
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroanalytical characterization of Np(VI)/Np(V) redox in a pentadentate ligand environment and stabilization of [NpVO2]+ by hydrogen bonding†
Emily R.
Mikeska
 ab,
T. Davis
Curry
a,
Richard E.
Wilson
*b and
James D.
Blakemore
*a
ab,
T. Davis
Curry
a,
Richard E.
Wilson
*b and
James D.
Blakemore
*a
aDepartment of Chemistry, University of Kansas, 1567 Irving Hill Road, Lawrence, KS 66045, USA. E-mail: blakemore@ku.edu
bChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: rewilson@anl.gov
First published on 28th October 2024
Abstract
The redox chemistry of the actinyl cations (AnO2n+) heavily influences their reactivity and speciation in solution, but the redox properties of the actinyls in non-aqueous media have received far less attention than they deserve. Here, the non-aqueous electrochemistry of a chemically reversible Np(VI)/Np(V) redox manifold is reported in both protic (CH3OH) and aprotic (CH3CN) organic media. Using a neutral Np(VI) complex supported by a chelating and strongly donating pentadentate ligand that was fully characterized in prior work, a clean Np(VI)/Np(V) redox couple was found to be accessible under ambient conditions. Coupled electrochemical and spectroscopic studies, as well as simulations of cyclic voltammetry data, confirm the 1e− nature of this couple and establish it to be chemically reversible and nearly electrochemically reversible as well. Bulk electrolysis of a solution of the neutral Np(VI) complex facilitated isolation of the corresponding anionic and monomeric Np(V) species. Data from X-ray diffraction analysis as well as optical and vibrational spectroscopies provide strong evidence in support of metal-centered reduction and the Np(V) oxidation state, findings that are in accord with the measured reduction potentials. Distinctive hydrogen bonding interactions between the terminal (yl) oxo groups and water molecules appear to stabilize the isolated Np(V) species in the solid state, providing insight into the features that afford the uncommon chemically reversible redox encountered in this system.
1 Introduction
Speciation of actinide elements is primarily dictated by oxidation state.1–5 In the high valent +VI and +V states under aerobic conditions, trans-dioxo ligands are typically encountered for uranium, neptunium, and plutonium with a more labile equatorial belt, giving rise to overall coordination numbers between 6–8.6 Lower valent species can adopt coordination numbers greater than 8, making control of speciation and speciation-dependent properties a challenge in the field of f-element chemistry due to the significant number of degrees of freedom associated with high coordination numbers.7,8 While a few of the lanthanides have accessible redox processes under ambient conditions, the early actinides are unique in that they feature several accessible oxidation states and these can often exist simultaneously in a single solution.2,5,9,10 Under a given set of conditions in solution, the various oxidation states of the actinides have been established to equilibrate with each other by way of ligand exchange/displacement, disproportionation reactions, or formation of cation–cation interactions (CCIs) that represent formation of oxo-bridged species via the terminal (yl) oxo ligands as first reported by Sullivan.6,11–13 This nomenclature, coined by Sullivan, describes the interaction of two overall cationic species where at least one species is an actinyl of the form AnO2n+ (n = 1 or 2) in which the terminal oxo moiety of one actinyl species acts as ligand to another cationic species, with either a linear or T-shaped geometry, or by formation of a diamond core motif involving two actinyl species. Though more recent work has distinguished between interaction pairs containing two actinyls ("actinyl–actinyl interactions”)14–16versus other pairs, the interaction is, in the simplest terms, a Lewis acid–base interaction between a Lewis acidic metal center and the Lewis basic oxo ligands of the actinyl moiety. The generality of the “cation–cation interaction” nomenclature was also demonstrated by Sullivan in reports of interactions between actinyl species and other non-actinide ions.17–19 Given the correlation between oxidation state and solution speciation, a fundamental understanding of the redox properties of the f-elements in both aqueous and non-aqueous environments can be concluded to be critical for controlling their chemistry.On the one hand, the redox properties of the transuranic actinyl ions in both acidic and basic aqueous media have been investigated extensively in prior work; their standard reduction potentials were tabulated decades ago.2,9,20Fig. 1 shows the Latimer diagram for neptunium 1.0 M in HClO4; this diagram represents a summary of the reduction potentials corresponding to the redox interconversion between this element's various observable oxidation states. For example, Np displays a (VI/V) redox couple at E° = +1.14 V vs. the standard hydrogen electrode (SHE) in 1 M acid.9 The Np(VI/V) couple has been demonstrated to be a reversible 1e− couple at low concentrations of neptunium over a range of acid concentrations for nitric, perchloric, and sulfuric acids, but irreversible in phosphoric acid.9,21
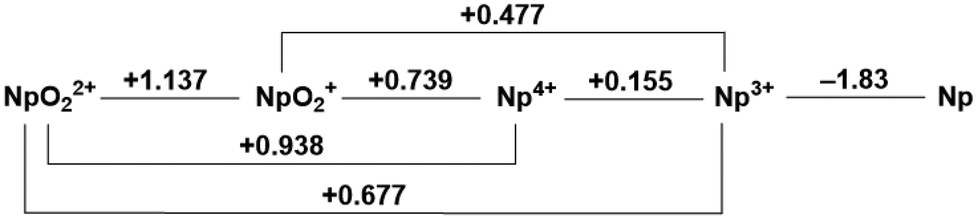 | ||
| Fig. 1 Latimer diagram of neptunium in 1.0 M HClO4.9 | ||
On the other hand, only a very limited number of reports on the redox properties of transuranic actinyls in non-aqueous media are available. An early report can be found in a PhD thesis that details the electrochemical properties of a putative Np(V) perchlorate in acetonitrile media.22 However, as the Np(V) material studied in this work was not fully characterized, An(VI/V) redox cycling at the electrode surface could not be rigorously established. More recently, the redox behavior of a Np(VI) coordination complex was reported in dimethylsulfoxide-based electrolyte,23 a NpV/IV couple was studied in the case of NpCl62− dissolved in room-temperature ionic liquids,24 and a NpV/IV reduction potential was measured in another non-actinyl system.25 Of these examples, none fully investigated the reversibility of the An(VI)/An(V) redox from both the chemical and electrochemical perspectives or provided full characterization of both the reduced and oxidized forms. Pursuit of information on these features underpinning the redox chemistry of neptunium in particular could disambiguate intrinsic features of the redox from secondary chemical reactions, particularly given the likelihood of ligand displacement following electron transfer to/from the actinyl species in the noted prior work. As the ligands used in the prior work with the neptunyl ion were not able to coordinatively saturate the neptunium metal center, there remains a significant possibility of follow-up reactivity.
Recently, we reported the neptunyl(VI) and plutonyl(VI) complexes of a pentadentate Schiff-base-type ligand26 for which the uranyl(VI) analogue was already known (see Fig. 2).27–29 All of these complexes display attractive solubility properties in the non-aqueous solvent acetonitrile (CH3CN); the uranium complex also displays a chemically reversible U(VI/V) redox couple in CH3CN-based electrolyte under inert atmosphere, a finding in accord with the high-yielding chemical synthesis of the corresponding uranyl(V) complex from the U(VI) species under air-free conditions.29 The clean chemical and electrochemical redox behavior of the U(VI)/U(V) manifold can be attributed in part to the dianionic and pentadentate chelating nature of the Schiff-base-type ligand supporting the actinyl core in this system; the ligand appears readily able to fully satisfy the natural bonding preferences of the actinyl unit in the equatorial plane, a concept underpinned by voltammetric studies of other uranyl complexes supported by pentadentate ligands30 as well as prior work on structurally diverse Schiff-base ligands.31–34 Coordinative saturation in the equatorial plane appears to alleviate some difficulties with speciation, in that neither labile ligands prone to exchange (e.g., solvent) nor potential bridging anions (e.g., chloride) are present. The Np(VI) oxidation state is particularly stabilized by the chosen pentadentate ligand, as judged by spectroscopic monitoring in acetonitrile solutions over time. In contrast, formation of lower Np(V) and Np(IV) oxidation states gives rise to significant reactivity, resulting in formation of unique di- and trinuclear complexes that we have characterized in other recent work.35 Given this situation, we anticipated that investigating the redox properties of our neptunyl(VI) complex26 in acetonitrile- and methanol-based electrolytes would represent an opportunity to characterize Np-centered redox in non-aqueous media with both electroanalytical and chemical approaches (see Fig. 2). Overcoming the established challenges of interrogating neptunium redox chemistry could afford new insights applicable to next-generation nuclear fuel reprocessing schemes as well as new approaches to preparation of neptunium complexes.36,37
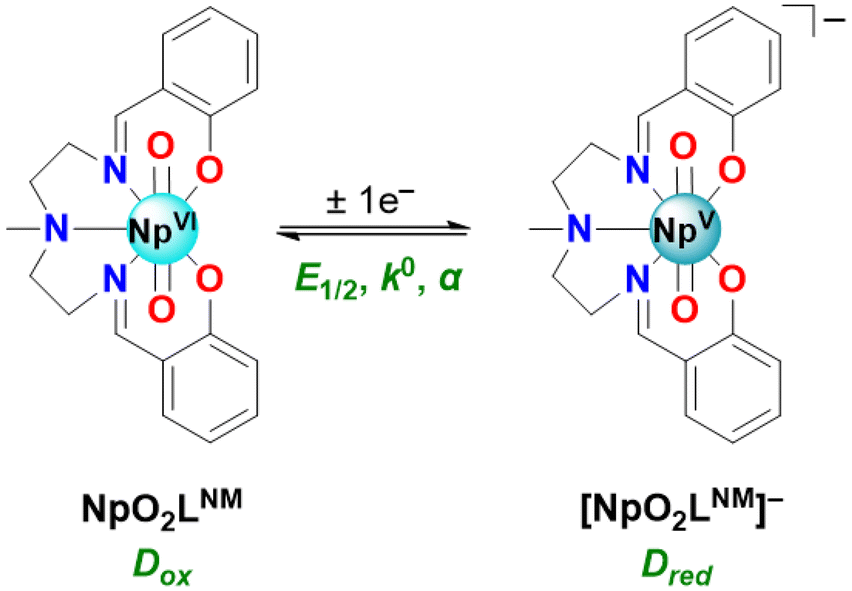 | ||
| Fig. 2 The Np(VI) and Np(V) species in this study, and the 1e− redox couple for their interconversion. The fundamental parameters describing this redox couple that were studied here include the Np(VI)/Np(V) half-wave potential (E1/2), the standard heterogeneous electron-transfer rate constant (k°), and the transfer coefficient or symmetry factor (α). The diffusion coefficients for the Np(VI) and Np(V) species are denoted Dox and Dred, respectively. | ||
Here, we report an electroanalytical investigation of redox chemistry starting with a neutral Np(VI) coordination complex (denoted NpO2LNM) in electrolytes based upon both a protic solvent (CH3OH) and an aprotic solvent (CH3CN). In the (CH3CN) case, a clean Np(VI)/Np(V) couple was measured at −0.48 V vs. Fc+/0 without the need for inert atmosphere or sparging with inert gas. Scan rate-dependent behavior in each solvent/electrolyte indicates that both the reduced and oxidized forms of the compounds are freely diffusional and soluble in solution on the timescale of the voltammetry; companion data from double-potential-step chronoamperometry and chronocoulometry experiments support a high degree of chemical reversibility in this system, apparently free from complications that could arise due to formation of Sullivan-type cation–cation interactions. The cyclic voltammetric data across all scan rates could be satisfactorily simulated with a model involving only chemically reversible 1e− transfer processes; the estimated diffusion coefficients for the Np(VI) and Np(V) species from the simulations are in line with similarly sized monometallic complexes from other work. Regarding electrochemical reversibility, the heterogeneous electron transfer rates appear fast based on the simulated k° values, in accord with the modest peak-to-peak separation values that were measured directly from the voltammetric data. Additional evidence for chemical reversibility in this system as well as generation of a single Np(V) species was developed in a spectrochemical titration and spectroelectrochemical experiments. Structural and spectroscopic data obtained for the reduced form of the compound, including three structures from XRD analysis obtained with crystals resulting from bulk electrolytic reduction of the starting NpO2LNM, enable new comparisons to the previously reported analogous isostructural U(V) and isoelectronic Pu(VI) complexes. In the Np(V) structures, hydrogen bonding appears to play a pivotal role in the stability of the neptunyl(V) core, suggesting a marked increase in actinyl oxo basicity upon reduction and suggesting future opportunities for studies of actinyl redox chemistry.
2 Results
2.1 Structure and spectroscopy of [NpO2LNM]−
Building on our recent report of the preparation of a homologous series of U, Np, and Pu complexes ligated by a Schiff-base ligand, H2LNM, we became interested in the electron transfer properties of such systems (H2LNM = 2,2′-[(methylimino)bis(2,1-ethanediylnitrilomethylidyne)]-bis(phenol)). Previously, cyclic voltammetry (CV) data for the uranyl complex of LNM, UO2LNM, in acetonitrile-based electrolyte (0.1 M TBAPF6 in CH3CN; denoted hereafter as CH3CN/TBAPF6) has been reported, which displays a 1e− couple at E1/2 = −1.55 V vs. ferrocenium/ferrocene (denoted hereafter as Fc+/0) under inert atmosphere and under ambient lab conditions with rigorous N2 sparging (see ESI, Fig. S1†).28,29 Under inert atmosphere, the reduced form of the complex could be isolated and fully characterized structurally, spectroscopically, and electrochemically.29 Here, we have extended this general type of electrochemical investigation to neptunium and found that NpO2LNM displays a Np(VI/V) couple in CH3CN/TBAPF6 which is accessible without the need for inert atmosphere or sparging. This couple can be measured at E1/2 = −0.48 V vs. Fc+/0 and its modest reduction potential, as well as apparent reversibility, encouraged us to pursue the isolation and characterization of the reduced form of the compound. In this endeavor, we have found that bulk electrolysis of NpO2LNM at a concentration of 0.5 mM in CH3OH electrolyte enables the isolation of [NpO2LNM][nBu4N] (denoted hereafter as [NpO2LNM]−). Crystalline [NpO2LNM]− was obtained from such bulk electrolyses on multiple occasions in the form of various hydrates: [NpO2LNM]−·3H2O (m60a and m61a) and [NpO2LNM]−·2.5H2O (m36a; see ESI, pp. S60–S81 and Table S5† for crystallographic details). For the purpose of the following structural and spectroscopic discussion, m60a will be used as the representative [NpO2LNM]−·3H2O structure and will be denoted as [NpO2LNM]−·3H2O in the main text, while discussion of m36a and m61a can be found in the ESI (pp. S67–S74 and S75–S81, respectively, as well as in Table S5†).The structure [NpO2LNM]−·3H2O (m60a) contains two crystallographically independent molecules of [NpO2LNM]− (denoted hererafter as molecules A and B) in the asymmetric unit; molecules A and B are located in chemically distinctive environments (Fig. 3). Two counter cations of tetrabutylammonium are also present in the asymmetric unit to balance the charges coming from the two monoanionic Np-containing molecules (see ESI, pp. S60–S66 and Table S5† for crystallographic details). The actinyl moiety is preserved upon conversion from NpO2LNM to [NpO2LNM]−·3H2O; the Np center remains ligated by the five donor atoms of the organic ligand, though the degree of ligand puckering is much less in [NpO2LNM]− (as quantified by the ligand fold angle χ in Table 1), consistent with the expected larger ionic radius of Np(V) compared to Np(VI). Overall, the An–Oyl bond distances (1.809(5)–1.838(5) Å) and bond valence sum (BVS) parameter (5.0) are consistent with typical Np(V) metrics (Table 1).38 Consistent with the presence of Np(V), the An–Ophenoxide bonds lengthen by more than 0.1 Å in [NpO2LNM]− (2.365(6) Å and 2.404(6) Å) compared to NpO2LNM (2.226(7) Å); this elongation can be ascribed to arise from additional electron density on the Np metal center that repels the anionic phenoxide donor atoms. The bond metrics for the imine groups of the ligand suggest that there may be a negligible contribution of ligand radical character (i.e., mixed Np-ligand reduction character) in [NpO2LNM]−. This can be judged based upon the very slight elongation of the imine bond of [NpO2LNM]− compared to NpO2LNM (Δ(N1–C1) = 0.009(13) Å and Δ(N2–C14) = 0.015(13) Å). For comparison, Δ(N1–C1) and Δ(N2–C14) for UO2LNM/[UO2LNM]− are 0.015(10) Å and 0.005(10) Å, respectively.29 In the case of the Np species discussed here, spectroscopic studies were used to examine the issue of radical anion delocalization in [NpO2LNM]−, and these studies demonstrate that the site of reduction is primarily Np-centered (vide infra). Finally, despite the isoelectronic nature of U(V)/Np(VI) and Np(V)/Pu(VI), the actinyl bond distances remain reflective of the respective formal oxidation states in all instances, where the An(VI) distances are shorter than the An(V) distances with the computed BVS parameters for each species also being supportive of the assigned oxidation states.
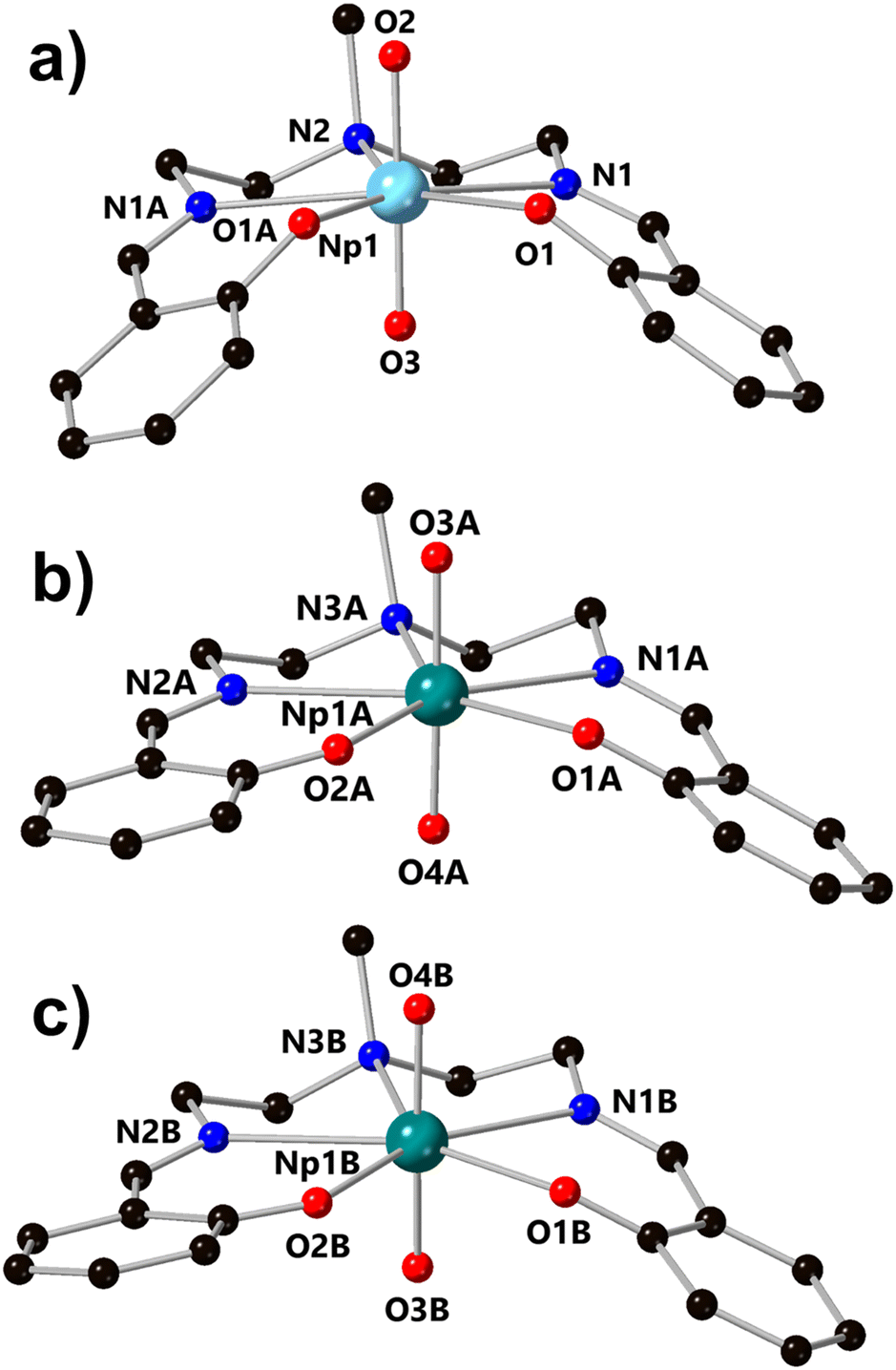 | ||
| Fig. 3 Structures of NpO2LNM (structure a) and the crystallographically independent molecules of [NpO2LNM]− in the asymmetric unit of [NpO2LNM]−·3H2O from X-ray diffraction analysis (structures b and c). All hydrogen atoms, minor components of disorder, outer-sphere co-crystallized solvent molecules, and counter cations (associated with [NpO2LNM]−) are omitted for clarity. | ||
| UO2LNM | [UO2LNM]− | NpO2LNM | [NpO2LNM]−·3H2O (m60a) | [NpO2LNM]−·2.5H2O (m36a) | PuO2LNM | |
|---|---|---|---|---|---|---|
| a Average values are shown; value in parentheses refers to the e.s.d that is the largest for an individual entry among the independent values used to compute the average. | ||||||
| An–Oyl (Å) | 1.784(3)a | 1.853(7) | 1.743(11) | 1.809(5) | 1.803(4) | 1.754(6) |
| — | — | — | 1.813(5) | 1.821(5) | — | |
| An–Oyl (Å) | 1.793(3)a | 1.847(7) | 1.733(9) | 1.819(5) | 1.811(4) | 1.743(5) |
| — | — | — | 1.838(5) | 1.836(4) | — | |
| Oyl–An–Oyl (°) | 173.6(1)a | 173.1(3) | 176.8(5) | 175.6(2) | 175.4(2) | 176.8(3) |
| An–Ophen (Å) | 2.223(3)a | 2.368(7)a | 2.226(7) | 2.404(6)a | 2.395(4)a | 2.246(4) |
| — | — | — | 2.365(6)a | 2.368(4)a | — | |
| N1–C1 (Å) | 1.290(6)a | 1.305(10) | 1.267(13) | 1.276(10)a | 1.273(8)a | 1.267(8) |
| N2–C14 (Å) | 1.287(5)a | 1.292(10) | — | 1.285(10)a | 1.279(8)a | — |
| χ (°) | 18.7(1)a | 0(4) | 64.4(5) | 34.0(3)a | 35.6(3)a | 61.4(3) |
| BVS | 5.8 | 5.0 | 5.8 | 5.0 | 5.0 | 5.7 |
| Ref. | 29 | 29 | 26 | This work | This work | 26 |
In the structure of the Np(VI) complex NpO2LNM from our prior work,26 the only hydrogen bonding interactions found in the structure involve H+ donation from a single disordered outer-sphere methanol molecule to the phenoxide moieties in the organic ligand (Fig. 4a). In contrast, [NpO2LNM]−·3H2O features three water molecules that engage in multiple hydrogen bonding interactions with the anionic Np-containing molecules, as well as weak electrostatic interactions that surround the two crystallographically inequivalent Np-containing sites in the asymmetric unit. Specifically, each [NpO2]+ motif engages in at least one moderate–strength hydrogen bond with an outer-sphere co-crystallized molecule of water (donor–acceptor distances of 2.925(17) Å for O4A⋯O2W and 2.815(8) Å for O4B⋯O3W) in addition to weak electrostatic interactions with the tetrabutylammonium cations.39 As indicated by the difference in the donor–acceptor interaction distances, there is a slight asymmetry to the nature of the water–oxo interaction where O3W interacts more strongly with the oxo O4B than O2W does with the oxo O4A. This could be the result of the half-occupancy of the water molecule associated with O2W; the occupancy of O2W was found to be related to disorder in one butyl chain of a nearby tetrabutylammonium counter cation (see ESI, p. S54 and Fig. S51†). Although this feature appears subtle in the crystallographic data, the slight asymmetry of the hydrogen bonding is clearly seen in the asymmetric elongation of the Np1A–O4A/B bond and is also reflected in the Raman spectroscopic results (vide infra). Stated another way, the Np–Oyl distances corresponding to the oxos that serve as acceptors for the moderate–strength hydrogen bonds are significantly longer (Np1A–O4A, 1.819(5) Å; Np1B–O4B, 1.838(5) Å) than the Np–Oyl distances to those that do not (Np1A–O3A, 1.809(5) Å; Np1B–O3B, 1.813(5) Å). The Np–Oyl distance associated with the fully occupied H-bond donor water molecule (Np1B–O4B, 1.838(5) Å) is also longer than the corresponding distance to the half-occupied H-bond donor H2O (Np1A–O4A, 1.819(5) Å).
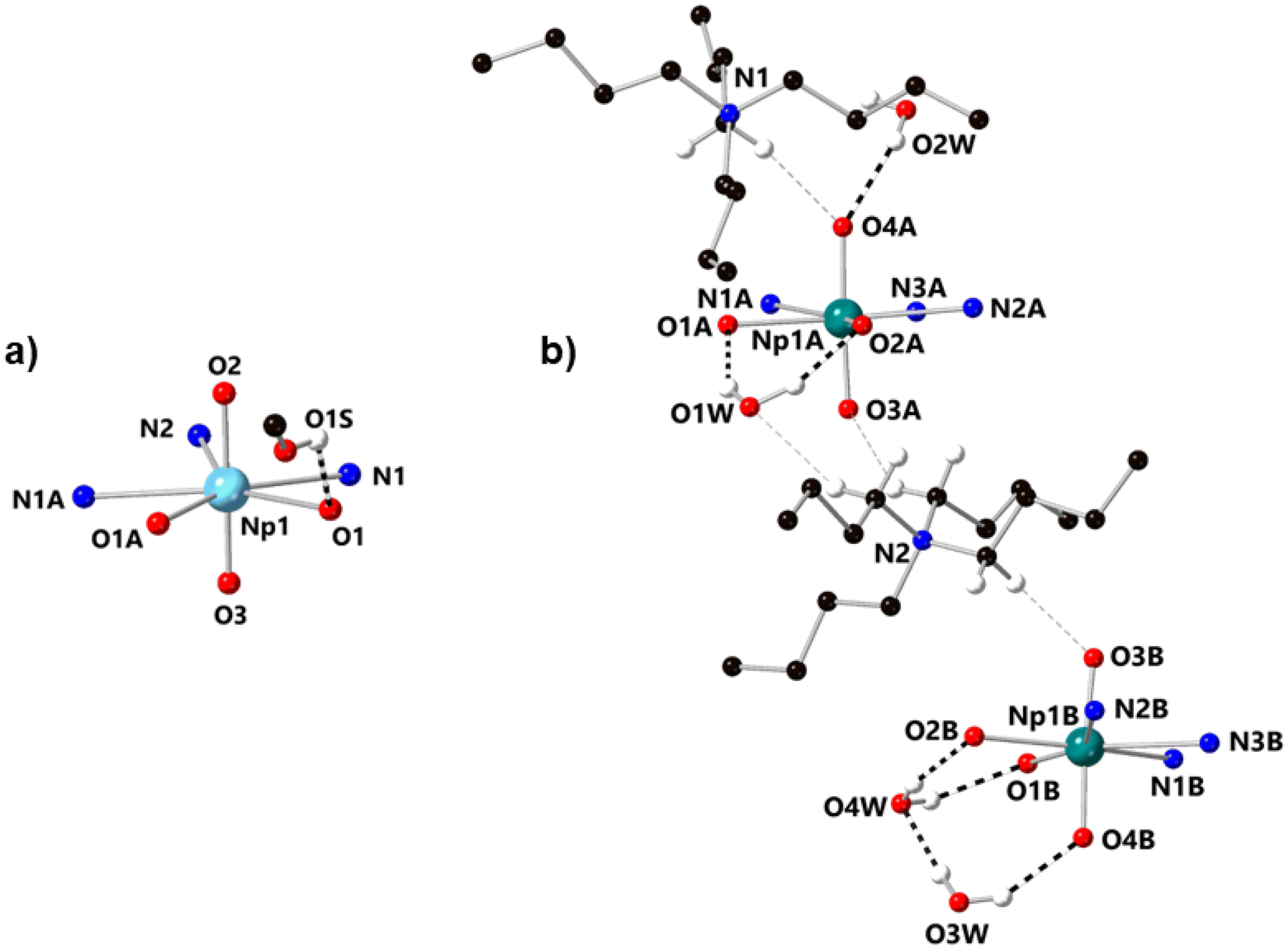 | ||
| Fig. 4 (a) Structure of NpO2LNM from X-ray diffraction analysis, showing the outer-sphere methanol found in the structure which serves as an H-bond donor to the phenoxides of the ligand.26 (b) Molecules A and B in the asymmetric unit of [NpO2LNM]−·3H2O from X-ray diffraction analysis. Each Np-containing molecule engages in hydrogen bonding interactions with outer-sphere water molecules (denoted with bold dashed lines), as well as weak electrostatic interactions with the tetrabutylammonium counter ions (denoted with grey dashed lines). All minor components of disorder and hydrogen atoms except those engaged in hydrogen bonding or weak electrostatic interactions are omitted for clarity. | ||
As mentioned above, crystals of [NpO2LNM]− were obtained from virtually identical conditions on multiple occasions (slow evaporation of a sample of 0.5 mM NpO2LNM following bulk electrolysis in methanol electrolyte). Notably, control bulk electrolysis experiments performed in CH3CN/TBAPF6 ([NpO2LNM] = 0.5 mM, analogous to the conditions that yield [NpO2LNM]− in CH3OH/TBAPF6) exclusively gave rise to the crystals of the previously reported [NpVNpIVNpV] trimeric species (see ESI, Fig. S45†).35 In addition to [NpO2LNM]−·3H2O, another structure, [NpO2LNM]−·2.5H2O, was found which also contains multiple co-crystallized water molecules, suggesting that the incorporation of water into the solid-state structure of [NpO2LNM]− is favored by generation on Np(V). The hydrogen bonding interactions that result from this water incorporation could be critical to stabilizing [NpO2LNM]− in the solid state, especially considering the direct H-bonding interactions between the co-crystallized water molecules and the neptunyl(V) motifs in the structures. [NpO2LNM]−·2.5H2O shows the same connectivity of the main Np-containing sites as [NpO2LNM]−·3H2O, but contains only two fully occupied water molecules and one half-occupied water molecule. In contrast to [NpO2LNM]−·3H2O, only one Np-containing site in [NpO2LNM]−·2.5H2O displays a hydrogen bonding interaction between an outer-sphere water and an actinyl (O2W⋯O4B; donor–acceptor distance of 2.817(6) Å); the impact of this asymmetry can be directly observed in the Raman spectroscopic results (vide infra). Despite the variable number of water molecules observed across the two structures, it appears that the hydrogen bonding motifs present in the structure of [NpO2LNM]− play a critical role in the stabilization and crystallization of this species in methanol over acetonitrile, in spite of the propensity of An(V) species to disproportionate in protic media.
The differences in the hydrogen bonding patterns present in the structures of NpO2LNM and [NpO2LNM]− afford insight into the changes in the Lewis basicity of the actinyl oxos that result from metal-centered reduction. In the structure of NpO2LNM, the most basic site which can engage in hydrogen bonding appears to be the phenoxides of the ligand; this is apparent in the H-bonding interactions of the disordered methanol molecule with the phenoxide O-atoms. However, upon reduction, the actinyl oxos seem to display a similar basicity to the phenoxides in that water molecules interact with both the phenoxides and the actinyl oxos. One could hypothesize that the phenoxides remain more basic than the actinyl oxos in [NpO2LNM]− because the two water molecules in [NpO2LNM]−·3H2O which hydrogen bond with the phenoxides (O1W and O4W) are fully-occupied, while one of the waters which hydrogen bonds with an actinyl is only half-occupied (O2W). This hypothesis can be set aside, however, when realizing that one of the phenoxide-interacting water molecules in [NpO2LNM]−·2.5H2O (O3W) is half-occupied, while the water molecules engaging in a hydrogen bond with the actinyl (O2W) is fully-occupied. Thus, despite a possible increase in the relative basicity of the phenoxides from the lengthening of the Np–Ophenoxide bonds in [NpO2LNM]−, the actinyl oxos in [NpO2LNM]− are at least similar in basicity to the phenoxides, on the basis of our close inspection of the hydrogen bonding in [NpO2LNM]−·3H2O and [NpO2LNM]−·2.5H2O. Moreover, the actinyl oxos for the Np(V) species are definitely more basic than those in the starting Np(VI) species NpO2LNM.
The solution-phase optical spectrum of [NpO2LNM]− is also consistent with Np in the +V oxidation state. The spectrum in CH3CN displays ligand-centered π–π* absorptions between 350–500 nm but is rather featureless between 500–1000 nm, with the exception of a sharp feature at 994 nm (Fig. 5). With a molar absorptivity of ca. 100 M−1 cm−1, this transition is diagnostic of Np(V) and matches well with the feature observed during a companion spectrochemical titration of NpO2LNM with Cp*2Fe (Fig. 12). Notably, the absence of broad features in the visible and near-infrared regions (from 500–1000 nm) strongly suggests that the reduction is principally centered on Np, as a ligand-centered reduction would be expected to display such broad absorptions in this range.40 For comparison, the U analog, [UO2LNM]−, features a broad absorption between 500–800 nm (ε ≥ 200 M−1 cm−1) in addition to its signature U(V) f–f transition, suggestive of mixed U-ligand radical character (see ESI, Fig. S38†).29 This observation, in turn, is consistent with the significantly more negative reduction potential of −1.55 V associated with U(VI)/U(V) redox cycling in comparison to the value of −0.48 V associated with Np(VI)/Np(V) cycling; the more negative potential for generation of U(V) promotes delocalization of electron density into ligand-centered orbitals that are empty in the U(VI) species.
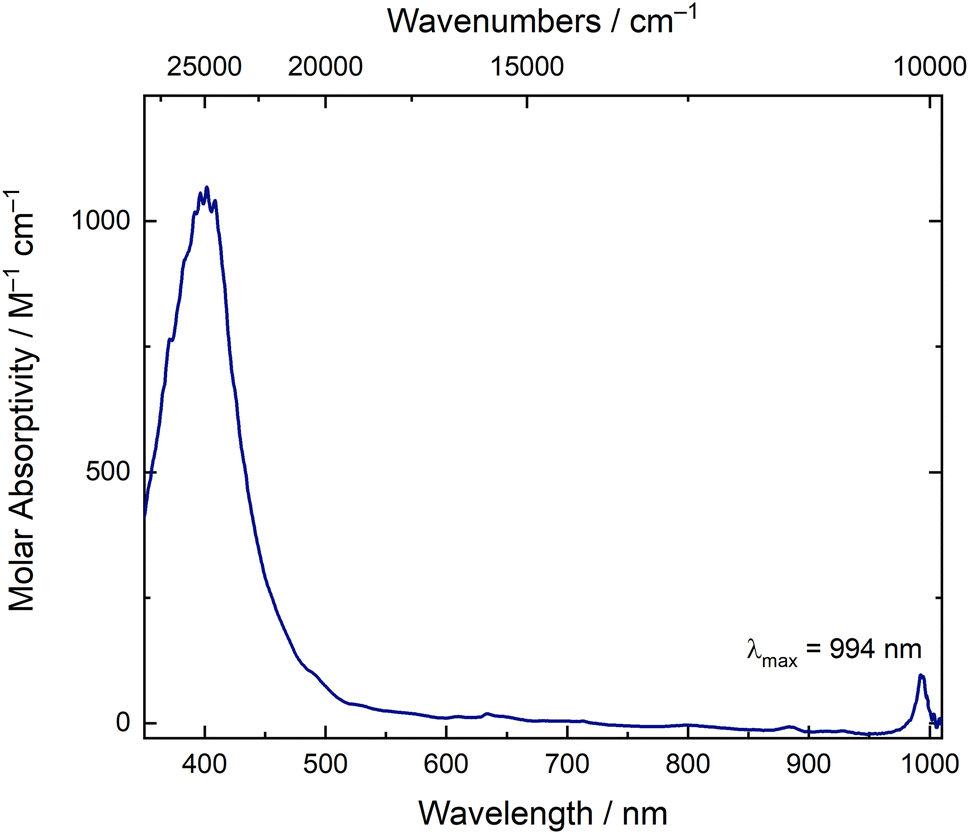 | ||
| Fig. 5 Electronic absorption spectrum of [NpO2LNM]− in 0.2 TBAPF6 in CH3CN ([Np] = 1.1 mM). | ||
The solid-state Raman spectra of [NpO2LNM]− (both [NpO2LNM]−·3H2O and [NpO2LNM]−·2.5H2O) support the presence of the +V oxidation state, but a deeper understanding of the influence of hydrogen bonding in these structures was also gained from these spectra by correlation with the companion structural information which are also reported here (vide supra). Overall, the ligand-based modes remain mostly unperturbed, further bolstering the claim that the Np is the principle site of reduction. The spectrum of [NpO2LNM]−·3H2O displays an symmetric [NpO2]+ stretch at 735 cm−1, which compares well with other Np(V) stretches (Fig. 6).41,42 Similarly, the spectrum of [NpO2LNM]−·2.5H2O features a stretch at 732 cm−1; both of these are shifted with respect to the ν1 of NpO2LNM to lower energy by 55 and 52 cm−1, respectively, consistent with the presence of a more electron-rich Np center in [NpO2LNM]−. A marked difference is observed, however, in the intensities of the ν1 stretches compared to the adjacent ligand band in [NpO2LNM]−·3H2O vs. [NpO2LNM]−·2.5H2O, with the intensity of the actinyl ν1 being much lower in [NpO2LNM]−·2.5H2O. Closer inspection of the ν1 feature in [NpO2LNM]−·2.5H2O reveals a primary feature at 732 cm−1 with a shoulder at 742 cm−1 (the feature at 762 cm−1 corresponds to a ligand-based feature; Fig. 7). As the structure of [NpO2LNM]−·2.5H2O contains one molecule of [NpO2LNM]− whose actinyl oxo serves as an H-bond acceptor to an outer-sphere water (located near molecule B) and one molecule that does not engage in a hydrogen bond through its actinyl oxo (molecule A) in its asymmetric unit, we ascribe the band at 732 cm−1 to ν1 of the [NpO2]+ unit in molecule B and the band at 742 cm−1 to ν1 of the [NpO2]+ unit in molecule A. This assignment is consistent with the longer actinyl bonds observed for molecule B (Table 1) and suggests that the presence of a single, moderate strength hydrogen bond interacting with an oxo group of a [NpVO2] core in the solid state imparts a shift of 10 cm−1 to the Raman shift of ν1.
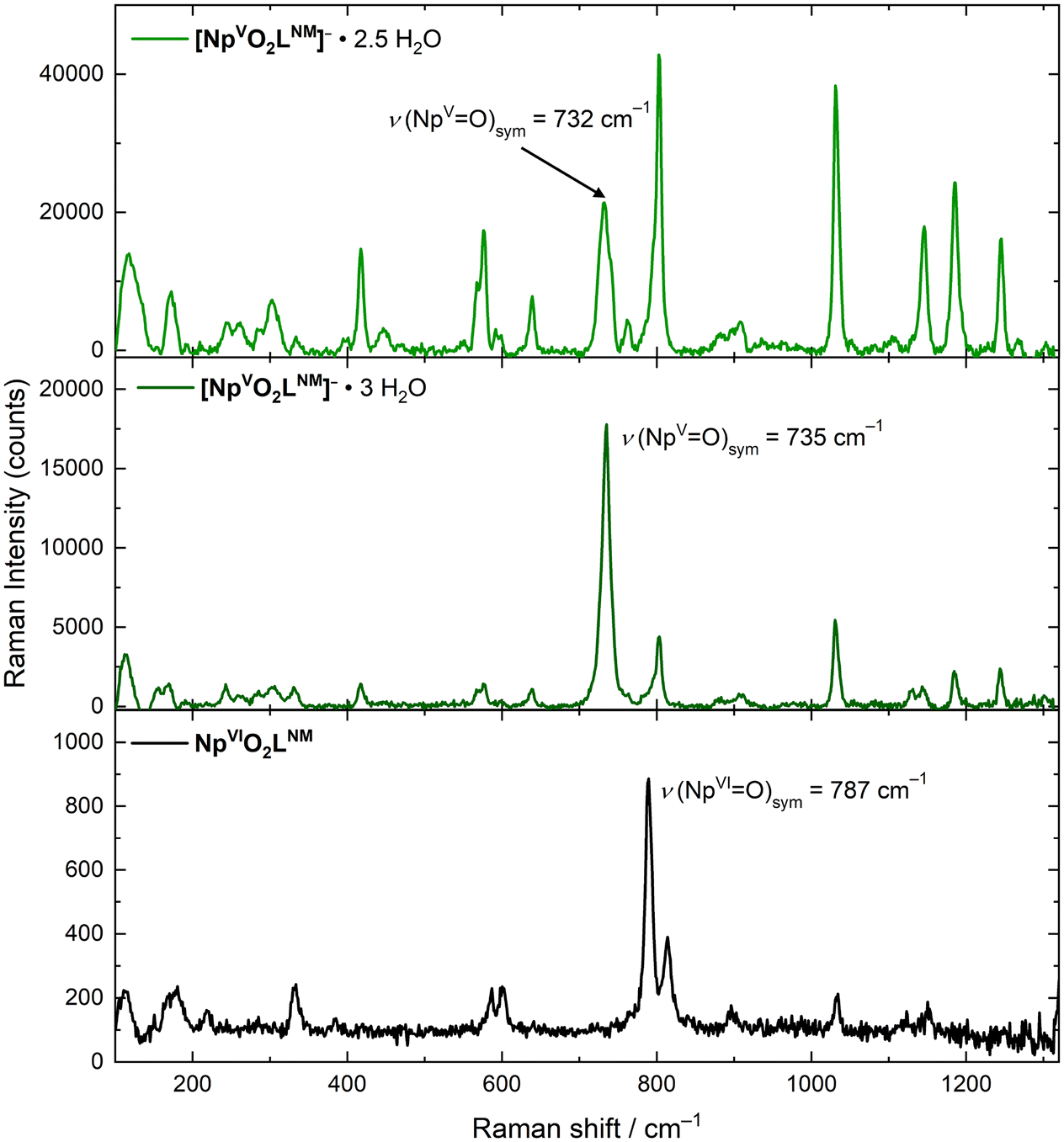 | ||
| Fig. 6 Solid state Raman spectra of [NpO2LNM]−·2.5H2O (upper panel), [NpO2LNM]−·3H2O (middle panel) and NpO2LNM (lower panel). | ||
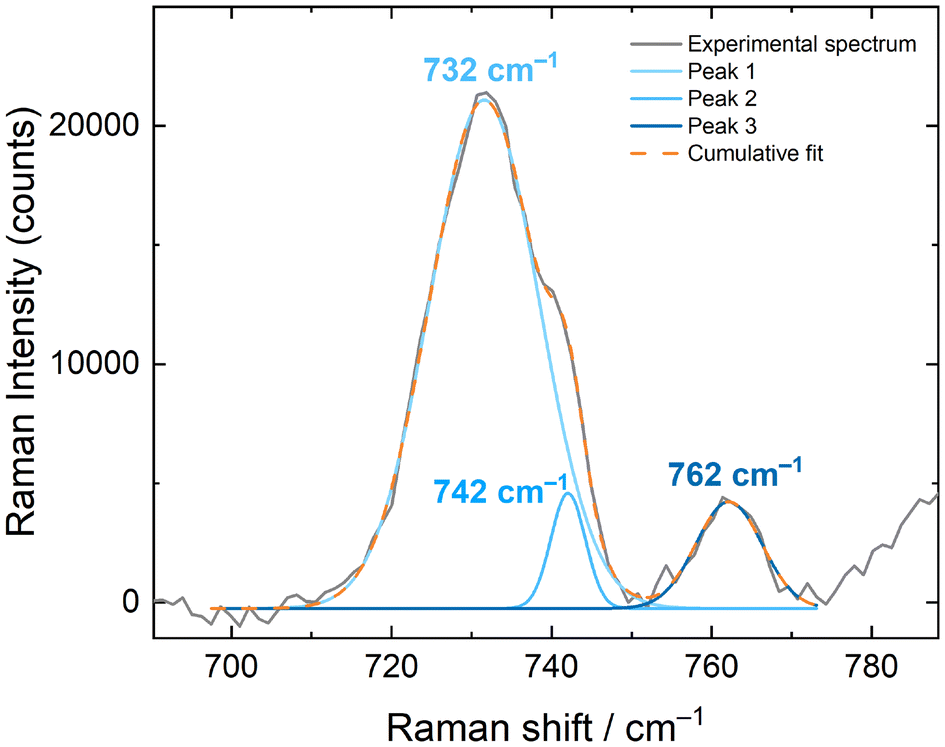 | ||
| Fig. 7 Fitting of the ν1 feature in the solid state Raman spectrum of [NpO2LNM]−·2.5H2O. | ||
Other compounds of Np(V) have been reported which feature hydrogen bonding interactions with the actinyl oxos and display multiple features in the typical range for ν1, resulting in unusually stretched or structured ν1 features. In those cases, the structure in the ν1 feature is often ascribed to a decrease in symmetry of the actinyl ion from D∞h to C∞v as a result of asymmetry in the actinyl bonds (0.023 Å).43,44 In several examples, however, the structure of the features is not accompanied by the drastic decrease in intensity which is observed here in the case of [NpO2LNM]−·2.5H2O. It is worth noting that the Δ(ν3 − ν1) is typically ca. 100 cm−1 for Np(VI) compounds,45 but ranges from 22–50 cm−1 for Np(V) compounds in the three available documented examples.46–48 Though asymmetry in actinyl bond distances comparable to literature examples is present in site B of [NpO2LNM]−·2.5H2O, the difference in between the main feature and shoulder is only 10 cm−1. In light of these prior reports and our observations, we therefore hypothesize that the structure in the actinyl feature in [NpO2LNM]−·2.5H2O is a result of the presence of two crystallographically distinct Np-containing sites in the asymmetric unit, giving insight into the magnitude of the hydrogen bonding influence on actinyl vibrational spectroscopy rather than ν3 stretching.
2.2 Electroanalytical studies of NpO2LNM and simulation of cyclic voltammetry data
We measured the voltammetric response of NpO2LNM in CH3CN/TBAPF6, which shows a Np(VI/V) couple at E1/2 = −0.48 V vs. Fc+/0 without the need for inert atmosphere or sparging. The couple appeared to be a quasi-reversible on the basis of its ΔEp of 97 mV (see ESI, Fig. S2;† ΔEp of the Fc+/0 couple under these same conditions was 78 mV). Data at various scan rates were collected across two orders of magnitude to enable an investigation of the degree of chemical reversibility and the diffusional nature of the compounds involved in the redox couple. A slight loss of the near-ideal behavior was seen at slower scan rates by inspecting the ratio of anodic peak current to cathodic peak current (see ESI, Fig. S3†). This is consistent, however, with a minor influence of convection in our electrochemical cell induced by background vibrations; our electrochemical cell was located in a radiochemical fume cupboard during experimentation and thus turbulent air flow or fan vibrations could shake the cell slightly, resulting in loss of apparently high chemical reversibility at slower scan rates. Despite this, the current response for both the anodic and cathodic peaks is consistent with the Randles–Ševčík equation in that it increases as a function of the square root of scan rate, indicating that both the oxidized and reduced forms of the complex are diffusional in solution as shown in panels a and b of Fig. 8. | ||
| Fig. 8 Panel (a) scan-rate-dependent cyclic voltammetry data for NpO2LNM in CH3CN/TBAPF6. Panel (b) plot of raw peak currents vs. (scan rate)1/2, demonstrating the diffusional nature of the reduced and oxidized species. Panel (c) scan-rate-dependent cyclic voltammetry data for NpO2LNM in CH3OH/TBAPF6. Panel (d) plot of peak currents vs. (scan rate)1/2, demonstrating the diffusional nature of the reduced and oxidized species. Conditions: [NpO2LNM] = 1 mM. | ||
Building on the behavior of NpO2LNM in CH3CN, as well as our recent report of multimetallic species derived from reactivity of neptunyl diacetate with methanol and H2LNM,35 we also carried out measurements of the voltammetry of NpO2LNM in methanol-based electrolyte (0.1 M TBAPF6 in CH3OH; denoted hereafter as CH3OH/TBAPF6). An accessible quasi-reversible redox couple was measured at E1/2 at −0.22 V vs. Fc+/0 with ΔEp = 121 mV. Similar to the case in CH3CN, a small degree of non-ideal behavior was observed at slow scan rates on the basis of the ratio of anodic to cathodic peak current at the rates below 150 mV s−1 or so (see ESI, Fig. S11†). However, both the oxidized and reduced forms of the complex are freely diffusional in solution as shown in panels c and d of Fig. 8, with the peak anodic and cathodic currents both displaying linear trends with respect to the square root of scan rate.
As Np has been documented to adsorb onto metallic surfaces,49,50 such as those used as electrodes in other electrochemical studies, we utilized double potential step chronoamperometry (DPSCA) and chronocoulometry experiments to probe the chemical reversibility of the Np(VI/V) couple and whether the oxidized or reduced species composing the Np(VI/V) manifold would display similar adsorption behavior towards the oriented graphitic electrodes utilized in this study. DPSCA can enable detection and quantification of follow-up chemical steps (as in an Electrochemical-Chemical, EC, process) via analysis of the distinctive time-dependent decay of current over time if such steps are operational.51 In the absence of a follow-up chemical step, the ratio of the anodic current at the end of the backward potential step to the cathodic current at the end of the forward potential step (denoted RDPS) can be used as a measure of chemical reversibility (see ESI, eqn (1) & (2)†). For the [NpO2LNM]0/− couple, RDPS was found to be 0.934 in acetonitrile electrolyte, which indicates a high degree of chemical reversibility as the value for a perfectly chemically reversible system is unity (assuming identical diffusion coefficients for the oxidized and reduced species). A linear fit of t−1/2vs. current from the DPSCA data allows for the experimental determination of the diffusion coefficient (see ESI, Fig. S4, S5, S12, and S13†). Given in Table 2, the experimental values from DPSCA agree well with the values obtained from the simulations. Finally, analysis of corresponding chronocoulometry data (obtained here by numerical integration of the DPSCA data) indicates that there is minimal evidence for adsorption of NpO2LNM or [NpO2LNM]− on the electrode surface in either CH3CN- or CH3OH-based electrolytes (see ESI, Fig. S6–S9 and S14–S17†).52
| D ox (cm2 s−1) | D red (cm2 s−1) | D ox (cm2 s−1) (exp) | D red (cm2 s−1) (exp) | k° (cm s−1) | |
|---|---|---|---|---|---|
| UO2LNM | 1.07 × 10−5 | 1.22 × 10−5 | — | — | 0.014 |
| NpO2LNM (CH3CN) | 1.27 × 10−5 | 2.64 × 10−5 | 2.28 × 10−5 | 4.35 × 10−5 | 0.008 |
| NpO2LNM (CH3OH) | 9.35 × 10−6 | 1.28 × 10−5 | 3.06 × 10−5 | 7.38 × 10−5 | 0.005 |
In an effort to quantify the fundamental parameters governing the apparent chemical and electrochemical reversibility of the [NpO2LNM]0/− couple, digital simulations of the CV data were employed.53 We simulated data across a range of scan rates which were experimentally measured for NpO2LNM in order to determine the diffusion coefficients of the oxidized and reduced forms (Dox and Dred, respectively), as well as the heterogeneous electron transfer rate constant for the Np(VI)/Np(V) system (k°). Each CV was simulated as a reversible 1e− system involving no other reactivity or chemical equilibria. Additionally, a sensitivity analysis was carried out on the values obtained for Dox, Dred, and k° to evaluate the validity of each parameter within the larger set of floating values on which the simulations depend; this was done by examining the goodness-of-fit of the simulation result in comparison with the experimental data (see ESI, Fig. S24–S26†). As shown in Fig. 9, the simulations agree well with the experimental data when the established and known parameters are used as inputs for the fits (see ESI, Fig. S21 and S22† for additional comparisons). The satisfactory agreement between the experimental and simulated data and the reasonable magnitudes of the diffusion coefficients extracted from the simulation results suggest that the concentration of neptunium probed experimentally by the voltammetry agrees well with the prepared solution concentration as measured by liquid scintillation counting prior to the experiment. This match also provides additional quantitative evidence that the measured redox event corresponds, as we have assigned, to the compounds prepared and studied in this report. In this context, anomalously small diffusion coefficients would in all likeliood be observed if chemical reactivity occurred in solution, for example by precipitation or deposition of electroinactive material on the electrode. In our case, the measured diffusion coefficients support that the Np-containing species which we prepared are indeed undergoing redox cycling in the electrochemical measurements.
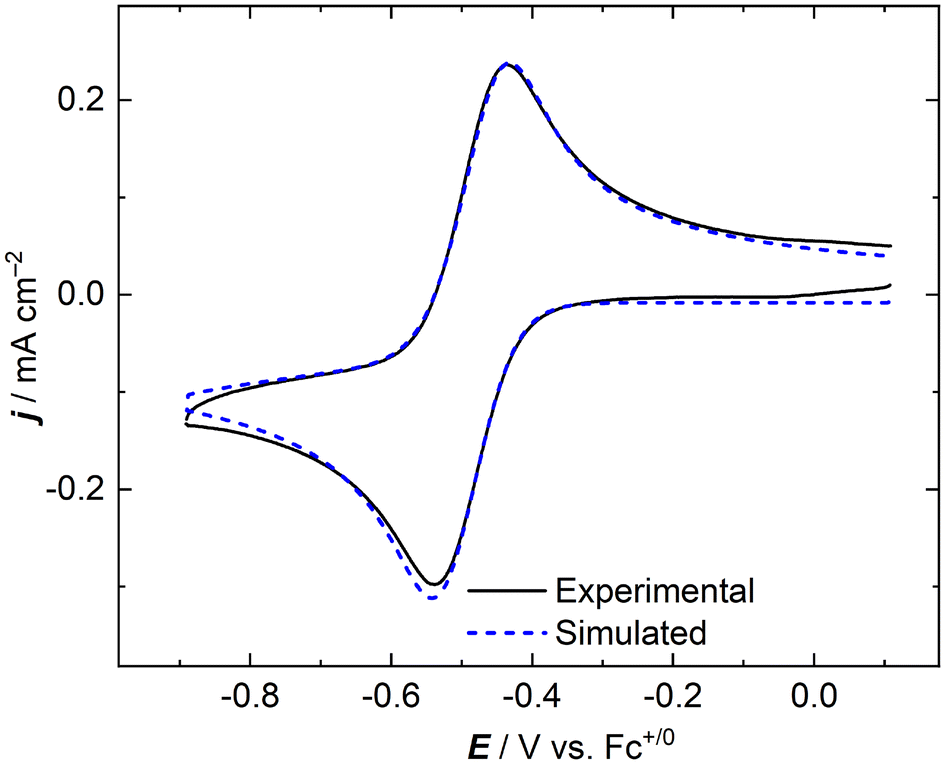 | ||
| Fig. 9 Experimental and simulated cyclic voltammograms of NpO2LNM in CH3CN/TBAPF6 (scan rate: 100 mV s−1). | ||
The diffusion coefficients for the oxidized and reduced forms that were extracted from the simulations are given in Table 2. Comparison of these values to those of the related complex UO2LNM (whose data was collected under similar conditions; see ESI, Fig. S27–S30†) shows good agreement. This finding is in line with the considerations that diffusion is typically governed by hydrodynamic radius and that these compounds both display virtually no solution speciation beyond electrode-induced changes in oxidation state. The diffusion coefficients of ferrocenium and ferrocene, corresponding as a pair to the redox manifold of the most commonly utilized non-aqueous reference potential, are both 2.6 × 10−5 cm2 s−1 in acetonitrile-based electrolyte. This value benchmarks our measured diffusion coefficients as reasonable under these experimental conditions.54,55
Across the whole set of voltammograms collected over a range of scan rates collected and subsequently simulated, the fitted values of E1/2, Dox, and Dred remain constant as expected for a chemically and electrochemically reversible couple (see ESI, Fig. S23–S25†). This invariance in E1/2 and the diffusion coefficients over the range of investigated scan rates spanning two orders of magnitude further confirms the distinct lack of follow-up chemical reactivity or electrode fouling and supports the interpretation of a clean 1e− couple. The lack of any correlation between the fitted diffusion coefficients and scan rate is particularly telling in this context, since apparent diffusion coefficients would be differentially impacted by follow-up chemical processes and/or alteration of the electrode surface (from electrode fouling) as a function of the time elapsed during cycles of voltammetry. Stated another way, our findings show that we are measuring the intrinsic diffusion characteristics of the oxidized and reduced compounds, free from reactivity-induced complications. By fixing the diffusion coefficients at their simulated values, k° could be extracted from the simulations as well Table 2. In CH3CN, k° was found to be near the fast limit of electron transfer at our electrode type, placing our system in the quasi-reversible regime.56 We thus conclude that our system here displays minimal effects in the voltammetry that are derived from impeded heterogeneous electron transfer.
With an analogous set of data in hand for NpO2LNM in CH3OH electrolyte, we simulated this data as well, defining it as a 1e− chemically reversible couple for the extraction of electrochemical parameters. Despite the closeness of the couple to the apparent solvent/electrolyte breakdown potential of CH3OH on the cathodic side and the contribution of background processes to the increased background current (see ESI, Fig. S10†), the values obtained for Dox and Dred for NpO2LNM in CH3OH/TBAPF6 are similar to those found for NpO2LNM in CH3CN/TBAPF6 (Table 2). This finding indicates that the hydrodynamic radii of the redox-active species are similar in both solvents. This observation suggests the absence of solution-phase neptunyl–neptunyl interactions of the classic Sullivan type in either solvent; the Np moieties appear to remain firmly ligated by LNM in both CH3CN and CH3OH media. Such species often occur for Np(VI) and Np(V) but these appear not to form on the time scale of the voltammetry experiment, as the presence of such interactions in solution would likely manifest as an increase the hydrodynamic radius, an accompanying decrease in the diffusion coefficient, and a change in the overall appearance of the voltammetry towards irreversibility. Additionally, k° extracted from simulations of the CV data in CH3OH/TBAPF6 is similar to that measured in CH3CN and near the fast limit of electron transfer for our system (thus in the quasi-reversible regime here as in CH3CN). Taken together, the voltammograms in both CH3CN and CH3OH and the simulations of both datasets support the assignment of the measured redox couple as chemically reversible and electrochemically quasi-reversible. The indication of good solubility and stability of both the oxidized and reduced forms of the NpO2LNM motivated, however, use of additional solution-phase techniques to probe these complexes’ properties and reactivity, since in this case complications associated with follow-up chemical reactivity could be avoided (vide infra).
To wrap up our electroanalytical work, with [NpO2LNM]− in hand from the isolation experiments described above, we also probed the electrochemical behavior of this species, anticipating that it would be identical to that of NpO2LNM. We found that [NpO2LNM]− displays a reversible couple at E1/2 = −0.43 V vs. Fc+/0 with a ΔEp of 87 mV, indicating quasi-reversible behavior. Both the oxidized and reduced forms of the complex appear diffusional according to the scan rate-dependent data (see ESI, Fig. S19†), showing overall excellent agreement with NpO2LNM. The slight differences in the measured E1/2 and ΔEp values of the two forms of the complex are likely due to the difference in electrolyte concentration (0.2 M TBAPF6 in CH3CN in the case of [NpO2LNM]− and 0.1 M TBAPF6 in the case of NpO2LNM) (Fig. 10).
 | ||
| Fig. 10 Comparison of cyclic voltammetry data for [NpO2LNM]− and NpO2LNM in CH3CN/TBAPF6. Conditions: scan rate, 100 mV s−1; [NpO2LNM] = 1.0 mM; [[NpO2LNM]−] = 1.1 mM; electrolyte for [NpO2LNM]− = 0.2 M TBAPF6 in CH3CN; electrolyte for NpO2LNM = 0.1 M TBAPF6 in CH3CN. | ||
2.3 Spectroelectrochemistry and spectrochemical titration of NpO2LNM
Additional evidence for the chemical reversibility of the Np(VI/V) redox couple measured for NpO2LNM was pursued through spectroelectrochemistry and a spectrochemical titration. The redox couple of NpO2LNM in both CH3CN and CH3OH is accessible under ambient conditions, as mentioned above, and does not require an inert atmosphere in order to facilitate access to the reduced form. In an effort to observe isosbestic reduction of NpO2LNM, we monitored the UV-visible absorption spectrum during electrochemical polarization at a potential sufficiently negative to accomplish Np(VI) reduction without the use of an additional chemical reductant. Fig. 11 shows the evolution of the spectra upon cathodic polarization, revealing an isosbestic point at 402 nm and a decrease in the features corresponding to the LMCT transitions of NpO2LNM consistent with the generation of the reduced complex. Subsequent anodic polarization returns the spectrum to the original profile, giving direct evidence for the chemical reversibility of the system. Unfortunately, the short pathlength and low concentration of Np precluded the observation of the signature Np(V) f–f transition, but the isosbestic behavior and nearly identical nature of the spectral profiles pre- and post-polarization are consistent with the reversibility of the redox process (similar behavior was observed in CH3OHelectrolyte; see ESI, Fig. S40†). | ||
| Fig. 11 Spectroelectrochemistry of NpO2LNM in CH3CN/TBAPF6 ([Np] = 0.5 mM; path length: 1 mm). Left panel: cathodic polarization at −0.6 V vs. Fc+/0. Right panel: anodic polarization at +0.3 V vs. Fc+/0. | ||
To interrogate the reduced form through a spectrochemical titration, we chose to use decamethylferrocene (Cp*2Fe where Cp* is pentamethylcyclopentadienyl); E1/2 = −0.48 V vs. Fc+/0 in CH3CN as a reductant, as it is a molecular, 1e− chemical reductant that is stable under ambient conditions.57 The profile of a solution of NpO2LNM prior to the addition of Cp*2Fe displayed the aforementioned broad ligand-to-metal charge transfer band with two maxima at 536 and 640 nm. As Cp*2Fe was added, these bands disappeared and two new transitions grew in at 781 and 992 nm (Fig. 12). The feature at 781 nm corresponds to the oxidized [Cp*2FeIII]+ which is co-generated as the Np is reduced from Np(VI) to Np(V),58 while the feature at 992 nm corresponds to an f–f transition for Np(V).59 Likely due to the close proximity of the Np(VI/V) redox potential to that of Fe(III/II) in Cp*2Fe, isosbestic conversion from Np(VI) to(V) was not observed, consistent with a concentration-dependent redox equilibrium rather than complete electron transfer from Fe(II) to Np(VI) upon each addition. Despite the estimated low driving force provided by Cp*2Fe, Np(V) can be generated upon addition of 1 equiv. of Cp*2Fe and persists in solution for at least 20 minutes, displaying minimal changes in the spectral signatures of Fe(III) and Np(V) upon addition of extra equivalents of Cp*2Fe (see ESI, Fig. S37 and S39†). This is consistent with the persistence of the Np(V) complex in solution as also supported by the electroanalytical studies.
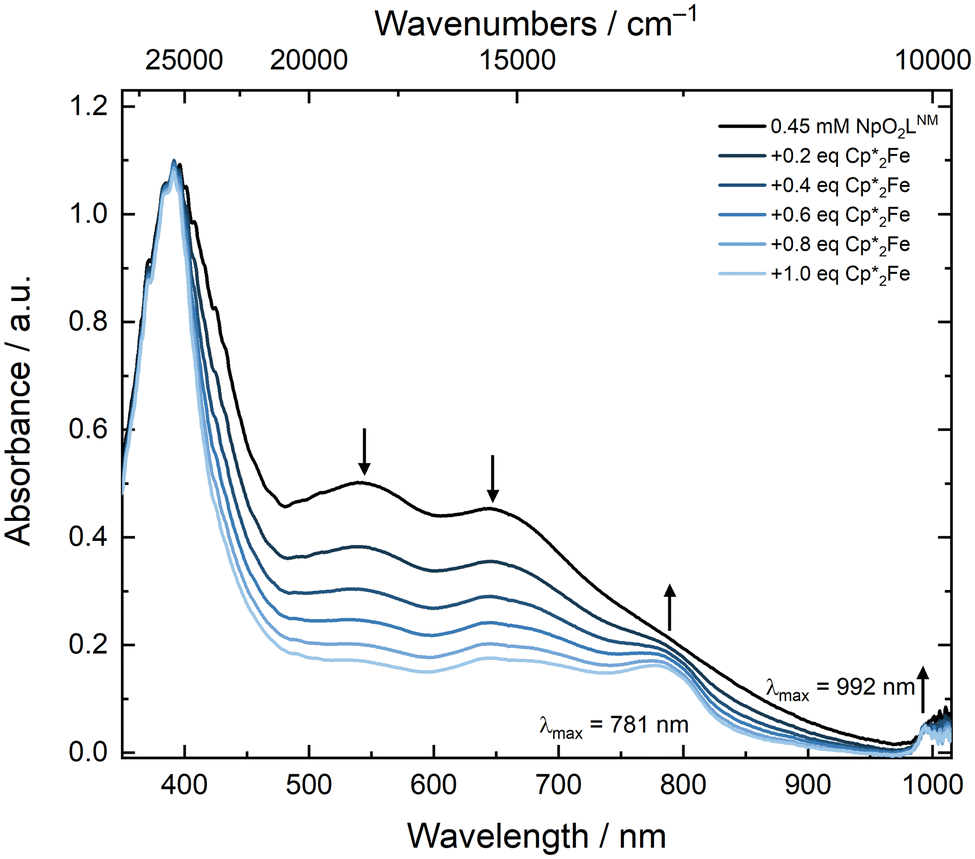 | ||
| Fig. 12 Spectrochemical titration of NpO2LNM with Cp*2Fe in CH3CN ([Np] = 0.45 mM; pathlength: 1 cm). | ||
3 Discussion
In this work, we have described the redox behavior of a neutral coordination compound of Np(VI) in non-aqueous media, including the structural and spectroscopic characterization of both the oxidized (NpVI) and reduced (NpV) forms of the complex which are related by 1e− transfer. Chemical and electrochemical reversibility of the accessible 1e− redox couple of NpO2LNM was established in two organic solvents (CH3CN and CH3OH) under ambient lab conditions. The Np(VI/V) reduction potential measured here is more positive than the U(VI/V) of the analogous complex in CH3CN/TBAPF6 in line with the established standard potentials in acidic aqueous media. Notably, the separation between the U(VI/V) and Np(VI/V) potentials in CH3CN/TBAPF6 measured in this work (ΔEMeCN = 1.07 V) is virtually identical to the difference between the potentials of the respective aquated species (ΔEaq = 1.08 V), suggesting that the organic ligand impacts the properties of the actinyl moieties in a virtually identical manner in both the cases of U and Np.60 This is reasonable when considering the more ionic nature of bonding in the equatorial girdle of actinyl cations compared to the axial interactions with the (yl) oxo ligands. Notably, this observation may suggest that reduction potentials should not be taken as a significant indicator of enhanced covalency of ligands in the equatorial plane surrounding U vs. Np.61 Rather, the change in potential in these systems may be driven primarily from ligand-driven electrostatic effects.Though CH3CN and CH3OH differ in their protic nature and other intrinsic properties, the metrics associated with electron transfer and diffusion for NpO2LNM are quite similar across the two solvents/electrolytes explored here. Despite this, crystallization of [NpO2LNM]− was only observed in methanolic solutions. Control electrolyses performed at 0.5 mM NpO2LNM in CH3CN/TBAPF6 only gave rise to the previously reported [NpV,NpIV,NpV] trimeric phase as a crystalline product (see ESI, Fig. S45†).35 Upon inspection, crystallization of [NpO2LNM]− appears to be encouraged by the presence of stabilizing, moderate–strength hydrogen bonding interactions between outer-sphere water molecules and the actinyl oxos directly. We suggest that acetonitrile, while capable of hydrogen bonding interactions, primarily serves as an acceptor since the only protons in acetonitrile are contained in C–H bonds; these moieties could serve, at best, as intrinsically weak H-bonding donors. Furthermore, classic “cation–cation interactions” of the Sullivan type appear to be disfavored in our system for two reasons: the equatorial coordinative saturation provided by the pentadentate ligand and the diffuse nature of the counter cation (tetrabutylammonium). Indeed when a tetradentate Schiff base ligand was employed with Np(V) in the presence of more charge-dense potassium ions, a tetrameric product was obtained which featured both Np(V)–Np(V) CCIs as well as interactions of the neptunyl(V) oxos with K+.62 Thus we hypothesize that Np(V) in [NpO2LNM]− is likely stabilized in its monomeric form by hydrogen bonding interactions with methanol and water in solution, but that the interactions with water are stronger and give rise to crystallization of the monomer in the solid state. Notably, hydrogen bonding has also been invoked as a stabilizing factor for Np(VI) in solutions containing high concentrations of NEt4Cl.63
The ability to generate and characterize the reduced, anionic form of NpO2LNM is a key highlight of this work. This ability, in turn, enabled both structural and spectroscopic insight into the oxidized and reduced species which are related through the redox couple measured by voltammetry. Despite studies of aqueous Np(VI/V) redox present in the literature, examination of Np(VI/V) redox in non-aqueous media has received far less attention than it deserves. In addition, though Np(V) is more stable than Np(VI) in acidic aqueous solutions, the dianionic organic ligand employed here appears to stabilize Np(VI) over Np(V). In line with this view, we have observed that solutions containing the Np(VI) species are stable over time on the basis of extended spectroscopic monitoring of NpO2LNM in non-aqueous solutions.35 Thus, this work represents a contribution toward understanding Np(VI/V) electron transfer behaviors and chemical reactivity patterns in non-aqueous media. The evidence from vibrational and optical spectroscopy assembled here suggests that the primary site of reduction is the Np center; there is virtually no evidence that would be suggestive of reduced ligand character in this system, although the analogous case with U appears more likely to be impacted by partial reduced ligand character. Notably, the structure of [NpO2LNM]− is distinctive from those reported in our recent study of PCET-driven reactivity of Np(VI) in that the Np(V) actinyl remains intact and monomeric.35 Though the dimeric [NpV,NV] and trimeric [NpV,NpIV,NpV] compounds can be generated by bulk electrolysis under conditions where the concentration of Np is higher, we have found that the monomeric [NpO2LNM]− species is selectively formed when the concentration of Np is low (0.5 mM) and when the electrolysis is performed in CH3OH-based electrolyte. The involvement of PCET as the mechanism of formation for the dimeric [NpV,NV] complex in particular may at first glance seem to counter the results presented here, although we would propose that a low concentration of Np favors for the generation of the monomeric Np(V) species over the dimer or trimer (i.e., concentration dependence is a hallmark of polymerization reactivity, further supporting this proposition). Additionally, we note that differences in reactivity between the CH3OH and CH3CN electrolytes are worthy of future study, particularly given the presence of adventitious H2O in both under our experimental conditions.
Electroanalytical work of the type presented here is attractive from the standpoint that it offers insights into both the thermodynamics and kinetics of electron transfer in transuranic compounds in non-aqueous systems. Through studies of this type, the redox processes that underpin selectivity in separations systems and reaction sequences can be better understood. This work represents a methodology or conceptual framework which can provide robust information on fundamental chemical and electrochemical properties. By rigorously characterizing individual features across both chemical and electrochemical work, information regarding speciation of actinyl species in different oxidation states in non-aqueous media can be pursued step by step. We have found, for example, that the diffusion coefficients of the oxidized and reduced forms, as well as heterogeneous electron transfer rate constants, can be readily extracted from the simulated data in a straightforward manner. The excellent agreement of these parameters with those of other similar compounds which display chemically and electrochemically reversible electron transfer illustrates the robust nature of our experimental methods as experimentally measured values of concentration, electrode area, and solution resistance served as direct inputs for the model. Along the same line, considering the high cost and experimental challenges associated with handling transuranium compounds, simulation of voltammetry data to extract fundamental parameters can “make the most” of even limited electroanalytical datasets.
In closing, we wish to foreground the modest value of the Np(VI/V) reduction potential measured in this work. The rather positive value of this potential not only enables the isolation of the reduced Np(V) species itself, but it also enables the direct comparison of structural and spectroscopic properties across the series of the isostructural U(V) and isoelectronic Pu(VI) analogues. This result is quite fortuitous, in that localization of the reduction potential within a convenient potential range affords openings for future work based on the intrinsically chemically reversible Np(VI)/Np(V) redox manifold measured here. Surely significant opportunities lie in electroanalytical work probing, for example, reduction-induced reactions with exogeneous reagents. Taken together, this study represents the elucidation of a molecular neptunyl-containing system that can be a platform for future work; as it displays well-behaved redox, it is a demonstration of the ability to develop a thorough understanding of actinyl redox chemistry by the strategic combination of electroanalytical experiments, voltammetric simulations, and parallel chemical work.
4 Conclusions
Here, we have reported an electroanalytical approach to outline the redox chemistry of a neutral Np(VI) complex in two different non-aqueous solvents. This report represents, in our view, perhaps the most thorough documentation of the electrochemical properties of a neptunyl complex under non-aqueous conditions that has been assembled so far. A modest reduction potential was measured for the Np(VI/V) couple in comparison to that of an analogous U(VI/V) system in acetonitrile, with an even more positive potential measured for Np(VI/V) in methanol. Diffusion coefficients of the oxidized and reduced forms of the Np complex are in good agreement with the control U species and confirm the consistent solubility and monomeric nature of the oxidized and reduced species across the timescale of the experiment. Additionally, simulations and spectroelectrochemical work provide complementary evidence that the Np(VI/V) couple is 1e− in nature; this manifold is also chemically reversible and electrochemically quasi-reversible, although we note that the simulated heterogeneous electron transfer rate constants are the fast limit of the formally quasi-reversible regime. Isolation of the Np(V) complex has enabled direct structural, spectroscopic, and electrochemical comparisons with the Np(VI) form and the isostructural U(V) and isoelectronic Pu(VI) forms. Quantitative analysis with parallel electrochemical and chemical methods have enabled characterization of the Np(VI/V) couplein this work, and similar analyses are now underway on an analogous plutonium system in our laboratories.5 Experimental
5.1 Materials and methods
Caution! 237Np is an α-emitting isotope with a specific activity of 660 μCi per gram. The primary decay product, 233Pa, is also a β- and γ-emitter. Consequently, samples containing 237Np pose significant health risks and should be handled only in dedicated laboratory facilities with strict radiological controls. Thus, all experiments were conducted using appropriate radiological controls in purpose-built laboratories for the safe handling of radionuclides. All reactions were performed under ambient laboratory conditions, and all materials, with the exception of 237Np, were obtained from commercial chemical sources and used as received. NpO2LNM was synthesized according to literature procedure.265.2 Single-crystal X-ray diffraction
Crystals suitable for single crystal X-ray diffraction were picked from the reaction mixture and affixed to thin glass fibers by using a quick setting epoxy. Data were collected on a Bruker APEX II diffractometer using Mo Kα radiation. All data manipulations for these structures, including data collection, integration, and scaling, were carried out using the Bruker APEX2 or APEX4 Software Suites.64,65 Absorption corrections were applied using the program SADABS.70 Intrinsic phasing methods were used for the structure solutions employing SHELXT with subsequent refinements of the structure solution using full-matrix least-squares refinements on F2 using SHELXL within the ShelXle and Olex2 GUIs.66–69 Crystallographic data reported here have been deposited with the Cambridge Crystallographic Data Centre under accession codes 2373058, 2379285, and 2379286.†The final structural model for each compound incorporated anisotropic thermal parameters for all nonhydrogen atoms; isotropic thermal parameters were used for all included hydrogen atoms. Hydrogen atoms associated with the ligand backbone in each complex were fixed at idealized riding model sp2- or sp3-hybridized positions with C–H bond lengths of 0.95–0.99 Å. Methyl groups were incorporated into the structural models either as sp3-hybridized riding model groups with idealized “staggered” geometry and a C–H bond length of 0.98 Å or as idealized riding model rigid rotors (with a C–H bond length of 0.98 Å) that were allowed to rotate freely about their C–C bonds in least-squares refinement cycles. The isotropic thermal parameters of idealized hydrogen atoms in all three structures were fixed at values 1.2 (non-methyl) or 1.5 (methyl) times the equivalent isotropic thermal parameter of the carbon or oxygen atom to which they are covalently bonded. With respect to the hydrogen atoms on the outer-sphere water molecules, an initial check was done for Q peaks with reasonable positions and magnitudes. If found, hydrogen atoms were placed there and fixed. When the hydrogen atoms were not found in the difference map, they positions were refined to idealized positions and fixed (see the Special Refinement Details for each structure in the ESI† for more information).
5.3 Spectroscopy
Raman spectra were collected on single crystals harvested directly from the reaction vessels and used for single-crystal X-ray diffraction measurements. Spectra were collected using a Renishaw in-via confocal Raman microscope with an excitation line of 785 nm and 10% laser power. Solid samples were placed on a microscope slide with a concave cavity and covered with a glass coverslip affixed with epoxy. UV-vis-NIR spectra of solution phase samples were collected using an Ocean Optics Flame spectrometer equipped with a Mikropack light source using quartz or plastic cuvettes.5.4 Electrochemistry
Electrochemical experiments were carried out under ambient conditions in a hood. Tetra(n-butylammonium) hexafluorophosphate (Sigma-Aldrich; 0.1 M in acetonitrile) served as the supporting electrolyte, unless otherwise specified. Measurements were made with a BAS100B Potentiostat/Galvanostat using a standard three-electrode configuration. The working electrode was the basal plane of highly-oriented pyrolytic graphite (A = 0.09 cm2), the counter electrode was a platinum wire, and a silver wire immersed in electrolyte served as a pseudo-reference electrode. The reference was separated from the working solution by a Vycor frit. Ferrocene (Sigma Aldrich, twice-sublimed; 0.1 M) was measured in the same electrolyte solution at the beginning of each experiment and the midpoint potential of the ferrocenium/ferrocene couple (denoted as Fc+/0) served as an external standard for comparison of the recorded potentials.Spectroelectrochemistry was carried out under ambient conditions as described above with electrolyte composed of 0.1 M [nBu4N][PF6] in CH3CN or CH3OH as noted. A thin layer quartz cell was used with a Teflon cap for housing the electrodes (BASi, West Lafayette, IN; path length: 1.0 mm). The working electrode was a platinum mesh flag electrode covered with a PTFE shrink tube up to the flag, the counter electrode was a platinum wire, and the silver pseudo-reference (as described above) served as the reference electrode.
Bulk electrolysis was carried out in a two-compartment H-cell in 0.1 M TBAPF6 in CH3CN or CH3OH with the same pseudo-reference as used in the cyclic voltammetry, a Pt coil for the working electrode, and a Pt wire for the counter electrode. The solutions were stirred in bulk electrolysis experiments.
Electrochemical simulations were carried out using DigiElch8.FD.
Data availability
Data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited with Cambridge Crystallographic Data Centre under accession numbers 2373058, 2379285, and 2379286.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted at Argonne National Laboratory, operated by UChicago Argonne LLC for the United States Department of Energy and supported by the U.S. Department of Energy Office of Science, Office of Basic Energy Sciences, Chemical Sciences Geological and Biosciences Division, Heavy Element Chemistry program under Contract DE-AC02-06CH11357. This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists, Office of Science Graduate Student Research (SCGSR) program. E. R. M. was supported by the SCGSR program, which is administered by the Oak Ridge Institute for Science and Education for the DOE under contract number DE-SC0014664. This work was also supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences through the Early Career Research Program (DE-SC0019169).References
- J. Sullivan and J. Hindman, The hydrolysis of neptunium(IV), J. Phys. Chem., 1959, 63, 1332–1333 CrossRef CAS.
- G. H. Coleman, The Radiochemistry of Plutonium, United States Atomic Energy Commission, Livermore, California, 1965 Search PubMed.
- A. B. Kersting, D. W. Efurd, D. L. Finnegan, D. J. Rokop, D. K. Smith and J. L. Thompson, Migration of plutonium in ground water at the Nevada Test Site, Nature, 1999, 397, 56–59 CrossRef CAS PubMed.
- A. P. Kovikov, S. N. Kalmyov, S. Utsunomiya, R. C. Ewing, F. Horreard, A. Merkulov, S. B. Slark, V. V. Takechev and B. F. Myasoedov, Colloid Transport of Plutonium in the Far-field of the Mayak Production Association, Russia, Science, 2006, 314, 638–641 CrossRef PubMed.
- L. S. Natrajan, A. N. Swinburne, M. B. Andrews, S. Randall and S. L. Heath, Redox and environmentally relevant aspects of actindie(IV) coordination chemistry, Coord. Chem. Rev., 2014, 266–267, 171–193 CrossRef CAS.
- H. Steele and R. J. Taylor, A theoretical study of the inner-sphere disproportionation reaction mechanism of the pentavalent actinyl ions, Inorg. Chem., 2007, 46, 6311–6318 CrossRef CAS PubMed.
- J. L. Hoard and J. V. Silverton, Stereochemistry of Discrete Eight-Coördination. I. Basic Analysis, Inorg. Chem., 1963, 2, 235–242 CrossRef CAS.
- E. L. Muetterties and C. M. Wright, Molecular polyhedra of high co-ordination number, Q. Rev., Chem. Soc., 1967, 21, 109–194 RSC.
- G. A. Burney and R. M. Harbour, The Radiochemistry of Neptunium, United States Atomic Energy Commission, Aiken, South Carolina, 1974 Search PubMed.
- J. E. Gindler, The Radiochemistry of Uranium, United States Atomic Energy Commission, Lemont, Illinois, 1992 Search PubMed.
- D. L. Clark, D. E. Hobart and M. P. Neu, Actinide carbonate complexes and their importance in actinide environmental chemistry, Chem. Rev., 1995, 95, 25–48 CrossRef CAS.
- J. C. Sullivan, J. C. Hindman and A. J. Zielen, Specific Interaction between Np(V) and U (VI) in aqueous perchloric acid media1, J. Am. Chem. Soc., 1961, 83, 3373–3378 CrossRef CAS.
- J. M. Cleveland, The Chemistry of Plutonium, Gordon and Breach, Science Publishers, Inc., New York, NY, 1970 Search PubMed.
- M. M. Pyrch, J. L. Bjorklund, J. M. Williams, D. L. Parr, S. E. Mason, J. Leddy and T. Z. Forbes, Impacts of hydrogen bonding interactions with Np(V/VI)O2Cl4 complexes: vibrational spectroscopy, redox behavior, and computational analysis, Dalton Trans., 2020, 49, 6854–6866 RSC.
- M. M. Pyrch, L. J. Augustine, J. M. Williams, S. E. Mason and T. Z. Forbes, Use of vibrational spectroscopy to identify the formation of neptunyl-neptunyl interactions: a paired density functional theory and Raman spectroscopy study, Dalton Trans., 2022, 51, 4772–4785 RSC.
- L. J. Augustine, M. M. F. Pyrch, D. V. Kravchuk, J. M. Williams, S. E. Mason and T. Z. Forbes, Density Functional Theory Guided Investigation of Ligand-Induced Neptunyl-Neptunyl Interactions, Eur. J. Inorg. Chem., 2023, 26, e202200693 CrossRef CAS.
- J. C. Sullivan, Complex Ion Formation between Cations. Spectra and Identification of a Np(V)-Cr(III) Complex, J. Am. Chem. Soc., 1962, 84, 4256–4259 CrossRef CAS.
- J. C. Sullivan, The Formation of a Neptunium(V)-Chromium(III) Complex. Kinetics and Equilibria in Perchlorate Solutions, Inorg. Chem., 1964, 3, 315–319 CrossRef CAS.
- R. K. Murmann and J. C. Sullivan, The formation of a neptunium(V)-rhodium(III) complex. Kinetics and equilibriums in acidic solutions, Inorg. Chem., 1967, 6, 892–900 CrossRef CAS.
- D. Palmer, R. Guillaumont, T. Fanghaenel, V. Neck, J. Fuger, I. Grenthe and M. Rand, Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium, and Technitium, Elsevier, 2003 Search PubMed.
- C. E. Plock, Voltammetric determination of neptunium at the glassy carbon electrode, J. Electroanal. Chem., 1968, 18, 289–293 CrossRef CAS.
- F. Couffin, Ph.D. thesis, L'Universite Paris XI, 1979.
- B. E. Klamm, C. J. Windorff, C. Celis-Barros, M. J. Beltran-Leiva, J. M. Sperling and T. E. Albrecht-Schönzart, Exploring the oxidation states of neptunium with Schiff base coordination complexes, Inorg. Chem., 2020, 59, 18035–18047 CrossRef CAS PubMed.
- A. J. Unger and M. P. Jensen, Spectroscopic and Electrochemical Investigation of Uranium and Neptunium in Chloride Room-Temperature Ionic Liquids, Inorg. Chem., 2023, 62, 5186–5199 CrossRef CAS PubMed.
- K. S. Otte, J. E. Niklas, C. M. Studvick, A. C. Boggiano, J. Bacsa, I. A. Popov and H. S. La Pierre, Divergent Stabilities of Tetravalent Cerium, Uranium, and Neptunium Imidophosphorane Complexes, Angew. Chem., Int. Ed., 2023, 62, e202306580 CrossRef CAS PubMed.
- E. R. Mikeska, R. E. Wilson, A. Sen, J. Autschbach and J. D. Blakemore, Preparation of Neptunyl and Plutonyl Acetates to Access Non-Aqueous Transuranium Coordination Chemistry, J. Am. Chem. Soc., 2024, 146, 21509–21524 CrossRef PubMed.
- V. Mougel, J. Pécaut and M. Mazzanti, New polynuclear U(IV)-U(V) complexes from U(IV) mediated uranyl(V) disproportionation, Chem. Commun., 2012, 48, 868–870 RSC.
- E. R. Mikeska and J. D. Blakemore, Evidence for Reactivity of Decamethylcobaltocene with Dichloromethane, Organometallics, 2023, 42, 1444–1447 CrossRef CAS.
- R. R. Golwankar, M. Z. Makos, N. Gajiao, M. S. Neidig, A. G. Oliver, C. S. Day, V. W. Day, V.-A. Glezakou and J. D. Blakemore, Electrochemical Activation and Functionalization of the Uranyl Ion, ChemRxiv, 2023. DOI:10.26434/chemrxiv-2023-gnbpd.
- K. Takao, M. Kato, S. Takao, A. Nagasawa, G. Bernhard, C. Hennig and Y. Ikeda, Molecular Structure and Electrochemical Behavior of Uranyl(VI) Complex with Pentadentate Schiff Base Ligand: Prevention of Uranyl(V) Cation–Cation Interaction by Fully Chelating Equatorial Coordination Sites, Inorg. Chem., 2010, 49, 2349–2359 CrossRef CAS PubMed.
- M. Augustin, H. J. Kerrinnes and W. Langenbeck, Chelates of β,β′-bis(salicylaldimino)diethylamine, J. Prakt. Chem., 1964, 26, 130–136 CrossRef CAS.
- M. N. Akhtar, E. D. McKenzie, R. E. Paine and A. J. Smith, Seven coordinate uranyl compounds of the planar quinquedentate ligand N,N′-disalicylidene-1,5-diamino-3-azapentane, Inorg. Nucl. Chem. Lett., 1984, 5, 673–677 CrossRef.
- D. E. Fenton, U. Casellato, P. A. Vigato and M. Vidali, Acyclic and macrocyclic Schiff base complexes of lanthanides and actinides, Inorg. Chim. Acta, 1984, 95, 187–193 CrossRef CAS.
- N. A. Bailey, D. E. Fenton, C. A. Phillips, U. Casellato, S. Tamburini, P. A. Vigato and R. Graziani, The preparation and crystal structure of a dioxouranium(VI) and of an oxovanadium(IV) complex with a hexadentate, compartmental Schiff base ligand, Inorg. Chim. Acta, 1984, 109, 91–98 CrossRef.
- E. R. Mikeska, R. E. Wilson and J. D. Blakemore, PCET-driven Reactivity of Neptunyl(VI) Yields Oxo-bridged Np(V) and Np(IV) Species, ChemRxiv, 2024. DOI:10.26434/chemrxiv-2024-3rrhz.
- N. Srinivasan, M. V. Ramaniah, S. K. Patil and V. V. Ramakrishna, Estimation of neptunium in a fuel reprocessing plant, J. Radioanal. Chem., 1971, 8, 223–229 CrossRef CAS.
- P. K. Verma and P. K. Mohapatra, Fate of Neptunium in nuclear fuel cycle streams: state-of-the art on separation strategies, Radiochim. Acta, 2022, 110, 527–548 CrossRef CAS.
- S. E. Gilson and P. C. Burns, The crystal and coordination chemistry of neptunium in all its oxidation states: An expanded structural hierarchy of neptunium compounds, Coord. Chem. Rev., 2021, 445, 213994 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, NY, 1997, p. 12 Search PubMed.
- E. R. Mikeska, A. C. Ervin, K. Zhang, G. M. Benitez, S. M. R. Powell, A. G. Oliver, V. W. Day, M. Caricato, C. G. Comadoll and J. D. Blakemore, Evidence for Uranium(VI/V) Redox Supported by 2,2′-Bipyridyl-6,6′-dicarboxylate, Inorg. Chem., 2023, 62, 16131–16148 CrossRef CAS PubMed.
- C. Madic, G. M. Begun, D. E. Hobart and R. L. Hahn, Raman spectroscopy of neptunyl and plutonyl ions in aqueous solution: hydrolysis of neptunium(VI) and plutonium(VI) and disproportionation of plutonium(V), Inorg. Chem., 1984, 23, 1914–1921 CrossRef CAS.
- D. L. Clark, D. W. Keogh, P. D. Palmer, B. L. Scott and C. D. Tait, Synthesis and Structure of the First Transuranium Crown Ether Inclusion Complex: Crown-6)ClO4, Angew. Chem., Int. Ed., 1998, 37, 164–166 CrossRef CAS.
- M. K. Payne, M. M. Pyrch, M. Jubinsky, M. C. Basile and T. Z. Forbes, Impacts of oxo interactions within actinyl metal organic materials: highlight on thermal expansion behaviour, Chem. Commun., 2018, 54, 10828–10831 RSC.
- M. M. Pyrch, J. L. Bjorklund, J. M. Williams, D. L. Parr IV, S. E. Mason, J. Leddy and T. Z. Forbes, Impacts of hydrogen bonding interactions with Np(v/vi)O2Cl4 complexes: vibrational spectroscopy, redox behavior, and computational analysis, Dalton Trans., 2020, 49, 6854–6866 RSC.
- M. Autillo, R. E. Wilson, M. Vasiliu, G. F. de Melo and D. A. Dixon, Periodic Trends within Actinyl(VI) Nitrates and Their Structures, Vibrational Spectra, and Electronic Properties, Inorg. Chem., 2022, 61, 15607–15618 CrossRef CAS PubMed.
- G. B. Andreev, A. M. Fedosseev, N. A. Budantseva and M. Y. Antipin, Structural and spectral investigation of a neptunium(V) complex with 2,2′-bipyridine, Mendeleev Commun., 2001, 11, 58–59 CrossRef.
- A. M. Fedosseev, G. B. Andreev, N. A. Budantseva and J. C. Krupa, Synthesis, Crystal Structure and Properties of Np(V) Compounds Formed with N-bearing Ligands, J. Nucl. Sci. Technol., 2002, 39, 414–417 CrossRef.
- G. H. John, I. May, M. J. Sarsfield, H. M. Steele, D. Collison, M. Helliwell and J. D. McKinney, The structural and spectroscopic characterisation of three actinyl complexes with coordinated and uncoordinated perrhenate: [UO2(ReO4)2(TPPO)3], [(UO2)(TPPO)32(μ2-O2)][ReO4]2 and [NpO2(TPPO)4][ReO4], Dalton Trans., 2004, 734–740 RSC.
- A. G. Samartseva and Z. A. Zhuravleva, Adsorption of Neptunium on a Polished Platinum Surface, Radiokhimiya, 1973, 15, 161–167 CAS.
- R. E. Meyer, W. D. Arnold, F. Case, S. Y. Shiao and D. A. Palmer, Valence Effects on Adsorption, U.s. nuclear regulatory commission technical report, 1983.
- J.-M. Savéant and C. Constentin, Elements of Molecular and Biomolecular Electrochemistry, John Wiley & Sons, Inc., Hoboken, NJ, 2nd edn, 2019, pp. 25–27 Search PubMed.
- F. C. Anson, Innovations in the Study of Adsorbed Reactants by Chronocoulometry, Anal. Chem., 1966, 38, 54–57 CrossRef CAS.
- D. Britz, Digital Simulation in Electrochemistry, Springer Cham, 2016 Search PubMed.
- A. M. Bond, K. B. Oldham and G. A. Snook, Use of the Ferrocene Oxidation Process To Provide Both Reference Electrode Potential Calibration and a Simple Measurement (via Semi-integration) of the Uncompensated Resistance in Cyclic Voltammetric Studies in High-Resistance Organic Solvents, Anal. Chem., 2000, 72, 3492–3496 CrossRef CAS PubMed.
- Y. Wang, E. I. Rogers and R. G. Compton, The Measurement of the Diffusion Coefficients of Ferrocene and Ferocenium and their Temperature Dependence in Acetonitrile Using Double Potential Step Microdisc Electrode Chronoamperometry, J. Electroanal. Chem., 2010, 648, 15–19 CrossRef CAS.
- D. K. Gosser Jr., Cyclic Voltammetry: Simulation and Analysis of Reaction Mechanisms, Wiley-VCH, Inc., New York, NY, 1993 Search PubMed.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- R. R. Golwankar, A. C. Ervin, M. Z. Makoś, E. R. Mikeska, V.-A. Glezakou and J. D. Blakemore, Synthesis, Isolation, and Study of Heterobimetallic Uranyl Crown Ether Complexes, J. Am. Chem. Soc., 2024, 146, 9597–9604 CrossRef CAS PubMed.
- P. G. Hagan and J. M. Cleveland, The Absorption Spectra of Neptunium Ions in Perchloric Acid Solution, J. Inorg. Nucl. Chem., 1966, 28, 2905–2090 CrossRef CAS.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, M. Dekker, New York, 1985 Search PubMed.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, Energy-Degeneracy-Driven Covalency in Actinide Bonding, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- R. Copping, V. Mougel, C. Den Auwer, C. Berthon, P. Moisy and M. Mazzanti, A tetrameric neptunyl(v) cluster supported by a Schiff base ligand, Dalton Trans., 2012, 41, 10900–10902 RSC.
- S. L. Estes, B. Qiao and G. B. Jin, Ion association with tetra-n-alkylammonium cations stabilizes higher-oxidation-state neptunium dioxocations, Nat. Commun., 2019, 10, 59 CrossRef CAS PubMed.
- SAINT, Ver 8.34A, Bruker Analytical X-ray Systems, Madison, WI, 2014 Search PubMed.
- APEX2, Version 2 User Manual, M86-E01078, Bruker Analytical X-ray Systems, Madison, WI, 2006 Search PubMed.
- G. M. Sheldrick, SHELXT - Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hubschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt Graphical User Interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, Olex2: a complete structure solution, refinement, and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SADABS, version 2.10, University of Göttingen, Germany, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical data, Raman and optical spectra, and detailed information about X-ray crystallographic data (PDF). Cartesian coordinates for the XRD structure (XYZ). CCDC 2373058, 2379285 and 2379286. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02557a |
| This journal is © The Royal Society of Chemistry 2024 |