
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing the mechanism of reductive, mechanochemical Li recycling from LiFePO4†
David
Geiß
 a,
Oleksandr
Dolotko
abc,
Sylvio
Indris
a,
Christian
Neemann
a,
Andrei
Bologa
a,
Thomas
Bergfeldt
d,
Michael
Knapp
a and
Helmut
Ehrenberg
ab
a,
Oleksandr
Dolotko
abc,
Sylvio
Indris
a,
Christian
Neemann
a,
Andrei
Bologa
a,
Thomas
Bergfeldt
d,
Michael
Knapp
a and
Helmut
Ehrenberg
ab
aKarlsruhe Institute of Technology (KIT), Institute for Applied Materials-Energy Storage Systems (IAM-ESS), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Karlsruhe, Germany. E-mail: david.geiss@kit.edu; sdolotko@yahoo.com; sylvio.indris@kit.edu; christian.neemann@student.kit.edu; andrei.basketball@hotmail.de; michael.knapp@kit.edu; helmut.ehrenberg@kit.edu
bHelmholtz-Institute Ulm for Electrochemical Energy Storage (HIU), P.O. Box 3640, D-76021 Karlsruhe, Germany
cNIO GmbH, Montgelasstraße 14, D-81679 Munich, Germany
dKarlsruhe Institute of Technology (KIT), Institute for Applied Materials-Applied Materials Physics (IAM-AWP), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Karlsruhe, Germany. E-mail: thomas.bergfeldt@kit.edu
First published on 18th May 2024
Abstract
In order to mitigate the risks associated with cobalt supply, a safe and affordable LiFePO4-based (LFP) cathode for Li-ion batteries can be a significant solution to meet the rapidly growing battery market. However, economical and environmentally friendly recycling of LFP is impossible with currently available recycling technologies. In this study, an acid-free mechanochemical approach is applied to reclaim Li from LFP using Al as a reducing agent. The reaction mechanism involved in reductive ball-milling followed by water leaching has been elucidated through the examination of various milling times and molar ratios of components, fostering a deeper understanding of the process. Assessing the yield and purity of the final products provides insights into potential enhancements for this technology. Utilizing Al as the material of the current collector eliminates the need for additional external additives, thereby simplifying the recycling workflow. Continued research into this process has the potential to facilitate efficient and economical recycling of LFP materials.
1. Introduction
Lithium-ion batteries (LIBs) greatly influence our daily lives, as they are used for various important applications. They are utilized for small mobile devices such as smartphones, digital cameras, and laptops, as well as for electric vehicles and stationary energy storage.1,2 The battery market is anticipated to expand by 25% annually, driven primarily by the surging demand for electric vehicles.3 In addition, the deployment of batteries in electricity grids is poised to gain heightened significance in the approaching future. This escalating demand for batteries naturally predicates a proportional increase in the requisite resources. The largest relative increase is expected for lithium, which is predicted to rise by almost 1000% by 2050.3,4 But as the global lithium resources are restricted to around 17 million tons, multiple agencies worldwide already forecast lithium shortages in the near future.4 Because of the huge consumption of LIBs, we will have to deal with almost half a million tons of waste batteries by 2025,4 but less than 5% of waste batteries get recycled at the moment, with a majority just ending up in landfills.5,6 All these developments now make battery recycling an important ecological and economic challenge.These days, layered transition metal oxides like LiCoO2 (LCO), LiNixMnyCo1−x−yO2 (NMC), and LiNixCoyAl1−x−yO2 (NCA) are employed as cathode materials as well as spinel type LiMn2O4 (LMO) and olivine structured LFP, while graphite is the commonly used anode material.7,8 NMC and NCA are offering high energy densities (>330 W h kg−1vs. graphite),9 but the cobalt content makes these materials toxic and expensive.9,10 LFP, on the other hand, is attractive because of its low costs, high thermal stability, environmental friendliness, and independence of the Co market.9,11,12 The low costs of LFP are resulting from Li being the primary valuable element, whereas in the case of NMC and NCA both Li and the transition metals are valuable components.2 However, the energy density (270 W h kg−1vs. graphite)9 of LFP is lower compared to NMC and NCA.
At present, the industry predominantly concentrates on recycling cathodes through the employment of pyrometallurgy and hydrometallurgy techniques.6–13 Initially, several pretreatment steps are necessary for both methods, which encompass sorting, discharging, dismantling, crushing, sieving, and thermal treatment. In the pyrometallurgical process, metals are reduced to obtain alloys and slag by smelting the batteries at temperatures above 1000 °C.13 To reclaim d-element salts, these alloys undergo further processing through multi-step leaching. Unfortunately, all the lithium is lost in the slag, which typically cannot be recycled further.14 Additionally, graphite cannot be reclaimed as it functions as a reducing agent, facilitating the formation of carbon monoxide.15 Despite the noted advantages of the pyrometallurgical method, including its capacity to leverage existing equipment and streamline the process by bypassing certain pretreatment steps, it falls short in recycling lithium iron phosphate (LFP) materials. This limitation arises because lithium, the sole valuable component in LFP, is not recoverable in the pyrometallurgical process.13
In the hydrometallurgical process, all valuable metals can be recycled with high purity, making it cost-efficient and effective, particularly for recycling LCO or NMC materials, which contain numerous valuable elements. However, this method produces a significant amount of corrosive wastewater during the complex multi-step leaching and precipitation phases.16 Additionally, it necessitates physical pretreatment, contributing to high operating costs. Consequently, despite its effectiveness with NMC materials, hydrometallurgy is generally considered uneconomical for recycling LFP cathodes.13
As both methods have a lot of disadvantages, an alternative needs to be found to make LIB recycling more environmentally friendly and at the same time profitable for industry.
Currently, there are only laboratory-scale technologies for recycling LFP because of the low economic value of Li compared to Ni & Co.17,18 Therefore, an assessment of the cost-benefit is important for LFP recycling methods.17,19–21 Dismantling and separation of the active material is performed manually, which is a challenge for upscaling.17,20,22 The major ways for reclaiming valuable materials from LFP are hydrometallurgical leaching, where the LFP structure is destroyed, and direct recycling, where the LFP structure is retained.17 Because spent LFP material is typically featured with iron occupying the lithium site over repeated cycles and partial loss of active lithium, the approach of direct recycling includes healing the LFP structure through a facile relithiation and annealing process.23 Therefore, this method is also effective for its usage for the non-destructive recycling of the battery production scrap. Direct regeneration of LFP is usually achieved by solid-state reactions, including ball-milling and heat treatment with the addition of new Li and carbon sources.17,22–24 Hydrometallurgical recovery of LFP can be implemented using acids (mostly H2SO4) in combination with oxidizing agents like O2 or H2O2 or by using mechanochemical reactions.17,19,25–28
An additional alternative recycling technology to consider is the mechanochemically induced method. This approach facilitates physical, structural, and chemical transformations in materials, potentially offering a novel route to more efficient recycling processes.29–31 For instance, this method can lead to an increase in the specific surface area, cause amorphization, oxidation or reduction, and break chemical bonds, which results in the formation of new compounds. Furthermore, cathode materials can be recycled mechanochemically through reactions with co-grinding agents. These agents can be categorized into various groups:
(1) Organic acids, chelates and polymers,29,30,32 (2) acidic, alkaline and neutral inorganic reagents,29,33 (3) oxidizing and reducing agents,29,31,34 (4) gaseous reagents,35 (5) Li and carbon sources for direct recycling.36,37 Incorporation of chemical conversion into the mechanical pretreatment by reactive milling can simplify the recycling process workflow.34 Another advantage of mechanochemical recycling is the reduced consumption of chemicals (especially corrosive) and water.29,33,38 Only low temperatures and ambient pressure are needed, which offers enhanced energy efficiency compared to other recycling techniques.29–31
In this study, pristine LFP was used as a model to examine the mechanochemical recycling process, isolating it from side reactions with other components typically present in cathode materials. This approach which facilitates a deeper understanding of the reaction behaviour of LFP. Past research has documented the use of ball-milling with Al as a co-grinding agent to induce reductive mechanochemical reactions, aiming to extract valuable elements from various cathode materials in an environmentally friendly and straightforward manner.34,39 Importantly, when Al is used as a reducing agent, removing the Al cathodic current collector is unnecessary, as it can be repurposed in the reaction, further simplifying the overall recycling process. While this recycling approach has proven to be very effective for various cathode materials, it has only achieved low lithium recovery rates when applied to LFP.39 Seeing that understanding of reaction mechanism and kinetics can foster process development of battery recycling,40,41 in our study we want to delve deeper into this process by investigating different ball-milling parameters to uncover the fundamental reaction mechanisms behind the reductive mechanochemical recycling of LFP. Additionally, the subsequent leaching process and Li recovery stages were analysed to explore potential avenues for making recycling of LFP effective and profitable.
2. Experimental section
2.1. Materials
LiFePO4 (∼98.5%, coated with carbon) was purchased from MTI Corporation, aluminium foil – from Novelis, aluminium powder (99.5%, 325 mesh) – from Alfa Aesar. All materials were used as received.2.2. Experimental procedure and analysis
The process to recover Li employed in this work is schematically illustrated in Fig. 1 and can be divided into three steps namely reactive milling, water leaching and purification.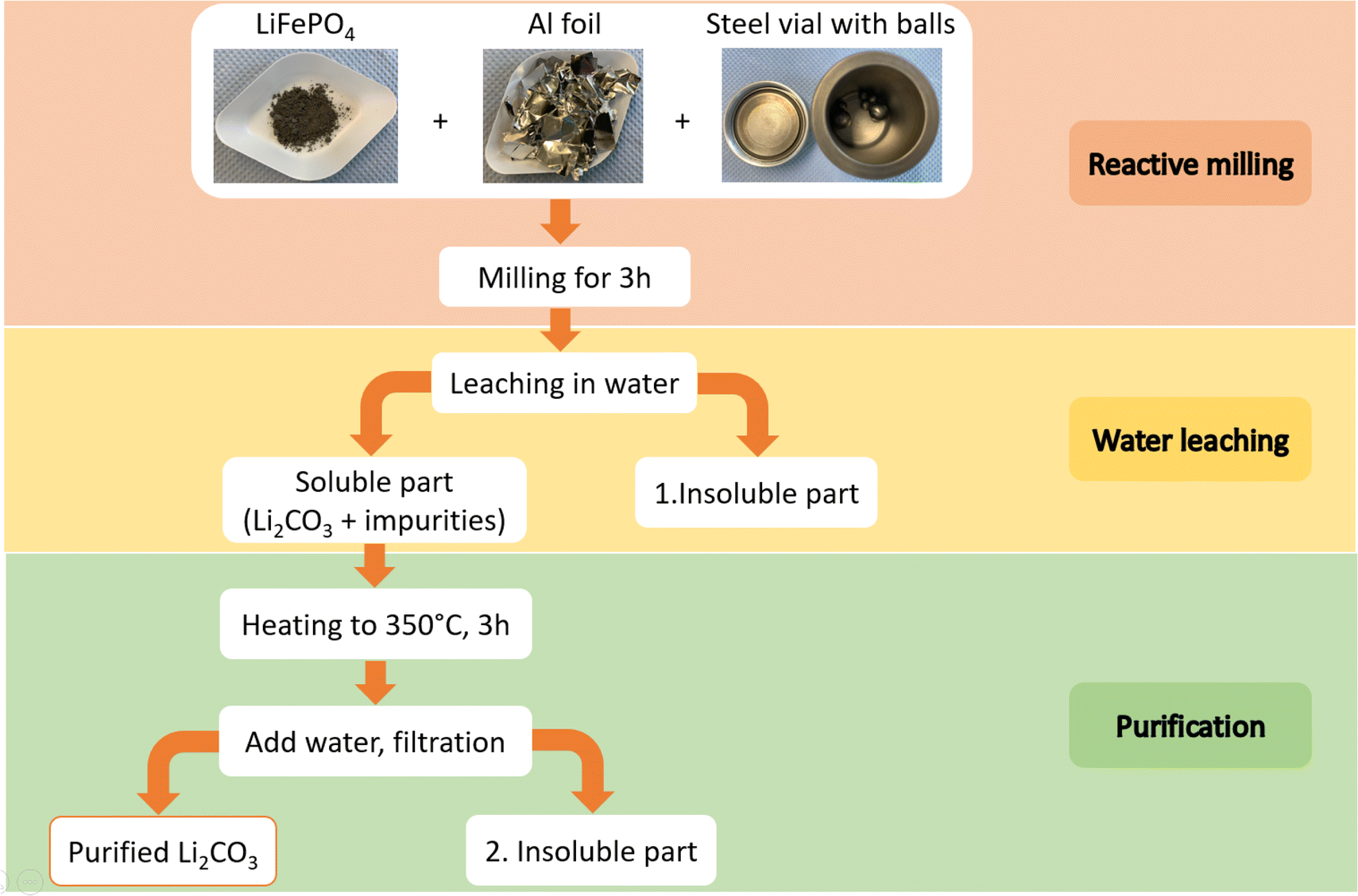 | ||
| Fig. 1 Schematic illustration of the process for mechanochemical recovery of lithium from LFP. | ||
First, about 2 g of a certain molar ratio of LFP and Al foil were ball-milled for various milling times in a 65 ml hardened-steel vial with 20 g of steel balls utilizing SPEX 8000 shaker mill. Prior to milling, the Al foil was cut into pieces of about 1 cm2. All experiments were performed under air and ambient temperature and pressure.
Afterward, the ball-milled material was mixed with deionized water and filtered through a paper filter using a vacuum pump. It is crucial for safety to introduce the ball-milled material to water gradually and in small portions because of the formation of toxic and flammable phosphine-gas. To mitigate environmental impact, the toxic PH3 gas can be oxidized to transform into phosphoric acid by reacting with oxygen and water. It can be achieved by installing a bubbler at the reactor's outlet. The insoluble residue was washed multiple times till the suspension reached pH = 7. The soluble part was concentrated by water evaporation, and both the soluble and insoluble part were dried at 70 °C overnight.
In the purification step, the soluble part was heated to 350 °C for 3 h in a muffle oven under air. The resulting solid was mixed with water, stirred for 1 h, and filtered. Again, recrystallization of the purified Li2CO3 solution was done by water evaporation. The resulting Li2CO3 and insoluble part were dried at 70 °C overnight.
In the experiments to calculate yield, approximately 1.25 g of material was milled in a 45 ml Silicon Nitride (Si3N4) vial containing 10 g of Si3N4 balls, following the previously described procedure. Si3N4 vials were employed in the ball milling process to prevent iron contamination from steel vials and guarantee accurate calculations. Moreover, aluminium was used in powdered form to accelerate the reaction kinetics, as Si3N4 vials deliver lower mechanical energy, demanding extended milling time.
2.3. Characterization of samples
X-ray powder diffraction (XRD) was utilized to characterize reaction products using a STOE Stadi P powder diffractometer with monochromatic Cu-Kα1 radiation (λ = 1.54056 Å) in transmission geometry. A 0.015° 2θ step between 10 and 70 degrees of 2θ was used at room temperature. The samples were prepared on a Kapton foil which adds an amorphous-like background to the XRD pattern between 10° and 17°.57Fe Mössbauer spectroscopy was performed in transmission mode at room temperature on a constant-acceleration spectrometer (WissEl) with a 57Co(Rh) source. A triangular velocity sweep was used and the velocity scale was calibrated with α-Fe metal foil (25 μm). 1024 channels were used to acquire the intensity as a function of source velocity (512 channels after folding). The sample mass was about 20 mg cm−2 and the measurement duration was 5 days. The α-Fe metal foil gives a sextet with Lorentzian lines with a width of 0.29 mm s−1. The spectra were fitted with WinNormos. All isomer shifts are given relative to that of α-Fe metal.
X-ray absorption spectroscopy (XAS) was performed at beamline P64 at PETRA III, Germany, at the Fe K-edge (7.1 keV) in continuous mode (6 min per spectrum). A Si(111) double-crystal monochromator was used to modulate the incoming photon energy and the beam size was 0.3 × 1.5 mm2. All XAS spectra were analysed and processed utilizing the ATHENA software package. The X-ray absorption near edge spectra (XANES) regions of the XAS spectra were acquired by subtracting the pre-edge background from the overall absorption and normalizing to the spline fit using the ATHENA software package.
Inductively coupled plasma optical emission spectroscopy (ICP-OES) was utilized to determine the mass percentage of Li, Al, Fe, and P using an iCAP 7600 DUO by Thermo-Fisher-Scientific. Oxygen was determined via carrier gas hot extraction with a G8 Galileo by Bruker AXS. Calculation of Li2CO3 purity and lithium lost in the insoluble part in different process steps is given in the ESI.†
2.4. Lithium recovery yield
To calculate the lithium recovery yield, the ball-milled sample with a well-known molar composition was weighed and the leaching and purification protocol was performed without taking samples in between. As no visual gas release was observed, the initial composition was considered to remain unchanged. Finally, the purified Li2CO3 was weighed and compared to the theoretical molar amount of Li calculated using the initial weight.3. Results and discussion
In previous studies, reactive milling of LFP with Al-foil as reducing agent and subsequent aqueous leaching to recover lithium was already investigated.39 This study showed that the reaction behaviour of LFP is unique compared to LCO, LMO and NMC, because phosphorous takes part in the reaction leading to different products. Most importantly, process 2 proposed in this study cannot be applied to LFP because of this deviating reaction behaviour.Our study delves deeper into the interactions between LFP and Al, employing a combination of analytical techniques such as XRD, Mössbauer, and XAS. We also investigate the milling and leaching behaviour of various molar ratios of components. While we have not yet achieved an increase in lithium recovery, our study provides valuable insights into the reasons behind the previously hindered effective leaching and offers a path forward for further investigations.
3.1. Influence of milling time on reactive milling
To investigate the first step of the recycling procedure illustrated in Fig. 1 and to learn more about the reductive milling of LFP, different ball-milling times of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar mixture of LFP and Al were analysed by XRD, as depicted in Fig. 2. While after 15 min and 30 min, only the starting materials can be observed, the initial structures are destroyed after 1 h of milling, and new products are forming. With prolonged milling above 2 hours, no significant changes in the XRD powder pattern are visible, and only a difference in relative intensities of the reflections occurs. The FePO4 compound, detected after 1 hour of milling, becomes undetectable upon prolonged mechanochemical treatment. This phenomenon could be related to its amorphization or further interactions, suggesting that FePO4 serves as an intermediate product in this reaction. The reflections labelled with a question mark cannot be identified because the intensities are too low for a reasonable assignment to a matching phase. Given that the XRD pattern showed no significant changes after 2–3 hours of milling—aside from a minor decrease in intensity attributed to microstructural changes—this time frame was identified as optimal. The products forming are Al2O3, FeP, Fe2P, and metallic iron (Fe0). Generally, all reflections are getting broader, and intensities are decreasing with more prolonged milling, which can be explained by the amorphization of the substances through mechanochemical treatment. Due to the broadness of the reflections in the XRD pattern, it is challenging to differentiate between the reflections of Fe2P and Fe, as their 2θ positions overlap. Consequently, Mössbauer spectroscopy proved to be a much more precise method for identifying their presence in the reaction products. Mössbauer spectroscopy was employed to analyse the products of the LFP-3Al mixture that had been ball-milled for varying durations. Fig. 3 shows the Mössbauer spectra of pristine LFP and the LFP-3Al mixtures milled between 0.25 and 5 h. The spectra of the products obtained after 0.25 h and 0.5 h of milling appear markedly different compared to those obtained after 1 h or longer, which fits well with the results obtained from XRD analysis. The spectrum of pristine LFP is dominated by a broad doublet with isomer shift IS = 1.3 mm s−1 and quadrupole splitting QS = 3.1 mm s−1 (Fig. 3 and Table S1†). These values are characteristic of Fe2+ in LFP.42–44 Two reasons for the large quadrupole splitting are the asymmetric local atomic environment of Fe and the asymmetric electronic charge distribution with the d6 configuration.30,45 A weak second doublet is visible after 0.25 h and 0.5 h of milling with IS = 0.8 mm s−1 and QS = 0.4 mm s−1 revealing the presence of a small amount of Fe3+.46 This might hint at the formation of FePO4 or rather a delithiation of LFP.45,47,48 The intensity of this Fe3+ doublet is increasing, reaching an area fraction of 24%, which shows that for these early stages of milling an oxidation of Fe2+ to Fe3+ occurs. During further milling, the spectra are getting more complex because various signals are overlapping. Therefore, it is hard to distinguish them reliably. The spectra can be described by additional contributions, marked with green colour. This reveals the formation of additional Fe-containing phases. In agreement with the XRD results in Fig. 2 and literature data, this contribution is assigned to FeP and Fe2P.49–51 Because of the overlapping of the doublets, the relative amount of phosphides cannot be evaluated. After 5 h of milling, a sextet contribution is visible (Fig. S1†) with isomer shift of 0 mm s−1 and a splitting corresponding to a hyperfine field of 31.3 T. This contribution thus clearly shows the formation of metallic Fe after 5 h, in agreement with the XRD results described above. Ball-milling generally leads to broad crystal size distributions, which is visible by comparing the broad peaks of Fe3+ and Fe0 components to the initial Fe2+ from LFP.
:3 molar mixture of LFP and Al were analysed by XRD, as depicted in Fig. 2. While after 15 min and 30 min, only the starting materials can be observed, the initial structures are destroyed after 1 h of milling, and new products are forming. With prolonged milling above 2 hours, no significant changes in the XRD powder pattern are visible, and only a difference in relative intensities of the reflections occurs. The FePO4 compound, detected after 1 hour of milling, becomes undetectable upon prolonged mechanochemical treatment. This phenomenon could be related to its amorphization or further interactions, suggesting that FePO4 serves as an intermediate product in this reaction. The reflections labelled with a question mark cannot be identified because the intensities are too low for a reasonable assignment to a matching phase. Given that the XRD pattern showed no significant changes after 2–3 hours of milling—aside from a minor decrease in intensity attributed to microstructural changes—this time frame was identified as optimal. The products forming are Al2O3, FeP, Fe2P, and metallic iron (Fe0). Generally, all reflections are getting broader, and intensities are decreasing with more prolonged milling, which can be explained by the amorphization of the substances through mechanochemical treatment. Due to the broadness of the reflections in the XRD pattern, it is challenging to differentiate between the reflections of Fe2P and Fe, as their 2θ positions overlap. Consequently, Mössbauer spectroscopy proved to be a much more precise method for identifying their presence in the reaction products. Mössbauer spectroscopy was employed to analyse the products of the LFP-3Al mixture that had been ball-milled for varying durations. Fig. 3 shows the Mössbauer spectra of pristine LFP and the LFP-3Al mixtures milled between 0.25 and 5 h. The spectra of the products obtained after 0.25 h and 0.5 h of milling appear markedly different compared to those obtained after 1 h or longer, which fits well with the results obtained from XRD analysis. The spectrum of pristine LFP is dominated by a broad doublet with isomer shift IS = 1.3 mm s−1 and quadrupole splitting QS = 3.1 mm s−1 (Fig. 3 and Table S1†). These values are characteristic of Fe2+ in LFP.42–44 Two reasons for the large quadrupole splitting are the asymmetric local atomic environment of Fe and the asymmetric electronic charge distribution with the d6 configuration.30,45 A weak second doublet is visible after 0.25 h and 0.5 h of milling with IS = 0.8 mm s−1 and QS = 0.4 mm s−1 revealing the presence of a small amount of Fe3+.46 This might hint at the formation of FePO4 or rather a delithiation of LFP.45,47,48 The intensity of this Fe3+ doublet is increasing, reaching an area fraction of 24%, which shows that for these early stages of milling an oxidation of Fe2+ to Fe3+ occurs. During further milling, the spectra are getting more complex because various signals are overlapping. Therefore, it is hard to distinguish them reliably. The spectra can be described by additional contributions, marked with green colour. This reveals the formation of additional Fe-containing phases. In agreement with the XRD results in Fig. 2 and literature data, this contribution is assigned to FeP and Fe2P.49–51 Because of the overlapping of the doublets, the relative amount of phosphides cannot be evaluated. After 5 h of milling, a sextet contribution is visible (Fig. S1†) with isomer shift of 0 mm s−1 and a splitting corresponding to a hyperfine field of 31.3 T. This contribution thus clearly shows the formation of metallic Fe after 5 h, in agreement with the XRD results described above. Ball-milling generally leads to broad crystal size distributions, which is visible by comparing the broad peaks of Fe3+ and Fe0 components to the initial Fe2+ from LFP.
 | ||
| Fig. 2 XRD patterns of a 1:3 molar mixture of LFP and Al measured after different ball-milling times. The most intense Bragg-reflections are assigned to different phases for analysis. The reflections labeled with a question mark cannot be identified because the intensities are too low for a reasonable assignment to a matching phase. 2–3 h are necessary to finish the reaction. | ||
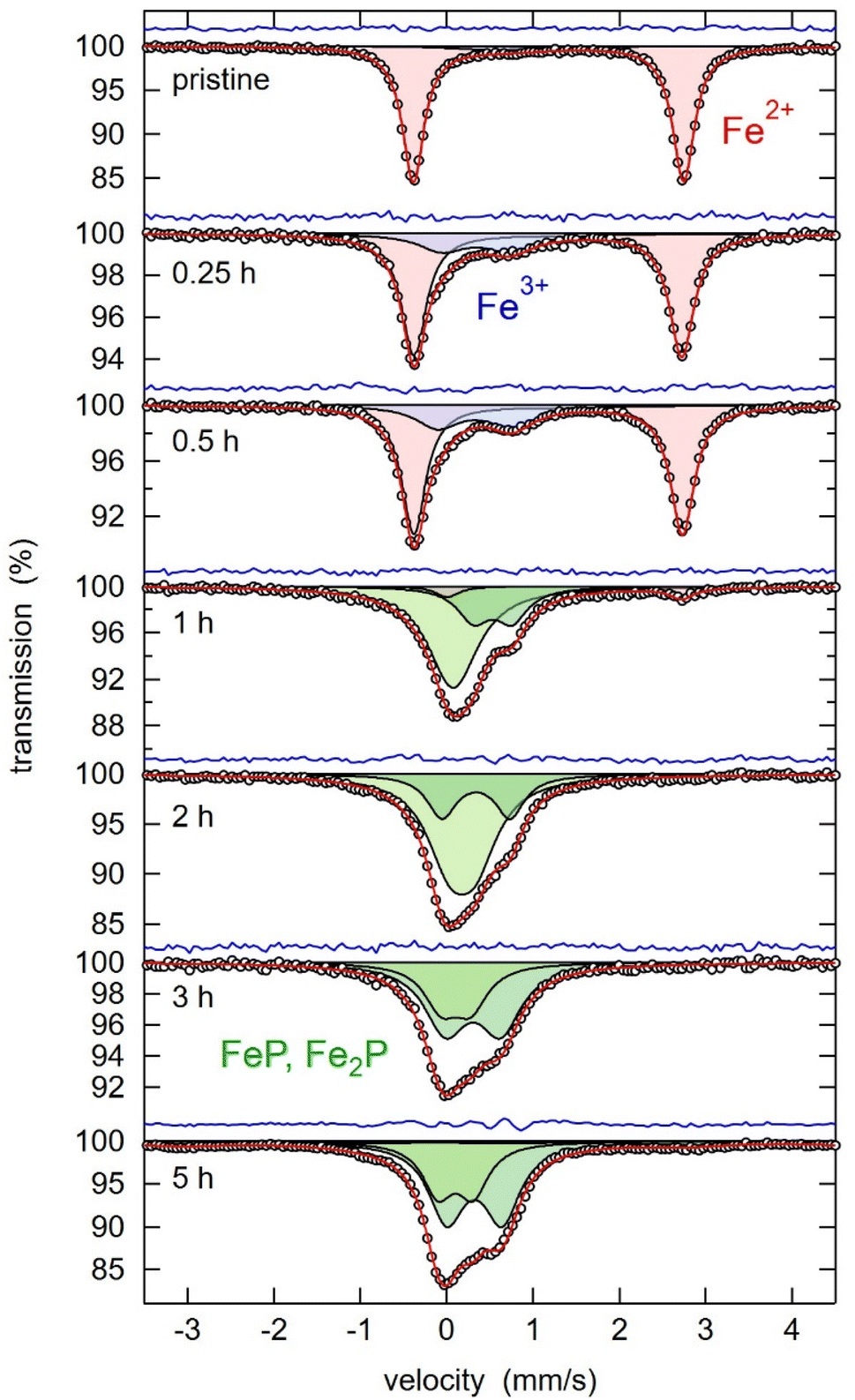 | ||
| Fig. 3 Mössbauer spectra of pristine LFP and LFP ball-milled with Al metal foil for 0.25 h to 5 h (1:3 molar ratio of LFP to Al). The experimental data points are shown as white spheres, the overall fit as red line, the difference as blue line, and the subspectra are shown as red, blue, and green singlets/doublets. | ||
In order to extend the investigation of reductive milling and get further insights, XAS of the LFP-3Al mixture ball-milled for varying durations was employed. The X-ray absorption near edge structure (XANES) spectra in Fig. 4 are showing, in good agreement with XRD analysis and Mössbauer spectroscopy, that the initial LFP structure is converted between 30 min and 1 h of milling and no significant changes are visible afterwards. After 1 h, an iron reduction with a near edge structure comparable to that of iron metal was observed. Besides iron metal, FeP and Fe2P are not visible in these spectra. The radial distributions obtained from the Fourier-transform of the extended X-ray absorption fine structure (EXAFS) are depicted in Fig. S2.† They are providing more information on the coexistence of different phases. For pristine LFP, oxygen was assigned as the nearest neighbour, which is in agreement with the octahedral oxygen surrounding of iron in the olivine structure. Second neighbours with longer radial distance are iron and phosphorus. After 1 h of milling, iron is reduced to its metallic form, as also observed in the XANES spectra. However, an additional peak at around 1.8 Å can be detected. This hints at the coexistence of other iron phases, which supports XRD and Mössbauer analysis.
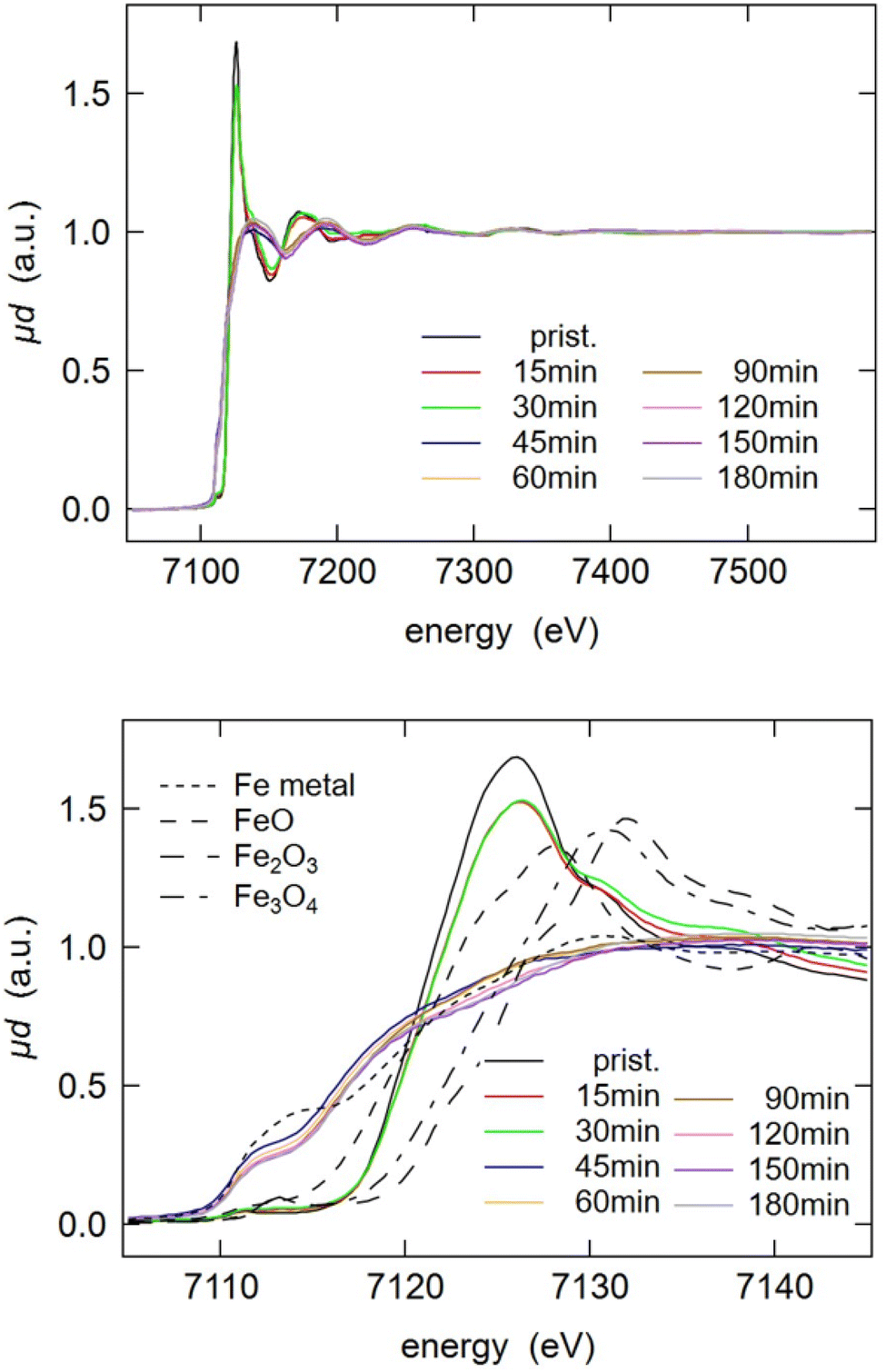 | ||
| Fig. 4 Fe K-edge X-ray absorption near edge structure (XANES) spectra of pristine LFP and LFP ball-milled with Al metal foil for 0.25 h to 5 h (1:3 molar ratio of LFP to Al). The spectra of iron metal, FeO, Fe2O3 and Fe3O4 were plotted for comparison. | ||
3.2. Influence of LFP to Al ratio on reactive milling
To identify the underlying reaction mechanism of mechanochemical interactions in the LFP-Al system, the screening of various milling times was extended to different molar ratios of starting materials. From the molar ratio of LFP to Al of 1:2 up to 1:5, the start of the reaction can be observed between 0.5 h and 1 h, as depicted in Fig. 2, S4, and S5.† For a 1:1 ratio, only a partial conversion of LFP is detectable between 1 h and 2 h, as evidenced in Fig. S3.† Reflections corresponding to the pristine LFP material remain visible on the XRD pattern even after 2 h of milling. Meanwhile, reflections of Al are absent, indicating that its conversion is completed and products have formed. In Fig. 5 the XRD patterns of different mixtures of components milled for 3 h are depicted. The 1:1 ratio shows that the Al amount is insufficient to convert all LFP, as it is still visible after 3 h of milling. Comparing the relative ratios of products for various compositions of the LFP-Al system, some trends can be observed. The reflections for Fe0, Al2O3, and AlP are getting more intense while the intensity of FeP and Fe2P decreases with higher molar amount of Al. For LFP-6Al, even an aluminium–iron-alloy AlxFe2 (with x ≈ 5.6 identified by lattice parameters) can be observed. Compared to previous studies,39 the analysis of different molar mixtures of components allows deeper insight into the mechanism of interaction between LFP and aluminium. For instance, AlP was proposed as a possible product because it was not detectable by XRD. As the amount of this compound increases with higher aluminium content, it now gets detectable by XRD. Additionally, FeP and Fe0 got visible in the XRD patterns. The proposed reaction mechanism derived from the combination of the XRD analysis and Mössbauer spectroscopy can be described by eqn (1)–(6). Li2O as a feasible reaction intermediate, is given in parentheses.| 7O2 + 4LiFePO4 + 8Al → 4Al2O3 + {2Li2O} + 4FePO4 | (1) |
| 3FePO4 + 8Al → 3FeP + 4Al2O3 | (2) |
| 2FeP + Al → AlP + Fe2P | (3) |
| Fe2P + Al → 2Fe + AlP | (4) |
| xAl + 2Fe → AlxFe2 | (5) |
| 4Al + 3O2 → 2Al2O3 | (6) |
 | ||
| Fig. 5 XRD patterns of various molar mixtures of LFP and Al measured after 3 h of ball-milling. The most intense Bragg-reflections are assigned to different phases for analysis. X ≈ 5.6 for AlxFe2, identified by lattice parameters. | ||
The detection of FePO4 as an intermediate product in Fig. 2, 3, and S4† leads to the assumption that first LiFePO4 is delithiated with the formation of Li2O, as described in eqn (1). Subsequently, the initial olivine structure is broken up as Al captures the oxygen from FePO4 leading to the formation of FeP as indicated in eqn (2). Following the removal of oxygen, Al continues to seize increasing amounts of phosphorus, resulting in the generation of Fe2P, and elemental iron, as described by eqn (3) and (4). Several competing reactions occur simultaneously during the mechanochemically induced reduction reaction between LFP and Al. XRD analysis reveals that an increased amount of Al in the mixture results in its interaction with Fe2P and FeP with the formation of AlP, as outlined in eqn (3) and (4). Once iron is fully reduced to its metallic form, the excess Al in the LFP-6Al system promotes the formation of the intermetallic AlxFe2 compound by the reaction shown in eqn (5). Furthermore, since the mechanochemical reactions take place in an air atmosphere, oxidation of the pulverized Al by atmospheric oxygen occurs also, as indicated in eqn (6).
3.3. Influence of LFP to Al ratio on water leaching
Building on understanding the behaviour and resulting products of the mechanochemical interaction in the LFP-Al system at various molar ratios, the water leaching and purification protocol shown in Fig. 1 was employed to recover lithium as purified Li2CO3. The intermediate products as well as the final Li2CO3, were monitored by XRD to develop an understanding of the whole process and to find optimum conditions. Optimizing Li extraction is crucial to establish a profitable recycling process. Fig. 6 depicts XRD patterns of the different process steps of leaching and purification of a 1:3 molar ratio of LFP to Al. The XRD pattern of the residue after the initial water leaching, termed “insoluble”, closely resembles that of the material milled for 3 h, as depicted in Fig. 2 for the LFP-3Al system. This is attributed to all reflections detectable by XRD originating from insoluble compounds. Given that the leaching process was conducted in an aqueous solution, bayerite (α-Al(OH)3) formed, and its reflections were evident in the XRD powder pattern. The relative intensities for both AlP and Al(OH)3 increase with a higher content of Al in the milled mixture, which is visible in Fig. 5, 6, and S3–S5.† A phosphine odor and self-igniting gas were detected when adding the ball-milled material to water. Therefore, it is crucial for safety to introduce the ball-milled material to water gradually and in small portions. This observation suggests the reaction outlined in eqn (7).| AlP + 3H2O → Al(OH)3 + {PH3}↑ | (7) |
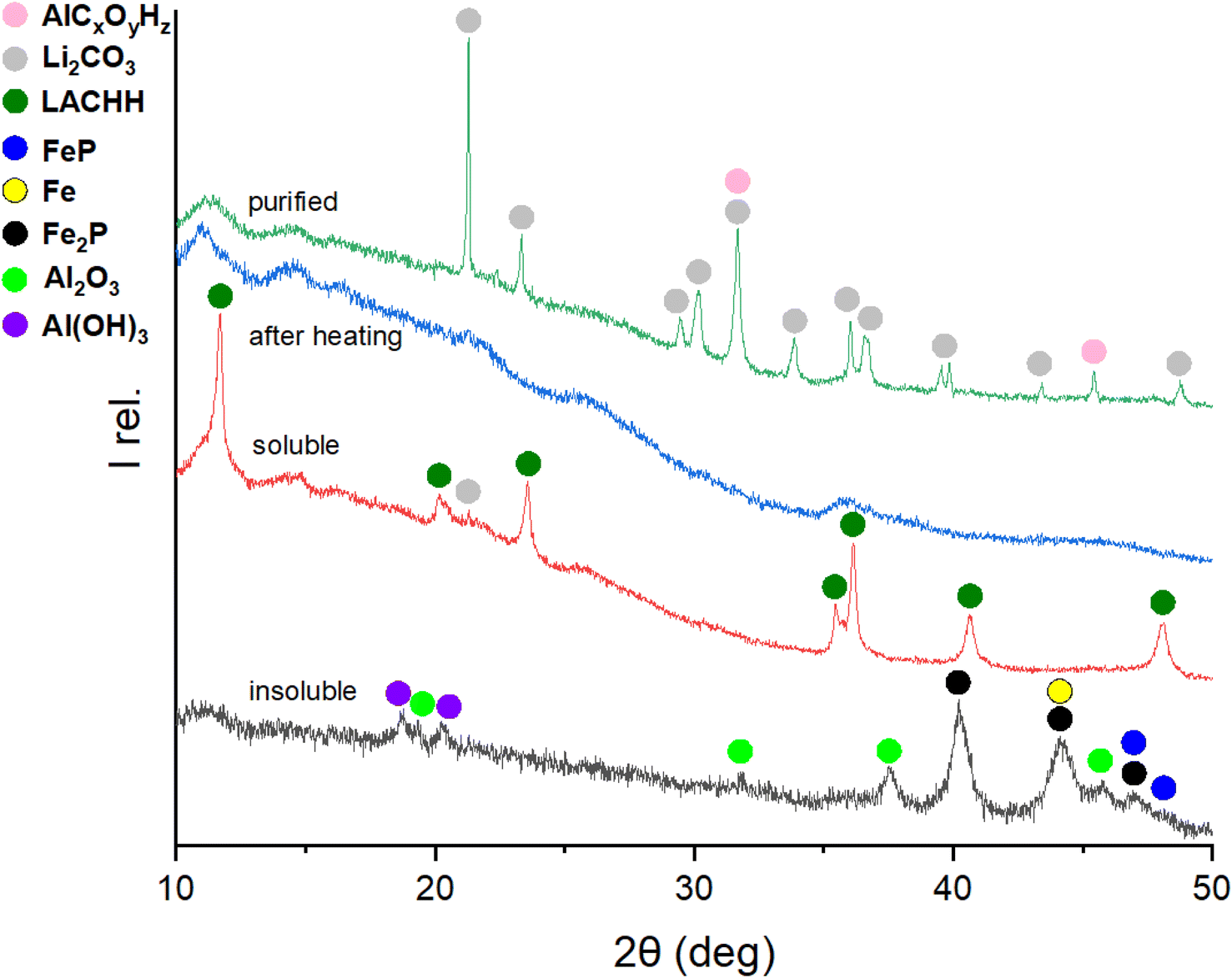 | ||
| Fig. 6 XRD patterns of the products after the leaching and purification process obtained from a 1:3 molar mixture of LFP and Al ball-milled for 3 h. The most intense Bragg-reflections are assigned to different phases for analysis. | ||
To mitigate environmental impact, the toxic PH3 gas can be oxidized to transform into phosphoric acid by reacting with oxygen and water. It can be achieved by installing a bubbler at the reactor's outlet. Li2O, as a feasible reaction intermediate from ball-milling, will react to LiOH when water is added, as shown in eqn (8).
| {Li2O} + H2O → {2LiOH} | (8) |
The formation of an alkaline solution with a measured pH of 9 lends support to this reaction. LiOH typically reacts with CO2 and converts to Li2CO3 when undergoing a drying process in open air as described by described by eqn (9).
| 2{LiOH} + CO2 → Li2CO3 + H2O | (9) |
In the soluble part (denoted “soluble”) in Fig. 6, lithium aluminium carbonate hydroxide hydrate, Li2Al4(CO3)(OH)12·3H2O (LACHH) can be identified as the primary compound in the XRD pattern. According to Simon and Gluth LACHH forms from a solution during recrystallization by water evaporation with CO2 provided by air.52 As LiOH·H2O and Al(OH)3 are used for the synthesis of this material, the formation of LACHH in this leaching process can be explained by eqn (10) and (11) where LixAlOx(OH)z represents a soluble precursor of LACHH.
| {LiOH} + Al(OH)3 → {LixAlOx(OH)z} | (10) |
| {LixAlOx(OH)z} + H2O + CO2 → LACHH | (11) |
It can be assumed that these reactions are competing with eqn (9), leading to only a barely visible reflection of Li2CO3 in the soluble part.
Literature studies show that LACHH can be decomposed by heating with a multistep decomposition mechanism.53,54 As intermediates between 250 °C and 600 °C LACHH transforms to Li2CO3 and amorphous Al2O3 as shown in eqn (12).53
| Li2Al4(CO3)(OH)12·3H2O → Li2CO3 + 2Al2O3 + 9H2O↑ | (12) |
Therefore, the dried soluble part was heated to 350 °C, and the XRD pattern “after heating” shows X-ray amorphous products. With a second water leaching of this amorphous product, purified Li2CO3 can be extracted in the soluble part and recrystallized by water evaporation. Besides Li2CO3 in the pattern “purified”, extra reflections can be characterized as AlCxOyHz resulting from water-soluble aluminium-containing impurities.
The purification of the soluble fraction from the LFP-5Al system yielded similar products, as depicted in Fig. S5.† In contrast, the 1:1 and 1:2 mixtures of LFP and Al (Fig. S3 and 4†) exhibited a slightly different behaviour. No LACHH compound was detected in the XRD pattern of the soluble fraction. Instead, Li3PO4 was identified as an additional Li-containing product. Even after heating the soluble fraction to 350 °C and performing subsequent second leaching, the co-presence of Li2CO3 and Li3PO4 persisted. This Li3PO4 formation was found in both systems with lower Al content. Given the reaction mechanism of the mechanochemical process in the LFP-Al system, it can be deduced that Li3PO4 arises due to the partial conversion of phosphate structures into FeP and Fe2P. Hence, to exclusively produce Li2CO3 as the sole lithium product after leaching, using a 1:3 molar ratio of LFP to Al is recommended as the most suitable condition for the reductive milling of the LFP-Al system.
3.4. Yield and purity of recovered Li
Having gained insight into the entire process and analysed the mechanisms of mechanochemical reaction and leaching, we investigated the yield and purity of obtained Li2CO3 to evaluate its economic viability. Various LFP to Al molar ratios were examined to determine the impact of the Al content on the final lithium yield. Fig. 7 shows that the Li-yield of purified Li2CO3 slightly increases with a higher content of Al in the reaction mixture. The LFP-5Al system exhibited the highest lithium yield, coming in at 28.8%, which is considerably lower than anticipated. This raised questions regarding the points of lithium loss throughout the process. Apart from the usual minor experimental losses, lithium is predominantly believed to be contained within the insoluble fractions from the two leaching stages highlighted in Fig. 1. As shown in Fig. 7, ICP-OES results revealed that a significant amount (more than 70%) of the lithium remains in the initial insoluble residue after the first leaching. This phenomenon can be attributed to Li-containing products that appear XRD-amorphous. It is plausible these compounds formed during milling are not readily soluble under the applied leaching conditions. Efforts to tackle this issue and enhance Li yield are currently in progress. The analysis of the ball-milling parameters revealed that with an increased content of Al, the reduction mechanism described by eqn (1)–(6) persists until elemental iron is finally produced. It is conceivable that intensifying the reduction could lead to the formation of products that allow for a more substantial leaching of lithium. This hypothesis gains traction from the noticeable reduction in lithium loss with a rise in Al content, as depicted in Fig. 7.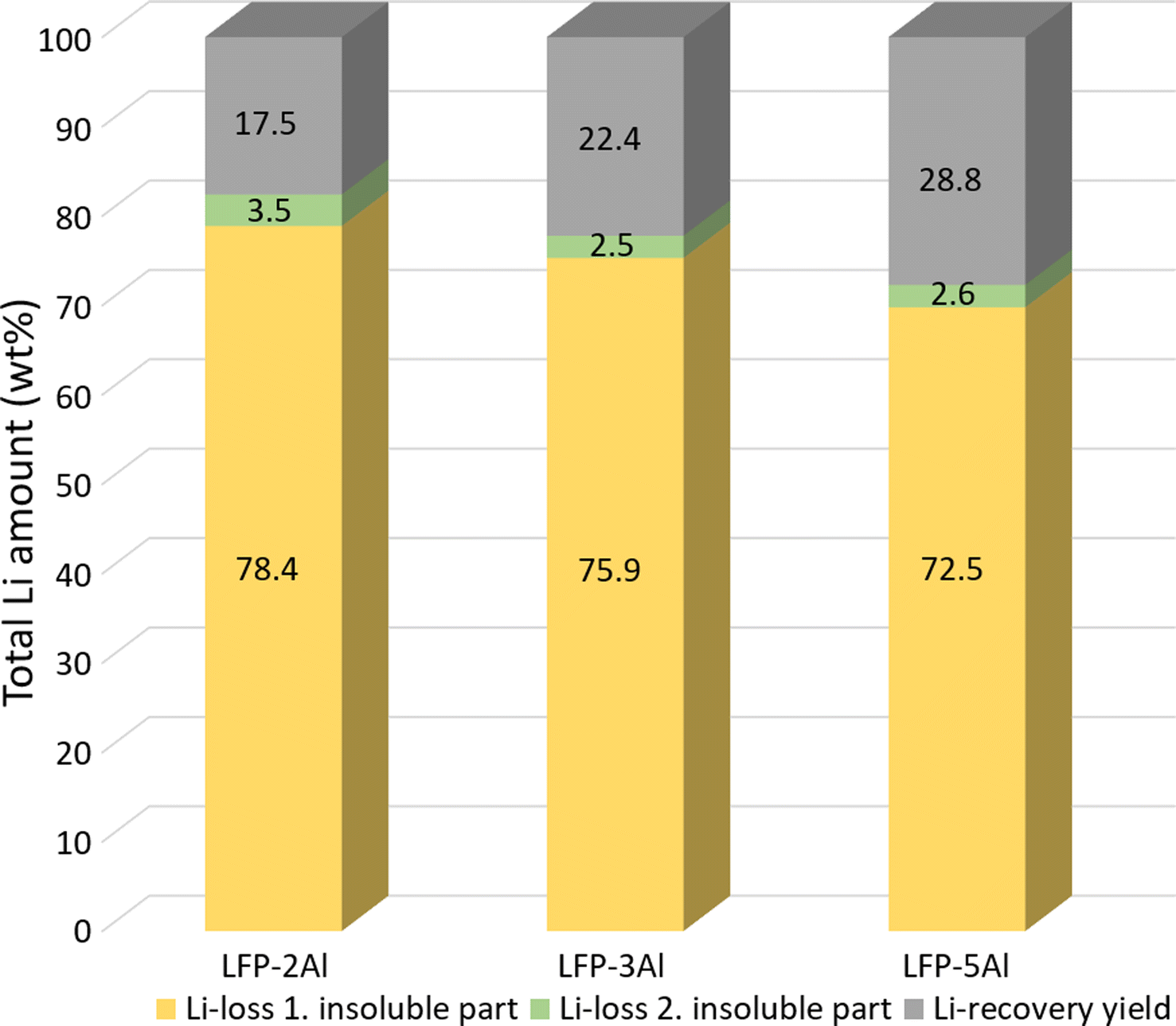 | ||
| Fig. 7 Comparison of Li-loss during the leaching process and Li-yield of purified Li2CO3 for 1:2, 1:3 and 1:5 molar mixtures of LFP and Al. Li-loss was calculated using ICP-OES as explained in the ESI† and lithium recovery yield was calculated by weighing initial mixture and final product. Because of two different methods applied, the total number can exceed 100%. | ||
Even though the insoluble LACHH compound prominently appears in the soluble fraction after the initial leaching (notably for LFP-3Al and LFP-5Al), a mere 3% of lithium persists in the insoluble fraction following the second leaching. This attests to the efficiency of LACHH breakdown at 350 °C and is in line with prior research conducted by Dolotko et al.39
For efficient Li recycling, both the recovery yield and the purity of the final product are critical, ensuring that the recycled products can be used to manufacture new cathode materials. Hence, ICP-OES was also utilized to calculate the purity of Li2CO3, as explained in the ESI.†Fig. 8 shows the wt%-purity of Li2CO3 and the amount of impurities for a LFP-3Al mixture. A purity of 74.6 wt% was achieved for the resultant Li2CO3. Considering that pure LFP was used in this study, the 7.3% impurities from other elements are likely attributed to carbon and hydrogen, possibly introduced during various filtration steps or from other sources. As previously highlighted, AlCxOyHz, as shown in Fig. 6, originates from water-soluble aluminium-containing impurities. Given its high solubility, rinsing the Li2CO3 at adjusted temperatures can further reduce this contaminant. Phosphorus impurities arise from minor quantities of Li3PO4, evident from the subtle reflection at ∼22° 2θ in Fig. 6. Ongoing efforts aim to further refine the purity of the Li2CO3.
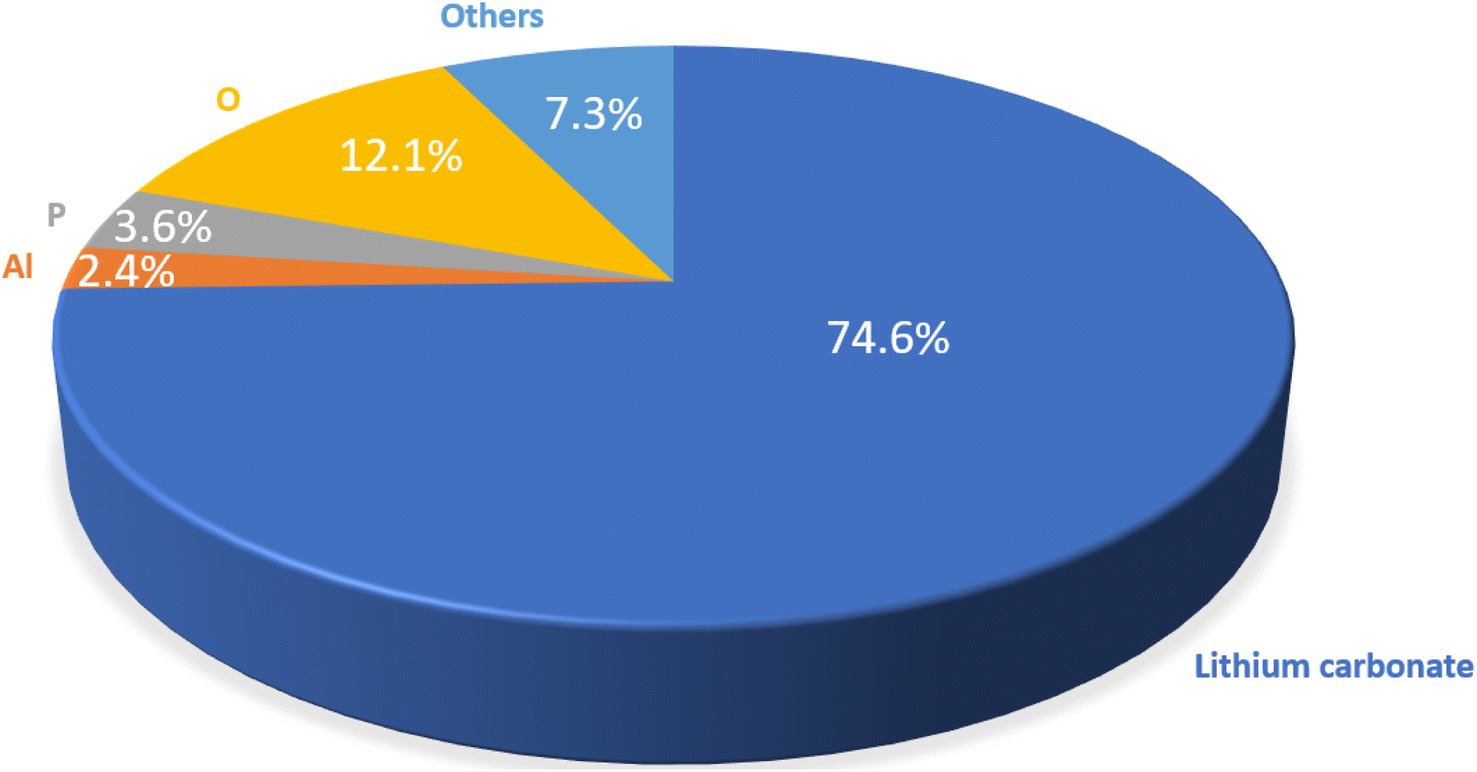 | ||
| Fig. 8 Wt%-purity of Li2CO3 final product calculated using ICP-OES for a 1:3 molar mixture of LFP and Al. 7.3% of other elements probably result from C and H. To calculate the Li2CO3 purity, the weight percentage (wt%) of Li was used to determine a stochiometric wt% of carbon and oxygen. Li2CO3 purity was then obtained by adding these wt% to the wt% of Li. Impurities were stated as received from the measurement except oxygen. For oxygen the oxygen contained in the Li2CO3 was subtracted. | ||
While purity and lithium recovery have not yet been fully optimized, mechanochemically induced recycling of LFP using Al-foil as a reducing agent shows the potential to advance environmental sustainability compared to the currently employed hydrometallurgical method for LFP recycling, with further development. At its current stage of technological development, industrial-scale hydrometallurgical methods are deemed uneconomical for recycling LFP waste cathodes due to the low quantities of recycled lithium and the associated high processing costs.13 These methods require complex pretreatment and multi-step leaching processes that involve various strong acids and bases.14
In contrast, our approach employs aluminium foil instead of fresh chemicals. In the battery recycling workflow, a significant amount of low-purity aluminium foil is generated, which is challenging to further purify and is typically considered waste. Hence, our method effectively utilizes waste to treat waste. Furthermore, our straightforward technology can streamline the process by incorporating chemical conversion into the pretreatment stage of recycling.34 The simplified leaching process also reduces wastewater production, as only water is used for leaching. This eliminates the need for expensive post-treatment of highly corrosive liquid wastes, which is common in hydrometallurgical methods. Additionally, the mechanochemically reduced LFP material enables fast leaching kinetics at room temperature within minutes, whereas the existing methods require several hours of stirring at elevated temperatures.13
Moreover, it is noteworthy that during the formation of Li2CO3, a product of our recycling process, atmospheric CO2 gas is incorporated. This aspect makes the process “negative emissions,” resulting in a reduction of the carbon footprint during its implementation.
However, it's important to acknowledge the challenges that our proposed approach faces at its current developmental stage. One significant challenge is the formation of toxic and flammable PH3 gas, as well as its transformation into phosphoric acid, which must be carefully controlled in an industrial-level process. Another challenge relates to the limited throughput for industrial application due to the currently available size of high-energy ball mills. Additionally, there is a need to address potential mechanochemical side reactions that may occur when dealing with end-of-life batteries. To overcome these challenges, we are actively working to further improve this high-risk, yet potentially highly rewarding, process.
4. Conclusions
The mechanochemical approach proposed by Dolotko et al.39 for recycling lithium from LFP was explored in this study. Pristine LFP was used as a model to delve deeper into the reaction behaviour. LFP typically retains its structural integrity even after undergoing fatigue, which makes our proposed method well-suited for its recycling. However, it's important to acknowledge that in the black mass or production scrap from batteries, other constituents such as graphite, binder, electrolyte salt, copper, or carbon black may also be present. While we do not anticipate these materials having a substantial impact on the process, conducting additional studies could provide valuable insights. Furthermore, it is worth noting that the formation of LiF or other impurities might be expected during the recycling process. Using aluminium as a reducing agent in this process offers an advantage to simplify the battery recycling workflow since Al is already present in the battery constituents as a current collector. Different ball-milling times and LFP to Al molar ratios were analysed to reveal the underlying reaction mechanism.After completing the mechanochemical reaction with 3 h of milling, a variety of products form, including aluminium- and iron phosphides, aluminium oxide, and elemental iron. Aqueous leaching facilitates the extraction of water-soluble Li-containing compounds from insoluble aluminium and iron products. Li-containing LACHH in the soluble fraction can be effectively decomposed by heating to 350 °C to obtain purified Li2CO3 after a second leaching step. Given that the 1:1 and 1:2 LFP to Al mixtures produce a mixture of Li2CO3 and Li3PO4, the LFP-3Al system is selected as the optimal composition for recovering Li in the Li2CO3 form.
Currently, the proposed process allows for the recovery of only approximately 28% of the initial lithium. Yet, ICP-OES analysis indicates that 72% of Li is retained in the insoluble fraction after first leaching. Consequently, optimizing the leaching parameters could be instrumental in extracting these insoluble Li-containing compounds.
Further refinement of this straightforward and energy-saving method has the potential to facilitate the economic recycling of LFP, for which no industrial solution currently exists. This research offers vital insights into the process, laying the groundwork for its continued advancement.
Abbreviations
| EXAFS | Extended X-ray absorption fine structure |
| ICP-OES | Inductively coupled plasma optical emission spectroscopy |
| LACHH | Li2Al4(CO3)(OH)12·3H2O |
| LCO | Lithium cobalt oxide |
| LFP | Lithium iron phosphate |
| LMO | Lithium manganese oxide |
| LIB | Lithium-ion batterie |
| NCA | LiNixCoyAl1−x−yO2 |
| NMC | LiNixMnyCo1−x−yO2 |
| RT | Room temperature |
| XANES | X-ray absorption near edge structure |
| XAS | X-ray absorption spectroscopy |
| XRD | X-ray powder diffraction |
Data availability
The research data is available free of charge at https://doi.org/10.35097/1912.Author contributions
D. G. guided the study, performed experiments, collected and visualized data and contributed to its interpretation, wrote the initial draft of the manuscript and conducted revisions; O. D. conceived the idea and methodology, supported coordination and management of the study and contributed to data interpretation and revision of the manuscript; S. I. performed and visualized Mössbauer and XAS measurements, wrote the corresponding experimental sections and contributed to their data interpretation; C. N. and A. B. performed experiments for calculation of the lithium recovery; T. B. performed ICP-OES measurements; M. K. supervised the study, contributed to data interpretation and revision of the manuscript; H. E. contributed to revision of the manuscript. All authors provided comments during preparation of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was facilitated through funding by the LiCORNE project. LiCORNE is funded by the European Union under Grant Agreement No. 101069644. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Climate, Infrastructure and Environment Executive Agency (CINEA). Neither the European Union nor the granting authority can be held responsible for them. This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe). We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank Akhil Tayal for assistance in using beamline P64. Beamtime was allocated for Proposal I-20220585.References
- O. K. Park, Y. Cho, S. Lee, H. Yoo, H. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621–1633 RSC.
- Y. Zhao, O. Pohl, A. I. Bhatt, G. E. Collis, P. J. Mahon, T. Rüther and A. F. Hollenkamp, Sustain. Chem., 2021, 2, 167–205 CrossRef CAS.
- Y. Zhao, O. Pohl, A. I. Bhatt, G. E. Collis, P. J. Mahon, T. Rüther and A. F. Hollenkamp, Sustain. Chem., 2021, 2, 167–205 CrossRef CAS.
- H. Bae and Y. Kim, Mater. Adv., 2021, 2, 3234–3250 RSC.
- BCC publishing staff, Am. Ceram. Soc. Bull., 2022, 102(1), 5 Search PubMed.
- Y. Bai, N. Muralidharan, Y. K. Sun, S. Passerini, M. S. Whittingham and I. Belharouak, Mater. Today, 2020, 41, 304–315 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561–1800584 CrossRef PubMed.
- M. Maisch, pv magazine, 2022, vol. 10, available online: https://www.pv-magazine.com/magazine-archive/cobalt-clings-on/(accessed on 3.10.2023) Search PubMed.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- R. Castellano, Seeking Alpha, 2017, available online: https://seekingalpha.com/article/4113417-how-to-minimize-teslas-cobalt-supply-chain-risk (accessed on 3.10.2023) Search PubMed.
- N. Mohamed and N. K. Allam, RSC Adv., 2020, 10, 21662–21685 RSC.
- Y. Ding, Cathode for Thin-Film Lithium-Ion Batteries, Intech Open, 2020, pp. 1–17 Search PubMed.
- A. Pražanová, V. Knap and D. Stroe, Energies, 2022, 15, 1086–1114 CrossRef.
- O. Velázquez-Martínez, J. Valio, A. Santasalo-Aarnio, M. Reuter and R. Serna-Guerrero, Batteries, 2019, 5, 68–100 CrossRef.
- G. Ren, S. Xiao, M. Xie, B. Pan, J. Chen, F. Wang and X. Xia, Trans. Nonferrous Met. Soc. China, 2017, 27, 450–456 CrossRef CAS.
- E. Mossali, N. Picone, L. Gentilini, O. Rodrìguez, J. M. Pérez and M. Colledani, J. Environ. Manage., 2020, 264, 110500–110511 CrossRef PubMed.
- F. Forte, M. Pietrantonio, S. Pucciarmati, M. Puzone and D. Fontana, Crit. Rev. Environ. Sci. Technol., 2021, 51(19), 2232–2259 CrossRef CAS.
- J. Kumar, R. R. Neiber, J. Park, R. A. Soomro, G. W. Greene, S. A. Mazari, H. Y. Seo, J. H. Lee, M. Shon, D. W. Chang and K. Y. Cho, Chem. Eng. J., 2022, 431(1), 133993 CrossRef CAS.
- X. Qiu, B. Zhang, Y. Xu, J. Hu, W. Deng, G. Zou, H. Hou, Y. Yang, W. Sun, Y. Hu, X. Cao and X. Ji, Green Chem., 2022, 24, 2506–2515 RSC.
- Y. Xu, X. Qiu, B. Zhang, A. Di, W. Deng, G. Zou, H. Hou and X. Ji, Green Chem., 2022, 24, 7448–7457 RSC.
- S. Zhou, J. Du, X. Xiong, L. Liu, J. Wang, L. Fu, J. Ye, Y. Chena and Y. Wu, Green Chem., 2022, 24, 6278–6286 RSC.
- J. Chen, L. Ye, Y. Tian, T. Fu, J. Min, D. Fu, W. Zhang and L. Zhang, CN110581323 A, 2019.
- X. Liu, M. Wang, L. Deng, Y.-J. Cheng, J. Gao and Y. Xia, Ind. Eng. Chem. Res., 2022, 61, 3831–3839 CrossRef CAS.
- Q. Sun, X. Li, H. Zhang, D. Song, X. Shi, J. Song, C. Li and L. Zhang, J. Alloys Compd., 2020, 818, 153292 CrossRef CAS.
- H. Jin, J. Zhang, D. Wang, Q. Jing, Y. Chen and C. Wang, Green Chem., 2022, 24, 152–162 RSC.
- Y. Yang, S. Song, M. Sun, W. Sun and H. Tang, CN113772649, 2021.
- L. Wu, F.-S. Zhang, Z.-Y. Zhang and C.-C. Zhang, J. Cleaner Prod., 2023, 396, 136504 CrossRef CAS.
- Q. Zhang, E. Fan, J. Lin, S. Sun, X. Zhang, R. Chen, F. Wu and L. Li, J. Hazard. Mater., 2023, 443(Part A), 130160 CrossRef CAS PubMed.
- M. Wang, K. Liu, J. Yu, C.-C. Zhang, Z. Zhang and Q. Tan, Circ. Econ., 2022, 1, 100012–100020 Search PubMed.
- E. Fan, L. Li, X. Zhang, Y. Bian, Q. Xue, J. Wu, F. Wu and R. Chen, ACS Sustainable Chem. Eng., 2018, 6, 11029–11035 CrossRef CAS.
- J. Guan, Y. Li, Y. Guo, R. Su, G. Gao, H. Song, H. Yuan, B. Liang and Z. Guo, ACS Sustainable Chem. Eng., 2017, 5, 1026–1032 CrossRef CAS.
- M.-M. Wang, C.-C. Zhang and F.-S. Zhang, Waste Manage., 2017, 67, 232–239 CrossRef CAS PubMed.
- M. Wang, Q. Tan, L. Liu and J. Li, J. Cleaner Prod., 2021, 279, 123612 CrossRef CAS.
- O. Dolotko, I. Z. Hlova, Y. Mudryk, S. Gupta and V. P. Balema, J. Alloys Compd., 2020, 824, 153876–153894 CrossRef CAS.
- M. Wang, Q. Tan, Q. Huang, L. Liu, J. F. Chiang and J. Li, J. Hazard. Mater., 2021, 413, 125222 CrossRef CAS PubMed.
- W. Wang, G. Hu, Z. Peng, K. Du, Y. Cao and J. Duan, Ceram. Int., 2018, 44(2), 1425–1431 CrossRef CAS.
- Q. Meng, J. Duan, Y. Zhang and P. Dong, J. Ind. Eng. Chem., 2019, 80, 633–639 CrossRef CAS.
- M.-M. Wang, C.-C. Zhang and F.-S. Zhang, Waste Manage., 2016, 51, 239–244 CrossRef CAS PubMed.
- O. Dolotko, N. Gehrke, T. Malliaridou, R. Sieweck, L. Herrmann, B. Hunzinger, M. Knapp and H. Ehrenberg, Commun. Chem., 2023, 6, 49–56 CrossRef CAS PubMed.
- J. Ren, Z. Zhang, Z. Chen, L. Wan, K. Shi, X. Zeng, J. Li and Q. Liu, Green Chem., 2023, 25, 9795–9804 RSC.
- F. Larouche, F. Voisard, K. Amouzegar, G. Houlachi, P. Bouchard, A. Vijh and G. P. Demopoulos, Ind. Eng. Chem. Res., 2023, 62, 903–915 CrossRef CAS.
- A. S. Anderson, B. Kalska, L. Häggström and J. O. Thomas, Solid State Ionics, 2000, 130, 41–52 CrossRef.
- A. Yamada, S. C. Chung and K. Hinokuma, J. Electrochem. Soc., 2001, 148, A224–A229 CrossRef CAS.
- M. Maccario, L. Croguennec, A. Wattiaux, E. Suard, F. Le Cras and C. Delmas, Solid State Ionics, 2008, 179, 2020–2026 CrossRef CAS.
- B. Hannoyer, A. A. M. Prince, M. Jean, R. S. Liu and G. X. Wang, Hyperfine Interact., 2007, 167, 767–772 CrossRef.
- F. Menil, J. Phys. Chem. Solids, 1985, 46, 763–789 CrossRef CAS.
- C. Piña, H. Arriola and N. Nava, J. Phys.: Conf. Ser., 2010, 217, 012037–012041 CrossRef.
- J. M. Millet, C. Virely, M. Forissier, E. Bussiere and J. C. Vedrine, Hyperfine Interact., 1989, 46, 619–628 CrossRef.
- R. Wäppling, L. Häggström, S. Rundqvist and E. Karlsson, J. Solid State Chem., 1971, 3, 276–292 CrossRef.
- V. V. Nemoshkalenko, N. A. Tomashevskii, V. B. Chernogorenko and L. Ya. Solomatina, Poroshk. Metall., 1982, 1, 61–63 Search PubMed.
- R. E. Bailey and J. F. Duncan, Inorg. Chem., 1967, 6, 1444–1447 CrossRef CAS.
- S. Simon and G. J. G. Gluth, Open Ceram., 2021, 5, 100060–100066 CrossRef CAS.
- S. J. Beckerman, R. B. Ford and M. T. Nemeth, Powder Diffr., 1996, 11, 312–317 CrossRef CAS.
- C. A. Drewien, D. R. Tallant and M. O. Eatough, J. Mater. Sci., 1996, 31, 4321–4325 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00014e |
| This journal is © The Royal Society of Chemistry 2024 |