
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reduction of [(C5H4SiMe3)2Ln(μ-OR)]2 (Ln = La, Ce) complexes: structural effects of bridging alkoxides†
Adrian N.
Brown
 a,
Jack N.
Kelleher
a,
Alexander M.
Brown
b,
Peter
Saghy
b,
Joshua J.
Bohl
a,
Jerome R.
Robinson
*b and
Daniel N.
Huh
*a
a,
Jack N.
Kelleher
a,
Alexander M.
Brown
b,
Peter
Saghy
b,
Joshua J.
Bohl
a,
Jerome R.
Robinson
*b and
Daniel N.
Huh
*a
aDepartment of Chemistry, University of Rhode Island, Kingston, RI 02881, USA. E-mail: danhuh@uri.edu
bDepartment of Chemistry, Brown University, Providence, RI 02912, USA. E-mail: jerome_robinson@brown.edu
First published on 20th August 2024
Abstract
Alcoholysis of Cp′3Ln (Ln = La, Ce; Cp′ = C5H4SiMe3) generate high-yielding (72–97%) bimetallic LnIII complexes of [Cp′2Ln(μ-OR)]2 [R = Et, iPr, or C6H4-4-tBu]. Single-crystal X-ray diffraction of these complexes reveal unexpected decreases in Ln⋯Ln distances, increasing Cpcent–Ln–Cpcent angles, and increasing intermolecular C⋯C contacts with bulkier bridging alkoxides, in line with structural control driven by significant dispersion forces. 1H NMR spectroscopy of [Cp′2Ce(μ-OEt)]2 and [Cp′2Ce(μ-OiPr)]2 revealed significantly upfield resonances assigned as methylene and methine moieties of −43.74 and −70.85 ppm, respectively. 2D 1H DOSY NMR experiments of [Cp′2Ce(μ-OiPr)]2 in C6D6 supported a dimeric structure in solution, including in the presence of a Lewis base (i.e., THF). Reduction of [Cp′2La(μ-OiPr)]2 using KC8 in the presence of 2.2.2-cryptand at −78 °C generated a purple solution and X-band EPR spectroscopy revealed an eight-line hyperfine pattern indicative of a LaII species.
Introduction
Bimetallic lanthanide (Ln) complexes have served as useful compounds for polymerization catalysis1–3 as well as research areas including single-molecular magnetism (SMM) and quantum information science (QIS).4–14 Reduction chemistry of bimetallic Ln complexes has recently emerged as a method to form mixed-valent LnIII/LnII species,15,16 with intervalence-charge transfer (IVCT) character (i.e. Robin-Day Class II or III categorization) and in some cases, yielding Ln–Ln bonds with large coercive magnetic fields.10 Reductive Ln chemistry has typically been examined for monometallic compounds. Low-valent Ln ions supported by cyclopentadienide derivatives, for example, have been shown to have interesting physical properties/performance including reinforced magnetic phase memory,17–20 and molecular qudit properties.21 Recently, there have also been several reports involving syntheses of hetero-multimetallic complexes including Ln/Al (Ln = Sm, Dy, Y, Yb) supported by tris(pyrazolyl)borates,22 Ln/V (Ln = La, Nd, Gd, Yb, Er) with bridging redox-active ligands,23 Y/M (M = Al, Ga) containing C5Me5 and a boryl moiety,24 Ln2/Al2 (Ln = La, Nd) isoprene polymerization catalyst,25 Ce2/Cu alkoxide complex,26 and Ln/Ni (Ln = Y, La, Lu) complexes supported by a crown-ether-like macrocycle.27Synthesis of homobimetallic complexes supported by cyclopentadienyl ligands and bridged by alkoxides or aryloxides, [CpR2Ln(μ-OR)]2, in particular, have historically been reported via two synthetic routes: (1) reductive cleavage of glycols,28–30 and (2) alcoholysis31–35 (Fig. 1). Due to their small steric profile compared to amido or cyclopentadienyl groups, alkoxide and aryloxides can frequently promote formation of bimetallic species. Of the reported [CpR2Ln(μ-OR)]2 complexes, only two La29,32 and four Ce28,31,32 have been published. Often these compounds are presented as side-products from reduction reactions in the presence of glycols or as an additional means to synthesize heteroleptic Cp/alkoxide compounds. However, assessing the structural impact of the bridging alkoxide moiety is often overlooked and revisiting this class of compounds could provide useful insights to subtle structural modulations.
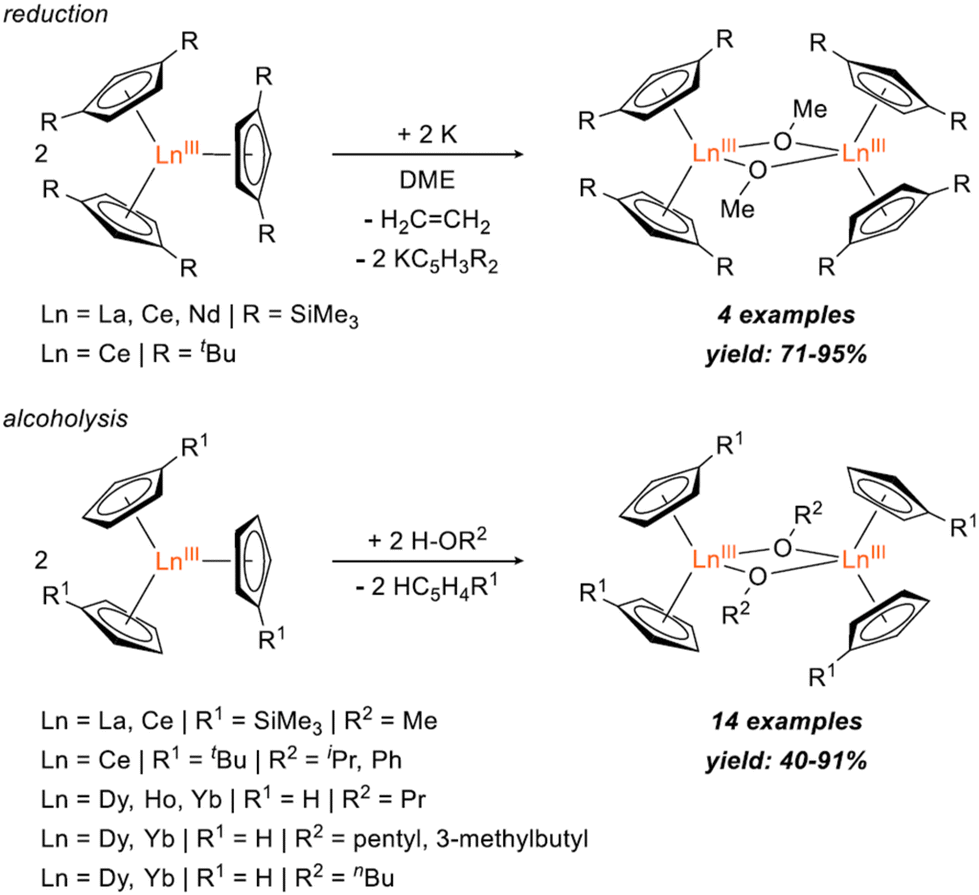 | ||
| Fig. 1 Reported syntheses of [CpR2Ln(μ-OR)]2 bimetallic complexes via reduction28,29 (top) and alcoholysis31–35 (bottom). | ||
Herein, we report the synthesis of [Cp′2Ln(μ-OR)]2 (Cp′ = C5H4SiMe3; Ln = La, Ce; R = Et, iPr, C6H4-4-tBu) complexes via alcoholysis of Cp′3Ln generating yields of 72–97%. 2D 1H DOSY NMR and SC-XRD unambiguously identify the dimer structure of these complexes. Structural analyses reveal unexpected decreases in Ln⋯Ln distances, increases in Cpcent–Ln–Cpcent angles, and increases in intermolecular C⋯C contacts with bulkier bridging alkoxides, in line with structural control driven by significant dispersion forces. Finally, reduction of [Cp′2La(μ-OiPr)]2 using KC8 in the presence of 2.2.2-cryptand generated a transient LaII species characterized by CW X-band EPR.
Results and discussion
Alcoholysis reactions of Cp′3Ln (1-Ln, Ln = La, Ce) using HO-R (R = Et, iPr, C6H4-4-tBu) in Et2O at room temperature form isolable bimetallic complexes [Cp′2Ln(μ-OR)]22-Ln/R in 72–97% yields (Fig. 2). Evans’ method of 2-Ce/R have magnetic moments of 1.28 (R = Et), 1.90 (R = iPr), and 1.34μB (R = C6H4-4-tBu) (Fig. S12–S15†). UV-vis data (Fig. S16†) of 2-Ce/R in toluene were also collected and have λmax values of 458 (R = Et), 452 (R = iPr), and 471 nm (R = C6H4-4-tBu). These values are similar to the reported UV-vis spectrum of 2-Ce/Me in toluene with a λmax of 475 nm.32 Crystals suitable for SC-XRD were structurally characterized for [Cp′2Ce(μ-OEt)]22-Ce/Et, [Cp′2La(μ-OiPr)]22-La/iPr, and [Cp′2Ce(μ-OiPr)]22-Ce/iPr. Despite numerous attempts, X-ray quality single crystals of [Cp′2Ln(μ-OC6H4-4-tBu)]2 (2-Ln/C6H4-4-tBu, Ln = La and Ce) were unable to be obtained. | ||
| Fig. 2 Top: synthesis of 2-Ln/R by alcoholysis of 1-Ln in Et2O. Bottom: crystal structures of 2-Ce/Et and 2-Ce/iPr with displacement ellipsoids at the 50% probability level. Hydrogen atoms and disorder were removed for clarity. | ||
The metrical parameters of 2-La/iPr, 2-Ce/Et, and 2-Ce/iPr are summarized in Table 1. Crystallographic data of 14 structures adopting the [Cp2Ln(μ-OR)]2 (R = alkyl) geometry have been reported.28–35 Of these compounds, only two La complexes and three Ce complexes were reported: [Cp′2La(μ-OMe)]2,32 {[C5H3(SiMe3)2]2La(μ-OMe)}2,29 [Cp′2Ce(μ-OMe)]2,32 [(C5H4tBu)2Ce(μ-OiPr)]2,31 and [(C5H3tBu2)2Ce(μ-OMe)]228 (Table 1). Despite the increased steric bulk offered by the isopropyl group, Ln⋯Ln distances of 2-Ln/iPr are significantly shorter than that of the reported 2-Ln/Me; [La: 3.8119(4) and 3.8428(4) vs. 3.8566(4) Å; Ce: 3.7977(8) and 3.7810(8) vs. 3.8286(6) Å].32 Upon closer inspection of the 2-Ln/iPr and 2-Ce/Et complexes, several short intramolecular C⋯C distances (dC⋯C) are present between the R groups (iPr, Et) and sp2 carbons of the Cp′ ligand, approaching 3.644(3) and 3.641(3) Å, respectively (Fig. S1†). These distances are much shorter than the tert-butyl-substituted trityl compound, [C(C6H3-3,5-tBu2)3]2, which is well-established to display significant dispersion forces [dC⋯C: 3.95(2)–4.15(2) Å].36–39 Furthermore, the Cpcent–Ln–Cpcent angles of 2-Ce/Et, 2-La/iPr, and 2-Ce/iPr range from 129.69 to 132.92° (Table 1), which are the largest angles observed for any [CpR2Ln(μ-OR)] architecture (123.9 to 128.9°).28,31–35,40,41 Taken together, the observed distortions for 2-Ce/Et, 2-La/iPr, and 2-Ce/iPr (i.e., shorter than expected Ln⋯Ln distances and larger Cpcent–Ln–Cpcent angles) are likely stabilized via significant inter-ligand dispersion forces.
| 2-La/Me 32 | 2-La/iPr | 2-Ce/Me 32 | 2-Ce/Et | 2-Ce/iPr | [(C5H3tBu2)2Ce(μ-OMe)]228 | [(C5H4tBu)2Ce(μ-OiPr)]231 | |
|---|---|---|---|---|---|---|---|
| a Shortest intramolecular C⋯C distance between a cyclopentadienide and –OR group. b Number of intramolecular C⋯C distances less than 4.0 Å between sp2 cyclopentadienide and –OR. | |||||||
| Ln–O | 2.375(1), 2.413(1) | 2.360(1), 2.364(1), 2.377(1), 2.403(1) | 2.350(3), 2.387(2) | 2.335(1), 2.360(1) | 2.339(4), 2.339(4), 2.347(4), 2.361(4) | 2.366(3), 2.386(4) | 2.369(3), 2.373(3) |
| Ln–Cpcent | 2.562, 2.572 | 2.572, 2.573, 2.578, 2.581 | 2.531, 2.539 | 2.532, 2.551 | 2.541, 2.542, 2.542, 2.546 | 2.564, 2.596 | 2.565, 2.577 |
| Ln⋯Ln | 3.8566(4) | 3.8119(4), 3.8428(4) | 3.8286(6) | 3.8052(6) | 3.7977(8), 3.7810(8) | 3.887(1) | 3.844(2) |
| Ln–O–Ln | 107.31(4) | 107.02(4), 107.56(5) | 107.85(9) | 108.28(4) | 107.59(2), 107.78(1) | 109.8(1) | 108.3(1) |
| Cpcent–Ln–Cpcent | 128.22 | 130.42, 130.5, 131.33, 132.92 | 127.67 | 131.38 | 129.69, 129.96, 130.13, 130.17 | 126.20 | 128.51 |
| Shortest C⋯Ca | 3.546(2) | 3.645(3) | 3.521(2) | 3.563(3) | 3.64(2) | 3.74(1) | 3.67(1) |
| sp2 C⋯C < 4.0 Åb | 4 | 17 | 4 | 10 | 17 | 2 | 18 |
All hydrogen signals of 2-Ln/R complexes were identifiable at RT using 1H NMR spectroscopy (Fig. 3). 2-Ln/R displayed effective D2h symmetry in solution, with the anticipated number of peaks present R = Et (5), iPr (5), C6H4-4-tBu (6). For the paramagnetic CeIII complexes, these resonances spanned a relatively large range covering ∼+20 to −70 ppm. Notably, methylene and methine hydrogens of 2-Ce/Et and 2-Ce/iPr, respectively, were observed at significantly negative shifts: OCH2Me, −43.74 and OCHMe2, −70.85 ppm. Other CeIII alkoxides have been reported along with their partial 1H NMR assignment, e.g., [(C5H4tBu)2CeIII(μ-OiPr)]231 [(tBuCHO)2CeIII(μ-OCHtBu)]2,42 [C5H3tBu2]2CeIII(OC6H11)(THF), however, this marks the first observation of CeIII alkoxide –CH resonances by 1H NMR spectroscopy.
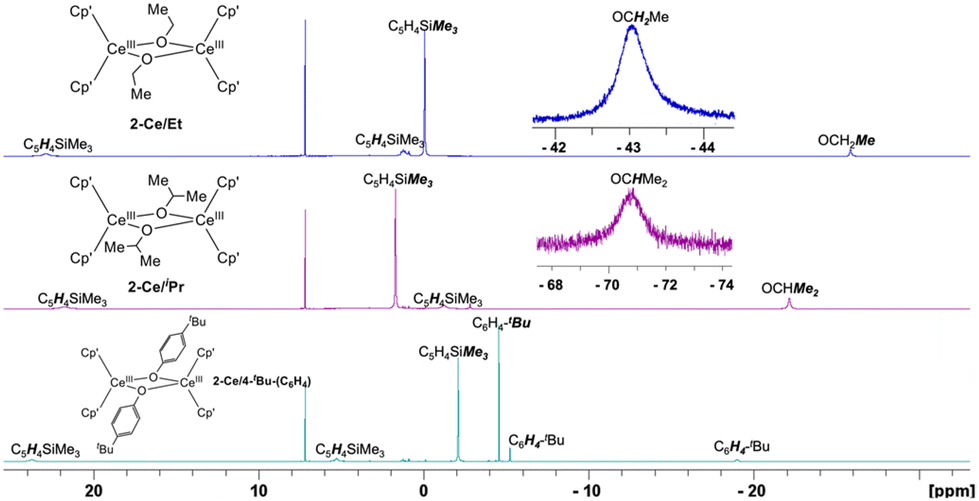 | ||
| Fig. 3 1H NMR 298 K spectra in C6D6 of 2-Ce/Et, 2-Ce/iPr, and 2-Ce/C6H4-tBu including insets of significantly upfield methylene and methine 1H signals of 2-Ce/Et and 2-Ce/iPr, respectively. | ||
Given the resolution and relatively long t1 relaxation time,43–451H DOSY NMR data was collected to establish whether the dimeric nature of 2-Ce/iPr in the solid-state was conserved in solution (Fig. 4). 2D 1H DOSY NMR experiments provided a diffusion coefficient used to calculate an effective molecular weight of 1072 g mol−1via the Stokes–Einstein Gierer–Wirtz Estimation (SEGWE) model46,47 and a hydrodynamic radius (rH) of 7.0 Å using the Stokes–Einstein equation.48,49 These were in good agreement with the expected molecular weight (948 g mol−1) and rH from the crystal structure (7.4 Å, Fig. S20†), in line with a dimeric structure of 2-Ce/iPr in C6D6. Furthermore, 1H NMR of 2-Ce/iPr in the presence of 2 equiv. of THF maintained an effective molecular weight of 1152 g mol−1 and rH of 7.4 Å, suggesting that the dimeric structure was also maintained in the presence of moderate-strength Lewis bases (Fig. S11 and S15†).
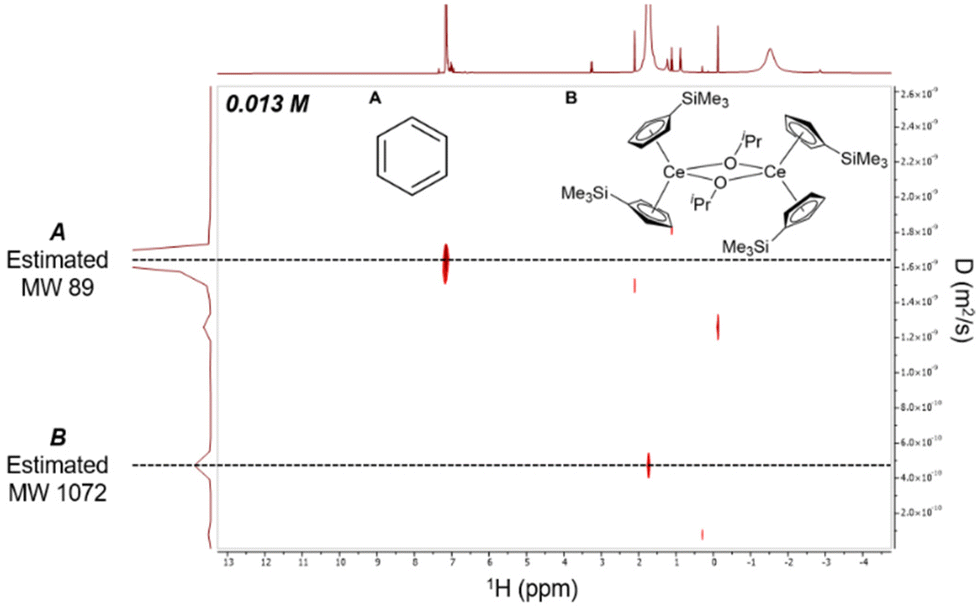 | ||
| Fig. 4 2D 1H DOSY NMR spectra of 0.013 M 2-Ce/iPr in C6D6 demonstrating preservation of dimer in solution. | ||
Electrochemistry of 2-Ln/iPr
With a comprehensive understanding of the solution-structures of 2-Ln/R, we evaluated the electrochemical behavior of 2-Ln/iPr using cyclic voltammetry (CV) and differential pulse voltammetry (DPV). CVs and DPVs of 2-La/iPr (∼2.5 mM) in THF using 100 mM [NBu4][PF6] collected from +0.5 to −3.5 V vs. Fc revealed two irreversible features. Starting at ∼–0.25 V, an irreversible reduction was observed around −3.3 V scanning cathodically, while an irreversible oxidation was observed with an onset of ∼0.2 V. These were tentatively assigned as an irreversible LaIII/II reduction and ligand oxidation, respectively. CVs and DPVs of 2-Ce/iPr revealed a similar irreversible reduction (Epc = –3.32 V), along with three irreversible oxidations (Epa = −0.20, −0.07, and +0.28 V). The oxidative event at +0.28 V generated a corresponding reductive feature at −1.25 V, which may be associated with the CeIII/IV couple (see ESI†). Notably, Evans and coworkers reported irreversible reductions for Cp′3LaIII and Cp′3Ce in THF around the same potential (−3.36 and −3.43 V vs. Fc),50 which suggest a relatively minor impact of ligand identity (Cp′ vs. OiPr) and nuclearity (monomer vs. dimer) on the electrochemical accessibility of the non-classical (4fn5d1) LaII and CeII ions. While evaluation of additional compounds would be needed to fully validate this observation, our results clearly indicate accessible reductive and oxidative events within the bimetallic 2-Ln/iPr complexes.Reduction of 2-La/iPr
Encouraged by the electrochemical behavior of 2-Ln/iPr, chemical reduction of 2-La/iPr was pursued. Addition of KC8 and 2.2.2-cryptand to THF solutions of 2-La/iPr at −78 °C immediately generated purple-colored mixtures. After removing graphite via filtration at −78 °C, a thermally-sensitive purple solution was obtained. The reduction product generated from 2-La/iPr + KC8 + 2.2.2-cryptand was significantly more sensitive than [Cp′3LaII]1− reported by Evans and co-workers, where the reduction of Cp′3LaIII was possible at room temperature.51 Upon warming to room temperature, the solution undergoes decomposition to an intractable yellow oil within 30 min. Attempts to remove solvent under reduced pressure or recrystallization at −35 °C also yielded intractable yellow oils. Alternatively, purple solutions of the reduction product were much more stable at −78 °C, where no decomposition was observed over the course of 4 h. The improved stability under these conditions enabled additional characterization by X-band EPR spectroscopy at 77 K and 298 K prior to decomposition (Fig. 5). At room temperature, the reduction product exhibited an isotropic, 8-line EPR spectrum [gave = 1.970 and Aave = 424 MHz (153 G)], due to clear hyperfine coupling with the 139La nucleus (I = 7/2). Alternatively, at 77 K, the reduction product exhibited an axial, 16-line EPR spectrum [g∥ = 2.005, g⊥ = 1.963; A∥ = 417 MHz (149 G), A⊥ = 431 MHz (157 G)]. Both spectra were consistent with the formation of a LaII species and were readily modelled using EasySpin.52 Although the A-value parameters are similar to [Cp′3La]1−, the g-values are distinct and only a single LaII species was observed (Table 2). These observations are inconsistent with ligand redistribution of 2-La/iPr + KC8 to form [Cp′3La]1−. Other examples of ligand redistribution after reduction of mixed ligand systems, however, have resulted in significant speciation.29,53 Additionally, UV-vis spectra of the reduction were collected (Fig. S17†) with absorption features located at 563, 859, and 938 nm. | ||
| Fig. 5 EasySpin 52 simulation and experimental CW X-band EPR spectra of the reduction of [Cp′2La(μ-OiPr)]2 (2-La/iPr) with KC8 and 2.2.2-cryptand dissolved in THF (∼5 mM) collected at 298 K (mode: perpendicular; gave = 1.970; Aave = 424 MHz (153 G); ν = 9.856 GHz) (top) and at 77 K (mode: perpendicular g∥ = 2.005, g⊥ = 1.963; A∥ = 417 MHz (149 G), A⊥ = 431 MHz (157 G); ν = 9.653 GHz) (bottom). | ||
| LaII species | T (K) | g | A (MHz) | A (G) | Ref. |
|---|---|---|---|---|---|
| a This work | |||||
| [Cp′3La]1− | 80 | g ∥ = 1.999 | A ∥ = 420 | A ∥ = 150 | 51 |
| g ⊥ = 1.956 | A ⊥ = 430 | A ⊥ = 157 | |||
| 295 | g ave = 1.994 | A ave = 430 | A ave = 154 | ||
| {[C5H3(SiMe3)2]3La}1− | 80 | g ∥ = 2.001 | A ∥ = 392 | A ∥ = 140 | 53–55 |
| g ⊥ = 1.950 | A ⊥ = 385 | A ⊥ = 141 | |||
| 298 | g ave = 1.97 | A ave = 366.2 | A ave = 133.7 | ||
| {[C5H4tBu]3La}1− | 77 | g ∥ = 1.995 | — | A ∥ = 197 | 56 |
| g ⊥ = 1.941 | A ⊥ = 208 | ||||
| 298 | g ave = 1.959 | — | A ave = 204 G | ||
| {[C5H3tBu2]3La}1− | 40 | g ∥ = 1.998 | A ∥ = 650 | A ∥ = 232 | 57 |
| g ⊥ = 1.934 | A ⊥ = 630 | A ⊥ = 233 | |||
| [(C5H4Me)3La]1− | 298 | g ave = 1.971 | — | A ave = 195 | 58 |
| [(C5Me4H)3La]1− | 298 | g ave = 1.970 | A ave = 802 | A ave = 291 | 59 |
| This work | 77 | g ∥ = 2.005 | A ∥ = 417 | A ∥ = 149 | |
| g ⊥ = 1.963 | A ⊥ = 431 | A ⊥ = 157 | |||
| 298 | g ave = 1.970 | A ave = 424 | A ave = 153 | ||
These absorbances are different than the reported [K(2.2.2-cryptand)][Cp′3LaII] of 554 nm.51 Taken together, the initial chemical reduction studies are consistent with the accessible reductive events observed by CV and DPV, and further investigation of the reductive and oxidative reactivity of 2-Ln/R and other bimetallic Ln are promising areas for future studies.
Conclusions
We have synthesized several new Ln metallocenes bridged by alkoxide or aryloxide via alcoholysis in good to excellent yields and characterized them by NMR and SC-XRD. X-ray crystal structures of 2-Ln/iPr and 2-Ce/Et reveal decreasing Ln⋯Ln distances, increasing Cpcent–Ln–Cpcent angles, and increasing intermolecular C⋯C contacts with bulkier bridging alkoxides. These suggest that significant dispersion forces mediated by the R groups (R = Et, iPr) could control the structure of the [Ln(μ-OR)]2 core. All hydrogens in 2-Ce/R complexes were identified using 1H NMR including methylene (R = Et) and methine (R = iPr) hydrogens which were observed as highly upfield signals. 2D 1H DOSY NMR confirmed that the dimeric structure of 2-Ce/iPr in the solid-state was maintained in solution, while electrochemical studies of 2-Ln/iPr suggested accessible reductive and oxidative events. Chemical reduction of 2-La/iPr using KC8 in the presence of 2.2.2-cryptand at −78 °C generated a purple solution and EPR spectroscopy supported the formation of a novel LaII species. Our studies indicate that heteroleptic cyclopentadienide/alkoxide systems readily support the formation of bimetallic complexes in the solid- and solution-state with accessible redox events. Further investigations of constructing bimetallic f element complexes containing non-covalent interactions and reduction chemistry to determine their ability to form mixed-valent and/or metal–metal bonds are ongoing.Experimental
General considerations
All synthesis techniques described below were conducted under nitrogen with exclusion of air using glovebox and Schlenk-line techniques. KC8,60 Ln[C5H4SiMe3]351 (Ln = La, Ce), and KC5H4SiMe361 were prepared using previously published procedures. Hexanes, toluene, THF, and Et2O were dried using a pure process technology solvent purification system and stored over activated 4 Å molecular sieves. EtOH and iPrOH were degassed with three freeze-pump-thaw cycles and stored over activated 4 Å molecular sieves. HOC6H4-4-tBu was degassed under vacuum overnight and prior to use. NMR solvent C6D6 was dried using NaK, degassed with three freeze-pump-thaw cycles, and vacuum-transferred prior to use. 1H, 13C NMR and Evans’ method data were obtained on a Bruker Avance III 300 MHz or Bruker AvanceHD 400 MHz spectrometer at 298 K (see ESI for spectra). UV-vis data were collected on a Shimadzu UV-3600 Plus using a sealed 5 mm quartz cuvette (see ESI spectra). Elemental analysis was data collected through the Center for Enabling New Technologies Through Catalysis (CENTC) at the University Rochester using a PerkinElmer 2400 Series II Analyzer.Synthesis of [Cp′2La(μ-OEt)]2 (2-La/Et)
In an N2-filled glovebox, EtOH (21 μL, 0.36 mmol) was added into a transparent Et2O (10 mL) solution of Cp′3La (1-La) (199 mg, 0.36 mmol), yielding an opaque, colorless mixture. The mixture was allowed to stir for 2 h, after which volatiles (HCp′ and Et2O) were removed in vacuo to yield a microcrystalline colorless solid (159 mg, 94%). 1H (400 MHz, C6D6, 25 °C, δ, ppm): 6.69 (t, 8H, C5H4SiMe3), 6.42 (t, 8H, C5H4SiMe3), 3.32 (q, 4H, OCH2CH3), 1.16 (t, 6H, OCH2CH3), 0.39 (s, 36H, C5H4SiMe3). 13C{1H} (100.6 MHz, C6D6, 25 °C, δ, ppm): 124.09 (C5H4SiMe3), 121.36 (C5H4SiMe3), 117.91 (C5H4SiMe3), 59.69 (OCH2Me), 21.13 (OCH2Me), 0.78 (C5H4SiMe3). Anal. calcd for C36H62O2Si4La2·OEt2: C, 48.47; H, 7.32. Found: C, 48.76; H, 7.19.Synthesis of [Cp′2La(μ-OiPr)]2 (2-La/iPr)
As described for 2-La/Et, iPrOH (36 μL, 0.45 mmol) was added to a colorless Et2O (10 mL) solution of Cp′3La (1-La) (251 mg, 0.45 mmol) and colorless solids were isolated (208 mg, 97%). Single crystal X-ray quality crystals were obtained from a concentrated Et2O solution at −35 °C. 1H (400 MHz, C6D6, 25 °C, δ, ppm): 6.73 (t, 8H, C5H4SiMe3), 6.40 (t, 8H, C5H4SiMe3), 3.13 (m, 2H, OCHMe2), 1.25 (d, 12H, OCHMe2), 0.43 (s, 36H, C5H4SiMe3). 13C{1H} (100.6 MHz, C6D6, 25 °C, δ, ppm): 123.23 (C5H4SiMe3), 122.02 (C5H4SiMe3), 117.59 (C5H4SiMe3), 64.70 (OCHMe2), 27.98 (OCHMe2), 1.08 (C5H4SiMe3). Anal. calcd for C38H66O2Si4La2: C, 48.29; H, 7.04. Found: C, 48.51; H, 7.20.Synthesis of [Cp′2La(μ-OC6H4-4-tBu)]2 (2-La/OC6H4-4-tBu)
In a N2-filled glovebox, Et2O (10 mL) was added to a combination of Cp′3La (212 mg, 0.38 mmol) and 4-tert-butylphenol (57 mg, 0.38 mmol), which yielded a colorless opaque solution seconds after stirring was initiated. The solution stirred for 24 h, after which the solvent was removed in vacuo to yield a colorless powder (191 mg, 88%). 1H (400 MHz, C6D6, 25 °C, δ, ppm): 7.25 (d, 4H, OC6H4-4-tBu), 6.93 (d, 4H, OC6H4-4-tBu), 6.73 (s, 8H, C5H4SiMe3), 6.65 (s, 8H, C5H4SiMe3), 1.36 (s, 18H, OC6H4-4-tBu). 0.101 (s, 36H, C5H4SiMe3). 13C{1H} (100.6 MHz, C6D6, 25 °C, δ, ppm): 153.69 (OC6H4-4-tBu), 144.47 (OC6H4-4-tBu), 128.11 (OC6H4-4-tBu), 124.89 (C5H4SiMe3), 123.34 (C5H4SiMe3), 119.50 (OC6H4-4-tBu), 118.60 (C5H4SiMe3), 34.29 (OC6H4-4-tBu), 31.66 (OC6H4-4-tBu), 0.20 (C5H4SiMe3). Anal. calcd for C52H78O2Si4La2: C, 55.50; H, 6.99. Found: C, 55.61; H, 6.91.Synthesis of [Cp′2Ce(μ-OEt)]2 (2-Ce/Et)
In an N2-filled glovebox, EtOH (16 μL, 0.26 mmol) was syringed into a royal blue Et2O (10 mL) solution of Cp′3Ce (146 mg, 0.26 mmol), immediately yielding a transparent, golden-yellow solution. The solution was stirred for 30 min and volatiles (HCp′ and Et2O) were removed in vacuo to yield a golden-yellow solid (133.8 mg, 72%). Single crystal X-ray quality yellow crystals were obtained after storing a concentrated hexanes solution at −35 °C in a freezer overnight. 1H (400 MHz, C6D6, 25 °C, δ, ppm): 23.12 (s, 8H, C5H4SiMe3), 0.97 (s, 8H, C5H4SiMe3), −0.05 (s, 36H, C5H4SiMe3), −26.24 (s, 6H, OCH2CH3), −43.74 (s, 4H, OCH2CH3). Evans method: 1.28μB. UV-vis (THF) λmax, nm (ε, M−1 cm−1): 458 (555). Anal. calcd for C36H62O2Si4Ce2·0.5OEt2: C, 47.72; H, 7.06. Found: C, 47.67; H, 6.71.Synthesis of [Cp′2Ce(μ-OiPr)]2 (2-Ce/iPr)
As described for 2-Ce/Et, iPrOH (48 μL, 0.63 mmol) was added into a royal blue Et2O (10 mL) solution of Cp′3Ce (346 mg, 0.63 mmol) and a bright yellow solid was isolated (274.3 mg, 92%). Single crystal X-ray quality crystals were obtained from a concentrated hexanes solution after storing in a −35 °C freezer overnight. 1H (300 MHz, C6D6, 25 °C, δ, ppm): 21.75 (s, 8H, C5H4SiMe3), 1.69 (s, 36H, C5H4SiMe3), −1.25 (s, 8H, C5H4SiMe3), −22.16 (s, 12H, OCHMe2), −70.85 (s, 2H, OCHMe2). Evans method: 1.90μB. UV-vis (THF) λmax, nm (ε, M−1 cm−1): 452 (650). Anal. calcd for C38H66O2Si4Ce2: C, 48.17; H, 7.02. Found: C, 48.49; H, 6.94.Synthesis of [Cp′2Ce(μ-OC6H4-4-tBu)]2 (2-Ce/OC6H4-4-tBu)
In an N2-filled glovebox, a diethyl ether (5 mL) solution of HOC6H4-4-tBu (14 mg, 0.091 mmol) was added to a royal blue Et2O (5 mL) solution of Cp′3Ce (50 mg, 0.091 mmol), yielding a dark yellow solution after 15 seconds of stirring. The solution was allowed to stir for 30 minutes, after which the product was dried in vacuo to yield a dark yellow solid (80.7 mg, 85%). 1H (300 MHz, C6D6, 25 °C, δ, ppm): 23.73 (s, 8H, C5H4SiMe3), 5.26 (s, 8H, C5H4SiMe3), −2.10 (s, 36H, C5H4SiMe3), −4.57 (s, 18H, OC6H4-4-tBu), −5.23 (s, 4H, OC6H4-4-tBu), −18.98 (s, 4H, OC6H4-4-tBu). Evans method: 1.34μB. UV-vis (THF) λmax, nm (ε, M−1 cm−1): 471 (731). Anal. calcd for C52H78O2Si4Ce2: C, 55.38; H, 6.97. Found: C, 55.57; H, 6.94.Reduction of 2-La/iPr
In a N2-filled glovebox, THF (10 mL) solution of 2-La/iPr (50 mg, 0.053 mmol) and a flask containing KC8 (8 mg, 0.059 mmol) and 2.2.2-cryptand (22 mg, 0.059 mmol) were chilled in a −78 °C coldwell for 1 h. The 2-La/iPr THF solution was added to the KC8 and 2.2.2-cryptand mixture and swirled vigorously for 5 min. A purple-solution with graphite immediately formed. While in the coldwell, the purple mixture was filtered through a dry Celite plug to remove graphite yielding a purple solution. UV-vis (THF) λmax, nm: 563, 859, 938.X-ray crystallography
Samples were removed from their mother liquor in an inert-atmosphere glovebox, covered in Paraton™ oil in a separate 20 mL vial, sealed with electrical tape and stored within a sealed jar, and transported to Brown University. These samples were transferred to a glass slide where it was evaluated and mounted with the assistance of an optical microscope. X-ray reflection intensity data were collected on a Bruker D8 Venture with a Photon III CPAD detector employing a IμS 3.0 Mo-Kα radiation source (λ = 0.71073 Å) at a temperature of 173(1) K. Rotation frames were integrated using SAINT,62 producing a listing of unaveraged F2 and σ(F2) values which were then passed to the SHELXT63 program package for further processing and structure solution. The intensity data were corrected for Lorentz and polarization effects and for absorption using SADABS.64 The structures were solved by using SHELXT,63 using Olex2 as the graphical interface.65 Refinement was by full-matrix least squares based on F2 using SHELXL.66 All reflections were used during refinements. Non-hydrogen atoms were refined anisotropically and hydrogen atoms were refined using a riding model. Disorders were refined with the help of similarity restraints using standard/default values on 1,2 and 1,3 distances (SADI) and rigid bond restraints (RIGU) of the disordered groups.67,68EPR spectroscopy
EPR spectra were collected on a Bruker EMX Premium-X spectrometer with a field strength of 9.65 GHz and a microwave power of 2.0 mW at 77 K using a liquid-nitrogen finger dewar (Wilmad, 50 mL Suprasil). Sample solutions (∼5 mM) were prepared as nitrogen-saturated, toluene solutions in 4 mm o.d. quartz EPR tubes. Samples were glassed by slowly lowering the sample into liquid nitrogen (∼2 mm s−1). The experimental spectra were simulated using EasySpin.52 In all cases, a representative fit was achieved through the use of the Nelder/Mead simplex model algorithm while assuming isotropic line broadening.Electrochemistry
All electrochemistry experiments were conducted using a CH Instruments (CHI) 700E series potentiostat and performed under inert atmosphere in a nitrogen-filled glovebox outfitted with electrical leads (KF-40 port). The electrochemical cells consisted of 4 mL vials using THF as solvent with 100 mM [NBu4][PF6] as the electrolyte, 1–5 mM analyte (2-Ce/iPr or 2-La/iPr), a glassy carbon electrode (3 mm, CHI) as the working electrode, a platinum wire as the counter electrode, and a Ag/Ag+ non-aqueous electrode (CHI, THF, 100 mM [NBu4][PF6],10 mM AgOTf) as the reference electrode. The working electrode was polished to a shiny-mirror like finish with 0.05 micron micropolish powder (CH Instruments) and washed with water and acetone outside of the glovebox, brought inside the glovebox, and inserted into the electrochemical cell. The working electrode was replaced periodically upon scanning irreversible oxidation or reduction features to prevent fouling of the electrode surface. All data were collected in a positive-feedback IR compensation mode; cell resistances measured with THF as a solvent were 150–250 Ω. Differential pulse voltammetry experiments (DPV) were performed using the same electrochemical cell and electrodes, as well as the same IR compensation procedure. All DPVs were collected at 10 mV s−1, with a 30 s quiet time before each scan.Author contributions
Adrian N. Brown: conceptualization, investigation, formal analysis, writing – original draft, writing – review & editing. Jack N. Kelleher: investigation, formal analysis, writing – original draft, writing – review & editing. Alexander M. Brown: investigation, formal analysis. Peter Saghy: investigation, formal analysis. Joshua J. Bohl: investigation. Jerome R. Robinson: investigation, formal analysis, writing – original draft, writing – review & editing. Daniel N. Huh: conceptualization, writing – review & editing, supervision, funding acquisition.Data availability
Crystallographic data for 2-Ce/Et, 2-La/iPr, and 2-Ce/iPr have been deposited at the CCDC under 2372725, 2372726, and 2372727, respectively and can be obtained from ccdc.cam.ac.uk/structures.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research was provided by the University of Rhode Island (DNH: start-up funds) and the National Science Foundation (EPR and electrochemistry accessories purchased from CHE-1900248). Instrumentation for the University of Rhode Island Chemistry 400 MHz AvanceHD was supported from a grant through the National Science Foundation (CHE-1531963). X-ray diffraction experiments were performed with a diffractometer purchased through a grant through the NSF-MRI program (CHE-2117549) located at Brown University. Elemental analysis data was collected by the CENTC at the University of Rochester (CHE-0650456).References
- J. Qin, B. Xu, Y. Zhang, D. Yuan and Y. Yao, Cooperative rare earth metal–zinc based heterometallic catalysts for copolymerization of CO2 and cyclohexene oxide, Green Chem., 2016, 18, 4270–4275 RSC.
- Q. Yao, Y. Wang, B. Zhao, X. Zhu, Y. Luo, D. Yuan and Y. Yao, Syntheses of Heterometallic Neodymium–Zinc Complexes and Their Performance in the Copolymerization of CO2 and Cyclohexene Oxide, Inorg. Chem., 2022, 61, 10373–10382 CrossRef CAS.
- K. Yin, L. Hua, L. Qu, Q. Yao, Y. Wang, D. Yuan, H. You and Y. Yao, Heterobimetallic rare earth metal–zinc catalysts for reactions of epoxides and CO2 under ambient conditions, Dalton Trans., 2021, 50, 1453–1464 RSC.
- F. Delano IV, F. Benner, S. Jang and S. Demir, Pyrrolyl-Bridged Metallocene Complexes: From Synthesis, Electronic Structure, to Single-Molecule Magnetism, Inorg. Chem., 2023, 62, 14604–14614 CrossRef CAS.
- C. A. Gould, E. Mu, V. Vieru, L. E. Darago, K. Chakarawet, M. I. Gonzalez, S. Demir and J. R. Long, Substituent Effects on Exchange Coupling and Magnetic Relaxation in 2,2′-Bipyrimidine Radical-Bridged Dilanthanide Complexes, J. Am. Chem. Soc., 2020, 142, 21197–21209 CrossRef CAS.
- F. Benner and S. Demir, From unprecedented 2,2′-bisimidazole-bridged rare earth organometallics to magnetic hysteresis in the dysprosium congener, Inorg. Chem. Front., 2023, 10, 4981–4992 RSC.
- F. Benner, L. La Droitte, O. Cador, B. Le Guennic and S. Demir, Magnetic hysteresis and large coercivity in bisbenzimidazole radical-bridged dilanthanide complexes, Chem. Sci., 2023, 14, 5577–5592 RSC.
- P. Zhang, R. Nabi, J. K. Staab, N. F. Chilton and S. Demir, Taming Super-Reduced Bi23− Radicals with Rare Earth Cations, J. Am. Chem. Soc., 2023, 145, 9152–9163 CrossRef CAS.
- F. Benner and S. Demir, Isolation of the elusive bisbenzimidazole Bbim3−˙ radical anion and its employment in a metal complex, Chem. Sci., 2022, 13, 5818–5829 RSC.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Ultrahard magnetism from mixed-valence dilanthanide complexes with metal-metal bonding, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- H. Kwon, K. R. McClain, J. G. C. Kragskow, J. K. Staab, M. Ozerov, K. R. Meihaus, B. G. Harvey, E. S. Choi, N. F. Chilton and J. R. Long, Coercive Fields Exceeding 30 T in the Mixed-Valence Single-Molecule Magnet (CpiPr5)2Ho2I3, J. Am. Chem. Soc., 2024, 146, 18714–18721 CrossRef CAS PubMed.
- C. Uhlmann, L. Münzfeld, A. Hauser, T. Ruan, S. K. Kuppusamy, C. Jin, M. Ruben, K. Fink, E. Moreno-Pineda and P. W. Roesky, Unique Double and Triple Decker Arrangements of Rare-Earth 9,10-Diborataanthracene Complexes Featuring Single-Molecule Magnet Characteristics, Angew. Chem., Int. Ed., 2024, 63, e202401372 CrossRef CAS PubMed.
- J. Long, F. Habib, P. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, Single-Molecule Magnet Behavior for an Antiferromagnetically Superexchange-Coupled Dinuclear Dysprosium(III) Complex, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed.
- P. Lin, T. Burchell, R. Clérac and M. Murugesu, Dinuclear Dysprosium(III) Single-Molecule Magnets with a Large Anisotropic Barrier, Angew. Chem., Int. Ed., 2008, 47, 8848–8851 CrossRef CAS.
- J. C. Wedal, L. Anderson-Sanchez, M. T. Dumas, C. A. Gould, M. J. Beltrán-Leiva, C. Celis-Barros, D. Páez-Hernández, J. W. Ziller, J. R. Long and W. J. Evans, Synthesis and Crystallographic Characterization of a Reduced Bimetallic Yttrium ansa-Metallocene Hydride Complex, [K(crypt)][(μ-CpAn)Y(μ-H)]2 (CpAn = Me2Si[C5H3(SiMe3)-3]2), with a 3.4 Å Yttrium–Yttrium Distance, J. Am. Chem. Soc., 2023, 145, 10730–10742 CrossRef CAS PubMed.
- M. D. Roy, T. P. Gompa, S. M. Greer, N. Jiang, L. S. Nassar, A. Steiner, J. Bacsa, B. W. Stein and H. S. La Pierre, Intervalence, Charge Transfer in Nonbonding, Mixed-Valence, Homobimetallic Ytterbium Complexes, J. Am. Chem. Soc., 2024, 146, 5560–5568 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Molecular magnetic hysteresis at 60 kelvin in dysprosocenium, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II), J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS PubMed.
- K. R. McClain, C. A. Gould, D. A. Marchiori, H. Kwon, T. T. Nguyen, K. E. Rosenkoetter, D. Kuzmina, F. Tuna, R. D. Britt, J. R. Long and B. G. Harvey, Divalent Lanthanide Metallocene Complexes with a Linear Coordination Geometry and Pronounced 6s–5d Orbital Mixing, J. Am. Chem. Soc., 2022, 144, 22193–22201 CrossRef CAS PubMed.
- A. Ariciu, D. H. Woen, D. N. Huh, L. E. Nodaraki, A. K. Kostopoulos, C. A. P. Goodwin, N. F. Chilton, E. J. L. McInnes, R. E. P. Winpenny, W. J. Evans and F. Tuna, Engineering electronic structure to prolong relaxation times in molecular qubits by minimising orbital angular momentum, Nat. Commun., 2019, 10, 3330 CrossRef PubMed.
- L. E. Nodaraki, A. M. Ariciu and D. N. Huh, Ligand Effects on the Spin Relaxation Dynamics and Coherent Manipulation of Organometallic La(II), Potential Qudits, J. Am. Chem. Soc., 2024, 146, 15000–15009 CrossRef CAS PubMed.
- T. Chowdhury, F. Murphy, A. R. Kennedy, C. Wilson, J. H. Farnaby and C. E. Weetman, Synthesis and Reactivity of Bis-tris(pyrazolyl)borate Lanthanide/Aluminum Heterobimetallic Trihydride Complexes, Inorg. Chem., 2024, 63, 9390–9394 CrossRef CAS PubMed.
- J. R. Hickson, S. J. Horsewill, C. Bamforth, J. McGuire, C. Wilson, S. Sproules and J. H. Farnaby, The modular synthesis of rare earth-transition metal heterobimetallic complexes utilizing a redox-active ligand, Dalton Trans., 2018, 47, 10692–10701 RSC.
- M. Bonath, D. Schädle, C. Maichle-Mössmer and R. Anwander, The Alkylaluminate/Gallate Trap: Metalation of Benzene by Heterobimetallic Yttrocene Complexes [Cp*2Y(MMe3R)] (M = Al, Ga), Inorg. Chem., 2021, 60, 14952–14968 CrossRef CAS PubMed.
- E. C. Moinet, P. Wetzel, O. Tardif, C. Maichle-Mössmer and R. Anwander, Ancillary Ligand-Free Mixed Chlorido/Isobutylaluminato Lanthanide Complexes in Isoprene Polymerization, Organometallics, 2024, 43, 611–616 CrossRef CAS.
- H. Tsurugi, Y. Ikeda, K. Shinohara, S. Shirase, N. Toya, S. Tanaka and K. Mashima, Synthesis and Characterization of Alkoxide-Bridged Heterometallic Clusters of Cerium and Copper, Inorg. Chem., 2019, 58, 12565–12572 CrossRef PubMed.
- J. P. Karnes, A. Kumar, J. A. Hopkins Leseberg, V. W. Day and J. D. Blakemore, Trivalent Cations Slow Electron Transfer to Macrocyclic Heterobimetallic Complexes, Inorg. Chem., 2024, 63, 8710–8729 CrossRef PubMed.
- Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, Activation of a C-O bond by reaction of a tris(cyclopentadienyl)lanthanide complex with an alkali metal in dimethoxyethane (DME); crystal structures of [Ndη-C5H3(SiMe3)2-1,32(μ-OMe)2Li(DME)] and [{Ce(η-C5H3tBu2-1,3)2(μ-OMe)2], J. Organomet. Chem., 1995, 499, 213–219 CrossRef.
- M. C. Cassani, M. F. Lappert and F. Laschi, First identification by EPR spectra of lanthanum(II) organometallic intermediates (and E½ for La3+ → La2+) in the C–O bond activation of dimethoxyethane, Chem. Commun., 1997, 1563–1564 RSC.
- A. V. Khvostov, V. K. Belsky, B. M. Bulychev, A. I. Sizov and B. B. Ustinov, ansa-Ytterbocenes(+3) with a short bridge and bulky substituents: synthesis and crystal structure of meso-(CH3)2Si[3-(CH3)3SiC5H3]2YbCl(THF), rac-(CH3)2C[3-tBuC5H3]2Yb(μ2-Cl)2Li(OEt2)2, and [meso-(CH3)2C[3-tBuC5H3]2Yb(μ2-OCH3)]2, J. Organomet. Chem., 1998, 571, 243–249 CrossRef CAS.
- S. D. Stults, R. A. Andersen and A. Zalkin, Chemistry of trivalent cerium and uranium metallocenes: reactions with alcohols and thiols, Organometallics, 1990, 9, 1623–1629 CrossRef CAS.
- C. T. Palumbo, L. E. Darago, C. J. Windorff, J. W. Ziller and W. J. Evans, Trimethylsilyl versus Bis(trimethylsilyl) Substitution in Tris(cyclopentadienyl) Complexes of La, Ce, and Pr: Comparison of Structure, Magnetic Properties, and Reactivity, Organometallics, 2018, 37, 900–905 CrossRef CAS.
- Z. Wu, Z. Xu, X. You, X. Zhou and Z. Jin, Synthesis and characterization of binuclear organolanthanide complexes involving cyclopentadienyl and n-propyloxide ligands. X-ray crystal structure of [(C5H5)2Yb(μ-OCH2CH2CH3)]2, Polyhedron, 1992, 11, 2673–2678 CrossRef CAS.
- Z. Wu, Z. Huang, R. Cai, Z. Xu, X. You and X. Huang, Synthesis and characterization of dinuclear organolanthanide alkoxides, X-ray crystal structures of [(C5H5)2Yb(μ-OCH2CH2CH2CH2CH3)]2 and [(C5H5)Yb(μ-OCH2CH2CH(Me)2]2, Polyhedron, 1996, 15, 13–22 CrossRef CAS.
- W. Zhongzhi, X. Zheng, Y. Xiaozeng, Z. Xigeng and H. Xiaoying, Dehydrogenation of organolanthanide alkoxides and X-ray crystal structures of the reaction product [{(C5H5)2Ln(μ-O(Me)C-CHCH3)}2] (Ln = Dy Yb), J. Organomet. Chem., 1994, 483, 107–113 CrossRef.
- E. Osawa, Y. Onuki and K. Mislow, The central bond length in hexaphenylethane and hexakis(2,6-di-tert-butyl-4-biphenylyl)ethane, J. Am. Chem. Soc., 1981, 103, 7475–7479 CrossRef CAS.
- W. D. Hounshell, D. A. Dougherty, J. P. Hummel and K. Mislow, Structure of hexaphenylethane and congeners as determined by empirical force field calculations, J. Am. Chem. Soc., 1977, 99, 1916–1924 CrossRef CAS.
- S. Grimme and P. R. Schreiner, Steric Crowding Can Stabilize a Labile Molecule: Solving the Hexaphenylethane Riddle, Angew. Chem., Int. Ed., 2011, 50, 12639–12642 CrossRef CAS PubMed.
- D. Liptrot and P. Power, London dispersion forces in sterically crowded inorganic and organometallic molecules, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- S. Pagano and W. Schnick, Di-μ-tert-butanolato-bis[bis(η5-cyclopentadienyl)erbium(III)], Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m473 CrossRef CAS PubMed.
- Y. Luo, Y. Yao, Q. Shen and Y. Xing, Synthesis and Crystal Structure of Bis(tert-butyl-cyclopentadienyl) Erbium Ethoxide,(t-BuCp)2ErOEt, J. Rare Earths, 2002, 20, 374 Search PubMed.
- H. A. Stecher, A. Sen and A. L. Rheingold, Synthesis, structure, and reactivity of cerium(III) alkoxides. 2. Thermal decomposition of Ce(OC-tert-Bu3)3 and the structure of [Ce(OCH-tert-Bu2)3]2, Inorg. Chem., 1989, 28, 3280–3282 CrossRef CAS.
- M. P. Crockett, H. Zhang, C. M. Thomas and J. A. Byers, Adding diffusion ordered NMR spectroscopy (DOSY) to the arsenal for characterizing paramagnetic complexes, Chem. Commun., 2019, 55, 14426–14429 RSC.
- J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, Tuning Reactivity and Electronic Properties through Ligand Reorganization within a Cerium Heterobimetallic Framework, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS.
- J. R. Robinson, Y. Qiao, J. Gu, P. J. Carroll, P. J. Walsh and E. J. Schelter, The role of dynamic ligand exchange in the oxidation chemistry of cerium(III), Chem. Sci., 2016, 7, 4537–4547 RSC.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Improving the Interpretation of Small Molecule Diffusion Coefficients, Anal. Chem., 2018, 90, 3987–3994 CrossRef CAS PubMed.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Quantitative Interpretation of Diffusion-Ordered NMR Spectra: Can We Rationalize Small Molecule Diffusion Coefficients?, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
- E. J. Cabrita and S. Berger, DOSY studies of hydrogen bond association: tetramethylsilane as a reference compound for diffusion studies, Magn. Reson. Chem., 2001, 39, S142–S148 CrossRef CAS.
- D. Li, G. Kagan, R. Hopson and P. G. Williard, Formula Weight Prediction by Internal Reference Diffusion-Ordered NMR Spectroscopy (DOSY), J. Am. Chem. Soc., 2009, 131, 5627–5634 CrossRef CAS PubMed.
- M. T. Trinh, J. C. Wedal and W. J. Evans, Evaluating electrochemical accessibility of 4fn5d1 and 4fn+1 Ln(ii) ions in (C5H4SiMe3)3Ln and (C5Me4H)3Ln complexes, Dalton Trans., 2021, 50, 14384–14389 RSC.
- M. E. Fieser, M. R. MacDonald, B. T. Krull, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, Structural, Spectroscopic, and Theoretical Comparison of Traditional vs Recently Discovered Ln2+ Ions in the [K(2.2.2-cryptand)][(C5H4SiMe3)3Ln] Complexes: The Variable Nature of Dy2+ and Nd2+, J. Am. Chem. Soc., 2015, 137, 369–382 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- S. A. Moehring and W. J. Evans, Evaluating Electron-Transfer Reactivity of Complexes of Actinides in +2 and +3 Oxidation States by using EPR Spectroscopy, Chem. – Eur. J., 2020, 26, 1530–1534 CrossRef CAS PubMed.
- D. N. Huh, J. W. Ziller and W. J. Evans, Chelate-Free Synthesis of the U(II) Complex, [(C5H3(SiMe3)2)3U]1−, Using Li and Cs Reductants and Comparative Studies of La(II) and Ce(II) Analogs, Inorg. Chem., 2018, 57, 11809–11814 CrossRef CAS PubMed.
- P. Hitchcock, M. Lappert, L. Maron and A. Protchenko, Lanthanum Does Form Stable Molecular Compounds in the +2 Oxidation State, Angew. Chem., Int. Ed., 2008, 47, 1488–1491 CrossRef CAS PubMed.
- M. A. Angadol, D. H. Woen, C. J. Windorff, J. W. Ziller and W. J. Evans, tert-Butyl(cyclopentadienyl) Ligands Will Stabilize Nontraditional +2 Rare-Earth Metal Ions, Organometallics, 2019, 38, 1151–1158 CrossRef CAS.
- J. Liu, L. E. Nodaraki, D. O. T. A. Martins, M. J. Giansiracusa, P. J. Cobb, J. Emerson-King, F. Ortu, G. F. S. Whitehead, G. K. Gransbury, E. J. L. McInnes, F. Tuna and D. P. Mills, Non-classical early lanthanide(II) di-tert- butylcyclopentadienyl complexes, Eur. J. Inorg. Chem., 2023, 26, e202300552 CrossRef CAS.
- D. H. Woen, D. N. Huh, J. W. Ziller and W. J. Evans, Reactivity of Ln(II) Complexes Supported by (C5H4Me)1− Ligands with THF and PhSiH3: Isolation of Ring-Opened, Bridging Alkoxyalkyl, Hydride, and Silyl Products, Organometallics, 2018, 37, 3055–3063 CrossRef CAS.
- T. F. Jenkins, D. H. Woen, L. N. Mohanam, J. W. Ziller, F. Furche and W. J. Evans, Tetramethylcyclopentadienyl Ligands Allow Isolation of Ln(II) Ions across the Lanthanide Series in [K(2.2.2-cryptand)][(C5Me4H)3Ln] Complexes, Organometallics, 2018, 37, 3863–3873 CrossRef CAS.
- D. E. Bergbreiter and J. M. Killough, Reactions of potassium-graphite, J. Am. Chem. Soc., 1978, 100, 2126–2134 CrossRef CAS.
- J. K. Peterson, M. R. MacDonald, J. W. Ziller and W. J. Evans, Synthetic Aspects of (C5H4SiMe3)3Ln Rare-Earth Chemistry: Formation of (C5H4SiMe3)3Lu via [(C5H4SiMe3)2Ln]+ Metallocene Precursors, Organometallics, 2013, 32, 2625–2631 CrossRef CAS.
- Bruker SAINT, v8.37a, Bruker AXS Inc., Madison, Wisconsin, 2012 Search PubMed.
- G. M. Sheldrick, SHELXT–Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- Bruker SADABS, v2014/5, Bruker AXS Inc., Madison, Wisconsin, 2001 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. Muller, R. Herbst-Irmer, A. Spek, T. Schneider and M. Sawaya, Crystal Structure Refinement: A Crystallographer's Guide to SHELXL, OUP, Oxford, 2006, vol. 8 Search PubMed.
- A. Thorn, B. Dittrich and G. M. Sheldrick, Enhanced rigid-bond restraints, Acta Crystallogr., Sect. A: Found. Crystallogr., 2012, 68, 448–451 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2372725–2372727. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02137a |
| This journal is © The Royal Society of Chemistry 2024 |