
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosystem I complexes form remarkably stable self-assembled tunneling junctions†
Nahid
Torabi
 a and
Ryan C.
Chiechi
*ab
a and
Ryan C.
Chiechi
*ab
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
bDepartment of Chemistry & Organic and Carbon Electronics Cluster, North Carolina State University, Raleigh, North Carolina 27695-8204, USA. E-mail: ryan.chiechi@ncsu.edu
First published on 26th September 2024
Abstract
This paper describes large-area molecular tunneling junctions comprising self-assembled monolayers (SAMs) of light-harvesting protein complexes using eutectic Ga–In (EGaIn) as a top contact. The complexes, which are readily isolable in large quantities from spinach leaves, self-assemble on top of SAMs of [6,6]-phenyl-C61-butyric acid (PCBA) on gold (Au) supported by mica substrates (AuMica), which induces them to adopt a preferred orientation with respect to the electron transport chain that runs across the short axis of each complex, leading to temperature-independent rectification. We compared trimeric protein complexes isolated from thermophilic cyanobacteria to monomeric complexes extracted from spinach leaves by measuring charge-transport at variable temperatures and over the course of at least three months. Transport is independent of temperature in the range of 130 to 310 K for both protein complexes, affirming that the likely mechanism is non-resonant tunneling. The junctions rectified current and were stable for at least three months when stored at room temperature in ambient conditions, with the yield of working junctions falling from 100% to 97% over that time. These results demonstrate a straightforward strategy for forming remarkably robust molecular junctions, avoiding the fragility that is common in molecular electronics.
Introduction
The field of Molecular Electronics is concerned with using molecules as building blocks to leverage self-assembly and exploit the properties of matter at the molecular scale.1 Potential applications of molecular electronics include sensors, displays, smart materials, molecule-scale transistors, and energy storage devices.2,3 Molecular junctions typically exploit the interactions between molecules and under-coordinated metal atoms on an electrode surface, forming, for example, gold–thiolate bonds.4 In previous works, various studies have explored the flow of charge through molecules in molecular junctions within molecular-scale devices.5–13 Biomolecules such as proteins are intriguing molecular building blocks because they naturally comprise a large number of precisely-ordered atoms, exceeding the complexity achievable by modern synthetic chemistry. The study and exploitation of their electronic-conductance properties is an emerging sub-field of Molecular Electronics that has attracted increasing attention over the past decade.14–23 A key challenges is the formation of reliable, high-conductance contacts between electrode and molecule, which requires both forming a strong electrode–molecule interaction to control orientation, and strong electronic overlap to enable efficient charge transport.Proteins, in general, can be considerably more robust and can support tunneling charge-transport over longer distances than small molecules designed for those functions, which is presumably a byproduct of evolutionary pressure.24,25 Solid-state electronic transport via azurin (Az) protein has been intensively investigated.26–29 Au-bound N42C Az mutant monolayers were shown to be sufficiently robust for solid-state electron transport measurements from room temperature down to 10 K over which these 3.5 nm thick junctions exhibited temperature-independent conductivity.20 Several groups have reported temperature-independent charge transport in solid-state device configuration for junctions where the charge carriers almost certainly interact strongly with the (bio)molecule due to the larger 10 nm thickness.30–33 Comparative long-range electron transport in organic small-molecules has been reported for bis-thienylbenzene molecular films in the range of 8 to 22 nm at low temperatures, which shows weak temperature dependence, but no thermal hopping.34 Remarkably, charge-transport through multilayers of bacteriorhodopsin has been shown to be nearly independent of temperature over distances of at least 50 nm, far beyond the expected limits for tunneling charge-transport through any medium.35
We investigated the photosystem I (PSI) protein complex from two disparate sources, one extracted from a thermophilic cyanobacteria (Cyb) Thermosynechococcus elongatus36 and the other extracted from spinach leaves (Sp-PSI). Both Sp-PSI and Cyb-PSI formed reproducible large-area molecular ensemble junctions in high yield that exhibit temperature-independent charge transport over a distance of approximately 9 nm. We describe the assembled junctions as AuMica/PCBA//PSI//EGaIn, where ‘/’ denotes interfaces defined by chemisorption and ‘//’ by physisorption, noting that the SAMs of PSI form on top of SAMs of [6,6]-phenyl-C61-butyric acid (PCBA) as depicted in Fig. 1. Following their extraction and further isolation, the PSI complexes formed monolayers spontaneously in the presence of SAMs of PCBA supported by AuMica substrates, in which the fullerene cages bind to Au through reversible non-covalent bonds.37,38 Both PSI complexes formed junctions that were stable under ambient conditions for at least three months, rectifying current and exhibiting temperature-independent charge-transport from 130 to 310 K, reflecting the intrinsic robustness and facile tunneling charge-transport of PSI, regardless of biological kingdom. However, despite being less stable in vivo and in solution, Sp-PSI maintained higher magnitudes of rectification and degraded even more slowly than Cyb-PSI.
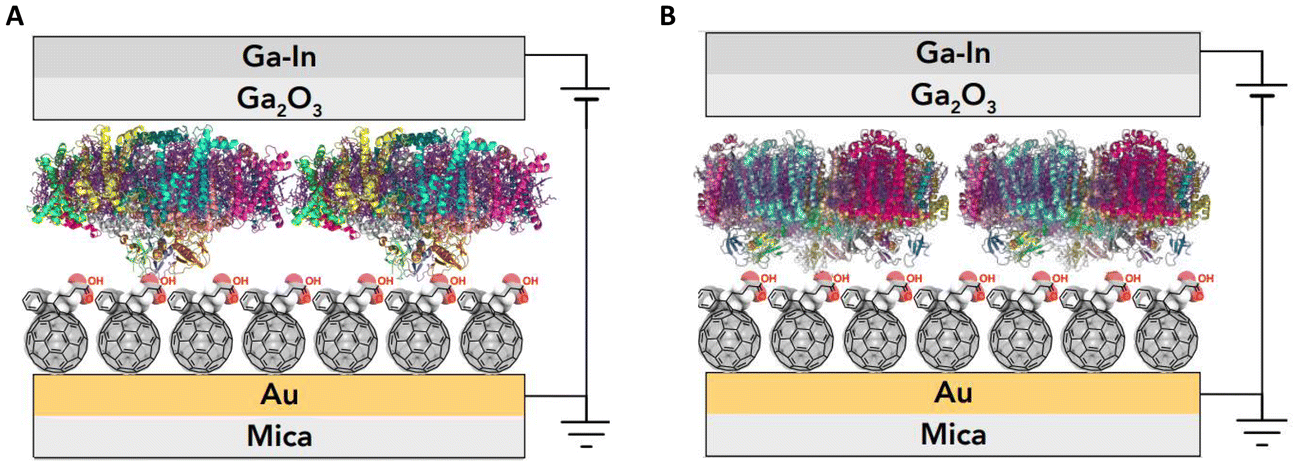 | ||
| Fig. 1 A schematic of an assembled (A) AuMica/PCBA//Sp-PSI//EGaIn and (B) AuMica/PCBA//Cyb-PSI//EGaIn junction showing the preferred orientation adopted by Sp-PSI and Cyb-PSI when self-assembled on top of SAMs of PCBA. The top EGaIn contact is depicted as bulk eutectic Ga–In presenting a sub-nm self-limiting Ga2O3 layer at the interface with Sp-PSI and Cyb-PSI. The schematic is scaled to emphasize the relative orientations and interfaces of the PSI complexes and PCBA. | ||
Results and discussion
Structure of PSI
Crystal structures of photosystem complexes in cyanobacteria39,40 and plants41,42 have provided valuable insights into the arrangement and functionality of the respective pigment networks. Each PSI complex includes a reaction center and an electron transfer chain encircled by an array of pigment molecules that serve as antennas for photons, all held together by a protein scaffold. However cyanobacteria photosystems exist as trimeric complexes of PSI, denoted Cyb-PSI, while plant light-harvesting complexes (LHCs) exist as monomers, denoted Sp-PSI; we will refer to both as PSI for clarity. The subtle differences between the bacterial PSI monomers and plant PSI are illustrated in Fig. 2. Plant complexes consist of a central reaction center, where exciton energy is converted to free charges, comprising up to 14 subunits, which is surrounded by a membrane-bound light-harvesting antenna complex (LHCI) that captures light energy and directs it toward the reaction center. Overall, approximately 200 pigment molecules (shown in purple) and 3 Fe4S4 clusters are associated with Sp-PSI. The monomeric cyanobacterial complexes are structurally similar, with each comprising 12 protein subunits and 127 cofactors as well as approximately 120 pigment molecules (also shown in purple) and 3 Fe3S4 clusters.39,40 The Cyb-PSI monomers are smaller in size compared with Sp-PSI, having a reaction center that resembles the one in plants but with no peripheral antennas.41 Cyanobacteria Cyb-PSI monomers assemble into trimeric structures, while trimers of Sp-PSI are not observed.43,44 The formation and stability of trimers in cyanobacteria are attributed to the subunits PsaM and PsaL39 as shown in Fig. 2A. The subunits PsaG and PsaH are present only in plants (Fig. 2B). The lack of the PsaM subunit, alterations in the PsaL subunit structure and the involvement of the PsaH subunit collectively prevent Sp-PSI from forming trimers.45–47 Furthermore, the subunit PsaL displays important structural differences between plants and cyanobacteria. As another difference in the organization of cyanobacteria and plants, Sp-PSI typically forms a supercomplex comprising the central core and an additional array of peripheral chlorophyll a/b binding LHCI complexes,41,48 designated Lhca1–Lhca4 and arranged in a semi-ring surrounding the PSI core. Within Sp-PSI, the PsaG subunit acts as an anchor point for Lhca subunits, illustrated in Fig. 2B.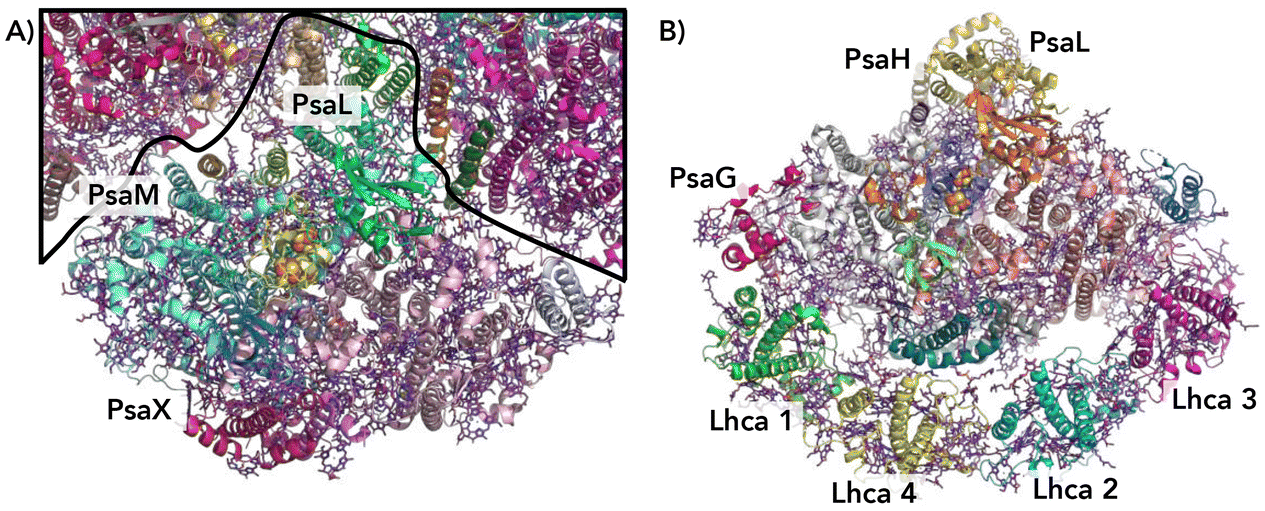 | ||
| Fig. 2 Comparison of cyanobacterial and plant PSI/LHC structures (not to scale).49 (A) A view from the cytoplasmic side of cyanobacterial PSI showing one monomer, with the borders between PSI monomers depicted as black lines. Subunits PsaM and PsaX are unique to cyanobacteria. (B) The stroma side of plant PSI. Subunits PsaG and PsaH are unique to plants. In the plant PSI, each Lhca1–4 subunit is colored differently, Lhca 1 and 2 (green and cyan) and Lhca 3 and 4 (magenta and yellow). | ||
The fundamental structural similarities of PSI complexes discussed above are due to the core function of converting light energy to chemical energy, which is common to all photosynthetic organisms. Distinct differences in the supramolecular arrangement between Sp-PSI and Cyb-PSI occur when exposed to iron-stress conditions. Cyanobacteria form a circular arrangement of iron-stress-induced (isiA) subunits encircling a trimeric PSI core, leading to an almost twofold increase in the light-absorbing area of the core complex.50–52 However, no corresponding isiA rings are detected in plants or algae. While there are similarities in how Lhca subunits surround a PSI core in plants and algae, a significant distinction lies in the attachment strength: the coupling between the PSI core and Lhca subunits is less robust than the connection between the PSI core and the isiA ring in cyanobacteria.53 Collectively, these properties endow Cyb-PSI with additional stability that makes them the preferred PSI for ex vivo studies.33,36,54–59 Such studies are conducted on PSI in a variety of contexts, but we are specifically interested in the charge-transport properties of surface-bound assemblies of PSI.
Molecular ensemble junctions comprising PSI
In order to inject charges and/or utilize the spatial separation of electron/hole pairs ex vivo, the individual PSI complexes must be oriented properly with respect to the electrodes. We previously investigated the ability of different SAMs to direct the orientation of Cyb-PSI complexes as they self-assemble into another layer on top of these linker-SAMs.33,36 This approach allows the anchoring of ordered arrays of oriented PSI (i.e. monolayers) to AuMica substrates with control over the orientation of PSI.60 The linker-SAMs, in turn, can be anchored to AuMica through thiol bonds or C60 cages.37 In that configuration, electrons are collected via the C60 anchors and holes are collected by an electrode/layer placed on top of the PSI to form biophotovoltaic devices;38 the ex vivo stability of PSI is an important factor in those devices. Several reports have shown that PSI-based biophotoelectrodes can retain their activity for up to several months,61,62 for example, Ciesielski et al. have demonstrated that biohybrid electrochemical cells fabricated using a dense multilayer of PSI complexes remained active for at least 280 d in ambient conditions.63 We have also shown that PSI can be replaced in operando using the aforementioned linker-SAM strategy in microfluidic biophotovoltaic devices.60 An underlying assumption in the use of PSI in solid-state devices is that Cyb-PSI is more stable than Sp-PSI, justifying the comparatively difficult process of preparing bacterial cultures and isolating Cyb-PSI. However, organic semiconductor devices typically comprise thin-films, while our interest in monolayers of PSI in which each complex resides on an electrode surface;64 the molecular ensemble junctions investigated in this work directly probe this architecture by measuring the charge-transport properties of the surface-bound complexes directly.Both Sp-PSI and Cyb-PSI are roughly cylindrical and approximately 1.5× as wide as it is tall, which defines three distinct surfaces, top, bottom and periphery. As with most membrane proteins, the top and bottom are polar and the periphery is nonpolar, which favors the formation of monolayers of PSI on surfaces randomly oriented with the top or bottom facing into the surface. The top and bottom interact differently with polar-protic and polar-ionic surfaces such that they will orient with a preferred orientation on surfaces decorated with linker-SAMs, as described above, that express either protic or ionic groups.33,36,59,60 We have previously shown that SAMs of PCBA (which is protic) perfectly orient Cyb-PSI on AuMica surfaces, while also facilitating both the extraction and injection of charge out of/into the PSI complexes.38,65Fig. 3 shows AFM height images of Cyb-PSI (Fig. 3A) and Sp-PSI (Fig. 3B) assembled on SAMs of PCBA on AuMica. The trimeric complexes of Cyb-PSI are clearly visible in the inset, while Sp-PSI monomers only show some random aggregation. The yellow insets are digital blowups of the regions indicated on the AFM images. Rendered PSI complexes from the same crystal structures as Fig. 2 are shown below the insets, highlighting the striking similarity of the AFM topography to Cyb-PSI and illustrating the difficulty resolving the monomeric PSI of Sp-PSI, though individual complexes are clearly present. The inset bars show the scaling of the renders, i.e. Cyb-PSI is only slightly larger than the size of Sp-PSI; Fig. 4 shows full-sized versions of the rendered PSI complexes with PCBA for comparison. The orientation of the complexes/trimers is not resolvable from AFM topography, but can be determined from the charge-transport properties of junctions comprising PSI on AuMica.
 | ||
| Fig. 3 AFM images of partial monolayers of (A) Cyb-PSI and (B) Sp-PSI on SAMs of PCBA with the scanned area of 200 × 200 nm2 and zoomed AFM images of (C) Cyb-PSI and (D) Sp-PSI on SAMs of PCBA. The zoomed AFM images are scanned over 100 × 100 nm2. In Cyb-PSI, the PSI complexes are organized as trimers, while Sp-PSI is monomeric. The Z-scales are 8.5 nm for Cyb-PSI and 9.5 nm for Sp-PSI. The yellow squares are digitally zoomed insets of the indicated regions and are shown above renders of the two PSI complexes. The blue bars show the relative scaling of the rendered complexes. | ||
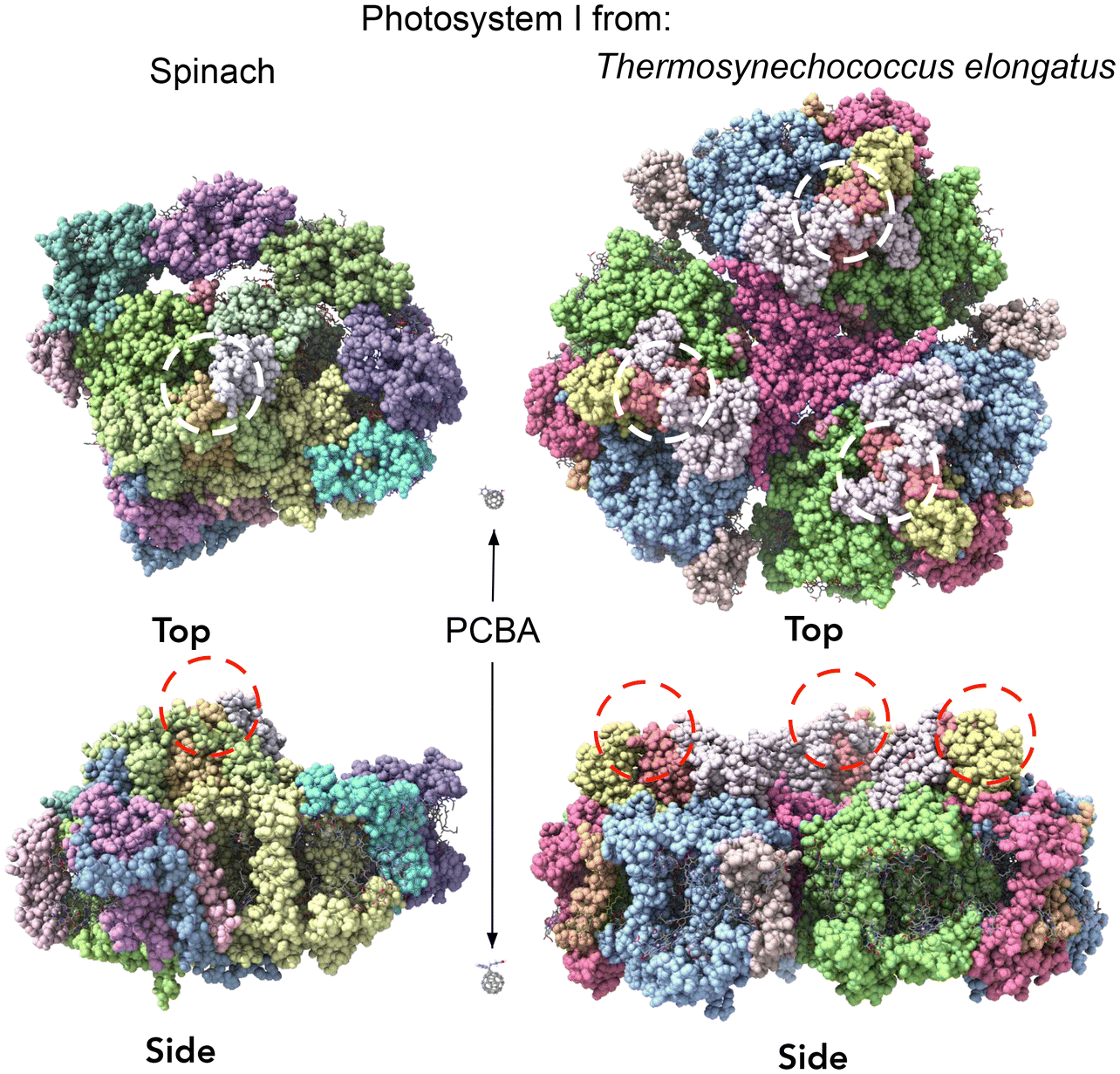 | ||
| Fig. 4 Views of PSI isolated from spinach (left) and Thermosynechococcus elongatus (right) from the top and side with the Fe4S4 clusters indicated with dashed circles. Bacterial PSI comprises a trimer of monomeric PSI that is structurally similar to plant PSI, which exists only as the monomer. The views are to scale, with PCBA shown as a reference. | ||
To form a junction, a top-contact must be applied. Different techniques can be used to form these junctions and perform current–voltage measurements, such as suspended nanowires22 and eutectic Ga–In (EGaIn).66 These techniques allow the junction to be stable over a wide temperature range without damaging the protein complexes.67–69 EGaIn top-contacts are also reversible, allowing the disassembly and interrogation of junctions after J–V measurements without damaging the protein, with high reproducibility and with high yields of working junctions. We formed junctions using EGaIn top-contacts66 and AuMica as conducting substrates supporting SAMs of PCBA that are anchored to the surface via their C60 cages, presenting carboxylic acid groups at the ambient interface (see Experimental). The monolayer of PSI forms through preferential, reversible interactions between the FB site and these carboxylic acid groups.38,65 Unlike small-molecule SAMs, monolayers of PSI do not have to be densely packed to prevent shorts because the high surface tension of EGaIn prevents it from penetrating the nm-size gaps between complexes; see Fig. 5 and the accompanying text for details.
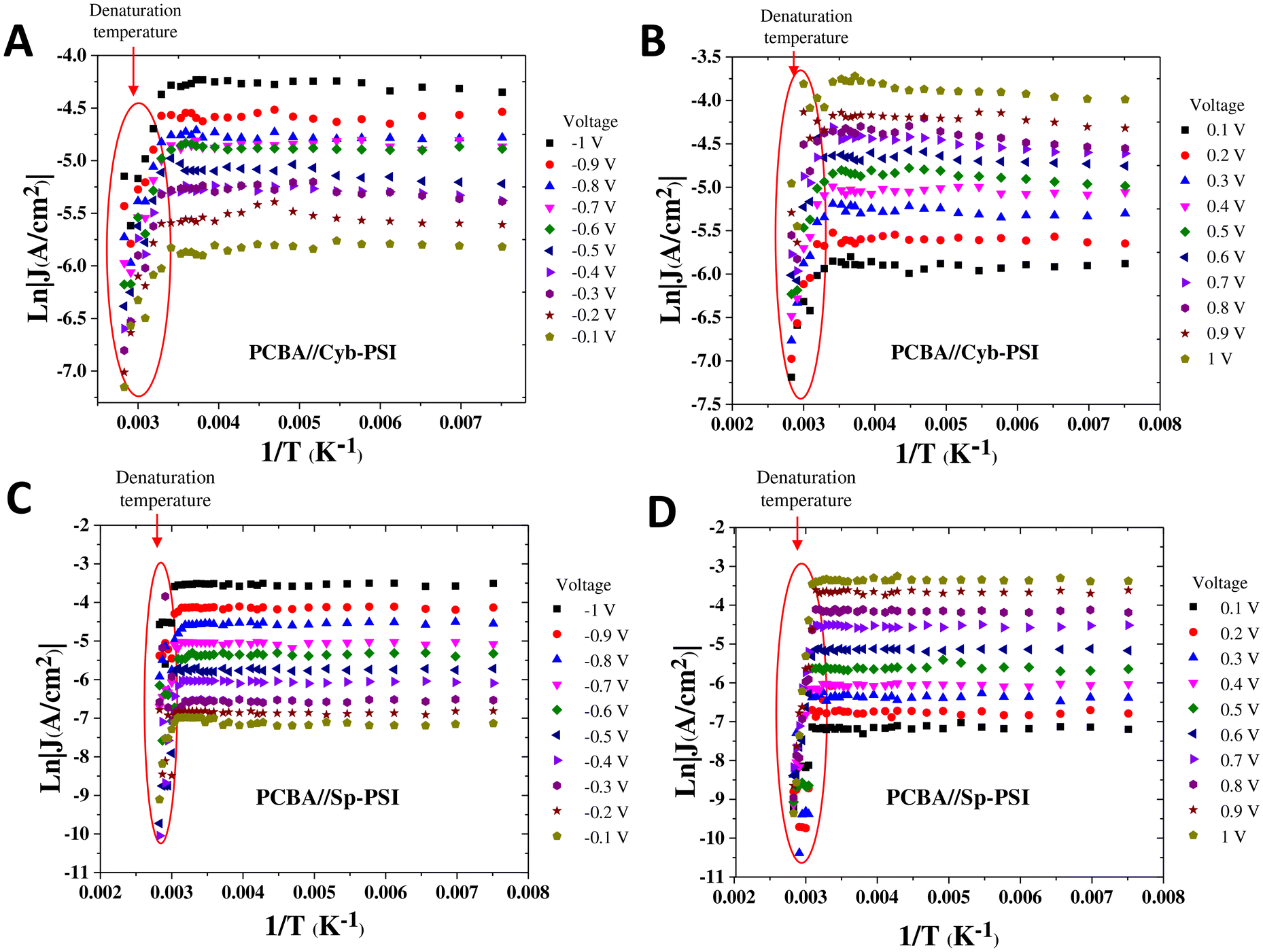 | ||
| Fig. 5 Arrhenius plots of bilayer comprising of AuMica/PCBA//Cyb-PSI//EGaIn (A–B) and AuMica/PCBA//Sp-PSI//EGaIn junctions (C–D) between temperature ranges 133–353 K from −0.1 to −1 V (A, C) and from 0.1 to 1 V (B, D). The red ovals show the temperatures corresponding to PSI protein denaturation. | ||
We compared two types of bilayer junctions: AuMica/PCBA//Cyb-PSI//EGaIn and AuMica/PCBA//Sp-PSI//EGaIn. A key feature of junctions comprising Cyb-PSI is that they exhibit orientation-dependent rectification, log|R| 6 ≠ 0 in eqn (1) when Cyb-PSI assembles with a preferred orientation. This rectification is independent from the director SAM70 because it originates from the collective influence of the oriented dipole moments in the protein complex.33,59,71 Moreover, the magnitude and sign of log|R| depend on the degree of preferential orientation as well as whether the top or bottom of Cyb-PSI is facing into the substrate as previously reported by us,33,36,65 and Cahen et al.59 This sensitivity of log|R| allows the degree of orientation to be determined using large-area EGaIn top-contacts as well as the absolute direction using conducting probe AFM to contact the protein complexes individually.
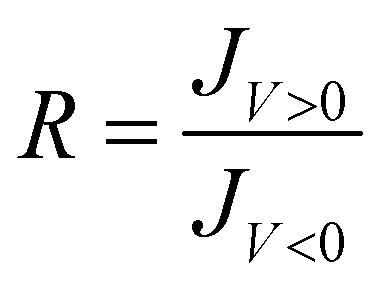 | (1) |
Mechanism of charge-transport
The mechanisms of charge transport through ensembles/SAMs can be characterized by two extremes. One is dominated by coherent tunneling, which describes temperature-independent coherent tunneling in solid-state junctions between electrodes via the molecular bridge. The other regime involves temperature-dependent charge transfer processes where the electron fully relaxes inside the junction, requiring thermal activation for traversal, which describes thermally activated incoherent tunneling (i.e. hopping).72,73 The most commonly used theoretical model to describe temperature-independent tunneling is the simplified Simmons model,74 which can be applied in the case of coherent tunneling (eqn (2)):| J = J0(V)e−βd | (2) |
| J = J0(V)e−Ea/kBT | (3) |
To elucidate the charge transport mechanism across the PSI complexes, we measured current density as a function of temperature and made Arrhenius plots. The temperature-dependent measurements were performed on a custom-built cryogenic probe station in which conical EGaIn top-contacts can be formed in a controlled atmosphere. We were able to measure J/V data from 130 to 353 K at intervals of 10 K. We only collected J/V data at each temperature after allowing the system to stabilize for at least 10 min. The purpose of measuring both above and below ambient temperatures is that rectification vanishes when PSI denatures and 353 K is above the denaturation temperature of both Cyb-PSI and Sp-PSI. Fig. 5A and B show the Arrhenius plots as a function of inverse temperature through the AuMica/PCBA//Cyb-PSI//EGaIn junctions measured across the aforementioned range of temperatures from 130–353 K. The results confirm that charge-transport through Cyb-PSI is independent of temperature until denaturation, at which point the internal structure of the proteins is lost. This behavior is consistent with non-resonant quantum mechanical tunneling, confirming that the mechanism of charge-transport through intact Cyb-PSI is not impacted by the presence of PCBA and is consistent with our prior studies using thiol-SAMs and peptides.33,36 At elevated temperatures, the yields of working junctions decreased from >90% to ∼5% as Cyb-PSI denatured. The non-shorting junctions also showed a sharp drop in the magnitude of J at the onset of denaturation, which occurs at ∼335 K for Cyb-PSI.
Fig. 5C and D show analogous Arrhenius plots for AuMica/PCBA//Sp-PSI//EGaIn. As with Cyb-PSI, these data show that charge-transport is independent of temperature for Sp-PSI until the onset of denaturation at ∼313 K. This behavior indicates that charge-transport is non-resonant quantum mechanical tunneling for AuMica/PCBA//Sp-PSI//EGaIn, moreover, the values of J are even more stable for Sp-PSI than Cyb-PSI across the full temperature range, implying, counterintuitively, that Sp-PSI forms more stable junctions than Cyb-PSI. The red ovals in Fig. 5 show the same drop in the magnitude of J (and yields of working junctions) for Sp-PSI that was observed for Cyb-PSI. Taken together, these results show that the differences in quaternary structure and self-assembly that are readily apparent in Fig. 3 have no measurable effect on the mechanism of charge-transport, which instead reflects the similarities in the tertiary structures summarized in Fig. 2. The apparent lack of thermal activation over long distances in junctions comprising proteins35 may indeed be a feature of highly structured nano-objects. Naaman et al. propose a model in which electronic reorganization occurs at the “entrance” and “exit” of a nano-object, the electronic structure of which polarizes in response to the applied field, necessitating the inclusion of electron–electron interactions in the tunneling mechanism.75 In any case, the lack of thermal activation in PSI is demonstrably unrelated to any properties intrinsic to Cyb-PSI, such as its trimeric structure or higher denaturation temperature.
Junction stability
Having established that the mechanism of charge-transport is the same for Cyb-PSI and Sp-PSI and that it is unaffected by the use of PCBA as a director SAM, we investigated the robustness of these complexes, e.g. the stability of their J/V characteristics over time. Junctions comprising thiol-based SAMs (e.g. alkanethiols) degrade quickly in air and require nontrivial packaging to remain stable for more than a day.76–78 These challenges have led to increased interest in non-thiol anchors for SAMs designed specifically for long-lived molecular junctions.37,79,80 Proteins are particularly interesting in this regard because they are structurally complex, synthesized with exact precision by organisms and simple proteins such as bovine serum albumin are very robust in biomolecular devices;81 however longevity studies on more complex proteins with more complex functions, particularly as tunneling media in junctions are rare enough that there is no established baseline against which to compare.We repeatedly measured junctions of AuMica/PCBA//Cyb-PSI//EGaIn and AuMica/PCBA//Sp-PSI//EGaIn from the same substrates at regular intervals over a period of 100 and 110 days respectively, during which they were stored on the bench top. Measurements were performed under ambient conditions in a custom-built Faraday cage with cone-shaped EGaIn top contact. The junctions remained remarkably stable, yielding 100% non-shorting junctions and stable J/V curves, with the yield dropping only to 97% for Sp-PSI and 95% for Cyb-PSI by the last day, after which we stopped acquiring data. Statistics of EGaIn junctions for both Sp-PSI and Cyb-PSI are summarized in Table S1.†Fig. 6 compares the J/V (A–B) and log|R| (C–D) data of both of Cyb-PSI and Sp-PSI every 10 d (the full dataset is shown in Fig. S2†). Each data point is an average of 12–15 different junctions from 3 different substrates. Both Cyb-PSI and Sp-PSI show an initial drop in the magnitude of J on Day 10 compared to Day 1, however, Sp-PSI remains almost invariant from Day 10 to Day 90, while Cyb-PSI declines steadily. When molecular junctions degrade, they tend to increase in current as pinholes form in the SAM, eventually shorting. The PSI junctions are clearly not forming pinholes, rather, the decrease in the magnitude of J in time suggests that the proteins are undergoing a loss of structure similar to the decrease in J observed in Fig. 5 as transport switching to hopping when heated above the denaturation temperature. In addition to having a higher denaturation temperature, the primary reason that Cyb-PSI is commonly used for ex vivo studies is that it loses its photoactivity much more slowly than Sp-PSI, presumably because it retains its structure longer. The comparison of studies of photoactivity to J/V properties in junctions is not entirely straightforward, as activity studies necessarily involve constant irradiation, but it is reasonable to assume that membrane proteins on surfaces exposed to ambient conditions will degrade over time. Nonetheless, the observation that the J/V characteristics of Sp-PSI are significantly more stable over 100 days than Cyb-PSI is counterintuitive because it shows that the longevity of Cyb-PSI in solution—determined by measuring photo-activity—compared to Sp-PSI does not carry over into stability on surfaces—determined by electrical properties.
 | ||
| Fig. 6 Room-temperature current density versus voltage characteristics of (A) AuMica/PCBA//Cyb-PSI//EGaIn and (B) AuMica/PCBA//Sp-PSI//EGaIn junctions every 10 days during 100 d and 110 d respectively. The conductance of devices fabricated of AuMica/PCBA//Cyb-PSI//EGaIn and AuMica/PCBA//Sp-PSI//EGaIn junctions decreased after 100 d and 110 d respectively. Plots of log|R|/|V| of (C) AuMica/PCBA//Cyb-PSI//EGaIn and (D) AuMica/PCBA//Sp-PSI//EGaIn junctions every 10 days during 100 d and 110 d respectively. The rectification ratio decreases for both AuMica/PCBA//Cyb-PSI//EGaIn and AuMica/PCBA//Sp-PSI//EGaIn junctions over time. | ||
As discussed above, the relationship between the structure and orientation of Cyb-PSI and R is well established, allowing R to serve as a proxy measure of structural degradation; i.e. if the decrease in the magnitude of J in time is due to a degradation process similar to thermal denaturation (which completely destroys the ability of Cyb-PSI to rectify), then R will also change in time. Although R is not commonly used to characterize changes to the structure of molecular junctions in time, that is simply because few studies examine junctions that are long-lived enough to evolve in time. However, R is directly related to the deliberate inclusion of defects that affect leakage current.82Fig. 6C and D shows plots of log|R|/|V| computed directly from the J/V plots shown in Fig. 6A and B for Cyb-PSI and Sp-PSI, respectively. These data show an initial drop in the magnitude of log|R| from Day 1 to Day 10, but then very little change after that and a consistently larger magnitude of log|R| for Sp-PSI than Cyb-PSI across the entire time period. Both of these trends are more clearly visible in Fig. 7, which compares log|R| at V = |1 V| and V = |1.5 V|, showing the initial drop between Day 1 and Day 10, the subsequent stability of log|R|, and the consistently larger magnitude of log|R| for Sp-PSI. Taken together, the J/V and log|R|/|V| data show that Cyb-PSI degrades at a faster rate than Sp-PSI, but that the degradation mechanism is a loss of structural order rather than desorption and/or the formation of pinholes. These disordered structures are more resistive than intact PSI, but have a negligible impact on the yield of non-shorting junctions. These observations are consistent with tunneling charge-transport; due to the exponential dependence of J on d, resistive defects tend to impact the magnitude of J, but not physical manifestations of non-resonant tunneling such as R and β.83 Treating large-area junctions as an array of nanoscopic diodes (intact PSI complexes) and resistors (degraded PSI complexes), the total current is shared between all resistors and diodes and the magnitude of log|R| is insensitive to the number of diodes in parallel, while the magnitude of log|J| scales with the resistance of each resistor. Thus, as each intact PSI complex degrades and increases its resistance, log|J| decreases for all values of V, while log|R| is only affected by the ratio of intact to degraded complexes as the fraction of the total current carried by resistors increases.
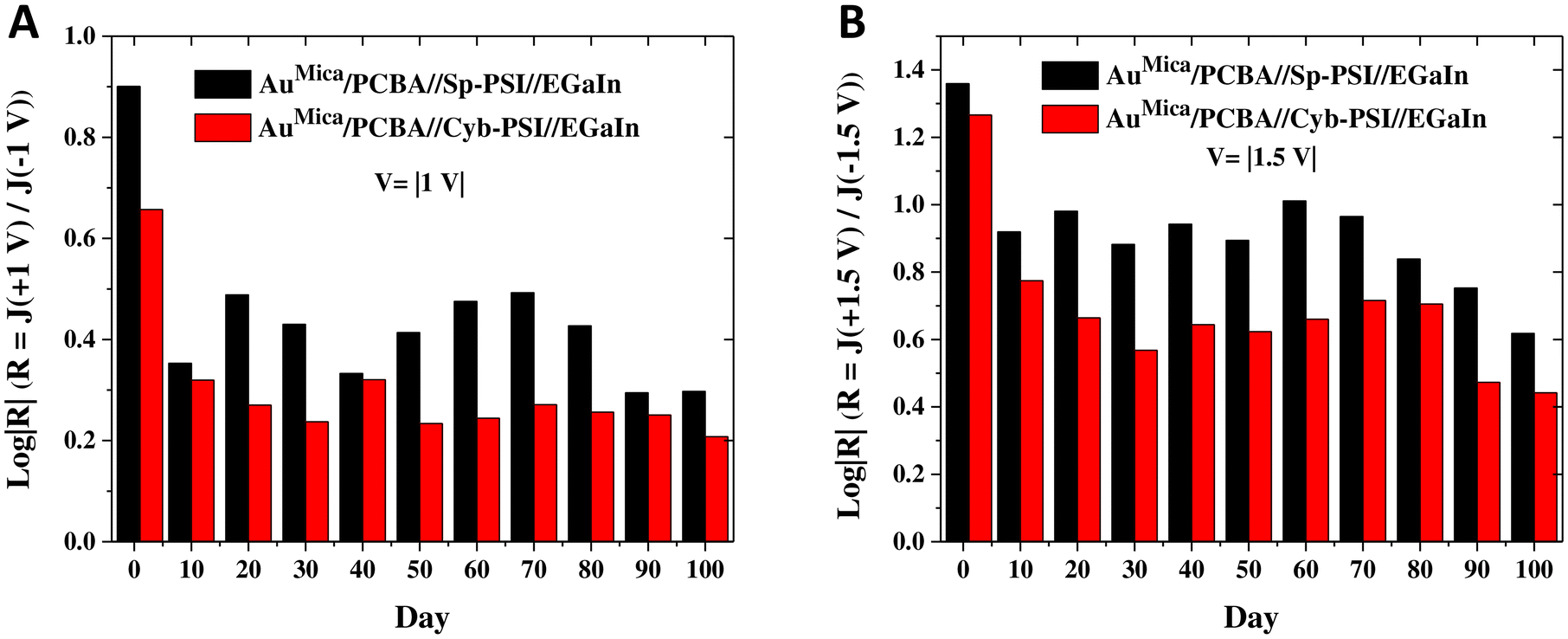 | ||
| Fig. 7 Comparison of the rectification ratios of PSI complexes (A) at V = |1 V| and (B) at V = |1.5 V| solid-state junctions. For both voltages, the rectification ratios of Cyb-PSI and Sp-PSI exhibit fluctuations over approximately 100 days, with a trend showing a decrease from day 1 to day 100. This decline in rectification ratios suggests a diminishing trend over the observed period. | ||
Conclusions
In the context of Molecular Electronics, large-area tunneling junctions comprising ensembles of molecules have the most immediate potential utility because they can be prepared in quantitative yields and wired together in device-like packages. These advantages are undermined by short-lived junctions and the tendency of junctions comprising small molecules to fail by catastrophic shorting. We have shown that ensembles of PSI complexes formed on top of SAMs anchored by fullerenes solve both of these problems; yields of non-shorting AuMica/PCBA//PSI//EGaIn junctions are still 97% after three months in ambient conditions. They retain their ability to rectify current through a non-resonant tunneling mechanism established by measuring their electrical properties over 130 to 353 K and do not precipitate shorts even when degraded or thermally denatured. The remarkable stability of ensembles of PSI on electrodes that form spontaneously and with a preferred orientation is broadly applicable in any context in which holes and electrons are injected/extracted (or photo-generated). Moreover, we found that PSI isolated from spinach leaves is significantly more robust than PSI isolated from Thermosynechococcus elongatus bacteria. Although this work focuses on empirical relationships between electrical properties and the structural differences between Sp-PSI and Cyb-PSI, future computational studies may be able to elucidate the self-assembly process with enough detail to ascribe the loss of log|R| to specific structural changes within the complexes themselves. Nonetheless, the useful and complex functionality of PSI complexes, the ease and reproducibility of their self-assembly on electrodes, and the abundance and availability of spinach-derived PSI make them ideal for applications and fundamental studies in Molecular Electronics, in general.Experimental
Photosystem I preparation and purification from spinach leaves
All the isolation procedures described for the extraction of PSI from spinach leaves were performed in a cold room at 4 °C under dim light. In this study, spinach leaves were purchased from the local market and then prechilled in a dark cold room overnight. Isolation of unstacked thylakoids was performed as described recently with some modifications.84 The leaves were blended in a buffer consisting of sucrose (0.3 M, Sigma-Aldrich, 99.5%), NaCl (15 mM, Sigma-Aldrich, >99%), and tricine (30 mM, Sigma-Aldrich, >90%) at pH 7.8. The slurry was first filtered through a layer of Whatman filter paper and then centrifuged for 2 min at 2000g. Unstacked membranes were isolated at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min. The resulting pellet was suspended in a buffer consisting of ethylenediaminetetraacetic acid (EDTA) (5 mM) and tricine (5 mM) at pH 7.8 and was homogenized for 30 min. The homogenized solution was centrifuged for 20 min at 15000g. Unstacked membranes were suspended in a buffer consisting of sucrose (0.3 M) and tricine (30 mM) at pH 7.8, homogenized for 30 min, and then centrifuged for 20 min at 27000g. The concentrations of Chlorophyll a and b were determined in buffered aqueous 80% acetone as described recently.85 For further separation and isolation of native PSI, spinach thylakoids were solubilized in 0.5% sodium dodecyl sulfate (SDS) (w/v). After homogenizing for 20 min, the sample was centrifuged at 15000g for 10 min. In the last step, for the purification, ion exchange chromatography was used with a diethylaminoethyl (DEAE) cellulose matrix as an elution buffer containing 20 mM tricine, 0.3 M NaCl, and 20 mM Tris at pH 7.8. The absorbance spectra for the solutions collected at different times from the chromatography column are shown in Fig. S1.† The collected solutions from the column containing PSI complexes were then dialyzed. The PSI suspension was stored at −80 °C.
000g for 20 min. The resulting pellet was suspended in a buffer consisting of ethylenediaminetetraacetic acid (EDTA) (5 mM) and tricine (5 mM) at pH 7.8 and was homogenized for 30 min. The homogenized solution was centrifuged for 20 min at 15000g. Unstacked membranes were suspended in a buffer consisting of sucrose (0.3 M) and tricine (30 mM) at pH 7.8, homogenized for 30 min, and then centrifuged for 20 min at 27000g. The concentrations of Chlorophyll a and b were determined in buffered aqueous 80% acetone as described recently.85 For further separation and isolation of native PSI, spinach thylakoids were solubilized in 0.5% sodium dodecyl sulfate (SDS) (w/v). After homogenizing for 20 min, the sample was centrifuged at 15000g for 10 min. In the last step, for the purification, ion exchange chromatography was used with a diethylaminoethyl (DEAE) cellulose matrix as an elution buffer containing 20 mM tricine, 0.3 M NaCl, and 20 mM Tris at pH 7.8. The absorbance spectra for the solutions collected at different times from the chromatography column are shown in Fig. S1.† The collected solutions from the column containing PSI complexes were then dialyzed. The PSI suspension was stored at −80 °C.
Photosystem I preparation and purification from cyanobacteria
Photosystem I was extracted from the thylakoid membranes of the thermophilic cyanobacterium Thermosynechococcus elongatus BP-1 in BG11 (plant culture media, Sigma-Aldrich) medium under continuous light at 55 °C.86 Thylakoid membranes were prepared using a protocol that combines elements from two previously established preparation method.87,88 The purified PSI sample was adjusted to a Chlorophyll a concentration of 300 μM and stored at −80 °C. Chlorophyll a concentration of the resulting PSI solutions was determined by the methods as described previously by Porra85 and Baba et al.89Substrate preparation
For all current–voltage measurements, AuMica substrates were used, which provide a mono-crystalline Au(111) surface. Thin films of Au were grown on mica substrates (Grade V1, 25 × 75 mm). The adhesion of gold to mica was accomplished by controlling the temperature. Before Au evaporation, the mica sheets were heated for 6 h under a vacuum pressure of 10−7 mbar at deposit temperature 375 °C. Then the gold evaporation onto a freshly 1 mm × 3 mm piece of mica was accomplished at an evaporation rate ranging from 0.3 Å s−1 to 0.5 Å s−1 at 375 °C, followed by 200 nm gold deposition. After deposition, the AuMica was heated for 2 h at 375 °C inside the vacuum chamber and then cooled slowly down to room temperature (10 °C min−1). For the preparation of samples, the monolayers of PCBA were formed by incubating the freshly cleaned AuMica substrates in a saturated solution of PCBA in THF for 24 h. The substrate was rinsed with THF and dried under gentle flow of N2. We then immersed them in two different PSI protein solutions, one extracted from cyanobacteria and the other extracted from spinach leaves, and incubated them for 8 h. After 8 h, the AuMica substrates on which protein was deposited were gently rinsed with DI water and dried with N2.Temperature-dependent measurements in cryogenic probe station
By using the EGaIn setup as the top contact in the cryogenic probe station, electrical measurements of samples were performed over a wide range of temperatures. This setup is specifically designed to create a thermal gradient across junctions by utilizing conical EGaIn tips. The upper electrode consists of a conical EGaIn tip, while the lower electrode is made of AuMica. These components are enclosed within a stainless-steel vacuum chamber, equipped with a glass window that allows observation of the electrodes from the outside using a camera. A Blackfly 5.0 BFS-U3-50S5C camera with a zoom lens was used to view the junctions. The J/V measurements of the junctions as a function of temperature were carried out in a cryogenic Lakeshore probe station under a vacuum. A code written in LabView was used to operate the Keithley source meter (Model 6430 SUB Femtoamp remote source meter) and record the J/V curves. In this setup, the change of pressure from ambient to vacuum and solidification of the bulk EGaIn between 243–248 K did not result in shorts, open circuits, or change the electrical characteristics of the devices notably in any other way. The devices were slowly cooled, and their J/V characteristics were measured at intervals of 5 K and 10 K, allowing the devices to stabilize before performing each scan. The method relies on EGaIn top electrodes stabilized in a syringe, providing stability and enabling measurements over a large range of temperatures (150–353 K). In each set of experiments, voltage sweep was 0 → 1 V → 0 → −1 V → 0. Data collection involved measuring a total of 12 junctions, with 10 scans performed per junction at each temperature.Stability measurements at room temperature
For stability measurements at room temperature, electrical measurements were performed in a custom-built Faraday cage using a Keithley 6430 source meter in ambient conditions with cone-shaped EGaIn top junctions. Bias was applied to a syringe filled with EGaIn. Data were obtained by sweeping the potential from −1.0 V to 1.0 V at a scan rate of 0.1 V. For all measurements, the bottom AuMica substrate was electrically grounded while the EGaIn top contact was biased. In each set of experiments, voltage sweep was (0 → 1.0 V → 0 → −1.0 V → 0).Atomic force microscopy (AFM)
The surface morphology of the samples was characterized using tapping mode AFM (Atomic Force Microscopy) with ScanAsyst-Air probes (Bruker), which have a spring constant of k = 0.4 N m−1 and a resonance frequency of 70 kHz. The AFM images were acquired at a scan rate of 0.7 Hz and a resolution of 512 samples per line. Subsequently, the obtained AFM images were analyzed using Bruker's NanoScope Analysis 1.5 software.Data availability
The data supporting this article have been included as part of the ESI.†The open-source software used to process raw I/V data can be downloaded via this https://doi.org/10.5281/zenodo.6422417.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professor Bert Poolman and Gea Schuurman at the Department of Biochemistry at the University of Groningen for their cooperation during the extraction of PSI from spinach leaves. N. T. acknowledges C. L. Mthembu and Dr N. Rotthowe for their technical assistance with the cryogenic probe station.References
- A. Vilan, D. Aswal and D. Cahen, Large-Area, Ensemble Molecular Electronics: Motivation and Challenges, Chem. Rev., 2017, 117, 4248–4286, DOI:10.1021/acs.chemrev.6b00595.
- A. Nitzan and M. A. Ratner, Electron Transport in Molecular Wire Junctions, Science, 2003, 300, 1384–1389, DOI:10.1126/science.1081572.
- D. M. Adams, L. Brus, C. E. D. Chidsey, S. Creager, C. Creutz, C. R. Kagan, P. V. Kamat, M. Lieberman, S. Lindsay, R. A. Marcus, R. M. Metzger, M. E. Michel-Beyerle, J. R. Miller, M. D. Newton, D. R. Rolison, O. Sankey, K. S. Schanze, J. Yardley and X. Zhu, Charge Transfer on the Nanoscale: Current Status, J. Phys. Chem. B, 2003, 107, 6668–6697, DOI:10.1021/jp0268462.
- Y. Liu, X. Qiu, S. Soni and R. C. Chiechi, Charge Transport Through Molecular Ensembles: Recent Progress in Molecular Electronics, Chem. Phys. Rev., 2021, 2, 021303, DOI:10.1063/5.0050667.
- X. Liu, H. Yang, H. Harb, R. Samajdar, T. J. Woods, O. Lin, Q. Chen, A. I. B. Romo, J. Rodríguez-López, R. S. Assary, J. S. Moore and C. M. Schroeder, Shape-Persistent Ladder Molecules Exhibit Nanogap-Independent Conductance in Single-Molecule Junctions, ChemRxiv, 2023.
- S. Li, H. Yu, J. Li, N. Angello, E. R. Jira, B. Li, M. D. Burke, J. S. Moore and C. M. Schroeder, Transition Between Nonresonant and Resonant Charge Transport in Molecular Junctions, Nano Lett., 2021, 21, 8340–8347, DOI:10.1021/acs.nanolett.1c02915.
- B. Lawson, E. Vidal, M. M. Haley and M. Kamenetska, Extreme anti-ohmic conductance enhancement in neutral diradical acene-like molecular junctions, 2024, DOI:10.48550/ARXIV.2403.04906.
- B. Lawson, E. Vidal, M. M. Haley and M. Kamenetska, Extreme anti-ohmic conductance enhancement in neutral diradical acene-like molecular junctions, 2024, DOI:10.48550/ARXIV.2403.04906.
- X. Pan, E. Montes, W. Y. Rojas, B. Lawson, H. Vázquez and M. Kamenetska, Cooperative Self-Assembly of Dimer Junctions Driven by Stacking Leads to Conductance Enhancement, Nano Lett., 2023, 23, 6937–6943, DOI:10.1021/acs.nanolett.3c01540.
- H. E. Skipper, C. V. May, A. L. Rheingold, L. H. Doerrer and M. Kamenetska, Hard–Soft Chemistry Design Principles for Predictive Assembly of Single Molecule-Metal Junctions, J. Am. Chem. Soc., 2021, 143, 16439–16943, DOI:10.1021/acs.nanolett.3c01540.
- H. E. Skipper, B. Lawson, X. Pan, V. Degtiareva and M. Kamenetska, Manipulating Quantum Interference Between and Orbitals in Single-Molecule Junctions via Chemical Substitution and Environmental Control, ACS Nano, 2023, 17, 16107–16114, DOI:10.1021/acsnano.3c04963.
- O. A. Al-Owaedi, S. Bock, D. C. Milan, M.-C. Oerthel, M. S. Inkpen, D. S. Yufit, A. N. Sobolev, N. J. Long, T. Albrecht, S. J. Higgins, M. R. Bryce, R. J. Nichols, C. J. Lambert and P. J. Low, Insulated Molecular Wires: Inhibiting Orthogonal Contacts in Metal Complex Based Molecular Junctions, Nanoscale, 2017, 9, 9902–9912, 10.1039/c7nr01829k.
- L. Herrer, S. Naghibi, I. Marín, J. S. Ward, J. M. Bonastre, S. J. Higgins, S. Martín, A. Vezzoli, R. J. Nichols, J. L. Serrano and P. Cea, Sheathed Molecular Junctions for Unambiguous Determination of Charge-Transport Properties, Adv. Mater. Interfaces, 2023, 10, 2300133, DOI:10.1002/admi.202300133.
- A. Operamolla, R. Ragni, F. Milano, R. R. Tangorra, A. Antonucci, A. Agostiano, M. Trotta and G. Farinola, Garnishing the Photosynthetic Bacterial Reaction Center for Bioelectronics, J. Mater. Chem. C, 2015, 3, 6471–6478, 10.1039/c5tc00775e.
- T. Kothe, N. Plumeré, A. Badura, M. M. Nowaczyk, D. A. Guschin, M. Rögner and W. Schuhmann, Combination of a Photosystem1-Based Photocathode and a Photosystem2-Based Photoanode to a Z-Scheme Mimic for Biophotovoltaic Applications, Angew. Chem., Int. Ed., 2013, 52, 14233–14236, DOI:10.1002/anie.201303671.
- M. T. Giardi and E. Pace, Photosynthetic Proteins for Technological Applications, Trends Biotechnol., 2005, 23, 257–263, DOI:10.1016/j.tibtech.2005.03.003.
- C. Romero-Muñiz, M. Ortega, J. G. Vilhena, I. Diéz-Pérez, J. C. Cuevas, R. Pérez and L. A. Zotti, Mechanical Deformation and Electronic Structure of a Blue Copper Azurin in a Solid-State Junction, Biomolecules, 2019, 9, 506, DOI:10.3390/biom9090506.
- N. L. Ing, M. Y. El-Naggar and A. I. Hochbaum, Going the Distance: Long-Range Conductivity in Protein and Peptide Bioelectronic Materials, J. Phys. Chem. B, 2018, 122, 10403–10423, DOI:10.1021/acs.jpcb.8b07431.
- N. Amdursky, D. Marchak, L. Sepunaru, I. Pecht, M. Sheves and D. Cahen, Electronic Transport via Proteins, Adv. Mater., 2014, 26, 7142–7161, DOI:10.1002/adma.201402304.
- J. A. Fereiro, T. Bendikov, I. Pecht, M. Sheves and D. Cahen, Protein Binding and Orientation Matter: Bias-Induced Conductance Switching in a Mutated Azurin Junction, J. Am. Chem. Soc., 2020, 142, 19217–19225, DOI:10.1021/jacs.0c08836.
- K. Garg, M. Ghosh, T. Eliash, J. H. van Wonderen, J. N. Butt, L. Shi, X. Jiang, F. Zdenek, J. Blumberger, I. Pecht, M. Sheves and D. Cahen, Direct Evidence for Heme-Assisted Solid-State Electronic Conduction in Multi-Heme-Type Cytochromes, Chem. Sci., 2018, 9, 7304–7310, 10.1039/c8sc01716f.
- J. A. Fereiro, X. Yu, I. Pecht, M. Sheves, J. C. Cuevas and D. Cahen, Tunneling Explains Efficient Electron Transport via Protein Junctions, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4577–E4583, DOI:10.1073/pnas.1719867115.
- M. W. Shinwari, M. J. Deen, E. B. Starikov and G. Cuniberti, Electrical Conductance in Biological Molecules, Adv. Funct. Mater., 2010, 20, 1865–1883, DOI:10.1002/adfm.200902066.
- S. K. Saxena, U. M. Tefashe and R. L. McCreery, Photostimulated Near-Resonant Charge Transport Over 60 Nm in Carbon-Based Molecular Junctions, J. Am. Chem. Soc., 2020, 142, 15420–15430, DOI:10.1021/jacs.0c06764.
- Z. Futera, I. Ide, B. Kayser, K. Garg, X. Jiang, J. H. van Wonderen, J. N. Butt, H. Ishii, I. Pecht, M. Sheves, D. Cahen and J. Blumberger, Coherent Electron Transport Across a 3 Nm Bioelectronic Junction Made of Multi-Heme Proteins, J. Phys. Chem. Lett., 2020, 11, 9766–9774, DOI:10.1021/acs.jpclett.0c02686.
- B. Kayser, J. A. Fereiro, R. Bhattacharyya, S. R. Cohen, A. Vilan, I. Pecht, M. Sheves and D. Cahen, Solid-State Electron Transport via the Protein Azurin Is Temperature-Independent Down to 4 K, J. Phys. Chem. Lett., 2019, 11, 144–151, DOI:10.1021/acs.jpclett.9b03120.
- M. P. Ruiz, A. C. Aragonès, N. Camarero, J. G. Vilhena, M. Ortega, L. A. Zotti, R. Pérez, J. C. Cuevas, P. Gorostiza and I. Díez-Pérez, Bioengineering a Single-Protein Junction, J. Am. Chem. Soc., 2017, 139, 15337–15346, DOI:10.1021/jacs.7b06130.
- H. B. Gray and J. R. Winkler, Long-Range Electron Transfer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3534–3539, DOI:10.1073/pnas.0408029102.
- Q. Chi, J. Zhang, J. U. Nielsen, E. P. Friis, I. Chorkendorff, G. W. Canters, J. E. T. Andersen and J. Ulstrup, Molecular Monolayers and Interfacial Electron Transfer of Pseudomonas Aeruginosa Azurin on Au(111), J. Am. Chem. Soc., 2000, 122, 4047–4055, DOI:10.1021/ja993174t.
- N. Amdursky, D. Ferber, I. Pecht, M. Sheves and D. Cahen, Redox Activity Distinguishes Solid-State Electron Transport From Solution-Based Electron Transfer in a Natural and Artificial Protein: Cytochrome C and Hemin-Doped Human Serum Albumin, Phys. Chem. Chem. Phys., 2013, 15, 17142, 10.1039/c3cp52885e.
- M. Koch, F. Ample, C. Joachim and L. Grill, Voltage-Dependent Conductance of a Single Graphene Nanoribbon, Nat. Nanotechnol., 2012, 7, 713–717, DOI:10.1038/nnano.2012.169.
- K. S. Kumar, R. R. Pasula, S. Lim and C. A. Nijhuis, Long-Range Tunneling Processes Across Ferritin-Based Junctions, Adv. Mater., 2015, 28, 1824–1830, DOI:10.1002/adma.201504402.
- O. E. C. Ocampo, P. Gordiichuk, S. Catarci, D. A. Gautier, A. Herrmann and R. C. Chiechi, Mechanism of Orientation-Dependent Asymmetric Charge Transport in Tunneling Junctions Comprising Photosystem I, J. Am. Chem. Soc., 2015, 137, 8419–8427, DOI:10.1021/jacs.5b01241.
- H. Yan, A. J. Bergren, R. McCreery, M. L. D. Rocca, P. Martin, P. Lafarge and J. C. Lacroix, Activationless Charge Transport Across 4.5 to 22 Nm in Molecular Electronic Junctions, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5326–5330, DOI:10.1073/pnas.1221643110.
- S. Bera, J. A. Fereiro, S. K. Saxena, D. Chryssikos, K. Majhi, T. Bendikov, L. Sepunaru, D. Ehre, M. Tornow, I. Pecht, A. Vilan, M. Sheves and D. Cahen, Near-Temperature-Independent Electron Transport Well Beyond Expected Quantum Tunneling Range via Bacteriorhodopsin Multilayers, J. Am. Chem. Soc., 2023, 24820–24835, DOI:10.1021/jacs.3c09120.
- P. Gordiichuk, D. Pesce, O. E. C. Ocampo, A. Marcozzi, G.-J. A. H. Wetzelaer, A. Paul, M. Loznik, E. Gloukhikh, S. Richter, R. C. Chiechi and A. Herrmann, Orientation and Incorporation of Photosystem I in Bioelectronics Devices Enabled by Phage Display, Adv. Sci., 2017, 4, 1600393, DOI:10.1002/advs.201600393.
- X. Qiu, V. Ivasyshyn, L. Qiu, M. Enache, J. Dong, S. Rousseva, G. Portale, M. Stöhr, J. C. Hummelen and R. C. Chiechi, Thiol-Free Self-Assembled Oligoethylene Glycols Enable Robust Air-Stable Molecular Electronics, Nat. Mater., 2020, 19, 330–337, DOI:10.1038/s41563-019-0587-x.
- N. Torabi, X. Qiu, M. López-Ortiz, M. Loznik, A. Herrmann, A. Kermanpur, A. Ashrafi and R. C. Chiechi, Fullerenes Enhance Self-Assembly and Electron Injection of Photosystem I in Biophotovoltaic Devices, Langmuir, 2021, 37, 11465–11473, DOI:10.1021/acs.langmuir.1c01542.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauß, Three-Dimensional Structure of Cyanobacterial Photosystem I at 2.5A Resolution, Nature, 2001, 411, 909–917, DOI:10.1038/35082000.
- P. R. Chitnis, Photosystem I: Function and Physiology, Annu. Rev. Plant Physiol. Plant Mol. Biol., 2001, 52, 593–626, DOI:10.1146/annurev.arplant.52.1.593.
- A. Ben-Shem, F. Frolow and N. Nelson, Crystal Structure of Plant Photosystem I, Nature, 2003, 426, 630–635, DOI:10.1038/nature02200.
- P. Wang, F. Zhao, A. Frank, S. Zerria, A. Lielpetere, A. Ruff, M. M. Nowaczyk, W. Schuhmann and F. Conzuelo, Rational Design of a Photosystem I Photoanode for the Fabrication of Biophotovoltaic Devices, Adv. Energy Mater., 2021, 11, 2102858, DOI:10.1002/aenm.202102858.
- M. K. Şener, S. Park, D. Lu, A. Damjanović, T. Ritz, P. Fromme and K. Schulten, Excitation Migration in Trimeric Cyanobacterial Photosystem I, J. Chem. Phys., 2004, 120, 11183–11195, DOI:10.1063/1.1739400.
- P. Fromme, E. Schlodder and S. Jansson, Light-Harvesting Antennas in Photosynthesis, Springer Netherlands, 2003, pp. 253–279, DOI:10.1007/978-94-017-2087-8_8.
- A. Kitmitto, A. O. Mustafa, A. Holzenburg and R. C. Ford, Three-Dimensional Structure of Higher Plant Photosystem I Determined by Electron Crystallography, J. Biol. Chem., 1998, 273, 29592–29599, DOI:10.1074/jbc.273.45.29592.
- R. Kouřil, N. van Oosterwijk, A. E. Yakushevska and E. J. Boekema, Photosystem I: A Search for Green Plant Trimers, Photochem. Photobiol. Sci., 2005, 4, 1091–1094, 10.1039/b505519a.
- Z. Gardian, L. Bumba, A. Schrofel, M. Herbstova, J. Nebesarova and F. Vacha, Organisation of Photosystem I and Photosystem II in Red Alga Cyanidium Caldarium: Encounter of Cyanobacterial and Higher Plant Concepts, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 725–731, DOI:10.1016/j.bbabio.2007.01.021.
- F. Klimmek, U. Ganeteg, J. A. Ihalainen, H. van Roon, P. E. Jensen, H. V. Scheller, J. P. Dekker and S. Jansson, Structure of the Higher Plant Light Harvesting Complex I: In Vivo Characterization and Structural Interdependence of the Lhca Proteins, Biochemistry, 2005, 44, 3065–3073, DOI:10.1021/bi047873g.
- M. K. Şener, C. Jolley, A. Ben-Shem, P. Fromme, N. Nelson, R. Croce and K. Schulten, Comparison of the Light-Harvesting Networks of Plant and Cyanobacterial Photosystem I, Biophys. J., 2005, 89, 1630–1642, DOI:10.1529/biophysj.105.066464.
- T. S. Bibby, J. Nield and J. Barber, Iron Deficiency Induces the Formation of an Antenna Ring Around Trimeric Photosystem I in Cyanobacteria, Nature, 2001, 412, 743–745, DOI:10.1038/35089098.
- T. S. Bibby, J. Nield and J. Barber, Three-Dimensional Model and Characterization of the Iron Stress-Induced CP43′-Photosystem I Supercomplex Isolated From the Cyanobacterium Synechocystis PCC 6803, J. Biol. Chem., 2001, 276, 43246–43252, DOI:10.1074/jbc.m106541200.
- E. J. Boekema, A. Hifney, A. E. Yakushevska, M. Piotrowski, W. Keegstra, S. Berry, K.-P. Michel, E. K. Pistorius and J. Kruip, A Giant Chlorophyll–protein Complex Induced by Iron Deficiency in Cyanobacteria, Nature, 2001, 412, 745–748, DOI:10.1038/35089104.
- A. N. Melkozernov, J. Kargul, S. Lin, J. Barber and R. E. Blankenship, Energy Coupling in the PSI-LHCI Supercomplex From the Green Alga Chlamydomonas Reinhardti, J. Phys. Chem. B, 2004, 108, 10547–10555, DOI:10.1021/jp049375n.
- L. Liu and S. Choi, Enhanced Biophotoelectricity Generation in Cyanobacterial Biophotovoltaics With Intracellularly Biosynthesized Gold Nanoparticles, J. Power Sources, 2021, 506, 230251, DOI:10.1016/j.jpowsour.2021.230251.
- M. Brecht, M. Hussels, E. Schlodder and N. V. Karapetyan, Red Antenna States of Photosystem I Trimers From Arthrospira Platensis Revealed by Single-Molecule Spectroscopy, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 445–452, DOI:10.1016/j.bbabio.2011.11.012.
- T. S. Bibby, I. Mary, J. Nield, F. Partensky and J. Barber, Low-Light-Adapted Prochlorococcus Species Possess Specific Antennae for Each Photosystem, Nature, 2003, 424, 1051–1054, DOI:10.1038/nature01933.
- P. Bombelli, T. Müller, T. W. Herling, C. J. Howe and T. P. J. Knowles, A High Power-Density, Mediator-Free, Microfluidic Biophotovoltaic Device for Cyanobacterial Cells, Adv. Energy Mater., 2014, 5, 1401299, DOI:10.1002/aenm.201401299.
- L. Liu and S. Choi, PEDOT:PSS/MnOCNT Ternary Nanocomposite Anodes for Supercapacitive Energy Storage in Cyanobacterial Biophotovoltaics, ACS Appl. Energy Mater., 2020, 3, 10224–10233, DOI:10.1021/acsaem.0c02054.
- J. A. Fereiro, T. Bendikov, A. Herrmann, I. Pecht, M. Sheves and D. Cahen, Protein Orientation Defines Rectification of Electronic Current via Solid-State Junction of Entire Photosystem-1 Complex, J. Phys. Chem. Lett., 2023, 14, 2973–2982, DOI:10.1021/acs.jpclett.2c03700.
- X. Qiu, O. Castañeda Ocampo, H. W. de Vries, M. van Putten, M. Loznik, A. Herrmann and R. C. Chiechi, Self-Regenerating Soft Biophotovoltaic Devices, ACS Appl. Mater. Interfaces, 2018, 10, 37625–37633, DOI:10.1021/acsami.8b11115.
- E. A. Gizzie, G. LeBlanc, G. K. Jennings and D. E. Cliffel, Electrochemical Preparation of Photosystem I–Polyaniline Composite Films for Biohybrid Solar Energy Conversion, ACS Appl. Mater. Interfaces, 2015, 7, 9328–9335, DOI:10.1021/acsami.5b01065.
- K. R. Stieger, D. Ciornii, A. Kölsch, M. Hejazi, H. Lokstein, S. C. Feifel, A. Zouni and F. Lisdat, Engineering of Supramolecular Photoactive Protein Architectures: The Defined Co-Assembly of Photosystem I and Cytochrome C Using a Nanoscaled DNA-matrix, Nanoscale, 2016, 8, 10695–10705, 10.1039/c6nr00097e.
- P. N. Ciesielski, F. M. Hijazi, A. M. Scott, C. J. Faulkner, L. Beard, K. Emmett, S. J. Rosenthal, D. Cliffel and G. K. Jennings, Photosystem I – Based Biohybrid Photoelectrochemical Cells, Bioresour. Technol., 2010, 101, 3047–3053, DOI:10.1016/j.biortech.2009.12.045.
- P. I. Gordiichuk, G.-J. A. H. Wetzelaer, D. Rimmerman, A. Gruszka, J. W. de Vries, M. Saller, D. A. Gautier, S. Catarci, D. Pesce, S. Richter, P. W. M. Blom and A. Herrmann, Solid-State Biophotovoltaic Cells Containing Photosystem I, Adv. Mater., 2014, 26, 4863–4869, DOI:10.1002/adma.201401135.
- X. Qiu and R. C. Chiechi, Printable Logic Circuits Comprising Self-Assembled Protein Complexes, Nat. Commun., 2022, 13, 2312, DOI:10.1038/s41467-022-30038-8.
- R. C. Chiechi, E. A. Weiss, M. D. Dickey and G. M. Whitesides, Eutectic Gallium-Indium (EGaIn): A Moldable Liquid Metal for Electrical Characterization of Self-Assembled Monolayers, Angew. Chem., Int. Ed., 2008, 47, 142–144, DOI:10.1002/anie.200703642.
- E. M. Freer, O. Grachev, X. Duan, S. Martin and D. P. Stumbo, High-Yield Self-Limiting Single-Nanowire Assembly With Dielectrophoresis, Nat. Nanotechnol., 2010, 5, 525–530, DOI:10.1038/nnano.2010.106.
- C. A. Nijhuis, W. F. Reus, J. R. Barber and G. M. Whitesides, Comparison of SAM-Based Junctions With EGaIn Top Electrodes to Other Large-Area Tunneling Junctions, J. Phys. Chem. C, 2012, 116, 14139–14150, DOI:10.1021/jp303072a.
- P. A. Smith, C. D. Nordquist, T. N. Jackson, T. S. Mayer, B. R. Martin, J. Mbindyo and T. E. Mallouk, Electric-Field Assisted Assembly and Alignment of Metallic Nanowires, Appl. Phys. Lett., 2000, 77, 1399–1401, DOI:10.1063/1.1290272.
- X. Qiu, S. Rousseva, G. Ye, J. C. Hummelen and R. C. Chiechi, In Operando Modulation of Rectification in Molecular Tunneling Junctions Comprising Reconfigurable Molecular Self-Assemblies, Adv. Mater., 2021, 33, 2006109, DOI:10.1002/adma.202006109.
- A. Kovalchuk, T. Abu-Husein, D. Fracasso, D. Egger, E. Zojer, M. Zharnikov, A. Terfort and R. Chiechi, Transition Voltages Respond to Synthetic Reorientation of Embedded Dipoles in Self-Assembled Monolayers, Chem. Sci., 2016, 7, 781–787 RSC.
- A. J. Kronemeijer, E. H. Huisman, H. B. Akkerman, A. M. Goossens, I. Katsouras, P. A. van Hal, T. C. T. Geuns, S. J. van der Molen, P. W. M. Blom and D. M. de Leeuw, Electrical Characteristics of Conjugated Self-Assembled Monolayers in Large-Area Molecular Junctions, Appl. Phys. Lett., 2010, 97, 173302, DOI:10.1063/1.3503607.
- T. Hines, I. Diez-Perez, J. Hihath, H. Liu, Z.-S. Wang, J. Zhao, G. Zhou, K. Müllen and N. Tao, Transition From Tunneling to Hopping in Single Molecular Junctions by Measuring Length and Temperature Dependence, J. Am. Chem. Soc., 2010, 132, 11658–11664, DOI:10.1021/ja1040946.
- J. G. Simmons, Generalized Formula for the Electric Tunnel Effect Between Similar Electrodes Separated by a Thin Insulating Film, J. Appl. Phys., 1963, 34, 1793–1803, DOI:10.1063/1.1702682.
- R. Naaman, D. H. Waldeck and J. Fransson, New Perspective on Electron Transfer Through Molecules, J. Phys. Chem. Lett., 2022, 13, 11753–11759, DOI:10.1021/acs.jpclett.2c03141.
- G. Puebla-Hellmann, K. Venkatesan, M. Mayor and E. Lörtscher, Metallic Nanoparticle Contacts for High-Yield, Ambient-Stable Molecular-Monolayer Devices, Nature, 2018, 559, 232–235, DOI:10.1038/s41586-018-0275-z.
- H. B. Akkerman, P. W. M. Blom, D. M. de Leeuw and B. de Boer, Towards Molecular Electronics With Large-Area Molecular Junctions, Nature, 2006, 441, 69–72, DOI:10.1038/nature04699.
- H. B. Akkerman, A. J. Kronemeijer, J. Harkema, P. A. van Hal, E. C. Smits, D. M. de Leeuw and P. W. Blom, Stability of Large-Area Molecular Junctions, Org. Electron., 2010, 11, 146–149, DOI:10.1016/j.orgel.2009.09.013.
- R. L. McCreery, Carbon-Based Molecular Junctions for Practical Molecular Electronics, Acc. Chem. Res., 2022, 55, 2766–2779, DOI:10.1021/acs.accounts.2c00401.
- N. Chen, S. Li, P. Zhao, R. Liu, Y. Xie, J.-L. Lin, C. A. Nijhuis, B. Xu, L. Zhang, H. Xu and Y. Li, Extreme Long-Lifetime Self-Assembled Monolayer for Air-Stable Molecular Junctions, Sci. Adv., 2023, 9, eadh3412, DOI:10.1126/sciadv.adh3412.
- E. D. Mentovich, B. Belgorodsky, I. Kalifa, H. Cohen and S. Richter, Large-Scale Fabrication of 4-Nm-Channel Vertical Protein-Based Ambipolar Transistors, Nano Lett., 2009, 9, 1296–1300, DOI:10.1021/nl802694k.
- L. Yuan, L. Jiang, D. Thompson and C. A. Nijhuis, On the Remarkable Role of Surface Topography of the Bottom Electrodes in Blocking Leakage Currents in Molecular Diodes, J. Am. Chem. Soc., 2014, 136, 6554–6557, DOI:10.1021/ja5007417.
- E. A. Weiss, R. C. Chiechi, G. K. Kaufman, J. K. Kriebel, Z. Li, M. Duati, M. A. Rampi and G. M. Whitesides, Influence of Defects on the Electrical Characteristics of Mercury-Drop Junctions: Self-Assembled Monolayers of N-Alkanethiolates on Rough and Smooth Silver, J. Am. Chem. Soc., 2007, 129, 4336–4349, DOI:10.1021/ja0677261.
- X. Qin, W. Wang, K. Wang, Y. Xin and T. Kuang, Isolation and Characteristics of the PSI-LHCI-LHCII Supercomplex Under High Light, Photochem. Photobiol., 2010, 87, 143–150, DOI:10.1111/j.1751-1097.2010.00830.x.
- R. Porra, W. Thompson and P. Kriedemann, Determination of Accurate Extinction Coefficients and Simultaneous Equations for Assaying Chlorophylls a and B Extracted With Four Different Solvents: Verification of the Concentration of Chlorophyll Standards by Atomic Absorption Spectroscopy, Biochim. Biophys. Acta, Bioenerg., 1989, 975, 384–394, DOI:10.1016/s0005-2728(89)80347-0.
- R. Rippka, R. Y. Stanier, J. Deruelles, M. Herdman and J. B. Waterbury, Generic Assignments, Strain Histories and Properties of Pure Cultures of Cyanobacteria, Microbiology, 1979, 111, 1–61, DOI:10.1099/00221287-111-1-1.
- E. El-Mohsnawy, M. J. Kopczak, E. Schlodder, M. Nowaczyk, H. E. Meyer, B. Warscheid, N. V. Karapetyan and M. Rögner, Structure and Function of Intact Photosystem 1 Monomers From the Cyanobacterium Thermosynechococcus Elongatus, Biochemistry, 2010, 49, 4740–4751, DOI:10.1021/bi901807p.
- D. Mukherjee, M. May, M. Vaughn, B. D. Bruce and B. Khomami, Controlling the Morphology of Photosystem I Assembly on Thiol-Activated Au Substrates, Langmuir, 2010, 26, 16048–16054, DOI:10.1021/la102832x.
- K. Baba, S. Itoh, G. Hastings and S. Hoshina, Photoinhibition of Photosystem I Electron Transfer Activity in Isolated Photosystem I Preparations With Different Chlorophyll Contents, Photosynth. Res., 1996, 47, 121–130, DOI:10.1007/bf00016175.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02554g |
| This journal is © The Royal Society of Chemistry 2024 |