
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePYTA: a universal chelator for advancing the theranostic palette of nuclear medicine†
Megan E.
Simms‡§
 a,
Zhiyao
Li‡
bc,
Megan M.
Sibley
a,
Alexander S.
Ivanov
a,
Caroline M.
Lara
d,
Timothy C.
Johnstone
e,
Vilmos
Kertesz
f,
Amanda
Fears
bc,
Frankie D.
White
*g,
Daniel L. J.
Thorek
*bchi and
Nikki A.
Thiele
*a
a,
Zhiyao
Li‡
bc,
Megan M.
Sibley
a,
Alexander S.
Ivanov
a,
Caroline M.
Lara
d,
Timothy C.
Johnstone
e,
Vilmos
Kertesz
f,
Amanda
Fears
bc,
Frankie D.
White
*g,
Daniel L. J.
Thorek
*bchi and
Nikki A.
Thiele
*a
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: thielena@ornl.gov
bDepartment of Radiology, Washington University in St. Louis School of Medicine, St. Louis, MO 63110, USA. E-mail: thorekd@wustl.edu
cProgram in Quantitative Molecular Therapeutics, Washington University in St. Louis School of Medicine, St. Louis, MO 63110, USA
dDepartment of Biological Sciences, University of Notre Dame, Notre Dame, IN 46556, USA
eDepartment of Chemistry and Biochemistry, University of California Santa Cruz, Santa Cruz, CA 95064, USA
fBiosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
gRadioisotope Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: whitefd@ornl.gov
hDepartment of Biomedical Engineering, Washington University in St. Louis, St. Louis, MO 63110, USA
iOncologic Imaging Program, Siteman Cancer Center, Washington University in St. Louis School of Medicine, St. Louis, MO 63110, USA
First published on 17th June 2024
Abstract
To clinically advance the growing arsenal of radiometals available to image and treat cancer, chelators with versatile binding properties are needed. Herein, we evaluated the ability of the py2[18]dieneN6 macrocycle PYTA to interchangeably bind and stabilize 225Ac3+, [177Lu]Lu3+, [111In]In3+ and [44Sc]Sc3+, a chemically diverse set of radionuclides that can be used complementarily for targeted alpha therapy, beta therapy, single-photon emission computed tomography (SPECT) imaging, and positron emission tomography (PET) imaging, respectively. Through NMR spectroscopy and X-ray diffraction, we show that PYTA possesses an unusual degree of flexibility for a macrocyclic chelator, undergoing dramatic conformational changes that enable it to optimally satisfy the disparate coordination properties of each metal ion. Subsequent radiolabeling studies revealed that PYTA quantitatively binds all 4 radiometals at room temperature in just minutes at pH 6. Furthermore, these complexes were found to be stable in human serum over 2 half-lives. These results surpass those obtained for 2 state-of-the-art chelators for nuclear medicine, DOTA and macropa. The stability of 225Ac–PYTA and [44Sc]Sc–PYTA, the complexes having the most disparity with respect to metal-ion size, was further probed in mice. The resulting PET images (44Sc) and ex vivo biodistribution profiles (44Sc and 225Ac) of the PYTA complexes differed dramatically from those of unchelated [44Sc]Sc3+ and 225Ac3+. These differences provide evidence that PYTA retains this size-divergent pair of radionuclides in vivo. Collectively, these studies establish PYTA as a new workhorse chelator for nuclear medicine and warrant its further investigation in targeted constructs.
Introduction
Radioactive isotopes of metal ions across the periodic table, from alkalis to actinides, are increasingly being explored in the clinic to diagnose and treat cancer.1–4 The use of radiometals in this context leverages the unique ionizing emissions afforded by nuclear decay. Radiometals that emit positrons (β+) or gamma (γ) rays can be used to image lesions via positron emission tomography (PET) or single-photon emission computed tomography (SPECT), respectively.5 By contrast, therapy can be achieved using radiometals that emit cytotoxic alpha (α) particles, beta (β−) particles, or Meitner–Auger electrons.6,7 When paired together, diagnostic and therapeutic radiometals with complementary properties can form powerful “theranostic” platforms to provide precise information about disease localization, dosimetry, and treatment response.8 Consequently, this information can be harnessed to develop treatment plans tailored to the individual patient.9Irrespective of emission type, a universal requirement for the implementation of radioactive metal ions in targeted nuclear medicine is their attachment to tumor-seeking carrier molecules, such as antibodies or peptides, via bifunctional chelators. This attachment ensures that each radiometal is selectively delivered to sites of disease, minimizing off-target radiotoxicity to healthy organs. In this regard, the formation of metal–chelator complexes of high thermodynamic and kinetic stability is crucial to prevent dissociation in vivo. To achieve this stability, many ligand design efforts have focused on matching and optimizing the structure of a chelator to the coordination preferences of a single radiometal ion of interest.10 This strategy has enabled significant advances over the last decade in the development of new metal-based radiopharmaceuticals.11–13 However, as the number and chemical diversity of medically useful radiometals continues to expand, ligand development using the conventional one-metal–one-chelator paradigm has struggled to keep pace. With a shortage of suitable chelators for the ever-growing nuclear chocolate box,1 the advancement of many emerging radiometals has been halted in the so-called “valley of death” between production and clinical implementation, unable to reach their full medicinal potential.
To address this challenge, new efforts have been directed towards developing chelators for nuclear medicine that exhibit more versatile binding properties. For example, the 18-membered macrocycles macrodipa,14 py-macrodipa15 and py2-macrodipa16 (see Fig. S1 in the ESI†) were developed recently as dual-size-selective chelators for binding both large and small radioactive rare-earth ions, such as the Meitner–Auger-electron emitter lanthanum-135 (135La3+, 8-coordinate ionic radius = 1.16 Å)17 and the β+ emitter scandium-44 (44Sc3+, 8-coordinate ionic radius = 0.87 Å),17 respectively. Several high denticity acyclic bispidine18,19 and picolinic acid20,21 chelators have also been identified to mutually stabilize the α-emitting radionuclide actinium-225 (225Ac) and either indium-111 (111In), a SPECT radionuclide, or lutetium-177 (177Lu), a therapeutic β− emitter with imageable γ emissions (Fig. S1†). With respect to metal-binding range, however, the 12-membered tetraaza macrocycle DOTA (Scheme 1) remains unsurpassed in its ability to stabilize a wide assortment of radiometals with disparate chemical and physical properties. DOTA plays a central role in theranostics as the bifunctional chelator in several clinically approved gallium-68 (68Ga) and 177Lu radiopharmaceuticals for PET imaging and targeted β− therapy of cancer, respectively.22–25 Targeted constructs comprising DOTA are also being investigated in clinical trials for use with copper-64 (64Cu) for PET26,27 and with 225Ac and other α emitters for targeted α therapy (TAT).28,29
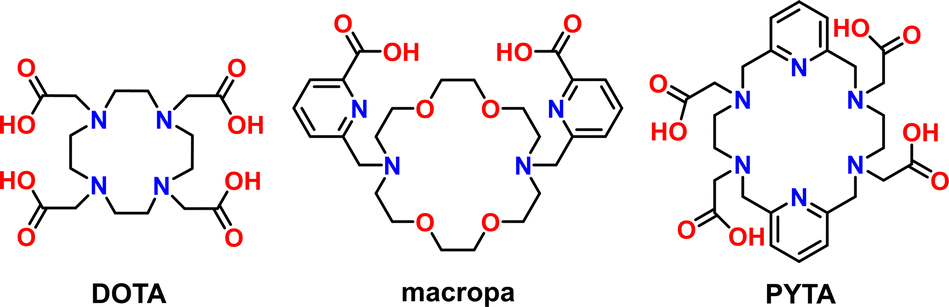 | ||
| Scheme 1 Chemical structures of the chelators used in this work. | ||
Despite its demonstrated versatility, one prominent shortcoming of DOTA is its inability to chelate most radiometals at room temperature. Although its slow complex formation kinetics can be overcome by heating these radiolabeling reactions, high temperatures are not compatible with the use of thermolabile biological targeting vectors, such as antibodies. As such, the development of alternative chelators that retain DOTA's broad chelation properties but radiolabel rapidly at low temperature would be highly valuable for use in nuclear medicine. Notably, there is a strong preference for peptide or antibody conjugates to use a single chelator for both imaging and therapy because it would have nearly identical pharmacokinetic properties and simplify regulatory filing, as only a single chemical entity would need to be advanced to production, quality control, and clinical evaluation.
Towards this end, we sought to investigate the radiometal chelation properties of a macrocyclic ligand called PYTA (Scheme 1). Like DOTA, PYTA possesses four tetraacetic acid pendent arms, but has a core 18-membered macrocycle into which two pyridine rings are embedded. Previous studies have demonstrated that PYTA and related pendent-arm derivatives form remarkably stable complexes with a variety of non-radioactive transition and rare-earth metal ions and may be useful as magnetic resonance imaging (MRI) contrast agents.30–38 While our studies were underway, a triacetic acid derivative of PYTA for the chelation of radio-yttrium was reported.39 However, the suitability of PYTA itself as a chelator for targeted radiopharmaceutical applications has remained largely unexplored.30 In this work, we evaluated the ability of PYTA to complex 225Ac3+, 177Lu3+, 111In3+, and 44Sc3+, a complementary suite of theranostic α-, β−-, γ-, and β+-emitting radiometal ions, respectively. These radiometals were selected because they are among the most intensely investigated for use in targeted radiopharmaceutical development and translation, yet have been extremely challenging to bind using a single chelator due to their significant differences in ionic radii (Table 1) and coordination chemistries. Through a combination of structural, radiolabeling, serum stability, and in vivo studies, we demonstrate that PYTA can rapidly chelate all four radiometals at room temperature, forming complexes that are highly stable under physiological conditions. These results establish PYTA as one of the most versatile chelators investigated to date for theranostic applications.
| Lab | Lub | Inc | Scc | |
|---|---|---|---|---|
| a Values in parentheses are one standard deviation (of the last significant figures) from the average of crystallographically independent values. In instances where there is only one crystallographically independent value, the standard uncertainty of the refined value is reported. b From ref. 32. c This work. d Eight-coordinate ionic radii from ref. 17. e Averages were taken according to the number of carboxylic acid functional groups bound to the metal center (La, 4; Lu and Sc, 3; In, 2). | ||||
| Ionic radiusd | 1.160 | 0.977 | 0.92 | 0.870 |
| Space group | P21/n | C2/c |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
C2/c |
| M–Npy | 2.637(1) | 2.508(2) | 2.3401(10) | 2.463(4) |
| M–Namine | 2.702(10) | 2.594(27) | 2.523(89) | 2.578(49) |
| M–Ocarbe | 2.604(69) | 2.250(5) | 2.1328(10) | 2.137(5) |
| Npy–M–Npy | 178.40(7) | 146.45(7) | 108.58(5) | 146.04(5) |
Results and discussion
PYTA was synthesized according to published procedures,32,40 with minor modifications (see Section 1 of the ESI†). As a first assessment of the suitability of PYTA for the chelation of 225Ac, 177Lu, 111In, and 44Sc, its solution-state chemistry with La3+, Lu3+, In3+, and Sc3+ was probed by 1H nuclear magnetic resonance (NMR) spectroscopy in D2O. Ac3+ was substituted with La3+ for this study because Ac3+ does not possess any stable isotopes, rendering the obtention of NMR spectra challenging.41 La3+ is often used as a surrogate for Ac3+ because of its similar chemical properties and only slightly smaller ionic radius (1.160 Å for 8-coordinate La3+, 1.220 Å for 9-coordinate Ac3+).17,42 Our density functional theory calculations and ab initio molecular dynamics simulations reveal similarities between La3+ and Ac3+ coordination by PYTA, supporting the notion that the La3+ complex approximates the Ac3+ complex in solution (Section S2, Fig. S2 and Table S1†). NMR samples were prepared at pH ∼ 7 using equimolar amounts of chelator and metal ion, and their spectra were acquired after 1 h at room temperature, except for the La3+–PYTA sample, which required further heating at 60 °C (unoptimized) to drive the complexation reaction to completion (Section S3†). High-resolution mass spectrometry confirmed complex formation for all samples (Fig. S3–S6†).The NMR spectra obtained for La3+ and Lu3+ (Fig. S7†) were similar to those reported previously.32 Specifically, complexation of La3+ by PYTA gave rise to 8 sharp signals, indicating the formation of a D2-symmetric complex in which all pendent arms are bound to the metal center in a rigid 10-coordinate configuration. By contrast, the Lu3+ spectrum was consistent with an asymmetric 9-coordinate complex. This configuration was evident from the presence of 2 triplets and 2 doublets for the 6 pyridyl protons of the chelator, which signals that the pyridyl groups experience different magnetic environments. Additionally, a singlet was observed at 4.22 ppm, which can be assigned to the methylene protons of one uncoordinated acetate arm.
The spectrum of the Sc–PYTA complex (Fig. S7†) matched closely to that of the Lu–PYTA complex, indicating a shared 9-coordinate structural conformation. By contrast, the spectrum of the In–PYTA complex was less straightforward to interpret (Fig. S7†). Broadening of the aliphatic resonances precluded any meaningful analysis of this region. However, two sets of signals can be discerned in the aromatic region in approximately a 65/35 ratio, one set belonging to a symmetric species (major) and the other to an asymmetric species (minor). Upon heating the sample to 90 °C, the two sets of aromatic signals were observed to coalesce (Fig. S8†). These findings are consistent with the existence of two configurational isomers in solution undergoing slow exchange on the NMR timescale. Another notable feature of the In–PYTA spectrum is that the aromatic resonances are shifted considerably downfield in comparison to those of the other complexes. This shift may signify that the pyridyl groups of PYTA are positioned closer to the electron-withdrawing metal center when chelating In3+versus La3+, Lu3+, and Sc3+. Collectively, the differences observed in the spectra of these complexes in aqueous solution point towards the flexibility of PYTA to adopt the configurations necessary to satisfy the coordination preferences of a range of metal ions.
To further interrogate the structural changes that PYTA undergoes upon binding these metals, we obtained single-crystal structures of its Sc3+ and In3+ complexes (Fig. 1) and compared them to the published crystal structures of its La3+ and Lu3+ complexes, [La(H2PYTA)](NO3)·3H2O and [Lu(HPYTA)]·3H2O.32 Select interatomic distances and angles are presented in Table 1, and additional details of the crystallographic refinement are provided in Section S4 and Table S2.† The Sc3+ complex crystallized in the C2/c space group as [Sc(HPYTA)]·3H2O (Fig. 1), with a conformation that is isostructural to that of the Lu3+ complex. Specifically, 3 of the 4 pendent arms are deprotonated and engaged in binding interactions with the metal center, giving rise to a 9-coordinate complex that is consistent with our NMR analysis. Additionally, the pyridine rings of the core macrocycle are twisted away from one another and folded inward towards the metal center (Fig. 2), giving rise to a Npy–M–Npy angle of 146.04(5)° (Table 1). This “twist-fold” conformation is typical for PYTA when complexing smaller lanthanide ions, whereas the macrocycle adopts a more planar structure when binding larger ions like La3+ (Fig. 2, Npy–M–Npy = 178.40(7)°).32
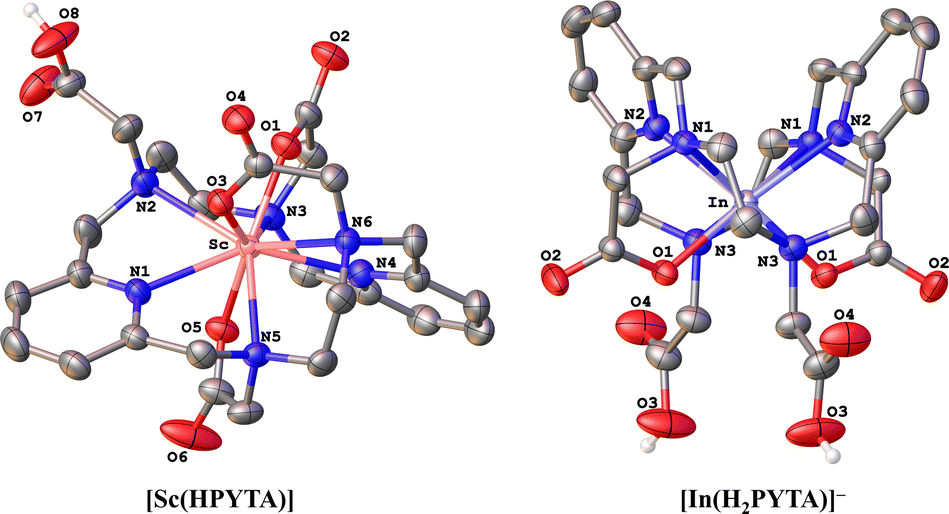 | ||
| Fig. 1 X-ray crystal structures of Sc3+ (left) and In3+ (right) complexes of PYTA. Ellipsoids for carbons and heteroatoms are drawn at the 50% probability level. Counteranions, outer-sphere solvent molecules, and hydrogen atoms attached to carbon centers have been omitted for the sake of clarity. | ||
 | ||
| Fig. 2 Configuration of the macrocyclic backbone of PYTA when bound to La3+, Lu3+, In3+, Sc3+, or Cu2+. 8-Coordinate ionic radii (6-coordinate for Cu2+) are provided below each metal ion. Pendent acetate arms have been omitted for clarity. | ||
Interestingly, In–PYTA crystallized in the Rc space group as the 8-coordinate C2-symmetric complex [In(H2PYTA)]ClO4 (Fig. 1). This result was somewhat unexpected because the ionic radius of In3+ is intermediate between that of Lu3+ and Sc3+ (Table 1). If the conformation of PYTA were driven exclusively by the size of the metal ion, the In–PYTA complex would be expected to crystallize in a configuration similar to that of Lu–PYTA and Sc–PYTA. Instead, for the In3+ structure, two acetate groups of PYTA remain protonated and uncoordinated, and the macrocyclic backbone is folded nearly in half (Fig. 2; Npy–M–Npy = 108.58(5)°). This complex represents an extreme example of the twist-fold conformation32 that has not been observed previously for py2[18]dieneN6 macrocycles, to the best of our knowledge. As the NMR data suggested, this conformation allows for the macrocycle to be drawn closer to the metal center, leading to shorter average interatomic metal-donor distances than expected based on the ionic radius of In3+ (Table 1). This divergent configuration in comparison to the other complexes likely arises from differences in the aqueous chemistry of In3+versus La3+, Lu3+, and Sc3+. Although all 4 ions are classified as relatively “hard” according to Pearson's hard-soft acid–base theory,43–45 the ionicity (IA) of In3+, taken as the ratio of the electrostatic and covalent contributions to bonding, is lower than that of La3+, Lu3+, and Sc3+ (6.30 versus 10.30, 10.07, and 10.49, respectively).46–49 Moreover, In3+ typically forms complexes with lower coordination numbers ranging from 4 to 8.50,51 These properties may be expected to direct the configuration of the In–PYTA complex to one that cannot be predicted on the basis of ion size. As a final demonstration of the extreme versatility of PYTA, its crystal structure was obtained with Cu2+, a chemically softer, divalent metal ion with a 6-coordinate ionic radius of only 0.73 Å17 and an IA of 2.68.43–45 In this structure (Fig. S9 and Table S2†), all pendent arms remain unbound, and the nitrogen atoms of the macrocycle are arranged in a pseudo-octahedral geometry around the Cu center. This “twist-wrap” conformation of PYTA,32 shown in Fig. 2, is likewise adopted by the unfunctionalized py2[18]dieneN6 macrocycle when bound to Cu2+.52
With these results in hand demonstrating that PYTA can accommodate a range of disparate metal ions by dramatically changing its conformation, we next sought to evaluate PYTA's ability to bind and stabilize 225Ac (t1/2 = 10.0 d), 177Lu (t1/2 = 6.65 d), 111In (t1/2 = 2.80 d), and 44Sc (t1/2 = 3.97 h). Concentration-dependent radiolabeling experiments were performed in pH 6 NH4OAc buffer at room temperature (RT). Radiochemical yields (RCYs) were assessed after 5 min and 60 min using radio-thin layer chromatography (Table S3 and Fig. S10–S23†). To benchmark the performance of PYTA, we also conducted additional radiolabeling experiments with DOTA and macropa (Table S3 and Fig. S10–S23†). Macropa (Scheme 1) was included for comparison because of its rare ability to rapidly form stable complexes with 225Ac and other large radiometals at room temperature.53–56 The results of the radiolabeling studies are collected in Fig. 3 and S24.†
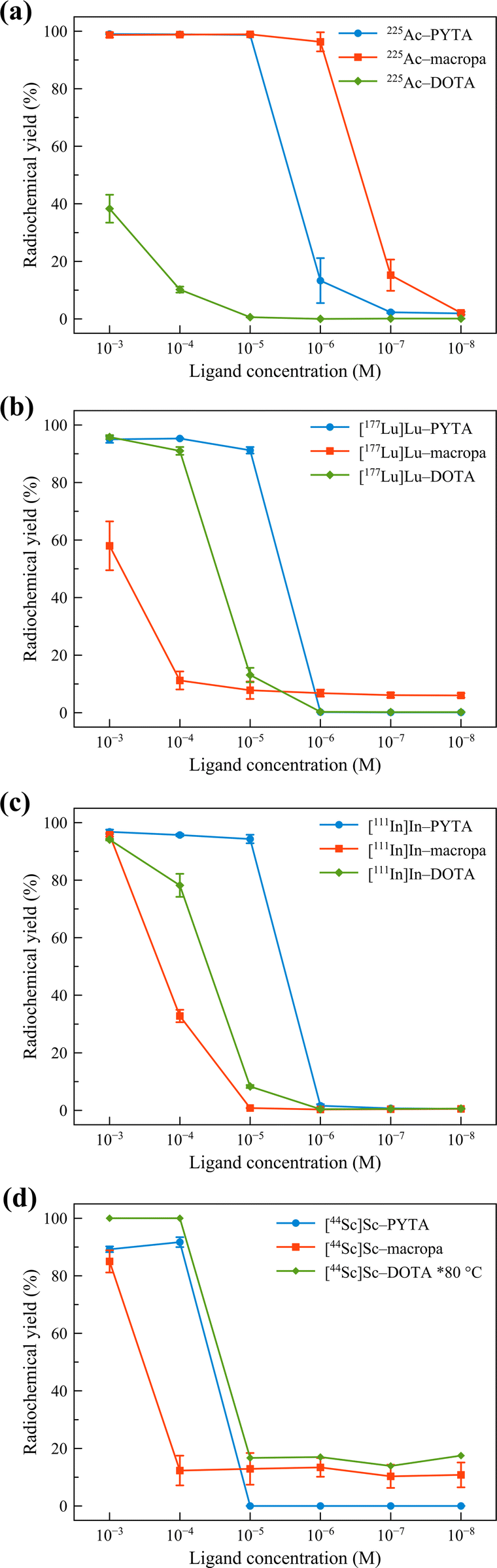 | ||
| Fig. 3 Concentration-dependent radiolabeling of PYTA, macropa, and DOTA with (a) 225Ac3+ (9.3–11.1 kBq), (b) [177Lu]Lu3+ (22–37 kBq), (c) [111In]In3+ (74–111 kBq), and (d) [44Sc]Sc3+ (185 kBq) after 5 min reaction time. All reactions were carried out at room temperature in NH4OAc (0.5 M, pH 6.0) except for [44Sc]Sc–DOTA, which was at 80 °C, VT = 150 μL. The error bars represent the standard deviations. Data for 225Ac–macropa and 225Ac–DOTA were taken from ref. 16. | ||
Remarkably, quantitative radiolabeling of PYTA was achieved with 225Ac, 177Lu, and 111In in just 5 min at chelator concentrations as low as 1 × 10−5 M. Likewise, PYTA was able to complex nearly all the 44Sc at a chelator concentration of 1 × 10−4 M (and down to 4 × 10−5 M, measured at 60 min only). We note that for all studies, a second, minor peak was observed in the radio-TLCs of 44Sc control samples regardless of the type of plate or mobile phase used. This small portion of activity, which remained unchelated even at the highest PYTA concentrations, has been attributed previously to the formation of insoluble (or difficult-to-chelate) oxide/hydroxide species.57 Increasing the mixing time to 60 min for 225Ac, 177Lu, 111In, and 44Sc reactions did not give rise to higher RCYs, indicating that complexation equilibrium was reached rapidly for each radionuclide. Under the same conditions, DOTA was found to be a less effective chelator than PYTA for 225Ac, 177Lu, and 111In, even after allowing the reactions to proceed for 60 min (Fig. S24†) and heating them to 80 °C (Fig. S25†). This inferior labeling efficacy is reflected by the low RCYs of 1%, 44%, and 24% obtained for 225Ac, 177Lu, and 111In at 10−5 M chelator concentration, 60 min, and RT, conditions under which PYTA is quantitatively radiolabeled. Radiolabeling of DOTA with 44Sc at 80 °C gave rise to RCYs on par with those obtained for PYTA (Fig. 3 and S24†) but was observed to be inconsistent at room temperature (Fig. S22†). Similarly, the RCYs of macropa were also substantially lower than those of PYTA for 177Lu, 111In, and 44Sc, but higher for 225Ac (Fig. 3 and S24†). These results are in line with macropa's exceptional ability to bind large metal ions like Ac3+ but correspondingly low affinity for smaller metal ions like Lu3+, In3+ and Sc3+.
Following these radiolabeling studies, the kinetic inertness of the resulting complexes of PYTA, DOTA, and macropa with 225Ac, 177Lu, 111In, and 44Sc (PYTA only) were investigated in 75% human serum at 37 °C. Human serum contains a variety of endogenous ligands and proteins that may compete with a chelator for metal binding, and therefore serves as a challenge to indicate the likelihood of transchelation in vivo. TLC systems and chelator concentrations used in these challenges, as well as representative radio-TLC traces, are provided in Table S4 and Fig. S26–S37.† The percentages of intact complexes over time are compiled in Table 2. Impressively, 225Ac–PYTA, [177Lu]Lu–PYTA, [111In]In–PYTA, and [44Sc]Sc–PYTA all remained ≥95% intact in serum over the course of two half-lives of the respective radionuclides. Similar results were also observed for DOTA, which retained 88%, 97%, and 96% of 225Ac3+, [177Lu]Lu3+ and [111In]In3+ over the course of the study. It should be noted, however, that due to DOTA's subpar radiolabeling efficacy, the concentration of DOTA required in the 177Lu and 111In studies (10−4 M) to ensure complex formation at the start of the experiment was an order of magnitude higher than the concentration used for PYTA (10−5 M). Like PYTA and DOTA, macropa also formed a highly stable complex with 225Ac, releasing <3% of bound 225Ac over 21 d. By contrast, [177Lu]Lu–macropa completely dissociated within the first hour of the experiment, and [111In]In–macropa underwent 75% decomplexation within 1 d. Interestingly, when a higher concentration of macropa (8.4 × 10−4 M vs. 1 × 10−4 M) was used in the 111In serum challenge, a dramatic rise in complex stability to 92% at 4 d was noted. This increase in intact complex with increasing chelator concentration is likely a thermodynamic effect arising from metal–ligand recombination in solution in the closed-tube system used for these studies, rather than a reflection of enhanced kinetic stability. Collectively, the radiolabeling studies and serum challenges reveal PYTA to be far superior to DOTA and macropa for rapidly binding and stabilizing this “superfecta” of medical radionuclides under mild, physiologically relevant conditions.
| Complex | Before serum addition | 1 h | 2 h | 8 h | 1 d | 2 d | 3 d | 4 d | 7 d | 14 d | 21 d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Standard deviations for all values are ≤2%. Bolded values indicate stability at the final time point. b ND = not determined. c Data taken from ref. 16. d C chelator = 1 × 10−5 M. e C chelator = 1 × 10−3 M. f C chelator = 1 × 10−4 M. g C chelator = 8.4 × 10−4 M. h Measured at t = 30 min. | |||||||||||
| 225Ac–PYTAd | 100 | 99 | ND | ND | 98 | 99 | 99 | 98 | 97 | 95 | 95 |
| 225Ac–macropac,d | 100 | 99 | ND | ND | 99 | 99 | 99 | ND | 99 | 98 | 97 |
| 225Ac–DOTAd | 99 | 98 | ND | ND | 98 | 97 | 96 | ND | 94 | 89 | 88 |
| [177Lu]Lu–PYTAd | 100 | 99 | ND | ND | 99 | 99 | 98 | 98 | 99 | 98 | ND |
| [177Lu]Lu–macropae | 82 | 0.6 | ND | ND | 0.9 | ND | ND | ND | ND | ND | ND |
| [177Lu]Lu–DOTAf | 99 | 98 | ND | ND | 99 | 99 | 98 | ND | 98 | 97 | ND |
| [111In]In–PYTAd | 97 | 97 | ND | ND | 97 | 95 | 96 | 97 | ND | ND | ND |
| [111In]In–macropag | 99 | 95 | ND | ND | 87 | 91 | 93 | 92 | ND | ND | ND |
| [111In]In–macropaf | 99 | 95 | ND | ND | 25 | ND | ND | ND | ND | ND | ND |
| [111In]In–DOTAf | 98 | 98 | ND | ND | 96 | 96 | 95 | 96 | ND | ND | ND |
| [44Sc]Sc–PYTAd | 94 | 94h | 95 | 93 | ND | ND | ND | ND | ND | ND | ND |
Finally, to establish the potential use of PYTA for in vivo diagnostic and therapeutic applications, we performed imaging and biodistribution studies in mice with [44Sc]Sc–PYTA and 225Ac–PYTA, the complexes having the most extreme disparity with respect to metal-ion size. For 44Sc studies, healthy female Swiss Webster mice were injected with [44Sc]Sc–PYTA (∼5.18 MBq) and monitored by dynamic PET imaging over the course of 55 min. Groups of mice were also sacrificed at 15 min, 1 h, and 4 h post injection, and the activity in their organs were quantified ex vivo. Frames across the dynamic PET acquisition (Fig. 4a) revealed uptake and localization of activity primarily in the kidneys and bladder of mice, indicative of rapid renal clearance of the intact complex. Additionally, a small amount of intestinal uptake was noted at these early time points (Fig. S38 and S39†), suggesting that some excretion also occurs through the hepatobiliary pathway. The biodistribution profile of [44Sc]Sc–PYTA (Fig. 4b and Table S5†) corroborated the results of the PET imaging, showing high kidney uptake at 15 min (10.3% ID per g) that diminished over time as the complex was excreted, and a spike in intestinal activity (7.2% ID per g) at 4 h post injection. Interestingly, this gut uptake is substantially less than that observed previously for other radioscandium complexes such as [43/47Sc]Sc–HOPO (∼19% ID per g at 4 h),57 highlighting the different pharmacokinetic properties of these complexes. Of significance, the biodistribution profile of [44Sc]Sc–PYTA differs dramatically from that of free 44Sc (administered as [44Sc]ScCl3), which exhibits a long circulation time and substantial uptake in the heart, lungs, liver, and spleen of mice (Fig. 4c).58 Thus, the absence of accumulation of activity in these organs upon administering [44Sc]Sc–PYTA to mice signifies that PYTA is able to retain 44Sc in vivo over the time course of the study. Similar biodistribution results were obtained upon administering 225Ac–PYTA (∼3.7 kBq) to mice (Fig. 5 and Table S6†). Specifically, the complex was rapidly cleared through the renal system, and no activity was observed to accumulate in any organ over 48 h. This distribution pattern contrasts with that of free 225Ac3+, which is taken up by the liver, spleen, and bone of mice.53 The predominant renal clearance of [44Sc]Sc–PYTA and 225Ac–PYTA observed here in mice is similar to the biodistribution of [153Sm]Sm–PYTA observed previously in rats.30 Collectively, the results of these in vivo studies establish proof of concept for PYTA as a robust chelator for a complementary set of medically important radionuclides.
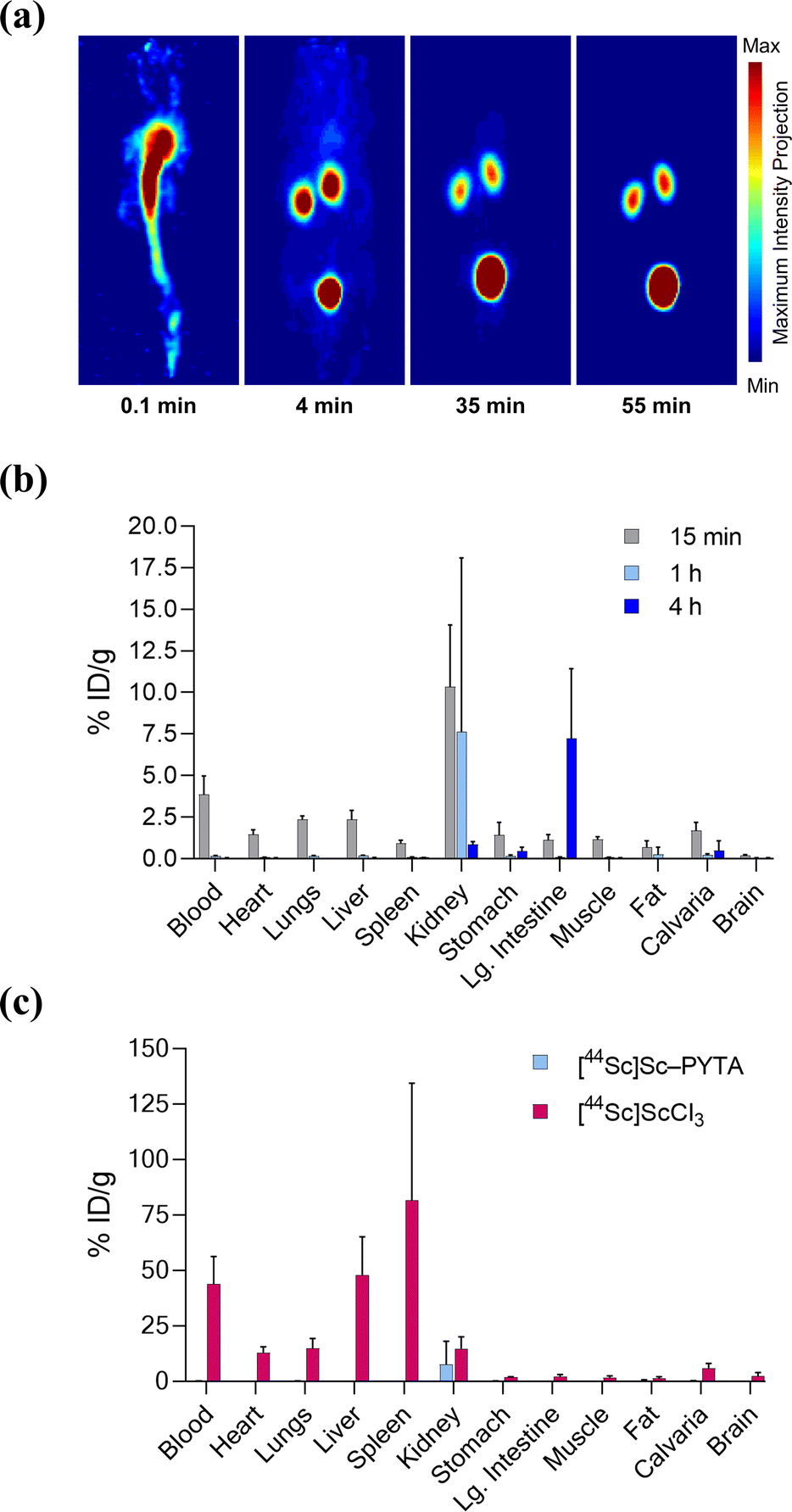 | ||
| Fig. 4 PET imaging and ex vivo biodistribution of [44Sc]Sc–PYTA in female Swiss Webster mice (6–8 weeks, n = 4 per time point) following intravenous injection. (a) Representative 3D maximum intensity projections of the PET acquisitions at 0.1, 4, 35, and 55 min post injection indicating rapid transit from the blood pool to the kidneys and bladder. (b) Biodistribution of [44Sc]Sc–PYTA for select organs after 15 min, 1 h, and 4 h post injection. (c) Comparison of biodistribution of [44Sc]Sc–PYTA and [44Sc]ScCl3 at 1 h post injection. Values for each time point are given as the mean % ID per g ± 1 SD. Data for [44Sc]ScCl3 are taken from ref. 58. | ||
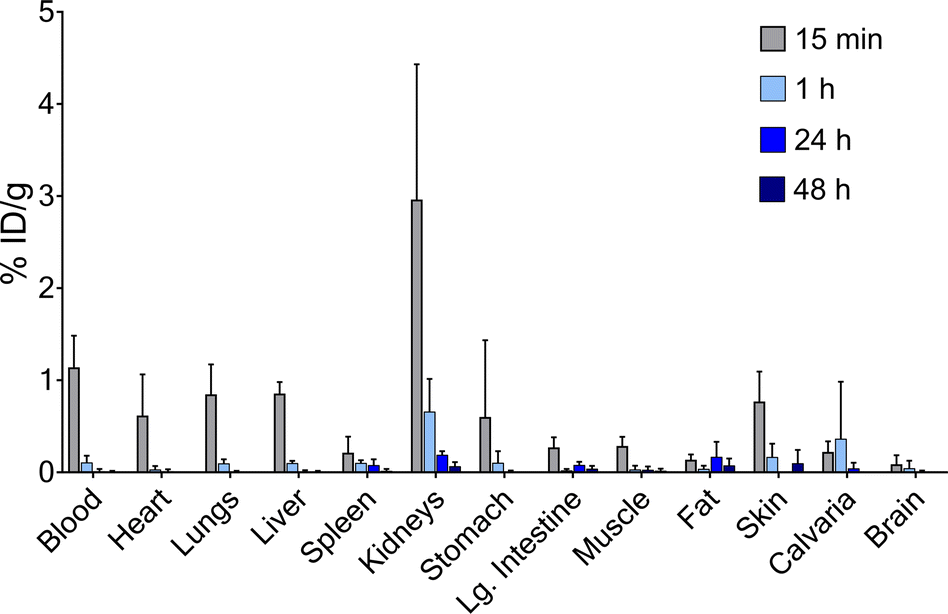 | ||
| Fig. 5 Ex vivo biodistribution of 225Ac–PYTA for select organs following intravenous injection in mice. Female Swiss Webster mice (6–8 weeks) were sacrificed 15 min, 1 h, 24 h, or 48 h post injection. Values for each time point are given as the mean % ID per g ± 1 SD (n = 4). | ||
Conclusions
In summary, we have reported a thorough investigation of the potential of PYTA for the chelation of 225Ac, 177Lu, 111In, and 44Sc, 4 radionuclides that are of rising importance in nuclear medicine for targeted α therapy, targeted β therapy, SPECT imaging, and PET imaging, respectively. Through a dramatic change in conformation revealed by NMR and X-ray studies, PYTA can optimally accommodate and form distinct complexes with La3+ (as a surrogate for Ac3+), Lu3+, In3+, and Sc3+. Our studies further showed that PYTA rapidly binds this set of radiometals under mild conditions, forming complexes that are stable in vitro and in vivo. Notably, these combined results were superior to those obtained with 2 current state-of-the art chelators, DOTA and macropa, revealing PYTA as a highly versatile and effective chelator for radiotheranostic development. Future efforts are focused on preparing a bifunctional derivative of PYTA for use with tumor-targeting biomolecules.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported in part by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory (ORNL; M. E. S., M. M. S., A. S. I., F. D. W., C. M. L., N. A. T.). Additionally, A. F., Z. L., and D. L. J. T. thank the National Cancer Institute at the National Institutes of Health for support under award numbers R01CA240711 and R01CA229893. C. M. L. was supported in part by an appointment to the Oak Ridge National Laboratory GEM Fellow internship program, sponsored by the U.S. Department of Energy (DOE) and administered by the Oak Ridge Institute for Science and Education. The 225Ac, 177Lu, and 44Ti/44Sc used in this research was supplied by the DOE Isotope Program, managed by the Office of Isotope R&D and Production. This research also used resources of the Oak Ridge Leadership Computing Facility at ORNL. This work has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the U.S. DOE. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for U.S. Government purposes. The DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan).References
- P. J. Blower, Dalton Trans., 2015, 44, 4819–4844 RSC.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- N. Herrero Álvarez, D. Bauer, J. Hernández-Gil and J. S. Lewis, ChemMedChem, 2021, 16, 2909–2941 CrossRef PubMed.
- M. Radzina, L. Saule, E. Mamis, U. Koester, T. E. Cocolios, E. Pajuste, M. Kalnina, K. Palskis, Z. Sawitzki, Z. Talip, M. Jensen, C. Duchemin, K. Leufgen and T. Stora, EJNMMI Radiopharm. Chem., 2023, 8, 27 CrossRef PubMed.
- S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149–162 RSC.
- J.-P. Pouget and J. Constanzo, Front. Med., 2021, 8, 692436 CrossRef.
- B. M. Bavelaar, B. Q. Lee, M. R. Gill, N. Falzone and K. A. Vallis, Front. Pharmacol., 2018, 9, 996 CrossRef.
- F. Roesch and M. Martin, J. Radioanal. Nucl. Chem., 2023, 332, 1557–1576 CrossRef CAS.
- L. Bodei, K. Herrmann, H. Schöder, A. M. Scott and J. S. Lewis, Nat. Rev. Clin. Oncol., 2022, 19, 534–550 CrossRef CAS PubMed.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- E. Boros and J. P. Holland, J. Label. Compd. Radiopharm., 2018, 61, 652–671 CrossRef CAS PubMed.
- I. Carbo-Bague and C. F. Ramogida, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd, 2021, pp. 1–34 Search PubMed.
- A. Hu and J. J. Wilson, Acc. Chem. Res., 2022, 55, 904–915 CrossRef CAS PubMed.
- A. Hu, S. N. MacMillan and J. J. Wilson, J. Am. Chem. Soc., 2020, 142, 13500–13506 CrossRef CAS PubMed.
- A. Hu, E. Aluicio-Sarduy, V. Brown, S. N. MacMillan, K. V. Becker, T. E. Barnhart, V. Radchenko, C. F. Ramogida, J. W. Engle and J. J. Wilson, J. Am. Chem. Soc., 2021, 143, 10429–10440 CrossRef CAS PubMed.
- A. Hu, M. E. Simms, V. Kertesz, J. J. Wilson and N. A. Thiele, Inorg. Chem., 2022, 61, 12847–12855 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Cieslik, M. Kubeil, K. Zarschler, M. Ullrich, F. Brandt, K. Anger, H. Wadepohl, K. Kopka, M. Bachmann, J. Pietzsch, H. Stephan and P. Comba, J. Am. Chem. Soc., 2022, 144, 21555–21567 CrossRef CAS PubMed.
- I. Kopp, P. Cieslik, K. Anger, T. Josephy, L. Neupert, G. Velmurugan, M. Gast, H. Wadepohl, S. A. Brühlmann, M. Walther, K. Kopka, M. Bachmann, H. Stephan, M. Kubeil and P. Comba, Inorg. Chem., 2023, 62, 20754–20768 CrossRef CAS PubMed.
- L. Wharton, M. d. G. Jaraquemada-Pelaéz, C. Zhang, J. Zeisler, C. Rodríguez-Rodríguez, M. Osooly, V. Radchenko, H. Yang, K.-S. Lin, F. Bénard, P. Schaffer and C. Orvig, Bioconjugate Chem., 2022, 33, 1900–1921 CrossRef CAS PubMed.
- L. Wharton, H. Yang, M. d. G. Jaraquemada-Pelaéz, H. Merkens, G. Engudar, A. Ingham, H. Koniar, V. Radchenko, P. Kunz, P. Schaffer, F. Bénard and C. Orvig, J. Med. Chem., 2023, 66, 13705–13730 CrossRef CAS PubMed.
- U. Hennrich and K. Kopka, Pharmaceuticals, 2019, 12, 114 CrossRef CAS PubMed.
- U. Hennrich and M. Benešová, Pharmaceuticals, 2020, 13, 38 CrossRef CAS PubMed.
- U. Hennrich and M. Eder, Pharmaceuticals, 2022, 15, 1292 CrossRef CAS PubMed.
- Society of Nuclear Medicine and Molecular Imaging, J. Nucl. Med., 2022, 63(5), 13N Search PubMed.
- C. B. Johnbeck, U. Knigge, A. Loft, A. K. Berthelsen, J. Mortensen, P. Oturai, S. W. Langer, D. R. Elema and A. Kjaer, J. Nucl. Med., 2017, 58, 451–457 CrossRef CAS PubMed.
- O. O. Krasnovskaya, D. Abramchuck, A. Erofeev, P. Gorelkin, A. Kuznetsov, A. Shemukhin and E. K. Beloglazkina, Int. J. Mol. Sci., 2023, 24, 9154 CrossRef CAS PubMed.
- B. J. B. Nelson, J. D. Andersson and F. Wuest, Pharmaceutics, 2021, 13, 49 CrossRef CAS PubMed.
- A. Jang, A. T. Kendi, G. B. Johnson, T. R. Halfdanarson and O. Sartor, Int. J. Mol. Sci., 2023, 24, 11626 CrossRef CAS PubMed.
- W. D. Kim, G. E. Kiefer, F. Maton, K. McMillan, R. N. Muller and A. D. Sherry, Inorg. Chem., 1995, 34, 2233–2243 CrossRef CAS.
- L. Miao, D. Bell, G. L. Rothremel Jr, L. H. Bryant Jr, P. M. Fitzsimmons and S. C. Jackels, Supramol. Chem., 1996, 6, 365–373 CrossRef CAS.
- L. Valencia, J. Martinez, A. Macías, R. Bastida, R. A. Carvalho and C. F. G. C. Geraldes, Inorg. Chem., 2002, 41, 5300–5312 CrossRef CAS PubMed.
- R. Bastida, D. E. Fenton, M. López-Deber, A. Macías, L. Valencia and M. Vicente, Inorg. Chim. Acta, 2003, 355, 292–301 CrossRef CAS.
- M. D. C. Fernández-Fernández, R. Bastida, A. Macías, P. Pérez-Lourido, C. Platas-Iglesias and L. Valencia, Inorg. Chem., 2006, 45, 4484–4496 CrossRef PubMed.
- C. Núñez, R. Bastida, A. Macías, M. Mato-Iglesias, C. Platas-Iglesias and L. Valencia, Dalton Trans., 2008, 3841–3850 RSC.
- G. Castro, M. Regueiro-Figueroa, D. Esteban-Gõmez, R. Bastida, A. Macías, P. Pérez-Lourido, C. Platas-Iglesias and L. Valencia, Chem.–Eur. J., 2015, 21, 18662–18670 CrossRef CAS PubMed.
- G. Castro, M. Regueiro-Figueroa, D. Esteban-Gómez, P. Pérez-Lourido, C. Platas-Iglesias and L. Valencia, Inorg. Chem., 2016, 55, 3490–3497 CrossRef CAS PubMed.
- G. Castro, G. Wang, T. Gambino, D. Esteban-Gómez, L. Valencia, G. Angelovski, C. Platas-Iglesias and P. Pérez-Lourido, Inorg. Chem., 2021, 60, 1902–1914 CrossRef CAS PubMed.
- C. Harriswangler, L. Caneda-Martínez, O. Rousseaux, D. Esteban-Gómez, O. Fougère, R. Pujales-Paradela, L. Valencia, M. I. Fernández, N. Lepareur and C. Platas-Iglesias, Inorg. Chem., 2022, 61, 6209–6222 CrossRef CAS PubMed.
- G. L. Rothermel Jr, L. Miao, A. L. Hill and S. C. Jackels, Inorg. Chem., 1992, 31, 4854–4859 CrossRef.
- B. W. Stein, A. Morgenstern, E. R. Batista, E. R. Birnbaum, S. E. Bone, S. K. Cary, M. G. Ferrier, K. D. John, J. L. Pacheco, S. A. Kozimor, V. Mocko, B. L. Scott and P. Yang, J. Am. Chem. Soc., 2019, 141, 19404–19414 CrossRef CAS PubMed.
- G. J.-P. Deblonde, M. Zavarin and A. B. Kersting, Coord. Chem. Rev., 2021, 446, 214130 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- R. S. Drago, G. C. Vogel and T. E. Needham, J. Am. Chem. Soc., 1971, 93, 6014–6026 CrossRef CAS.
- R. D. Hancock and F. Marsicano, Inorg. Chem., 1978, 17, 560–564 CrossRef CAS.
- R. D. Hancock and F. Marsicano, Inorg. Chem., 1980, 19, 2709–2714 CrossRef CAS.
- A. E. Martell and R. D. Hancock, Metal Complexes in Aqueous Solutions, Plenum Press, New York, 1996 Search PubMed.
- H. R. Maecke, A. Riesen and W. Ritter, J. Nucl. Med., 1989, 30, 1235–1239 CAS.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- L. Branco, J. Costa, R. Delgado, M. G. B. Drew, V. Félix and B. J. Goodfellow, J. Chem. Soc., Dalton Trans., 2002, 3539–3550 RSC.
- N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. H. Robertson, C. Rodríguez-Rodríguez, P. Schaffer, C. Williams Jr, J. W. Babich, V. Radchenko and J. J. Wilson, Angew. Chem., Int. Ed., 2017, 56, 14712–14717 CrossRef CAS PubMed.
- E. Aluicio-Sarduy, N. A. Thiele, K. E. Martin, B. A. Vaughn, J. Devaraj, A. P. Olson, T. E. Barnhart, J. J. Wilson, E. Boros and J. W. Engle, Chem.–Eur. J., 2020, 26, 1238–1242 CrossRef CAS PubMed.
- F. Reissig, D. Bauer, M. Ullrich, M. Kreller, J. Pietzsch, C. Mamat, K. Kopka, H.-J. Pietzsch and M. Walther, Pharmaceuticals, 2020, 13, 272 CrossRef CAS PubMed.
- D. S. Abou, N. A. Thiele, N. T. Gutsche, A. Villmer, H. Zhang, J. J. Woods, K. E. Baidoo, F. E. Escorcia, J. J. Wilson and D. L. J. Thorek, Chem. Sci., 2021, 12, 3733–3742 RSC.
- M. D. Phipps, S. Cingoranelli, N. V. S. D. K. Bhupathiraju, A. Younes, M. Cao, V. A. Sanders, M. C. Neary, M. H. Devany, C. S. Cutler, G. E. Lopez, S. Saini, C. C. Parker, S. R. Fernandez, J. S. Lewis, S. E. Lapi, L. C. Francesconi and M. A. Deri, Inorg. Chem., 2023, 62, 20567–20581 CrossRef CAS PubMed.
- N. Benabdallah, H. Zhang, R. Unnerstall, A. Fears, L. Summer, M. Fassbender, B. E. Rodgers, D. Abou, V. Radchenko and D. L. J. Thorek, EJNMMI Res., 2023, 13, 17 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and X-ray characterization data, DFT calculations, animal protocols, and representative radio-TLC chromatograms. CCDC 2297429, 2297430, and 2339314. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06854d |
| ‡ These authors contributed equally to this work. |
| § Present address: Radioisotope Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, United States. |
| This journal is © The Royal Society of Chemistry 2024 |