
Synergism between copper and silver nanoclusters induces fascinating structural modifications, properties, and applications
Priyanka
Sharma
a,
Mainak
Ganguly
 *a and
Ankita
Doi
b
*a and
Ankita
Doi
b
aSolar Energy Conversion and Nanomaterials Laboratory, Department of Chemistry, Manipal University Jaipur, Dehmi Kalan, Jaipur 303007, India. E-mail: mainak.ganguly@jaipur.manipal.edu
bDepartment of BioSciences, Manipal University Jaipur, Dehmi Kalan, Jaipur 303007, India
First published on 10th September 2024
Abstract
Among the group 11 transition metal elements, Cu and Ag are widely studied due to their cost effectiveness and easy availability. However, the synergism between copper and silver is also very promising, exhibiting intriguing structures, properties, and applications. Nanoclusters, which are missing links between atoms and nanoparticles, are highly fluorescent due to their discrete energy levels. Their fluorescence can be efficiently tuned because of the synergistic behaviour of copper and silver. Furthermore, their fluorescence can be selectively altered in the presence of various analytes and sensing platforms, as reported by various groups. Moreover, copper clusters can be utilized for sensing silver while silver nanoclusters can be utilized for sensing ionic copper due to the strong interaction between copper and silver. Furthermore, DFT studies have been performed to understand the structural modification due to CuAg synergism. A concise summary of the synergism between copper and silver can open a new window of research for young scientists venturing into the field of environmental nanoscience.
 Priyanka Sharma | Priyanka Sharma obtained her B.Sc. and M.Sc. degrees from Maharaja Brij University in India. She is now pursuing her Ph.D. degree at Manipal University Jaipur, India, under the guidance of Dr Mainak Ganguly. Her area of interest is materials science and environmental science. |
 Mainak Ganguly | Dr Mainak Ganguly received his Ph.D. degree from the Indian Institute of Technology, Kharagpur, India in 2014. He was a Postdoctoral Researcher up to 2019 at Furman University (USA) and McGill University (Canada). He is currently working as an Asst. Professor in the Department of Chemistry, Manipal University Jaipur (India). His research interests include nanoparticles, clusters, biophysical chemistry, environmental remediations, etc. He has published more than 65 papers and two book chapters. |
 Ankita Doi | Ankita Doi received her graduate degree from Alankar P.G. Mahavidyalaya (Jaipur, Rajasthan, India) and postgraduate degree from Alankar P.G. Mahavidyalya (Jaipur, Rajasthan). Currently, she is pursuing a Ph.D. from Manipal University, Jaipur under the supervision of Dr Mainak Ganguly. Her research area is nanoparticles. |
1. Introduction
According to researchers, nanoparticles (NPs) possess several novel and exciting properties not seen in bulk materials, as revealed in the last 20 years.1 Accordingly, research has focused on the creation of metal NPs (>2 nm) because of their promising application in optics, sensing, drug administration, and catalysis.2–4 The finding of metal nanoclusters (MCNs) (<2 nm) (e.g., platinum, gold, silver, and copper) has substantially expanded the scope of nanomaterials (NMs) science. MCNs, which are made up of a few to a few hundred atoms, are a novel type of luminous NMs that have received significant interest in recent years.5,6 In these nanosystems, as their size approaches the Fermi-wavelength of e−, they display molecule-like properties, endowing them with an important function: the missing link between solitary metal-atoms and plasmonic metal NPs.7–9 MCNs have significantly different electrical, optical, and chemical capabilities than larger metal nanoparticles due to their distinct energy levels.10–14 Their intense photoluminescence, easy synthesis, good quantum yields, controllable fluorescence emission, significant Stokes shift, and great photostability distinguish them from other types of particles.15–18 MCNs can be passivated by an organic ligand monolayer, producing high fluorescence intensity.19–23Wei et al.24 indicated that the Ag29(SSR)12 framework is made up of an Ag13kernel, which is icosahedral and supported by an Ag12(SSR)12 shell. Four bare Ag atoms with a tetrahedral pattern cap the resulting Ag25(SSR)12 structure.
The CN state of matter has long piqued the interest of chemists and physicists, given that it answers many fundamental questions such as metallic bond formation, the evolution of electronic and magnetic properties, and new assembly structures, among many other emergent phenomena. Thus, significant scientific research is being conducted on transition MCNs.25–28 This is partly due to the novelty of nanoclusters and the difficulties in comprehending their formation,29–31 stabilisation,32–34 aggregation,35 and features that rely on size and shape.36 Additionally, nanoclusters have the potential to be used in a variety of fields, such as catalysis (including catalysts for fuel cells),37–41 quantum computing,42 photochemistry,43 optics,44 nanoelectronics,45,46 and chemical sensors.47–49 Over the past few decades, monodisperse and structurally distinct coinage MCNs have attracted increasing research interest. This is because they offer a viable platform to link structures to characteristics and their remarkable structural diversity, unique luminescence, catalytic activity, and potential bio applications.50–59
In the previous decade, the definition of nanoclusters was unclear. For example, Ang et al.60 reported the preparation of AgCu nanoclusters with a size of ∼4–6 nm. Wang et al.61 stated that the size of the nanocluster was ∼3–10 nm. Pellarin et al.62 showed that the size of nanoclusters was ∼5 nm. Ganguly et al.63 reported the synthesis of AuAg giant clusters, where tiny silver clusters (Ag2, Ag3) were decorated on the surface of gold nanospheres.
Copper and CuCNs are the most promising NMs among the noble metals given that metallic Cu has good electrical conductivity and is inexpensive. Furthermore, CuCNs are important components in contemporary electrical circuits. Cu-based nanomaterials are widely used because of their superior conductivity and biocompatibility, particularly as a crucial component in future nanodevices. However, the synthesis of CuCNs is a greater challenge than noble metals such as Ag and Au due to their increasing instability in aqueous solutions. Specifically, their exposure to air leads to rapid surface oxidation and aggregation. Scientists mitigate this by employing reduction methods to produce CuCNs in an inert environment using organic solvents with the aid of protective polymers to prevent oxidation.64–67 CuCNs can act as a cost-effective material for a variety of applications. Moreover, the majority of known water-soluble CuCNs emit blue or green light at short wavelengths.68–75 However, due to their susceptibility to oxidation or difficulty in controlling their size during their synthesis, poor stability and low quantum yield (QY) are the major challenges associated with CuCNs, limiting their practical applications and inspiring researchers to develop simpler and more effective methods for their synthesis.76–86 In this case, the use of larger capping agents and employing innovative reaction conditions such as microwave-aided or sonochemical synthesis have shown promise in enhancing the performance of CuCNs. Furthermore, the aggregation-induced emission (AIE)86 and charge repulsion87 strategies have been expanded to enhance the optical characteristics of CuCNs.
AgCNs are particularly appealing among the noble MCNs identified thus far due to their unusual physical features, such as high fluorescence and ultra-small size. These qualities make it possible to build luminous probes for bioimaging and sensing applications. The Ag(0) valent state is more reactive and simpler to oxidize than Au, making it more difficult to make AgCNs and explore their characteristics compared to their gold equivalents, which have been extensively investigated. As a result, access to high-quality AgCNs with well-defined sizes, structures, and surfaces is critical for both basic and applied science.88,89
When two or more entities have a greater combined impact than the sum of their individual effects, this is known as a synergistic effect. The synergistic behavior of gold and silver bimetallic nanoclusters, as illustrated in Fig. 1, which also demonstrates the significance of the synergistic impact.90 In contrast to other elements in the periodic table, copper and silver, both belonging to Group 11, often demonstrate a synergistic interaction. Rout et al. investigated a bimetallic Ag–Cu alloy nanoparticle as a highly active and reusable catalyst for the reaction of 1,3-dicarbonyl with various amines.91 Hao et al. demonstrated the antibacterial mechanism between bimetallic Ag and copper nanoparticles.92 Ponrani et al. investigated the structural and optical behavior of bimetallic copper and silver nanoparticles.93 Sharma et al.94 demonstrated the complexation–reduction mechanism for the evolution of NPs for the synergistic detection of AgCu. Kukushkina et al.95 reviewed a silver-based synergistic antimicrobial composite and provided a systematic review of the current state of research in the development of hybrid Ag-based nanoantimicrobials in the presence of Cu.
 | ||
| Fig. 1 Synergistic effect of bimetallic nanocluster. Reproduced from ref. 90 with permission from RSC Adv., Copyright 2016. | ||
However, there is no review article in the literature on the synergistic behavior between copper and silver nanoclusters. Thus, in this review article, we review the synergism between copper and silver nanoclusters together with their applications. Also, the quenching mechanism with CuAg nanoclusters is discussed and the synergistic behavior of CuAg presented.
2. Structural modification with AgCu synergism
Structural modification of copper and silver nanoclusters involves altering the arrangement and composition of the atoms in these tiny clusters. Nanoclusters are aggregates of atoms that exhibit unique properties due to their small size, often on the nanometer scale.Sharma et al.96 examined the chemistry and synthesis of dichalcogenolato silver and copper clusters, detailing their precise structures. Among them, the [Cu13{S2CNR}6{CCR′}4]+ (denoted as A) cluster stood out as the only copper cluster featuring Cu13-centered cuboctahedra, mimicking the bulk fcc structure on a miniature scale. Through galvanic exchange, the central copper could be substituted with silver or gold, resulting in a similar structure for the formed bimetallic [Ag/Au@Cu12(S2CN′′Bu2)6(C = sCPh)4][CuCl2] species. The other corresponding M (13) cores in Group 11 super atomic chemistry typically exhibited compact icosahedral structures. As shown in Fig. 2, Hayton's clusters 1 and 2 are considered to consist of a central Cu13 icosahedron with a formal charge of +1, which contains the two jellium electrons. This icosahedron is shielded by an outer sphere composed of peripheral copper(I) ions. In cluster 4, 4 of the 8 tri-angular faces of the cube octahedron are bound by RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CM groups in a μ3 manner, and each of the 6-square faces is bridged by an R2C2S2 ligand in a (μ2, μ2) fashion.
CM groups in a μ3 manner, and each of the 6-square faces is bridged by an R2C2S2 ligand in a (μ2, μ2) fashion.
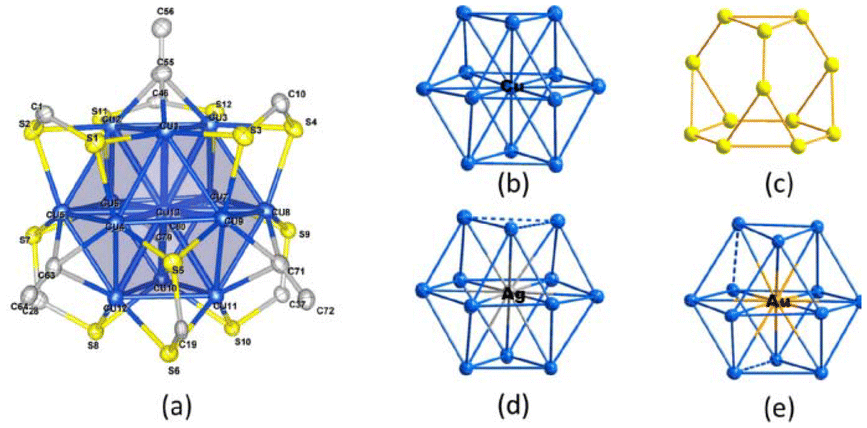 | ||
| Fig. 2 Structure of A. Reproduced from ref. 96 with permission from Acc. Chem. Res., Copyright 2018. | ||
Yan et al.97 utilized chiral ammonium cations to synthesize optically active, (−) charged chiral MNPs. They identified a racemic mixture, an RS-chiral three homocentric shell CN known as [Ag28Cu12(RS)24] (1), through XRD. TEM studies showed that the produced AgCu NPs possessed a uniform size of 2.06 ± 0.22 nm. As shown in Fig. 3, structure 1 exhibited a two-shell silver-4@silver-24 core shielded by four nearly planar Cu3(RS)6 (denoted by 2) moieties. The silver-4@silver-24 core consisted of an inward Td silver-4 core with four triangular silver-6 facets significantly twisted on each of its four faces. The Ag24 shell was formed by the interconnection of the four Ag6 facets, while the outer surface layer was composed of a tetrahedral cap of four Cu3(SR)6 moieties. Thiolates coordinated the Cu atoms in three dimensions. On the surface of structure 1, pairwise pi–pi interactions between the six-thiolates of 2-patterns were observed.
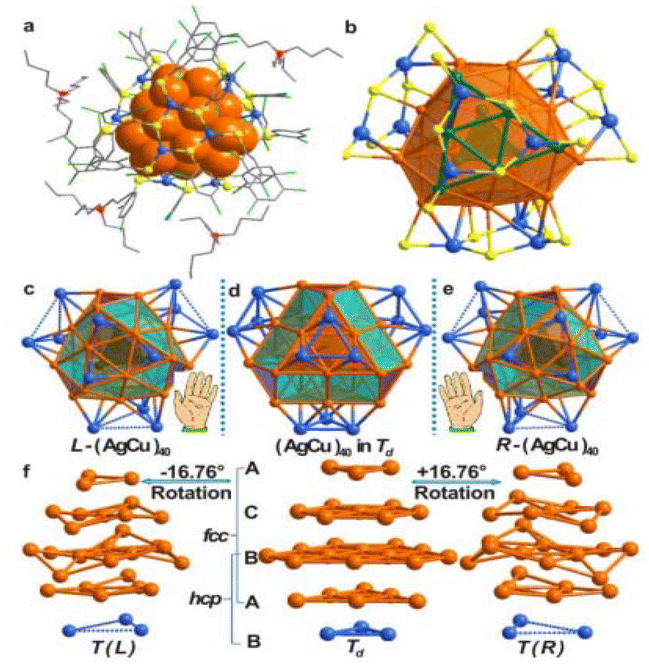 | ||
| Fig. 3 Structure of (nBu4N)4[Ag28Cu12(SR)24] (1·TBA) and CNs with the R- and L-groups. Reproduced from ref. 97, with permission from J. Am. Chem. Soc., Copyright 2016. | ||
Zou et al.98 investigated the structure of a bi-metallic [Ag61Cu30(Adms)38S3]BPh4 (Ag61Cu30) CNs. The Ag61Cu30 CNs were comprised of an exterior Ag48(AdmS)38S3 shell surrounding an Ag13@Cu30 kernel. It featured shell-by-shell structure, with 30 Cu atoms forming the center layer within Ag13@Cu30@Ag48(SAdm)38S3. An inherent characteristic of the Au/Ag-based nanoclusters was the presence of an icosahedral Ag13kernel, where typical Agkernel–Agkernelshell and Agkernelshell–Agkernelshell distances were measured to be 2.74 and 2.88 Å, respectively, which are slightly shorter than the bond length of 2.89 Å in bulk silver. The Cu–Cu distance within the Cu30 shell ranged from 2.82 to 2.89 Å, with an average distance of 2.87 Å, which is longer than the 2.6 Å bond length in bulk copper (Fig. 4). The silver–copper distance averaged 2.71 Å in the Ag13 kernel shell connected with 5 pentagonal copper atoms, which is less than the gold–copper bond length of 2.78 Å in Au19Cu3.99 The Ag/Cu atomic ratio was determined by XPS to be 8.99/3.94, which is consistent with the theoretical ratio of 61/30. The XPS data for Ag3d exhibited lower binding energies (BE) (368.87 eV) compared to silver(I) and higher BE (368.34 eV) compared to bulk silver. This indicated that the Ag in the Ag61Cu30 CNs had a valence state ranging from 0 to +1. There was evidence for the presence of copper(I) and copper(0) in the Cu30 layer based on the deconvolution of the BE of copper 2p3/2.100
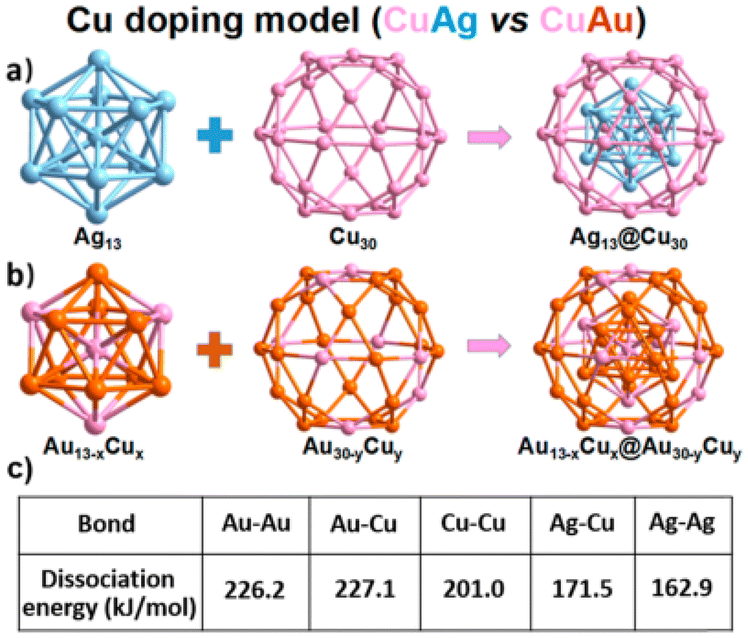 | ||
| Fig. 4 (a) Shell by shell alloying mode (AgCu alloying) and (b) mixed alloying mode (AuCu alloying). Reproduced from ref. 98, with permission from J. Phys. Chem. Lett., Copyright 2020. | ||
Yao et al.101 described the unusual structural transition of a Cu(I) cluster into two distinct forms of Cu–silver (CuAg) alloy nanoclusters. Ag-doping caused a significant expansion of the initial Cu15, and an Ag13Cu20 nanocluster was formed by the unexpected insertion of an Ag13 kernel within the Cu(I)–S shell. Ag13Cu20 exhibited high activity for initiating the photopolymerization of previously difficult-to-print inorganic polymers in 3D laser microprinting. Modifying the reaction conditions created an Ag18−xCuxS (8 ≤ x) nanocluster with a core S2− anion. This nanocluster possessed a distinct electronic structure compared to the CuAg nanoclusters without templates.
Silalahi et al.102 demonstrated the fluorescence behaviour of bimetallic copper–silver and copper–gold CNs. These CNs featured a centered-cuboctahedral M@Cu12 core surrounded by dithiocarbamate/dithiophosphate and alkynyl ligands, with a counter anion to balance the charge (Fig. 5). The clusters exhibited the ideal Td symmetry and contained 2 ns valence electrons, categorizing them as pseudo-spherical 2e− super atoms with a 1S2 configuration. The method for their synthesis involved treating excess C6H8 with dithiolato-stabilized Cu hydrides and the addition of gold(I) and silver(I) salts, differing from the earlier templated galvanic replacement procedures. These CNs displayed enhanced stability and luminescence properties, with the luminescence in all states at room temperature. The luminescence originated from a spin-forbidden triplet excited state, with the emission lifetimes in the ms range. The incorporation of gold and silver atoms into the polyhydrido CuCNs significantly improved their photoluminescence QY.
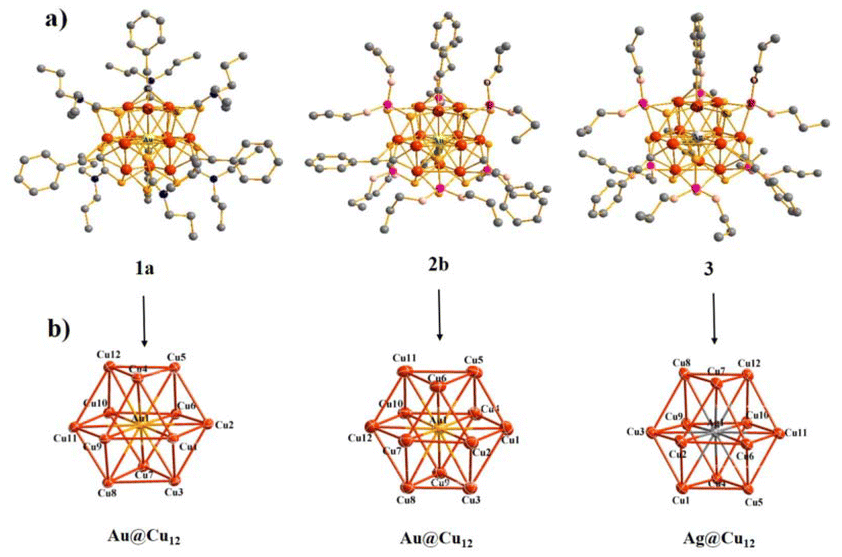 | ||
| Fig. 5 Structures of CN cations 1a+, 2b+, and 3+ with hydrogen atoms excluded. Reproduced from ref. 102, with permission from Inorganic Chemistry, Copyright 2021. | ||
Baksi et al.103 showed that Ag29(BDT)12(PPh3)4 (BDT = 1,3 benzenedithiol) CNs could efficiently exchange multiple copper atoms into semiconducting Cu12S6(DPPPT)4 (DPPPT = bis-(diphenylphosphino)pentane) CNs without altering their structure. High-resolution trapped ion mobility spectrometry analyzed the structural changes caused by silver/copper exchange in 29-atom CNs. The instrument had a mobility determination of 201–251 and an accuracy of 0.3%.104,105 Although Cu atoms have a 12% smaller van der Waals radius than Ag, the M–M and M–S bond lengths in the coinage MCNs were highly dependent on their bonding conditions and electronic structure (Fig. 6). The structure of the Ag29 CNs had 4 distinct locations for silver atoms. The icosahedron consisted of a central atom, 12 other atoms, 12 cluster shell atoms bonded to S atoms, and 4 undercoordinated silver atoms.
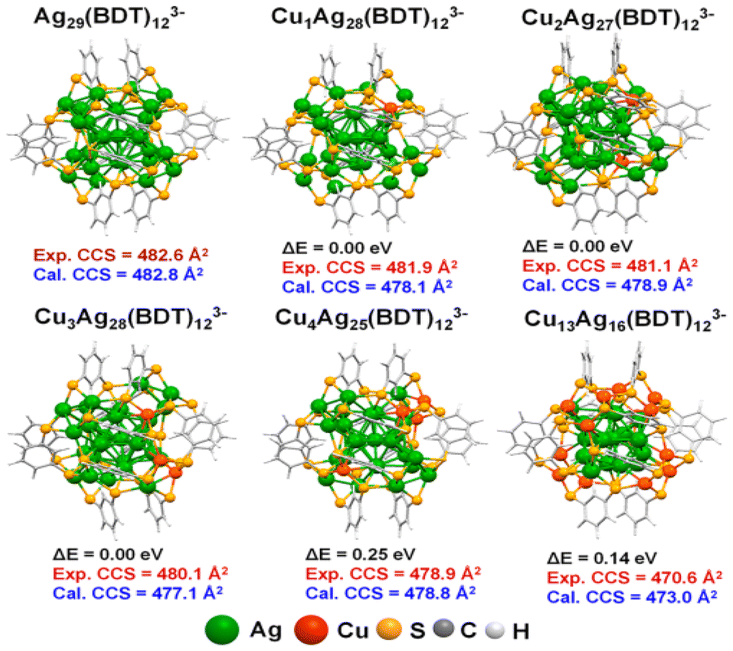 | ||
| Fig. 6 DFT-optimized structure of the probable isomer. Reproduced from ref. 103, with permission from ACS Nano, Copyright 2020. | ||
Ang et al.60 conducted the synthesis of dodecanethiol-protected Ag/Cu bimetallic CNs. These CNs were created using a liquid-phase method with varying ratios of copper to silver. TEM analysis revealed that the CNs were about 4–6 nm in size. Various spectroscopic techniques indicated that the surfaces of the CNs were predominantly enriched with copper atoms. Two melting transitions were observed by differential scanning calorimetry in some alloy samples, indicating distinct dodecanethiol chain packing tendencies on the surfaces rich in copper and silver. A galvanic exchange process was shown to be responsible for the surface enrichment of copper atoms during the preparation of the CNs. The degree of enrichment was found to be related to the initial copper feed ratio. This copper surface enrichment was significant for the catalytic properties and surface behavior of the bimetallic alloy. The behavior of dodecanethiol 3D self-assembled monolayers (SAMs) was influenced by the copper atom surface enrichment. Thiolated Ag/Cu bimetallic alloy CNs with copper-enriched surfaces were synthesized, offering insights into the surface properties and potential catalytic applications of these bimetallic alloys.
Shahzad et al.104 demonstrated the use of GSH-protected copper–silver bimetallic CNs as sensitizers in MCN sensitized solar cell (SSC). They employed a spray-coating technique to deposit these CNs onto photoanodes. Titanium dioxide modified with copper-rich alloyed CNs exhibited a short circuit photocurrent (Jsc) of 2.87 mA cm−2 and a voltage (Voc) of 691 mV. Monometallic CNs composed of silver, copper, AgCu alloys were synthesized using metal salt reduction procedures. The particle sizes of the alloyed CNs were in the range of 2 to 6 nm, with an average size of 4.1 nm. The SAED pattern revealed rings and dots, indicating two distinct species, a characteristic pattern for polycrystalline NPs, with the first ring corresponding to the silver and copper (111) planes. Titanium dioxide treated with Ag CNs showed the highest absorption, while copper-coated titanium dioxide showed the lowest absorption compared to bare titanium dioxide. The incorporation of metal CNs into titanium dioxide photoanodes reduced the HOMO–LUMO band gap. TiO2 treated with Ag CNs had the lowest band gap value of 2.9 eV, enhancing the absorption of Ag CNs (Fig. 7). As the [Cu] in the copper–silver alloyed CNs increased, the band gaps of the modified titanium dioxide increased from 2.8 to 3 eV. Increasing the silver content in copper–silver alloyed CNs decreased the band gap of the modified titanium dioxide from 3 eV to 2.8 eV. Ag reduced the band gap of titanium dioxide by absorbing low-energy photons and extending the light absorption spectrum into the visible range.
 | ||
| Fig. 7 Spectra of bare titanium dioxide and titanium modified with monometallic and alloyed CNs. Reproduced from ref. 104, with permission from Journal of Power Sources, Copyright 2015. | ||
Ag–Cu alloy CN composites were formed and their optical absorbent properties were compared in the time range of fs at 790 nm by Wang et al.61 In the case of the silver–copper 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and silver–copper 1:3 samples, the SPR peaks were 442 nm and 558 nm, respectively, which is similar to that of Ag and Cu CNs (approximately 401 and 571 nm, respectively). The peak position of the AgCu1:3 sample was 558 nm, which is close to that of Cu nanoclusters. The AgCu alloy exhibited a strong red shift in the SPR peak band with an increase in the content of Cu. The SAED pattern of the AgCu-implanted samples corresponded to the fcc phase, with the lattice constants (LC) of silver–copper (1:1 = 0.396 ± 0.002 nm) and silver–copper (1:3 = 0.37 ± 0.002 nm). The LC of silver (Ag = 0.40 nm) and Cu (Cu = 0.37 nm) were situated between them, suggesting the formation of silver and copper AgCu alloys.
:1 and silver–copper 1:3 samples, the SPR peaks were 442 nm and 558 nm, respectively, which is similar to that of Ag and Cu CNs (approximately 401 and 571 nm, respectively). The peak position of the AgCu1:3 sample was 558 nm, which is close to that of Cu nanoclusters. The AgCu alloy exhibited a strong red shift in the SPR peak band with an increase in the content of Cu. The SAED pattern of the AgCu-implanted samples corresponded to the fcc phase, with the lattice constants (LC) of silver–copper (1:1 = 0.396 ± 0.002 nm) and silver–copper (1:3 = 0.37 ± 0.002 nm). The LC of silver (Ag = 0.40 nm) and Cu (Cu = 0.37 nm) were situated between them, suggesting the formation of silver and copper AgCu alloys.
2.1. Copper core and silver shell
In the study by Pellarin et al.,62 an alumina matrix was used to disperse Cu(1 − x)Agx clusters, which had a diameter of approximately 5 nm and generated in a laser vaporization source with a precisely determined stoichiometry (x = 0.0%, 25.0%, 50.0%, 75.0%, and 100%). CuAgNPs could be generated using simultaneous atomic vapor deposition and XPS on copper–silver CNs from a sputtering source. Their core–shell structure was exhibited by the Moiré pattern at the center of the particle and numerical dark-field image produced from the inverse FFT, showing the location of the copper and silver fringe. The mean cluster sizes were in the range of 4.5–5.0 nm.2.2. Kasha's rule
Anumula et al.105 synthesized tiny Ag3Cu2 CNs shielded by six ligands of 2,4-dimethylbenzene thiol. The Ag3Cu2 nanocluster was composed of a triangular Ag3 and two copper atoms on either side of a trigonal bipyramid metallic core. This alloy cluster displayed peculiar dual fluorescence, whereby its emission band at 823 nm was ascribed to the S1–S0 transition state according to Kasha's rule. Although the fluorescence peaks at 534/574 nm was ascribed to the high-excited states related to anti-Kasha's rule, which are commonly observed in organic luminophores. The Ag–Cu clusters were formed by embedding an [Ag3Cu2(SPhMe2)6] motif in a tetraphenylphosphonium ion and a dichloromethane solvent molecule. The PPh4+ cation works as a counter ion, stabilizing the cluster together with the solvent. As shown in Fig. 8, the six AgCu side edges of the Ag3Cu2 trigonal bipyramid were stabilized and shielded by six 2,4-dimethylbenzenethiol ligands. This allowed the methyl groups on the ligands to be separated, reducing their steric repulsion and boosting the stability of the cluster (Fig. 8). The emission at 830 nm was caused by the S1–S0 de-excitation (DE) process, which occurred during the e− transition from LUMO to HOMO (L to H). The higher emission band was caused by the Sn–S0 (n = 7–2) DE processes, which occurred when an e− jumps from L to H-m. The occupied orbitals, H and H-m (m = 1–6), were influenced by the 3d orbitals of the copper atoms, 3p orbitals of the S atoms, and some π orbitals of the phenyl groups. The insets showed the optimized ground-state structure of Ag3Cu2 and its molecular orbitals, which were connected to the e− transition from H-5 to L.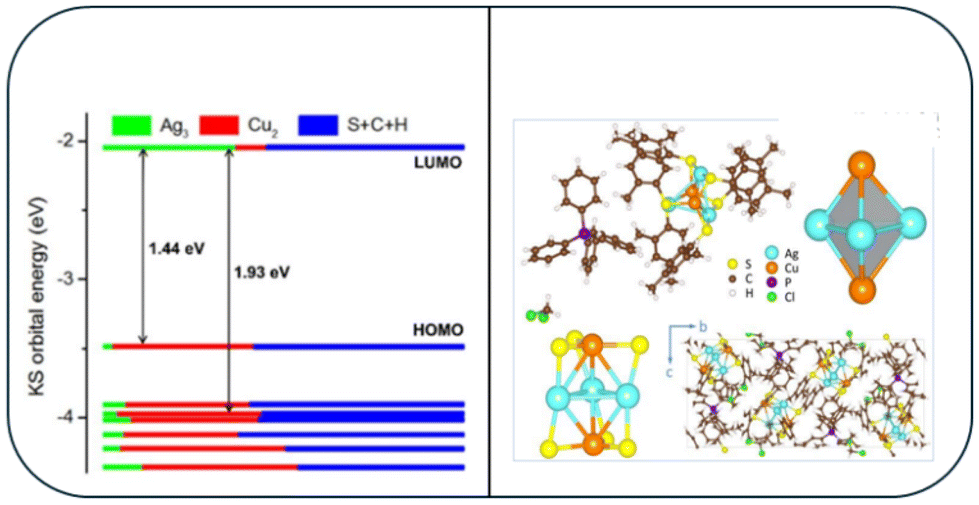 | ||
| Fig. 8 X-ray structure of the [Ag3Cu2(SPhMe2)6]− and DFT-calculated molecular orbital levels. Reproduced from ref. 105, with permission from Nanoscale, Copyright 2020. | ||
2.3. Ag/Cu-based hydride clusters
Lv et al.106 evaluated Ag/Cu ACNs and their alloys, which have become popular in recent decades due to their ease of synthesis, separation, structure determination, and low cost compared to noble MCNs. They show great potential in mediating reduction reactions and H2 storage. Single-crystal X-ray diffraction, 1H nuclear magnetic resonance, electrospray ionization mass spectrometry, density functional theory, and single-crystal neutron diffraction provided crucial information about the structural properties of Ag/Cu hydride clusters, allowing a deeper understanding of their inherent bonding principles. [Ag25Cu4(H)8X6(CCAr)12(PPh3)12]3+ (denoted as Ag25Cu4H8, where X = Cl and Br; Ar = Ph, 4-ethynyl toluene) was prepared by Mizuta et al. The co-reduction of AgI and CuI complexes with NaBH4, following the addition of NEt4X enabled the formation of different Ag25Cu4H8 clusters (Ag25Cu4H8–Cl and Ag25Cu4H8–Br). The orientation of the encapsulated μ3-hydrides was consistent, whereas the μ4-hydrides were postulated to be placed within the Ag4 tetrahedra, instead of the capping Cu atoms. Zheng et al. created a new reductant, (PPh3)2CuBH4, by reacting PPh3, CuCl, and NaBH4. This reductant effectively controlled the size and formed specific metal nanoclusters. For example, [Ag25Cu4(H)8Cl6(CCPh)12(PPh3)12]3+ (Ag25Cu4H8–Cl) could be readily produced with a high yield (∼55%) by reducing PhCCAg and PPh3AgCl with (PPh3)2CuBH4.
2.4. Trimetallic nanoclusters
The structure of CNs with three different types of atoms in different proportions was estimated using the EAM formula and microstructure simulations (MC). Utilizing the Metropolis MC method, the lowest energy configuration at 300 K was attained. MC simulation methods based on the Metropolis algorithm were used to perform surface-segregation calculations for different alloys. Using these techniques, equilibrium alloy configurations were developed, and the thermodynamic ensemble average profile was produced. The equilibrium microstructures of the silver–copper, silver–nickel, and copper–nickel binary alloys were examined in the first MC simulations. The lower surface energy components (Ag in Ag–Cu and Ag–Ni and Cu in Cu–Ni) remained on the surface, whereas the higher surface energy components (copper in silver–copper, nickel in silver–nickel, and nickel in copper–nickel) were in the core. The MC simulations for the ternary silver–copper–nickel alloy CNs revealed that the most energetically favorable configurations exhibited surface-segregated structures, with the lower surface energy Ag atoms preferentially occupying the low coordination sites such as corners, edges, and surfaces. The surface energies of the constituent elements followed the order of Ag, Cu, and Ni. The potential energy of the 4 nm-diameter Ag–Cu–Ni ternary alloy nanoclusters varied as a function of temperature and composition (Fig. 9). The total potential energy curve showed a transition from solid to liquid phase, corresponding to a melting point of 1100 K for 50% Cu, 50% Ni, and 850 K for an alloy of 34% Ag, 33% Cu, and 33% Ni.107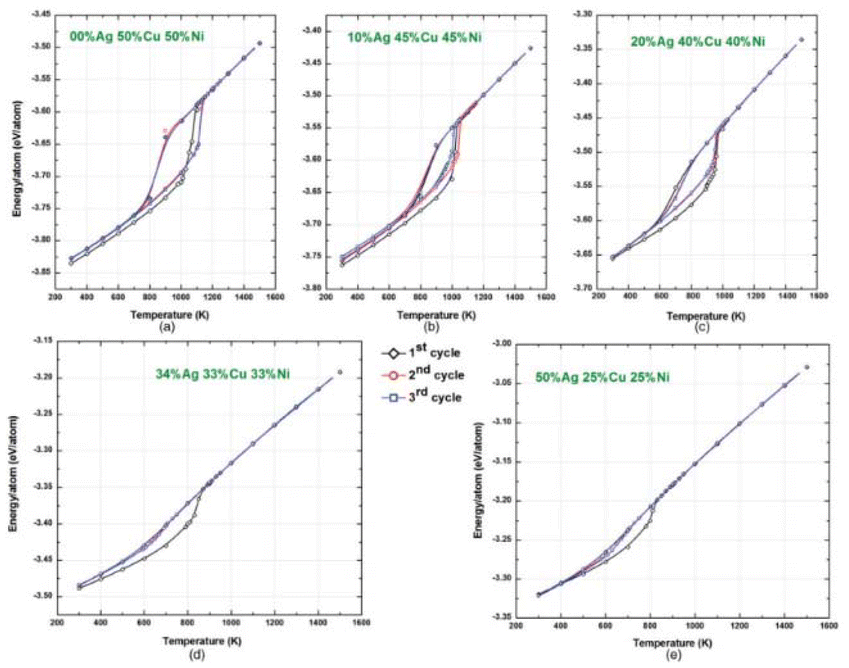 | ||
| Fig. 9 Variations in potential energy with temperature for various compositions of Ag–Cu–Ni ternary alloy CNs. Reproduced from ref. 107, with permission from Phys. Rev. B, Copyright 2011. | ||
Datta et al.108 examined the alloying, electrical, optical, and antibacterial properties of 25-atom CumAgn (m + n = 25) CNs with Mg doping at the surface Ag sites. They studied seven compositions of CNs (Cu4Ag21, Cu6Ag19, Cu9Ag16, Cu12Ag13, Cu15Ag10, Cu19Ag6, and Cu21Ag4), including both copper-rich and silver-rich variants, as well as an almost 50:50 mix. The introduction of Mg doping on CuAg CNs was analyzed by investigating their structural, alloying, electronic, optical, and bactericidal properties through computational simulations using DFT. This study employed the generalized gradient approximation (GGA) for the exchange–correlation energy function, utilizing copper, silver and magnesium valence electrons. A simple cubic supercell with periodic boundary conditions and a vacuum separation of 12 Å was employed. The electronegativity of the atoms affected the charge redistribution during alloying, and the Mg doping intensified the mixing tendencies in the CNs. Also, their optical properties were assessed through absorption spectral calculations, and the phonon spectrum confirmed the dynamical stability of the optimized structures.
3. Evolution of aggregation-induced fluorescence via CuAg synergism
Aggregation-induced emission (AIE) is a phenomenon where certain molecules, known as AIE luminogens (AIEgens), exhibit enhanced fluorescence when they aggregate.109 This is different from the more common aggregation-caused quenching (ACQ), where the fluorescence is diminished upon aggregation. Typically, AIEgens possess molecular structures that allow significant intramolecular rotation when in solution. This free rotation enables these molecules to dissipate energy non-radiatively, preventing fluorescence. However, when these molecules aggregate, their intramolecular motions are restricted.110 This restriction of intramolecular motion (RIM) prevents non-radiative decay pathways, allowing the molecules to emit fluorescence efficiently. The unique properties of AIEgens make them useful for a variety of applications. In biological imaging, AIEgens provide bright fluorescence in the aggregated state and are highly biocompatible, making them ideal for cellular and tissue imaging. They can effectively track cellular processes and detect specific biomolecules. In chemical sensing, AIEgens can detect analytes such as metal ions, explosives, and pollutants. The presence of specific analytes induces their aggregation, triggering fluorescence, thus and signal detection.111Kong et al.112 demonstrated the self-assembly of copper CNs (S1) and silver(I)-doped silver/copper CNs (S2 and S3), which were coated with D-penicillamine. These CNs (S1–S3) exhibited significant luminescent properties, with a QY ranging from 11.3% to 14.1%. When exposed to UV light, their solid-state emissions were strong, changing color from reddish to orange for S1–S3. Their electronic absorption spectra revealed broad bands at 295–371 nm for S1 and 279–348 nm for S2 and S3. S2 and S3 showed a hypsochromic shift compared to S1, indicating silver ion doping, which resulted in a wider energy gap between the HOMO and LUMO in the CNs. The average luminescence lifetimes of S1, S2, and S3 at 615 nm were 24.7 μs, 13 μs, and 10.7 μs, respectively. With an increase in the amount of silver(I)-doped copper/silver CNs in S2 and S3, the ligand-related non-radiative relaxation decreased, resulting in shorter lifetimes. The structure of the silver(I)-doped copper/silver CNs S2 and S3 became more compact than that of CuCNs S1 as the feed amount of silver ions increased. The luminescence originated from the triplet excited states, which are attributed to LMCT or LMMCT, as evidenced by the significant Stokes shift (about 290 nm) and extended lifetimes in the tenths of microseconds. The TEM images showed a clear transition from an irregular assembly structure (S1) to larger spherical particles (S2 and S3) with average sizes of 0.19 μm and 0.48 μm, respectively. With an increase in the D-penicillamine levels, the interactions among the D-penicillamine molecules intensified, leading to the quicker formation of a turbid colloid. These changes impacted the self-assembly structure of copper/silver CNs and resulted in reduced luminosity. The importance of the interactions between the D-penicillamine molecules in AIE was demonstrated by substituting D-penicillamine with other structural capping agents such as cysteine and N-acetyl D-penicillamine.
4. Ag nanoclusters for Cu2+ sensing
Although our focus is on the synergism between copper and silver in the cluster regime, we also discuss individual copper and silver nanoclusters. In the case of Ag+ sensing, CuCNs were used. Thus, the effect of the synergism between copper and silver in the cluster regime was noteworthy for ionic silver sensing. Likewise, in the case of Cu2+ sensing, AgCNs were used. Thus, the effect of synergism between copper and silver in the cluster regime was noteworthy for ionic copper sensing. To illustrate all these aspects, we mention individual Cu or Ag nanoclusters, together with bimetallic AgCu CNs.5. Fluorometric detection
A substance that has absorbed light or other electromagnetic radiation emits light when it undergoes fluorescence, which is a type of light reflection.113 Typically, this process involves the absorption of high-energy photons, which excites electrons in the substance to a higher energy state. When these electrons return to their ground state, they emit photons of lower energy, resulting in the characteristic glow of fluorescence. Fluorescence quenching refers to any process that decreases the fluorescence intensity of a given substance.114 Quenching can occur through various mechanisms, such as dynamic quenching, static quenching, and energy transfer. Dynamic quenching involves a collisional interaction between the fluorescent molecule and a quencher, leading to non-radiative energy loss. Static quenching occurs when a non-fluorescent complex forms between the fluorophore and the quencher before excitation. Energy transfer quenching, such as Förster resonance energy transfer (FRET), happens when energy is transferred from the excited fluorophore to another nearby molecule without the emission of a photon.1155.1. Amino acid capping
Li et al.116 reported the use of blue-fluorescent tryptophan-coated silver nanoclusters (Ag–TrpCNs) for multicolor bioimaging and Cu2+ detection utilizing a straightforward one-step method. Ag–TrpCNs exhibited strong blue fluorescence, a high QY of 35.7%, and a long lifespan of 18.5 ns. With the introduction of Cu2+, the luminescence was reduced by static quenching and charge transfer. The generated luminescence probe for the detection of copper(II) showed great selectivity and sensitivity, with an LOD of 0.029 μM.A fluorescent oligonucleotide-stabilized (DNA/AgCNs) probe was developed by Peng et al.117 to detect mercury and copper ions with high sensitivity. This probe had two customized DNA sequences. A signal probe consisted of a cytosine-rich sequence template for the synthesis of AgCNs and a link sequence on both ends. The other was a guanine (G)-rich sequence for signal enhancement and a link sequence that complements the signal probe. After hybridization, the luminescence of the hybridized double-strand DNA/AgCNs was 200-fold enhanced due to the luminescence enhancement effect of DNA/AgCNs in the proximity of the G-rich DNA sequence. When 6 nM of copper(II) was introduced in the double-strand DNA/AgCNs, their emission at the λem of 565 nm began to decrease. At 250 nM of copper(II), up to 75% of the fluorescence was quenched. Double-stranded DNA/AgCNs with increased fluorescence from G-rich DNA were effective for copper(II) sensing. The [Cu2+] was 6 nM–240 nM. The R2 for copper(II) was 0.99 and the LOD was 2.4 nM.
Sarkar et al.118 developed red-emitting lysozyme-protected silver CNs using a dithiothreitol-reduced LYS scaffold. The CNs had a uniform size distribution with a QY of 6%, together with a massive (280 nm) Stokes shift. The pH stability enhanced their applicability in dual-mode sensing platforms for copper(II) and vitamin B12 (VB12). The sensing was accomplished through two opposing luminescence-quenching mechanisms. Copper-induced quenching occurs through a multifaceted mechanism, which includes both static and dynamic quenching, whereas the inner filter effect and Förster resonance energy transfer primarily caused VB12-induced quenching.
5.2. DNA capping
Zhang et al.119 presented a Cu2+ sensor utilizing DNA–AgCNs. The DNA–AgCN solution was extremely luminescent, but it was shown that copper(II) quenched its luminescence. DNA–AgCNs exhibited strong red luminescence with an λem at 624 nm, showing that the C-rich DNA template T1 could capture more silver through the C–Ag+–C complexes and reduction with NaBH4. The luminescence quenching of DNA–AgCNs was caused by Cu2+ reacting with the phosphate or base groups in the DNA. The fluorescence quenching by M–M interaction and EDTA, a strong metal ion chelator, was used in competition with DNA–AgCNs for Cu2+. The addition of 10 mM Cu2+ to room-temperature DNA–AgCNs resulted in a rapid decline in fluorescence, reaching the minimum value in under 10 min. The fluorescence was sensitive to [Cu2+] and R2 was 0.99.Lan et al.120 demonstrated a technique for detecting Cu2+ ions that uses water-soluble DNA–Ag AgCNs. The presence of Ag+ ions caused a considerable change in the ellipticity and non-planar and tilted orientations of the bases due to their interactions with silver(I). When the silver(I) ions were reduced with NaBH4 to form silver atoms, the ellipticity at 265 nm decreased due to the lower interactions of the DNA strands with silver(0) than with silver(I). After the addition of Cu2+ to generate DNA–Cu/AgCNs, the ellipticity became more negative, showing the more rigid structure of the DNA template. In the Cu2+ concentration range of 10–200 nM, the R2 = 0.99 and the LOD was 8 nM.
A luminescent DNA/AgCN probe for sensing mercury and copper ions was demonstrated by Peng et al.117 This probe was comprised of 2 customized DNA sequences. Firstly, a signal probe with a link sequence at both ends and a cytosine-rich sequence template for the synthesis of AgCNs was used. The other was a G-rich sequence for signal amplification and a link sequence complementary to the link sequence of the signal probe. Due to the luminescence enhancement effect of DNA/AgCNs in proximity to the G-rich DNA sequence, the luminescence of the hybridized double-strand (DS) DNA/AgCNs was enhanced by 200-fold after hybridization. Hg2+ and Cu2+ could effectively inhibit the emission of both the as-prepared DNA and AgCNs. The λem of the DS DNA/AgCNs at 565 nm started to be quenched when 6 nM of copper(II) was added, and up to 75% of the luminescence was quenched when 250 nM of copper(II) was applied. The R2 was 0.9952 with a detection limit of 3.4 nM for copper(II).
Liu et al.121 showed a highly sensitive and selective ratiometric fluorescent nanoprobe using DNA-stabilized AgCNs to analyze Hg2+ and Cu2+ levels. An easy approach was used to generate the ratiometric luminescence nanoprobe by reducing silver nitrate with sodium borohydride (NaBH4) and a customized FAM-labeled DNA sequence. The luminescence of the oligonucleotides remained constant before and after the preparation of AgCNs. The λem of AgCNs was quenched by mercury(II) and copper(II), serving as a responsive signal. At 1.0 M of [Cu2+], the F519/F625 ratio plateaued. The LOD was 2.8 nM in the [Cu2+] range of 0 to 1.0 M (R2 = 0.9923).
Using single-standard DNA–AgCNs as a light-scattering probe, Liu et al. presented the sensitive and selective detection of copper ions.122–126 The DNA–AgCN probe was developed using ssDNA as a template. It exhibited a high fluorescence signal, and cysteine (cyst) was discovered to reduce its emission strength drastically. The light-scattering signal of the DNA–Ag CN/cyst probe could be activated by copper ions. For the creation of the silver nanocluster, a single-stranded oligonucleotide sequence (50-(CCCTAA)3CCCTA-30) was used as the scaffold. The emission was quenched when the cyst was added to the DNA–AgCN solution because the additional cysteine was coupled to the DNA–AgCNs via the coordination interaction between silver and S atoms, and the luminescence of DNA–AgCNs was quenched in the presence of cyst. The pH of 7.0 produced the best conditions for the increase in light scattering in the presence of Cu2+ ions. The TEM results showed that the addition of copper(II) ions produced particles with greater diameters. Rayleigh scattering theory states that this process results in an increase in the light scattering signal.127–129
As a major environmental contaminant and a crucial trace element in biological systems, the detection and monitoring of Cu2+ are in high demand, particularly when using techniques that enable sensitive and selective assays in aqueous conditions. According to Shang et al.,130 copper(II) quenched the luminescence of AgCNs templated with poly(methacrylic acid) (PAAM). The fluorescence emission spectra of PMAA–AgCNs exposed to different [Cu2+] were recorded. The intensity of the light was steadily reduced as the quantity of copper(II) increased. The sensing of copper(II) was in the range of 1 × 10−8 M to 6 × 10−6 M with a detection limit as low as 8 nM. An increase in [Cu2+] led to an increase in the absorbance band of AgCNs at 514 nm. Also, the addition of copper(II) led to an increase in the absorbance in the range of 600–750 nm. The absorption band of AgCNs was highly sensitive to the adsorbed molecules, making the direct binding of copper(II) to the surface impossible. Fluorescence quenching was minimized by Cu2+ in the PAAM–AgCN solution containing free PAAM. Cu2+ interacts with the free PAAM carboxylic groups, reducing the fluorescence quenching. The carboxylic group binding sites were shown to be highly selective for Cu2+ compared to the other metal ions studied. Carboxylic groups preferentially bond with hard acid metals such as Cu2+, making them a popular ligand for metal ion sensors.
Yuan et al.131 reported the preparation of very stable hPEI–AgCNs with bluish-green emission. The AgCNs were viable for copper(II) detection at a concentration of 10 nM. The intense bluish-green emission was significantly reduced after adding Cu2+ to the hPEI–AgCN solution. The luminescence response of the hPEI–AgCNs towards various metal ions to investigate the specificity of the fluorescence of copper(II) was quenching. The [Cu2+] ranged from 10 nM to 7.7 nM (R2 = 0.993). Due to the IFE of cupric amine complexes, which may cause the cupric amine coordinates to decrease the luminescence of dye molecules, the luminescence of hPEI-capped copper(II) could quench carbon quantum dots. The binding of copper(II) to the hPEI protective layer and subsequent energy transfer from the AgCNs to the cupric amine locations caused the copper(II)-mediated fluorescence quenching of hPEI–AgCNs. When copper(II) was added to the AgCN solution, the solution turned bluish and had a broad absorption band in the range of 500–1000 nm and a minor shoulder at 300 nm, which showed the formation of cupric amine coordination. Due to the interactions between some copper(II) and the amine motifs in the free hPEI molecules, the efficacy of quenching may have been reduced, indicating that the distance between the AgCNs and cupric amine locations affected the quenching ability. Additionally, the copper(II)-mediated hPEI–AgCN fluorescence quenching could be reversed.
Sun et al.132 reported that the GSH ligand was utilized to produce and passivate AgCNs with intense red fluorescence and excellent water stability. In the GSH passivation procedure, the AgCNs were exposed to mercury(II) ions and copper(II) ions, and it was observed that the fluorescence of AgCNs could be muted by the peculiar GSH–Cu2+ chelation reaction. The [Cu2+] ranged from 0.0001 to 1 μM, with an LOD of 0.05 nM. Furthermore, even at high concentrations, the GSH-passivated AgCNs did not cause appreciable toxicity in live cells. Fluorescence imaging of yeast cells was utilized in the absence and presence of copper(II) ions. The GSH-passivated AgCNs with red fluorescence lit up the yeast cells, while the fluorescence was shut off when copper(II) ions were injected into the cell cultivation medium.
5.3. Amino black (AB) capping
Using AB as a stabilizing agent, Ma et al.133 created fluorescent AgCNs for the detection of copper(II). AB@AgCNs had an average diameter of around 1.3 nm and showed brilliant blue emission. The [Cu2+] range was 0.01 to 1.1 μmol L−1. The ability of AB to stabilize AgCNs was likely due to its functional groups, which are also present in tiny organic molecules such as thioflavin.134 The QY of AB@AgCNs was 0.20, which was calibrated with quinine bisulfate (QY = 0.55). The intensity of AB@AgCNs in the presence of copper(II) varied between pH 2.56–7.54, with the optimum pH of 6.80 detected. The selectivity of AB@AgCNs for sensing copper(II) ions was demonstrated, where copper(II) still caused a similar quenching toward AB@AgCNs in the presence of other ions. The R2 for copper(II) was 0.99. AB@AgCNs were dispersed in an aqueous solution and observed to possess a spherical form. The inclusion of copper(II) ions increased the average diameter of AB@AgCNs to around 5.26 nm. The quenching process can be attributed to the aggregation of AB@AgCNs caused by copper(II) binding to the functional groups of AB on the surface of AgCNs, leading to the formation of larger aggregates.135–137 Also, the remarkable selectivity of this sensor for Cu2+ over other species is due to a strong interaction between Cu2+ and AB.5.4. Visual detection
Chen et al.134 demonstrated a dual-emission fluorescent nanocomposite based on AgCNs. Using DHLA as a ligand, they created positively charged His–AgCNs, which emitted bright bluish light. After that, they created AgCNs with a stable reddish emission. The electrostatic self-assembly was utilized to effectively produce a dual-emission fluorescent nanoprobe with strong water solubility and outstanding stability.In the water-soluble luminescent DPA@AgCNs, D-penicillamine (DPA) was used as a stabilizer. However, the QY of DPA@AgCNs was low, and copper(II) was used to improve its fluorescence quantum yield, generating DPA@Ag/CuCNs after doping with copper. With the addition of copper(II), the QY increased fourfold, and the emission of CNs changed from reddish to yellowish. The addition of copper(II) to the reaction system changed the self-assembled structures, enhancing the band gap (Eg) between the HOMO and LUMO in CNs and produced the AIEE effect, which was the source of the change in fluorescence. DPA@AgCNs and DPA@Ag/CuCNs possessed an aggregation state and DPA@AgCNs could self-assemble to form fibrous structures. After the introduction of copper(II), the assembly structure of the CNs changed to lamellar, which was attributed to the metallophilicity caused by doping copper(II). The alteration in the self-assembly structure was induced by the promotion of the LMCT and/or LMMCT and the increase in the M–M contact between copper and silver after the doping of copper(II). The shift from looser to compact assemblies coincided with the transition from fibrous to lamellar self-assembled structures, giving rise to the AIEE effect. The enlarged HRTEM pictures demonstrated that small nanoparticles were the source of both the DPA@AgCN and DPA@Ag/CuCN assembled structures, suggesting that the AIEE effect was the source of the CN emission. The average lattice distances of DPA@AgCNs and DPA@Ag/CuCNs were 2.8 Å and 3.2 Å, respectively, indicating that the microscopic arrangement was more compact.134
Chen et al.135 developed a single-stranded DNA sequence (A31) to produce A31@templated@AgCNs. Copper(I)/copper(II) promoted the aggregation of A31@templated@AgCNs, significantly increasing their luminescence emission. The signal-on fluorescence sensing platform could be utilized to detect copper(I)/copper(II) within 30 min with a detection limit of 0.1 μM, as well as to perform intracellular imaging of copper(I)/copper(II) in cells with high cell permeability (Fig. 10).
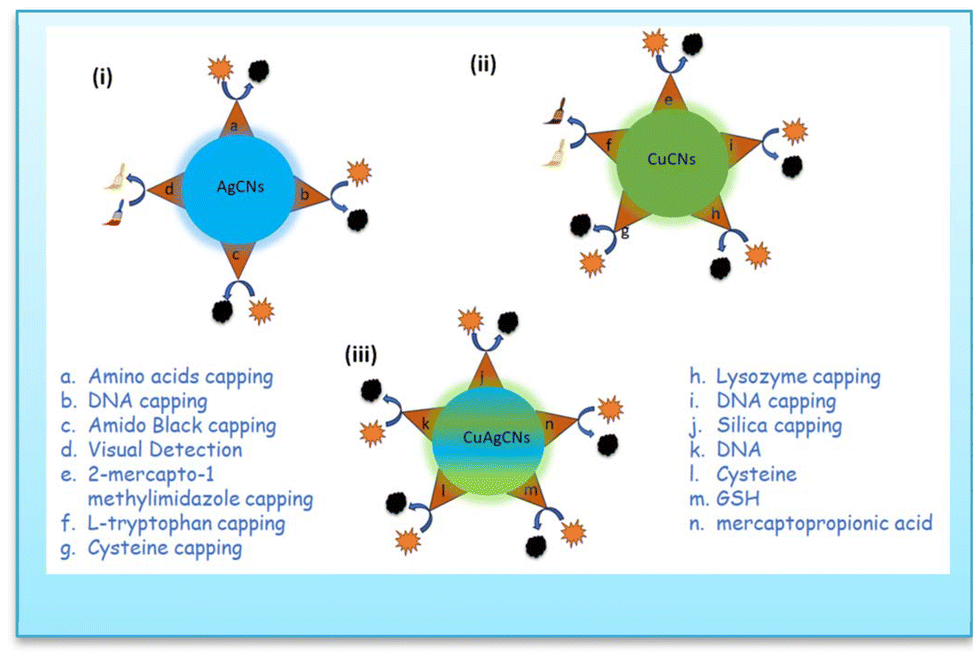 | ||
| Fig. 10 Schematic diagram of (i) AgCNs for copper ion sensing; (ii) CuCNs for silver ion sensing and (iii) CuAgCNs for sensing of analytes. | ||
6. Copper nanoclusters for Ag+ sensing
CuCNs with intense luminescence were manufactured utilizing a technique with 2-mercapto-1-methylimidazole (MIM) as a stabilizer. MIM@CuCNs showed a QY of 19.2%, a long luminescence duration (10.57 μs), and high photostability. The luminescence intensity of MIM@CuCNs was effectively suppressed by the addition of Ag+ ions. The silver(I) concentration range was 0.025–50 μM with a detection limit of 6.7 nM. The agglomeration-induced effect and static quenching were identified as the underlying mechanisms for the fluorescence quenching.136Aarya et al.137 produced greenish-emitting (λex = 380 nm, λem = 500 nm) Trp@CuCNs. The strong green photoluminescence intensity of CNs was dramatically reduced by the introduction of silver(I), resulting in the production of larger NPs and the loss of energy quantization. Upon the introduction of silver(I), the color changed from pale yellowish to reddish-brown. Small molecules such as Trp were employed as reducing agents to reduce the metal core during the development of stable CNs. When silver(I) was added to Trp@CuCNs, it could engage metallophilically with the copper core before being rapidly reduced to silver(0) by the Trp and hydrazine in the system. XPS studies confirmed the zero oxidation state of silver. Subsequently, silver-(0) was deposited on the surface of the copper CNs, increasing their size. The TEM image confirmed the increase in the size of Trp@CuCNs after the introduction of silver(I). The TEM image showed a considerable increase in size (∼9 fold), indicating a shift from CNs to NPs.
JingJing et al.138 demonstrated the effective synthesis of fluorescent CuCNs using N-acetyl-cyst (CAN) as a reducing and stabilizing reagent. Red luminescence was observed in CAN@CuCNs at λem 630 nm. The fluorescence quenching of CAN@CuCNs could be dramatically induced by silver ions. The [Ag+] range was 1.7 × 10−10–1.2 × 10−8 M with the LOD 7.8 × 10−11 M.
Lys@CuCNs scaffolded by a photoluminescent (pL) protein with dimensions of λex = 365 nm and λem = 437 nm could be utilized as an affordable dual metal sensor to detect Fe(II) and silver(I) ions with high sensitivity and selectivity using two separate pL turn-off mechanisms. Using a size-induced pL quenching approach including both static and dynamic mechanisms, the CNs could preferentially detect silver(I) (LOD = 4 nM) when generated in the addition of hydrazine (N2H4), a mild reducing agent.139
7. AgCu nanoclusters for environmental applications
The outstanding aggregation and plasmonic behavior of copper and silver particles leads to the manifestation of synergistic behavior. Copper and silver bimetallic nanoparticles demonstrated a variety of applications, including antibacterial activity, sensing, and dye degradation, which are all utilized for environmental remediation. Alternatively, copper and silver nanoclusters exhibited sensing behavior. There is no other significant application available concerning these nanoclusters. Due to their ultra-small size, CNs are highly sensitive to the environment. However, due to their separable energy levels, CNs have significant fluorescence with an interband transition. Thus, the application of nanoclusters is limited to analyte sensing.7.1. C–C Coupling
Ethylene plays an important role in the environment. It fosters plant growth,140 balancing the factors in plant stress,141etc. Thus, the laboratory synthesis of ethylene is an active area of research.AgCu alloy sub-nanoclusters (ASCNs) were loaded onto TiO2 by a simple stepwise photo-deposition technique developed by Yu et al.,142 allowing CO2 photoreduction to yield C2H4. DFT calculations showed that Cu and Ag synergistically impacted AgCu ASCNs, promoting the C2H4 desorption property due to Cu and enabling a good C–C coupling capacity due to Ag, hence improving the efficient and selective synthesis of C2H4. In Ag/TiO2, the size of the Cu–Ag alloy on the surface of TiO2 gradually decreased with an increase in the Cu content, and in the case of Cu0.8Ag0.2/TiO2 and Cu/TiO2, only metal sub-nanoclusters were found distributed on the surface of TiO2 according to the HAADF images. Ag nanoparticles with an inhomogeneous particle size (∼5 nm) were also distributed on the surface of TiO2. This was because the reduction potential of Ag (Ag+/Ag, E° = +0.80 V) was much higher than that of Cu (Cu2+/Cu, E° = +0.34 V), which resulted in a much faster growth rate of Ag than Cu in the photo-deposition process. The Cu0.8Ag0.2/TiO2 metal sub-nanoclusters were uniformly anchored over the surface, as demonstrated by the HAADF-STEM images. The metal sub-nanocluster sizes were very tiny, with a narrow size distribution of 0.71 ± 0.16 nm. The C–C coupling between CH2* and CH2* or CO* and CO* was an important step in producing C2H4 (Fig. 11). To find the best C–C coupling pathway, the C–C coupling of CO* was estimated, even if no CO* intermediate was present in the in situ FT-IR spectra. The C–C coupling of CO* with Cu/TiO2, Ag/TiO2, and Cu–Ag alloy/TiO2 demanded energy expenditures of 1.89, 1.40, and 1.19, respectively. The synergistic impact of Cu and Ag in AgCu ASCNs decreased the energy expenditure of the C–C coupling of CO*. However, the high energy expenditure made it difficult for CO* to execute C–C coupling, suggesting that the C–C coupling of CH2* was the crucial step for C2H4 generation.
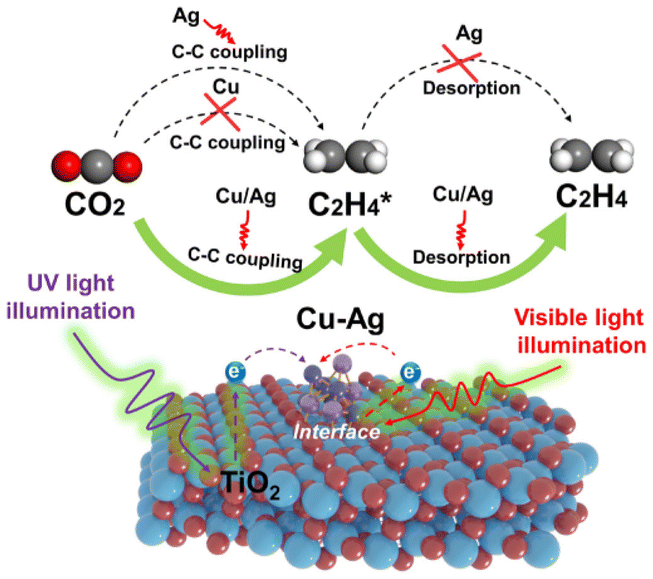 | ||
| Fig. 11 Photoreduction of ethylene via C–C coupling. Reproduced from ref. 142, with permission from PNAS, Copyright 2023. | ||
8. Turn-off fluorometric sensing
8.1. Temperature sensing
Based on weakly luminous glutathione (GSH)-capped Cu CNs, Han et al.143 created highly luminescent AgCu CNs. The GSH-protected AgCu CNs showed a 9-fold increase in luminescence intensity upon the addition of a specific quantity of Ag+. The AgCu CNs revealed a new shoulder peak at 390 nm and an even more intense absorption peak at 300 nm. The blue light emission in the luminescence band shifted from 620 to 604 nm. The AgCu CNs were monodispersed and approximately spherical. Furthermore, the possessed an average diameter of 1.4 ± 0.2 nm. As the temperature increased from 4 °C to 55 °C, the light intensity continued to decline (Fig. 12). The observed behavior was explained by the thermal agitation of the nonradiative processes. Specifically, the intensity of the emission from the excited state was reduced due to the increase in the nonradiative transition rate and molecular collision frequency at high temperatures. After the addition of ethanol, the AgCu CN solution showed solvent-induced aggregation. When the volumetric percentage of ethanol increased, the luminescence intensity of AgCu CNs increased substantially and peaked at 40 vol%, which coincided with the existence of more gel. The AgCu CNs showed strong orange-yellow luminescence under UV light after drying, and they became pale yellow under visible light.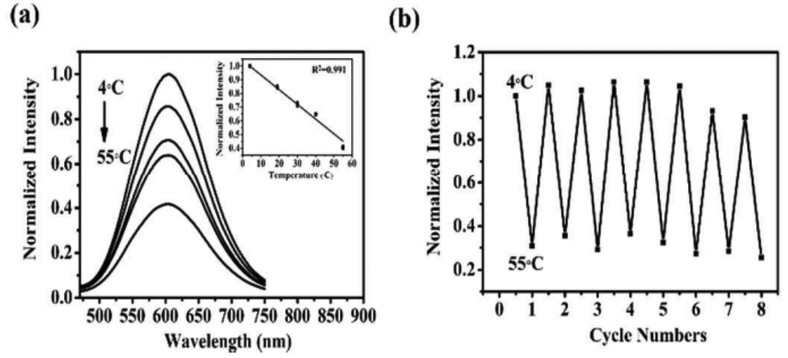 | ||
| Fig. 12 Fluorescence intensity-dependent temperature sensing. Reproduced from ref. 143, with permission from Anal. Methods, Copyright 2017. | ||
8.2. Heavy metal sensing
Huang et al.144 employed conventional C-rich DNA sequences as templates for generating AgCu CNs. The impact of neighboring and inserted G sequences in the DNA templates on the luminescence intensity of DNA–AgCu CNs was investigated. The metal ion sensing performance of two DNA–AgCu CNs and the mechanism for their quenching by mercury(II) and copper(II) were studied. DNA2–AgCu CNs exhibited a 2-fold increase in intensity compared to DNA1–Cu@AgCNs and a 3-fold increase over DNA9–AgCu CNs. DNA1 is a commonly used C-rich ssDNA for creating AgCN templates. In the DNA2–DNA8 strand, 5 or 10 G-nucleotides were extended at the end of DNA1 or inserted into two to three positions on the DNA1 strand. DNA2–AgCu CNs had a more intense maximum emission wavelength (λem) than DNA1–AgCu CNs and DNA9–AgCu CNs, shifting from 566 nm to 583 nm. The red shift in λem indicates a strong interaction between Cu/AgCNs and the G5-nucleotide at the 5′ end of DNA1. The DLS analysis indicated that the particles had an average hydrodynamic diameter of 5.6 nm. DLS yielded larger sizes due to the presence of hydration in the surface DNA strands and CNs. The sensing selectivity for common metallic ions was investigated. The concentration of ions was 10 μM, except for calcium(II), magnesium(II) and mercury(II), which was 10 mM, 10 mM, and 1.0 μM, respectively. Among them, only mercury(II) ions caused considerable quenching of DNA1–AgCu CNs. In DNA2–AgCu CNs, both copper(II) and mercury(II) ions significantly reduced the fluorescence. The expanded G5-sequence effectively covered the AgCu CNs, probably leading to the increase. The TEM images showed well-dispersed DNA2–AgCu CNs with an average size of 3 nm. However, their size increased to 15 nm in the presence of mercury(II) and 10 nm in the presence of copper(II). Aggregation-induced quenching (AIQ) could be another quenching mechanism for mercury sensing. The fluorescence intensity of DNA2–AgCu CNs decreased with an increase in [Cu2+] and the addition of 0.1 μM mercury(II) and 30 μM NaBH4. A linear range of 0.01–5 μM Cu2+ (R2 = 0.99), with a detection limit of 5 nM was observed. Masking Cu2+ ions with EDTA resulted in a linear range of 5 nM to 40 nM (R2 = 0.99) for Hg2+, with a detection limit of 1.0 nM [Table 2].Su et al.145 detected Cu2+ ions using DNA–AgCu CNs and 3-mercaptopropionic acid (AMP). Upon the addition to copper(II) ions, the AMP-induced fluorescence quenching of DNA–AgCu CNs was inhibited, resulting in the detection of [Cu2+] ions of 2.8 nM. The [thiol] of three compounds was vital in triggering the quenching of the DNA–AgCu CNs. Upon the addition of 2.5 μM AMP molecules, the luminescence of the DNA–AgCu CNs was completely suppressed (96%). This probe gave a broader dynamic range for detecting copper(II) ions by using 2.5 μM AMP.112 The abundant thiol compounds in AgCu CNs could decrease the binding between the DNA templates and MCNs. The thiol-induced fluorescence quenching of DNA–AgCu CNs occurred due to the insufficient stability provided by the DNA templates. When thiol compounds such as AMP interacted with DNA–AgCu CNs, charge transfer occurred, resulting in partial oxidation of the metal atoms and reduction of the thiols.143 AMP inhibited the fluorescence of DNA–AgCu CNs at 480 nm. AMP (2.5 μM) was quenched, which was regained by 150 nM copper(II) ions. The fluorescence of DNA–AgCu CNs was fully recovered in the presence of 0.5 μM copper(II) ions. Also, the presence of interfering metal ions did not affect the fluorescence signals of DNA–AgCu CNs, AMP, and copper(II) ions in solution. Increasing [Cu2+] ions to 500 nM boosted the amplification of fluorescence. An R2 of 0.988 was found for [Cu2+] in the range of 5–200 nM and LOD for Cu2+ of 2.7 nM [Table 1].
| Probe | λ em | Linear range | Detection range | Size |
|---|---|---|---|---|
| DNA–Cu@AgCNs | 566 nm | 0.01–5 μM Cu2+ | 7.0 nM for Cu2+ | 3 nm |
| 5 nM to 40 nM Hg2+ | 1.0 nm for Hg2+ | |||
| DNA–Ag@CuCNs and –mercaptopropionic acid | 480 nm | 5–200 nM range | 2.7 nM | — |
| DNA–scaffolded | 550 nm | 0.25 to 100 μM | 112.69 nm | 3 nm |
| Cu@Ag CNs | — | 20 to 180 μM | 7.0 μM | — |
| Cu@Ag CNs + GSH | — | 0.1–700 nM | 0.05 nm | — |
| Cu/AgNC samples | DNA strands | Sequence | Length |
|---|---|---|---|
| 1 | 1 | 5′-CCTTAATCCCC-3′ | 12 bases |
| 2 | 2 | 5′-GGGGGCCCTTAATCCCC-3′ | 17 bases |
| 3 | 3 | 5′-CCCTTAATCCCCGGGGG-3′ | 17 bases |
| 4 | 4 | 5′-CCCGGGTTAATGGCCCC-3′ | 17 bases |
| 5 | 5 | 5′-GGGGGCCCGGGTTAATGGCCCC-3′ | 22 bases |
| 6 | 6 | 5′-CCCGGGTTAATGGCCCCGGGGG-3′ | 22 bases |
| 7 | 7 | 5′-GGGGGCCCTTAATCCCCGGGGG-3′ | 22 bases |
| 8 | 8 | 5′-GGGGGCCCGGGTTAATGGCCCCCGGGGG-3′ | 27 bases |
| 9 | 9 | 5′-TTTTTTTTTTTTTTTTTTTTTCCCTTAATCCCC-3′ | 32 bases |
| 10 | 9 + A20 | 5′-TTTTTTTTTTTTTTTTTTTTCCCTTAATCCCC-3′ | 52 bases |
| 3′-AAAAAAAAAAAAAAAAAAAA-5′ | |||
| 11 | 10 | 5′-AAAAAAAAAAAAAAAAAAAACCCTTAATCCCC-3′ | 32 bases |
| 12 | 10 + T20 | 5′-AAAAAAAAAAAAAAAAAAAACCTTAATCCCC-3′ | 52 bases |
| 3′-TTTTTTTTTTTTTTTTTTTTT-5′ | |||
| 13 | 11 + A20 | 5′-CCCTTAATCCCCTTTTTTTTTTTTTTTTTTTT-3′ | 32 bases |
| 14 | 11+ | 5′-CCCTTAATCCCCTTTTTTTTTTTTTTTTTTTT-3′ | 52 bases |
| 3′-AAAAAAAAAAAAAAAAAAAA-5′ | |||
| 15 | 12 | 5′-CCCTTAATCCCCAAAAAAAAAAAAAAAAAAA-3′ | 32 bases |
| 16 | 12 + T20 | 5′-CCCTTAATCCCCAAAAAAAAAAAAAAAAAAAA-3′ | 52 bases |
| 3′-TTTTTTTTTTTTTTTTTTTT-5′ |
With a DNA scaffold (DNA@AgCNs) templated by a monolayer-stranded sequence with a 15-nt polythymine spacer between two distinct emitters, Ma et al.146 established a Cu-mediated turn-on and off switch for detecting pyrophosphate (PPi) or alkaline phosphatase (ALP). The switch was based on three positive characteristics, i.e., the capability of Cu2+ to quench DNA/AgCNs excited at 550 nm, the great affinity of PPi for Cu2+, and the capacity of ALP to convert PPi into orthophosphate (Pi). A maximum quenching efficiency of up to 99.52% was attained compared to the initial fluorescence intensity (before adding copper).
The highly sensitive fluorescence quenching of DNA AgCu CNs by sulfhydryl molecules was demonstrated by Li et al.147 The substrate for AChE was acetylthiocholine (ATCh), an analogue of ACh. AChE could hydrolyze ATCh to form thiocholine, whose sulfhydryl group could interact with DNA–AgCu CNs to cause DNA–AgCu CNs to lose their fluorescence. Compared to DNA1 (5′-GGGTGAGGGTCCCCCCACCCTCACCC-3′)-, DNA2 (5′CCCACGGGCCCC CCTCCTACCC-3′) and DNA3 (5′-GGGTAGGACCCGTGGG-3′)-AgCu CNs, DNA0–AgCu CNs had a more sensitive fluorescence response to the activity of AChE. Thiocholine could more easily combine with DNA0–Cu@AgCNs than DNA1– and DNA2–3–AgCu CNs, quenching the fluorescence of DNA0–AgCu CNs. The hairpin DNA and loop-containing dsDNA have more rigid structures, whereas ssDNA has a relatively flexible structure.
9. Turn-on fluorometric sensing
9.1. Cysteine sensing
Alshatteri et al.148 showed a special dual-emission fluorescence probe, where AgCNs served as the response signal in the sensing platform, while CuCNs served as the reference signal. To gauge the quantity of cysteine as a typical biothiol, copper/silver bimetallic CNs were employed as a composite. The addition of cysteine resulted in a discernible fluorescence color change from reddish to blueish under UV light. Different color photons were observed before and after the addition of cysteine using a smartphone and the Colour Grab app. Cysteine was observed as a sensing platform with an LOD of 7.0 mM.9.2. Mercury sensing
Silver/copper nanoclusters (Ag/CuCNs) were used to provide a straightforward colorimetric sensing approach for mercury(II) ions with outstanding selectivity, as proven by Cai et al.149 Sodium borohydride was used as a reducing agent and GSH as a template for producing bimetallic Ag@CuCNs The addition of mercury(II) considerably increased the peroxidase activity of silver and copper CNs. The [Hg2+] was detected in the range of 0.2–700 nM with a detection limit of 0.05 nM (Fig. 13).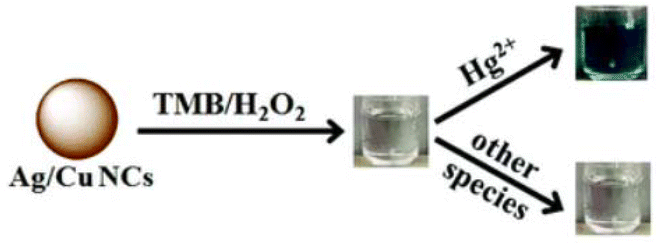 | ||
| Fig. 13 Schematic representation of mercury(II) sensing with Ag/CuCNs. Reproduced from ref. 149, with permission from Anal. Methods, Copyright 2021. | ||
Mao et al.150 synthesized several DNA–Ag CNs using eight single-stranded C-rich DNA templates and investigated their optical characteristics. The accompanying G-rich sequences could create complementary G-quadruplex structures to the templates. The pH preferences of DNA–Ag CNs varied depending on the amount of consecutive C bases, resulting in the detection of green, red, or orange emissions at varying excitation wavelengths. The orange emission of DNA–Cu/Ag CNs was noticeably lower than that of DNA–Ag CNs, but their green-emitting species were more stable and required a shorter incubation time. Hg2+ was detected using the Cu/Ag CNs. DNA–Cu/Ag CNs were less susceptible to metal ions than DNA–Ag CNs, except Hg2+. The addition of Cu2+ had a significant effect on DNA–Ag CNs but only a slight effect on DNA–Cu/Ag CNs. Ag CNs and Cu2+ elicited a comparable reaction to Hg2+; however, DNA–Cu/Ag CNs exhibited superior selectivity towards Hg2+. To detect Hg2+, Cu/Ag CNs templated by C-T30695 were further chosen. After Hg2+ interacted with the Cu/Ag CNs for two minutes, the fluorescence intensity did not change. The fluorescence intensity progressively decreased with the increasing Hg2+ titration into the DNA–Cu/Ag CN solution in the range of 40–550 nM (LOD 2.4 nM) with the R2 of 0.9905.
10. Synergism of AgCu (pros and cons)
The stability of atomically precise Cu CNs is a major concern due to the high susceptibility of the metal to oxidation. In this case, capping agents play pivotal role in their stability. The interactions between surfactants and nanoparticles decrease the interfacial tension, contributing to an increase in their stability.The distinctive physical characteristics of Ag CNs, such as their intense emission and their tiny size, make them especially alluring. These characteristics offer suitable foundations for building luminous probes for applications in bioimaging and sensing. Silver is more reactive and simpler to oxidize in its zero-valent state than the highly researched gold equivalents, making the preparation of Ag CNs and research into their characteristics more challenging. Therefore, the availability of high-quality Ag CNs with well-defined surfaces, sizes, and structures is essential for basic and applied science.
Several synthetic techniques have been devised to produce bimetal CNs with high monodispersity or luminescence to overcome the limitations of their individuals components. These traits are due to the synergistic impact of their physicochemical properties compared to their monometallic counterparts. Han et al.143 found modest red luminescence in a Cu CN solution under UV light at 365 nm. Alternatively, the addition of a specific quantity of Ag+ to the aqueous solution caused the light yellow Cu CNs to become brilliant yellow. The reaction solution produced a bright orange-yellow emission, which increased the luminescence intensity of AgCu BCNs by about 9-fold. Cu/Ag BCNs produced a stronger absorption peak at 300 nm and a new shoulder peak at 390 nm. The luminescence emission blue band shifted from 620 to 604 nm (Fig. 14). The Cu/Ag BCNs exhibited an aggregation-induced emission enhancement effect in the presence of ethanol through the solvent-induced mechanism. The Cu/Ag BCNs were monodispersed and approximately spherical. Moreover, their average diameter was 1.4 ± 0.2 nm, fitting the size of the parent Cu CNs. XPS verified the valence states of the Cu/Ag BCNs. Two prominent peaks were detected at 953.4 and 933.4 eV, which corresponded to the 2p3/2 and 2p1/2 characteristics of Cu0, respectively. Because the 2p3/2 binding energy of Cu0 was only around 0.1 eV different from that of Cu+, the valence state of Cu was most likely in the range of 0 to +1. The binding energies for the luminous Cu/Ag BCNs for Ag 3d5/2 and Ag 3d3/2 were found to be 367.8 and 373.8 eV, respectively. This indicated that Ag+ (367.9 eV) and Ag0 (367.7 eV) coexisted in the resulting Cu/Ag BCNs.
 | ||
| Fig. 14 Absorbance spectra of CuCNs and AgCuCNs. Reproduced from ref. 143 with permission from Anal. Methods, Copyright 2017. | ||
Tapfer et al.151 demonstrated the preparation of pure and/or mixed Ag and Cu CN-doped silica films via the sol–gel process. They prepared silver-co-doped silica films with Cu@Ag molar ratios of 1, 2, and 3 and a constant (silver + copper)/SiO2 molar ratio of 0.18. After annealing in a reducing environment, the SPR absorption bands for silver and copper were present in the optical absorption spectrum at 388 and 562 nm, respectively. Surface plasmon bands were observed in the pure Ag and Cu samples at 403 ± 1 nm and about 562 nm, respectively. The SPR peaks for Ag and Cu in the co-doped samples shifted compared to the single-metal doped silica. The composition of the film determined the size and distribution of the clusters. The 1Ag1Cu sample contained small (5 nm) and larger (40–50 nm) clusters. The size distribution of the 1Ag2Cu film was narrower, with diameters ranging from 5 to 35 nm. The significantly larger clusters were spherical. In the 1Ag3Cu sample, the clusters became spherical and had a more uniform distribution, measuring approximately 5–20 nm in diameter. Subsequently, Z-scan measurements were performed with a mode-locked, cavity-dumped dye laser powered by an Nd:YAG laser. The pulse duration was 6 ps, and the wavelength range was 570–596 nm. At 596 nm with a pulse repetition rate of 15.2 MHz, 1Ag1Cu and pure Cu had n2 values of around 10−13 m2/W. The analysis wavelength was far from the SPR of Ag particles. Thus, presence of copper drove the nonlinear response.
In the study by De et al.,151 three different sets of homogeneous coatings of high quality were placed on silica glass. These coatings corresponded to the molar ratio of copper to silver of 1, 2, and 3. The molar ratio of Ag1Cu1/SiO2 was 0.175. 1Ag1Cu, 1Ag2Cu, and 1Ag3Cu were the names given to the samples that have a molar composition of 1, 2, and 3, respectively, concerning the ratio of copper to silver. The optical absorption spectra of the pure Ag, pure Cu, and Ag–Cu co-doped silica coatings annealed in an H2–N2 environment were recorded. The pure Ag and Cu samples showed surface plasmon bands at around 403 ± 61 nm and approximately 562 nm, respectively. The reducing environment produced blue shifting of the Ag SPR band in contrast to the films annealed in the air, where the Ag SPR band was located at 410 ± 61 nm (Fig. 15). Compared to the SPR of single-metal doped silica, the SPR peaks of the co-doped samples exhibited a shift in both the Ag and Cu peaks.
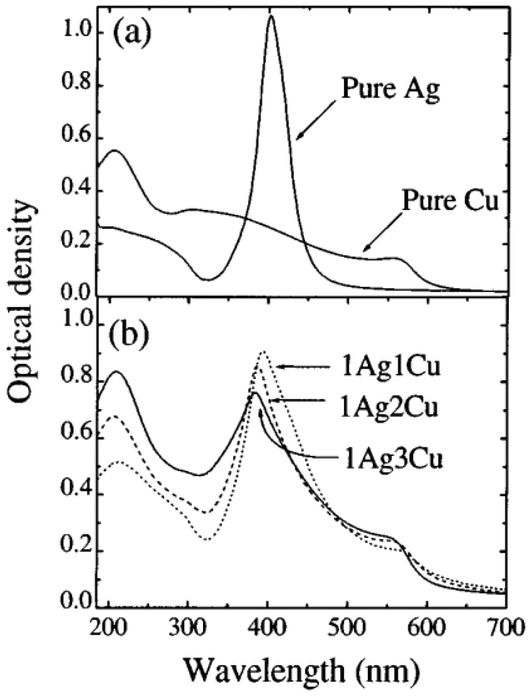 | ||
| Fig. 15 Molar atomic ratio of AgCu. Reproduced from ref. 151 with permission from Appl. Phys. Lett., Copyright 2017. | ||
Thus, the molar ratio of the atoms is important for obtaining successful applications. However, the optimization of the composition is a great challenge. Moreover, the introduction of Ag in Cu clusters makes the system expensive for practical applications. Also, the toxicity and cell viability must be gauged for bimetallic AgCu clusters before their biological applications.
Mao et al.150 demonstrated, as discussed above, that Hg2+ was detected using Cu/Ag CNs. DNA–Cu/Ag CNs were less susceptible to metal ions than DNA–Ag CNs, except Hg2+. They observed that DNA–Cu/Ag CNs were more suitable for heavy metal sensing than DNA–Ag CNs.
There are several uses for AgCu nanoparticles, such as antibacterial,152 anti-cancer,153 electrocatalytic, and dye degradation154 activity. However, AgCu nanoclusters do not possess these diverse applications due to their stability issues under altered experimental conditions. In this case, the inherent emissive behavior of AgCu nanoclusters makes them excellent candidates for sensing applications.
The major mechanisms of the synergy of Cu and Ag NPs were identified as the generation of Cu+ ions, quicker dissolution of Ag+ from Ag NPs, and decreased binding of Ag+ by proteins in the incubation media in the presence of Cu2+. The better dissolution of Ag in the presence of Cu ions due to their oxidation in the redox reaction, the production of more antibacterial Cu+ ions during the same redox reaction, and less favorable binding of Ag ions to the medium proteins in the presence of Cu ions are also important aspects. According to Malik et al.,155 Cu–Ag bimetallic nanoparticles were used for the catalytic activity with the degradation of dye. The bimetallic nanoparticles acted as an electron transfer redox catalyst for the degradation of dye. After the aromatic phenolic –OH groups of the extract were utilized as reducing agents, the biofabrication process was completed. The biosynthesis of Ag–Cu clusters was also driven by the presence of potential (NH)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of phytocomposition, which may be adsorbed onto the surface of Ag–Cu metal atoms. These groups were sensitive to following electronic conduction, and thus responsible for the process. In addition, functional groups, such as –CC and –CO, were accountable for the stable passivation of the surface and acted as stabilizers to prevent the aggregation of Ag–Cu nanoclusters simultaneously.156
O groups of phytocomposition, which may be adsorbed onto the surface of Ag–Cu metal atoms. These groups were sensitive to following electronic conduction, and thus responsible for the process. In addition, functional groups, such as –CC and –CO, were accountable for the stable passivation of the surface and acted as stabilizers to prevent the aggregation of Ag–Cu nanoclusters simultaneously.156
11. Future perspective
The synergism between Cu and Ag is being used to create commercially accessible CuAg bimetallic clusters at a low cost. These clusters can easily overcome the weak fluorescence of Cu clusters. Ag clusters exhibit higher fluorescence than Cu clusters but are unstable due to their oxidation and aggregation. In this case, cluster science tackles photostability as a key factor for biological value. Bimetallic clusters offer enhanced emission and quantum yield, as well as photo-stability and resistance to varying environmental conditions. The Cu and Ag contents in bimetallic clusters significantly affect their activation color and emission intensity. The ratio of Cu to Ag in bimetallic clusters affects both their emissivity and toxicity. Although Ag is more toxic than Cu, scientists have identified the optimal ratio of Cu and Ag for maximum toxicity. Few-atom bimetallic clusters and giant clusters are equally important in terms of applications. Solvents have a significant impact on the fluorescence of bimetallic clusters. However, non-aqueous water-miscible solvents only boost the quantum yield of large clusters, while maintaining their emission wavelength. In this case, the integration of Ag into Cu clusters causes major electronic and band structure changes, resulting in the unique characteristics of bimetallic clusters.Data availability
All the figures have been taken from different papers with proper permission except Fig. 10. Fig. 10 was prepared using Microsoft PowerPoint.No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.References
- S. H. Yau, O. Varnavski and T. Goodson, Acc. Chem. Res., 2013, 46, 1506 CrossRef CAS PubMed.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256 RSC.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469 CrossRef CAS PubMed.
- R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943 RSC.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409 CrossRef CAS PubMed.
- H. Xu and K. S. Suslick, Adv. Mater., 2010, 22, 1078 CrossRef CAS PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261 CrossRef CAS PubMed.
- J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35, 1162 RSC.
- L. Cademartiri and V. Kitaev, Nanoscale, 2011, 3, 3435 RSC.
- R. W. Murray, Chem. Rev., 2008, 108, 2688 CrossRef CAS PubMed.
- S. W. Chen, J. Electroanal. Chem., 2004, 574, 153 CrossRef CAS.
- H. Xu and K. S. Suslick, ACS Nano, 2010, 4, 3209 CrossRef CAS PubMed.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS PubMed.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386 CrossRef CAS PubMed.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207 CrossRef CAS PubMed.
- C. M. Ritchie, K. R. Johnsen, J. R. Kiser, Y. Antoku, R. M. Dickson and J. T. Petty, J. Phys. Chem. C, 2007, 111, 175 CrossRef CAS PubMed.
- E. G. Gwinn, P. O'Neill, A. J. Guerrero, D. Bouwmeester and D. K. Fygenson, Adv. Mater., 2008, 20, 279 CrossRef CAS.
- J. Sharma, H.-C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2010, 46, 3280 RSC.
- H. Yang, Y. Wang, J. Lei, L. Shi, X. Wu, V. Mäkinen, S. Lin, Z. Tang, J. He, H. Häkkinen, L. Zheng and N. Zheng, J. Am. Chem. Soc., 2013, 135, 9568 CrossRef CAS PubMed.
- Y. Song, K. Liu and S. Chen, Langmuir, 2012, 28, 17143 CrossRef CAS PubMed.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509 CrossRef CAS PubMed.
- M. De Nardi, S. Antonello, D.-E. Jiang, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, ACS Nano, 2014, 8, 8505 CrossRef CAS PubMed.
- X. Wang, B. Yin, L. Jiang, C. Yang, Y. Liu, G. Zou, S. Chen and M. Zhu, Science, 2023, 381, 784–790 CrossRef CAS PubMed.
- X. Wei, X. Kang, Z. Zuo, F. Song, S. Wang and M. Zhu, Natl. Sci. Rev., 2020, 8, nwaa077 CrossRef PubMed.
- G. Schmid, M. Baumle, M. Geerkens, I. Heim, C. Osemann and T. Sawitowski, Chem. Soc. Rev., 1999, 28, 179 RSC.
- J. D. Aiken III and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 CrossRef.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS PubMed.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382 CrossRef CAS.
- C. Besson, E. E. Finney and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 8179 CrossRef CAS PubMed.
- C. Besson, E. E. Finney and R. G. Finke, Chem. Mater., 2005, 17, 4925 CrossRef CAS.
- S. Ozkar and R. G. Finke, J. Am. Chem. Soc., 2002, 124, 5796 CrossRef PubMed.
- S. Ozkar and R. G. Finke, Langmuir, 2002, 18, 7653 CrossRef.
- S. Ozkar and R. G. Finke, Langmuir, 2003, 19, 6247 CrossRef.
- B. J. Hornstein and R. G. Finke, Chem. Mater., 2004, 1, 139 CrossRef.
- For a recent review, see: C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed.
- Y. Lin and R. G. Finke, J. Am. Chem. Soc., 1994, 116, 8335 CrossRef CAS.
- J. D. Aiken III and R. G. Finke, Chem. Mater., 1999, 11, 1035 CrossRef.
- G. A. Somorjai and Y. G. Borodko, Catal. Lett., 2001, 76, 1 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed.
- H. Bonneman and R. M. Richards, Eur. J. Inorg. Chem., 2001, 10, 2445 Search PubMed.
- J. Glanz, Science, 1995, 269, 1363 CrossRef CAS PubMed.
- A. Henglein, Chem. Rev., 1989, 89, 1861 CrossRef CAS.
- W. Caseri, Macromol. Rapid Commun., 2000, 21, 705 CrossRef CAS.
- G. Schmid and U. Simon, Chem. Commun., 2005, 6, 697 RSC.
- G. Schmid, Adv. Eng. Mater., 2001, 3, 737 CrossRef CAS.
- A. P. Alivisatos, K. P. Johnson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez Jr. and P. G. Schultz, Nature, 1996, 382, 609 CrossRef CAS PubMed.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078 CrossRef CAS PubMed.
- L. S. Ott and R. G. Finke, Coord. Chem. Rev., 2007, 251, 1075–1100 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS PubMed.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS PubMed.
- J. H. Yu, S. Choi and R. M. Dickson, Angew. Chem., Int. Ed., 2009, 48, 318–320 CrossRef CAS PubMed.
- W. Guo, J. Yuan, Q. Dong and E. Wang, J. Am. Chem. Soc., 2010, 132, 932–934 CrossRef CAS PubMed.
- J. P. Xie, Y. G. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- T. P. Ang and W. S. Chin, J. Phys. Chem. B, 2005, 47, 22228–22236 CrossRef PubMed.
- Y.-H. Wang, C. Z. Jiang, F. Ren and Q. Wang, J. Mater. Sci., 2007, 17, 7294–7298 CrossRef.
- M. Pellarin, I. Issa, C. Langlois, M.-A. Lebeault, J. Ramade, J. Lermé, M. Broyer and E. Cottancin, J. Phys. Chem., 2015, 9, 150204154540009 Search PubMed.
- M. Ganguly, A. Pal, Y. Negishi and T. Pal, Langmuir, 2013, 6, 2033–2043 CrossRef PubMed.
- S. Ghosh, N. K. Das, U. Anand and S. Mukherjee, J. Phys. Chem. Lett., 2015, 6, 1293–1298 CrossRef CAS PubMed.
- J. Nikhil, L. W. Zhong, K. S. Tapan and P. Tarasankar, Curr. Sci., 2000, 79, 1367–1370 Search PubMed.
- X. Song, W. Zhang and Z. Yin, J. Colloid Interface Sci., 2004, 273, 463–469 CrossRef CAS PubMed.
- S. Kapoor and T. Mukherjee, Chem. Phys. Lett., 2003, 370, 83–87 CrossRef CAS.
- R. Ghosh, A. K. Sahoo, S. S. Ghosh, A. Paul and A. Chattopadhyay, ACS Appl. Mater. Interfaces, 2014, 6, 3822–3828 CrossRef CAS PubMed.
- M. L. Cui, G. Song, C. Wang and Q. J. Song, Microchim. Acta, 2015, 182, 1371–1377 CrossRef CAS.
- X. J. Zhao and C. Z. Huang, New J. Chem., 2014, 38, 3673–3677 RSC.
- X. F. Jia, J. Li, L. Han, J. T. Ren, X. Yang and E. K. Wang, ACS Nano, 2012, 6, 3311–3317 CrossRef CAS PubMed.
- H. Chen, L. Lin, H. F. Li, J. Z. Li and J. M. Lin, ACS Nano, 2015, 9, 2173–2183 CrossRef CAS PubMed.
- H. Huang, H. Li, A. J. Wang, S. X. Zhong, K. M. Fang and J. J. Feng, Analyst, 2014, 139, 6536–6541 RSC.
- Y. Ling, J. J. Wu, Z. F. Gao, N. B. Li and H. Q. Luo, J. Phys. Chem. C, 2015, 119, 27173–27177 CrossRef CAS.
- X. F. Jia, J. Li and E. K. Wang, Small, 2013, 9, 3873–3879 CrossRef CAS PubMed.
- J. S. Shen, Y. L. Chen, Q. P. Wang, T. Yu, X. Y. Huang, Y. Yang and H. W. Zhang, J. Mater. Chem. C, 2013, 1, 2092–2096 RSC.
- R. Ghosh, U. Goswami, S. S. Ghosh, A. Paul and A. Chattopadhyay, ACS Appl. Mater. Interfaces, 2015, 7, 21626–21626 CrossRef CAS PubMed.
- H. Miao, Z. Zhou and X. Yang, Nanoscale, 2015, 7, 19066–19072 RSC.
- C. Wang, C. X. Wang, L. Xu, H. Cheng, Q. Lin and C. Zhang, Nanoscale, 2014, 6, 1775–1781 RSC.
- C. Wang, H. Cheng, Y. Sun, Q. Lin and C. Zhang, ChemNanoMat, 2015, 1, 27–31 CrossRef CAS.
- C. Wang, L. Ling, Y. G. Yao and Q. J. Song, Nano Res., 2015, 8, 1975–1986 CrossRef CAS.
- D. Li, Z. Chen, Z. Wan, T. Yang, H. Wang and X. Mei, RSC Adv., 2016, 6, 34090–34095 RSC.
- J. R. Bhamore, S. Jha, A. K. Mungara, R. K. Singhal, D. Sonkeshariya and S. K. Kailasa, Biosens. Bioelectron., 2016, 80, 243–248 CrossRef CAS PubMed.
- C. Wang, H. Cheng, Y. Huang, Z. Xu, H. Lin and C. Zhang, Analyst, 2015, 140, 5634–5639 RSC.
- S. Yang, X. Sun and Y. Chen, Mater. Lett., 2017, 194, 5–8 CrossRef CAS.
- D. Li, Y. Zhao, Z. Chen, X. Mei and X. Qiu, Mater. Sci. Eng., C, 2017, 78, 653–657 CrossRef CAS PubMed.
- B. Han, X. Hu, M. Yu, T. Peng, Y. Li and G. He, RSC Adv., 2018, 8, 22748–22754 RSC.
- T. Udayabhaskararao and T. Pradeep, J. Phys. Chem. Lett., 2013, 4, 1553–1564 CrossRef CAS PubMed.
- K. Zheng, X. Yuan, N. Goswami, Q. Zhang and J. Xie, RSC Adv., 2014, 4, 60581–60596 RSC.
- M. Ganguly, J. Jana, A. Pal and T. Pal, RSC Adv., 2016, 6, 17683–17703 RSC.
- L. Rout, A. Kumar, R. S. Dhaka and P. Dash, RSC Adv., 2016, 6, 49923–49940 RSC.
- Z. Hao, M. Wang, L. Cheng, M. Si, Z. Feng and Z. Feng, Front. Bioeng. Biotechnol., 2024, 11, 1337543 CrossRef PubMed.
- R. S. M. Ponrani, D. E. Nancy, S. G. Rejith and S. C. V. Durai, Phys. Chem. Org. Solid State, 2024, 25, 79–84 CrossRef.
- P. Sharma, M. Ganguly and A. Doi, Appl. Nanosci., 2024, 14, 739–751 CrossRef CAS.
- E. A. Kukushkina, S. I. Hossain, M. C. Sportelli, N. Ditaranto, R. A. Picca and N. Cioffi, Nanomaterials, 2021, 7, 1687 CrossRef PubMed.
- S. Sharma, K. K. Chakrahari, J.-Y. Saillard and C. W. Liu, Acc. Chem. Res., 2018, 10, 2475–2483 CrossRef PubMed.
- J. Yan, H. Su, H. Yang, C. Hu, S. Malola, S. Lin, B. K. Teo, H. Häkkinen and N. Zheng, Am. Chem. Soc., 2016, 39, 12751–12754 CrossRef PubMed.
- X. Zou, Y. Li, S. Jin, X. Kang, X. Wei, S. Wang, X. Meng and M. Zhu, J. Phys. Chem., 2020, 6, 2272–2276 Search PubMed.
- X. K. Wan, X. L. Cheng, Q. Tang, Y. Z. Han, G. Hu, D. E. Jiang and Q. M. Wang, J. Am. Chem. Soc., 2017, 139, 9451–9454 CrossRef CAS PubMed.
- W. Du, S. Jin, L. Xiong, M. Chen, J. Zhang, X. Zou, Y. Pei, S. Wang and M. Zhu, J. Am. Chem. Soc., 2017, 139, 1618–1624 CrossRef CAS PubMed.
- Y. Yao, W. Hao, J. Tang, K. Kirschbaum, C. G. Gianopoulos, A. Ren, L. Ma, L. Zheng, H. Li and Q. Li, Angrew Chem., 2024, 63, e202407214 CrossRef CAS PubMed.
- R. P. B. Silalahi, T.-H. Chiu, J.-H. Kao, C.-Y. Wu, C.-W. Yin, Y.-C. Liu, Y. J. Chen, J.-Y. Saillard, M.-H. Chiang and W. Liu, Inorg. Chem., 2021, 14, 10799–10807 CrossRef PubMed.
- A. Baksi, E. K. Schneider, P. Weis, I. Chakraborty, O. Fuhr, S. Lebedkin, W. J. Parak and M. M. Kappes, ACS Nano, 2020, 11, 15064–15070 CrossRef PubMed.
- N. Shahzad, F. Chen, L. He, W. Li and H. Wang, J. Power Sources, 2015, 294, 609–619 CrossRef CAS.
- R. Anumula, P. Xiao, C. Cui, H. Wu, G. Cui, W.-H. Fang, Z. Luo and J. Yao, Nanoscale, 2020, 12, 7864–7869 RSC.
- Y. Lv, T. Jiang, Q. Zhang, H. Yu and M. Zhu, Polyoxometalates, 2024, 3, 9140050 CrossRef.
- R. Subbaraman and S. K. R. S. Sankaranarayanan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 075434 CrossRef.
- C. Datta, U. Pal and T. Saha-Dasgupta, J. Phys. Chem. C, 2021, 20, 11066–11074 CrossRef.
- G. R. Suman, M. Pandey and A. S. Jeevan Chakravarthy, Mater. Chem. Front., 2021, 5, 1541–1584 RSC.
- X. Ma, R. Sun, J. Cheng, J. Liu, F. Gou, H. Xiang and X. Zhou, J. Chem. Educ., 2016, 93, 345–350 CrossRef CAS.
- S. Li, J. He and Q.-H. Xu, ACS Omega, 2020, 1, 41–48 CrossRef PubMed.
- L. Kong, X. Chu, C. Wang, H. Zhou, Y. Wu and W. Liu, Nanoscale, 2018, 10, 1631–1640 RSC.
- M. Swierczewska, S. Lee and X. Chen, Phys. Chem. Chem. Phys., 2011, 13, 9929–9941 RSC.
- J. R. Lakowicz, Anal. Biochem., 2004, 324, 153–169 CrossRef CAS PubMed.
- S. Lee, K. Park, K. Kim, K. Choi and I. C. Kwon, Chem. Commun., 2008, 4250–4260 RSC.
- S. Li, G. Li, H. Shi, M. Yang, W. Tan, H. Wang and W. Yang, Microchem. J., 2022, 2, 107222 CrossRef.
- J. Peng, J. Ling, X.-Q. Zhang, H.-P. Bai, L. Zheng, Q.-E. Cao and Z.-T. Ding, Spectrochim. Acta, Part A, 2015, 137, 1250–1257 CrossRef CAS PubMed.
- P. Sarkar, M. Saha, N. Nandi and D. K. Sahu, ACS Appl. Nano Mater., 2022, 5, 6 Search PubMed.
- M. Zhang and B.-C. Ye, Analyst, 2011, 136, 5139–5142 RSC.
- G. Y. Lan, C. C. Huang and H. T. Chang, Chem. Commun., 2010, 8, 1257–1259 RSC.
- X. Liu, L. Wang, N. Zhang and D. Shangguan, Anal. Methods, 2015, 7, 8019–8024 RSC.
- G. Liu, D.-Q. Feng, T. Chen, D. Li and W. Zheng, J. Mater. Chem., 2012, 22, 20885–20888 RSC.
- R. Janaky, V. Varga, A. Hermann, P. Saransaari and S. S. Oja, Neurochem. Res., 2000, 25, 1397–1405 CrossRef CAS PubMed.
- O. Rusin, N. N. St Luce, R. A. Agbaria, J. O. Escobedo, S. Jiang, I. M. Warner, F. B. Dawan, K. Lian and R. M. Strongin, J. Am. Chem. Soc., 2004, 126, 438–439 CrossRef CAS PubMed.
- J. M. Zen, A. S. Kumar and J. C. Chen, Anal. Chem., 2001, 73, 1169–1175 CrossRef CAS.
- Y. V. Tcherkas and A. D. Denisenko, J. Chromatogr., A, 2001, 913, 309–313 CrossRef CAS PubMed.
- R. F. Parternack, C. Bustamante, P. J. Collings, A. Giannetto and E. J. Gibbs, J. Am. Chem. Soc., 1993, 115, 5393–5399 CrossRef.
- C. Z. Huang, K. A. Li and S. Y. Song, Anal. Chem., 1996, 68, 2259–2263 CrossRef CAS PubMed.
- M. M. Sung, K. Sung, C. G. Kim, S. S. Lee and Y. J. Kim, Phys. Chem. B, 2000, 104, 2273–2277 CrossRef CAS.
- L. Shang and S. Dong, J. Mater. Chem., 2008, 18, 4636–4640 RSC.
- Z. Yuan, N. Cai, Y. Du, Y. He and E. S. Yeung, Anal. Chem., 2014, 1, 419–426 CrossRef PubMed.
- Z. Sun, S. Li, Y. Jiang, Y. Qiao, L. Zhang, L. Xu, J. Liu, W. Qi and H. Wang, Sci. Rep., 2016, 6, 20553 CrossRef CAS PubMed.
- F. Ma, S. Liang, Y. Peng, Y. Kuang, X. Zhang, S. Chen, Y. Long and R. Zeng, Anal. Bioanal. Chem., 2016, 12, 3239–3246 CrossRef PubMed.
- M. Chen, W. Ao, J. Bai, P. Li, W. Wei, S. Pang and X. Yang, Nanotechnology, 2022, 34, 345501 CrossRef PubMed.
- Z. Chen, W. Lv, C. Yang, M. Ping and F. F. Fu, Spectrochim. Acta, Part A, 2022, 283, 121734 CrossRef CAS PubMed.
- C. Zhang, C. Shao, J. Wang, Z. Li, M. Liang, Y. Wang, D. Liu and S. Lu, ACS Appl. Nano Mater., 2022, 5, 7449–7459 CrossRef CAS.
- Aarya, T. Thomas, B. R. Sarangi and S. S. Mojumdar, ACS Omega, 2023, 8, 14630–14640 CrossRef CAS PubMed.
- K. Jingjing, G. Pengfei, Z. Guomei, S. Lihong, Z. Ying, W. Jianlin, S. Shaomin and Z. Yan, Microchem. J., 2022, 1, 107370 Search PubMed.
- A. Sebastian, P. Kavya and S. S. Mojumdar, Chem. Phys. Impact, 2023, 6, 100249 CrossRef.
- M. Fatma, M. Asgher, N. Iqbal, F. Rasheed, Z. Seha, A. Sofo and N. A. Khan, Plants, 2022, 11, 2211 CrossRef CAS PubMed.
- H. Chen, D. A. Bullock, J. J. M. Alonso and A. N. Stepanova, Plants, 2022, 1, 33 Search PubMed.
- Y. Yu, Y. He, P. Yan, S. Wang and F. Dong, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2307320120 CrossRef CAS PubMed.
- B. Han, X. Hou, R. Xiang and G. He, Anal. Methods, 2017, 9, 4028–4032 RSC.
- X.-F. Huang, B.-X. Ren, C.-F. Peng, X.-L. Wei and Z.-J. Xie, Microchem. J., 2020, 158, 105214 CrossRef CAS.
- Y.-T. Su, G.-Y. Lan, W.-Y. Chen and H.-T. Chang, Anal. Chem., 2010, 20, 8566–8572 CrossRef PubMed.
- J.-L. Ma, B.-C. Yin, X. Wu and B.-C. Ye, Anal. Chem., 2016, 18, 9219–9225 CrossRef PubMed.
- W. Li, W. Li, Y. Hu, Y. Xia, Q. Shen, Z. Nie, Y. Huang and S. Yao, Biosens. Bioelectron., 2013, 47, 345–449 CrossRef CAS PubMed.
- A. H. Alshatteri and K. M. Omer, Microchem. J., 2023, 187, 108385 CrossRef CAS.
- Y. Cai, J. Wang, L. Niu, Y. Zhang, X. Liu, C. Liu, S. Yang, H. Qi and A. Liu, Analyst, 2021, 146, 4630–4635 RSC.
- A. Mao and C. Wei, Microchim. Acta, 2019, 186, 541 CrossRef PubMed.
- G. De, L. Tapfer, M. Catalano, G. Battaglin, F. Caccavale, F. Gonella, P. Mazzoldi and R. F. Haglund, Appl. Phys. Lett., 1996, 68, 3820–3822 CrossRef CAS.
- S. Kunwar, A. Roy, U. Bhusal, A. Gacem, M. M. S. Abdullah, P. Sharma, K. K. Yadav, S. Rustagi, N. Chatterjee, V. K. Deshwal, H.-K. Park and B.-H. Jeon, Catalysts, 2023, 13, 891 CrossRef CAS.
- V. Mayur, M. Shefaly, P. Angshuman and T. Sonal, Mater. Res. Bull., 2011, 46, 384–389 CrossRef.
- H. R. Tantawy, A. A. Nada, A. Baraka and M. A. Elsayed, Appl. Surf. Sci. Adv., 2021, 3, 100056 CrossRef.
- M. A. Malik, S. S. R. Albeladi, S. M. Al-Maaqar, A. A. Alshehri, S. A. Al-Thabaiti, I. Khan and M. R. Kamli, Life, 2023, 13, 653 CrossRef CAS PubMed.
- J.-Z. Guo and H. Cui, J. Phys. Chem. C, 2007, 111, 12254–12259 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |