (dd, 3JHH = 8.0 Hz, 4JHH = 2.3 Hz, 1 H, 6-H), 8.00 (td, 3JHH = 8.0 Hz, 4JHH = 1.5 Hz, 1 H, 3′-H), 8.10 (dt, 3JHH = 8.0 Hz, 4JHH = 1.2 Hz, 1 H, 2′-H), 8.31 (dd, 3JHH = 13.5 Hz, 4JHH = 1.6 Hz, 1 H, 2-H), 8.76 (d, 3JHH = 5.7 Hz, 4JHH = 0.7 Hz, 1 H, 5′-H) ppm. 13C{1H} NMR (100.6 MHz, CD3CN/CD2Cl2): δ = 9.0 (s, Cp*–Me), 18.8 (d, 2JCP = 6.0 Hz, C2′′ and C3′′), 19.8 (d, 2JCP = 2.0 Hz, C5′′ and C6′′), 25.6 (d, 1JCP = 35.0 Hz, C1′′), 25.7 (d, 1JCP = 35.0 Hz, C4′′), 92.3 (s, Cp*), 119.0 (s, C–NTf2), 121.5 (s, C2′), 122.2 (s, C1), 124.9 (d, 1JCP = 10.0 Hz, C6), 125.8 (s, C4′), 129.0 (d, 2JCP = 12.9 Hz, C2), 140.4 (s, C3′), 144.8 (d, 3JCP = 4.0 Hz, C5), 149.6 (s, C4), 153.3 (s, C5′), 157.5 (d, 3JCP = 6.6 Hz, C3), 165.9 (s, C1′) ppm. 31P{1H} NMR (161.97 MHz, CD3CN/CD2Cl2): δ = 61.6 ppm.
HR-MS (CD3CN/CD2Cl2): (C31H36AuF12IrN3O8PS4; [M − NTf2]+, pos. ESI) calcd: 1075.1046, found: 1075.1053.
Synthesis of 8
The triphosphane ligand L2H (0.578 g, 1.01 mmol, 1 equiv.) was dissolved in DCM (20 ml) and [AuCl(tht)] (0.324 g, 1.01 mmol, 1 equiv.) was added, resulting in a colour change to deep red. The solution was stirred for 1 h; then, all volatile compounds were removed in vacuo. The product was obtained quantitatively after recrystallisation from a mixture of 10 ml DCM and 50 ml n-hexane and washing with n-pentane. The compound is not light-sensitive and can be stored in a DCM solution for weeks. Single crystals (orange prisms) suitable for X-ray diffraction could be obtained by dissolving 10 mg in DCM and layering with n-hexane.1H NMR (400.1 MHz, CDCl3): δ = 8.66 (d, 3JHH = 4.8 Hz, 1 H, 5′-H), 8.02–7.94 (m, 2 H, 6′′-H), 7.81 (dd, 3JHH = 8.4, 4JPH = 2.1 Hz, 2 H, 3-H), 7.74 (td, 3JHH = 7.7, 4JHH = 1.9 Hz, 1 H, 3′-H), 7.67 (d, 3JHH = 7.9 Hz, 1 H, 2′-H), 7.64–7.60 (m, 2 H, 3′′-H), 7.49 (pseudo p, N = 7.2 Hz, 4 H, 4′′- and 5′′-H), 7.22 (dd, 3JHH = 7.3, 5.0 Hz, 1 H, 4′-H), 7.06 (dd, 3JHH = 8.3, 3JPH = 6.6 Hz, 2 H, 2-H), 2.84–2.64 (m, 4 H, 1′′′-H), 1.50–1.41 (m, 6 H, 2′′′-H), 1.37 (pseudo q, 3JHH/HP = 8.1 Hz, 6 H, 2′′′-H), 1.25 (pseudo q, 3JHH/HP = 6.9 Hz, 6 H, 2′′′-H), 0.82 (pseudo dt, 3JPH = 9.3 Hz, 3JHH = 6.9 Hz, 6 H, 2′′′-H) ppm. 13C{1H} NMR (100.6 MHz, CDCl3): δ = 156.7 (C1′), 149.5 (C5′), 145.3 (pseudo q, 1JCP = 12.5 Hz, C2′′), 142.3 (C1) 138.4 (m, C4/C1′′), 137.0 (C3′), 135.9 (C6′′), 131.9 (m, C2/C3′′), 130.7 (C4′′), 129.8 (C5′′), 126.8 (d, 4JCP = 4.6 Hz, C3), 122.2 (C4′), 120.6 (C2′), 27.2 (pseudo t, 1JCP = 13.3 Hz, C1′′′), 23.6 (m, C1′′′), 20.0 (m, C2′′′), 19.8 (m, C2′′′), 19.1 (C2′′′), 17.0 (C2′′′) ppm. 31P{1H} NMR (161.97 MHz, CDCl3): δ = 58.3 (d, 2JPP = 114.8 Hz, 2 P, Pterminal), −5.0 (t, 2JPP = 114.4 Hz, 1 P, Pcentral) ppm.
HR-MS (CH3OH): (C35H44AuClNP3; [M − Cl]+, pos. ESI) calcd: 768.2347 found: 768.2299.
Synthesis of heterobimetallic complexes 9 and 10
The gold complex 8 (0.5 mmol, 2 equiv.) was suspended in ACN (30 ml) together with [Cp*MCl2]2 (M = Rh or Ir, 0.25 mmol, 1 equiv.) and NaOAc (1.5 mmol, 6 equiv.) and stirred at room temperature for 48 h. The suspension was filtered over Celite, and then extracted with DCM. Removal of the volatiles in high vacuum gave the product quantitatively. Single crystals (orange needles) suitable for X-ray diffraction could be obtained by layering a frozen DCM solution with n-hexane (9) or from a hot saturated solution in ACN (10).9: 1H NMR (400.1 MHz, CD2Cl2): δ = 8.69 (d, 3JHH = 5.6 Hz, 1 H, 5′-H), 8.19 (m, 1 H, 6′′-H), 8.03 (m, 1 H, 6′′-H), 7.75 (dd, 3JHH = 4.8 Hz, 4JHH = 1.1 Hz, 2 H, 2′- and 3′-H), 7.68 (m, 2 H, 3′′-H), 7.65–7.60 (m, 2 H, 4′′- and 5′′-H), 7.58 (m, 2 H, 4′′- and 5′′-H), 7.50 (d, 3JHH = 8.1 Hz, 1 H, 5-H), 7.39 (d, 4JHH = 1.6 Hz, 1 H, 2-H), 7.21–7.17 (m, 1 H, 4′-H), 6.72 (dd, 3JPH,HH = 8.4 Hz, 4JHH = 1.7 Hz, 1 H, 6-H), 2.82 (m, 2 H, 1′′′-H), 2.67 (m, 2 H, 1′′′-H), 1.44 (m, 6 H, 2′′′-H) 1.42 (s, 15 H, Cp*–Me), 1.36 (m, 6 H, 2′′′-H), 1.28 (m, 6 H, 2′′′-H), 0.83 (ddd, 3JPH = 18.1 Hz, 3JHH = 6.7, 4.6 Hz, 6 H, 2′′′-H) ppm. 13C{1H} NMR (100.6 MHz, CD2Cl2): δ = 179.9 (d, 1JRhC = 33 Hz, C3), 164.8 (C1′), 151.9 (C5′), 145.2 (m, C1′′/C2′′), 144.2 (C4), 143.5 (m, C1′′/C2′′), 142.0 (m, C1), 139.9 (d, 2JCP = 11 Hz, C2), 137.7 (C3′), 137.3 (m, C6′′), 133.4 (d, 2JCP = 9.6 Hz, C3′′), 133.2 (d, 2JCP = 9.1 Hz, C3′′), 132.1 (d, 1JCP = 10.2 Hz, C4′′/C5′′), 131.1 (d, 2JCP = 14.2 Hz, C4′′/C5′′), 126.6 (d, 2JCP = 20 Hz, C6), 123.3 (d, 3JCP = 7 Hz, C5), 122.8 (C4′), 119.7 (C2′), 96.4 (d, 1JRhC = 6 Hz, Cp*), 28.2 (m, C1′′′), 24.3 (m, C1′′′), 20.3 (m, C2′′′), 19.4 (C2′′′), 17.9 (d, 2JCP = 5 Hz, C2′′′), 9.3 (Cp*–Me) ppm. 31P{1H} NMR (161.97 MHz, CD2Cl2): δ = 63.4 (dd, 2JPP = 98.6, 15.2 Hz, 2 P, Pterminal), 5.9 (t, 2JPP = 98.7 Hz, 1 P, Pcentral) ppm.
HR-MS (CH3OH): (C45H58AuCl2NP3Rh; [M − Cl]+, pos. ESI) calcd: 1040.2186 found: 1040.2214.
FT-IR (KBr): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 3046 (w), 2957 (s), 2923 (s), 2866 (m), 1719 (m), 1601 (s), 1560 (s), 1468 (s), 1422 (m), 1382 (m), 1314 (w), 1239 (m), 1160 (w), 1107 (m), 1025 (m), 881 (m), 775 (m), 721 (w), 661 (m), 630 (w), 583 (w), 526 (m), 453 (w), 434 (m).
(cm−1) = 3046 (w), 2957 (s), 2923 (s), 2866 (m), 1719 (m), 1601 (s), 1560 (s), 1468 (s), 1422 (m), 1382 (m), 1314 (w), 1239 (m), 1160 (w), 1107 (m), 1025 (m), 881 (m), 775 (m), 721 (w), 661 (m), 630 (w), 583 (w), 526 (m), 453 (w), 434 (m).
10: 1H NMR (CDCl3): δ (ppm) = 8.67 (dt, 3JHH = 5.6 Hz, 4JHH = 1.1 Hz, 1 H, 5′-H), 8.13 (m, 1 H, 6′′-H), 7.92 (m, 1 H, 6′′-H), 7.74 (d, 3JHH = 8.2 Hz, 1 H, 2′-H), 7.69–7.59 (m, 3 H, 3′- and 3′′-H), 7.59–7.41 (m, 4 H, 4′′- and 5′′-H), 7.49 (dd, 3JHH = 8.1, 3JPH = 2.4 Hz, 1 H, 5-H), 7.46 (dd, 3JPH = 6.8 Hz, 4JHH = 1.6 Hz, 1 H, 2-H), 7.07 (ddd, 3JHH = 7.3, 5.7 Hz, 4JHH = 1.4 Hz, 1 H, 4′-H), 6.68 (td, 3JPH,HH = 8.4 Hz, 4JHH = 1.7 Hz, 1 H, 6-H), 2.82–2.65 (m, 4 H, 1′′′-H), 1.48 (s, 15 H, Cp*–Me), 1.47–1.37 (m, 12 H, 2′′′-H), 1.28 (ddt, 3JPH = 10.4 Hz, 3JHH = 7.0, 3.4 Hz, 6 H, 2′′′-H), 0.82 (dq, 3JPH = 14.0 Hz, 3JHH = 7.3 Hz, 6 H, 2′′′-H). 13C{1H} NMR (CDCl3) δ (ppm) = 166.6 (C1′), 163.4 (C3), 151.3 (C5′), 146.2 (m, C1′′/C2′′), 144.6 (m, C1′′/C2′′), 143.8 (m, C1), 138.6 (d, 2JCP = 10.6 Hz, C2), 137.0 (C3′), 136.9 (m, C6′′), 132.2 (m, C3′′), 132.0 (C4), 131.1 (m, C4′′/C5′′), 129.9 (C4′′/C5′′), 126.1 (d, 2JPC = 20.4 Hz, C6), 123.5 (d, 3JCP = 8.2 Hz, C5), 122.5 (C4′), 119.1 (C2′), 88.6 (C, Cp*), 27.4 (m, C1′′′), 23.8 (m, C1′′′), 20.4 (m, C2′′′), 20.0 (m, C2′′′), 19.2 (m, C2′′′), 17.5 (C2′′′), 17.4 (C2′′′), 8.9 (Cp*–Me). 31P{1H} NMR (161.97 MHz, CD2Cl2): δ = 63.4 (dd, 2JPP = 98.6, 15.2 Hz, 2 P, Pterminal), 5.9 (t, 2JPP = 98.7 Hz, 1 P, Pcentral) ppm.
HR-MS (CH3OH): (C45H58AuCl2IrNP3; pos. ESI) calcd for [M − 2Cl + CN]+: 1121.3105, found: 1121.3150; calcd for [M − Cl]+: 1130.2754, found: 1130.2795.
FT-IR (KBr): (cm−1) = 3046 (w), 2957 (s), 2923 (s), 2866 (m), 1719 (m), 1601 (s), 1560 (s), 1468 (s), 1422 (m), 1383 (m), 1314 (w), 1239 (m), 1159 (w), 1107 (m), 1025 (m), 881 (w), 775 (m), 721 (w), 662 (m), 630 (w), 583 (w), 526 (m), 454 (w), 434 (m).
Anion exchange reaction: preparation of 11
AgNTf2 (77.5 mg, 0.2 mmol, 2 equiv.) was added to an orange solution of the gold–iridium–triphosphane complex 10 (116.4 mg, 0.1 mmol, 1 equiv.) in ACN (10 ml) resulting in a colour change to yellow and the formation of a white precipitate (AgCl). After 15 min of stirring in the dark, the solution was filtered over Celite and the filtrate was checked by 31P NMR spectroscopy. Due to instability upon removal of solvent or crystallisation, the complex was subjected to in situ characterisation and no yield was determined.1H NMR (CD3CN): δ (ppm) = 8.70 (ddd, 3JHH = 5.8, 4JHH = 1.5, 5JHH = 0.8 Hz, 1 H, 5′-H), 8.30 (bs, 2 H, 6′′-H), 7.99 (ddd, 3JHH = 8.3, 4JHH = 1.7, 5JHH = 0.8 Hz, 1 H, 2′-H), 7.97–7.91 (m, 1 H, 3′-H), 7.87 (m, 2 H, H3′′-H), 7.82–7.73 (m, 4 H, 4′′- and 5′′-H), 7.71 (dd, 3JHH = 8.1, 4JHH = 2.3 Hz, 1 H, 5-H), 7.36 (ddd, 3JHH = 7.3, 5.7, 4JHH = 1.6 Hz, 1 H, 4′-H), 7.26 (dd, 3JHP = 7.0, 4JHH = 1.7 Hz, 1 H, 2-H), 6.79 (td, 3JHH/HP = 8.2, 1.7 Hz, 1 H, 6-H), 3.12 (m, 2 H, 1′′′-H), 2.72 (m, 2 H, 1′′′-H), 1.49 (s, 15 H, Cp*–Me), 1.48–1.40 (m, 6 H, 2′′′-H), 1.32–1.19 (m, 12 H, 2′′′-H), 0.87 (pseudo-ddd, N = 19.0, 12.2, 6.6 Hz, 6 H, 2′′′-H). 13C{1H} NMR (CD2Cl2): δ (ppm) = 166.0 (C1′), 157.0 (C3), 152.5 (C5′), 145.4 (C4), 143.3 (d, 1JCP = 21.6 Hz, C1), 143.2 (m, C1′′/C2′′), 139.5 (C3′), 137.6 (C6′′), 137.5 (m, C2), 134.3 (d, 2JCP = 9.6 Hz, C3′′), 134.2 (1C, C4′′/C5′′), 133.1 (1C, C4′′/C5′′), 132.7 (m, 2C, C4′′/C5′′), 126.5 (d, 2JCP = 18.1 Hz, C6), 124.3 (C4′), 124.2 (d, 3JCP = 6.7 Hz, C5), 121.5 (C2′), 120.1 (C), 92.2 (C, Cp*), 26.8 (pseudo-td, N = 18.2, 10.0 Hz, C1′′′), 22.6 (m, C1′′′), 19.6 (bs, C2′′′), 19.1 (m, C2′′′), 19.0 (bs, C2′′′), 18.6 (bs, C2′′′), 17.0 (bs, C2′′′), 8.0 (Cp*–Me). 31P{1H} NMR (CD3CN) δ (ppm) = 71.9 (d, JPP = 70.0 Hz, 1 P, Pterminal), 71.5 (d, JPP = 70.5 Hz, 1 P, Pterminal), 17.0 (pseudo-t, JPP = 70.3 Hz, 1 P, Pcentral).
Testing of catalytic activity
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support from the German Israeli Foundation for Scientific Research and Development (GIF) and the Graduate School Building with Molecules and Nano-objects (BuildMoNa). We thank Manuela Roßberg for the elemental analysis, Ramona Oehme for the measurement of the mass spectra.References
- P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497 CrossRef CAS.
- H. Steinhagen and G. Helmchen, Angew. Chem., Int. Ed. Engl., 1996, 35, 2339 CrossRef CAS.
- J. A. Tainer, E. D. Getzoff, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 284 CrossRef CAS PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149 CrossRef CAS PubMed.
- J. H. H. Ho, J. Wagler, A. C. Willis and B. A. Messerle, Dalton Trans., 2011, 40, 11031 RSC.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev. - Sci. Eng., 2012, 54, 1 CrossRef CAS.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef CAS PubMed.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS PubMed.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750 CrossRef CAS PubMed.
- G. Bauer and X. Hu, Inorg. Chem. Front., 2016, 3, 741 RSC.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11 CrossRef CAS.
- M. S. Balakrishna, Polyhedron, 2018, 143, 2 CrossRef CAS.
- L. Maser, L. Vondung and R. Langer, Polyhedron, 2018, 143, 28 CrossRef CAS.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959 RSC.
- C. M. Jensen, Chem. Commun., 1999, 2443–2449 RSC.
- D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201 CrossRef CAS PubMed.
- Z. B. G. Fickenscher, P. Lönnecke, A. K. Müller, O. Hollóczki, B. Kirchner and E. Hey-Hawkins, Molecules, 2023, 28, 2574 CrossRef CAS PubMed.
- Z. B. G. Fickenscher, L. Torres-Texeira, P. Lönnecke and E. Hey-Hawkins, Inorg. Chim. Acta, 2023, 558, 121721 CrossRef CAS.
- Z. B. G. Fickenscher, P. Lönnecke, A. K. Müller, W. Baumann, B. Kirchner and E. Hey-Hawkins, Inorg. Chem., 2023, 62, 12750 CrossRef CAS PubMed.
- L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492 CrossRef CAS.
- W. Voskuil and J. F. Arens, Org. Synth., 1973, 5, 211 Search PubMed.
- Rigaku Corporation, CrysAlisPro Software System, Rigaku Oxford Diffraction, Wroclaw, Poland, 1995–2023 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- K. Brandenburg, DIAMOND 4, version 4.6.8, Crystal Impact GbR, Bonn, Germany Search PubMed.
- L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414 CrossRef CAS PubMed.
- M. Brasse, J. Cámpora, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2013, 135, 6427 CrossRef CAS PubMed.
- A. I. VanderWeide, W. W. Brennessel and W. D. Jones, J. Org. Chem., 2019, 84, 12960 CrossRef CAS PubMed.
- D. L. Davies, K. Singh and N. Tamosiunaite, Dalton Trans., 2021, 50, 13505 RSC.
- R. Romeo, L. A. Wozniak and C. Chatgilialoglu, Tetrahedron Lett., 2000, 41, 9899 CrossRef CAS.
- D. V. Griffiths, H. J. Groombridge, P. M. Mahoney, S. P. Swetnam, G. Walton and D. C. York, Tetrahedron, 2005, 61, 4595 CrossRef CAS.
- M. Hayashi, Chem. Lett., 2021, 50, 1 CrossRef CAS.
- Y. Hu, L. Li, A. P. Shaw, J. R. Norton, W. Sattler and Y. Rong, Organometallics, 2012, 31, 5058 CrossRef CAS.
- S. P. C. Dunstan, P. C. Healy, A. N. Sobolev, E. R. T. Tiekink, A. H. White and M. L. Williams, J. Mol. Struct., 2014, 1072, 253 CrossRef CAS.
- J. A. Muir, M. M. Muir, L. B. Pulgar, P. G. Jones and G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1174 CrossRef.
- K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994, 472, 371 CrossRef CAS.
- J. A. Muir, S. I. Cuadrado and M. M. Muir, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1072 CrossRef PubMed.
- M. Böhmer, F. Kampert, T. T. Y. Tan, G. Guisado-Barrios, E. Peris and F. E. Hahn, Organometallics, 2018, 37, 4092 CrossRef.
- J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 2493 CrossRef.
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS PubMed.
- C. N. Kona and C. V. Ramana, Chem. Commun., 2014, 50, 2152 RSC.
- C. N. Kona, M. N. Patil and C. V. Ramana, Org. Chem. Front., 2016, 3, 453 RSC.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391 CrossRef CAS PubMed.
- O. A. Egorova, H. Seo, Y. Kim, D. Moon, Y. M. Rhee and K. H. Ahn, Angew. Chem., Int. Ed., 2011, 50, 11446 CrossRef CAS PubMed.
- Y. Liu, P. Liu, B. Ling, G. Chen, T. Chen, Y. Li, S. Bi and D. Zhang, Eur. J. Org. Chem., 2019, 6822 CrossRef CAS.
- J. Segato, W. Baratta, P. Belanzoni, L. Belpassi, A. Del Zotto and D. Zuccaccia, Inorg. Chim. Acta, 2021, 522, 120372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and mass spectra of L1H, 1–3, 5–10, and crystallographic data of L1H, 1–3, 5, 6, 8–10. CCDC 2322723–2322731. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01247j |
| This journal is © The Royal Society of Chemistry 2024 |