
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, characterization, and release profile of a cannabidiol (CBD)-rich polyvinyl alcohol hydrogel†
Shujun
Cui‡
ab,
Maryam
Bahraminia‡
ab,
Mahmoud
Rouabhia
 *a,
Abdelhabib
Semlali
a,
François
Béland
c and
Ze
Zhang
b
*a,
Abdelhabib
Semlali
a,
François
Béland
c and
Ze
Zhang
b
aGroupe de Recherche en Écologie Buccale, Faculté de Médecine Dentaire Université Laval, Quebec, QC G1V 0A6, Canada. E-mail: Mahmoud.Rouabhia@fmd.ulaval.ca
bAxe Médecine Régénératrice Centre de Recherche du CHU de Québec – Université Laval, Département de Chirurgie Faculté de Médecine, Université Laval, Quebec, QC G1L 3L5, Canada
cSiliCycle® Inc. 2500, Parc-Technologique Blvd, Québec, QC G1P 4S6, Canada
First published on 20th August 2024
Abstract
Cannabidiol (CBD) has been reported to have a plethora of therapeutic opportunities in many diseases. Current CBD delivery systems include suspension, emulsion, or nanoparticles, which enable the rapid release of CBD. However, in some cases, long-term exposure to drugs is preferred, such as in controlling inflammation. This study aimed to prepare a CBD-rich hydrogel that exhibits controlled and sustained release of CBD while maintaining CBD bioactivity. Herein, PVA mixed with propylene glycol (PG) or vegetable glycerine (VG) in the presence of CBD were used to fabricate hydrogels through a cyclic freeze–thaw process. The successful loading of CBD inside the PVA gel was macroscopically demonstrated by the uniform pink color of the gel and chemically identified by the characteristic absorptions of CBD at 1630 and 1585 cm−1 in Fourier transform infrared spectroscopy (FTIR). Scanning electron microscopy (SEM) revealed small, fine pores dispersed throughout the gels when PG or VG was added. Differential scanning calorimeter (DSC) curves showed that the gels containing VG were more amorphous than those without VG. Thermogravimetric analyzer (TGA) results showed that the CBD-containing gels were stable up to 100 °C when they started to lose water and up to about 200 °C when they started to lose other molecules such as PG, indicating a good shelf-life. A compression test showed that the hydrogel was soft and could tolerate more than 60% deformation and 45 kPa pressure. CBD in vitro release assessment through ultraviolet-visible (UV-vis) spectrophotometer and UPLC-MS/MS analysis demonstrated that the hydrogel can continually release CBD for at least 24 h. Adding VG could significantly reduce PVA crystallinity and promote the release of CBD from the hydrogel into an aqueous solution; adding PG and VG together can regulate the release rate of CBD. Finally, 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay proved that the CBD released from the hydrogels through 24 h had good antioxidant activity. Considering the biocompatible nature of PVA, the CBD-enriched PVA hydrogels developed in this study can be used to topically deliver CBD to tissues or the body to treat inflammation and promote wound healing.
1. Introduction
Cannabidiol (CBD) is the second most prevalent active ingredient in cannabis (marijuana), and according to the World Health Organization report, CBD exhibits no effects indicative of abuse or dependence potential in humans.1,2 Real-world evidence suggests that CBD-rich treatment has a beneficial impact on pain, anxiety, and depression symptoms as well as overall well-being.3–6 What's more, studies have shown that CBD has direct and indirect antioxidant and anti-inflammatory effects.7 The human body is subjected to various conditions (stress, autocrine/endocrine changes, exposure to exogenous stimuli, etc.) leading to organ and tissue inflammatory disorders, such as those in the skin and the oral cavity.8,9 CBD products are therefore expected to become promising choices in controlling such tissue inflammation.Due to their distinctive characteristic properties, such as biocompatibility, hydrophilicity, viscoelasticity, softness, and permeability to oxygen and nutrients, hydrogels are very attractive in drug delivery.10–12 A CBD-rich hydrogel could be appropriate for a better-controlled delivery of CBD.13
Poly(vinyl alcohol) (PVA), a non-toxic and water-soluble polyhydroxy polymer, offers a hydrophilic surface compatible with living tissues. Furthermore, the porosity of PVA hydrogels can be tailored by physical or chemical cross-linking. These properties have prompted PVA to be used in biomedical applications such as drug delivery, wound dressing, and tissue engineering.14–17
This study investigates CBD-rich hydrogels by embedding CBD into a physically cross-linked PVA hydrogel matrix to control the release profile. The hydrogels retained CBD and released it into an aqueous solution stably. The prepared CBD-rich PVA hydrogels may have the potential as a drug carrier for the treatment of tissue inflammation.
2. Materials and methods
2.1 Reagents
PVA (#34584, Sigma-Aldrich). Sodium dodecylbenzenesulfonate (DBS, #289957, Sigma-Aldrich). Propylene glycol (PG, #398039, Sigma-Aldrich). Vegetable glycerine (VG, #1.04057, Sigma-Aldrich). High-purity grade CBD was extracted from cannabis biomass and purified by SiliCycle Inc. (Quebec, Canada).2.2 Preparation of hydrogels
The composition of the hydrogels is shown in Table 1. For the PVA-PG-VG, the ratio between PG and VG was 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 to make 5% in total, which is comparable with the other two formulations containing PG or VG. Firstly, PVA, DBS, PG, VG, or PG-VG were added to distilled water to generate a mixture in a conical centrifuge tube. The mixture was autoclaved under 121 °C for 20 minutes, stirred vigorously, and then cooled to room temperature. Afterward, 0.5 mL of the mixture was supplemented with 7.3 μL of CBD (3 mg), homogenized, and transferred to the wells of the 12-well tissue culture plates. The plates were subjected to 8 cycles of freeze–thaw. Each cycle includes a one-hour incubation at −80 °C to freeze the solution and a thawing period at room temperature. The prepared hydrogels were then used for subsequent experiments.
:2.5 to make 5% in total, which is comparable with the other two formulations containing PG or VG. Firstly, PVA, DBS, PG, VG, or PG-VG were added to distilled water to generate a mixture in a conical centrifuge tube. The mixture was autoclaved under 121 °C for 20 minutes, stirred vigorously, and then cooled to room temperature. Afterward, 0.5 mL of the mixture was supplemented with 7.3 μL of CBD (3 mg), homogenized, and transferred to the wells of the 12-well tissue culture plates. The plates were subjected to 8 cycles of freeze–thaw. Each cycle includes a one-hour incubation at −80 °C to freeze the solution and a thawing period at room temperature. The prepared hydrogels were then used for subsequent experiments.
| Samples | PVA | DBS | PG | VG | CBD |
|---|---|---|---|---|---|
| % (w/v) | % (w/v) | % (v/v) | % (v/v) | mg mL−1 | |
| PVA | 10 | 5 | — | — | 6 |
| PVA-PG | 10 | 5 | 5 | — | 6 |
| PVA-VG | 10 | 5 | — | 5 | 6 |
| PVA-PG-VG | 10 | 5 | 2.5 | 2.5 | 6 |
2.3 Morphological structure of the hydrogels
Morphological and structural characters are important because they are directly associated with hydrogel functions such as drug-releasing behaviors, and will shed light on the other properties such as visual shape, color, shrinking or not, water absorption, etc.18Following the freeze–thaw cycles, the specimens were taken out of the wells and examined manually to verify the formation and handleability of the hydrogels. They were also inspected visually to record the color, knowing that the solutions supplemented with CBD were pinkish. The hydrogels were dried at room temperature to a constant weight to evaluate how hydrogel formulations affected hydrogel shrinkage. The dried specimens were then incubated in phosphate-buffered saline (PBS) for 24 h, to assess the possibility of rehydrating them. For microscopic observations, the freeze-dried hydrogels were sputter-coated with gold (Fison Instruments, Polaron SC500, Uckfield, UK), and then subjected to scanning electron microscopy (SEM, model JSM-133 6360LV, JEOL, Tokyo, Japan) analysis at 15 kV of accelerating voltage. The samples were snap-frozen in liquid nitrogen to avoid creep deformation at cross section before being fractured. After careful observations, photomicrographs of various magnifications were taken from the representative locations of the surfaces and cross sections.
2.4 Water content and rehydration ratio of the hydrogels
Diverse medical applications require versatile hydrogels, including the one that can be dehydrated and hydrated. Dehydration allows easy storage, while hydration permits a soft tissue-like hydrogel, minimizing the frictional irritation with surrounding soft tissue.19 Furthermore, the drug release rate is highly dependent on the network parameters as well as the water content of the swollen hydrogel.20Because we intend to design a CBD-rich hydrogel for possible use in clinics, the analysis of the water content and rehydration of our hydrogels are important.
The water content of the hydrogels was determined gravimetrically. The wet weight (Ww) was measured after removing the surface moisture of the hydrogel by wiping it with a lens cleaning paper. The samples were then dried at atmosphere (25 °C, 50% humidity) until constant weight, then the dry weight (Wd) was recorded. The dried gels were finally placed in PBS for 24 h for rehydration, and the rehydrated weight (Wrhy) was recorded after wiping out the surface water. The water content was calculated according to the formula (1), and the rehydration ratio was calculated according to the formula (2). Measurements were performed 6 times for each type of hydrogel, and the results were expressed as the means ± standard deviations, shown in Table 2.
| Water content (wt%) = 100 × (Ww − Wd)/Ww | (1) |
| Rehydration ratio (wt%) = 100 × (Wrhy − Wd)/Wrhy | (2) |
| Hydrogel | Water content (%) | Rehydration ratio (%) | Swelling ratio (%) |
|---|---|---|---|
| PVA | 83.4 ± 0.2 | 77.6 ± 0.3 | 346.6 ± 5.1 |
| PVA-PG | 79.9 ± 0.2 | 76.6 ± 2.7 | 331.8 ± 25.6 |
| PVA-VG | 77.8 ± 0.3 | 72.0 ± 0.8 | 257.0 ± 6.0 |
| PVA-PG-VG | 78.9 ± 0.2 | 75.0 ± 1.2 | 300.5 ± 7.0 |
2.5 Mechanical properties of the hydrogels
Hydrogels should have the appropriate mechanical properties (compressibility/rigidity) of soft tissue to mimic mammalian soft tissue.21 Thus, we carried out compression tests to evaluate the mechanical properties of the hydrogels. To perform the test, hydrogels of 25 mm in diameter (Fisherbrand™ Traceable™ Digital Calipers) and 1 mm in thickness (MTG-DX2, Rex Gauge Company, Buffalo Grove, IL, USA) were compressed through dynamic mechanical analyzer (DMA) (RSA-3, TA Instruments, New Castle, DE) at a rate of 0.01 mm s−1 until sample failure or maximum load of the DMA. The force was measured using a 3500 g load cell. All the tests were done at room temperature with a relative humidity of 50%. The Young's modulus, which represents material rigidity, was obtained from the slope of the initial linear part of the stress–strain curves.2.6 FTIR characterization
Fourier-transform infrared (FTIR) spectroscopy can identify multiple substances in a drug sample. FTIR also provides a fast, easy, accurate, and economical way to ascertain the presence of laboratory-grade cannabis products inside a drug carrier.22 As our hydrogels contain cannabis derivatives, we performed FTIR analysis to demonstrate the presence of CBD in the hydrogels. Briefly, the hydrogels with and without CBD were air-dried and subjected to infrared spectral characterization. The Fourier transform infrared (FTIR) spectra of the hydrogels were recorded with a Nicolet Magna-IR 550 spectrophotometer (Nicolet Instrument, Madison, USA) in attenuated total internal reflectance (ATR) mode. The specimens were pressed against a hemispherical silicon crystal and scanned 64 times between 400 and 4000 cm−1 at a resolution of 4 cm−1. Considering the thickness of the membrane (ca. 300 μm) and the sampling depth of the silicon crystal (ca. 2–15 μm and is wavelength dependent), this analysis can be considered a surface analysis.232.7 Thermal analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) are routinely used in material analysis, providing essential information about material thermal behaviours and how the substances in a composite interact.The thermogravimetric analyzer TGA/SDTA 851e (Mettler-Toledo, Mississauga, Ontario, Canada) and differential scanning calorimeter (DSC 823, Mettler Toledo, Mississauga, Ontario, Canada) were used to characterize the thermal behaviors of the hydrogels. Briefly, 5–10 mg of dried hydrogel was heated at a rate of 10 °C min−1 from 25 to 800 °C for TGA and 0 to 260 °C for DSC in the atmosphere of nitrogen flowed at a rate of 20 mL min−1. The TGA and DSC tests were run in triplicate for each sample. Data analysis was performed using the instrument manufacturer's STARe software.
2.8 CBD release test
Hydrogels are cross-linked three-dimensional networks that can effectively release drugs sustainably.24 The use of hydrogels as a depot for local delivery of drugs is an approach that has gained momentum in the past decade, particularly when pathology is contained and localized.25 As our hydrogel intends to deliver CBD to soft tissue, it is critical to evaluate the release profile of the CBD. Briefly, a PBS solution was prepared as the release buffer. Each hydrogel was transferred to a well of a sterile 12-well plate containing 0.5 mL of PBS per well and incubated continuously at 37 °C for 1, 2, 4, 6, 12, and 24 h. At each time point, the PBS solutions were collected and replaced with another 0.5 mL of fresh PBS solutions. With this procedure, we can measure the release at 1, 2, 4, 6, 12, and 24 h. After the last incubation in PBS, each hydrogel was incubated for 2, 4, 6, and 24 h in 0.5 mL methanol, with a collection of the methanol and its replacement by fresh (0.5 mL) methanol at each time point. The incubation of the hydrogels in methanol allowed the residual CBD to be extracted from the hydrogels after PBS incubation. All solutions were analyzed using an ultraviolet-visible (UV-vis) spectral photometer (iMark™ Microplate Absorbance Reader, Bio-Rad, Canada). Methanol solutions containing 0, 0.166, 0.332, 0.581, 0,747, and 0.996 μg mL−1 of CBD were used to plot the CBD calibration curve. The CBD release was confirmed by UPLC-MS/MS analysis.2.9 UV-vis spectral measurement of CBD
The amount of CBD was measured using an iMark™ Microplate Absorbance Reader (Bio-Rad, Canada) in absorbance mode. Briefly, three replicates of 100 μL of PBS or methanol solutions collected in the CBD release experiment were transferred to a Sarstedt 3924 96-well standard microplate (polystyrene, flat-base, SARSTEDT Company, Nümbrecht, Germany), and the absorbance between 200 and 600 nm was recorded. The MPM6 software (Bio-Rad, Canada) was used to adjust parameters and record data. The experiment was repeated three times. The CBD standard curve was used to quantify CBD in the solutions. The total amount of CBD refers to the CBD released in PBS plus the CBD released in methanol. The percentage of release was calculated by dividing the CBD released into a solution with the total amount of CBD and multiplying it by 100.2.10 UPLC-MS/MS analysis of the CBD from the hydrogels
Supernatant collected from each hydrogel was used to measure the levels of released CBD. The measurements were performed by TransBioTech (Quebec, Qc, Canada). The following chemicals were used: methanol (MeOH), ethyl acetate, formic acid, acetonitrile, and isopropanol (Fisher Scientific). A CBD standard curve was generated using a CBD solution at 1.0 mg mL−1 (Sigma Aldrich Canada Ltd). To measure the levels of CBD in our samples, 100 μL of each supernatant was mixed with 400 μL of H2O, plus 500 μL of ethyl acetate, mixed for 20 s, and then centrifuged for 5 min in a microfuge. The supernatant was transferred to a reduced surface activity (RSA) vial. The extraction with ethyl acetate was repeated once more, then the supernatants were combined in the RSA vial and evaporated in a dry atmosphere under nitrogen at 30 °C. After drying, 100 μL of MeOH/H2O (80%/20%) solution was added to the RSA tube, mixed then incubated in an ultrasound bath for 5 min. The solution was then injected in Ultra-high performance liquid chromatography-MS/MS (UPLC-MS/MS) (UPLC Waters Xevo TQ-S micro). A standard curve was prepared using various concentrations (0.5 up to 500 ng mL−1) of CBD. All samples were analyzed using a Phenomenex Luna OMEGA C18, 2.1 × 150 mm, 1.6 μm reversed-phase column, at 40 °C. The mobile phases and gradient were as follows: mobile phase A: 0.1% formic acid in H2O; mobile phase B: 0.1% formic acid in acetonitrile/isopropanol (80/20). Flow rate: 0.3 mL min−1. The programmed step gradient was 55% B over 2 min, 55–100% B over 7 min, and 100–55% B over 0.1 min. MS parameters in positive mode: desolvation temperature: 450 °C; desolvation gas flow: 800 L h−1; source temperature: 150 °C; impactor: 2 kV; cone: 40 V, collision: 20 V. Collected values were used to determine the level of CBD in each supernatant, based on the standard curve.2.11 Antioxidant activity test
The free radical scavenging activity of the CBD released from the hydrogels was evaluated by 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay. Briefly, 0.2 mM fresh solution of DPPH in methanol was prepared, and an aliquot of 150 μL of the DPPH solution was mixed with 50 μL of sample solution collected from the CBD release experiment in a 96-well microplate. The mixture was incubated at room temperature for 30 min. Then, the absorbance was measured at 515 nm using the UV-vis spectrophotometer. Sample solutions collected from the hydrogels without CBD were used as the blank (A0). Sample solutions collected from the hydrogels that contained CBD were recorded as A1. A lower absorbance value indicates a higher free radical scavenging activity. The capability of scavenging the DPPH radicals was calculated using the following formula. All the tests were performed in triplicates, and the results were averaged.| DPPH scavenging capacity (%) = 100 × (A0 − A1)/A0 | (3) |
2.12 Statistical analysis
All experiments were repeated at least 3 times. The obtained results were expressed as the mean ± standard deviation. The significance in the difference between the two groups of data was assessed via the Student's t-test. P values < 0.05 were denoted as statistical significance.3. Results and discussion
3.1 Gross observation and morphological structure of the hydrogels
Through primary experiments, we observed a non-homogenous distribution of the CBD throughout the PVA hydrogel (data not shown). This can be explained by the oily structure of the CBD, which is immiscible with water. Previous studies have suggested the use of PG or VG to dissolve the CBD better in water-based hydrogels.26–28 When we add PG, VG or PG-VG to the CBD-rich PVA hydrogel, we get a homogeneous distribution throughout the hydrogel. Furthermore, all hydrogels appeared smooth and similar in size. All the hydrogels appeared light pink (Fig. 1) due to the presence of CBD. The uniform pink color indicates that the CBD oil was finely dispersed in the PVA matrix. The hydrogels were soft, elastic, and easy to handle.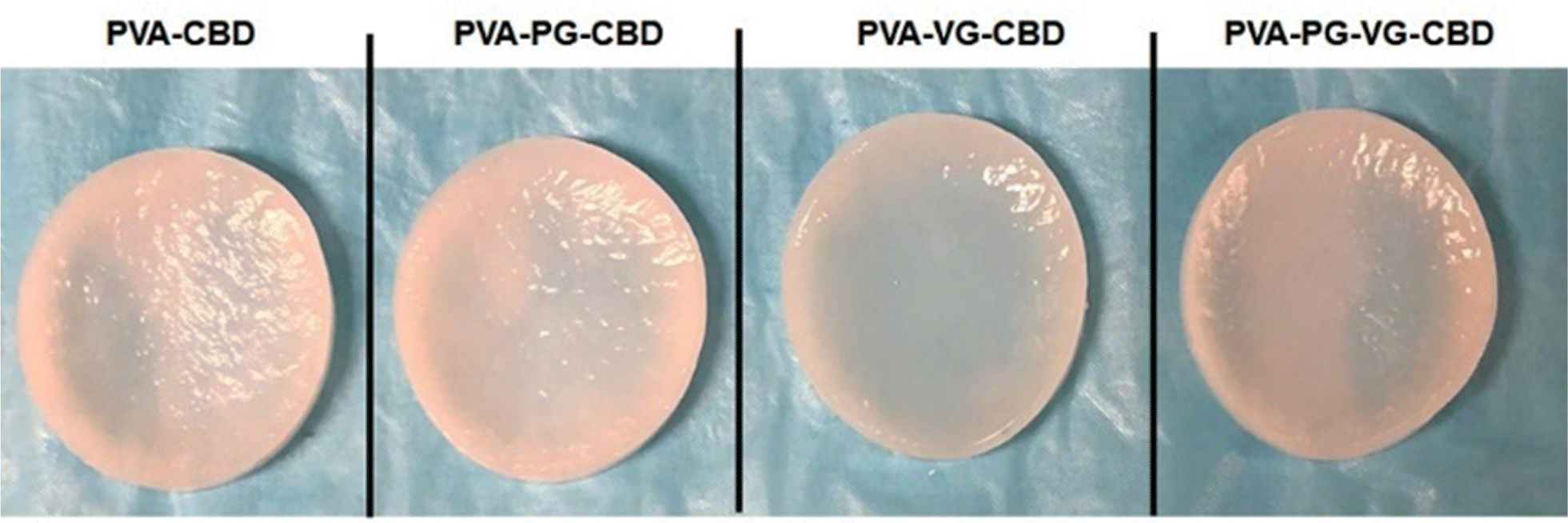 | ||
| Fig. 1 CBD-rich hydrogels produced through 8 freeze–thaw cycles. The softness, slight elasticity and toughness make the hydrogels can be comfortably handled. The uniform distribution of CBD renders the hydrogels pinkish. | ||
SEM images at cross-section (Fig. 2) showed that the PVA-PG-VG is more porous than others. The pore size and pore distribution in these three gels are more uniform than those in the PVA. The latter shows fewer pores and a wider variation in pore size. The surface of all gels is essentially non-porous.
 | ||
| Fig. 2 SEM of CBD-rich hydrogels, showing that the interiors (cross sections) of the gels are sub microporous and the surface of the gels is nonporous. The addition of PG and VG can affect the pore size and porosity inside the materials, with the PVA-PG-VG showing the highest porosity. | ||
The solutions used to prepare these hydrogels were oil-in-water emulsions. It is reasonable to believe that the pores inside the hydrogels were formed not only by ice crystals but also by the CBD-containing DBS micelles. Large pore size and more pores would favour the diffusion of CBD out of the hydrogel. Because of its non-porous nature and lower permeability, the surface layer served as a barrier that controls release speed. The SEM observations demonstrate that the presence of PG and VG affects the gel's microstructure, which is crucial for the future engineering of the products.
3.2 Water content and swelling property of the hydrogels
Table 2 shows that the hydrogel can absorb up to 83% of water, which corresponds to about 350% of the swelling ratio (Table 2). Such a water content gives the hydrogels a degree of softness and flexibility, like a human tissue. The presence of PG or VG decreased the water content, roughly about their percentages in the formula (Table 1), and there was no synergy when both PG and VG were added. The same trend was observed for the swelling ratio (Table 2). The swelling ratio is a measure of cross-link degree. Table 2 shows that the PVA-VG has the highest cross-link degree, followed by the PVA-PG-VG. It appears that adding VG can increase the cross-link degree in PVA gel.When the solutions were frozen at −80 °C, the distance between some PVA chains was reduced because of the formation of water ice crystals that occurred firstly in low PVA concentration regions and because ice crystals occupied more space than liquid water did. Some PVA molecules were eventually pushed closer enough to form hydrogen bonds, i.e., physical cross-links, between their hydroxyl groups. When the specimens were thawed at room temperature, the non-crosslinked polymer chains became unfrozen and could move freely again. The repeated freeze–thaw treatments caused more and more cross-link points to be formed, finally leading to a cross-linked stable 3D PVA network.29 Owing to a large number of hydroxyl groups and voids within the hydrogel when the dried PVA sponge was put into water, water penetrated the matrix by diffusion, capillary action, and hydrogen bonding, leading to a high volume of free and bound water integrated into the 3D network of PVA. The effect of PG and VG on water absorption ability and rehydration ratio of the hydrogels has been explained in our previous paper, and it was thought to be related to the ability of VG and PG to reduce PVA–water interactions.30
In the formulas (Table 1), PG or VG occupied 5%. In the final product (Table 2), only the PVA-VG gel showed 5% less water than the PVA gel, while the PVA-PG and PVA-PG-VG gels were 3% and 4% less, respectively. This is, probably because VG formed more trapped water inside the gel, which couldn’t be completely removed by drying it in the air. It is reasonable because VG has more hydroxyl groups than PG and can form more hydrogen bonds with water, leading to more trapped water molecules inside the gel.
3.3 Mechanical properties of the hydrogels
In the compressive strength test, owing to the excellent deformability of the PVA hydrogels, it was unable to reach sample failure at 45 kPa, as shown in Fig. 3.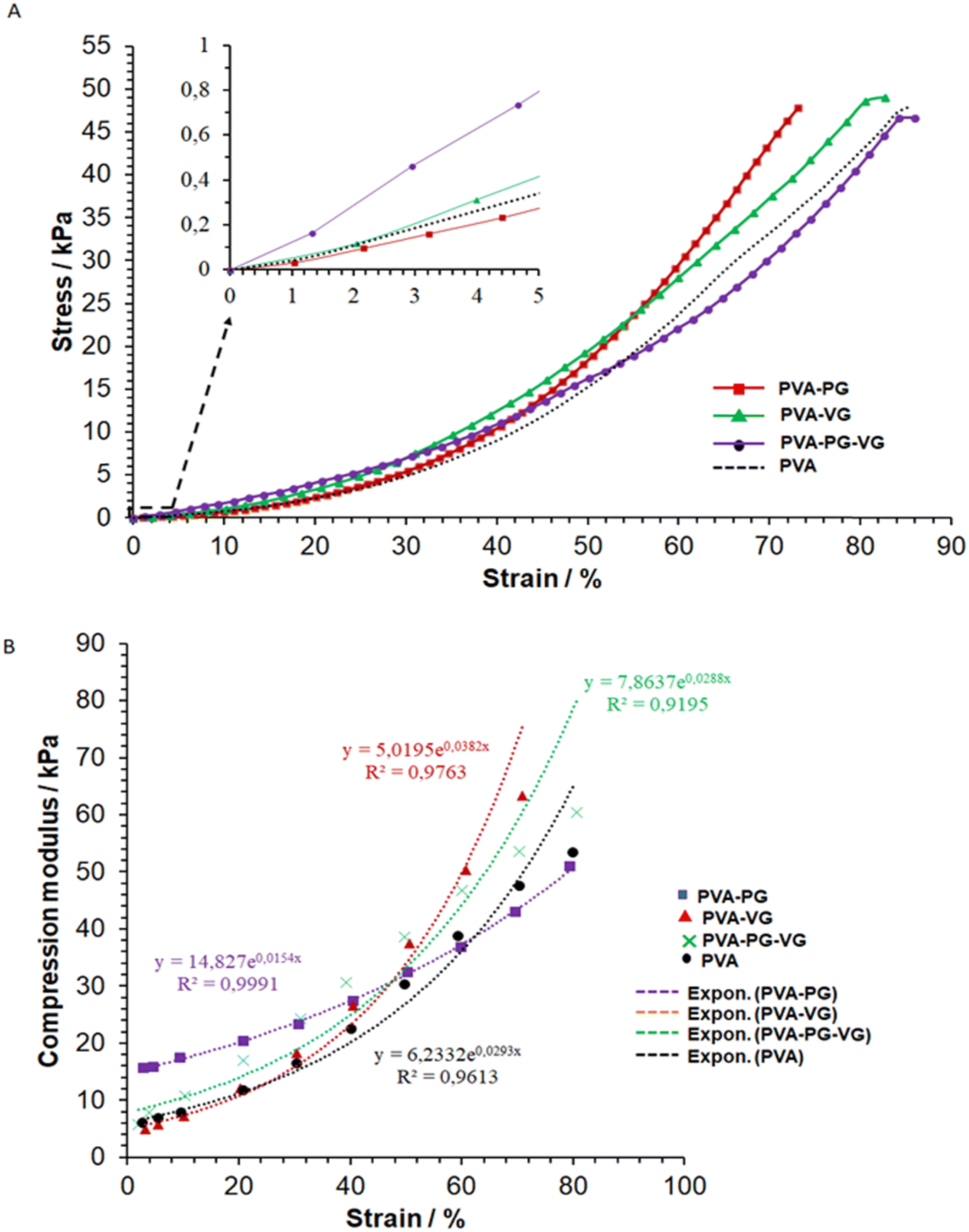 | ||
| Fig. 3 Typical stress–strain (A) and modulus–strain (B) curves of the hydrogels and the curve fittings (dot lines, (B)). | ||
Several factors contribute to the mechanical properties of the hydrogels, e.g., crystallinity, molecular entanglement, free/bound water content, and hydrogen bonding formed between PG-VG/water and PVA polymer chains. And each factor may contribute to the mechanical properties at different scales. Yong's moduli were calculated and listed in Table 3.
It shows that at a low compressive deformation, PG can increase the stiffness of the hydrogel, while VG may have the opposite effect. However, when it reached a high strain, i.e., about 45%, the PVA-VG began to show a high stress (Fig. 3A).
The fact that the PVA-PG gel was more resistant to small compressive deformation may be explained by its high crystallinity (see DSC data), which is known to increase material modulus. At high strain, however, a high degree of cross-link can reduce molecular disentanglement, leading to high stress at large deformation, which was what happened for the PVA-VG hydrogel.
Fig. 3B shows the relationship between hydrogel modulus and compression ratio, which is nonlinear. It demonstrates that the behaviors of all the gels fit an exponential relationship, with a correlation coefficient R2 of more than 0.9. Such an exponential relationship is typical of viscoelastic materials such as cross-linked hydrogels, polymeric elastomers, and natural polymers.29
Our data demonstrate that by changing the ratio of PG and VG, it is possible to prepare hydrogels mimicking the viscoelasticity of the skin.31–33
3.4 FTIR analysis
The characteristic peaks of CBD in the hydrogels are found at 1630 and 1585 cm−1 (Fig. 4), which are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching (phenyl ring) and C–H.34 In particular, the strong absorption at 1630 cm−1 is characteristic of the substituted phenyl ring, confirming the substitution of –OH groups in CBD. The CBD was, therefore, successfully preserved in the hydrogels. It can be noticed that the addition of VG and PG significantly reduced the signal of CBD on the gel surface. This could be either because the presence of VG or PG diluted CBD on the surface, or they drove CBD to the interior of the hydrogel.
C stretching (phenyl ring) and C–H.34 In particular, the strong absorption at 1630 cm−1 is characteristic of the substituted phenyl ring, confirming the substitution of –OH groups in CBD. The CBD was, therefore, successfully preserved in the hydrogels. It can be noticed that the addition of VG and PG significantly reduced the signal of CBD on the gel surface. This could be either because the presence of VG or PG diluted CBD on the surface, or they drove CBD to the interior of the hydrogel.
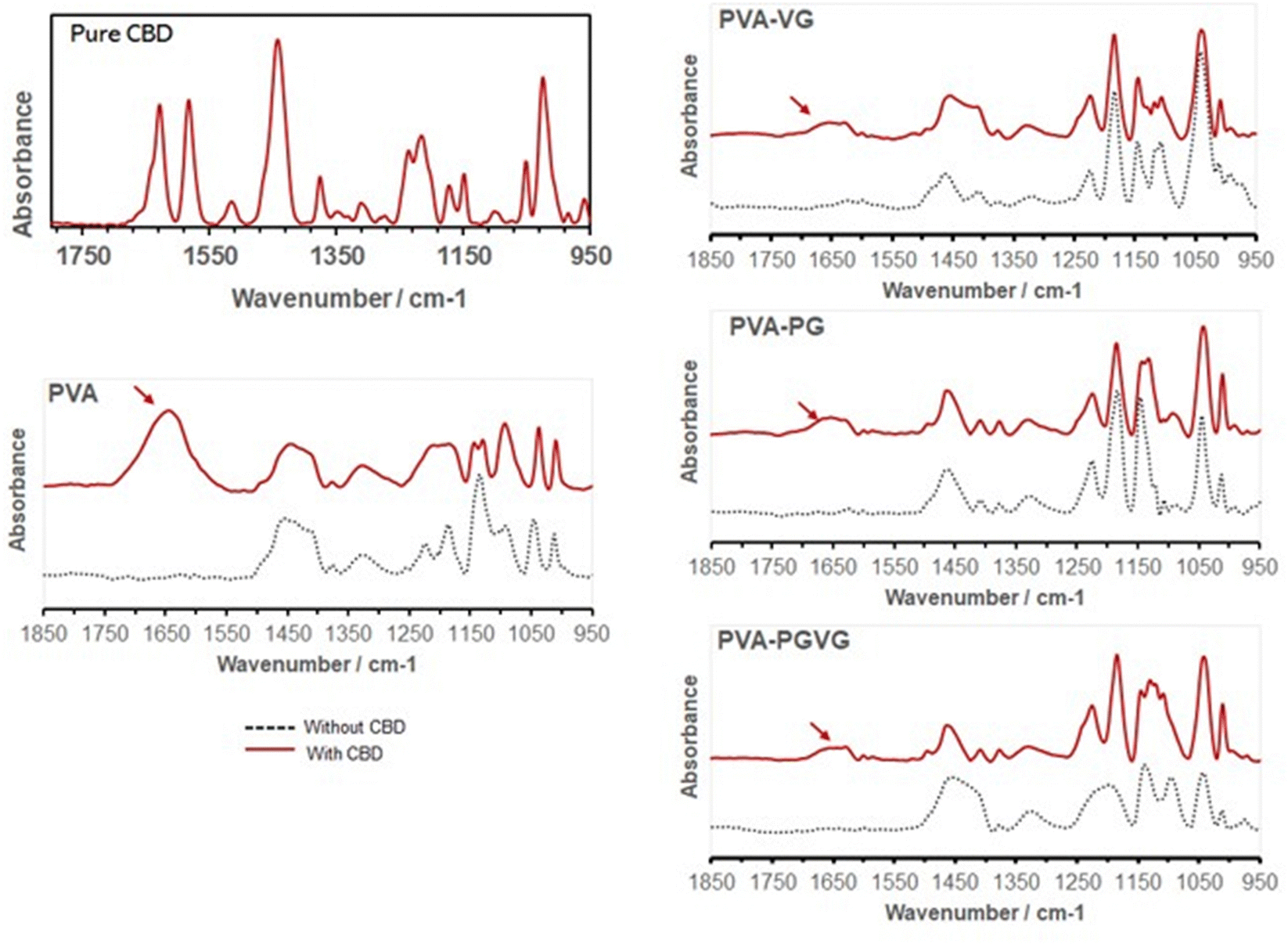 | ||
| Fig. 4 FTIR spectra confirming the presence of CBD in the hydrogels (arrows). Adding PG and VG attenuated the absorption of CBD on the hydrogel surface. | ||
3.5 TGA and DSC analysis
Table 4 shows the percentage of weight loss and the temperature at a maximum rate of weight change. Similar to what we reported previously, the weight loss in TGA took place in four stages, i.e., the evaporation of low molecular weight volatiles that may include moisture, PG, VG, and probably CBD as well, roughly below 225 °C, the thermal degradation of amorphous and crystalline PVA between 225 and 375 °C, decomposition of DBS around 450 °C, and finally the decomposition of carbon at about 730 °C. The initial weight loss is most pronounced for the PVA-PG, followed by PVA-CBD and PVA-VG. According to our previous report,30 there was roughly 10% weight loss in dried PVA hydrogel and that was because of hydrogen-bonded water. Therefore, it is safe to assign the 10% weight loss in PVA and PVA-VG in Fig. 5 to moisture instead of CBD and VG. The boiling point of PG and VG is about 188 and 290 °C, respectively, which explains the higher weight loss of the two PG-containing hydrogels in the first stage and the higher weight loss of the VG-containing hydrogels in the second stage. Important information can be obtained in the 2nd stage of weight loss, i.e., PVA decomposition. Because crystalline domains are more ordered structures than amorphous regions, the decomposition occurred firstly in amorphous PVA and then in crystalline PVA. Among the four gels, the PVA-VG showed the lowest amount of crystalline domains (the shoulder in PVA peak, DTG). This means that the PVA-VG is more amorphous than other gels and should be more permeable to CBD. Finally, the addition of PG slightly increased the temperatures of the two peaks associated with PVA, an indication of a marginally high thermostability.35| Samples | Peaks in DTG curve | Peak temperature (°C) | Weight loss (%) |
|---|---|---|---|
| PVA-CBD | 1st | 151.1 ± 3.1 | 9.2 ± 1.9 |
| 2nd | 293.1 ± 0.8 | 26.5 ± 2.7 | |
| 3rd | 319.6 ± 0.6 | 28.9 ± 1.8 | |
| 4th | 456.3 ± 3.0 | 18.6 ± 0.5 | |
| 5th | 743.5 ± 18.9 | 8.1 ± 1.8 | |
| Residue | — | 8.7 ± 0.9 | |
| PVA-PG-CBD | 1st | 214.8 ± 2.3 | 25.3 ± 0.9 |
| 2nd | 297.2 ± 4.4 | 20.1 ± 1.6 | |
| 3rd | 337.8 ± 6.1 | 24.7 ± 1.0 | |
| 4th | 457.3 ± 2.6 | 17.1 ± 1.2 | |
| 5th | 716.6 ± 17.5 | 5.5 ± 2.2 | |
| Residue | — | 7.3 ± 0.8 | |
| PVA-VG-CBD | 1st | 230.3 ± 3.0 | 9.4 ± 0.5 |
| 2nd | 275.7 ± 2.6 | 45.7 ± 2.5 | |
| 3rd | 329.8 ± 4.9 | 17.4 ± 2.2 | |
| 4th | 450.9 ± 0.8 | 15.7 ± 1.1 | |
| 5th | 722.3 ± 19.2 | 4.8 ± 1.4 | |
| Residue | — | 7.0 ± 0.5 | |
| PVA-PG-VG-CBD | 1st | 213.1 ± 2.4 | 18.5 ± 1.9 |
| 2nd | 290.9 ± 3.4 | 30.0 ± 0.6 | |
| 3rd | 340.8 ± 2.3 | 23.0 ± 0.6 | |
| 4th | 453.1 ± 1.1 | 15.4 ± 0.8 | |
| 5th | 737.8 ± 9.5 | 6.3 ± 1.3 | |
| Residue | — | 6.9 ± 0.9 |
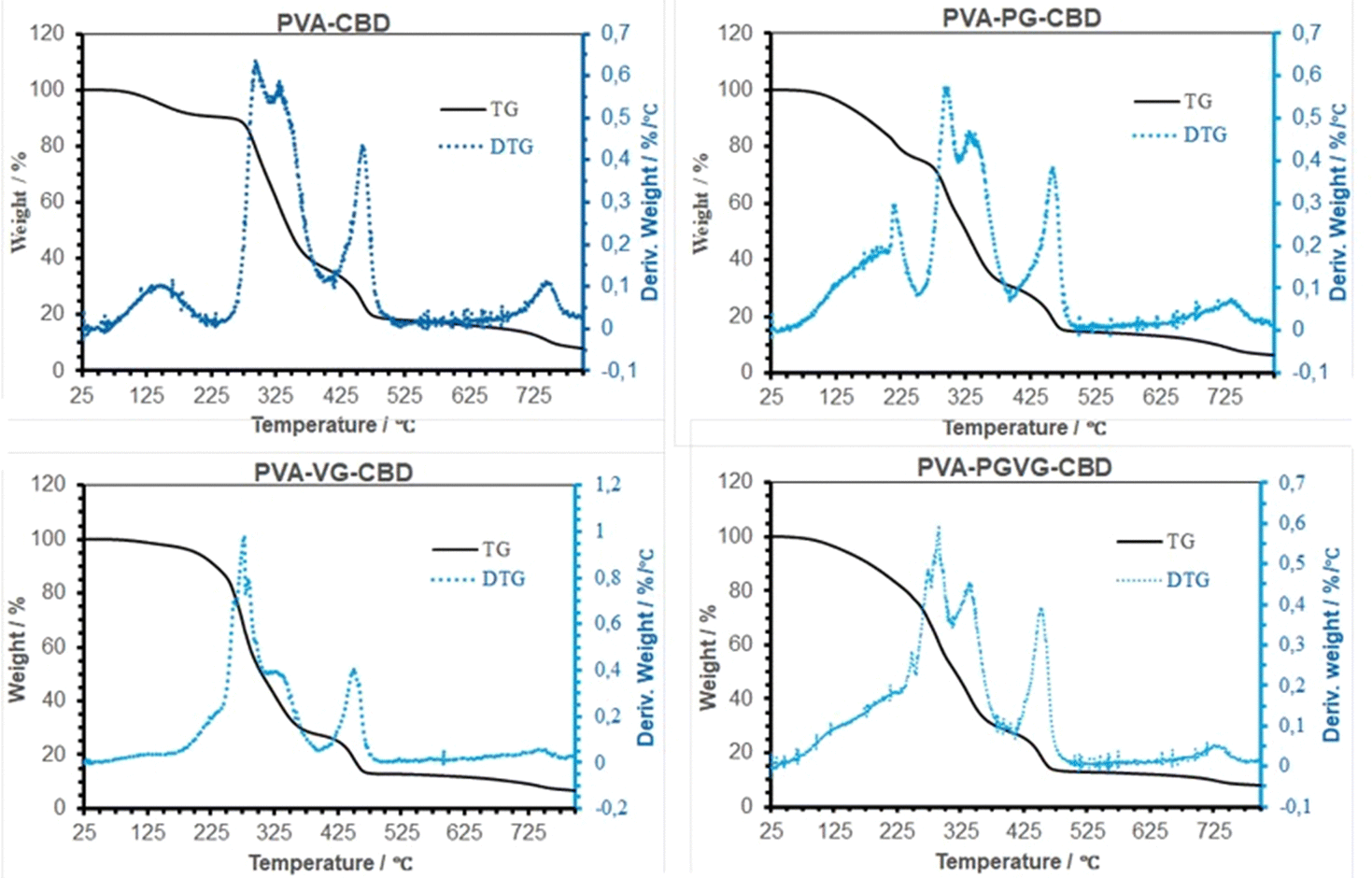 | ||
| Fig. 5 TG and DTG Curves of the hydrogels that were dried at room temperature. The maximum in DTG shows where the highest rate of weight loss occurred. | ||
Fig. 6 shows the DSC curves of different hydrogels, and the data are listed in Table 5. Both PVA and PVA with CBD in the formulation are almost identical in DSC spectrum, showing the melting of PVA crystalline domains started at about 210 and peaked at 228 °C (Tm), and the glass transition temperature (Tg) at about 40 °C. This means that the addition of a small amount of CBD didn’t affect the hydrogel crystallinity. With the addition of PG and VG, the melting temperature shifted to the low-temperature side, and Tg disappeared, probably out of range (lower than zero degree). PG and VG are small molecules that may act as plasticizers to increase PVA molecule mobility, leading to a lower Tg. PG and VG may also trap water molecules, bringing more plasticity to the gel resulting in lower Tg.36,37 Their effects on PVA crystalline structure are more complicated. Their interactions with PVA molecules made the PVA crystals less perfect, as testified by the lower Tm. On the other hand, the higher mobility of PVA molecules may also help them to organize into ordered structures. This can explain why the PVA-PG showed the highest crystallinity. VG, which has one more hydroxyl group than PG, may have had a stronger interaction with PVA, shifting the balance towards destroying the crystals rather than forming more ordered structures.
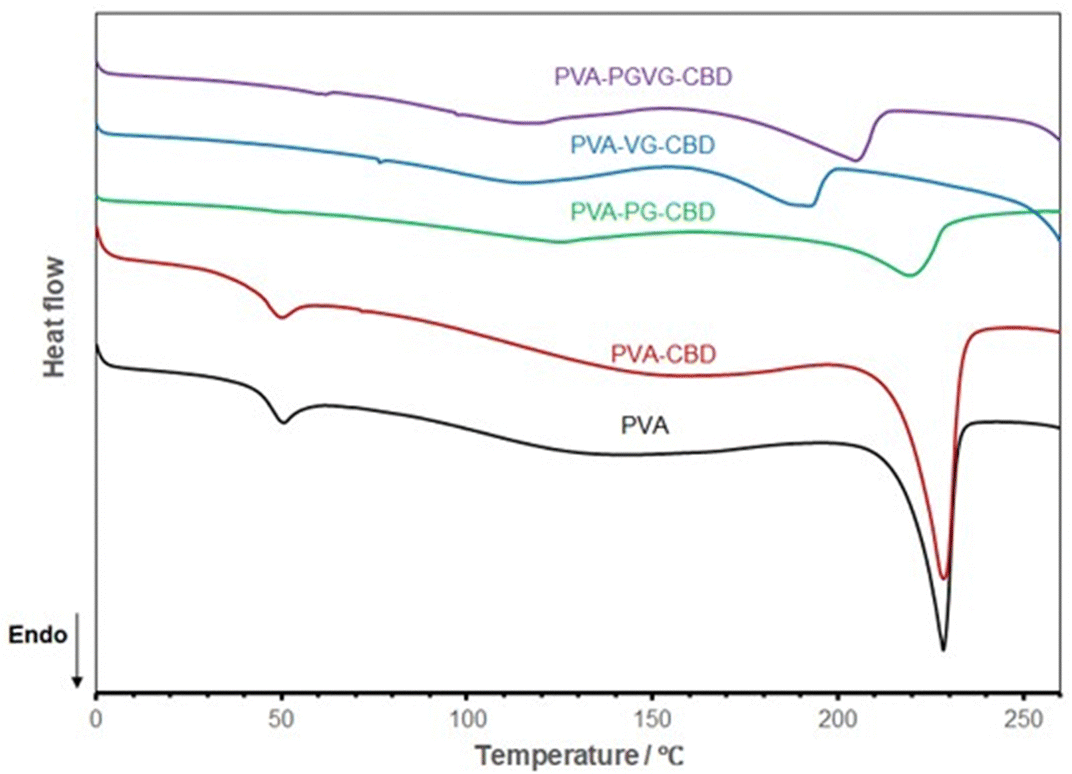 | ||
| Fig. 6 DSC curves of the hydrogels dried at room temperature show VG and PG's significant effect on PVA aggregation structures. The PVA sample is the hydrogel produced without PG, VG, and CBD. | ||
| Sample | T g/°C | T m/°C | ΔH@Tm/J g−1 | X c/% |
|---|---|---|---|---|
| T g: glass transition temperature; Tm: melting temperature; ΔH: heat of fusion; Xc: weight fraction of crystallinity; nd: not detected. | ||||
| PVA | 40.0 ± 1.9 | 228.0 ± 0.2 | 44.9 ± 0.2 | 0.32 ± 0.01 |
| PVA-CBD | 39.8 ± 0.9 | 227.9 ± 0.8 | 44.4 ± 3.3 | 0.32 ± 0.02 |
| PVA-PG-CBD | nd | 221.4 ± 2.3 | 60.2 ± 7.7 | 0.43 ± 0.05 |
| PVA-VG-CBD | nd | 193.0 ± 2.3 | 25.0 ± 0.5 | 0.18 ± 0.00 |
| PVA-PG-VG-CBD | nd | 204.7 ± 1.1 | 41.3 ± 4.7 | 0.30 ± 0.03 |
The weight percentage of crystalline domains in PVA hydrogels was calculated according to the following formula:
| Xc = ΔH@Tm/ΔH0@T0m | (4) |
The temperature range used in material analysis does not have to be the same as the material service temperature. The former is to adequately reveal the structure–property relationship, and the latter is only the limit when the material can be effectively used.
3.6 Measurement of CBD released from the hydrogels
CBD exhibits a single strong absorption in 280–295 nm region and a maximum at 287 nm that increases linearly with the concentration of CBD (Fig. 7B). This linear relationship allows us to use the regression line as the standard curve to calculate the concentration of CBD molecules in a solution (Fig. 7A). Some papers have reported the UV-vis spectroscopy of CBD. The small difference in the maximum may be related with the solvent used to dissolve CBD and the photodegradation during storage.39,40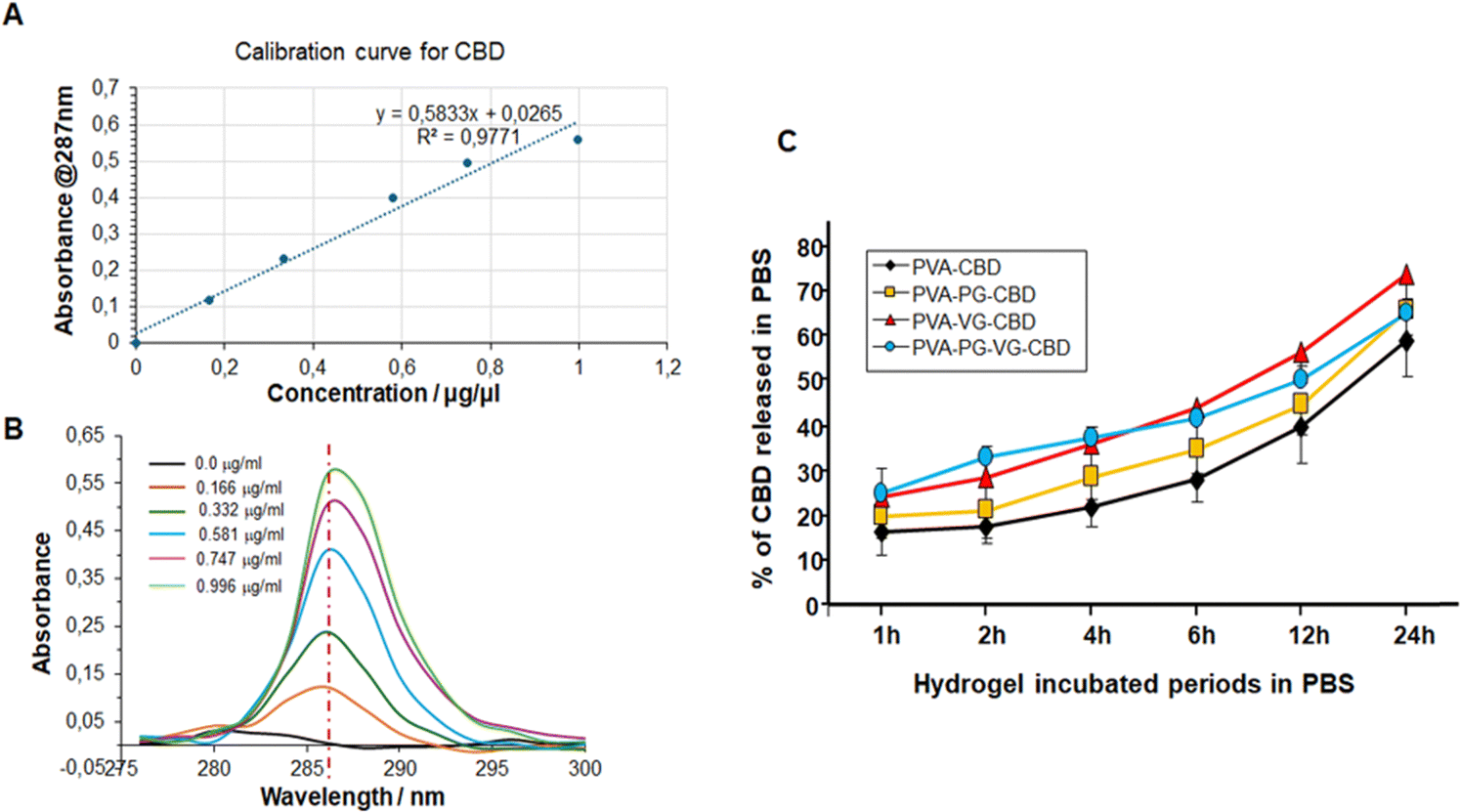 | ||
| Fig. 7 Cumulative release of CBD from hydrogels into PBS calculated from UV-vis data and calibration curve. A significant difference was found between PVA-VG or PVA-PG-VG and PVA hydrogels at 2, 4 and 6 h. Compared with PVA hydrogels, significant difference was found at 1 h for PVA-PG-VG (p = 0.045), at 2 h for both PVA-VG (p = 0.045) and PVA-PGVG (p = 0.002), at 4 h for both PVA-VG (p = 0.028) and PVA-PG-VG (p = 0.004), at 6 h for both PVA-VG (p = 0.017) and PVA-PGVG (p = 0.008), at 12 h for both PVA-VG (p = 0.049) and PVA-PGVG (p = 0.048), and at 24 h for PVA-VG (p = 0.048). (A) Calibration curve, (B) absorbance of pure CBD, and (C) CBD release from the hydrogels. | ||
As shown in Fig. S1 (ESI†), CBD was continuously released from the hydrogels to PBS for 24 h. The highest concentration was at 1 h for the PVA-PG-VG, probably because of its high porosity, and the lowest concentration was at 2 h for the PVA that happened to have the lowest porosity. And not all CBD was released in 24 h. The methanol extraction experiment showed (Fig. S2, ESI†) that after 24 h incubation in PBS, the residual CBD in the PVA, PVA-PG, PVA-PG-VG, and PVA-VG gels were 40, 35, 35, and 25 percent, respectively (Fig. 7). According to this figure, around 20% of CBD was released in the first hour, then the increment per hour decreased and was fairly linear. It needs to be pointed out that in the first 12 h, around 50% CBD was released from the gel, and in the second 12 hours, only 10–20% CBD was released. Because the release was driven by the concentration gradient of CBD inside the hydrogels, it is normal that CBD was released slowly in the late stage.
Fig. 7 also demonstrates clearly that adding VG and PG to the hydrogels accelerated CBD release. In particular, the presence of VG is critical and can significantly increase CBD release (Fig. 7C). While PG alone didn’t significantly increase the release of CBD, its presence might have accelerated the release in the early hours. Changing the VG and PG ratio might possibly adjust the release rate further. These observations were confirmed by UPLC-MS/MS, as shown in Table 6. Indeed, the release of CBD over 24 h from the PVA alone ranged from 366 ± 10 μg mL−1 at 1 h to 184 ± 7 at 24 h. With the PVA-PG hydrogels, the release of CBD ranged from 329 ± 15 to 216 ± 10 μg mL−1. However, with PVA-VG, the level of released CBD at an early time was higher than PVA-PG and PVA-PG-VG. The release level with PVA-VG ranged from 430 ± 12 μg mL−1 at 1 h, to 281 ± 13 at 24 h (Table 6). The high amount of CBD measured at 1 h is due to the burst release of the CBD from the top layer of the hydrogels, which is a common phenomenon in passive drug release. After that, the release is controlled by the diffusion from the inner to the surface of the hydrogels. The increased release between 4 h and 24 h could be due to the hydrogel adaptation to the PBS and incubation temperature, which was 37 °C, leading to changes in material aggregation states such as crystallinity. Further studies are needed to confirm such a hypothesis. Several factors control the release of CBD in a given system, including the microstructure of the hydrogel matrix, the behavior of the matrix in the selected environment, van der Weal's forces between CBD and other components in the gel, the aqueous solution, temperature, etc.41 Since PVA is stable in PBS and no other environmental stimuli exist, the releasing mechanism of CBD from the PVA gels should be diffusion-controlled.42,43 Our data shows an inverse correlation between PVA crystallinity and release rate in the early release periods. In fact, the hydrogels containing VG showed a lower crystallinity, confirmed by TGA and DSC. It is well known that the diffusion coefficient is lower in crystalline regions of polymer.44
| Incubation time | ||||||
|---|---|---|---|---|---|---|
| 1 h | 2 h | 4 h | 6 h | 12 h | 24 h | |
| PVA-CBD | 366 ± 10 | 110 ± 7 | 257 ± 10 | 173 ± 9 | 122 ± 3 | 184 ± 7 |
| PVA-PG-CBD | 529 ± 15 | 218 ± 8 | 214 ± 9 | 161 ± 8 | 191 ± 6 | 216 ± 10 |
| PVA-VG-CBD | 430 ± 12 | 202 ± 10 | 220 ± 6 | 240 ± 7 | 238 ± 8 | 281 ± 13 |
| PVA-PG-VG-CBD | 173 ± 7 | 92,6 ± 4 | 137 ± 3 | 145 ± 5 | 309 ± 9 | 322 ± 12 |
3.7 Antioxidant activity of the hydrogels
DPPH free radicals (DPPH˙) can be reduced to their non-radical form DPPH-H in an alcoholic solution with a hydrogen-donating antioxidant.45 It has been reported that CBD is an effective radical scavenger and has high antioxidant activities.46Fig. 8 demonstrated the 30% to 45% antioxidant activity of the PBS solutions used to incubate the CBD-containing PVA hydrogels, compared with the PVA hydrogels without CBD showing zero antioxidant activity. Fig. 8 showed that the PVA-VG hydrogel has the highest value in early time, and PVA has the highest value at 24 h, supporting that the antioxidant activity is roughly correlated with the amount of CBD released in PBS. This result also supports the idea that the addition of VG and PG, VG in particular, helps the release of CBD.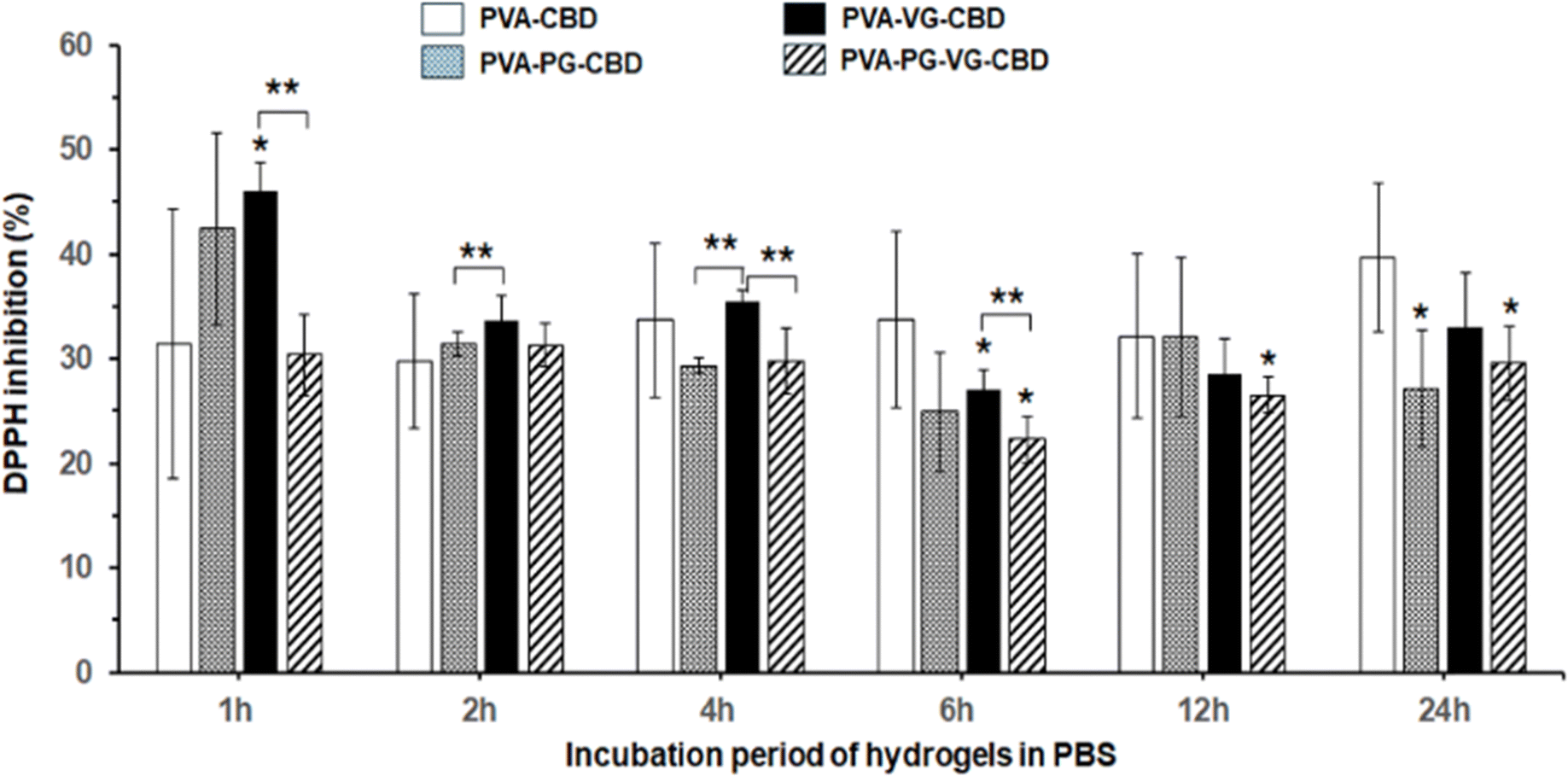 | ||
| Fig. 8 Collected solutions from Hydrogels incubated in PBS show DPPH inhibition capacity, confirming the presence of CBD with antioxidant activity. PVA without CBD showed no DPPH inhibition (data not shown). | ||
Oxidative stress caused by free radicals is often associated with various diseases and wound-healing inflammations.47,48 The antioxidant activity of the CBD-containing hydrogels, therefore, may find medical applications in controlling inflammation.
4. Conclusions
CBD-enriched PVA hydrogels were successfully prepared with sufficient mechanical strength and stiffness for easy manipulation. The CBD in the hydrogels can be continuously released into an aqueous environment and show radical scavenger activity for at least 24 h. Adding VG to PVA hydrogels reduced PVA crystallinity and consequently accelerated CBD release. High porosity in the PVA-PG-VA also augmented CBD release. Therefore, the PVA hydrogels containing VG demonstrated the best releasing profile, and the releasing profile can be regulated by adding PG. This CBD-enriched PVA hydrogel may be useful in reducing inflammation and helping wound healing.Author contributions
Conceptualization, M. R. and Z. Z.; methodology, S. C., M. B., M. R., Z. Z.; formal analysis, S. C., M. B., M. R. and Z. Z.; resources, M. R., Z. Z. A. S. and F. B.; writing – original draft preparation, S. C., Z. Z. and M. R.; writing – review and editing, M. B. A. S. and F. B.; supervision, M. R. and Z. Z. and A. S.; project administration, M. R. and Z. Z.; funding acquisition, M. R., Z. Z and A. S. All authors have read and agreed to the published version of the manuscript.Data availability
All data used for this manuscript were already included. We have no data that were not included in this manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. SiliCycle Inc., the company involved in the NSERC-Alliance grant, does not impact the work or the results presented in this manuscript.Acknowledgements
M. B. benefits from a studentship from the Natural Sciences and Engineering Research Council of Canada (NSERC)-training program (NSERC CREATE EvoFunPath). This research was funded by an NSERC-Alliance grant (ALLRP 561197) and the NSERC-Discovery grant to MR (RGPIN-2019-04475). The funding agencies do not influence the work or results of this manuscript.References
- Z. Atakan, Ther. Adv. Psychopharmacol., 2012, 2(6), 241–254 CrossRef CAS PubMed
.
- M. A. Huestis, R. Solimini, S. Pichini, R. Pacifici, J. Carlier and F. P. Busardò, Curr. Neuropharmacol., 2019, 17(10), 974–989 CrossRef CAS PubMed
- L. Rapin, R. Gamaoun, C. El Hage, M. F. Arboleda and E. Prosk, J. Cannabis Res., 2021, 3(1), 1–10 CrossRef PubMed
- J. Maghfour, H. R. Rietcheck, C. W. Rundle, T. M. Runion, Z. A. Jafri, S. Dercon and H. Yardley, J. Drugs Dermatol., 2020, 19(12), 1204–1208 CrossRef PubMed
- M. P. Chelliah, Z. Zinn, P. Khuu and J. M. Teng, Pediatr. Dermatol., 2018, 35(4), e224–e227 CrossRef PubMed
- T. Sheriff, M. J. Lin, D. Dubin and H. Khorasani, J. Dermatol. Treat., 2020, 31(8), 839–845 CrossRef CAS PubMed
- S. Atalay, I. Jarocka-Karpowicz and E. Skrzydlewska, Antioxidants, 2019, 9(1), 21 CrossRef PubMed
- M. W. Preteroti, H. Traboulsi, P. Greiss, O. Lapohos, G. J. Fonseca, D. H. Eidelman and C. J. Baglole, Immunol. Cell Biol., 2023, 101(2), 156–170 CrossRef CAS PubMed
- D. Thapa, E. A. Cairns, A. M. Szczesniak, J. T. Toguri, M. D. Caldwell and M. E. Kelly, Cannabis Cannabinoid Res., 2018, 3(1), 11–20 CrossRef CAS PubMed
- S. Bashir, M. Hina, J. Iqbal, A. H. Rajpar, M. A. Mujtaba, N. A. Alghamdi and S. Ramesh, Polymer, 2020, 12(11), 2702 CAS
- S. Jacob, S. A. B. Nair, J. Shah, N. Sreeharsha, S. Gupta and P. Shinu, Pharmscitech, 2021, 13(3), 357 CAS
- N. H. Thang, T. B. Chien and D. X. Cuong, Gels, 2023, 9(7), 523 CrossRef CAS PubMed
- Z. Zheng, J. Qi, L. Hu, D. Ouyang, H. Wang, Q. Sun and B. Tang, Biomater. Adv., 2022, 134, 112560 CrossRef CAS PubMed
- M. Wang, J. Bai, K. Shao, W. Tang, X. Zhao, D. Lin and J. Ye, Int. J. Polym. Sci., 2021, 2021, 1–16 CAS
- Y. Chen, J. Li, J. Lu, M. Ding and Y. Chen, Polym. Test., 2022, 108, 107516 CrossRef CAS
- S. Jiang, S. Liu and W. Feng, J. Mech. Behav. Biomed. Mater., 2011, 4(7), 1228–1233 CrossRef CAS PubMed
- F. Wu, J. Gao, Y. Xiang and J. Yang, Polymer, 2023, 15(18), 3782 CAS
- K. Laslo, A. Guillermo, A. Fluerasu, A. Maussaid and E. Geissler, Langmuir, 2010, 26, 4415–4420 CrossRef PubMed
- T. C. Ho, C. C. Chang, H. P. Chan, T. W. Chung, C. W. Shu, K. P. Chuang, T. H. Duh, M. H. Yang and Y. C. Tyan, Molecules, 2022, 27(9), 2902 CrossRef CAS PubMed
- A. T. Lin C-CMetters, Adv. Drug Deliv. Rev., 2006, 58(12), 1379–1408 Search PubMed
- M. Jamadi, P. Shokrollahi, B. Houshmand, M. D. Joupari, F. Mashhadiabbas, A. Khademhosseini and N. Annabi, Macromol. Biosci., 2017, 17(8), 1600479 CrossRef PubMed
- V. de la Asunción-Nadal, S. Armenta, S. Garrigues and M. de la Guardia, Talanta, 2017, 167, 344–351 CrossRef PubMed
-
L. Peter, Infrared and Raman Spectroscopy: Principles and Spectral Interpretation, Elsevier, 2011, pp. 27–54 Search PubMed
- R. Narayanaswamy and V. P. Torchilin, Molecules, 2019, 24(3), 603 CrossRef PubMed
- N. Oliva, S. Shitreet, E. Abraham, B. Stanley, E. R. Edelman and N. Artzi, Langmuir, 2012, 28, 15402–15409 CrossRef CAS PubMed
- K. Momenzadeh, D. Yeritsyan, N. Kheir, R. M. Nazarian and A. Nazarian, J. Cannabis Res., 2023, 5(1), 24 CrossRef PubMed
- B. Stella, F. Baratta, C. Della Pepa, S. Arpicco, D. Gastaldi and F. Dosio, Drugs, 2021, 81(13), 1513–1557 CrossRef CAS PubMed
- S. Johne, M. van der Toorn, A. R. Iskandar, S. Majeed, L. O. Torres, J. Hoeng and M. C. Peitsch, Food Chem. Toxicol., 2023, 175, 113708 CrossRef CAS PubMed
- W. Wan, A. D. Bannerman, L. Yang and H. Mak, Adv. Polym. Sci., 2014, 263, 283–321 CrossRef CAS PubMed
- S. Cui, Z. Zhang, D. Rodrigue, F. Beland and M. Rouabhia, Front. Drug Deliv., 2024, 4, 1303812 CrossRef
- W. Li, D. Wang, W. Yang and Y. Song, RSC Adv., 2016, 6(24), 20166–20172 RSC
- J. Oh, Y. H. Yoo, I. S. Yoo, Y. I. Huh, T. K. Chaki and C. Nah, J. Appl. Polym. Sci., 2014, 131(2), 39795 CrossRef
- J. L. Valentín, D. López, R. Hernández, C. Mijangos and K. Saalwachter, Macromol., 2009, 42(1), 263–272 CrossRef PubMed
- Y. Yordanov, D. Stefanova, I. Spassova, D. Kovacheva, V. Tzankova, S. Konstantinov and K. Yoncheva, Pharmaceutics, 2022, 14(12), 2625 CrossRef CAS PubMed
- F. Reguieg, L. Ricci, N. Bouyacoub, M. Belbachir and M. Bertoldo, Polym. Bull., 2020, 77(2), 929–948 CrossRef CAS
- N. A. Peppas and P. J. Hansen, J. Appl. Polym. Sci., 1982, 27(12), 4787–4797 CrossRef CAS
- P. Pirahmadi and M. Kokabi, AIP Conf. Proc., 2018, 1920, 1 CrossRef
- O. W. Guirguis and M. T. Moselhey, Nat. Sci., 2012, 4, 57–67 CAS
- M. Dirksen, T. A. Kinder, T. Brändel and T. Hellweg, Molecules, 2021, 26(11), 3181 CrossRef CAS PubMed
- P. Seccamani, C. Franco, S. Protti, A. Porta, A. Profumo, D. Caprioglio and D. Merli, J. Nat. Prod., 2021, 84(11), 2858–2865 CrossRef CAS PubMed
- L. Lei, Y. Bai, X. Qin, J. Liu, W. Huang and Q. Lv, Gels, 2022, 8(5), 301 CrossRef CAS PubMed
- M. Wang, J. Bai, K. Shao, W. Tang, X. Zhao, D. Lin and J. Ye, Int. J. Polym. Sci., 2021, 2021, 1–16 CAS
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1(12), 1–17 Search PubMed
- A. Hasimi, A. Stavropoulou, K. G. Papadokostaki and M. Sanopoulou, Eur. Polym. J., 2008, 44(12), 4098–4107 CrossRef CAS
- T. Ak and I. Gülçin, Chem. – Biol. Interact., 2008, 174(1), 27–37 CrossRef CAS PubMed
- E. M. Saint Martin, C. Marrassini, F. M. Silva Sofrás, I. N. Peralta, L. C. Cogoi, C. M. Van Baren and C. A. Anesini, Int. J. Pharm. Sci. Rev. Res., 2022, 77(2), 1–10 Search PubMed
- S. J. Chang and B. C. Yu, J. Bioenerg. Biomembr., 2010, 42, 457–459 CrossRef CAS PubMed
- F. Azam, M. V. V. Prasad and N. Thangavel, Curr. Top. Med. Chem., 2012, 12(9), 994–1007 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00398e |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |