
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe allure of targets for novel drugs
Colin J.
Suckling

Department of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1Xl, Scotland, UK. E-mail: c.j.suckling@strath.ac.uk
First published on 13th December 2023
Abstract
The challenges of bringing new medicines to patients have been extensively discussed and debated, including consideration of the contribution that academic laboratories can make. At the University of Strathclyde, drug discovery has been a continuing focal activity since the 1960s, and in the past 30 years, the author has led or contributed to many projects of different character and for diverse diseases. A feature common to these projects is the extension of concepts of molecular and biological targets in drug discovery research. In mechanistic terms, these have included compounds that are activators and not inhibitors, and in particular multitargeted compounds. With respect to relevance to disease, schizophrenia, pulmonary disfunction, autoimmune, and infectious disease are most relevant. These projects are discussed in the context of classical medicinal chemistry and more recent concepts in and approaches to drug discovery.
 Colin Suckling | Colin Suckling joined the University of Strathclyde in 1972 and, having held academic posts at all levels up to Vice Principal, is now a research professor. Over the years his research has moved between enzyme chemistry, in both synthetic and mechanistic aspects, heterocyclic chemistry, and in the past 20 years especially, medicinal chemistry. Colin holds the RSC Adrien Albert medal and, is a Fellow of RSC, the Royal Society of Edinburgh, the Indian Chemical Society, and medical and surgical Royal Colleges in Scotland. He was awarded the OBE for services to science and higher education in Scotland (2006). |
Introduction and challenge
Bringing a new drug to the market is perhaps the most challenging task in molecular science when all the necessary stages from identifying the new active pharmaceutical ingredient to manufacture are considered. The great therapeutic and commercial success of the global pharmaceutical industry from the 1960s for the next 40 years has led to high expectations for new medicines and for greater profits. To deal with this, the industry has substantially restructured so that there remain few global discovery and development companies but there are many more small and medium sized companies focused largely on discovery and early-stage development in the anticipation that a large company with the capacity to market a new discovery would take it on. The question is often asked whether small academic laboratories should seriously tackle drug discovery in view of the power of the industry. Historical trends as noted above provide an answer: industry can do great things but invention, innovation, and iconoclasm are well suited to the academic environment where protocols and paradigms can be challenged. This article is about a wide range of drug discovery projects, many with some of these characteristics, associated with the Department of Pure & Applied Chemistry at the University of Strathclyde. The selection is limited to projects that have achieved a successful experimental proof of concept outcome in an animal model of disease. It includes CNS active compounds, immunomodulators, and anti-infective compounds amongst others making use of a wide range of discovery strategies running often in parallel with mechanism of action studies. The partnership between chemical biology and drug discovery is ideal for the academic laboratory.Connecting with classical medicinal chemistry
Technological and methodological fashions have come and gone. Methods of synthesising and identifying leads such as combinatorial synthesis arose in the 1980s. Vast numbers of compounds were needed to feed the hungry robots of high throughput screening, the hope being that simply by evaluating more compounds more good leads would be obtained. However, because the chemistry applied in combinatorial synthesis offered limited genuine diversity, hope did not turn into reality in terms of the flow of new drugs down the pipeline. Attention was drawn to these limitations at the time and practitioners encouraged the view that ‘cleverness and creativity can provide active compounds in drug discovery’.1 The conclusion that more than sheer numbers of compounds are needed for successful drug discovery was drawn many times.2 Broadening creativity is at the heart of this article, as is appropriate from an academic laboratory. Responding to limitations of what might be called first generation combinatorial chemistry there have been efforts to improve the technology for the preparation of libraries of compounds and for the reactions themselves such that it has been possible to write ‘We are now in a better position to truly leverage the power of combinatorial technologies for the discovery and development of next-generation drugs’.3 Moreover powerful new technologies such as DNA coded libraries have become practical and useful.4,5An enduring and important methodology is structure-based drug design. In embryonic form structure-based design has been around since the discoveries of dihydrofolate reductase inhibitors such as trimethoprim, first used in 1962, when enzyme crystal structures first became widely available.6 Hitchings and Elion showed with pleasure visitors to Burroughs Wellcome Laboratories at Research Triangle Park, North Carolina, a wire model of the structure of dihydrofolate reductase to show how trimethoprim (1) worked and hence how other compounds might be invented.
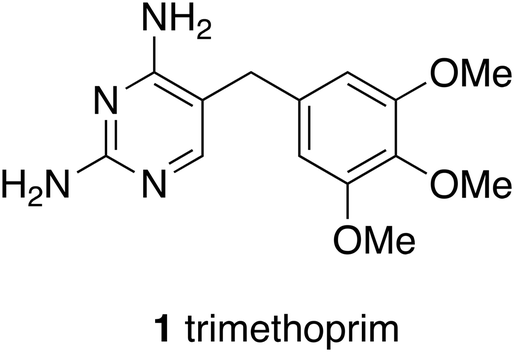
Fragment based drug discovery is more recent and brings together several technologies for measuring association of a compound to a target and several strategies for reaching the desired property space.8,9 Thus affinity of a molecule for a protein target can be measured, for example, by thermal shifts, and surface plasmon resonance, and details of binding by NMR spectroscopy and crystallography. In the context of the present discussion the application of several technologies to a central concept for drug discovery is significant. Thoughts are moving away from a single preferred or even specified approach to using the best relevant techniques available to support a general philosophical approach. It has been shown that there are several equally valid routes from initial hits to compounds with suitable profiles for progression. For example, in an investigation of antituberculosis drugs it was reported that ‘Optimization strategies include scaffold-hopping, synthesis, and evaluation of fragments of the lead compounds and property-focused optimization’.10
It is easy to look back now and to say that all of these technologies have value used in the right place and can contribute. However, at the time the technologies were the thing and they forced a narrow focus onto many drug discovery teams.
Focusing on a biological target
Underlying the above there was and remains another narrow focus, namely focus on a target for the drug. This focus is entirely rational but has been so powerful that thinking about anything else has become almost anathema. Taking a balanced view, it is clear that without the concept and the identification of a target relevant to the disease to be treated there would not have been the range of effective medicines we have today. On the WHO list of about 250 essential medicines for a basic health care system (vaccines and biologics excluded), roughly 95% have a recognised molecular target.11 This is perhaps unsurprising because the list comprises established drugs for which mechanisms of action are likely to be known. It has been said that these are the easy targets from the point of view of drug discovery. When the targets run out, the drug pipeline dries up. This narrow focus at the discovery, not use, stage is a shame because it limits inventiveness and creativity at the very start of a programme where these two features are most needed. Moreover, a pure reductionist approach does not accommodate the use of traditional medicines from many cultures in the world, which are undoubtedly effective when used properly and typically have several molecular targets in their mechanisms of action. There is a discontinuity here that has been widely recognised but it is hard to bridge.12None of the foregoing is to say that considering single molecular targets for drugs is wrong. It is simply pointing to an inevitable conclusion that a standard drug discovery paradigm of single molecule – single target – single effect is limited. In case anyone is upset by this, I was once told by a company in the face of overwhelming proof-of-concept and safety evidence in animal models words to the effect that ‘We are a target-driven company and we don't consider anything else whatever the evidence and evident need’. Well, this company can go its own way. What is clear now is that there should be alternative approaches, suited to the particular therapeutic need so that the strategic decisions are based upon the science and not upon any particular commercial or managerial dogma. Such approaches can be called ‘off-paradigm’. It would be wrong to call them ‘anti-paradigm’ because the standard paradigm has its place and shares the same goals. This is not a competition and a merging of approaches in a drug discovery programme is likely at some point because after the initial discovery and innovation, mechanisms and targets will be found and they can be used to improve the profile of new drugs. So that these arguments are not simply philosophical, several relevant examples of ancient and modern drugs are presented below before describing the Strathclyde projects.
Taking a broader view
In case it is thought that traditional medicines are only relevant in Indian and Chinese cultures where traditional medicine systems meet industrial pharmaceuticals in the same population, it is worth remembering that there is a substantial body of traditional medicine in Europe even in Anglo-Saxon England before the Norman conquest notably based upon native plants. The description of an anti-inflammatory medicine, for example, includes not only the extraction of the active compound, betonicine (2), from the betony plant (Stachys officinalis), but also its formulation suitable for specific ailments such as earache.13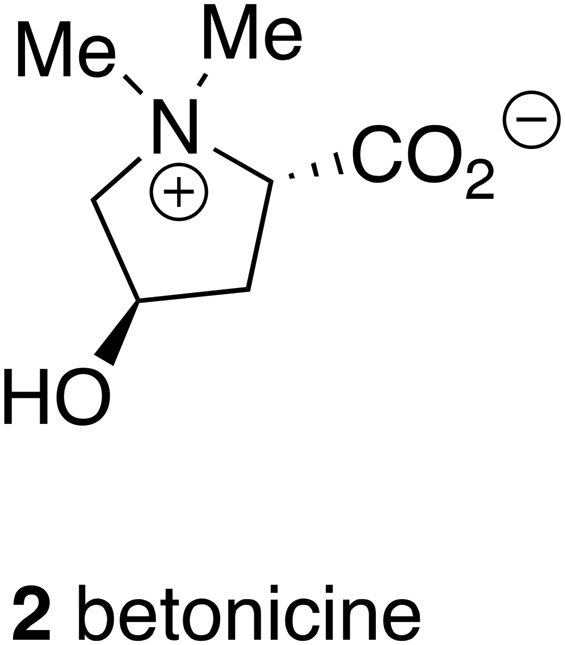
CNS active drugs
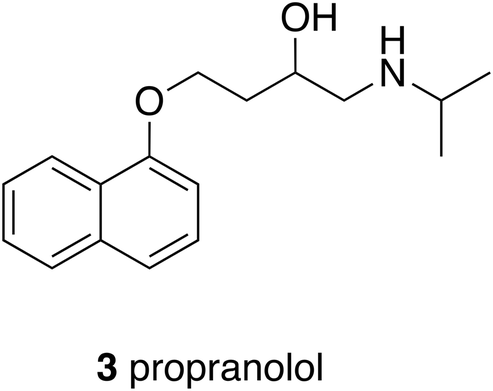
It is probably fair to say that focus on biological targets for drugs sharpened with the parallel development of precision pharmacology, of which the an early example that led to a block-buster drug was Black's innovation with β-blockers.14 The discovery of propranolol (3), which was first used in 1964, depended exquisitely upon tissue pharmacology that could distinguish between the different adrenergic receptors and then upon some very original chemical modifications of a natural product lead to obtain a selectively acting β-antagonist with respect to the α-receptor. A well-defined target was in mind at the time but as the science has developed, the target has changed to reflect increased knowledge. A drug that was once first line for treating hypertension is now relegated to fourth line because propranolol is a non-selective β-blocker; it has little selectivity for β-receptor subtypes and is also a weak 5-HT antagonist. A concept generalised from these developments, now widely understood, is that the profile of a drug as well as its specific activity at a target determines its utility. Depending upon the therapeutic circumstances then, it would be reasonable to avoid over insistence upon a narrow target; knowledge and requirements change.Profile in CNS active drugs, especially those active at G-protein coupled receptors is an essential consideration because many diseases of imbalance cannot be successfully treated by a single drug with, arguably, a single target. A case in point is schizophrenia and a project from the Universities of Glasgow and Strathclyde that was known as the Serominic Project.15 The biologists leading the project were convinced from clinical advice and their own investigations of animal models of schizophrenia that the therapies then available, typically D2-receptor antagonists or partial agonists such as haloperidol and clozapine, were inadequate. They postulated that a dual action compound that was an M4 agonist and a 5HT7 antagonist, which was called a serominic compound, would be more effective. The challenge for the medicinal chemistry team was to find a single compound with opposite effects on two different receptors. Moreover, it was important to avoid antagonism at the D2-receptor because this led to undesired side effects associated with existing anti-psychotic drugs. The required three-point profile was best achieved using an isoquinoline, to provide a 5HT-antagonist component, and an aminoindane to as the source of M4 activity (4), the two components overlapping (Fig. 1). 4 was able to reverse the symptoms of a drug-induced schizophrenia model in mice, suppressing hyperactivity in a dose dependent manner with and ED50 of 8 mg kg−1 ip. The successful proof-of-concept outcome for a novel dual-targeted drug was not taken further by the partner company. Underlying the need for multiple targeting in such cases is the complexity of biological networks that control many important functions including CNS activity and, as will be described later, immunological responses.
 | ||
| Fig. 1 Multitargeted CNS active drugs. | ||
Multiple targeting is now becoming frequent in drug discovery, in particular in the neurosciences, but it's still target driven. Usually, a single active substructure is linked in some way with a non-active substructure, an aromatic ring or aliphatic chain, to a partner with a different but necessary activity. This is slightly different from the serominic compounds in which the two active substructures overlapped at an essential amidine. In the field of Alzheimer's disease there are several examples of up to tetratargeting and the abandonment of the standard paradigm has been strongly urged.16 In one approach, cholinesterase inhibition, monoamine oxidase inhibition, and H3R antagonism have been combined with S1R agonism in a remarkably simple compound to give a molecule, contilisant (5), that is effective in animal model relevant to Alzheimer's disease restoring the cognitive deficit induced by Aβ1–42 in the radial maze assay.17 Following the same philosophy an alternative combination of targets centred on an anti-oxidant compound to which have been grafted cholinesterase inhibition and other activities has been reported (6).18 The thought cannot be escaped that if multitargeted molecules are acceptable for development in mainstream medicinal chemistry, what barrier is there in principle to using traditional medicines that act at several targets? The same regulatory principles for safety and efficacy should apply for access to markets.
This is not to say that single target approaches have been neglected at Strathclyde in projects to which I have contributed. The focal point of these projects was the fused pyrimidine ring system, of which the essential natural cofactor, the pteridine, tetrahydrofolate, was the seed structure. The importance of this compound has already been noted in the context of antibacterial compounds such as trimethoprim. Because of their central role in biology, cofactors and nucleotides are relevant to the investigations of many diseases. In our case, we have cardiovascular, antiparasitic, and anticancer applications (Fig. 2).
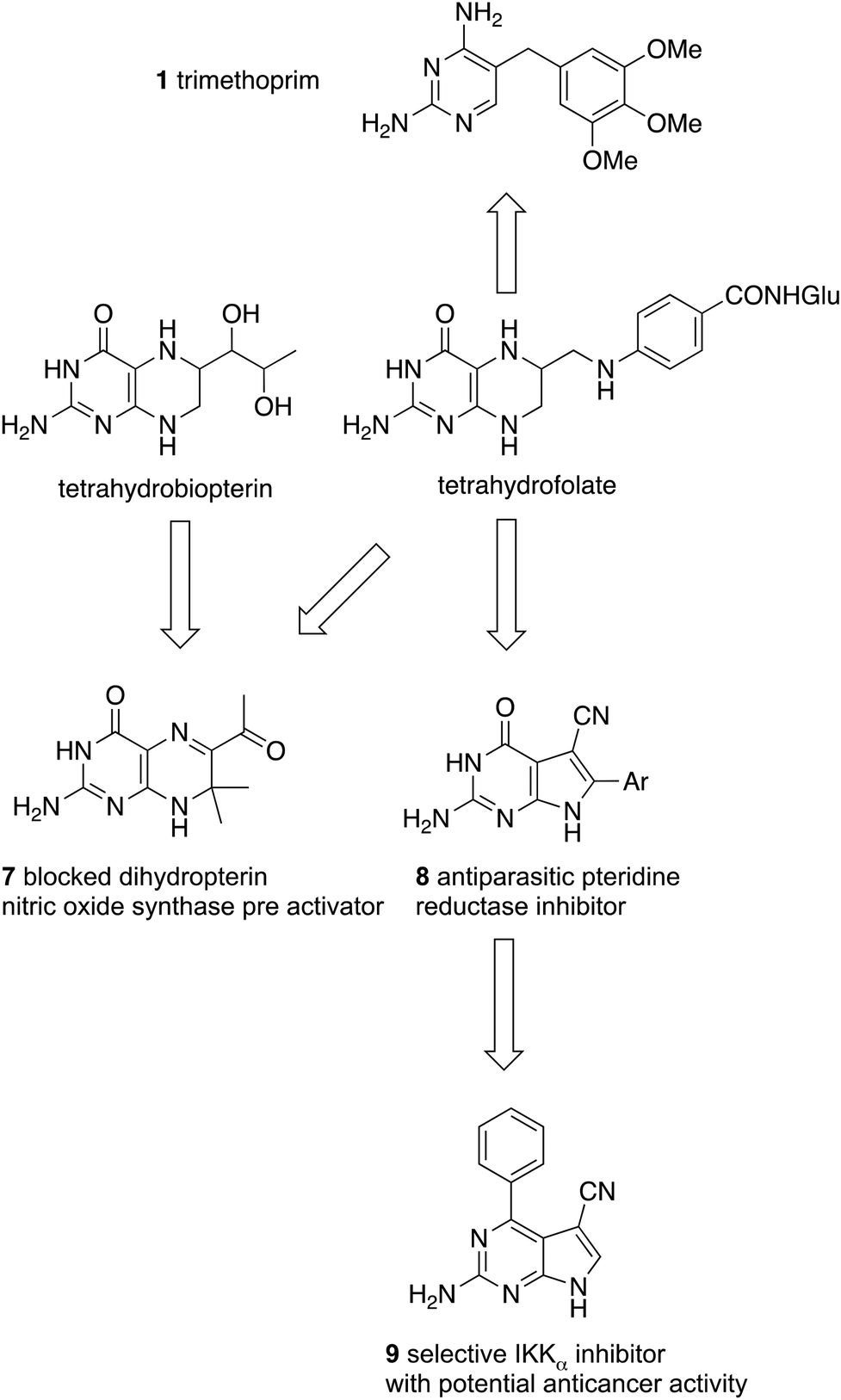 | ||
| Fig. 2 Structural relationships between the focal natural cofactor, tetrahydrofolate, and fused pyrimidines with various in vivo activities. | ||
Fused pyrimidines in cardiovascular and antiparasitic applications
The pteridine ring system itself is represented by blocked dihydropterins, which were introduced by Professor Hamish Wood, stimulated by the work of Hitchings and Elion.19 Nitric oxide is a hormone synthesised by the dimeric haem-containing enzyme, nitric oxide synthase, which oxidises arginine to nitric oxide and citrulline. Oxidation occurs through a cytochrome-P450 like cycle in which the reducing agent required to activate the iron is tetrahydrobiopterin, a 6-dihydoxypropyl substituted pterin. Although the blocked dihydropterin, 7, and several closely related compounds have some antibacterial properties linked to their inhibition of dihydropteroate synthase, the oxygen substituted side chain suggested a closer similarity with tetrahydrobiopterin. In this example, it is interesting that an enzyme activator, not inhibitor, was discovered and found to have potentially useful therapeutic properties.The tetrahydropterin formed by intracellular reduction of 7, a tetrahydrobiopterin analogue, was shown to be an activator of endothelial (eNOS) and inducible nitric oxide (iNOS) synthases.20 In the vascular system, nitric oxide functions as a vasodilator and an obvious question was whether the dihydropterin 7 could alleviate impaired vasodilation in pulmonary hypotension. Initially using arterial rings from hypotensive rats and then in vivo7 was shown to support production of nitric oxide with potential beneficial outcomes.21 The molecular justification for these experiments relied upon the reasonable assumption that 7, and its tetrahydro derivative, actually bind to NOS at a position in which it could contribute to electron transfer in the oxidation cycle. This was confirmed by a crystallographic study using bovine eNOS as the representative enzyme (Fig. 3),22 a study that also revealed significant information about the communication between the dimers of nitric oxide synthase in its mechanism of action. The commercial therapeutic potential of this discovery was recognised by a major multinational company which undertook significant preclinical development before it was dropped when the company moved out of the cardiovascular field.
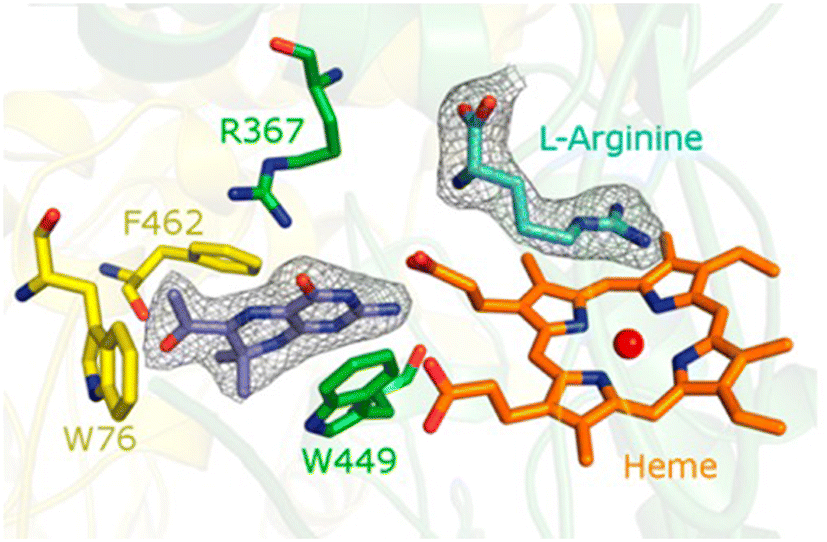 | ||
| Fig. 3 Location of blocked dihdydropterin 7 in the active site of eNOS as determined by X-ray crystallography. Reproduced from Biochemistry, 2014, 53, 4216.22 | ||
Dihydrofolate reductase is well known as the target for antibacterial drugs but in kinetoplastid parasites, there is a second reductase, pterindine reductase, which supports the cycling of tetrahydrobiopterin in the parasites. This enzyme too must be inhibited if a folate-dependent treatment for parasitic disease is to be obtained.23 A strongly structure-based enzyme inhibitor design project which was directly supported by X-ray crystallographic analysis of inhibitor–enzyme complexes by Professor William Hunter at the University of Dundee (Fig. 4).
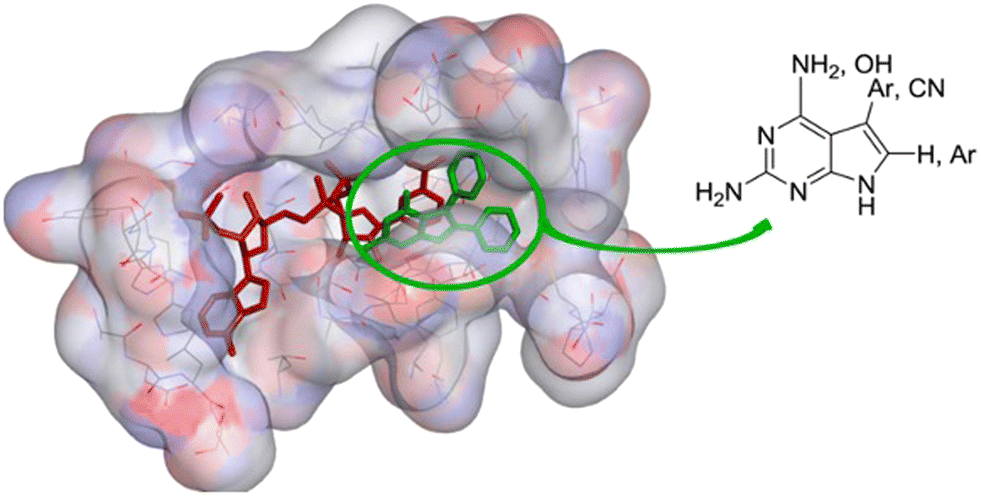 | ||
| Fig. 4 Location of pyrrolopyrimidine inhibitors (green) at the active site of PTR1 adjacent to the reducing cofactor, NADPH (red). Reproduced from J. Med. Chem., 2014, 57, 6479.24 | ||
Screening a library of pyrrolopyrimidines identified a number of compounds active against pteridine reductases, one of which was synergistic with methotrexate in challenging pteridine reductase 1. In a second study, 61 new pyrrolopyrimidines were synthsised, designed with the aid of crystal structures of which 23 were obtained, and eight pyrrolopyrimidines were sufficiently active to take forward to in vivo evaluation. From these eight, four were sufficiently potent to clear trypanosomal infection but were too toxic themselves to merit further development (Fig. 5). Other groups have since built upon these results with the discovery of improved compounds with different chemotypes.25 Because of the potential involvement of two reductases, dihydrofolate reductase and pterin reductase 1, it is notable that the multitargeted inhibitor strategy has been invoked.26
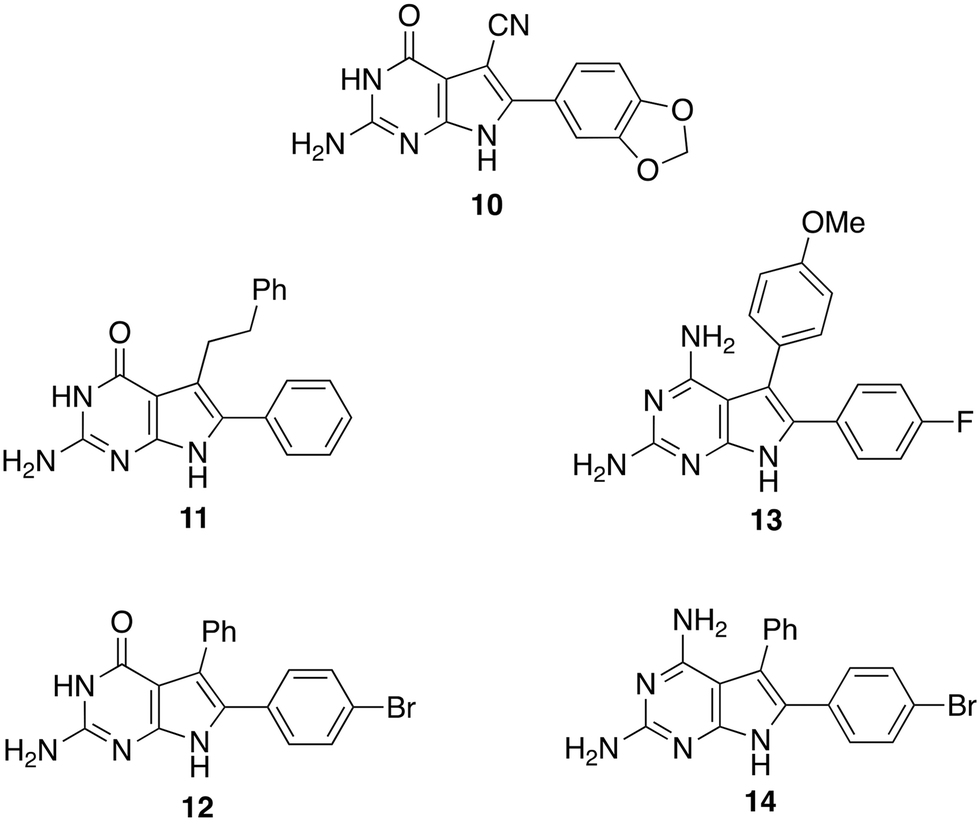 | ||
| Fig. 5 Pyrrolopyrimidines with antiparasitic activity. Seed compound (10 top) and four derivatives (11–14) that substantially cleared infection. | ||
In passing, a project led by Professor Simon Mackay that has significant implications for anticancer therapy but has not yet reached full proof of concept stage is worth mentioning. The connection is in the fused pyrimidine family of compounds, of which 9 (Fig. 2) was notable in this context. It was characterised as a selective IKKα inhibitor capable of blocking the non-canonical NF-κB pathway.27 Such compounds could have value in cancer therapy, of which prostate cancer is a prime target. In order to discover this compound, it was necessary to build and validate a homology model of IKKα so that design could avoid producing inhibitors of the closely related enzyme, IKKβ, which are known to lead to significant side effects in vivo. The first significant hits for the IKKα target were compounds of general structure 8. The nucleobase like structure of 9 pointed to binding in the ATP site of IKKα, emphasising the value of naturally occurring structures as a basis for drug discovery (see below).
No known target? Anti-inflammatory and immunomodulatory compounds
Targeted approaches, both single and multiple, have a contribution to make but still look at established targets and coupling individually active molecules together with a few covalent bonds. Can we not go further in a philosophical sense and find significant molecules with good reason for looking but no defined target? Having a reason for looking in the biological or chemical fields distinguishes this approach from blind phenotypic screening of libraries of compounds, typically from combinatorial chemistry sources, a strategy that has not lived up to initial expectations. This question, of course, refers to the discovery phase because as development and eventual use, hopefully, advance knowledge of targets and mechanism of action will undoubtedly accrue. It is also widely recognised that many if not most drugs have unexpected biological effects, which sometimes can be associated with physicochemical properties and other times with specific structural features such as potentially reactive functional groups.28,29An example of a molecule that is in the clinic for which at the time of regulatory approval there was no defined mechanism of action or target is pirfenidone (15, Fig. 6). Pirfenidone was identified as an anti-inflammatory and immunomodulatory compound in 1995 in evaluation in hamsters using the bleomycin-induced model of lung fibrosis.30 It subsequently progressed successfully to the market in 2008 in Japan; it has been licensed in several regulatory regimes for the treatment of idiopathic pulmonary fibrosis without a unique molecular target being identified. Pirfenidone is an example of where a genuine therapeutic need overcame the normal pressures to have a mechanism of action defined. Although important biological effects have been identified, including the downregulation of growth factor production and anti-inflammatory activity in cell and animal models, little is known about a molecular mechanism of action. Most recent studies support a view that pirfenidone modulates fibrogenic growth factors by suppressing TGF-β expression but no molecular mechanism has yet been proposed.31 What has emerged is that some idiosyncratic adverse reactions to pirfenidone might be mediated by cytochrome P450 oxidation to form a quinone methide which then reacts with thiol-containing compounds such as glutathione and N-acetyl cysteine and potentially also proteins32 (Fig. 6). The last of these possibilities could indeed be relevant to its anti-inflammatory activity as will be suggested below. Pirfenidone, then, stands to show that it is not futile to engage in the development of a new drug for which no target is known if the compound can be reasonably expected to have significant therapeutic benefits.
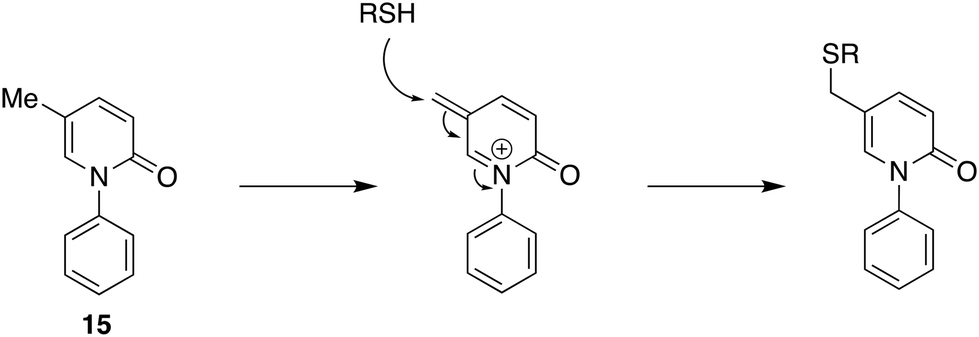 | ||
| Fig. 6 Pirfenidone 15, its quinone methide formed by oxidation in vivo, and its interaction with thiols in peptides and proteins. | ||
A further case in our laboratories lacking target information was stimulated by the properties of nematode parasitic worms, in particular a gerbil parasite (Acanthacheilomena viteae) which secretes a 62 kDa protein (known as ES-62) that has immunomodulatory properties.33 ES-62 and other related proteins can be considered to have evolved to suppress their hosts' immune system sufficiently to prevent it from attacking the parasite whilst not succumbing to opportunistic infection. ES-62 was shown to suppress cytokine output from macrophages and dendritic cells stimulated by PAMPs (pathogen associated molecular patterns) typical of bacterial and viral infection and was perceived to have potential therapeutic uses. Being a protein ES-62 was not suitable as a drug and it was also only available in microgram quantities. It was found, however, that small molecule analogues of ES-62 (SMAs, 16 and 17) are able to reproduce its immunomodulatory properties in a way that lends them to many therapeutic uses in inflammatory disease. Several have been investigated in the laboratories of Professors Maggie and Billy Harnett at the Universities of Glasgow and Strathclyde. The design of these SMAs was based upon a phosphoryltyrosine-containing peptide that had some ES-62-like properties. To avoid zwitterionic compounds and common biological activities associated with phosphate groups, the sulfone isostere was introduced.
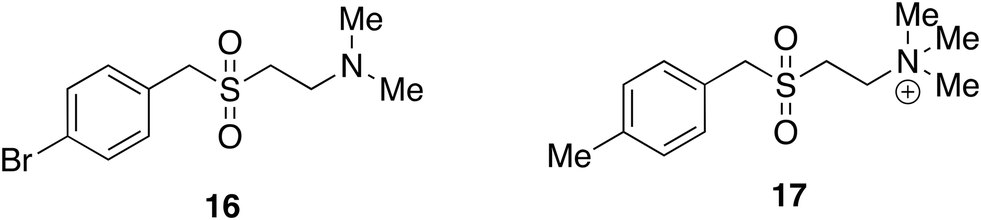
The SMAs replicate the function of phosphoryl choline surface post-translational modifications to ES-62 but at the time of their discovery, the mechanism of action was not known. Since SMAs are effective in so many disease models, a common biological signalling pathway was a probable location for targets (Fig. 7). Following the observation that levels of the adaptor protein, MyD88, were lowered in cells treated with ES-62 or SMAs the suggestion arose that the potential therapeutic effects were caused by the prevention of the dimerization of MyD88 which normally couples stimulation of TLR4 receptors to the intracellular signalling cascade that ultimately promotes pro-inflammatory cytokine synthesis and release. Using a combination of molecular modelling, that supported binding of 16 and 17 at the same site as known ligands, and pharmacological experiments, that showed that 17 by binding to the TIR domain inhibited the necessary dimerization of MyD88, it was shown that one mechanism of action of SMAs is indeed engagement with MyD88.43
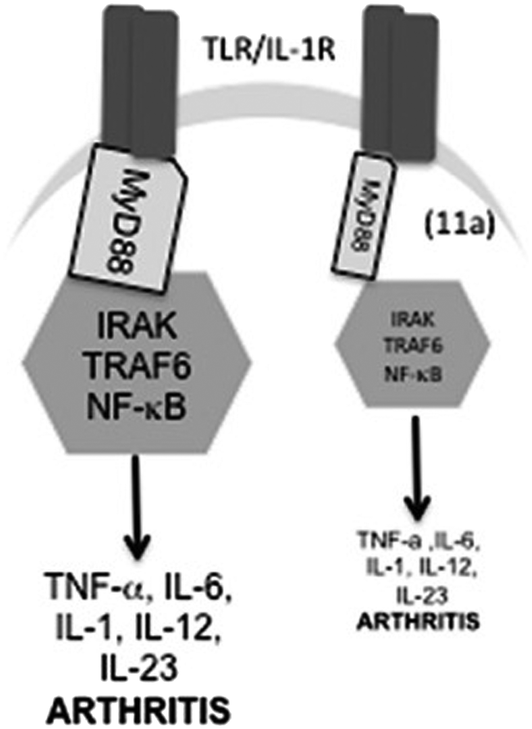 | ||
| Fig. 7 Schematic of model of action of 16. 16 downregulates MyD88 expression and hence induces a partial uncoupling of TLR/IL-1R from NF-κB activation and consequent pro-inflammatory cytokine production. Reproduced from J. Med. Chem., 2013, 56, 9982.34 | ||
This mechanistic insight followed many years after the original discovery of the SMAs. It is, however, not the whole story. In a study of the anti-allergic properties of SMAs it was found that one type of SMA, the quaternary ammonium salt 17 also inhibits inflammatory processes by inactivating the NRF2 inflammasome system probably through binding to the activator protein, Keap44 (Fig. 8). In molecular terms this could take place by covalent bonding to an essential cysteine residue, a mechanism known to be used by anti-inflammatory steroids (Fig. 9).45 It is plausible that the quinone methide derived from pirfenidone mentioned above might act in a similar way.
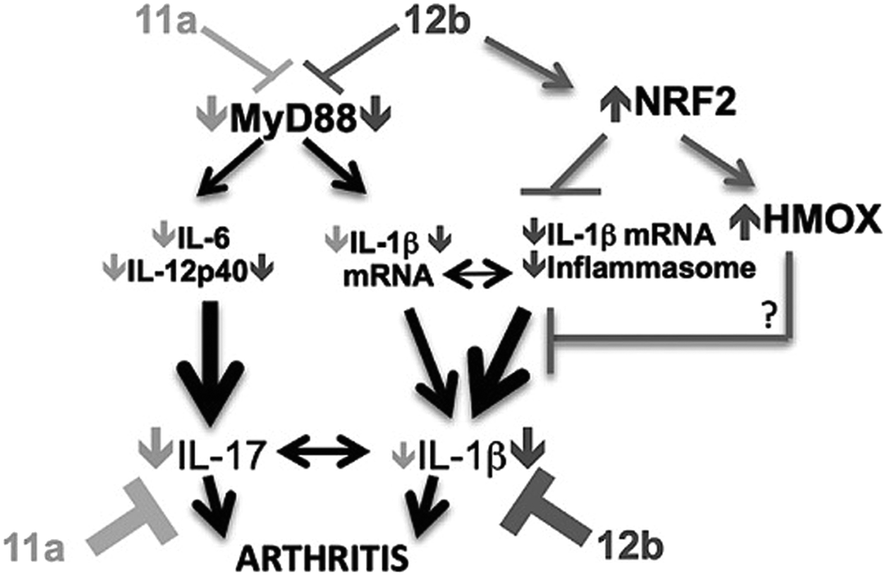 | ||
| Fig. 8 Model of SMAs' action in CIA: 12b = 17 protection predominantly reflects activation of NRF2 signaling to counteract MyD88-integrated inflammasome mediated IL-1β production. 11a = 17 preferentially targets MyD88-driven induction of the IL-17 inflammatory axis. Reproduced from J. Autoimmunity, 2015, 60, 59.44 | ||
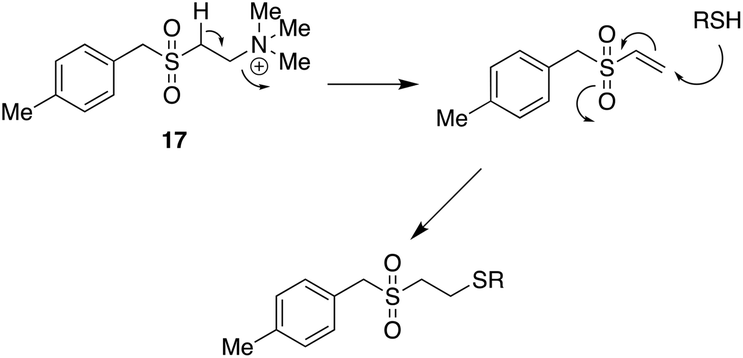 | ||
| Fig. 9 Possible mechanism for the interaction of 17 with Keap (RSH). | ||
The essence of the SMAs' effectiveness, then, is that they rebalance the immune system when inflammation has become excessive though activation of the NF-kB pathway (Fig. 7). Prevention of dimerization causes decoupling of MyD88 from its partner proteins leading to its degradation in the proteasome. Unless there is excess inflammation, the SMAs have no effect. The SMAs are neither activators nor inhibitors but act as control agents to rebalance the whole inflammatory cascade. In the many proof of concept and biological mechanism experiments that have been carried out in the Harnetts' laboratories, tiny doses, typically 10 μg/animal, have been required to afford beneficial effects, which have been both curative and prophylactic. It is also interesting that the effects of ES-62 are often best mimicked by treating with both the tertiary amide, 16, and the quaternary ammonium salt, 17, together. Adverse effects have not been observed. On the contrary, anecdotally, animal welfare colleagues have reported that mice treated with the SMAs seem sleeker, more active, and healthier than the control animals. Be that as it may, SMAs such as 16 and 17 have a remarkable property profile that argues strongly for their development towards the clinic. This challenge has been taken up by a new company, Vimelea Inc.
The discovery of the SMAs is a significant example of off-paradigm drug discovery because it was built solely upon the potential beneficial effects of the protein, ES-62. The only thing known at the time of discovery was a probable biological locus of action (TLR4); there was no information about the biological mechanism and still less about a molecular target. Although in the course of investigations of the therapeutic uses of SMAs some answers to these questions have been found, it is likely that there are many more features of mechanism to be discovered. There is a general point, however, that parallels the situation in the neuroscience field, namely that multifactorial diseases, of which those involving the immune system are also examples, require multiple functions for treatment. Far from having a single target, multiple targets are likely to be beneficial in a successful drug. The challenges for safety still remain but there's no point in developing a safe drug with constitutively limited effectiveness because it has one target only.
Anti-infective drugs
If there's one field in drug discovery where the standard paradigm is least suitable, and indeed has been argued to be failing,46,47 it is in anti-infective drugs and in challenging antimicrobial resistance (AMR). This is because infectious agents including bacteria, fungi, parasites, and, most topically, viruses become resistant to the action of established antibiotics through a variety of well-established mechanisms of which target mutation is most significant in the context of off-paradigm drug discovery. It is obvious that an anti-infective drug designed to act at a single target will instantly become useless if that target mutates such that the drug no longer binds. What is needed, therefore, for resilience to the development of resistance is a multitargeted drug. An opportunity arose at the University of Strathclyde to develop the field of minor groove binders (MGBs) for DNA based upon the natural products, distamycin and netropsin.The fundamental concept of this project, which has been reviewed in more detail,48 was that a short MGB can bind to a number of sites on DNA and thereby inhibit potentially several key biological processes in the infecting agent. Although a single molecule, DNA is a multi-target or perhaps more rigorously, multi-site in the context of off-target drug discovery. Not only should this strategy mitigate the development of resistance because of the multitargeting, but it should also be applicable to many types of infectious agent, bacteria, fungi, parasites, and even viruses. Provided that selectivity for the parasite can be obtained and avoidance of major challenges such as genotoxicity can be managed through careful medicinal chemical optimisation, MGBs offer an exceptional opportunity to discover drugs to combat AMR. The principal properties of S-MGBs as anti-infective compounds that attract attention are their resilience to the emergence of resistance (because of multitargeting), their activity against strains resistant to commonly used drugs (because of their distinct mechanism of action), and their selectivity (because of differential uptake into cells). These features can be illustrated by the following examples.
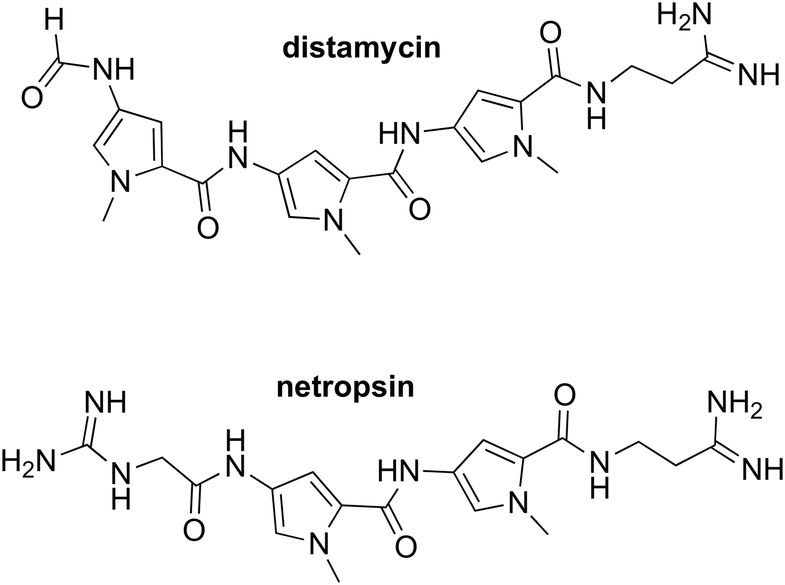
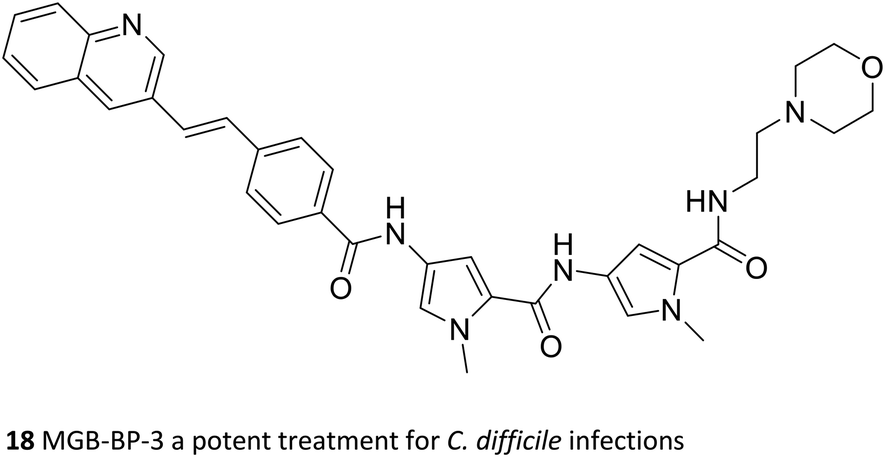
The key medicinal chemical modification that led to the developable Strathclyde MGB (S-MGB) family was to replace an amide link with an alkene.49 Within a short time successful proof of concept experiments were carried out in a mouse model of Staphyllococcus aureus infection. Subsequently there have been successful in vivo proof of concept experiments in mice for tuberculosis,50,51 animal African trypanosomiasis,52 aspergillosis,53 and most recently anti-COVID-19 activity.54 The most advanced S-MGB is known as MGB-BP-3 (18) and it has successfully completed a phase 2a clinical trial (NCT03824795; 2019–2020) for the treatment of Clostridioides difficile infection promoted by MGB Biopharma.55 The outcomes of the clinical trial were outstanding: at a dose of 125 mg twice daily, complete cures without recurrence were obtained, a result that, from an informal comparison, no other antibacterial drug available today can match for this condition. MGB Biopharma is now planning a pivotal phase 3 trial head-to-head comparing with the current standard of care, vancomycin. It is worth noting that the multi-target approach has been elegantly demonstrated by Boger through structural modifications to vancomycin tailoring it to fit several targets.56
In antifungal applications, the idea of mitigating resistance by dual targeting has also been investigated through an azole-COX inhibitor hybrid.57 Such hybrids remain vulnerable to the resistance, which of course already exists to azoles. The multitargeting mechanism of action of S-MGBs should still further reduce susceptibility to resistance in antifungal applications. S-MGB-363 (21) has broad activity against both animal and plant pathogens with levels of clinical significance (IC50 between 0.8 and 6.25 mg L−1) for Rhizopus arrhizus, Fusarium oxysporum, Aspergillus flavus, Scedosporium prolificans, Candida albicans, Cryptococcus neoformans, Candida glabrata, Aspergillus fumigatus and Candida auris. In an efficacy model, S-MGB-363 was as active as the standard of care drug, posaconazole, in combating infection in a mouse model of invasive aspergillosis.58 Through its fluorescent properties, it was possible to observe that S-MGB-363 associates with the fungal cell wall before transporting to the nucleus. Association at the cell membrane was not observed in a lung epithelial cell line (A549), nor was uptake, suggesting that entry into the target cell is a primary reason for the selectivity of S-MGB-363. Remarkably, 20 was also able to reduce the viral load and lung tissue damage in a hamster model of Sars-Cov-2 infection, opening new opportunities for discovery with S-MGBs.54 It is notable that in addition to being a therapeutically active compound in the model, S-MGB-363 acts as a probe of biological mechanism through its fluorescence. The provision of probe compounds to investigate biological mechanisms is an important contribution from academic laboratories and is at the heart of the field now known as chemical biology.
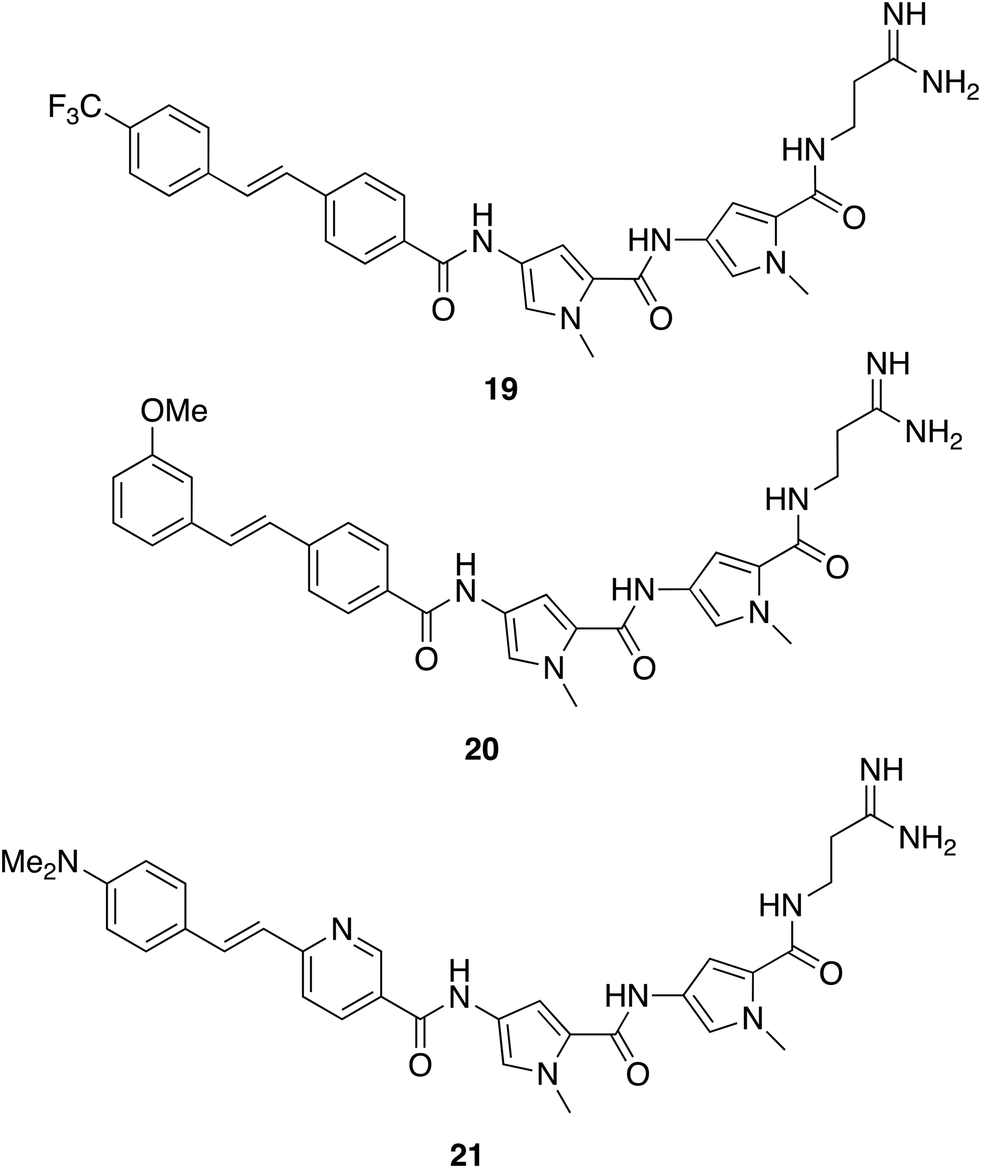
Finding an effective S-MGB against an intracellular parasite, such as in the case of Mycobacterium tuberculosis, that causes TB, is also a particular challenge. Not only must the drug be active against M. tuberculosis, but it must also be able to reach the bacterium inside the host macrophage and not kill that host cell. Add to that the difficulties of formulation and in vivo dosing, the effectiveness of 19 is remarkable. To reinforce the importance of selective behaviour at the cell wall or membrane, the properties of the antifungal S-MGB, 21, can be recalled.
The profiles and behaviours of the S-MGBs outlined above all hint at important components of mechanism of action, which continue to be investigated. MGB-BP-3, however, as a compound progressing well through clinical development, has been studied in much more detail. Overall, it behaves just as it should as an antibacterial drug according to its design: it is very resilient to the evolution of resistance; it inhibits some DNA modifying enzymes including topoisomerases;57 it binds to multiple sites in a bacterial genome (S. aureus) leading to the suppression of essential genes.59 For example, at the level of control, DNase I and permanganate footprinting showed that binding to SigA promoters and inhibition of promoter isomerization by RNA polymerase holoenzyme took place. SigA promoters control essential functions in S. aureus. Specifically, MGB-BP-3 was found to bind to the dnaD and mraY promoter regions thereby, interfering with transcriptional initiation of these genes. These binding sites were found by DNAse and permanganate footprinting of the relevant regions of genomic DNA. There are, of course, other possibilities. A crucial question for S-MGBs is the emergence of resistance, which multitargeting of DNA should prevent. S. aureus was repeatedly challenged by MGB-BP-3 in multiple passages at sub-MIC80 concentrations of either MGB-BP-3 or rifampicin as a control for 80 generations. Only resistance to rifampicin was observed. This is strong evidence for the resilience of MGB-BP-3 to drug induced resistance. Similar features have been found in other applications mentioned, in particular in antitrypanosomal activity. There is little doubt that a standard paradigm approach would not have led to such wide-ranging and significant outcomes. These important properties, activity against resistant strains and resilience to the emergence of resistance, commend the S-MGB family to commercial development, in which the new company, Rostra Therapeutics, is actively engaged.
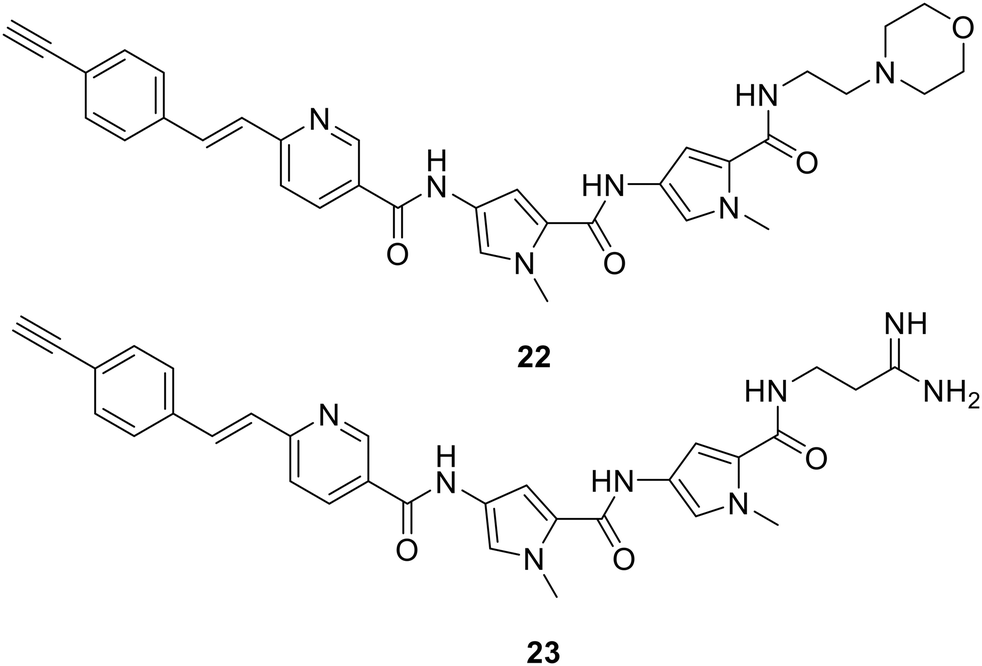
With respect to mechanism of action, modern technologies offer new experimental opportunities. Just as fluorescent probe S-MGBs have proved extremely valuable in pointing to key events in antifungal and antiparasitic applications, so Raman active S-MGBs have made it possible to examine the effect of tail group structure on uptake and localisation into mammalian cells as a pilot study.60 S-MGBs 22 and 23 containing the Raman active alkyne in the context of two different tail groups, the weakly basic morpholine, and the strongly basic amidine, were specially synthesised. Using PNT2 and HeLa cells as examples, amongst other things it was found that the morpholine containing S-MGB 22 accumulates more than the amidine 23, and that an active transport mechanism is not involved in this case. Such information is helpful to expand the scope of knowledge of S-MGBs so that improved compounds can be designed. This is the first time that Raman technology has been used in such cell-based studies.
It has been recognised for a very long time that the function of cells depends on objects of very different scales, at the smallest, molecules, and at the largest, organelles. Indeed, the Raman spectroscopic studies have already shown differences in localisation within cells between S-MGBs-22 and 23 notably in lysozomes. Between the two extremes are many molecular assemblies with important functions that are not bounded by membranes and therefore have a dynamic existence. Collectively, these assembles are known now as biomolecular condensates.61 Recently, the relevance of biomolecular conjugates to drug discovery has been highlighted, particularly from the point of view of modulating side effects and toxicity.62,63 It has been suggested, for example, that small molecules could interact with condensates in ways that improve their therapeutic profiles and that such structures should be designable. The fact that S-MGBs readily self-associate can be easily understood through their largely planar, amphiphilic structure.64 This same property would suggest that they should engage with biomolecular condensates that have an amphiphilic component and that the nature and extent of engagement might differ between different S-MGBs (and indeed other similar classes of MGB). The S-MGBs highlighted here, 19 and 21, have quite different head group structures, whilst sharing electron deficient characteristics. Investigations of these possibilities might not only help to understand the different profiles of the various S-MGBs but also help subsequently to design improved compounds as suggested by the recent reviews.62,63
Conclusion
The above examples argue for a broader approach to drug discovery in the earlier stages, expanding the standard paradigm in several possible ways, all of which have been shown to be effective in appropriate cases. The choice of the comparative word ‘broader’ is important because there is no single, omniapplicable strategy; intelligent selection of opportunities and approaches is especially important. Philosophical, commercial, and academic factors all contribute in different circumstances to different extents. A reviewer suggested that in parallel with in silico drug design, an in cerebro approach should be recognised as important, if not essential. This leaves open for the future to see what contribution AI will make!A recent contribution to the debate has been the suggestion that drugs and anti-infective drugs in particular are most likely to be successful if they have a basis in the structure of a natural product.65 It is argued that in this situation, nature is more likely to be able to take up the compound and let it do what it can. A particular point that was made is that the structural characteristics of anti-infective drugs in terms of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and molecular mass are different from those of most developed drugs, the molecular mass being higher than average. A further suggestion is the identification of pseudo natural products, that is compounds that contain a substructure very closely related to a natural product. This is obvious in the case of the S-MGBs but can also be seen specifically in the serominic compounds and by analogy in the fused pyrimidines. Thus, all examples cited in this article in their different ways fall back onto natural products, the fused pyrimidines onto folates, pteridines and nucleobases, the SMAs onto phosphorylcholine, and the S-MGBs onto the natural polypyrrole amides, distamycin and netropsin. This was certainly not in mind when the projects were chosen. Yes, we have to have primary targets but not, I would suggest, single targets in isolation to discover new and effective drugs. Reaching the target and doing the job there is the end game for a drug molecule.
P and molecular mass are different from those of most developed drugs, the molecular mass being higher than average. A further suggestion is the identification of pseudo natural products, that is compounds that contain a substructure very closely related to a natural product. This is obvious in the case of the S-MGBs but can also be seen specifically in the serominic compounds and by analogy in the fused pyrimidines. Thus, all examples cited in this article in their different ways fall back onto natural products, the fused pyrimidines onto folates, pteridines and nucleobases, the SMAs onto phosphorylcholine, and the S-MGBs onto the natural polypyrrole amides, distamycin and netropsin. This was certainly not in mind when the projects were chosen. Yes, we have to have primary targets but not, I would suggest, single targets in isolation to discover new and effective drugs. Reaching the target and doing the job there is the end game for a drug molecule.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work could not have been undertaken without the kind and enthusiastic participation of many colleagues at the University of Strathclyde and elsewhere. The full list of names can be read in the references, but I would particularly like to thank Michael Barrett, Colin Gibson, Reto Guler, Billy and Maggie Harnett, Alan Harvey, Iain Hunter, William Hunter, Simon Mackay, Fraser Scott, and Roger Waigh. I also appreciate the time reviewers took to make helpful suggestions. Funding for the research discussed in this review has come from many sources over many years. I am pleased to thank the following organisations for their support. Arthritis Research Council, Bioscience and Biotechnology Research Council UK, Cancer Research UK, Chief Scientist's Office Scotland, GALVMed, Greater Glasgow Health Board, Knowledge Transfer Partnerships UK, Medical Research Council UK, MGB Biopharma Ltd, PysRING Japan, Scottish Enterprise, Scottish Universities Life Sciences Alliance, Talon Inc. USA, University of Cape Town, University of Glasgow, University of Strathclyde, Welfide Corporation, Japan.Notes and references
- P. Seneci and S. Miertus, Mol. Diversity, 2000, 5, 75 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188 CrossRef CAS PubMed.
- R. Liu, X. Li and K. S. Lam, Curr. Opin. Chem. Biol., 2017, 38, 117 CrossRef CAS PubMed.
- B. Thomas, X. Lu, W. R. Birmingham, J. Huang, P. Both, J. E. Y. Martinez, R. J. Young, C. P. Davie and S. L. Flitsch, ChemBioChem, 2017, 18, 858 CrossRef CAS PubMed.
- G. H. Hitchings Jr., https://www.nobelprize.org/uploads/2018/06/hitchings-lecture.pdf.
- G. B. Elion, https://www.nobelprize.org/uploads/2018/06/elion-lecture.pdf.
- M. A. Olson, M. S. Lee, T. L. Kissner, S. Alam, D. S. Waugh and K. U. Saikh, Sci. Rep., 2015, 5, 14246 CrossRef PubMed.
- P. Kirsch, A. W. Hartman, A. K. H. Hirsch and M. Empting, Molecules, 2019, 24, 4309 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605 CrossRef CAS PubMed.
- J. A. Borthwick, C. Alemparte, I. Wall, B. C. Whitehurst, A. Argyrou, G. Burley, P. de Dios-Anton, L. Guijarro, M. C. Monteiro, F. Ortega, C. J. Suckling, J. C. Pichel, M. Cacho and R. J. Young, J. Med. Chem., 2020, 63, 2557 CrossRef CAS PubMed.
- World Health Organization Model List of Essential Medicines, 21st List, 2019, World Health Organization, Geneva, 2019, Licence: CC BY-NC-SA 3.0 IGO Search PubMed.
- B. Patwardhan and R. A. Mashelkar, Drug Discovery Today, 2009, 14, 804 CrossRef PubMed.
- Old English illustrated herbal and other medical remedies, MS Cotton Vitellius C, London, British Library Search PubMed.
- J. Black, https://www.nobelprize.org/uploads/2018/06/black-lecture.pdf.
- C. J. Suckling, J. A. Murphy, A. I. Khalaf, S.-Z. Zhou, D. E. Lizos, A. N. van Nhien, B. J. Morris, J. A. Pratt, A. McVie, H. Yasumatsu, A. L. Harvey, L. C. Young and C. McCraw, Bioorg. Med. Chem. Lett., 2007, 17, 2649 CrossRef CAS PubMed.
- J. Marco-Contelles, ACS Chem. Neurosci., 2019, 10, 1127 CrossRef CAS PubMed.
- Ó. M. Bautista-Aguilera, J. Budni, F. Mina, E. B. Medeiros, W. Deuther-Conrad, J. M. Entrena, I. Moraleda, I. Iriepa, F. López-Muñoz and J. Marco-Contelles, J. Med. Chem., 2018, 61, 6937 CrossRef PubMed.
- F. J. Pérez-Areales, M. Garrido, E. Aso, M. Bartolini, A. De Simone, A. Espargaró, T. Ginex, R. Sabate, B. Pérez, V. Andrisano, D. Puigoriol-Illamola, M. Palla, F. J. Luque, M. I. Loza, J. Brea, I. Ferrer, F. Ciruela, A. Messeguer and D. Muñoz-Torrero, J. Med. Chem., 2020, 63, 9360 CrossRef PubMed.
- S. S. Al Hassan, R. J. Cameron, A. W. C. Curran, W. J. S. Lyall, S. H. Nicholson, D. R. Robinson, A. Stuart, I. Stirling, C. J. Suckling and H. C. S. Wood, J. Chem. Soc., Perkin Trans. 1, 1985, 1645 RSC.
- C. J. Suckling, C. L. Gibson, J. K. Huggan, R. R. Morthala, B. Clarke, S. Kununthur, R. M. Wadsworth, S. Daff and D. Papale, Bioorg. Med. Chem. Lett., 2008, 18, 1552 CrossRef PubMed.
- S. Kunuthur, P. H. Milliken, C. L. Gibson, C. J. Suckling and R. M. Wadsworth, Eur. J. Pharmacol., 2011, 650, 37 CrossRef PubMed.
- G. Chreifi, H. Li, C. McInnes, C. L. Gibson, C. J. Suckling and T. L. Poulos, Biochemistry, 2014, 53, 4216 CrossRef CAS PubMed.
- L. B. Tulloch, V. P. Martini, J. Iulek, J. K. Huggan, J. H. Lee, C. L. Gibson, T. K. Smith, C. J. Suckling and W. N. Hunter, J. Med. Chem., 2010, 53, 221 CrossRef CAS PubMed.
- A. I. Khalaf, J. K. Huggan, C. J. Suckling, C. L. Gibson, K. Stewart, F. Giordani, M. P. Barrett, P. E. Wong, K. L. Barrack and W. N. Hunter, J. Med. Chem., 2014, 57, 6479 CrossRef CAS PubMed.
- P. Linciano, C. Pozzi, L. dello Iacono, F. di Pisa, G. Landi, A. Bonucci, S. Gul, M. Kuzikov, B. Ellinger, G. Witt, N. Santarem, C. Baptista, C. Franco, C. B. Moraes, W. Müller, U. Wittig, R. Luciani, A. Sesenna, A. Quotadamo, S. Ferrari, I. Pöhner, A. Cordeiro-da-Silva, S. Mangani, L. Costantino and M. P. Costi, J. Med. Chem., 2019, 62, 3989 CrossRef CAS PubMed.
- I. Pöhner, A. Quotadamo, J. Panecka-Hofman, R. Luciani, M. Santucci, P. Linciano, G. Landi, F. Di Pisa, L. dello Iacono, C. Pozzi, S. Mangani, S. Gul, G. Witt, B. Ellinger, M. Kuzikov, N. Santarem, A. Cordeiro-da-Silva, M. P. Costi, A. Venturelli and R. C. Wade, J. Med. Chem., 2022, 65, 9011 CrossRef PubMed.
- N. G. Anthony, J. Baiget, G. Berretta, M. Boyd, D. Breen, C. Gamble, A. I. Gray, A. L. Harvey, S. Hatziieremia, K. H. Ho, J. K. Huggan, S. Lnag, S. Llona-Minguez, K. McIntosh, A. Paul, R. J. Plevin, M. N. Robertson, R. Scott, C. J. Suckling, O. B. Sutcliffe, J. Edwards, J. L. Luo, L. C. Young and S. P. Mackay, J. Med. Chem., 2017, 60, 7043 CrossRef CAS PubMed.
- S. J. Chakravorty, J. Chan, M. N. Greenwood, M. I. Popa-Burke, K. S. Remlinger, S. D. Pickett, D. V. S. Green, M. C. Fillmore, T. W. Dean, J. I. Luengo and R. Macarron, SLAS Discovery, 2018, 23, 532 CrossRef CAS PubMed.
- D. G. Brown, G. F. Smith and H. J. Wobst, J. Med. Chem., 2020, 63, 6251 CrossRef CAS PubMed.
- S. N. Iyer, J. S. Wild, M. J. Schiedt, D. M. Hyde, S. B. Margolin and S. N. Giri, J. Lab. Clin. Med., 1995, 125, 779 CAS.
- S. M. Ruwanpura, B. J. Thomas and P. G. Bardin, Am. J. Respir. Cell Mol. Biol., 2020, 62, 413 CrossRef CAS PubMed.
- S. Zhou, W. Li, M. Tian, N. Zhang, X. Yang, W. Li, Y. Peng and J. Zheng, J. Med. Chem., 2020, 63, 8059 CrossRef CAS PubMed.
- W. Harnett, M. J. Worms, A. Kapil, M. Grainger and R. M. Parkhouse, Parasitology, 1989, 99, 229e239 CrossRef PubMed.
- L. Al-Riyami, M. A. Pineda, J. Rzepecka, J. K. Huggan, A. I. Khalaf, C. J. Suckling, F. J. Scott, D. T. Rodgers, M. M. Harnett and W. Harnett, J. Med. Chem., 2013, 56, 9982 CrossRef CAS PubMed.
- J. C. Coltherd, D. T. Rodgers, R. E. Lawrie, L. Al-Riyami, C. J. Suckling, W. Harnett and M. M. Harnett, Sci. Rep., 2016, 6, 19224 CrossRef CAS PubMed.
- C. J. Suckling, S. Mukherjee, A. I. Khalaf, A. Narayan, F. J. Scott, S. Khare, S. Dhakshinamoorthy, M. M. Harnett and W. Harnett, Acta Trop., 2018, 185, 212 CrossRef CAS PubMed.
- D. T. Rodgers, M. A. Pineda, C. J. Suckling, W. Harnett and M. M. Harnett, Lupus, 2015, 23, 1437 CrossRef PubMed.
- L. Janicova, J. Rzepecka, D. T. Rodgers, J. Doonan, K. S. Bell, F. E. Lumb, C. J. Suckling, M. M. Harnett and W. Harnett, Parasite Immunol., 2016, 38, 340 CrossRef CAS PubMed.
- F. E. Lumb, J. Crowe, J. Doonan, C. J. Suckling, C. Selman, M. M. Harnett and W. Harnett, Mol. Biochem. Parasitol., 2019, 234, 111232 CrossRef CAS PubMed.
- M. M. Harnett, J. Doonan, F. E. Lumb, J. Crowe, R. O. Damink, G. Buitrago, J. Duncombe-Moore, D. I. Wilkinson, C. J. Suckling, C. Selman and W. Harnett, Front. Immunol., 2022, 13, 953053 CrossRef CAS PubMed.
- J. Goran, M. McGear and R. McGough, The Shadows, Parlophone, 1968 Search PubMed.
- J. Doonan, D. Thomas, M. H. Wong, H. J. Ramage, L. Al-Riyami, F. E. Lumb, K. S. Bell, K. J. Fairlie-Clarke, C. J. Suckling, K. S. Michelsen, H.-R. Jiang, A. Cooke, M. M. Harnett and W. Harnett, Molecules, 2018, 23, 2669 CrossRef PubMed.
- C. J. Suckling, S. Alam, M. A. Olson, K. U. Saikh, M. M. Harnett and W. Harnett, Sci. Rep., 2018, 8, 2123 CrossRef PubMed.
- J. Rzepecka, M. A. Pineda, L. Al-Riyami, D. T. Rogers, J. K. Huggan, F. E. Lumb, A. I. Khalaf, P. J. Meakin, M. Corbet, M. L. Ashford, C. J. Suckling, M. M. Harnett and W. Harnett, J. Autoimmun., 2015, 60, 59 CrossRef CAS PubMed.
- M. Yu, H. Li, Q. Liu, F. Liu, L. Tang and C. Li, et al. , Cell. Signalling, 2011, 23, 883e892 Search PubMed.
- R. Tommasi, D. G. Brown, G. K. Walkup, J. I. Manchester and A. A. Miller, Nat. Rev. Drug Discovery, 2015, 14, 529 CrossRef CAS PubMed.
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29 CrossRef CAS PubMed.
- C. J. Suckling, I. S. Hunter and F. J. Scott, Future Drug Discovery, 2022 DOI:10.4155/fdd-2022-0001.
- C. J. Suckling, D. Breen, A. I. Khalaf, E. Ellis, I. S. Hunter, G. Ford, C. G. Gemmell, N. Anthony, J.-J. Helsebeux, S. P. Mackay and R. D. Waigh, J. Med. Chem., 2007, 50, 6116 CrossRef PubMed.
- L. Hlaka, M.-J. Rosslee, M. Ozturk, K. Santosh, S. Parihar, F. Brombacher, A. I. Khalaf, K. C. Carter, F. J. Scott, C. J. Suckling and R. Guler, J. Antimicrob. Chemother., 2017, 72, 3334 CrossRef CAS PubMed.
- N. S. Kieswetter, M. Ozturk, L. Hlaka, J. E. Chia, R. J. O. Nichol, J. M. Cross, L. M. C. McGee, I. Tyson-Hirst, R. Beveridge, F. Brombacher, K. C. Carter, C. J. Suckling, F. J. Scott and R. Guler, J. Antimicrob. Chemother., 2022, 77, 1061 CrossRef CAS PubMed.
- F. Giordani, A. I. Khalaf, K. Gillingwater, J. Munday, H. de Koning, C. J. Suckling, M. P. Barrett and F. J. Scott, J. Med. Chem., 2019, 62, 3021 CrossRef CAS PubMed.
- R. J. O. Nichol, C. Zhao, A. I. Khalaf, J. May, C. J. Suckling, F. J. Scott and M. Bromley, 2nd SCI/RSC Symposium on Antimicrobial Drug Discovery, 2018, London, England, 12th and 13th November 2018 Search PubMed.
- F. J. Scott, Chief Scientists Office (Scotland) https://www.cso.scot.nhs.uk/wp-content/uploads/COVSCL2001-1.pdf.
- MGB Biopharma, https://www.mgb-biopharma.com/mgb-biopharma-announces-successful-outcome-from-phase-ii-clinical-study-with-mgb-bp-3-a-potential-new-gold-standard-first-line-treatment-for-clostridium-difficile-infection-cdi/.
- A. Okano, N. A. Isley and D. L. Boger, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5052 CrossRef CAS PubMed.
- R. Elias, P. Basu and M. Fridman, J. Med. Chem., 2022, 65, 2361 CrossRef CAS PubMed.
- C. Hind, M. Clifford, C. Woolley, J. Harmer, L. M. C. McGee, I. Tyson-Hirst, H. J. Tait, D. P. Brooke, S. J. Dancer, I. S. Hunter, C. J. Suckling, R. Beveridge, J. A. Parkinson, J. M. Sutton and F. J. Scott, ACS Infect. Dis., 2022, 8, 2552 CrossRef CAS PubMed.
- L. Kerr, D. Browning, K. Lemonidis, T. Salih, I. Hunter, C. Suckling and N. Tucker, bioRxiv, 2020, preprint, arxiv:12.31.424950, DOI:10.1101/2020.12.31.424950.
- C. Tentellino, W. J. Tipping, L. M. C. McGee, L. M. Bain, C. Wetherill, S. Laing, I. Tyson-Hirst, C. J. Suckling, R. Beveridge, F. J. Scott, K. Faulds and D. Graham, RSC Chem. Biol., 2022, 3, 1403 RSC.
- A. S. Lyon, W. B. Peeples and M. K. Rosen, Nat. Rev. Chem. Biol., 2021, 22, 215 CrossRef CAS PubMed.
- D. M. Mitrea, M. Mittasch, B. F. Gomes, I. A. Klein and M. A. Murcko, Nat. Rev. Drug Discovery, 2022, 21, 841 CrossRef CAS PubMed.
- H. R. Kilgore and R. A. Young, Nat. Chem. Biol., 2022, 18, 1298 CrossRef CAS PubMed.
- J. A. Parkinson, F. J. Scott, C. J. Suckling and G. Wilson, Med. Chem. Commun., 2013, 4, 1105 RSC.
- R. J. Young, S. L. Flitsch, M. Grigalunas, P. D. Leeson, R. J. Quinn, N. J. Turner and H. Waldmann, JACS Au, 2022, 2, 2400 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |