
Autonomous generation of single photon emitting materials
Robert
Tempke
and
Terence
Musho
 *
*
Department of Mechanical, Materials and Aerospace Engineering, West Virginia University, P.O. Box 6106, Morgantown, WV, USA. E-mail: tdmusho@mail.wvu.edu
First published on 19th April 2024
Abstract
The utilization of machine learning in Materials Science underscores the critical importance of the quality and quantity of data in training models effectively. Unlike fields such as image processing and natural language processing, there is limited availability of atomistic datasets, leading to biases in training data. Particularly in the domain of materials discovery, there exists an issue of continuity in atomistic datasets. Experimental data sourced from literature and patents is usually only available for favorable data, resulting in bias in the training dataset. This study focuses on developing a SMILES-based model for generating synthetic datasets of quantum materials using a variational autoencoder. This study centers on the generation of a synthetic dataset of quantum materials specifically for quantum sensing applications, with a focus on two-level quantum molecules that exhibit a dipole blockade. The proposed technique offers an improved sampling algorithm by incorporating newly generated data into the sampling algorithm to create a more normally distributed dataset. Through this technique, the study was able to generate over 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 candidate quantum materials from a small dataset of only 8000 materials. The generated dataset identified several iodine-containing molecules as promising single photon emitting materials for potential quantum sensing applications.
000000 candidate quantum materials from a small dataset of only 8000 materials. The generated dataset identified several iodine-containing molecules as promising single photon emitting materials for potential quantum sensing applications.
1. Introduction
In recent years, the use of machine learning techniques in chemistry has become increasingly prevalent. Despite this, it has become apparent that many available chemical and materials science datasets suffer from bias.1–4 This is primarily because these datasets are often composed of a collection of patents and research articles that exist on the internet, rather than representing a continuous material space.4–6 In response to this issue, the present study seeks to leverage the predictive abilities of deep learning to generate a chemically diverse dataset that is less biased and more robust than those currently available.To achieve this goal, this study employs a deep learning technique known as a variational autoencoder (VAE), which is capable of synthesizing chemical species with specific chemical properties.7–10 The VAE forms a custom chemical compression intelligence that provides efficient generation of new specific chemical species by sampling the latent space of the VAE, which can be thought of as a representation of compressed chemical information.11–13 By training the neural network to learn the chemical and structural similarities of species with specific physical properties, we enable it to identify patterns in a higher dimensionality. See Fig. 1 for an illustration of this concept in application to this research.
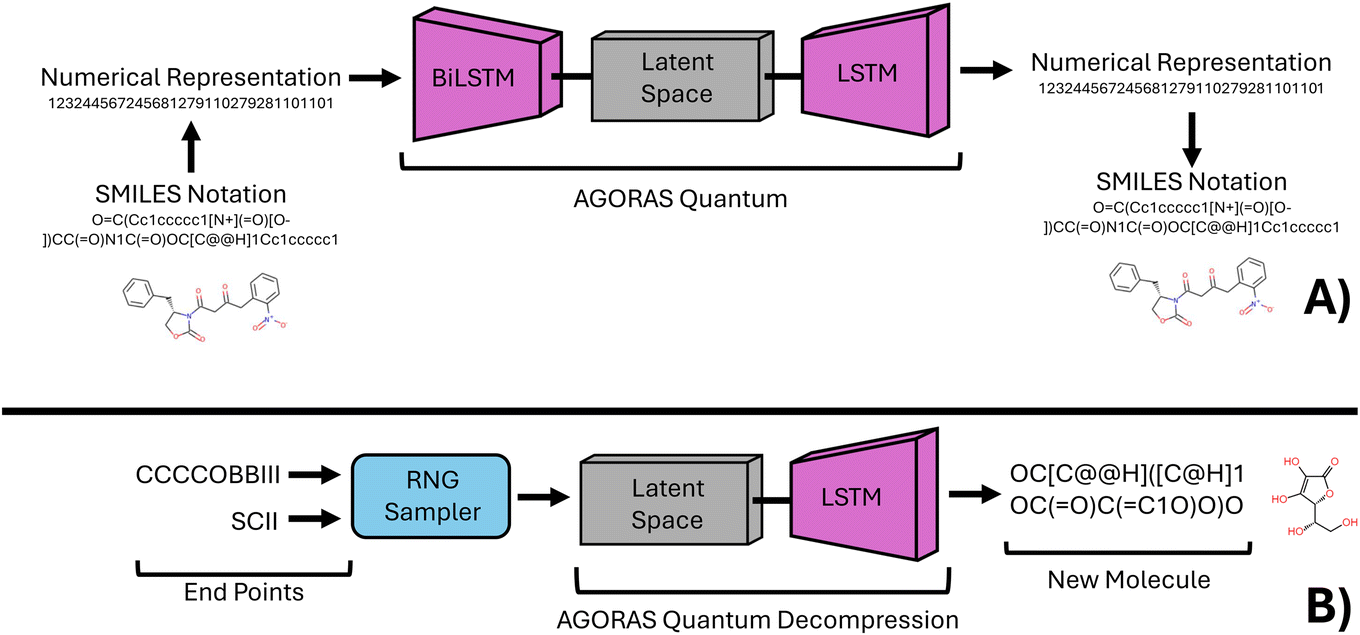 | ||
| Fig. 1 Subfigure A (top half) is an illustration of the AGoRaS network illustrating how a chemical species is encoded and used as training data for the network. Chemical database information is compressed and decompressed to form a high-dimensional latent space. Subfigure B (bottom half) is an illustration of how the trained latent space can be sampled to generate new single-photon emitting materials. | ||
This research is focused on the generation of candidate quantum materials, specifically single-photon source (SPS) materials, also referred to as UV/vis materials.14–18 These SPS materials rely on a two-level system of electronic states with the added complexity a secondary interaction to form a resonant behavior. While it is beyond the scope of this paper to discuss all of the potential quantum material frameworks the focus of this study is on the discovery of a material that exhibit SPS behavior with a strong dipole interaction. The creation of a chemically diverse dataset is critical to the development and training the furture of accurate machine learning algorithms. In the context of machine learning, the phrase “garbage in, garbage out” highlights the importance of high-quality data.19,20 The machine learning algorithm must be trained on a range of inputs and outputs, as the space is continuous, and it must learn everything to know everything.
Several studies have emphasized the issue of bias in experimental design and data collection, which ultimately leads to skewed and unreliable data. Griffiths et al. investigated biases in the natural sciences, focusing on the impact of data splitting, noisy datasets, and contextual variables on the outcome of experiments.21 Similarly, Kovács et al. highlighted the direct effects that biased and unbiased datasets can have on the quality of machine learning outputs.22 Glavatskikh et al. demonstrated how the lack of diversity in data limits the machine learning's potential to predict.23
The creation of an experimentally unbiased, continuous dataset is a costly and challenging task. However, our use of deep learning techniques, specifically VAEs, shows promise in generating a chemically diverse dataset with specific chemical properties.8,24,25 Through our research, we aim to contribute to the development of more accurate and reliable datesets to train machine learning algorithms in chemistry and materials science.
In the field of SPS materials, it is common to encounter incomplete or inaccurate data in the literature, making it unsuitable for machine learning algorithms. Zakutayev et al. have highlighted the significance of having sufficiently large and diverse datasets for the training of advanced machine learning algorithms in materials science26 emphasizes the importance of having a robust and extensive dataset to develop machine learning algorithms that can predict the structure, stability, and properties of various materials. Furthermore, it illustrates that existing machine learning algorithms can be quickly and easily adopted to address material science problems, provided there is a suitable dataset for training purposes.5,27,28
In the context of UV/vis research in the context of SPS materials, the research of Beard et al. stands out for their comprehensive collection of available materials and corresponding relevant calculations.29 The authors conducted an extensive search of over 400000 scientific documents to extract a database of just over 8000 unique compounds. Despite the use of state-of-the-art tools such as ChemDataExtractor, the process of creating a database for quantum materials is challenging. This is due to the wide variety of formatting among different scientific journals, discrepancies within the tools being used, and the lack of a standard set of ground truth rules for representing materials using the SMILES notation. Nevertheless, the database created by Beard et al. is currently the most complete UV/vis material dataset available.
SPS materials have been proposed for a variety of quantum applications, such as quantum communication, quantum computing, quantum information, and quantum precious metrology.16,17,30 SPS materials have found demonstrated application to date in quantum communication, remote sensing, and dipole gates.2,31,32 These materials exhibit a two-level system behavior, where a resonance is formed between a ground state and a excited state or between two excited states. In the application of quantum sensing or computing, the defining metric is the coherence time or the lifetime of the resonance. Often this involves several aspects of the material, which are deeply rooted in the atomic coordination on the atoms that make up the molecules. One of the targeted metrics in these organic SPS based quantum sensing materials, is discovering a material that exhibits both a strong photo absorption strength and a strong dipole interaction. This type of molecule, when interacting with neighboring molecules, will exhibit a quantum resonance in which a single photon can exist on only one of the molecules state at a time. The dipole interaction will shift or change the neighboring molecule's excited state energy level. This single photon–dipole interaction will give rise to a resonance with a precise energy that can be exploited for quantum sensing and other quantum applications. This two level system can be described by a Hamiltonian, which is beyond the scope of this research but will be required for complete understanding and control of this quantum system.
One potential application that seems more near term is remote sensing, which has gained significant attention in recent years as quantum materials technology has improved. These remote sensing methods have been used to monitor contaminants in water, air quality trends, dissolved nutrients in surface water, and many other advanced techniques.33–35 For example, Spangenberg et al. demonstrated how quantum materials could be combined to detect relative concentrations of mixtures within water in real-time.34 Fei et al. demonstrated that the right combination of machine learning algorithms and SPS material, the monitoring of groundwater contamination could be achieved.35 However, the limited dataset of 1665 materials used in Fei et al.'s work highlights the need for much larger datasets. Moreover, Mamede et al. further demonstrated the potential of machine learning to be applied with quantum materials by focusing on finding the UV/vis absorption spectrum of organic molecules using fingerprints generated from 2D chemical structures. Their work yielded a sample size of approximately 75000 molecules using only information about the chemical structures.36
Recent studies by De Leonardis et al. and Richter et al. have demonstrated the potential of quantum materials in overcoming phase-matching challenges in remote sensing applications.31,37 These recent research studies demonstrate the growing demand for new quantum materials application that can advance different fields. This highlights the need for more extensive datasets to facilitate machine learning algorithms in the discovery of new SPS materials.
The hypothesized approach in this study is to create a material compression intelligence via the latent space representation of an existing SPS materials experimental database, using a VAE with a similar structure as AGoRaS.7 To differentiate this network from this previous study we will refer to the network as AGoRaS-Quantum. The latent representation of the materials can be sampled at various points to generate new SPS materials with desirable characteristics. The latent space can be viewed as a representation of the compressed structural and chemical information inherent in the species used to train the network. By populating the latent space with structurally and chemically similar SPS materials, the network can learn the underlying similarities between the materials, resulting in the generation of new materials that share the desired characteristics.38
The VAE's ability to represent data in n-dimensions and fit input nodes to probabilistic distributions, typically multivariate Gaussian distributions, offers a significant advantage over traditional methods of data representation.8,12,13,39 The methodology proposed in this study demonstrates the practicality and flexibility of the AGoRaS network in creating SPS materials for quantum applications, with the generation of single materials replacing the balanced chemical reactions generated in the original AGoRaS study.7
The generation of SPS materials has to be rigorously validated, following a similar methodology to the one outlined by Beard et al., using a workflow that facilitates data collection, network training, network testing, material generation, and material testing.29 The modular nature of this workflow enhances code quality and robustness while also enabling non-data scientists to employ the methodology with ease, thereby expanding the network's utility to researchers in different fields who lack data science expertise. This methodology has been successfully utilized in other generative networks such as ChatGPT, enabling non-data scientists to generate text with background knowledge beyond their expertise. The developed network aims to have the same utility, but more narrow in application, with the focus of generating of SMILES representation of molecules.
2. Methodology
2.1. Processing the database
In this study, the chemical species used to train the AGoRaS-Quantum network were obtained from the dataset created by Beard et al. The data manipulation and network were written in Python. An illustration of the workflow can be found in the original AGoRaS publication for chemical reaction generation.7 The dataset, which contains approximately 8000 different species, was downloaded in JSON format.29 This is relatively small dataset compared to the generated data, which will be in the millions. To provide a scale, the input data is approximately 0.08% the size of the output generated. To ensure the quality and consistency of the dataset, each species was validated using RDKit to verify that its provided SMILES string matched its IUPAC name.40–42 RDKit is an open-source cheminformatic software that has a series of tools for checking the validity of SMILES strings. It is necessary to check the SMILES notation to avoid training on invalid SMILES strings. There are a set of standards that can be checked by RDKit and an error code is returned if the SMILES is invalid. All species were then read into a Pandas dataframe with relevant information, including SMILES string, excitation wavelength, intensities, and dipole moments.To simplify the network's predictions and improve reproducibility, the AGoRaS-Quantum network focused solely on predicting SMILES species and not on their associated properties, such as dipole moments and excitation wavelength.43 This decision allowed the network to focus on learning the underlying physical and chemical structural patterns rather than extending the prediction to properties, which could be calculated afterwards using quantum chemistry tools such as density functional theory (DFT).43–45 To generate new species, the network continued to use character-level embedding due to its advantages over word-level embedding in natural language processing. This allowed the generative network to use the information learned during training to generate new species based on the universal alphabet created from all the species in the dataset.46,47
The use of molecule embedding can improve the predictive power of machine learning models by formulating inputs into sequence embeddings.8,48–50 In this study, TensorFlow's built-in embedding techniques were used to create embeddings based on the universal alphabet created from the chemical species.51 This approach was inspired by Gaspar et al.'s work, which demonstrated that molecule embedding can be similar to NLP embeddings.49 By using sequence embeddings, the network can capture more of the structural and chemical similarities between the species, allowing it to generate new species with similar properties. The use of embedding techniques and a universal alphabet enables the AGoRaS-Quantum network to accurately represent chemical species and generate new species, making it a useful tool for materials science research.
2.2. AGoRaS-Quantum structure for quantum materials
The AGoRaS-Quantum algorithm is designed to generate new chemical species based on a vector representation of the longest SMILES string in the training dataset. The vector is passed through an Embedding layer in TensorFlow, which projects the input into a higher dimensionality space. This is a critical step as the intrinsic values of the numeric values are removed in the higher-dimensional space. The projected vectors are then passed through a bidirectional LSTM layer, with a recurrent dropout of 0.2.52,53 The mean and log variance of the output are used to sample the solution space using a sampling function.The sampled solution space is then decoded using a RepeatVector layer wrapped around the output of the latent space, which turns the data into a tensor vector that an LSTM layer can read. The LSTM layer's output is projected into a vector of length n, and this projection is used to calculate the loss of the network. AGoRaS-Quantum uses a sequence-to-sequence style loss function typical of variation autoencoders, and the kl loss is used as the monitoring metric during training. The network was trained for 500 epochs using a batch size of 25, an embedding dimensionality of 500, and a latent dimensionality of 350. The kl weight used was 0.1, and the activation function was SoftMax. The optimizer function was Adam, and the learning rate was set at 1 × 10−5. This structure closely mimics that of the original AGORAS network for chemical reaction prediction, except for the input vector's length.
The model takes in a vector representation of the longest SMILES string in the training dataset, which is then projected into a higher dimensionality space. The projected vectors are passed through a bidirectional LSTM layer with a recurrent dropout to extract the mean and log variance, which are then used to sample the solution space. A sequence-to-sequence style loss function is used to calculate the loss of the network, with the kl loss serving as the monitoring metric during training. The model's performance is governed by several hyperparameters, including batch size, embedding and latent dimensionality, kl weight, activation function, optimizer function, and learning rate. The AGoRaS-Quantum algorithm's combination of deep learning techniques and chemical domain knowledge allows it to generate new chemical species accurately and efficiently.
2.3. Training AGORAS for quantum materials
Once the chemical data had been pre-processed and converted into a numerical format suitable for neural network architecture, it was divided into three separate datasets: the training set, the validation set, and the test set. The training set comprised 70 percent of the available data, the validation set comprised 20 percent, and the remaining 10 percent was used for the test set. A k-folding approach was used to cross-validate the data over the dataset. Although AGoRaS-Quantum is a generative model, it can be validated using traditional methods. The VAE used in this study was evaluated by its ability to encode the validation sets, and decode it back to the original string construction with no loss of information.During the training process, a sequence-to-sequence loss function was employed to score the reconstructed string versus the original string. This approach enabled the validation of the VAE's ability to reconstruct the chemical equations with zero loss of information, which is indicative of a stable latent space. Given the small size of the data used in this study, it was essential to validate the stability of the latent space as much as possible. After it had been demonstrated that the network could reconstruct the test data, the remaining 90 percent of the data were also tested to further validate the stability of the latent space. Although the network should be able to reconstruct all the data used in training, this additional test served as a further validation of the latent space's stability.
Overall, validating the stability of the latent space is critical for this study. By demonstrating that the VAE can encode and decode the original chemical equations with zero loss of information, it is possible to confirm that the latent space is stable. This validation is especially important given the small size of the data used in this study.
2.4. Autonomously sample the latent representation for quantum materials
After a neural network has been trained, it is possible to create a sampler that can interface with the latent representation using real species representation. This can be achieved by selecting two materials and using them to access the latent space. The sampler then returns a new species located at some equidistant point between the two selected materials. Another way to sample the latent representation is to randomly select a species and have the decoder part of the network construct new species based on the equidistant points between the two materials. This directed sampling approach has a significant advantage over continuous sampling of the latent space. It enables researchers to focus on areas of the latent space where the decoded species possess characteristics of interest.Due to the probabilistic nature of the latent representation, an almost unlimited number of sample points can be taken to generate new species. However, this approach has diminishing returns as there are only a limited number of chemically feasible species that can be generated. Nevertheless, the directed sampling approach can still be a powerful tool for researchers to generate new species with specific characteristics of interest.
2.5. Validating generated species
The methodology for determining the chemical validity of species was extremely like that of the data cleaning process. The first step was to check duplicate species were eliminated. The second step was to check each species for chemical validity using RDKit, where any physically or chemically unstable species should be rejected by the software.41 This is a common practice when using a neural network with generated SMILES species. The ability of the network to generate valid chemical species helps to further prove that the latent space is stable and representative of the original dataset.The performance of the AGoRaS-Quantum networks is determine by comparing the number of generated species to the number of unique species. It was determined that 10% of the generated species are unique on the first iteration. That means nearly 10 million species need to generate in order to discover 1 million unique species on the first interation. Success was around 10% at first because the latent space was limited, since we started with only 8000 species. As the latent space was sampled more and more stable species were added to the list of stable species, the future predictions became better. This increased the stability of the sampling. In the end, approximately 45% of generated species were stable per interation. Approximately 5% of those were repeats of previously generated species. This theoretically would continue to improve as we sampled more of the latent space as values can be sampled between known species in latent space.
2.6. Preform semi-empirical methods on generated species
Using the SMILES notion provided in the generated datasets from AGoRaS-Quantum, a custom Pipeline Pilot protocol was written that would take the SMILES entry and convert it to an atomistic description. Once the data was converted to an atomistic description, a semi-empirical quantum chemistry calculation was conducted. Pipeline Pilot is a high-throughput framework capable of manipulating and analyzing large quantities of scientific data through open source and commercial quantum chemistry tools. This framework is developed by Dassault Systems.54,55The semi-empirical quantum chemistry model that was implemented in the automated Pipeline Pilot script was based on the Dassault Systems' Materials Studio VAMP software package.56 Geometry optimization was conducted with a diatomic differential overlap (NDDO) and PM6 Hamiltonian, with auto multiplicity, and a spin state starting with the most rigorous; an unrestricted Hartree–Fock (UHF), restricted Hartree–Fock (RHF), or annihilated unrestricted Hartree–Fock (A-UHF).57,58 Several spin states were tested based on convergence. A Paulay/IIS convergence scheme was selected with a convergence energy tolerance of 2 × 10−4. The thermodynamics information and total dipole moment were output from VAMP output. Thermodynamic information is based on the optimized atomistic description, semiemperical molecular information, and electronic and phonon calculation.
The Pipeline Pilot script conducted a series of data preparation steps prior to the semi-empirical calculation. After data was read using SMILES format the SMILES was checked for consistency, followed by making and cleaning of the molecule. The cleaning steps included centering the molecule, adding hydrogen, and conducting a quick empirical elastic relaxation of the structure to refine the initial geometry. The structure was provided to a programmed series of VAMP calculations starting with the most rigorous spin state and relaxing the spin state in the case of failure and retrying the calculation. In the event that the semi-empirical calculation fails for each spin state, the molecule was assumed unstable and removed from the dataset.
The semi-empirical calculation was chosen because it throughput and robustness of the calculation. Compared to all-electron density functional theory calculations, the semi-empirical calculations take between one to two orders of magnitude less time to provide a prediction. This is critical to this approach where we are aiming to predict the properties of hundreds of thousands of molecules. The reason to use the semi-empirical approach was to provide a quick estimate of the molecule stability and properties, which could be later investigated using higher fidelity models after the initial screening.
The semi-empirical model did provide an estimate of several SPS properties of the molecules, not limited to the formation energy, dipoles, and UV/vis properties. VAMP use the calculated molecular wavefunctions to derive the dipole moment and associated excited state properties. This is done using the LCAO method of molecular orbitals rather than the standard MNDO Hamiltonian calculation.56,59 VAMP is also able to calculate accurate dipole moments using the Natural Atomic Orbital-Point Charge model for molecular electrostatic properties. All of the structures generated along with their properties predicted with the semi-empirical model can be found on the NOMAD materials science database, see Data Availability Section.
2.7. Compare training data with generated data
The semi-empirical calculations for the UV/vis spectra, dipole moments, and total energy is compared for both the original dataset and the generated data to quantify the diversity of the generated data. To determine if our generated species offer a diverse generation, these three properties were compared and contrasted. In addition, this study also compared the number of atoms for both the generated materials and the original materials. Due to the disparity of the dataset sizes, a normalized percentage comparison between the larger generated dataset and the original dataset containing approximately 8000 materials, was taken into account. By normalizing the dataset size between the two, it was possible to compare the total number of atoms, dipole moments, UV/vis spectra, and total energy directly between the datasets. A histogram was used to get a view of the distribution of values for each dataset. A histogram for the dipole moments with a comparison between the original species (blue) and the generated species (red) is provided in Fig. 2.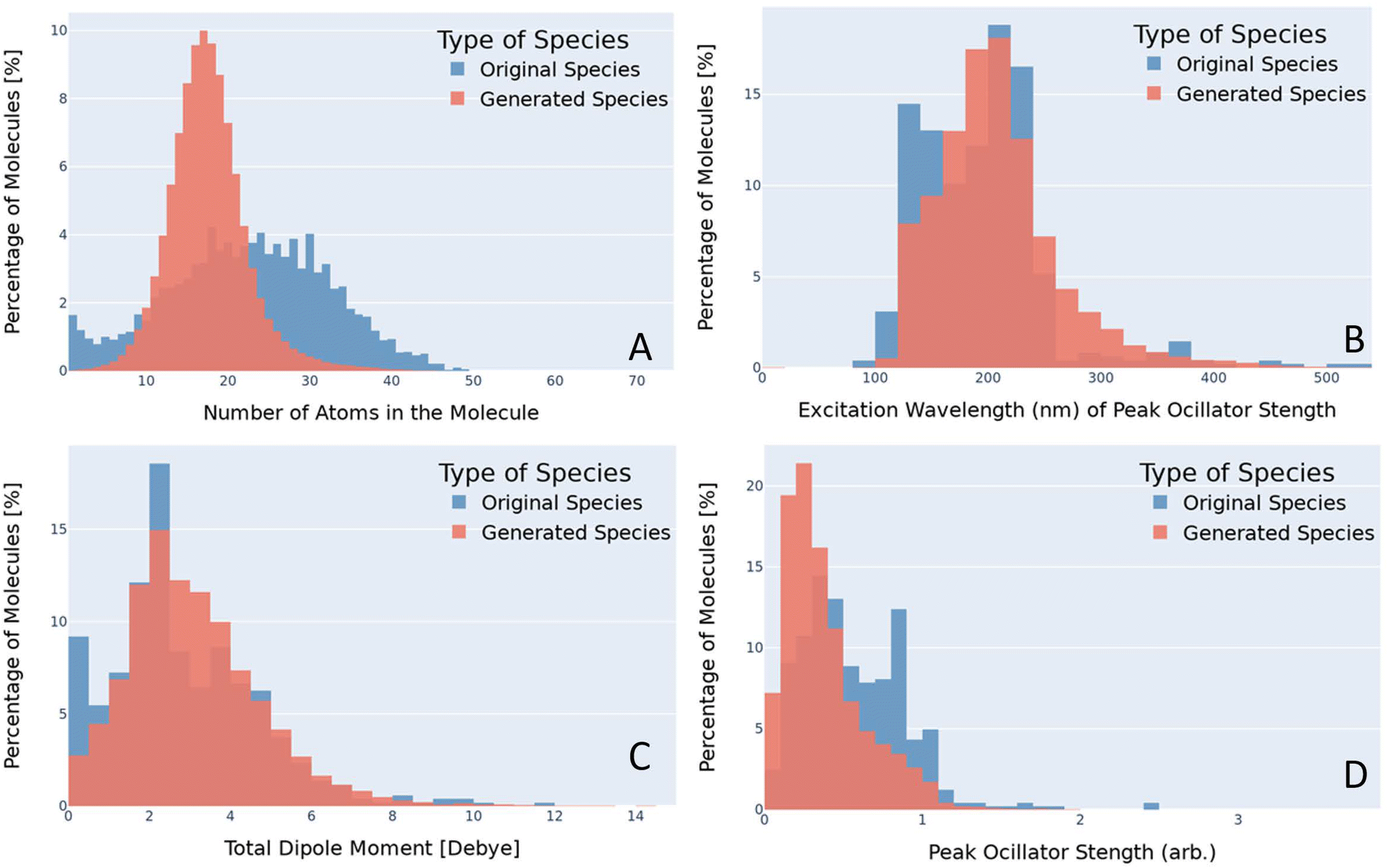 | ||
| Fig. 2 Histogram comparing the number atoms (A), excitation wavelength (B), the total dipole moment (C), and the peak oscillator strength (D). The blue bins are the training (original) species and the red bins are the generated species from the VAE generated in this study. The goal is to identify small molecules with an excitation wavelength near 500 nm, a strong dipole moment, and strong oscillator strength. | ||
2.8. Identify promising material
Once the dataset has been proven to contain diverse, realistic values, it was possible to sort the data for promising SPS materials. The data is sorted by the three criteria discussed in the previous sections. For this study we have identified two materials with strong dipoles and a single frequency characteristics in the 500–600 nm range with a strong peak compared to other peaks in the UV/vis spectrum. These criteria are selected due to their direct interest to researchers in the field of quantum sensing, where SPS materials that exhibit a strong dipole moments with a strong optical absorption peak are opportunistic materials for quantum sensors.2.9. Validate material with TDDFT
Based on the seven candidate materials outlined in the table in Fig. 3, the remainder of this study will focus on the two most promising, which are FIII and C[I][I]II. These two materials warrant experimental consideration, however, before this, it deemed necessary to put these molecules through more rigorous quantum chemistry computational validation. This involves conducting a time-dependent density functional theory (TDDFT) calculation. The higher accuracy of the TDDFT method than that of the semi-empirical method, provides greater confidence in the predicted properties. This higher level of validation also allows for experimentation with these molecules to help identify what is the molecular origin or mechanism of these strong optical absorption peaks and associated oscillator strength. The molecule's emission spectra can be investigated by adding or removing elements to molecule to investigate the response. Moreover, the TDDFT approach allows the researcher to visualize the wavefunctions and local density of states as shown in Fig. 4. In this figure for FIII (A and B) and C[I][I]II (C and D), the ground state density of states (A and C) and the excited states (B and D) can are shown. The yellow is associated with the spin-up states and the blue isosurfaces are associated with the spin-down states. An important aspect is that the overlap of the wavefunction is similar to provide good coherence of the excited electron. Table 1 provides an outline of the most promising excited state transitions for both of these molecules. As can be associated between this Table 1 and Fig. 4, a strong oscillator strength (f-osc) is desirable with a large overlap of the wavefunctions. While these may not be the best optical absorbers within the 500–600 nm range, this demonstrates that there is potential in using the AGoRaS-Quantum network to find SPS materials. As seen here for these two molecules the C[I][I]II molecule provides slightly better oscillator strength when compared to FIII, at the expense of a larger molecule. While it is not the scope of this research to discover the best performing SPS materials, it is interesting to point out that both these materials are iodine containing molecules and there were only a handful of iodine containing molecules in the original dataset. | ||
| Fig. 3 Example of the ability to search the generated solution space for molecules of potential interest. The inset table within the figure outline seven molecules in the 500–600 nm wavelengths with an oscillator strength (pronounced optical peak) above 2.0. These seven correspond to the black box in the histogram. Note, most materials exhibit low excitation wavelength and oscillator strength. | ||
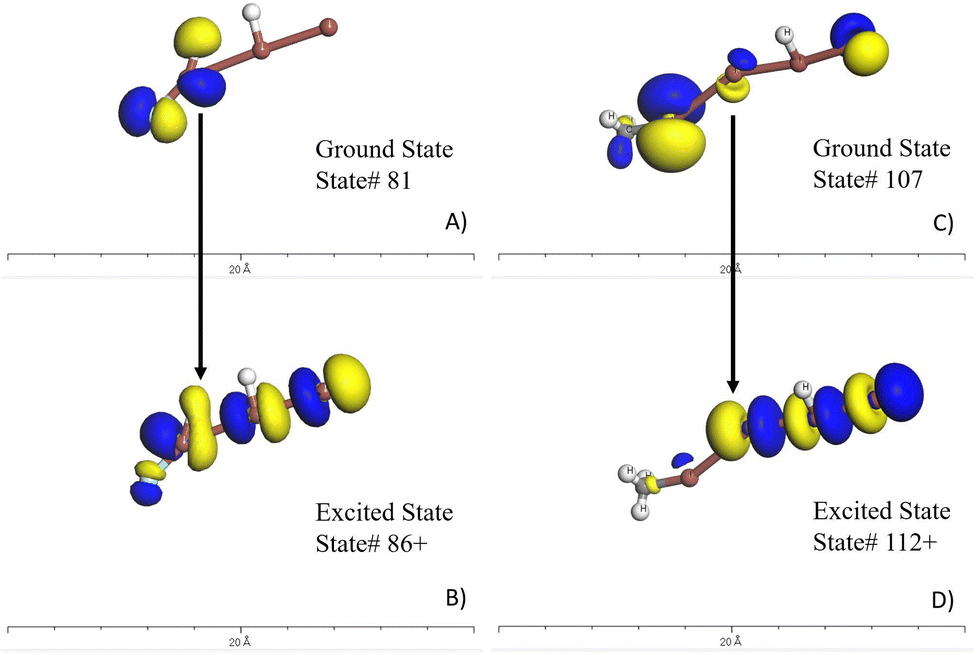 | ||
| Fig. 4 TDDFT results for the FIII (H2FI3) molecule (A and B) and C[I][I]II (C and D). Subfigure A and C illustrate the ground states local density of states surfaces. The yellow is associated with spin-up and blue is associated with spin-down electrons. Subfigure B and D are the excited states local density of states. Comparing the ground state to the exited states provide spatial information of the electron transition outlined in Table 1. It is desirable to have a high overlap of these states to avoid decoherence of the states. | ||
| From | To | TD-ex [eV] | TD-ex [nm] | f-osc | Overlap |
|---|---|---|---|---|---|
| 85 | 86+ | 1.31 | 950 | 0.000026 | 0.50 |
| 83 | 86− | 1.46 | 851 | 0.000110 | 0.87 |
| 81 | 86+ | 2.17 | 571 | 0.001437 | 0.39 |
| 80 | 86+ | 2.76 | 450 | 0.000796 | 0.44 |
| From | To | TD-ex [eV] | TD-ex [nm] | f-osc | Overlap |
|---|---|---|---|---|---|
| 111 | 112+ | 1.83 | 676 | 0.001886 | 0.61 |
| 109 | 112− | 1.99 | 622 | 0.000295 | 0.59 |
| 107 | 112+ | 2.25 | 550 | 0.082783 | 0.56 |
| 106 | 112+ | 2.32 | 535 | 0.003263 | 0.46 |
3. Results
After the SMILES strings had been embedded and the VAE was trained, the latent space was sampled to generate new chemical species. The approach of sampling the latent is a unique approach that saw success in a previous study7 where bias in the training dataset was corrected. Sampling was stopped once approximately a million valid species had been disseminated. This arbitrary stopping criterion was selected because it was greater than 10 times the size of the original dataset. All species were run through the data consistency checker to check their uniqueness and stability. The new species contained species with atoms ranging from 1 to 74 with an average of 18 atoms while the original dataset had molecules with atoms between 1 and 49 with an average of 22. A comparison of these distributions can be seen in Fig. 2A. The number of atoms within the molecule was calculated using RDKit. It should be noted that the ability for AGoRaS-Quantum to predict larger molecules than trained on demonstrates the benefits of this approach over other approaches such as retrosynthesizing. We can see statistically most of the larger molecules are outliers when compared to the rest of the molecules. It is hypothesized that if the latent space were to continue to be sampled, especially if the large molecule species were targeted sampling the area could produce many large molecules.The distribution of these atom counts can be seen in Fig. 2, which allows for a more in-depth analysis. Again, the training dataset is pictured in blue in the background with the generated dataset in the foreground in red. The y-axis in all the plots is the percentage of all molecules within that bin and the x-axis is the associated chemical property. It can be seen from Fig. 2 that the generated species and original species share an approximately normal distribution with two main differences. The first is the high percentage of species in the first two bins and the second is that the distributions are not centered around the same number of atoms. Both of these differences are due to the original dataset containing single-element molecules. Since they are already included in the original species and AGoRaS-Quantum cannot come up with new elements, it is impossible for it to share that feature. However, due to the existence of these small molecules, it biased the network into creating species that were on average smaller than the median number of atoms for the original dataset.
Of course, for these molecules to be useful as either a dataset for machine learning or as a database for potential experimentalists, it had to be proven that these generated species shared the same properties as the original species. It was decided to look at molecules containing no more than 10 atoms. This was due to the computational complexity and cost associated with the semi-empirical calculations. The criteria that were deemed the most important to compare between the datasets were those that can aid in identifying if a material is a SPS material. This criteria included whether or not the calculated emission spectra of the molecule exhibit a single strong peak (high oscillator strength) at an optical wavelength.
Using semi-empirical calculations it was possible to calculate the wavelength and emission strength of excited electrons. It is important to note that the intensity of the photon being emitted is difficult to compare between molecules but can be compared between other peaks in the spectrum for a given material to provide a normalized intensity. This is due to the Franck–Condon Principle, which explains the relative intensities of vibronic transitions. These intensities are the relation between the probability of a vibrational transition to the overlap of the vibrational wave functions. These calculations at each energy level led to the calculated emission spectra that will be used as validation of these materials. All electronic levels of each molecular species were determined at the standard state using the semi-empirical computational technique described earlier. Other properties of interest, but secondary to the peak strength, were the total dipole moment, vibrational spectrum, and vibrational strength.
Once it was confirmed that the generated data exhibited single peak behavior it was necessary to perform further analysis to confirm that the generated data shared similar value ranges as the original data. For this overlapping histograms were determined to be an ideal way to show that the generated data had properties similar to that of the real data. Fig. 2D illustrates a histogram of the original and generated species’ peak oscillator strength. It can be seen that both the generated and original species have a semi-normal distribution with a slightly left skew to the values. However, it appears that on average the generated materials have a slightly weaker peak oscillator strength.
In this type of behavior, both the lower average peak strength and the identical distribution of values are expected due to the bias in the training data. Since the network uses all of the original data as a starting point for sampling the latent space it will always return data of a similar distribution. This problem could easily be overcome by sampling only data from the underrepresented regions until a uniform distribution was created.
The high percentage of molecules being generated that produce weaker strengths is also a byproduct of the inherited bias. Due to the training data being sourced from experimental results, only the best material i.e., the strongest emitters are reported. This leaves a lot of materials for AGoRaS-Quantum to be able to generate that still meet the chemical and physical requirements but do not produce as strong of a peak. Simply put the area of the latent space that generates strong peak materials is crowded, while the rest of the latent space is sparsely populated. As shown in study two, however, if the latent space were to continue to be sampled until we reached 100 times the number of generated materials to the original material. Then the generated distributions would be exactly that of the original materials.
Another important aspect of these types of materials is at which frequency these peaks occur. Fig. 2B depicts the excitation wavelength at which the molecule's peak oscillator strength occurs for the original and generated species. Once again it can immediately be seen that the original and generated materials follow a similar distribution of semi-normal with a left skew. Like with the previous figure, this could be corrected with a more directed sampling methodology. Another factor in the similarity of these distributions is that, unlike the other histograms that have been shown in this study, their values could theoretically be anything. The excitation wavelengths are calculated between 100 and 1400 nm, which helps to enforce an equal distribution of values within that range. An interesting find from Fig. 2B is that the original data has a disjointed distribution of values when the excitation wavelength is greater than 250 nm. The generated data shows a much more normal distribution as the values tail out to 1200 nm. This helps to suggest that even if the training data has a disjointed distribution that a VAE will be able to generate a smooth distribution of the data.
The total dipole moments of the real and generated materials were also calculated, which was important in selecting a molecule that could potentially be operated in the dipole blockade quantum sensing application. The dipole is based on the partial charge and positions of the atoms. The overlaid histogram for the total dipole moments of the original and generated species can be seen in Fig. 2C. As we have seen previously it is a semi-normal distribution with a left skew. It should be noted that there is a the high percentage of dipole values around 0 Debye for the original species. This is due to the original dataset containing single atom species which would have zero dipole moment. It is interesting to note, in Fig. 2B, where the original data has a bit of an uneven distribution, however, the generated data is a single peak distribution. The network cannot generate any more single element materials with new elements from the periodic table so the zero dipole materials are limited.
The filling in of the generated data represents an extremely important aspect of VAEs and especially of the AGoRaS-Quantum network. This stems from the ability of the network to map the latent probabilistic solution space of these materials to the real space. By sampling the latent space, the network is able to fill in the gaps between points and discovery new spaces. It is the aim to demonstrate that the new materials are filling in the solution space and therefore, effectively removing the bias. To visualize this a t-Distributed Stochastic Neighbor Embedding (t-SNE) was generated as shown in Fig. 5. The t-SNE algorithm is used primarily to be able to explore and visualize high-dimensional data such as text. At its most simple level, it allows a user to get an understanding of how data is arranged in high-dimensional space. The algorithm accomplishes this through an unsupervised learning method of stochastic neighbor embedding to give high-dimensional data a single point on a two-dimensional grid.
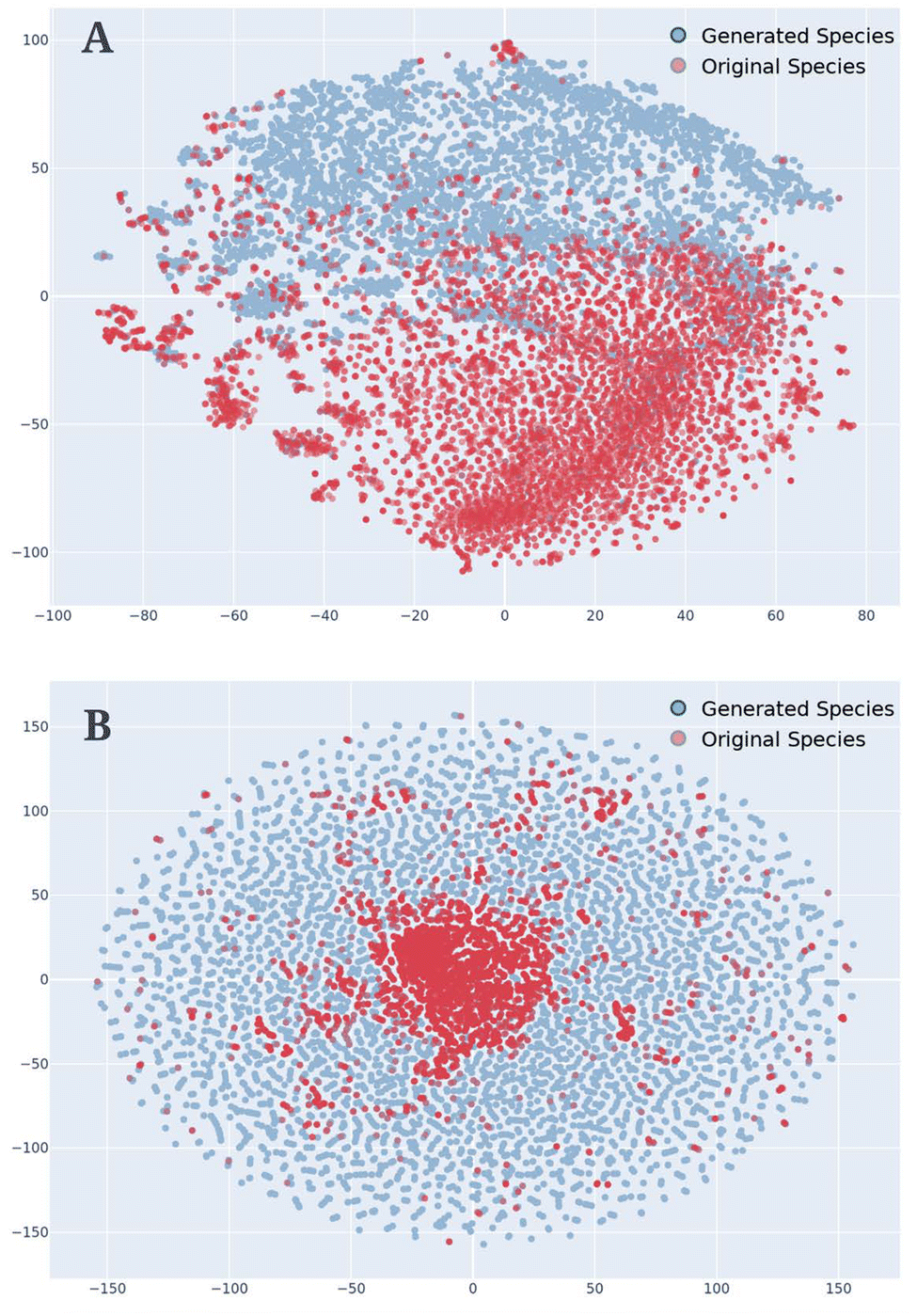 | ||
| Fig. 5 t-SNE plot of the training dataset and the generated dataset with the oscillator strength representative of the size of the points. Subfigure A has all of the 8000 original species and a randomly sampled 8000 generated species. Subfigure B has all of the 8000 original species and 80000 randomly sampled generated species. | ||
For this t-SNE algorithm, the only input was the SMILES representations of the molecules embedded as numbers just as in the original training for the AGoRaS-Quantum network. The blue circles represent the generated data set and the red circles represent the training data. Fig. 5A has all of the original data 8000 species while only showing a randomly selected 8000 of the generated species. Meanwhile, Fig. 5B also illustrates the 8000 generated species but has 80000 randomly selected generated species. This is done to illustrate how as we sample more species we can fill in the latent space. It can be seen from Fig. 5 that AGoRaS-Quantum is starting to fill in the blank spaces in the latent space. It is interesting to note that most of the original species are concentrated within a small area in the latent space. Fig. 5B clearly illustrates how the network is beginning to fill in all of the available space with generated materials. It appears the areas around the original species are the most densely populated with generated materials. This would make sense as species were used as entry points into the latent space to for the initial sampling. Therefore, a high proportion of the early generated species would be located near the original species. Due to the memory cost, it was not possible to show how using 800000 species would look visually, but imagine an even more densely packed latent space that is expanding outward.
3.1. Candidate quantum materials
While this study focused on the AGoRaS-Quantum framework for the generation and discovery of new materials, it was interesting to point out that there were a few species that show promise for the targeted application of quantum sensing. The most impressive of the species, are two iodine containing structure shown in Fig. 4. There two iodine containing molecules were identify as FIII and C[I][I]II. Both of these molecule had two of the strongest oscillator strengths that was confirmed with TDDFT calculations. These molecules also exhibited a strong dipole moment. The peak wavelength of FIII was 851 nm, which is in the infrared. But C[I][I]II was 550 nm, which is a yellow color in the visible spectrum. This is close to the wavelengths used in fiber optic communication. A brief literature reveals that iodine has extensive use in the field of photochemistry, which is encouraging and supports the predictive capability of AGoRaS-Quantum and further investigation into this material space.4. Conclusions
In this study, the AGoRaS network7 was extended from the generation of gas-phase chemical reactions to the generation of quantum materials. This study was designed to demonstrate how an AGoRaS-Quantum VAE could be used in other applications of nanoscale materials science. The primary purpose of this study was to demonstrate a small dataset of materials can be used to synthetically generate a large number of new materials. The focus materials system for this study was single photon emitting materials. But the developed approach is application agnostic. AGoRaS-Quantum was trained on a core dataset containing 8000 molecular species. A sampling of the latent space was stopped after 1000000 new molecular species were created. This was an arbitrary stopping point and sampling could have continued until the latent space was saturated. The utility of the generated data was demonstrated how the generated species decrease the bias present in the original dataset. The generated dataset or synthetic dataset can be used to train other, more application networks for material discovery.
The novel aspect of the AGoRaS-Quantum network was its ability to generate a large quantity of new molecular species that were both stable and shared the same defining feature as the training dataset. This was an improvement of the previous AGoRaS sampling method in the ability to use the SMILES representations of the molecular species as starting points in sampling the latent space. This allowed for targeted sampling of the latent space to generate materials with specific types of properties. This is possible due to the ability of the VAE to gather knowledge of physics and chemistry from the dataset it is trained on and to generate new molecular species beyond the size and descriptions contained in the training data. We were able to acheive nearly 40 percent generation of new species on each sampling interation.
This developed SMILES language modeling approach opens the possibilities for more in-depth analysis of these generative models for chemistry and materials science. For example, there is potential for using a traditional machine learning analysis to be performed on the AGoRaS-Quantum network in order to gain a better understanding of the underlying processes. The covariant estimates of the different parameters within the network would be one approach. This would also help quantify the overfitting of the latent space via the network's variance. Another interesting study to improve the network speed and efficiency, is the autonomous design of the network parameters. While this study was based on hand-tuned parameters until a stable network could be created. This leaves a great opportunity for the design of a more memory-efficient network. Both the source code and datasets are availible (see Data Availability Section) for this study in hopes for others to expand upon the network and apply this approach to other application.
Data availability
Input and Output Data, which includes generated structures will be available on NOMAD online materials repository at the following address: https://nomad-lab.eu/.The source code corresponding to the machine learning model and the Pipeline Pilot script can be downloaded on GitHub at the following address: https://github.com/Dr-Musho-Research-Group/AGORAS_QUANTUM.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. T. would like to acknowledge the U.S. Department of Energy ORISE Fellowship for support in completing this research, which is funded in part by DE-SC0014664. T. M. would like to acknowledge his attendance to the inaugural 2023 U.S. Quantum Information Science School that was hosted by the SQMS Center and U.S. Department of Energy's (DOE's) Fermi National Accelerator Laboratory (Fermilab). T. M. would like to acknowledge the help of NOMAD materials database team on helping write a custom scheme to import json data into their database. R. T. and T. M. would like to acknowledge that the computational resources for this research were provided by the WVU Research Computing Dolly Sods HPC cluster, which is funded in part by NSF OAC-2117575. This manuscript is being submitted to celebrate the 150th anniversary of Vanderbilt University.References
- Y. Zhang and C. Ling, npj Comput. Mater., 2018, 4, 28–33 CrossRef
.
- S. Go, J. Kim, S. S. Park, M. Kim, H. Lim, J. Y. Kim, D. W. Lee and J. Im, Remote Sens., 2020, 12, 1–34 CrossRef
- M. Shepperd and M. Cartwright, IEEE Trans. Softw. Eng., 2001, 27, 987–998 CrossRef
- A. Amini, W. Schwarting, G. Rosman, B. Araki, S. Karaman and D. Rus, IEEE International Conference on Intelligent Robots and Systems, 2018, pp. 568–575.
- K. K. Yalamanchi, M. Monge-Palacios, V. C. Van Oudenhoven, X. Gao and S. M. Sarathy, J. Phys. Chem. A, 2020, 124, 6270–6276 CrossRef CAS PubMed
- H. A. Carroll, Z. Toumpakari, L. Johnson and J. A. Betts, PLoS One, 2017, 12, 1–19 Search PubMed
- R. Tempke and T. Musho, Nat. Commun. Chem., 2022, 1–10 Search PubMed
- A. B. L. Larsen, S. K. Sønderby, H. Larochelle and O. Winther, Icml, 2016, vol. 4, pp. 2341–2349.
- R. Burks, K. A. Islam, Y. Lu and J. Li, 2019 IEEE 10th Annual Ubiquitous Computing, Electronics and Mobile Communication Conference, UEMCON 2019, 2019, pp. 0660–0665.
- S. Semeniuta, A. Severyn and E. Barth, EMNLP 2017 - Conference on Empirical Methods in Natural Language Processing, Proceedings, 2017, pp. 627–637.
- W. Jin, R. Barzilay and T. Jaakkola, arXiv, 2018.
- L. Yu, W. Zhang, J. Wang and Y. Yu, 31st AAAI Conference on Artificial Intelligence, AAAI 2017, 2017, pp. 2852–2858.
- S. Rajeswar, S. Subramanian, F. Dutil, C. Pal and A. Courville, arXiv, 2017.
- C. L. Degen, F. Reinhard and P. Cappellaro, Rev. Mod. Phys., 2017, 89, 1–39 CrossRef
- S. Alexander, O. Zachariah, S. Peana, D. Sychev, X. Xu, A. S. Lagutchev, A. Boltasseva and V. M. Shalaev, Sci. Adv., 2021, 7, 50 Search PubMed
- M. D. Eisaman, J. Fan, A. Migdall and S. V. Polyakov, Rev. Sci. Instrum., 2011, 82, 071101 CrossRef CAS PubMed
- A. Slachter, PhD Thesis, University of Groningen, 2005.
- A. B. D. al jalali wal ikram Shaik and P. Palla, Sci. Rep., 2021, 11, 1–27 CrossRef PubMed
- L. T. Rose and K. W. Fischer, Measurement, 2011, 9, 222–226 Search PubMed
- H. Sanders and J. Saxe, Proceedings of Blackhat 2017, 2017, p. 6.
- R. R. Griffiths, P. Schwaller and A. A. Lee, ChemRxiv, 2018.
- D. P. Kovács, W. McCorkindale and A. A. Lee, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed
- M. Glavatskikh, J. Leguy, G. Hunault, T. Cauchy and B. Da Mota, J. Cheminf., 2019, 11, 1–15 Search PubMed
- M. A. Kayala, P. Baldi, S. Rajeswar, S. Subramanian, F. Dutil, C. Pal, A. Courville, J. Zhao, Y. Kim, K. Zhang, A. M. Rush, Y. LeCun, H. Catherine, M. L. Cook, E. Mckone, R. R. Griffiths, P. Schwaller, A. A. Lee, M. Glavatskikh, J. Leguy, G. Hunault, T. Cauchy, B. Da Mota, T. F. Cova, A. A. Pais, A. C. Mater, M. L. Coote, M. J. Kusner, J. M. Hernández-Lobato, R. D. Camino, C. A. Hammerschmidt, R. State, E. Choi, S. Biswal, B. Malin, J. Duke, W. F. Stewart, J. Sun, P. Potash, A. Rumshisky, R. D. Camino, C. A. Hammerschmidt, R. State, M. A. Kayala, C. A. Azencott, J. H. Chen, P. Baldi, C. McCutchen, J. Schmidt, M. A. M. R. Marques, S. Botti, M. A. M. R. Marques, D. P. Kovács, W. McCorkindale, A. A. Lee, P. L. Kang, Z. P. Liu, M. A. Kayala, P. Baldi, L. Yu, W. Zhang, J. Wang and Y. Yu, arXiv, 2019, 68, 1–9.
- M. Ryo and M. C. Rillig, Ecosphere, 2017, 8(11), e01976 CrossRef
- A. Zakutayev, N. Wunder, M. Schwarting, J. D. Perkins, R. White, K. Munch, W. Tumas and C. Phillips, Sci. Data, 2018, 5, 1–12 CrossRef PubMed
- D. C. Elton, Z. Boukouvalas, M. S. Butrico, M. D. Fuge and P. W. Chung, Sci. Rep., 2018, 8, 1–12 CAS
- M. Hirohara, Y. Saito, Y. Koda, K. Sato and Y. Sakakibara, BMC Bioinf., 2018, 19, 83–88 CrossRef PubMed
- E. J. Beard, G. Sivaraman, Á. Vázquez-Mayagoitia, V. Vishwanath and J. M. Cole, Sci. Data, 2019, 6, 1–11 CrossRef PubMed
- J. Q. Jiandong Qiao, F. M. Fuhong Mei and Y. Y. Yu Ye, Chin. Opt. Lett., 2019, 17, 020011 CrossRef
-
A. Richter and T. Wagner, Solar Backscattered Radiation: UV, Visible and Near IR - Trace Gases, 2011, pp. 67–122 Search PubMed
- K. K. Kärkkäinen, A. H. Sihvola and K. I. Nikoskinen, IEEE Trans. Geosci. Remote Sens., 2000, 38, 1303–1308 CrossRef
- S. I. Ahmad, P. Koteshwar Rao and I. A. Syed, J. Taibah Univ. Sci., 2016, 10, 381–385 CrossRef
- M. Spangenberg, J. I. Bryant, S. J. Gibson, P. J. Mousley, Y. Ramachers and G. R. Bell, Sci. Rep., 2021, 11, 1–8 CrossRef PubMed
- C. Fei, X. Cao, D. Zang, C. Hu, C. Wu, E. Morris, J. Tao, T. Liu and G. Lampropoulos, Proc. SPIE 11703, AI and Optical Data Sciences II, 2021, 117031D Search PubMed
- R. Mamede, F. Pereira and J. Aires-de Sousa, Sci. Rep., 2021, 11, 1–11 CrossRef PubMed
- F. De Leonardis, R. A. Soref, M. Soltani and V. M. Passaro, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed
- T. Mikolov, K. Chen, G. Corrado and J. Dean, 1st International Conference on Learning Representations, ICLR 2013 - Workshop Track Proceedings, 2013, pp. 1–12.
- Y. Dan, Y. Zhao, X. Li, S. Li, M. Hu and J. Hu, npj Comput. Mater., 2020, 6, 1–16 CrossRef
- J. He, H. You, E. Sandström, E. Nittinger, E. J. Bjerrum, C. Tyrchan, W. Czechtizky and O. Engkvist, J. Cheminf., 2021, 13, 1–17 Search PubMed
- Rdkit.org, RDKit:Open-source cheminformatics, https://www.rdkit.org/.
- A. I. Sarker, A. Aroonwilas and A. Veawab, Energy Procedia, 2017, 114, 2450–2459 CrossRef CAS
- J. Jornet-Somoza and I. Lebedeva, J. Chem. Theory Comput., 2019, 15, 3743–3754 CrossRef CAS PubMed
-
F. Sottile, F. Bruneval, A. G. Marinopoulos, L. K. Dash, S. Botti, V. Olevano, N. Vast, A. Rubio and L. Reining, TDDFT from molecules to solids: The role of long-range interactions, 2005 Search PubMed
- J. S. Smith, O. Isayev and A. E. Roitberg, Chem. Sci., 2017, 8, 3192–3203 RSC
- M. J. Kusner, B. Paige and J. Miguel Hernández-Lobato, arXiv, 2017.
-
A. Nigrin, Neural networks for pattern recognition, MIT Press, Massachusetts, 1st edn, 1993, p. 413 Search PubMed
- S. Jaeger, S. Fulle and S. Turk, J. Chem. Inf. Model., 2018, 58, 27–35 CrossRef CAS PubMed
- H. A. Gaspar, M. Ahmed, T. Edlich, B. Fabian, Z. Varszegi, M. Segler, J. Meyers and M. Fiscato, ChemRxiv, 2021, 1–10.
- J. D. Prusa and T. M. Khoshgoftaar, J. Big Data, 2017, 4, 1–16 CrossRef
- Google, ImageDataGenerator, https://www.tensorflow.org/api_docs/python/tf/keras/preprocessing/image/ImageDataGenerator.
- S. Metlek, K. Kayaalp, I. B. Basyigit, A. Genc and H. Dogan, Int. J. RF Microw. Comput.-Aided Eng., 2021, 31, 1–10 Search PubMed
- S. Gajendran, D. Manjula and V. Sugumaran, J. Biomed. Inform., 2020, 112, 103609 CrossRef PubMed
- A. Jinich, B. Sanchez-Lengeling, H. Ren, R. Harman and A. Aspuru-Guzik, ACS Cent. Sci., 2019, 5, 1199–1210 CrossRef CAS PubMed
- H. Catherine, M. L. Cook and E. Mckone, Iclr, 2017, vol. 15, pp. 401–437.
- VAMP is a Semiempirical Molecular Orbital Package [Computer software], 2021. Retrieved from https://www.3ds.com/.
- J. J. Goings, F. Ding, M. J. Frisch and X. Li, J. Chem. Phys., 2015, 142(15), 154109 CrossRef PubMed
- K. K. Ni, T. Rosenband and D. D. Grimes, Chem. Sci., 2018, 9, 6830–6838 RSC
- Agilent Technologies, Models in the 85071E Materials Measurement Software, https://na.support.keysight.com/materials/docs/85071Emodels.pdf.
| This journal is © The Royal Society of Chemistry 2024 |