
Sulfur-regulated CoSe2 nanowires with high-charge active centers for electrochemical nitrate reduction to ammonium†
Wuyong
Zhang
,
Yingjie
Wen
,
Haocheng
Chen
,
Minli
Wang
,
Caihan
Zhu
,
Yunan
Wang
* and
Zhiyi
Lu
 *
*
Key Laboratory of Advanced Fuel Cells and Electrolyzers Technology of Zhejiang Province, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, P. R. China. E-mail: luzhiyi@nimte.ac.cn; wangyunan@nimte.ac.cn
First published on 22nd June 2024
Abstract
Developing high-efficiency electrocatalysts for nitrate-to-ammonia transformation holds significant promise for the production of ammonia, a crucial component in agricultural fertilizers and as a carbon-free energy carrier. In this study, we propose a viable strategy involving sulfur doping to modulate both the microstructure and electronic properties of CoSe2 for nitrate reduction. This approach remarkably enhances the conversion of nitrate to ammonia by effectively regulating the adsorption capability of nitrogenous intermediates. Specifically, sulfur-doped CoSe2 nanowires (S-CoSe2 NWs) exhibit a peak faradaic efficiency of 93.1% at −0.6 V vs. RHE and achieve the highest NH3 yield rate of 11.6 mg h−1 cm−2. Mechanistic investigations reveal that sulfur doping facilitates the creation of highly charged active sites, which enhance the adsorption of nitrite and subsequent hydrogenation, leading to improved selectivity towards ammonia production.
New conceptsIn this study, we demonstrate a new concept that regulates the nanodimensions of transition-metal dichalcogenides (TMDs) by introducing sulfur. Our sulfur doping method differs from the normal electronic regulation of materials and further modulates the morphology of the CoSe2 materials from 2D to 1D by changing the supersaturation and ionization of the synthetic system. The 1D ternary S-CoSe2 NWs can drive nitrate electroreduction to ammonia (NARR) with high activity and selectivity due to their advanced one-dimensional structure and high-charge active center. This is the first time that a morphology and electron-control method has been proposed to provide an advanced structure for NARR in TMDs. The significance of an advanced microstructure and electronic structure in NARR has been proven, and we are sure this is a highly innovative approach from the perspective of materials chemistry and from electrochemistry, which opens up an interesting field of research. |
Introduction
Ammonia is a cornerstone of human existence, primarily fueling fertilizer production.1–3 However, the dominant method for large-scale ammonia synthesis, the Haber–Bosch process, heavily relies on elevated temperatures and pressures, consumes nearly 2% of global energy, and contributes to approximately 1% of greenhouse gas emissions.4–8 The electrochemical nitrogen reduction reaction (NRR) has emerged as a promising alternative, offering the potential for ammonia production under ambient conditions.9 Nonetheless, in aqueous systems, the faradaic efficiency (FE) and partial current density typically fall below 50% and 10 mA cm−2, respectively.10–13 This significant gap between the current NRR performance and industrial requirements, demanding a partial current density of 300 mA cm−2 with 90% FE, remains a challenge.14The primary obstacle to advancing NRR lies in the inertness of nitrogen (941 kJ mol−1 for N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and its low water solubility (20 mg L−1).15 In contrast, nitrate (NO3−) boasts a more readily activated chemical bond (204 kJ mol−1 for N–O) and greater solubility, facilitating nitrate reduction reactions (NARRs) at the solid–liquid interface with a lower energy barrier.16 Moreover, nitrate is abundant in the Earth's environment and is extensively used as a nitrogen-containing fertilizer.17 Also, precisely because of this, its widespread use has led to pervasive nitrogen contamination, significantly impacting biodiversity.18 Additionally, many individuals are exposed to water environments exceeding regulated nitrate limits (50 mg L−1, WHO, 2004), potentially leading to health issues such as non-Hodgkin's lymphoma and methemoglobinemia.19–21 Therefore, reversing nitrate conversion is of substantial importance for both industrial ammonia production and environmental remediation.22
N) and its low water solubility (20 mg L−1).15 In contrast, nitrate (NO3−) boasts a more readily activated chemical bond (204 kJ mol−1 for N–O) and greater solubility, facilitating nitrate reduction reactions (NARRs) at the solid–liquid interface with a lower energy barrier.16 Moreover, nitrate is abundant in the Earth's environment and is extensively used as a nitrogen-containing fertilizer.17 Also, precisely because of this, its widespread use has led to pervasive nitrogen contamination, significantly impacting biodiversity.18 Additionally, many individuals are exposed to water environments exceeding regulated nitrate limits (50 mg L−1, WHO, 2004), potentially leading to health issues such as non-Hodgkin's lymphoma and methemoglobinemia.19–21 Therefore, reversing nitrate conversion is of substantial importance for both industrial ammonia production and environmental remediation.22
In addition to the aforementioned merits, the NARR process generates undesirable byproducts such as dinitrogen, nitrite, and nitric oxide due to multiple electron–proton transfer processes.23–25 The formation of these byproducts depends on the nitrate concentration and electrolyte pH, highlighting the need for novel materials with abundant active sites to facilitate efficient ammonia production.26–28 While traditional efficient NARR processes often rely on noble metals like Ru-based materials, their high cost and natural scarcity pose significant barriers to widespread applications.29–32 In contrast, transition-metal dichalcogenides (TMDs) such as CoSe2 and CoS2 have emerged as compelling candidates due to their abundance and cost-efficient production processes.33–35 Moreover, given that electrochemical reactions predominantly occur at material surfaces or interfaces, the high specific surface area of TMDs, akin to MoS2 for hydrogen evolution, holds promise for electrochemical energy conversion.36–38 However, the common 2D TMDs suffer from aggregation or restacking that severely limits their potential performance, necessitating the construction of advanced structures from the molecular level to enhance the intrinsic activity of TMDs.39,40 Furthermore, the application of TMDs in the NARR has been scarcely reported to date, highlighting the untapped potential in this domain.41,42
Herein, we present a novel ternary TMD, S-CoSe2 nanowire, designed and synthesized for the NARR. By introducing sulfur to modulate the ionization and supersaturation of the synthetic system, the microstructure of the original binary CoSe2 transitions from 2D to 1D, coinciding with the generation of high-charge cobalt centers. The resulting 1D S-CoSe2 nanowire with an advanced structure exhibits pronounced nitrate-to-ammonia activity and selectivity, achieving a peak faradaic efficiency of 93.1% and the highest NH3 yield rate of 11.6 mg h−1 cm−2. Further theoretical studies suggest that the high-charge cobalt center enhances the adsorption of NARR intermediates, such as nitrite, for further hydrogenation.
Results and discussion
Ternary ultrathin sulfur-doped CoSe2 nanowires (S-CoSe2 NWs) were synthesized via a solvothermal method.43–45 Scanning electron microscopy (SEM) images at low magnification revealed the homogeneity of the obtained materials, devoid of any other byproducts, thereby confirming the purity of the synthesized nanowires. Additionally, the SEM images indicate that the material consisted of nanowires with lengths spanning in the micrometer range, suggesting the successful fabrication of elongated nanostructures (Fig. S1, ESI†). The evolution of the porous structure of S-CoSe2 NWs by the overlapping and interweaving of nanowires was corroborated by nitrogen physisorption experiments (Fig. S2, ESI†). It shows a type I(b) isotherm with a high uptake of nitrogen at high relative pressure, which indicates the adsorption of macropores according to the IUPAC classification.46 Such a porous architecture is crucial for facilitating mass transport and increasing the active surface area, thereby enhancing the performance of electrochemical applications. Further investigation of the microstructure of S-CoSe2 NWs was conducted using transmission electron microscopy (TEM). The TEM images revealed that the S-CoSe2 NWs exhibited a nerve-like morphology, characterized by a flexible, smooth, and even transparent appearance (Fig. 1a). Bright-field high-resolution TEM (HRTEM) was employed to further elucidate the ultrathin nature of S-CoSe2 NWs. The interplanar distance was measured to be 0.26 nm (Fig. 1b). This can be attributed to the (210) plane of the CoSe2 lattice fringes, which implies that the growth direction of the S-CoSe2 NWs was [210]. The polycrystalline nature of the S-CoSe2 NWs was further confirmed by selected area electron diffraction (SAED) pattern analysis (Fig. S3a, ESI†), and the diffraction rings can be indexed to (210) and (311), which is in good agreement with the powder X-ray diffraction (XRD) patterns (Fig. S3b, ESI†). All observed diffraction peaks are broadening and some reflections of CoSe2 are missing, which should be attributed to the ultrathin structure of the S-CoSe2 NWs.44 Under the inspection of aberration-corrected high-angle dark-field scanning transmission electron microscopy (AC HAAD-STEM) with sub-angstrom resolution, it was clearly observed that the fasciculate nanowire had an order arrangement of atoms along the growth direction (Fig. 1c). The Raman spectrum of the S-CoSe2 NWs shows an obvious peak at around 220 cm−1, which can be assigned to the Se–Se stretching mode of CoSe2 (Fig. S4, ESI†).47 The thickness of S-CoSe2 NWs was measured using an atomic force microscopic (AFM, Fig. 1d), and the corresponding height profile clearly shows a single S-CoSe2 NWs average thickness of around 1.5 nm (Fig. 1e), which is almost equal to that of two single-unit-cell CoSe2 slab.45 SEM energy-dispersive X-ray (EDX) spectra confirm the presence of cobalt, selenium and sulfur, and the Se/Co atomic ratio was close to 2 (Fig. S5, ESI†). The corresponding EDX mapping images and line scanning clearly demonstrate the homogenous distribution of the elements (Fig. 1f and Fig. S6, ESI†). The sulfur content in S-CoSe2 NWs was determined to be 4.3 at% by elemental analysis (Table S2, ESI†). Considering this, the above results clearly demonstrate the successful fabrication of ternary S-CoSe2 NWs.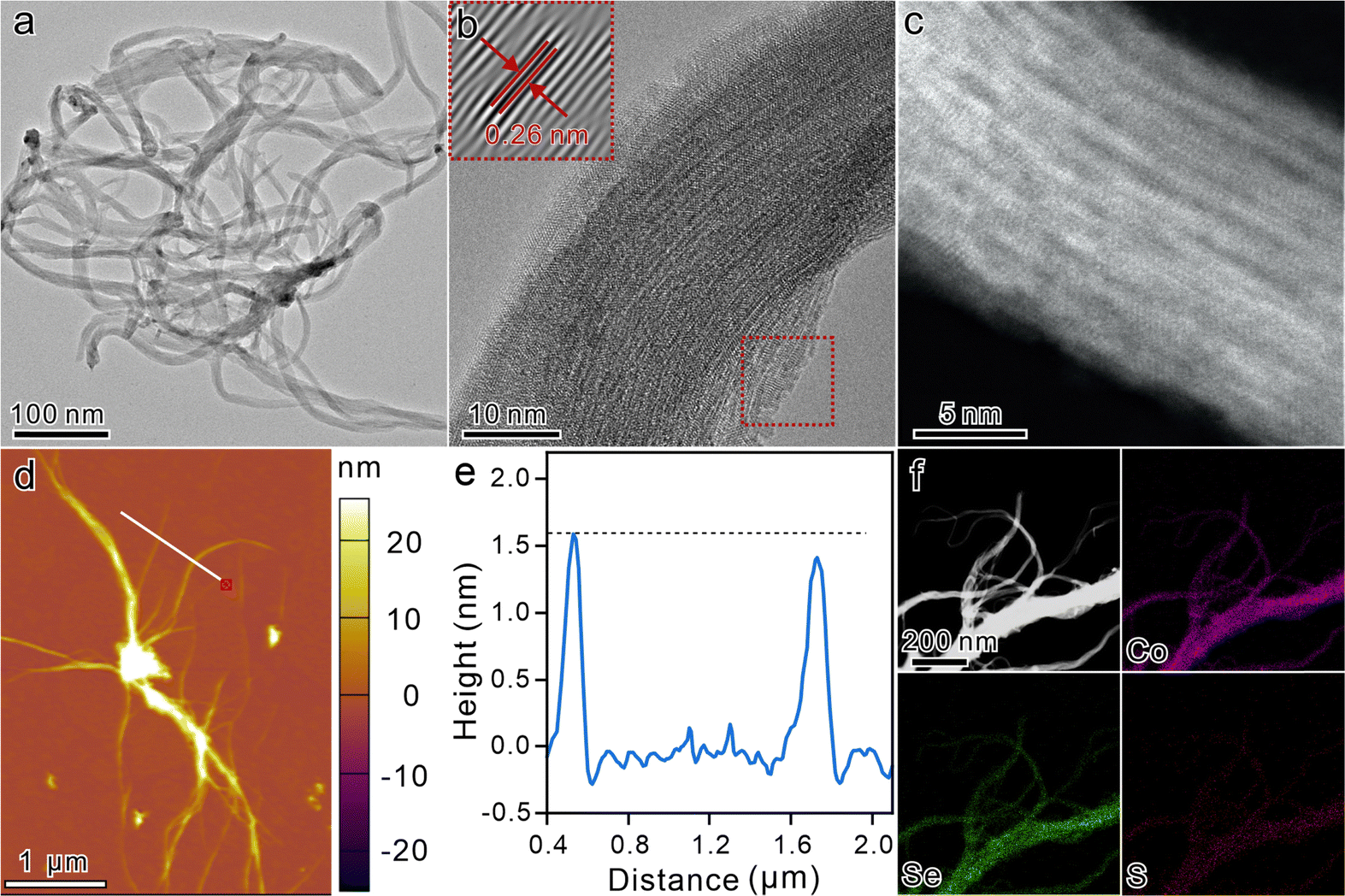 | ||
| Fig. 1 Microstructure characterization of S-CoSe2 NWs: (a) TEM image; (b) HRTEM image (inset image shows the lattice fringe spacing); (c) AC HAAD-STEM image; (d) and (e) AFM image and the corresponding height profile; and (f) HAAD-STEM and EDX-mapping images of all elements. | ||
The fabrication process of the S-CoSe2 NWs was also further investigated. First, the formation of the S-CoSe2 NWs highly relies on the reaction temperature and time. Sufficient temperature is required to generate enough solvent pressure to drive the formation of the desired morphology. By regulating the reaction temperature from 120 °C to 190 °C, it was observed that temperatures below 170 °C resulted in the formation of unexpected bulk materials, and the materials obtained individual fine nanowires at temperatures over 170 °C (Fig. S7, ESI†). With an increase of the reaction time, the material is gradually stretched to obtain a flexible and smooth nanowire morphology (Fig. S8, ESI†). Furthermore, it was found that the addition of sulfur was pivotal to forming a unique nanowire morphology. When sulfur is absent in the synthesis process, a totally different nanosheet-like material (CoSe2 NSs) can be obtained (Fig. S9, ESI†). With the regulation of sulfur usage, it can be clearly observed that the material has an obvious evolutionary morphology change from nanosheets to nanowires (Fig. 2b–f and Fig. S10, ESI†). Meanwhile, the XRD patterns of CoSe2 also broadened and weakened, indicating that the crystallinity of the material was poor. The slightly negative shift of the XRD peaks indicates the lattice expansion of the as-prepared materials with sulfur doping (Fig. S11, ESI†). This intriguing phenomenon drives us to elucidate the intrinsic formation mechanism of S-CoSe NWs. Ethylenediamine (EDA) was employed as the solvent and sequestrant in this system. Co2+, Se, S can coordinate with EDA to form a series of complexes [Co(EDA)2]2+, [Sex-EDA]− and [Sx-EDA]−, respectively.48 The addition of sulfur can replace the counterion for [Co(EDA)2]2+, which results in a blue shift in the UV-vis spectra (Fig. S12, ESI†). The distinct precursor complex also has a different supersaturation (S) of the solution and electrolytic dissociation (α) of the reactants.49S can influence the surface engineering of the resulting materials, and α can determine whether the growth of materials is isotropic or anisotropic.50,51 There is no doubt that EDA is a weak electrolyte with low α (10−4.07), which results in the anisotropic growth of CoSe2 to be 2D nanosheets or a 1D nanowires, as the EDA can provide a constant and equilibrium-driven force to support the orientated growth. Moreover, the new counterion of [Sx-EDA]− in the system significantly decreases the S of the final products, which further results in a small, saturated region with a slow growth rate to obtain regularly shaped S-CoSe2 NWs (Fig. 2a). Besides the morphology regulation, sulfur doping also affects the electronic states of the resulting materials. With more electronegative sulfur doping, the oxidation state of cobalt in the S-CoSe2 NWs is significantly different from that of CoSe2 NSs, which can be observed from the positive binding energy shift from Co 2p X-ray photoelectron spectroscopy (Fig. S13, ESI†). The local structure and coordination information of the CoSe2 materials were further studied by X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy. The main adsorption Co K-edge XANES peak shifts to higher energy after sulfur doping, which further consolidates the higher oxidation state of the S-CoSe2 NWs (Fig. 2g). Moreover, CoSe2 NSs and S-CoSe2 NWs both show a pre-edge peak at around 7709.8 eV for the 1s to eg intra-atomic transition in cobalt, which indicates the sulfur doping has a negligible influence on the octahedral configuration of CoSe2.52 However, it is worth mentioning that the intensity of the pre-edge peak has a perceptible decrease with sulfur incorporating, indicating the enhancive eg filling of cobalt (inset in Fig. 2g).52 Further extended X-ray absorption fine structure (EXAFS, Fig. 2h) spectroscopy discloses an obvious decrease of the Co–Se scattering path due to the shorter bond distance of Co–S than that of Co–Se, and the emergent Co–S scattering path in S-CoSe2 NWs is clearly observed in wavelet transform analysis with the high-k direction extension (Fig. 2i and j). The corresponding EXAFS fitting also implies a monotonic decrease of the coordination number (CN) of Co–Se and increase of the CN of Co–S (Fig. S14 and Table S2, ESI†). Generally, the mechanistic investigation and spectroscopic studies on CoSe2 materials reveal that sulfur doping can readily regulate not only the microstructure from 2D to 1D but also controllably tailor the electronic structure and local chemical environment of CoSe2.
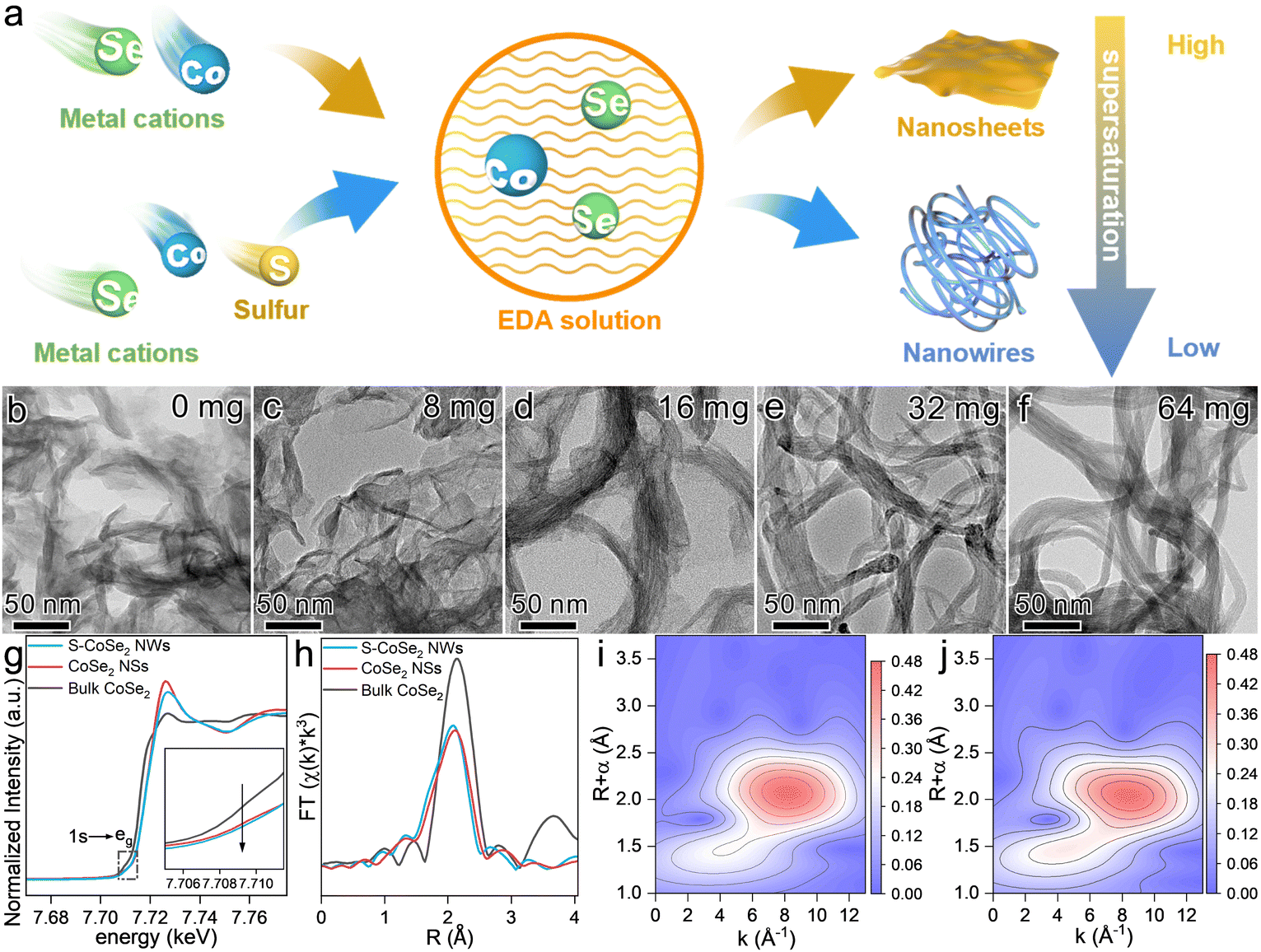 | ||
| Fig. 2 (a) Formation mechanism of CoSe2 NSs and S-CoSe2 NWs. (b)–(f) Morphological evolution of CoSe2 materials with different sulfur contents. (g) Co K-edge XANES spectra and (h) corresponding Fourier transforms of k3-weighted EXAFS spectra for CoSe2 materials. Wavelet transforms of k3-weighted EXAFS spectra of the Co K-edge for (i) CoSe2 NSs and (j) S-CoSe2 NWs. | ||
Based on the above analysis, CoSe2 NSs and S-CoSe2 NWs with distinct microstructures and electronic structures could serve as promising research platforms to explore the structure–property relationship of nitrate reduction in TMDs, and further unveil the fundamental mechanisms involved in the NARR. As doping strategy can effectively regulate the catalytic activity, and sulfur can combine with hydrogen with the p-orbital to form active hydrogen, which is considered an important species to combine with NARR intermediates to modulate the reaction behavior.53–57 Based on these considerations, the self-supported working electrode of the in situ growth S-CoSe2 NWs on 1 × 1 cm2 carbon cloth (S-CoSe2 NWs@CC, Fig. S15, ESI†) was employed for the NARR test, and CoSe2 NSs@CC was used as a reference. The electrodes were further dwelled in an H-cell filled with 0.5 M Na2SO4 and 0.5 M KNO3 as electrolytes. Linear sweep voltammetry (LSV) was initially conducted on the utilized catalysts, while both CoSe2 NSs@CC and S-CoSe2 NWs@CC exhibited pronounced current responses in the presence of NO3−. However, S-CoSe2 NWs@CC had a more positive onset potential and higher current response (Fig. 3a), which can be attributed to the higher intrinsic activity and advanced porous structure with a higher BET surface area(Fig. S16, ESI†).58 The ammonia concentration of the electrolytes for the above catalysts was investigated after chronoamperometry at −0.5 V vs. RHE using the indophenol blue method. Apparently, S-CoSe2 NWs@CC can produce more ammonia under the same potential compared to CoSe2 NSs@CC and bare CC (Fig. 3b), as indicated by the sharp peak of indophenol blue centered at 655.5 nm in the UV spectra. The NH3 yield and the corresponding FE of the electrodes at all given potentials are further calculated from the standard curve recorded at different concentrations of NH4+–N (Fig. S17, ESI†). S-CoSe2 NWs@CC can produce increasing amounts of ammonia with the cathodic potential boosting to be more negative, reaching the highest faradaic efficiency (FE) of 93.1% at −0.6 V vs. RHE and the highest rate of 11.6 mg h−1 cm−2 at −0.8 V vs. RHE (Fig. 3c). This can also be confirmed by the corresponding UV-vis spectra of the electrolytes after coloring (Fig. S18, ESI†). It is worth mentioning that the FE remains consistently above 90% at high potentials, indicating the remarkable selectivity exhibited by the S-CoSe2 NWs. The ammonia production of the S-CoSe2 NWs was additionally quantified using 1H nuclear magnetic resonance (NMR) spectroscopy with maleic acid as an internal standard (Fig. S19, ESI†). An FE of 91.5% at −0.6 V vs. RHE was calculated, which aligns well with the results obtained from the UV-vis analysis, further corroborating the high selectivity towards ammonia (Fig. S20, ESI†). The source of nitrogen in the produced ammonia was unequivocally confirmed by 1H NMR experiments employing 15N isotopic labeling. As 15N is a spin-½ nucleus that results in two symmetric signals for 1H-15N, in contrast to the three symmetric signals observed for 1H–14N interactions, which involve a spin-1 nucleus.59 With standard ammonium salt as a reference, the electrolyte after NARR with 15NO3− exhibited distinct double peaks, while triple peaks were observed with 14NO3− as the nitrogen source (Fig. S21, ESI†). The NARR byproducts of S-CoSe2 NWs and CoSe2 NSs, nitrite (NO2−) and the competitive HER product of H2 are also detected, and corresponding FEs are calculated (Fig. 3d and Fig. S22, ESI†). In the case of the S-CoSe2 NWs, as the potential is progressively increased to the positive range, the FEs of nitrite gradually decrease, falling below 5%, with ammonia becoming the dominant product. Conversely, for the CoSe2 NSs, a similar trend is observed with nitrite, albeit with a higher FE share. The superior selectivity towards ammonia exhibited by the S-CoSe2 NWs further validates the role of sulfur doping in providing higher charge active centers, which enhances the adsorption of intermediates. We further investigated the NARR performance of S-CoSe2 nanomaterials with different sulfur doping contents ranging from 0.86% to 4.3% (Fig. S23, ESI†). The FE collections clearly show an increase in the FE for the target ammonia, accompanied by a decrease in the FE for the byproduct nitrite, which strongly confirms the pivotal role of sulfur in enhancing the NARR performance. Moreover, bulk CoSe2 was also synthesized as a reference (Fig. S24, ESI†). It exhibited inferior activity and undesirable selectivity towards ammonia, underscoring the superiority of the nanostructured S-CoSe2 NWs (Fig. S25, ESI†). Besides activity, the stability of the S-CoSe2 NWs is also evaluated using a long-term chronopotentiometry test in a self-organized device (Fig. S26, ESI†). We fabricated this flow cell device by coupling the S-CoSe2 NWs@CC as the cathode and NiFe LDH@ Ni foam as the anode, which was separated by a Nafion membrane (Fig. S27, ESI†), under a high current of 200 mA cm−2; this device could continuously produce ammonia over 500 h without an obvious surge in the cell voltage. The structural integrity and electronic structure of the S-CoSe2 NWs were further examined using TEM and XPS after a long-term reaction. The analyses revealed that S-CoSe2 NWs nearly retained their original microstructures and electronic configuration, underscoring their robustness (Fig. S28, ESI†). After the operation, the ammonia produced can be further fixed in the solid form of struvite ((NH4)MgPO4·6H2O) by adding a coprecipitator of Mg2+ and PO43−, which can be further used as fertilizer. Using this struvite-precipitation method, we obtained over 150 g of struvite with high purity (Fig. S29, ESI†). It also provides a promising way to capture nitrite and phosphate in eutrophicated water and is further convert to a value-added fertilizer.
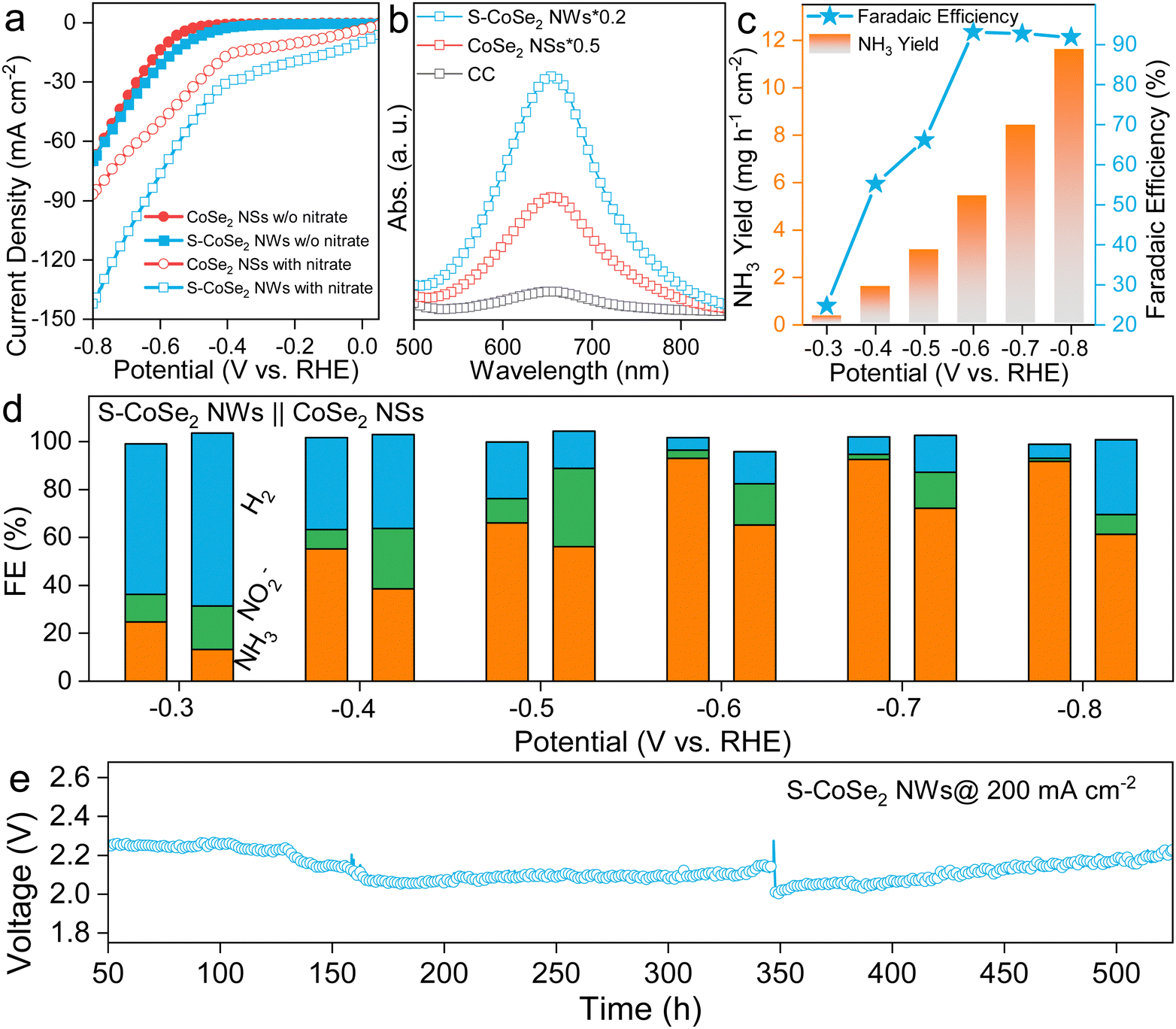 | ||
| Fig. 3 (a) LSV curves of CoSe2 NSs and S-CoSe2 NWs in 0.5 M KOH and 0.5 M KOH + 0.5 M KNO3, respectively. (b) UV-vis spectra of the electrolytes tested using the indophenol blue method after chronoamperometry at −0.6 V vs. RHE. (c) FE and NH3 yield rates of S-CoSe2 NWs at all potentials. (d) FE comparison between CoSe2 NSs and S-CoSe2 NWs at given potentials. (e) Chronopotentiometry curve of S-CoSe2 NWs with a two-electrode system in a self-organized device (the voltage jump at 350 h is due to the replenishment of fresh electrolyte to the cell). | ||
This superior performance motivated us to investigate the reaction mechanism and uncover the reason for the distinct selectivity difference between CoSe2 NSs and S-CoSe2 NWs. First, in situ electrochemical spectroscopy was employed to identify the nitrogen-containing intermediates during NARR. Attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR, Fig. S30, ESI†) was performed to detect the species adsorbed on the electrode surface, and the reference spectrum obtained under open circuit potential was collected as the background. Then, the potential in the operating range was stepped from −0.3 V to −0.7 V vs. RHE. With increasing potential, several downward peaks emerged that were independent of the working potential, indicating the generation of NARR intermediates. The downward bands at around 1150 cm−1 and 1210 cm−1 can be, respectively, attributed to the –N–O– stretching vibration of NH2OH and the N–O antisymmetric stretching vibration of NO2−, which are both important intermediates during NARR. Furthermore, the upward band at around 1350 cm−1, assigned to the asymmetric stretching vibration of NO3−, indicates the consumption of nitrate at the electrode interface.60 For CoSe2 NSs, the FTIR spectra collected were similar to those of S-CoSe2 NWs, indicating the analogous NARR route on both catalysts. However, the adsorption band center of NO2− on the CoSe2 NSs has an obvious red shift to 1224 cm−1, indicating the adsorption attenuation of NO2− on the materials, which could be difficult for further hydrogenation (Fig. S31, ESI†). Besides ATR-FTIR, in situ electrochemical Raman spectroscopy was performed to obtain adminicular information about the intermediates during the NARR. The Raman shifts at 979 cm−1 and 1046 cm−1 can be attributed to the sulfate and nitrate groups. The peaks observed at 1373, 1535, and 1598 cm−1 are associated with the symmetric N–O stretches of adsorbed NO2−, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches of NOH, and H–N–H of NH2, respectively.61,62 The possible NARR intermediates were also captured by online differential electrochemical mass spectrometry (DEMS) with consecutive cycles for uncovering the integrated mechanism.63 The major mass–charge ratio (m/z) signals of NH(15), NH2(16), and NH2OH (33) were definitely captured as reaction intermediates, and NH3(17) with a strong signal intensity indicates the formation of the NARR product NH3. It should be noted that the m/z signal of 46 represents the possible byproduct of NO2−, and its weak intensity also reversely validates the high selectivity of S-CoSe2 NWs. Combining the information provided by the above electrochemical in situ technologies, the nitrate reduction pathway for the given materials can be proposed as follows: NO3− → NO3* → NO2* → NO* → NOH* → NOH* → N* → NH* → NH2* → NH3.
O stretches of NOH, and H–N–H of NH2, respectively.61,62 The possible NARR intermediates were also captured by online differential electrochemical mass spectrometry (DEMS) with consecutive cycles for uncovering the integrated mechanism.63 The major mass–charge ratio (m/z) signals of NH(15), NH2(16), and NH2OH (33) were definitely captured as reaction intermediates, and NH3(17) with a strong signal intensity indicates the formation of the NARR product NH3. It should be noted that the m/z signal of 46 represents the possible byproduct of NO2−, and its weak intensity also reversely validates the high selectivity of S-CoSe2 NWs. Combining the information provided by the above electrochemical in situ technologies, the nitrate reduction pathway for the given materials can be proposed as follows: NO3− → NO3* → NO2* → NO* → NOH* → NOH* → N* → NH* → NH2* → NH3.
We further conducted density functional theory (DFT) calculations to determine the possible reaction mechanism based on the structural model of sulfur-doped CoSe2 and observed the experimental evidence. First, the (210) facet was selected as the NARR surface due to its lower surface energy than that of the exposed (311) facet (Fig. S32, ESI†). As NARR is a multiple electron–proton transfer process with many intermediates and adsorption models on active sites, the most possible pathway is illustrated (Fig. 4d).64 In terms of thermodynamics, the nitrate adsorption on sulfur-doped CoSe2 is unfavorable with an uphill free energy of 0.47 eV during the entire catalytic processes, which is lower than that on CoSe2 (0.55 eV, Fig. S33, ESI†). The first hydrogenation on the *NO3 and *NO2 (*NO3 to *NO2OH, *NO2 to *NOOH) without electron transfer shows uphill free energies of 0.08 eV and 0.17 eV, respectively. The rate-determining step (RDS) on S-CoSe2 NWs appeared on the hydrogenation of *NO (*NO to *NOH) with a free energy increase of 1.21 eV, and CoSe2 shows the same RDS but with a lower free energy increase of 1.08 eV. This result contradicts the fact that S-CoSe2 NWs with lower onset potential and overpotential have higher apparent activity. This inconformity could be explained by the strong competition of H* and nitrogen-containing species produced during NARR. Sulfur has a higher *H adsorption capability, which can promote hydrogenation of NARR; however, excess *H accumulation will also lead to the decrease of NARR selectivity, and the adjacent active sites of cobalt can in situ utilize the produced *H to balance the kinetics of production and utilization of *H. Moreover, as mentioned before, the structural advantage of S-CoSe2 NWs with a higher BET surface area and more abundant active sites cannot be ignored; it also contributes significantly towards apparent activity. The reduction steps of *NOH to NH3* on sulfur-doped CoSe2 are nearly downhill in free energy, which contributes to the remarkable catalytic performance of S-CoSe2 NWs. CoSe2 follows similar reaction steps and trends with sulfur-doped CoSe2, but the selectivity towards NO2− between them is distinct at all given potentials. This could be explained by the difference in the free energy between the hydrogenation and desorption of *NO2; this value is 0.22 eV for CoSe2, which is higher than that of sulfur-doped CoSe2 with a high-charge active center (0.12 eV), indicating a higher tendency of hydrogenation on the latter, which also reflects the better ammonia selectivity (Fig. S34, ESI†). The electronic structure analysis showed the higher charge state of cobalt on the S-CoSe2 NWs. Combined with the above DFT calculation results, the high activity and selectivity of ammonia can be attributed to the high-charge active sites.
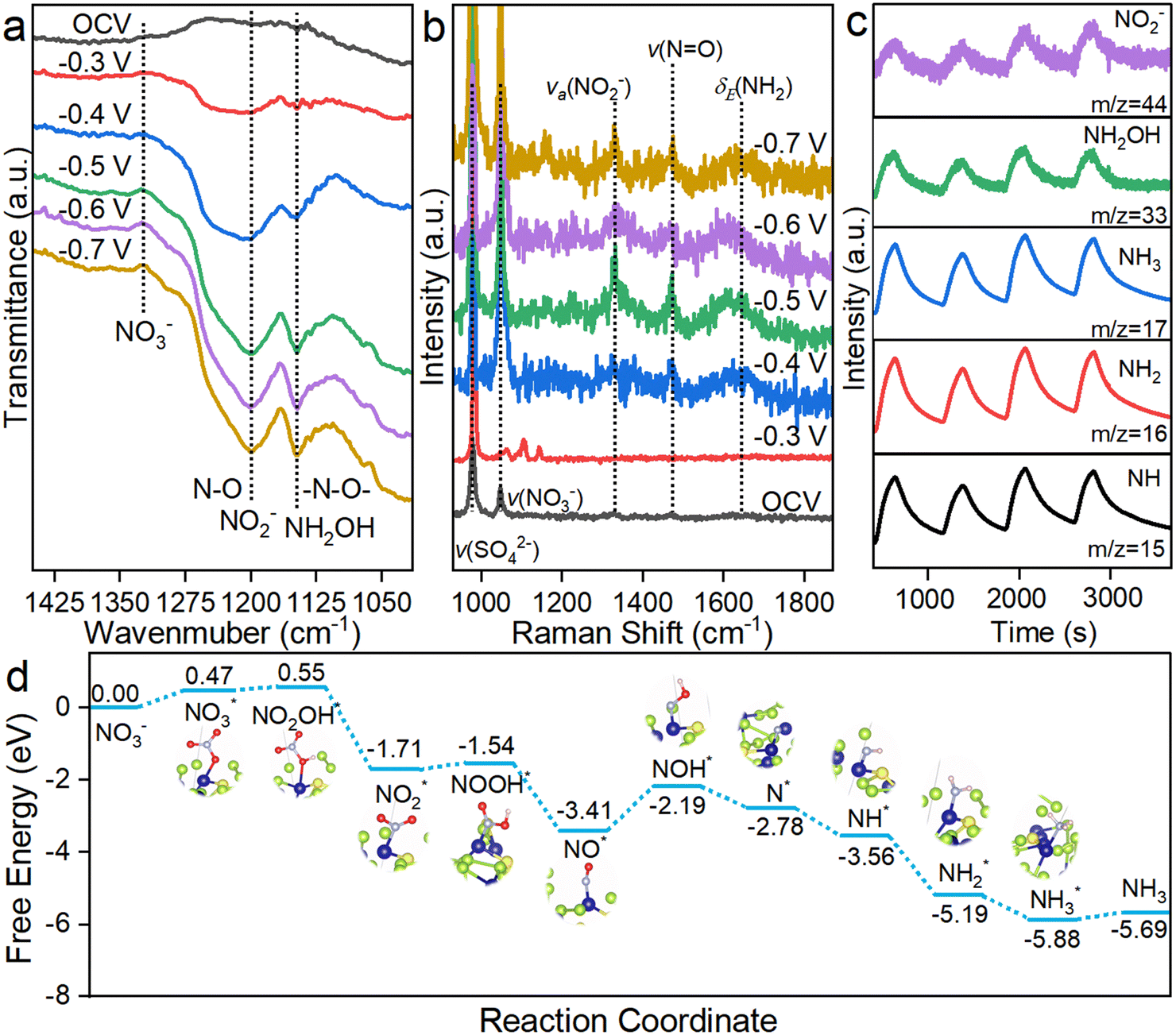 | ||
| Fig. 4 (a) Electrochemical in situ FTIR spectra and (b) in situ Raman spectra of S-CoSe2 NWs. (c) Online DEMS of S-CoSe2 NWs under a potential of −0.35 V vs. RHE. (d) Reaction-free energies for different intermediates of NARR based on the model of sulfur-doped CoSe2. | ||
Conclusion
In summary, sulfur-doped CoSe2 NWs were fabricated using a solvothermal method. By regulating the supersaturation and ionization degree of the synthetic system with sulfur addition, the morphology of the resulting CoSe2 materials can gradually transition from 2D to 1D with the generation of a high-charge cobalt center. Owing to their structural advantages, S-CoSe2 NWs contribute to their pronounced activity and selectivity in the electrochemical NARR to ammonia. A series of in situ electrochemical techniques combined with DFT calculations were employed to uncover the reaction pathway and mechanism. It was found that the high-charge active center of cobalt can stabilize NO2* for further hydrogenation rather than desorption to nitrite, which increases the reaction selectivity to ammonia. This work expands the application range of TMDs for electrochemical nitrate reduction in ammonia synthesis and provides fundamental insights into the reaction mechanisms of high-charge Co active centers.Author contributions
Wuyong Zhang: conceptualization, methodology, data curation, and writing – reviewing and editing; Yingjie Wen: data curation; Haocheng Chen: data curation; Minli Wang: data curation; Caihan Zhu: data curation; Yunan Wang: supervision; Zhiyi Lu: funding acquisition and supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declared that they have no conflicts of interest in this work.Acknowledgements
This research has been supported by the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB0450401. This research is also supported by the Bellwethers Project of Zhejiang Research and Development Plan (2022C01158), Ningbo Yongjiang Talent Introduction Programme (2021A-036-B), and the Hundred Talents Programs in Chinese Academy of Sciences (Y.N.W).References
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, ACS Energy Lett., 2021, 6, 3676–3685 CrossRef CAS
.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS
- H. Kobayashi, A. Hayakawa, K. D. Kunkuma, A. Somarathne and E. C. Okafor, P. Combust. Inst., 2019, 37, 109–133 CrossRef CAS
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS
- M. Capdevila-Cortada, Nat. Catal., 2019, 2, 1055 CrossRef
- N. Lehnert, B. W. Musselman and L. C. Seefeldt, Chem. Soc. Rev., 2021, 50, 3640–3646 RSC
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C. P. Chen, M. R. Smith, 3rd and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed
- S. J. Li, D. Bao, M. M. Shi, B. R. Wulan, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1700001 CrossRef PubMed
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS
- M. Nazemi, S. R. Panikkanvalappil and M. A. El-Sayed, Nano Energy, 2018, 49, 316–323 CrossRef CAS
- W. Zhang, S. Zhan, Q. Qin, T. Heil, X. Liu, J. Hwang, T. H. Ferber, J. P. Hofmann and M. Oschatz, Small, 2022, 18, e2204116 CrossRef PubMed
- G. Soloveichik, Renewable energy to fuels through utilization of energy dense liquids (REFUEL) Program Overview, 2016 Search PubMed.
- L. Hu, Z. Xing and X. F. Feng, ACS Energy Lett., 2020, 5, 430–436 CrossRef CAS
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, 2304021 CrossRef CAS PubMed
- F. T. Wakida and D. N. Lerner, Water Res., 2005, 39, 3–16 CrossRef CAS PubMed
- M. Scherer-Lorenzen, C. Palmborg, A. Prinz and E. D. Schulze, Ecology, 2003, 84, 1539–1552 CrossRef
- D. S. Powlson, T. M. Addiscott, N. Benjamin, K. G. Cassman, T. M. de Kok, H. van Grinsven, J. L. L'Hirondel, A. A. Avery and C. van Kessel, J. Environ. Qual., 2008, 37, 291–295 CrossRef CAS PubMed
- J. P. van der Hoek and A. Klapwijk, Water Res., 1987, 21, 989–997 CrossRef CAS
- M. H. Ward, S. D. Mark, K. P. Cantor, D. D. Weisenburger, V. Correa and S. H. Zahm, Epidemiol., 1996, 7, 465–471 CrossRef CAS
- V. Rosca, M. Duca, M. T. de Groot and M. T. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed
- H. Xu, Y. Ma, J. Chen, W.-X. Zhang and J. Yang, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC
- J. Xu, S. Zhang, H. Liu, S. Liu, Y. Yuan, Y. Meng, M. Wang, C. Shen, Q. Peng, J. Chen, X. Wang, L. Song, K. Li and W. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308044 CrossRef CAS PubMed
- W. Zhang, S. Zhan, J. Xiao, T. Petit, C. Schlesiger, M. Mellin, J. P. Hofmann, T. Heil, R. Muller, K. Leopold and M. Oschatz, Adv. Sci., 2023, 10, e2302623 CrossRef PubMed
- J. F. E. Gootzen, P. G. J. M. Peeters, J. M. B. Dukers, L. Lefferts, W. Visscher and J. A. R. vanVeen, J. Electroanal. Chem., 1997, 434, 171–183 CrossRef CAS
- M. Ahmadi and M. Nazemi, Ind. Eng. Chem. Res., 2024, 63, 9315–9328 CrossRef CAS
- D. Liu, L. Qiao, S. Peng, H. Bai, C. Liu, W. F. Ip, K. H. Lo, H. Liu, K. W. Ng, S. Wang, X. Yang and H. Pan, Adv. Funct. Mater., 2023, 33, 2303480 CrossRef CAS
- O. Peng, Q. Hu, X. Zhou, R. Zhang, Y. Du, M. Li, L. Ma, S. Xi, W. Fu, Z.-X. Xu, C. Cheng, Z. Chen and K. P. Loh, ACS Catal., 2022, 12, 15045–15055 CrossRef CAS
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S. X. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS
- Z. Zhang, D. Y. Li, Y. C. Tu, J. Deng, H. T. Bi, Y. C. Yao, Y. Wang, T. S. Li, Y. S. Luo, S. J. Sun, D. D. Zheng, S. A. C. Carabineiro, Z. Chen, J. J. Zhu and X. P. Sun, Susmat, 2024, 4, e193 CrossRef CAS
- Q. Fu, J. Han, X. Wang, P. Xu, T. Yao, J. Zhong, W. Zhong, S. Liu, T. Gao, Z. Zhang, L. Xu and B. Song, Adv. Mater., 2021, 33, 1907818 CrossRef CAS PubMed
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed
- A. Mondal and A. Vomiero, Adv. Funct. Mater., 2022, 32, 2208994 CrossRef CAS
- Y. Huang, Y. Sun, X. Zheng, T. Aoki, B. Pattengale, J. Huang, X. He, W. Bian, S. Younan, N. Williams, J. Hu, J. Ge, N. Pu, X. Yan, X. Pan, L. Zhang, Y. Wei and J. Gu, Nat. Commun., 2019, 10, 982 CrossRef CAS PubMed
- B. Li, K. Nie, Y. Zhang, L. Yi, Y. Yuan, S. Chong, Z. Liu and W. Huang, Adv. Mater., 2023, 35, 2303285 CrossRef CAS PubMed
- Y. Li, M. Wang and J. Sun, Adv. Energy Mater., 2022, 12, 2202600 CrossRef CAS
- K. Belay Ibrahim, T. Ahmed Shifa, S. Zorzi, M. Getaye Sendeku, E. Moretti and A. Vomiero, Prog. Mater. Sci., 2024, 144, 101287 CrossRef CAS
- M. Yu, H. Huang, J. Hu, S. Wang, J. Li and D. Wang, J. Mater. Chem. A, 2022, 10, 23990–23997 RSC
- L. Yang and W. Zhu, Appl. Surf. Sci., 2022, 596, 153624 CrossRef CAS
- M. R. Gao, W. T. Yao, H. B. Yao and S. H. Yu, J. Am. Chem. Soc., 2009, 131, 7486–7487 CrossRef CAS PubMed
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed
- L. Liang, H. Cheng, F. Lei, J. Han, S. Gao, C. Wang, Y. Sun, S. Qamar, S. Wei and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 12004–12008 CrossRef CAS PubMed
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS
- D. Kong, H. Wang, Z. Lu and Y. Cui, J. Am. Chem. Soc., 2014, 136, 4897–4900 CrossRef CAS PubMed
- J. Lu, Y. Xie, F. Xu and L. Zhu, J. Mater. Chem., 2002, 12, 2755–2761 RSC
- W.-H. Lai, Y.-X. Wang, Y. Wang, M. Wu, J.-Z. Wang, H.-K. Liu, S.-L. Chou, J. Chen and S.-X. Dou, Nat. Chem., 2019, 11, 695–701 CrossRef CAS PubMed
- X. Zhang and Y. Xie, Chem. Soc. Rev., 2013, 42, 8187–8199 RSC
- H. X. Lin, Z. C. Lei, Z. Y. Jiang, C. P. Hou, D. Y. Liu, M. M. Xu, Z. Q. Tian and Z. X. Xie, J. Am. Chem. Soc., 2013, 135, 9311–9314 CrossRef CAS PubMed
- M. Sano, Inorg. Chem., 1988, 27, 4249–4253 CrossRef CAS
- F. Lei, W. Xu, J. Yu, K. Li, J. Xie, P. Hao, G. Cui and B. Tang, Chem. Eng. J., 2021, 426, 131317 CrossRef CAS
- X. Chen, Z. Qiu, H. Xing, S. Fei, J. Li, L. Ma, Y. Li and D. Liu, Appl. Catal., B, 2022, 305, 121030 CrossRef CAS
- K. Wang, R. Mao, R. Liu, J. Zhang and X. Zhao, Appl. Catal., B, 2022, 319, 121862 CrossRef CAS
- N. Xue, Z. Lin, P. Li, P. Diao and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 28288–28297 CrossRef CAS PubMed
- Z. Li, L. G. Yu, C. X. Bi, X. Y. Li, J. Ma, X. Chen, X. Q. Zhang, A. B. Chen, H. T. Chen, Z. R. Zhang, L. Z. Fan, B. Q. Li, C. Tang and Q. Zhang, Susmat, 2024, 4, e191 CrossRef CAS
- Y. Li, Z. Kan, L. Jia, D. Zhang, Y. Hong, J. Liu, H. Huang, S. Li and S. Liu, Trans. Tianjin Univ., 2023, 29, 313–320 CrossRef CAS
- D. Cremer and J. Grafenstein, Phys. Chem. Chem. Phys., 2007, 9, 2791–27816 RSC
- J.-Y. Fang, Q.-Z. Zheng, Y.-Y. Lou, K.-M. Zhao, S.-N. Hu, G. Li, O. Akdim, X.-Y. Huang and S.-G. Sun, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed
- S.-E. Bae, K. L. Stewart and A. A. Gewirth, J. Am. Chem. Soc., 2007, 129, 10171–10180 CrossRef CAS PubMed
- D. P. Butcher and A. A. Gewirth, Nano Energy, 2016, 29, 457–465 CrossRef CAS
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, Angew. Chem., Int. Ed., 2020, 59, 10479–10483 CrossRef CAS PubMed
- Y. C. Zeng, C. Priest, G. F. Wang and G. Wu, Small Methods, 2020, 4, 2000672 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00593g |
| This journal is © The Royal Society of Chemistry 2024 |