
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemi-crystalline polymers with supramolecular synergistic interactions: from mechanical toughening to dynamic smart materials
Chen-Yu
Shi
,
Wen-Yu
Qin
and
Da-Hui
Qu
 *
*
Key Laboratory for Advanced Materials, Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China. E-mail: dahui_qu@ecust.edu.cn
First published on 11th May 2024
Abstract
Semi-crystalline polymers (SCPs) with anisotropic amorphous and crystalline domains as the basic skeleton are ubiquitous from natural products to synthetic polymers. The combination of chemically incompatible hard and soft phases contributes to unique thermal and mechanical properties. The further introduction of supramolecular interactions as noncovalently interacting crystal phases and soft dynamic crosslinking sites can synergize with covalent polymer chains, thereby enabling effective energy dissipation and dynamic rearrangement in hierarchical superstructures. Therefore, this review will focus on the design principles of SCPs by discussing supramolecular construction strategies and state-of-the-art functional applications from mechanical toughening to sophisticated functions such as dynamic adaptivity, shape memory, ion transport, etc. Current challenges and further opportunities are discussed to provide an overview of possible future directions and potential material applications.
 Chen-Yu Shi | Chen-Yu Shi received her B.S. and PhD degrees from the East China University of Science and Technology (ECUST), Shanghai, China, under the supervision of Prof. Da-Hui Qu. After 2023, she joined the group of Prof. He Tian as a postdoc. Her research focuses on dynamic chemistry, especially disulfide-based dynamic polymers for smart materials. |
 Wen-Yu Qin | Wen-Yu Qin received his B.S. degree from ECUST in 2021. He is currently a PhD student in the group of Prof. Da-Hui Qu. His current research focuses on dynamic polymer materials based on disulfide and host–guest chemistry. |
 Da-Hui Qu | Da-Hui Qu obtained his B.S. degree from Qingdao University in 2000 and received his PhD degree in applied chemistry in 2006 from ECUST, under the supervision of Prof. He Tian. From 2006 to 2009, he worked with Prof. Ben L. Feringa as a postdoctoral fellow. Since 2009, he has been working at ECUST and is now the dean of the School of Chemistry and Molecular Engineering, ECUST. His current research focuses on the use of supramolecular and dynamic chemistry to develop a series of smart dynamic molecular, supramolecular and polymeric systems and materials. |
1. Introduction
SCPs with alternating amorphous and crystalline domains as the basic skeleton are omnipresent in nature, such as protein, starch, cellulose, etc.1–3 For instance, ordered protein structures originate from adjacent amino acid chain-folding as crystalline motif configuration, which can be facilitated and stabilized by the thermodynamically driven assembly process based on a series of weak interactions.4 For synthetic polymers, materials science has developed rapidly since the proposal of the concept of a “macromolecule” by Staudinger in 1920, i.e., long chain molecules composed of structural units connected by covalent bonds.5 Nowadays, more than two-thirds of commercial polymers are semi-crystalline, such as polyolefin (PO),6 polycaprolactone (PCL),7 polyurethane (PU),8 and polyamide (nylons)9 are extensively utilized as commercial products (such as packaging and electronics) and industrial materials (such as aerospace and gas pipelines).The anisotropic alignment of polymer chains is the fundamental mechanism for SCPs. In the amorphous soft matrix, crystalline hard domains as physical crosslink sites can not only ensure dimensional stability and solvent resistance, but also improve network toughness, thus contributing to unique thermal and mechanical properties of SCPs.10 Nevertheless, for conventional covalent polymers which are usually composed of thermodynamically incompatible blocks, the formation of the crystalline domain is attributed to chain mobility driven by thermodynamic equilibrium and weak interchain van der Waals forces. The resulting SCPs undergo unavoidable external force-induced network cavitation and damage over time, which causes degradation in mechanical properties and eventually results in material failure.11,12 To overcome the performance limitation and irreversible resource waste, the construction of dynamic polymer materials based on supramolecular chemistry is an effective strategy.13,14 Besides weak van der Waals force, diverse supramolecular interactions (such as H-bonds, π–π stacking, and hydrophobic associations) with tunable bonding association and directionality can be introduced into polymer networks and participate in the formation of SCPs in two ways: (1) as soft dynamic crosslinking sites among rigid covalent chain crystal phases; (2) forming noncovalently interacting crystal phases in a soft polymeric matrix. Therefore, the introduction of supramolecular chemistry into crystalline interlayers and two-phase interfaces enables effective energy dissipation and rearrangement by exchange dynamics into superstructures of a hierarchical nature, which extends supramolecular synergistic SCPs (SSCPs) from mechanical toughening to dynamic smart applications including self-healing, shape memory, ion transport, recyclability, etc.15–17
In recent years, SCPs have gained ever-growing interest due to their simple synthesis chemistry and tailoring process compared to liquid crystalline elastomers (LCEs). Although numerous promising SCPs have been developed to achieve material performance improvement and multi-field applications, there have been few systematic reviews to summarize the structure, mechanism, and application of SCPs besides several engineering papers.11,18 Herein, we focus on the supramolecular synergic construction strategies of SCPs and the underlying chemistry mechanism (Fig. 1). Then, state-of-the-art functional applications for mechanical toughening and dynamic smart materials are summarized and highlighted. Finally, future opportunities and challenges are proposed for SSCPs with multidisciplinary applications. Notably, we focus on the recently emerged construction strategies and a fraction of representative applications as high-performance dynamic materials instead of a comprehensive review of SCPs. Organic crystals and metal–organic frameworks that are generally considered high crystalline static structures, LCEs, and multiphase composites are beyond the scope of this review.
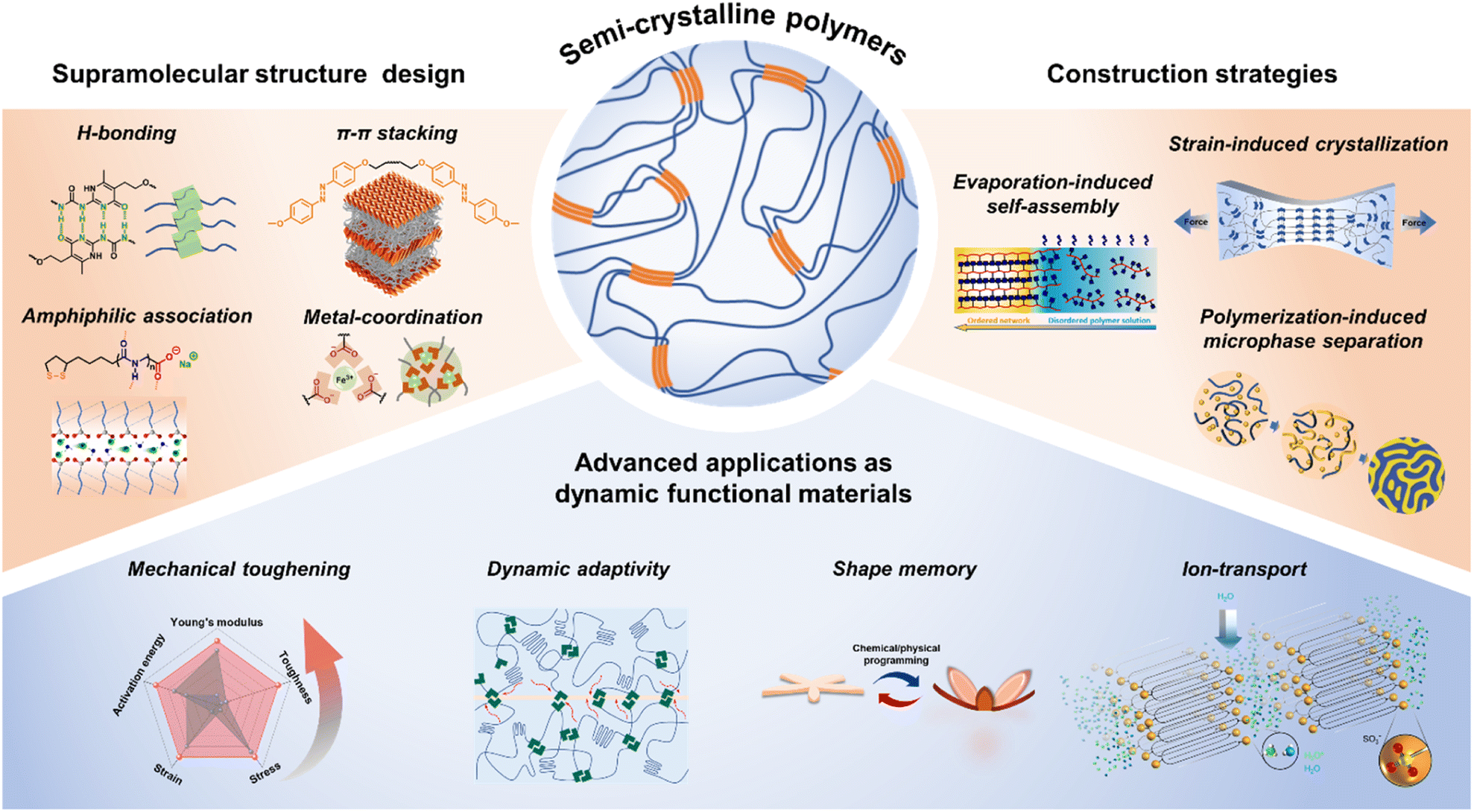 | ||
| Fig. 1 Schematic illustration of the structure design and construction strategies of SSCPs and their advanced applications as dynamic functional materials. | ||
2. Molecular design principles
In nature, semi-crystalline structures are normally formed spontaneously via the self-assembly of nanoscale molecular precursors driven by van der Waals interactions, H-bonding, electrostatic forces, hydrophobic interactions, solvation effects, etc.19 For example, spider silk with a two-phase structure is considered one of the toughest natural materials, in which the anisotropic β-sheet crystalline phase embedded in amorphous regions plays a key role in mechanical properties.20,21 Similar to the formation mechanism of natural polymers, the nanostructures of synthetic SCPs are composed of at least two chemically independent domains, i.e., robust crystalline domains as a crosslinker and flexible chains as a soft matrix.22,23For representative linear semi-crystalline polymers such as PO and PCL,24,25 driven by enthalpic and entropic contributions to thermodynamic equilibrium, flexible chains with high symmetry and regularity tend to be close together and shorten the interchain distance, thus promoting the formation of crystalline domains via increased van der Waals forces. The polymer chain length is critical to the formation of the crystalline phase and affects the topology, e.g., shorter polymer chains act as soft domains, while the introduction of rigid structures induces interchain crystallization. Long polymer chains tend to form intrachain crystallization through chain-folding.
Another typical class of hybrid structures comes from the block copolymerization of alternating thermodynamically incompatible segments, which has emerged as a major topic in polymer chemistry due to the resulting fascinating equilibrium and metastable morphologies over the past 60 years.23,26 Block incompatibility (Flory–Huggins interaction parameter χ) and the polymerization degree (N) are crucial for microphase separation. The soft chain mobility allows the block copolymers to arrange the microdomains in an ordered fashion. The size, distribution and strength of crystalline domains can be tailored by modifying the structure and proportion of hard/soft blocks, as well as crosslinking types (including covalent crosslinking and supramolecular crosslinking), thus generating ordered nanostructures such as lamellar, gyroid, cylindrical, and spherical morphologies.27
Although the crystalline domain can act as a physical crosslink site, conventional linear and covalent crosslink structures may cause irreversible damage to the polymeric network during stretching. Therefore, the introduction of additional weak interactions can synergize with the crystalline domains for crystalline configuration regulation and effective energy dissipation, thus endowing the polymer with enhanced mechanical and dynamic properties.1 Noncovalent bonding motifs are commonly incorporated in polymer mainchains and sidechains (including brush and branched polymers), or functionalized as end groups. Herein, molecular design principles will be discussed according to the representative supramolecular interactions, which assemble into dynamic crystalline motifs or trigger the nanoscale crystalline domains of polymer chains in a synergic way, thus promoting efficient energy dissipation as well as dynamic rearrangement.
2.1. H-bonding
As one of the simplest and most versatile noncovalent interactions, H-bonds are ubiquitous in natural semi-crystalline structures, such as proteins, cellulose, etc.28,29 Compared to other weak interactions, H-bonds are easily introduced from polar groups such as carboxyl, urethane, urea, etc. Therefore, it has emerged as a common approach to construct high-performance synthetic polymers such as nylon and polyurethane.9,30 For instance, Liu and coworkers designed poly(urethane-urea) polymers containing multiple acylsemicarbazide (ASCZ) and urethane moieties linked with alicyclic hexatomic and aromatic spacers (Fig. 2a).22 The stiff segments bearing abundant H-bond donors and acceptors as well as alicyclic hexatomic spacers with higher flexibility contributed to higher density of distinctive hard domains that exhibit smaller sizes but involve denser H-bonds. The high-density H-bond arrays vigorously strengthened the polymeric networks and dissipated substantial energy via dynamic rupture and reformation, thus enabling exceptional mechanical robustness comparable to natural spider silk.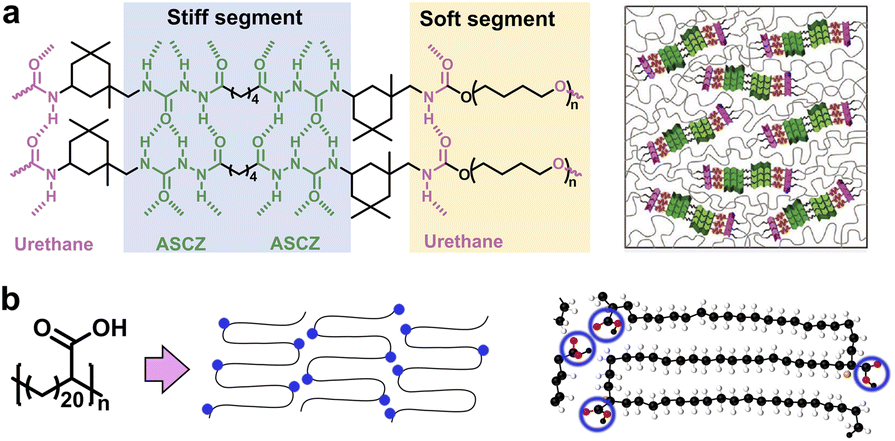 | ||
| Fig. 2 (a) Typical nanostructure of synthetic poly(urethane-urea) composed of stiff and soft segments. Copyright 2021, Wiley-VCH. Reproduced with permission from ref. 22. (b) Polyethylene brush polymer with precise hairpin architecture. Copyright 2017, American Chemical Society. Reproduced with permission from ref. 32. | ||
Unlike conventional linear polymers, the steric congestion of the sidechain enables suppression of the backbone entanglement, which promotes the self-assembly of well-ordered structures with characteristic nano-domains.31 Winey and co-workers designed a functional polyethylene having a precise architecture with pendant carboxylic acid sidechain groups precisely at every 21st backbone carbon atom (Fig. 2b).32 The mainchains were folded in a hairpin manner near each carboxylic acid group to obtain a semi-crystalline morphology with multiple embedded layers of functional groups that have an interlayer distance of 2.5 nm, which was roundly verified by atomistic molecular dynamics simulations with experimental X-ray scattering and Raman spectroscopy. The intriguing multilayered adjacent-reentry structure may be caused by the H-bond template provided by carboxyl groups between the crystal–amorphous interface. The resulting layers of acid groups could act as two-dimensional pathways for effective ion or molecular transport within polymeric crystals.
Despite the low bonding energy of single H-bonds, the binding affinity, directionality and multivalency can be tunable in a wide range via controlling the H-bonding sites and donor (D)/acceptor (A) sequence.33 The most representative example is 2-ureido-4[1H]-pyrimidinone (UPy), a complementary quadruple H-bond pioneered by Meijer et al.34 The AADD H-bonding array endows UPy with a high association constant up to 6 × 107 M−1 in chloroform. Because of its high binding affinity and modifiability, UPy-based noncovalent moieties have been widely introduced in polymer chains to provide multiple H-bonding crystalline domains.35 For example, Meijer and coworkers designed multiblock poly(ethylene glycol) (PEG)-based amphiphilic copolymers containing UPy moieties within the backbone and investigated the evolution of the crystal phase during hydration (Fig. 3a).36 Cryo-transmission electron microscopy (cryo-TEM) and small-angle X-ray scattering (SAXS) measurements verified the persistence of the nanometer-scale phase separation across a broad spectrum of conditions. Specifically, the dry polymer network exhibited a lamellar morphology composed of alternating crystalline and amorphous PEG segments. Throughout the hydration process, PEG crystalline regions preferentially absorbed water and underwent gradual dissolution, ultimately becoming an amorphous matrix. Meanwhile, the hydrophobic segments segregated into nanoscale domains to form small spherical compartments with a ∼2–5 nm diameter, which consist of UPy dimers flanked by the dense oligo-methylene hydrophobic segments to shield the water from disrupting the H-bonding. The UPy dimers embedded in segregated hydrophobic domains dispersed within the PEG matrix offered nanoscopic physical crosslink sites, jointly driving the cumulative mechanical properties of both dry polymers and hydrogels.
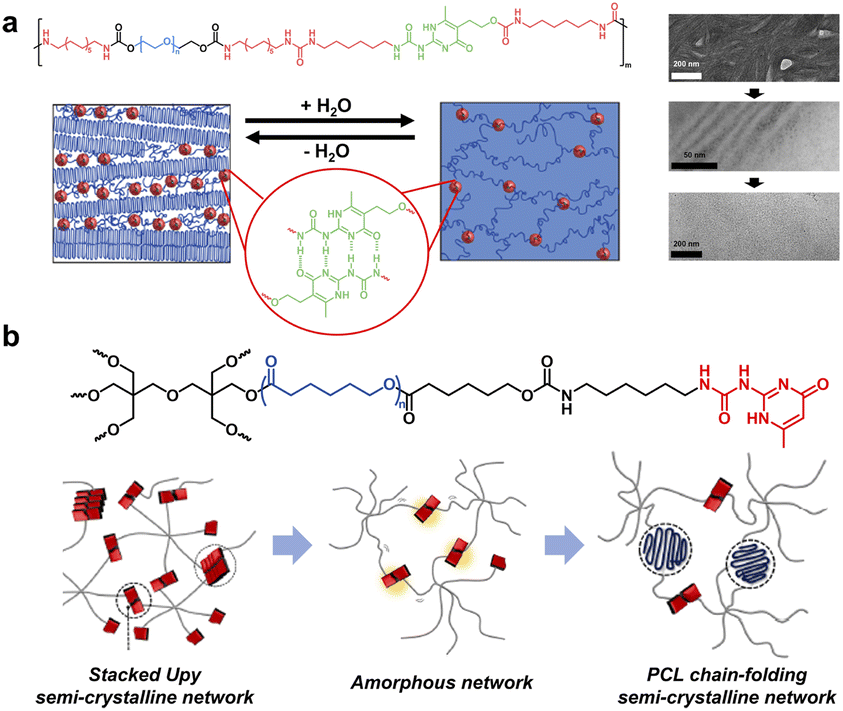 | ||
| Fig. 3 (a) Hydration triggered crystal-phase evolution of multiblock PEG-based amphiphilic copolymers containing UPy moieties. Copyright 2014, American Chemical Society. Reproduced with permission from ref. 36. (b) Dual dynamic networks composed of quadruple H-bonds and crystalline physical interactions, whose morphology is determined using the arm-length. Copyright 2020, Elsevier. Reproduced with permission from ref. 40. | ||
For branched structures, a high degree of cross-linking tends to discourage chain stacking and inhibit crystallization, resulting in amorphous networks instead of ordered structures.37–39 Interestingly, Chung and co-workers constructed dual dynamic networks composed of quadruple H-bonds and crystalline physical interactions using UPy terminated semi-crystalline branched PCLs (Fig. 3b).40 The arm-length was crucial for the phase transition and cross-linking density of the network; specifically, the network changed from UPy-stacked crystals to an amorphous phase and further to chain-folding polymeric crystals with increasing arm-length, along with the enhanced healing capability at the same time. In addition, well-controlled arm-length could provide a mechanically rigid semi-crystalline supramolecular network with efficient healing properties due to reversible dual dynamic features, associated with the re-association of UPy quadruple H-bonds and restoration of crystalline physical bonds during healing.
2.2. π–π stacking
π–π stacking is a kind of non-covalent interaction arising from the intermolecular overlap of p orbitals in aromatic group-based conjugated systems.41 The intrinsic rigid plane structures of aromatic compounds (e.g. benzene, naphthalene, and pyrene) are commonly used to construct highly crystalline conjugated systems with excellent photoelectric properties.42 For most conjugated polymers, the amorphous phase (α) typically coexists with aggregated crystalline regions originating from strong intermolecular interactions between functional units or aromatic rings in the polymer backbone. However, it's challenging to efficiently tune the polymorphism of conjugated polymers in aggregated, semi-crystalline phases due to their conformational freedom and anisotropic nature. To solve the above issue, Janssen and coworkers designed a diketopyrrolopyrrole-alt-quaterthiophene polymer (D-PDPP4T-HD), which could generate two aggregated, semi-crystalline phases (β1 and β2) by precisely tuning the solvent quality (Fig. 4).43 To be specific, with the addition of 1,2,4-trichlorobenzene (TCB) into chloroform (CF) from 0 vol% to 100 vol%, D-PDPP4T-HD showed a gradual β1–α–β2 phase transition. The phase evolution was ascribed to more extended configuration of D-PDPP4T-HD in TCB, which facilitated polymer crystallization via π–π interaction. Therefore, the β2 polymorph exhibited a lower optical band gap, enhanced photoluminescence, a reduced π-stacking distance, a higher hole mobility in field-effect transistors and improved photocurrent generation in polymer solar cells. Moreover, emerging D–A π-conjugated polymers have been developed to tune the configuration and size of the π–π stacked crystalline phase for tailored mechanical and photoelectric properties.44,45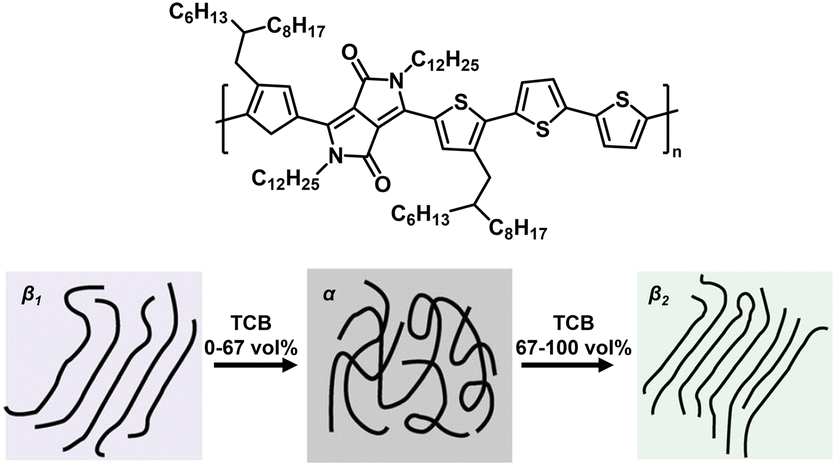 | ||
| Fig. 4 The gradual β1–α–β2 phase transition of D-PDPP4T-HD by adding TC into CF from 0 vol% to 100 vol%. Copyright 2019, Springer Nature. Reproduced with permission from ref. 43. | ||
On the other hand, introducing rigid aromatic motifs into the soft matrix offers extra opportunities for semi-crystalline polymers. For example, azobenzene (azo), as a typical photo-responsive compound, shows reversible photoisomerization characters.46 The trans isomers with a higher degree of symmetry tend to form tightly packed crystal phases via π–π stacking, while UV irradiation induced cis isomerization breaks the symmetrical structure, thus regulating the crystalline domains via reversible cis–trans transitions. Azo-terminated flexible polymer chains provide sufficient free volume for photoisomerization in the solid state, which may trigger the melting point (Tm) or glass transition temperature (Tg) change of the resulting polymers and the subsequent solid–liquid transition and other dynamic responsive behaviors.47,48 Meijer and coworkers designed a series of azo-functionalized siloxane oligomers of discrete length and explored substituent effects on aggregated structures (Fig. 5a).49 Specifically, well-ordered lamellar semi-crystalline morphology in which monolayers of crystalline azobenzene were exfoliated using liquid siloxane oligomers was obtained due to synergistic phase segregation and azo crystallization. UV irradiation could cause a rapid solid–liquid transition, which was reversible under blue light. In contrast, the introduction of terminal hydroxyl H-bonds inhibited the formation of crystalline domains, which resulted in more complex morphologies based on nanophase segregation alone.
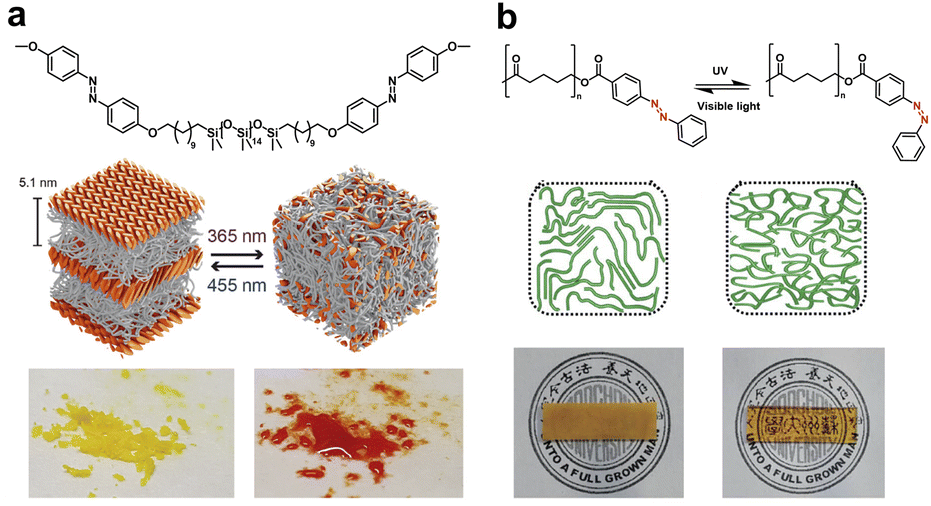 | ||
| Fig. 5 (a) Isomerism induced solid–liquid transition of azo-functionalized siloxane oligomers under UV/vis irradiation. Copyright 2018, Wiley-VCH. Reproduced with permission from ref. 49. (b) Photo-switchable Tm and transparency of azo-terminated PCL. Copyright 2021, Royal Society of Chemistry. Reproduced with permission from ref. 50. | ||
Later, Dong and coworkers reported an azo-terminated PCL semi-crystalline polymer with photo-switchable Tm and transparency (Fig. 5b).50 Under UV irradiation, the Tm of azo-PCL reduced from 58 °C to 48 °C due to the trans-to-cis isomerization of azo groups. Meanwhile, the photothermal effects induced the temperature of the azo-PCL film to increase to 44 °C, which resulted in polymer chain mobility and partial melting. Therefore, the resulting polymer exhibited both the semicrystalline-to-amorphous and opacity-to-transparency transformations relying on the synergy between the photo-isomerization and the photothermal effect of the azo groups, which promoted advanced applications as optically healable materials, security tags and smart windows.
2.3. Hydrophilic/hydrophobic association
Hydrophilic/hydrophobic association derived from thermodynamically incompatible constituents can drive the microphase separation and the ordered morphology at the nanoscale via spontaneous hierarchical assembly.51,52 For example, hydrophobic side chains in proteins aggregate internally to avoid the interaction with water molecules, which is essential for the stability of the protein and affects the maintenance of the folding and helical configuration. Amphiphilic structures can be constructed via covalent bonding or physical doping. The crystalline domain spacing and the uniformity of the assembled morphology are jointly determined using the proportion and sequence of hydrophilic and hydrophobic segments, polymerization degree, solvent environment, etc.Recently, Wu and coworkers selected polyvinyl alcohol (PVA) with abundant pendent hydroxyl groups as the hydrophilic hard crystalline phase and poly(2-methoxyethylacrylate) (PMEA) as the hydrophobic soft phase to fabricate high-strength amphibious polymer materials (Fig. 6a).53 The combination of solvent exchange and thermal annealing strategies led to nanophase separation. The resulting semicrystalline PVA/PMEA interpenetrating networks exhibited outstanding mechanical performance both in the hydrogel and solvent-free plastic states.
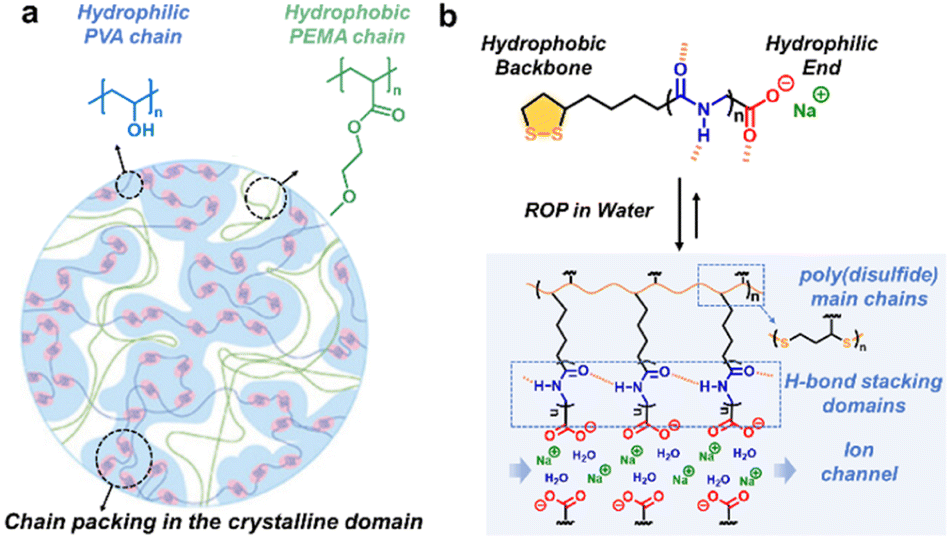 | ||
| Fig. 6 (a) Semicrystalline PVA/PMEA interpenetrating networks constructed by solvent exchange and thermal annealing strategies. Copyright 2024, Wiley-VCH. Reproduced with permission from ref. 53. (b) Ordered ion networks via the self-assembly of amphipathic small molecules. Copyright 2023, Chinese Chemical Society. Reproduced with permission from ref. 55. | ||
Besides the interpenetration strategies of thermodynamically incompatible polymeric chains, the hierarchical self-assembly of amphipathic precursors offers another effective chemical pathway.54 Qu's group designed an amphipathic small molecule via coupling oligopoly(glycine) units with thioctic acid (TA). The hydrophobic poly(disulfide) skeleton, β-sheet-inspired H-bonding and terminal ionic bonding were combined to produce sidechain-engineerable amphiphilic semi-crystalline ordered networks (Fig. 6b).55 Notably, the interlayer distances could be readily engineered with nanometer accuracy by varying the length of the oligopeptide sidechain and exhibited an odd–even effect within a certain range.
2.4. Metal coordination
Metal complexes are composed of ligands as electron donors and metal ions/atoms as electron acceptors. Because of the same nature of the coordination bonds as that of the covalent bonds, i.e., the sharing of electron pairs between atoms, metal coordination has high strength and directivity similar to the covalent bonds.56 Numerous ligands (such as catechol, histidine, pyridinyl and carboxylate) have been proven to coordinate effectively with metal ions, whose strength can be adjusted in a wide range by the tailored combination of soft and hard acid and base.57–60 For instance, 2,6-bis(10-methylbenzimidazolyl)pyridine (Mebip), as a polydentate pyridine ligand, can strongly bind with metals due to the chelation effect. The resulting supramolecular network exhibits high mechanical strength but brittleness and an inferior dynamic nature. To overcome the inherent tradeoff, Schrettl and coworkers designed two kinds of Mebip functionalized monomers, i.e., the rigid low-molecular-weight trifunctional building block and the flexible telechelic poly(ethylene-co-butylene) bifunctional macromonomer (Fig. 7a).59 The mixture of the two components coordinated with Zn2+ ions led to a phase-separated microstructure where the trifunctional components acted as hard crystalline domains. The resulting semicrystalline metallo-supramolecular networks exhibited tailored material properties including strength, stiffness, toughness, and repairability via simply varying the ratio of the two constituents.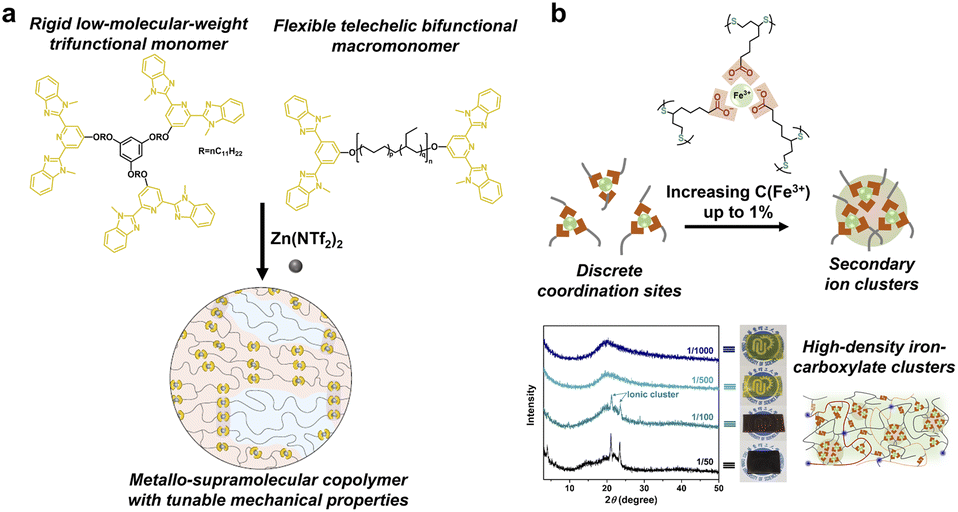 | ||
| Fig. 7 (a) The combination of rigid trifunctional and flexible bifunctional building blocks led to a phase-separated microstructure. Copyright 2022, Springer Nature. Reproduced with permission from ref. 59. (b) High-performance supramolecular networks with high density Fe3+–carboxylic acid complexes as secondary ion clusters. Copyright 2020, Wiley-VCH. Reproduced with permission from ref. 60. | ||
On the other hand, the discrete distribution of metal complexes in the polymerization network is unfavorable to the formation of hard domains and reduces the mechanical modulus of the material. Qu and coworkers added high content of Fe3+ ions (1% molar ratio to carboxylate) into solvent-free supramolecular networks to form high density Fe3+–carboxylic acid complexes as secondary ion clusters (Fig. 7b).60 The resulting dynamic polymer showed dramatically enhanced mechanical strength over 60-fold maintaining high stretchability and self-repairability. The superior performance was attributed to the hierarchical distribution of four dynamic interactions in the network, including dynamic covalent disulfide bonds, H-bonds, iron–carboxyl coordination, and ion cluster interactions.
3. Construction strategies
The material properties stem fundamentally from both their physical and chemical structures, which are commonly formed through spontaneous assembly of nanoscale molecular precursors on multiple length scales.61,62 The self-assembly process is driven by the driving force for a lower Gibbs free energy state in thermodynamics. However, it is significantly challenging to control the structures and dimensions when pushing towards smaller building blocks and periodic arrangements via structural engineering alone. The competitive self-assembly may produce a metastable equilibrium state of resultant morphology instead of the lowest Gibbs free energy state due to the barrier energy for forming the self-assembled state. Therefore, the induction process is equally important for the final network morphology in addition to intrinsic thermodynamic factors, where the kinetic pathway controls the self-assembly behaviors in principle.63–65 Some typical process-dependent construction strategies are discussed as follows.3.1. Polymerization-induced microphase separation (PIMS)
PIMS is an advantageous strategy to develop unique nanostructures with highly useful morphologies through the microphase separation of emergent block copolymers during polymerization.23,66 This strategy, i.e. combining polymerization-induced phase separation with in situ block polymer formation, has been proposed by Hillmyer since 2012 to overcome the lack of long-ranged continuity in hard and soft domains.67 Reversible addition–fragmentation chain transfer (RAFT) polymerization is the most common approach to promote chain extension and form block copolymers.68 For instance, Motokawa and co-workers reported the phase transition of homogeneous solution–disordered–lamellar–cylindrical–spherical during continual chain extension via living-radical bulk block copolymerization of poly(methyl methacrylate)-block-polystyrene (Fig. 8a).69,70 The crosslinking process induces the kinetic arrest of block segments as well as independent control over domain sizes and total domain volume to access highly structured co-continuous multiphase morphology with percolating domains, resulting in a vast inventory of porous materials with well-defined pore sizes, narrow pore distributions and distinct channels for mass transfer.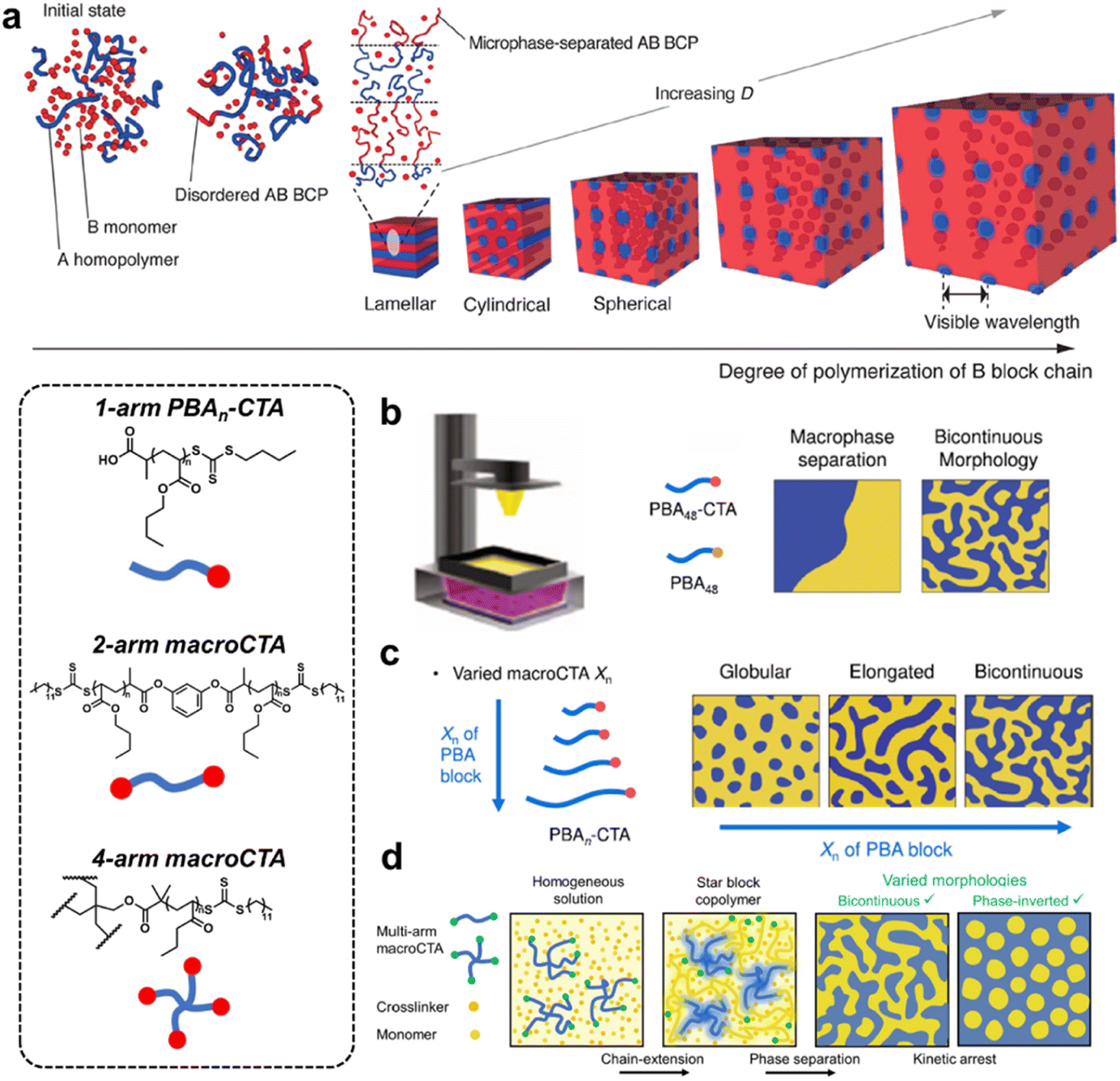 | ||
| Fig. 8 (a) The phase transition of homogeneous solution–disordered–lamellar–cylindrical–spherical during continual chain extension via living-radical bulk block copolymerization of poly(methyl methacrylate)-block-polystyrene. Copyright 2016, American Chemical Society. Reproduced with permission from ref. 70. (b) Controlled nanoscale morphology transition using light mediated 3D printing. Copyright 2022, Wiley-VCH. Reproduced with permission from ref. 74. (c) Precise control over the microphase separation behavior by varying the chain length of the macroCTA. Copyright 2022, Springer Nature. Reproduced with permission from ref. 75. (d) A variety of equilibrium microphase separated structures achieved with multi-arm block copolymers. Copyright 2022, Wiley-VCH. Reproduced with permission from ref. 76. | ||
The integration of the PIMS mechanism and a variety of processing techniques provides a facile chemical strategy to precisely control the nanoscale structural features. For example, the compatibility with 3D printing techniques bridges the gap between the high nanostructural tunability of PIMS and the simplified and versatile production methods of 3D printing for advanced materials manufacturing across multiple size scales.71–73 With the advancements in RAFT photochemistry, Boyer and co-workers reported a novel PIMS approach for the fabrication of materials with controlled nanoscale morphologies across all relevant length scales using light mediated 3D printing without specialized equipment or process conditions.74 The tunable phase separation process drove the material morphologies from disordered macrophase separation, to discrete elongated nanoscopic domains, and further to bicontinuous nanodomains (Fig. 8b). Despite successful 3D printing, limited control over the material nanostructure was demonstrated with a lack of ability to finely tune nanoscale features. Boyer's group further achieved a high level of control over the microphase separation behavior by varying the chain length of the macroCTA (Fig. 8c).75 A scaling law was well identified to describe the nano-morphology transition from globular and discrete elongated domains to bicontinuous domains with increasing macroCTA chain length and volume fraction. Importantly, the transition displayed a correlation with the bulk mechanical properties, where bicontinuous interpenetrating domains resulted in enhanced mechanical properties due to the increased interfacial interaction between soft and hard domains. Besides conventional linear macroCTA architectures, multi-arm block copolymers can lead to a variety of equilibrium microphase separated structures unachievable with linear block copolymers. The same group designed multi-arm (1-, 2-, and 4-arms) macroCTAs to mediate PIMS and prepare nanostructured materials via photoinduced 3D printing (Fig. 8d).76 As evidenced by atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS), the characteristic length scale of microphase-separated domains is determined using the macroCTA arm length, while nanoscale morphologies are controlled by the macroCTA architecture. To be specific, varying the macroCTA architecture, arm length, and weight percentage led to the resulting materials with tunable domain spacing and nanoscale bicontinuous and phase-inverted morphologies, which have not been previously observed in PIMS systems.
3.2. Strain-induced crystallization
The above molecular engineering strategy focuses on introducing the energy dissipation mechanism via refined chemical modification of block structures, which lacks universality and extensibility in large-scale industrial production. For disorderedly entangled polymer chains, strain-induced crystallization provides an alternative general approach of facile morphology programming for the transformation from randomly distributed isotropic nanostructures into hierarchically oriented crystalline domains, which is usually accompanied by visible material whitening.77–79 The underlying mechanism is the slide and alignment of linear polymer chains along the tensile direction and the rearrangement of interchain weak interactions (e.g. H-bonding). Tang and co-workers constructed a class of ultra-strong elastomers from biomass-derived long-chain polyamide copolymers with pendant polar hydroxyl or non-polar butyrate groups (Fig. 9a).80 The butyrate pendant groups facilitated an elastic amorphous matrix instead of a highly crystalline structure. The packing of linear alkyl chains as well as H-bonding among amide/hydroxyl side groups jointly contributed to the formation of nanocrystalline domains for high mechanical strength. The crystallization properties of resultant polyamides could be precisely tuned via changing the copolymerization ratios. More importantly, the mechanical strength could be exponentially enhanced to over 210 MPa via uniaxial step-cycle tensile deformation. The underlying mechanism of mechanical enhancement is the rearrangement and alignment of crystalline microstructures through van der Waals interactions and H-bonds (Fig. 9b). Moreover, densely clustered electron-rich amide groups led to strong luminescence of aliphatic polyamides due to the aggregation-induced emission (AIE) effect.81,82 In addition to high-density crosslinked solvent-free polymers, strain-induced crystallization also provided a robust toughening strategy for hydrogels with high-water content (50–70%), low polymer concentration and inhomogeneous network structures.83 Wang and co-workers fabricated tough poly(N-acryloylsemicarbazide-co-acrylic acid) hydrogels with high hydrogen bond energy via strain-induced phase conversion and multiple H-bond reconstruction. Notably, strain-induced microphase separated domains at the crack point can effectively hinder crack growth during the tearing process, leading to high tear resistance (Fig. 9c).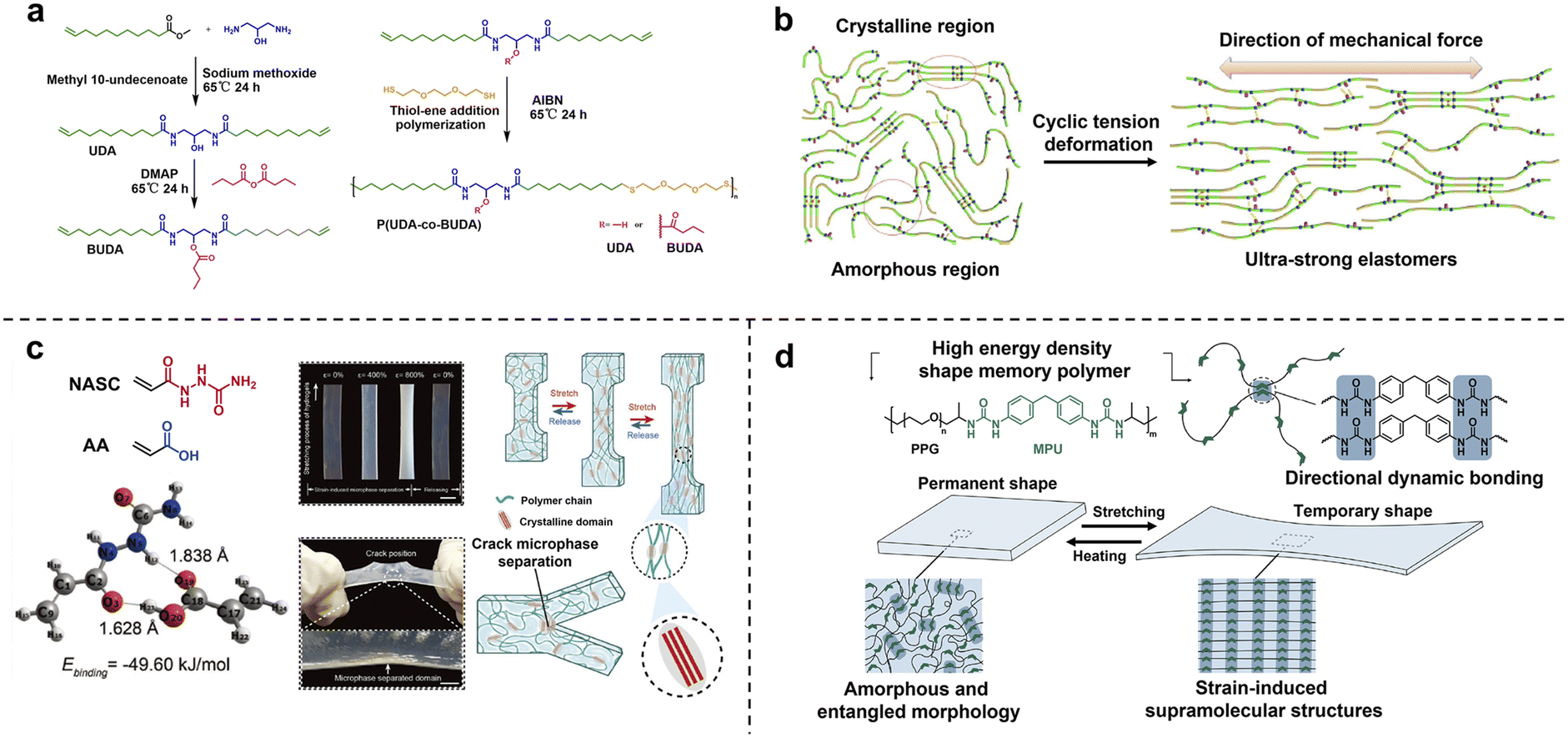 | ||
| Fig. 9 (a) The molecular structures of long-chain polyamide copolymers with pendant polar hydroxyl or non-polar butyrate groups. (b) The rearrangement and alignment of crystalline microstructures through van der Waals interactions and H-bonds during cyclic tension deformation. Copyright 2019, Springer Nature. Reproduced with permission from ref. 80. (c) The strain-induced microphase separation of hydrogels at the crack point, effectively hindering crack growth during the tearing process. Copyright 2023, Wiley-VCH. Reproduced with permission from ref. 83. (d) Strain-induced SMPs with high energy density. Copyright 2021, American Chemical Society. Reproduced with permission from ref. 85. | ||
A nanocrystallite in a highly stretched state efficiently stores entropic energy,84 which greatly enhances toughness and damage resistance and further expands the cutting-edge application as an actuator. Bao and co-workers reported one-way shape memory polymers (1W-SMPs) with a high energy density of 19.6 MJ m−3 and shape fixity/recovery above 90% based on the formation of strain-induced supramolecular nanostructures.85 The alignment of polymer chains during strain led to the formation of strong directional dynamic bonds, creating stable supramolecular nanostructures and trapping stretched chains in a highly elongated state. Upon heating, the dynamic bonds break and stretched chains contract to their initial disordered state (Fig. 9d). This work addressed the challenge of producing high energy density SMPs that simultaneously possess high recovery stress and large recoverable strain.
3.3. Evaporation-induced self-assembly
Hierarchical self-assembly is a fundamental topic in both biological and artificial supramolecular systems as the resulting ordered polymeric materials are of great significance in next-generation functional materials, such as ion-transport films, soft actuators, etc.1,86 Ordered self-assembly normally involves two key issues: (i) multiple interactions with different binding affinities for thermodynamically driven distinct spatial distribution;87 (ii) the kinetically driven self-assembly process in time scales.88 In this case, the evaporation-induced self-assembly of thermodynamically incompatible building blocks provides a facile construction strategy to precisely direct the ordered assembly pathway.89,90 Ho and co-workers reported a new approach to thermodynamically and kinetically regulate self-assembled morphologies by introducing the competitive interactions of π–π junctions.91 Specifically, block incompatibility, intermolecular interactions between chain ends or junctions, and kinetic control jointly regulate the final morphologies. In regard to the kinetic study, the process-dependent kinetics for morphological evolution is thus systematically investigated from the aspect of polymer chain dynamics with the control of the solvent evaporation rate (Fig. 10a). As shown in TEM micrographs and the corresponding 1D SAXS profiles, fast solvent evaporation (1 mL h−1) promoted the microphase-separated phase but with irregular microdomain texture at a low long-range ordering, and moderate evaporation (0.1 mL d−1) gave rise to hexagonally packed helices with long-range order, while slow evaporation (0.03 mL d−1) led to a disordered structure. The above results indicated the important role of solvent evaporation rate for resultant morphology, and the fast evaporation-induced concentration increase resulted in immediate microphase separation once reaching the critical micelle concentration (CMC). In contrast, slow evaporation offered enough time for junction stacking, thus overcoming the thermodynamic driving force for microphase separation and leading to the decay of structural ordering. | ||
| Fig. 10 (a) The process-dependent kinetics for the morphological evolution with the control of the solvent evaporation rate. Copyright 2017, American Chemical Society. Reproduced with permission from ref. 91. (b) The highly ordered supramolecular layered networks derived from amphiphilic small molecular “sodium thioctate”. Copyright 2019, American Chemical Society. Reproduced with permission from ref. 95. (c) Multilayered lamellar thin films of random copolymers. Copyright 2021, American Chemical Society. Reproduced with permission from ref. 99. | ||
The hierarchical self-assembly of small molecules represented a desirable but challenging pathway to introduce sophisticated structural information into a simple molecular backbone.92–94 Qu and co-workers designed an amphiphilic dynamic small molecule “sodium thioctate” as an ideal precursor building block towards highly ordered supramolecular layered networks.95 During the slow evaporation process, the synergy of multiple supramolecular interactions and evaporation-induced interfacial confinement effect contributes to layered structures with long-range order at both macroscopic and molecular scales (Fig. 10b). The resulting supramolecular layers are able to bind water molecules as structural water, which acts as an interlayer lubricant to modulate the material properties, including mechanical performance, self-healing capability, and actuating function. Moreover, dynamic disulfide backbones can be degraded into monomers and reformed through a water-mediated route, exhibiting full recyclability in a facile, mild, and environmentally friendly way.96–98 Moreover, Terashima and co-workers reported the first example of multilayered lamellar thin films of random copolymers which provided a general strategy for nanostructured functional materials (Fig. 10c).99 Random copolymers bearing octadecyl groups and hydroxyethyl groups induced crystallization-driven microphase separation via simple solution evaporation to form lamellar structures, in both bulk and thin film states, with controlled crystalline domain space at the 0.1 nm level.
In addition to the above representative examples, some more general pathways have been reported. For example, a facile and large-area fabrication method containing the diffusion-driven mixing process followed by freezing-induced gelation and microphase separation can inspire the design of materials with various functionalized surfaces.100 Specifically, in the diffusion-driven mixing process, polystyrene (PS) substrates at the contact interface were dissolved in the poly(vinyl alcohol) (PVA)/N-methylpyrrolidone (NMP) solution, leading to the interpenetration of PVA and PS chains to form a diffusion layer. The subsequent freezing induced a microphase separation in the diffusion layer, i.e., the collapsed PS chain at the interface separated from the crosslinked PVA/NMP gel to form a multiscale structure, whose surface was fixed with further gelation. The novel strategy of freezing-induced microphase separation addressed the challenge of complicated manufacturing processes and specific material modeling for the design and large-scale fabrication of nano-structured hydrogel surfaces. Overall, the chemical tunability of precursor structures, the diversity of the induction pathway, and interfacial supramolecular interactions jointly inspire semi-crystalline morphologies towards dynamic materials with sophisticated functions.
4. Advanced applications as dynamic functional materials
4.1. Mechanical toughening
Mechanical properties are the fundamental material properties to determine practical applications, containing multiple key parameters including strength, modulus, elongation, toughness, etc.101 Strength refers to the maximum external force at fracture, and modulus reflects the resistance ability of elastic deformation, which can be facilitated by the stable and dense covalent cross-linking structure of polymeric networks, while the restriction of polymer segment movement reduces the stretchability and toughness of the materials.102 Elongation reflects the stretchability of the material at fracture, and it can be enhanced via replacing strong covalent crosslinking with weak sacrificial bonds but it also usually decreases the mechanical strength.103 That is to say, there is always a tradeoff between mechanical strength and stretchability, and thus it's challenging to construct highly tough materials which are in substantial demand for various applications. Well-designed SCNs provide a multiscale strategy to overcome the above inherent compromise. The synergy of rigid phase-separated nanostructures and weak interactions crosslinked in a soft matrix can balance the mechanical strength and stretchability, thus fabricating ultra-strong polymers with high strength and toughness, satisfactory stretchability, and elasticity.104–107Inspired by natural load-bearing materials (e.g. tendons) with anisotropic hierarchical structures across multiple length scales,108 He and co-workers presented a toughening strategy to produce a multi-length-scale hierarchical hydrogel architecture via a freezing-assisted salting-out synergic treatment (Fig. 11a).106 Directional freezing caused PVA to be aligned and highly concentrated, and subsequent salting out strongly induced the aggregation and crystallization of PVA by phase separation to form nanofibrils. The HA–PVA hydrogels demonstrated an ultimate toughness of 210 ± 13 MJ m−3, stress of 11.5 ± 1.4 MPa and strain of 2900 ± 450%. The strengthening mechanism was mainly structural densification caused by H-bonds and crystalline domain formation, and the toughening mechanisms were pull-out, bridging and energy dissipation by the fibrils, which are typical for highly anisotropic materials. Moreover, the dynamic breakage and reformation of sacrificial H-bonds during deformation enabled the reversibility and reusability of the HA–PVA hydrogels, and the highly fatigue-resistance over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tensile cycles was ascribed to the crystalline domains and networks of fibres as strong barriers to cracks.
000 tensile cycles was ascribed to the crystalline domains and networks of fibres as strong barriers to cracks.
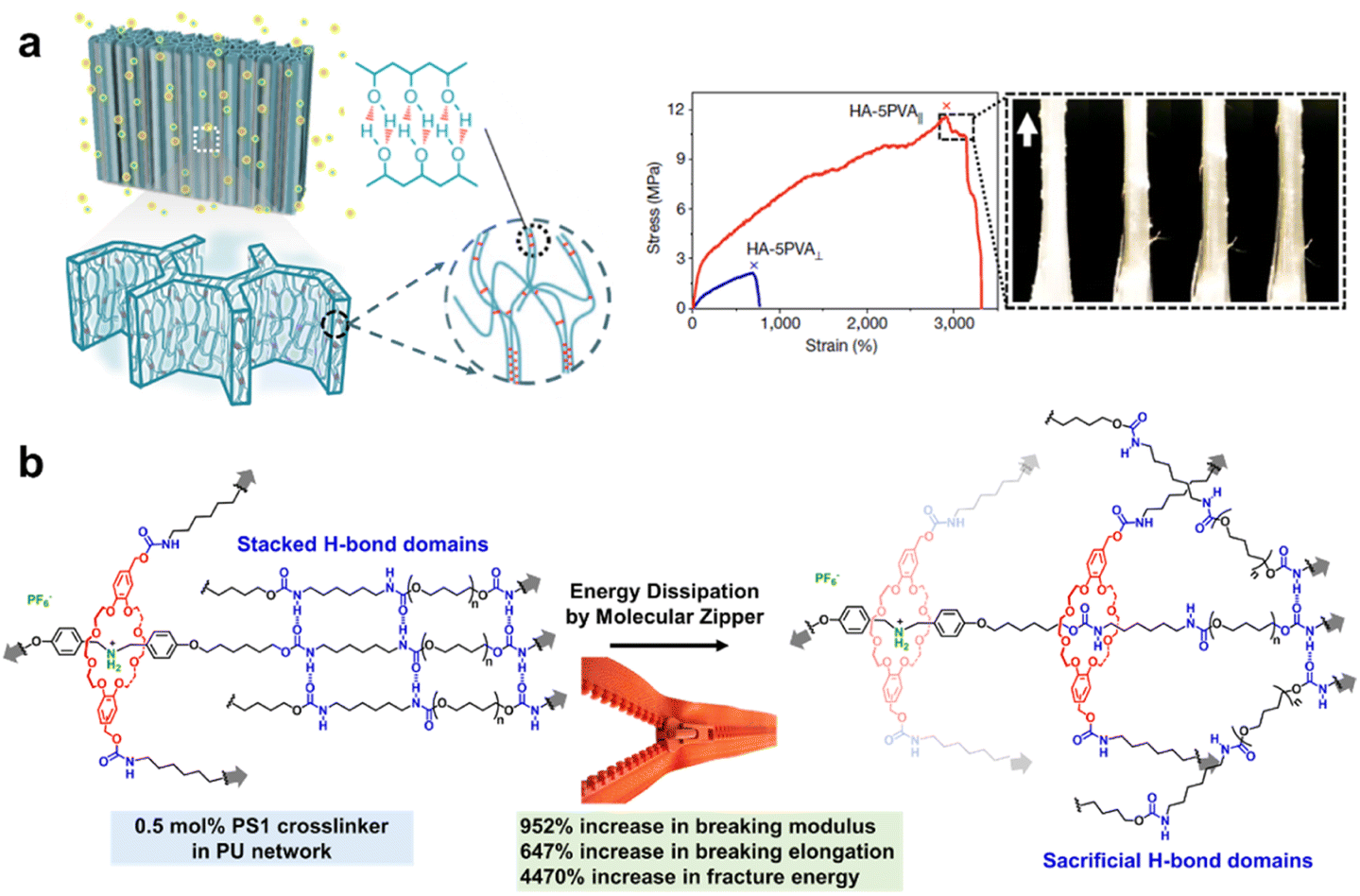 | ||
| Fig. 11 (a) A multi-length-scale hierarchical hydrogel architecture toughened via a freezing-assisted salting-out synergic treatment. Copyright 2021, Springer Nature. Reproduced with permission from ref. 106. (b) A H-bond-crosslinked PU elastomer toughened by a molecular zipper strategy. Copyright 2020, Wiley-VCH. Reproduced with permission from ref. 107. | ||
Furthermore, the effective synergy of multiple dynamic bonds in a single system can achieve a significant improvement in the material performance.109,110 Qu's group proposed a supramolecular strategy of introducing a zipper-like sliding-ring mechanism in a H-bond-crosslinked PU network.107 A very small amount (0.5 mol%) of pseudo[2]rotaxane crosslinker could dramatically increase both the mechanical strength and elongation of this H-bond-crosslinked PU network by nearly one order of magnitude (Fig. 11b). As demonstrated by the decreased scattering peaks in the X-ray diffraction (XRD) profile, the enhancement was ascribed to a unique molecular-level zipper-like ring-sliding motion, during which the breakage of the H-bonding crystalline domain efficiently dissipated mechanical work in the solvent-free network. The novel molecular zipper strategy not only provides a distinct and general strategy for the construction of high-performance elastomers but also paves the way for the practical application of artificial molecular machines toward solvent-free PU networks.
4.2. Dynamic adaptivity
For the development of a sustainable economy, it's highly demanded to fabricate dynamic high-performance polymers to improve the reliability of materials and prolong their service life.111,112 However, mechanical and dynamic properties, as key performance parameters, are intrinsically opposite. For most supramolecular designs, strong interactions result in rigid but less dynamic networks and weak interactions afford dynamic behaviors, but yield soft materials. Multiphase hierarchical assembly is an efficient supramolecular synergy strategy,17 where the program of dynamic motifs (e.g. H-bonding and π–π stacking) in hard–soft multiple systems can dissipate energy via dynamic fracture and rearrangement, thus bringing about not only enhanced toughness, but also dynamic reversibility for self-healing, reprocessability, and recyclability.113–115For instance, Sun and co-workers designed a high-strength reversible elastomer containing polyimide (PI) as an ideal rigid segment and poly(urea-urethane) (PUU) as a H-bonded soft segment.114 The resultant PI–PUU copolymer exhibited a record-high tensile strength of ≈142 MPa and an extremely high toughness of ≈527 MJ m−3, as well as excellent scratch and puncture resistance, which enhanced the practical durability of the elastomers. Meanwhile, accelerated by the mobility and diffusion of polymer chains, the H-bonds on the fractured surface were broken and reformed in solvent and under heat, thereby endowing the PI–PUU elastomers with efficient self-repairability and recyclability to restore the mechanical properties, prolong service life and reduce materials consumption.
Nevertheless, external interventions are usually required to promote intrinsic healing behaviors, such as heat, light, solvents, external pressure, etc.116–119 Even so, intrinsic self-healing materials under ambient conditions are generally soft and deformable hydrogels or elastomers. It's a formidable challenge to fabricate high-strength polymeric materials that could heal damage intrinsically under ambient conditions, due to the significantly hindered diffusion of polymer chains and incompact contact between fractured interfaces. To address the above challenges, Sun and co-workers designed an intermediary healing agent with the same composition as the target material to repair rigid poly(acrylic acid)–poly(allylamine hydrochloride) (PAA–PAH) copolymers under ambient conditions based on reversible supramolecular interactions.115 Commercially available PAA and PAH were tunably complexed to produce two distinct states, i.e., the rigid glassy PAA–PAH copolymer with a tensile stress of ∼67 MPa and an elastic modulus of ∼2 GPa, strengthened by phase-separated nanostructures, and viscoelastic PAA–PAH copolymer as a healing agent via electrostatic and H-bonding. Therefore, this rigid semi-crystalline copolymer could be fully repaired via inner noncovalent complexation under ambient conditions without external assistance (Fig. 12a).
 | ||
| Fig. 12 (a) Rigid self-healing PAA–PAH copolymers repaired using the intermediary healing agent with the same composition under ambient conditions. Copyright 2023, Chinese Chemical Society. Reproduced with permission from ref. 115. (b) The phase-separated nanostructures of the PI–PUU copolymer. Copyright 2021, American Chemical Society. Reproduced with permission from ref. 15. | ||
In addition to self-healing and recyclability as well-known dynamic properties, more adaptive behaviors such as softness-stiffness switchability are available via the dynamic aggregation of the nanoscale crystalline segments. Learning from sea cucumber dermis, Yan's group designed an impact-protective supramolecular polymer material (SPM) containing a poly(ethylene glycol/propylene glycol) triblock copolymer as a flexible soft matrix and UPy dimer as a supramolecular hard motif (Fig. 12b).15 The UPy dimer can not only promote the aggregation of rigid segments as cross-linked domains, but also provide enhanced energy-dissipation due to rapid kinetic reversibility. Therefore, the multiscale energy-dissipation pathways (i.e., breakage of the H-bonding, remodeling of hard domains, and realignment of soft strands) endowed the semi-crystalline SPM with unique impact-hardening and reversible stiffness switchability.
4.3. Shape memory
As a class of representative dynamic smart materials, SMPs can transform between temporary and permanent shapes under a specific stimulus, which is jointly determined using the block structures, reversible mobility changes, conformational entropy, and programming.120 SMPs normally contain two components: (i) permanent network crosslinks consisting of chemical crosslinks, physical crosslinks or crystals to maintain material stability; (ii) reversible segments that regulate molecular mobility to fix/release temporary shapes via crystallization/melting or vitrification transitions. Two-way SMPs (2W-SMPs) exhibit reversible and programmable shape-shifting behaviors superior to 1W-SMPs, thus promoting advanced applications such as actuators, artificial muscles, drug delivery, etc.121 SCN is one of the main molecular strategies towards the preparation of 2W-SMPs, which was first proposed by Mather in 2008,122 and has aroused increasing interest due to its universal structural design and simple tailoring process compared to liquid crystalline polymers (LCPs).123–126 The SMP networks consist of netpoints, crystalline crosslinking domains, and amorphous switching domains. Similar to LCPs, crystallization-induced elongation (CIE) and melting-induced contraction (MIC) under constant stress or stress-free conditions contribute to the two-way shape memory behaviors of SCN, and another requirement is that the stresses formed by structure change must be equivalent to those caused by entropy elastic recovery.Xie and co-workers created a single-component robot without macroscopic component assembly using a programmable semi-crystalline SMP with thermo- and photo-reversible bonds (Fig. 13).124 The semi-crystalline PU networks comprised ample esters and cinnamates, where the cinnamates undergo photo-reversible dimerization, and both urethane and ester linkages can be triggered thermally by the catalyst (dibutyltin dilaurate), used in the network synthesis, for reversible bond exchanges. These two kinds of noninterfering reversible bonds synergistically programmed the shape-morphing behaviors, i.e., the thermo-reversible bonds created the 3D shaped structural support via plastic deformation, and photo-reversible bonds can be activated in a spatio-selective manner to create network anisotropy, leading to precisely controlled local actuation via the reversible shape memory mechanism, thereby producing more sophisticated soft robots and reversible actuators. More recently, the same group constructed a zero-set reversible shape memory material via introducing a transesterification catalyst into a network containing two crystalline phases: poly(ε-caprolactone) (PCL) and poly(ω-pentadecalactone) (PPDL).125 The network can be programmed by two mechanisms: (i) the transesterification catalyzed by [1,8-diazabicyclo[5.4.0]undec-7-ene] (DBU) at elevated temperatures provided chemical mechanisms allowing permanent shape reconfiguration; (ii) the high melting transition PPDL phase allows physical programming for network anisotropy, which was reversibly actuated by the low melting PCL phase. The synergic mechanism expanded the scope for future multifunctional shapeshifting devices.
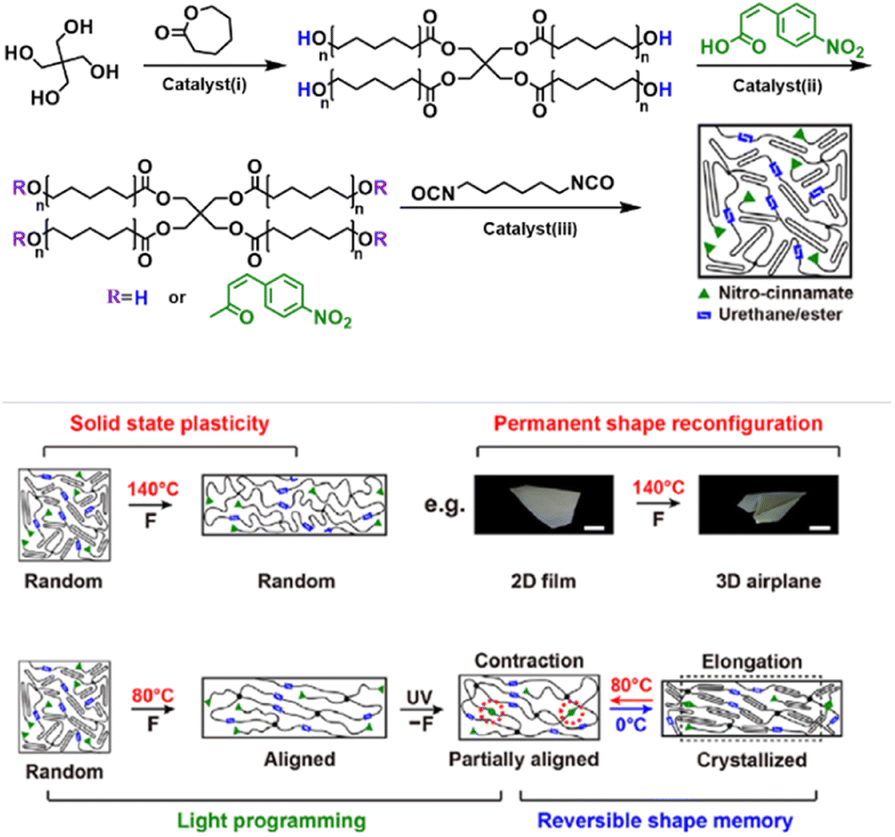 | ||
| Fig. 13 A single-component robot using a programmable semi-crystalline SMP with thermo-and photo-reversible bonds. Copyright 2018, AAAS. Reproduced with permission from ref. 124. | ||
4.4. Ion-transport
Soft ionic conductors have been widely used in wearable ionotronic devices, stretchable touch panels, actuators, etc., due to their ion-conducting and sensory functions, as well as outstanding environmental adaptability.126,127 Ion transport relies on polymer polarity and the segmental motions, often accompanied by the existence of a large amount of liquid. However, the mechanical strength and stability of materials are usually based on a stable covalent crosslinked network, thereby generating an inherent conflict between ionic conductivity and mechanical performance. The semi-crystalline multiphase structures afford an eminent chemistry strategy to improve the compatibility between ionic conductivity and mechanical properties in polymer electrolytes.128–130 In this case, mobile soft segments are responsible for ion transport and hard crystalline domains contribute to the mechanical stability. The dual-phase design performs its own functions in a synergic manner, thus defeating the conflict between ionic conductivity and mechanical compatibility. Ding et al. designed a novel ionic conductive elastomer via the phase-lock strategy,131 wherein locking the soft phase polyether backbone conducted Li+ transport and the synergy of dynamic disulfide metathesis and quadruple H-bonds in the hard phase contributed to the self-healing capacity and mechanical performance. Huang et al. addressed the conflict between mechanical strength and ionic conductivity in ionogels via the adoption of lithium ions,132 which not only promoted ionic conductivity through interchain transport but also formed a microphase-separated microstructure by forming lithium bonds with carbonyl oxygens on the polymer chains (Fig. 14a).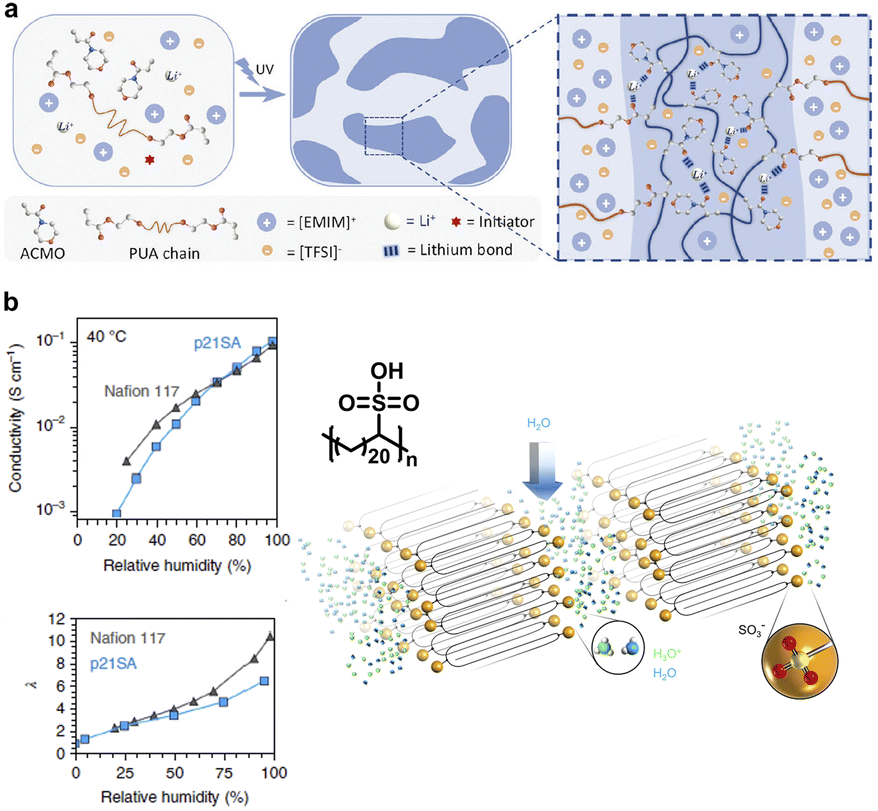 | ||
| Fig. 14 (a) A tough ionogel with high ionic conductivity promoted by lithium ions. Copyright 2023, Wiley-VCH. Reproduced with permission from ref. 132. (b) Layered sulfonated polyethylene with well-controlled methylene chain segment folding for crystallization and highly uniform hydrated acid layers for high proton conductivity. Copyright 2018, Springer Nature. Reproduced with permission from ref. 133. | ||
Furthermore, the precise control of chain folding morphology can produce ordered crystalline layers and ion channels with controllable thickness, thus achieving efficient and selective transport of protons, ions and small molecules with appropriate chemistry selection. Winey and co-workers designed sulfonated polyethylene with sulfonic acid groups pendant to precisely every twenty-first carbon atom, thereby inducing well-controlled methylene chain segment folding for crystallization and highly uniform hydrated acid layers of subnanometre thickness.133 The resultant high proton conductivity is on par with that of Nafion 117, the benchmark for fuel cell membranes (Fig. 14b). This layered polyethylene structure provided an innovative and versatile design paradigm for functional polymer membranes for efficient and selective ion transport. Furthermore, Shi et al. combined disulfide-mediated ring-opening polymerization with β-sheet-like H-bonding self-assembly to construct an ordered layer ionic network as ion-transport membranes.55 The stacked H-bonding crystalline domain synergizing hydrophilic terminal groups facilitated the formation and ordering of interlayer water channels, endowing the resulting membranes with high efficiency in transporting ions. Moreover, intrinsic dynamic poly(disulfide) backbones allowed chemical recycling to monomers under mild conditions, thus leading to a novel example of high-performance polyelectrolytes with closed-loop chemical recyclability.
5. Conclusions and outlook
Throughout this review, we have summarized the design strategies of SSCPs and their state-of-the art advances as dynamic functional materials. Learning from nature, the synergy of alternating amorphous and crystalline domains in multiphase skeletons can balance the tradeoff among mechanical strength, network flexibility and dynamic properties based on rational chemical design, thereby bringing more opportunities for dynamic high-performance polymers as potential candidates for next-generation smart applications.Despite numerous encouraging advances, the exploration of SSCPs is still in its infancy. A few issues remain unsolved and could potentially hinder further scientific development and valorization. Although SSCPs have emerged with increasing interest owing to their simple synthetic chemistry and tailorability, their material performance needs to be further improved (e.g. in regard to shape memory behaviors, SC networks exhibit lower elongations and required higher external tension than LCEs). Therefore, rational structural design, an accurate assembly program, and clear characterization of structure-property relationships are urgently required to expand the realms of functionality and application for SSCP materials.
The crystalline interlayers and the soft/hard domain interfaces are mainly crosslinked by H-bonds which are the most representative supramolecular interactions due to definite directivity, reversible bonding associations and broadly tunable binding affinities. However, the well-established dynamic chemistry toolbox should not be ignored. Host–guest interactions, electrostatic forces, dynamic covalent bonds, and even multiple synergies can be further used for molecular block design to expand the programmable dynamic behaviors.
Unlike sophisticated life and nature systems, the simplicity of artificial molecular structures and the limitation of the assembly pathways make it difficult to achieve accurate control of the semi-crystalline phase morphology (such as the size, distribution and configuration of the crystalline domains) for desired functions. Although some precise nanostructures have been designed at the microscopic level and in solution systems, there are still great challenges to achieve the scale-span regulation of macroscopic materials (such as bulk polymers with great practical engineering application prospects). This is because complex nanostructures formed by molecular assembly or polymerization are determined by a delicate balance of block structures, hard/soft segment ratio, intermolecular interactions, and interchain topological entanglement. Therefore, advanced characterization methodologies as well as theoretical models will become increasingly important in the exploration of the nanostructure dynamics of bulk SSCPs.
Overall, opportunities and challenges coexist in the advancement of SSCPs. The joint collaboration of theoretical chemists, analytical chemists, and synthetic chemists is significantly important for deeply digging into the underlying molecular behaviors and expanding integrated and well-structured architectures, thus pointing in the right direction for future development of next-generation smart SSCPs.
Author contributions
Conceptualization: C.-Y. Shi and D.-H. Qu; writing – original draft: C.-Y. Shi and W.-Y. Qin; writing – review and editing: D.-H. Qu.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22220102004 and 22025503), the Shanghai Municipal Science and Technology Major Project (grant no. 2018SHZDZX03), the Innovation Program of Shanghai Municipal Education Commission (2023ZKZD40), the Fundamental Research Funds for the Central Universities, the Programme of Introducing Talents of Discipline to Universities (grant no. B16017), the Science and Technology Commission of Shanghai Municipality (grant no. 21JC1401700), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (grant no. SN-ZJU-SIAS-006), the Postdoctoral Fellowship Program of CPSF (GZB20230210), and the China Postdoctoral Science Foundation (2023TQ0114).References
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- R. F. Tester, J. Karkalas and X. Qi, J. Cereal Sci., 2004, 39, 151–165 CrossRef CAS.
- F. L. Dri, L. G. Hector, R. J. Moon and P. D. Zavattieri, Cellulose, 2013, 20, 2703–2718 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53, 1073–1085 CrossRef.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- P. Krol, Prog. Mater. Sci., 2007, 52, 915–1015 CrossRef CAS.
- M. Varghese and M. W. Grinstaff, Chem. Soc. Rev., 2022, 51, 8258–8275 RSC.
- W. Hu, Phys. Rep., 2018, 747, 1–50 CrossRef CAS.
- Y. Lu and Y. Men, Macromol. Mater. Eng., 2018, 303, 1800203 CrossRef.
- Y. Zhang, P.-Y. Ben Jar, S. Xue and L. Li, J. Mater. Sci., 2019, 54, 62–82 CrossRef CAS.
- J. M. Lehn, Science, 1985, 227, 849–856 CrossRef CAS.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- K. Liu, L. Cheng, N. Zhang, H. Pan, X. Fan, G. Li, Z. Zhang, D. Zhao, J. Zhao, X. Yang, Y. Wang, R. Bai, Y. Liu, Z. Liu, S. Wang, X. Gong, Z. Bao, G. Gu, W. Yu and X. Yan, J. Am. Chem. Soc., 2021, 143, 1162–1170 CrossRef CAS.
- M. Wang, P. Zhang, M. Shamsi, J. L. Thelen, W. Qian, V. K. Truong, J. Ma, J. Hu and M. D. Dickey, Nat. Mater., 2022, 21, 359–365 CrossRef CAS PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 20, 467–472 CrossRef CAS PubMed.
- Y. Lin, E. Bilotti, C. W. M. Bastiaansen and T. Peijs, Polym. Eng. Sci., 2020, 60, 2351–2376 CrossRef CAS.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- S. Keten, Z. Xu, B. Ihle and M. J. Buehler, Nat. Mater., 2010, 9, 359–367 CrossRef CAS PubMed.
- S. M. Lee, E. Pippel, U. Gösele, C. Dresbach, Y. Qin, C. V. Chandran, T. Bräuniger, G. Hause and M. Knez, Science, 2009, 324, 488–492 CrossRef CAS PubMed.
- Z. Li, Y.-L. Zhu, W. Niu, X. Yang, Z. Jiang, Z.-Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, 2101498 CrossRef CAS.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2023, 62, e202307329 CrossRef CAS PubMed.
- D. N. Vaccarello, K. S. O'Connor, P. Iacono, J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2018, 140, 6208–6211 CrossRef CAS PubMed.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- J. Lequieu and A. J. D. Magenau, Polym. Chem., 2021, 12, 12–28 RSC.
- L. D. McIntosh, M. W. Schulze, M. T. Irwin, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2015, 48, 1418–1428 CrossRef CAS.
- Y. Deng, Q. Zhang and D.-H. Qu, ACS Mater. Lett., 2023, 5, 480–490 CrossRef CAS.
- L. J. Karas, C. Wu, R. Das and J. I. Wu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1477 CAS.
- R. H. Aguirresarobe, S. Nevejans, B. Reck, L. Irusta, H. Sardon, J. M. Asua and N. Ballard, Prog. Polym. Sci., 2021, 114, 101362 CrossRef CAS.
- Y. Chen, X. Zhang and Y. Jiang, Soft Matter, 2020, 16, 8047–8056 RSC.
- E. B. Trigg, M. J. Stevens and K. I. Winey, J. Am. Chem. Soc., 2017, 139, 3747–3755 CrossRef CAS.
- J. Pranata, S. G. Wierschke and W. L. Jorgensen, J. Am. Chem. Soc., 1991, 113, 2810–2819 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- J. Verjans and R. Hoogenboom, Prog. Polym. Sci., 2023, 142, 101689 CrossRef CAS.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- W. Jiang, Y. Zhou and D. Yan, Chem. Soc. Rev., 2015, 44, 3874–3889 RSC.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61–75 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS.
- W. Lee, S.-Y. Kwak and J. W. Chung, Eur. Polym. J., 2020, 138, 109976 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- Y. Hu, X. Cao and H. Fan, Polymers, 2022, 14, 4612 CrossRef CAS.
- M. Li, A. H. Balawi, P. J. Leenaers, L. Ning, G. H. L. Heintges, T. Marszalek, W. Pisula, M. M. Wienk, S. C. J. Meskers, Y. Yi, F. Laquai and R. A. J. Janssen, Nat. Commun., 2019, 10, 2867 CrossRef.
- P. Wang, J. Yang, Y. Zhang, W. Hu and H. Dong, ACS Mater. Lett., 2022, 4, 1112–1123 CrossRef CAS.
- Y. Zhang, C. Xu, P. Wang, C. Gao, W. Li, Z. Ni, Y. Han, Y. Zhao, Y. Geng, Z. Wang, W. Hu and H. Dong, Angew. Chem., Int. Ed., 2024, e202319997 CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- Z. Zhang, Z. Xie, C. Nie and S. Wu, Polymer, 2022, 256, 125166 CrossRef.
- W. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS.
- R. H. Zha, G. Vantomme, J. A. Berrocal, R. Gosens, B. De Waal, S. Meskers and E. W. Meijer, Adv. Funct. Mater., 2018, 28, 1703952 CrossRef.
- Y. Guo, J. Xiao, Y. Sun, B. Song, H. Zhang and B. Dong, J. Mater. Chem. A, 2021, 9, 9364–9370 RSC.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS.
- C. S. Patrickios and T. K. Georgiou, Curr. Opin. Colloid Interface Sci., 2003, 8, 76–85 CrossRef CAS.
- H. Wan, B. Wu, L. Hou and P. Wu, Adv. Mater., 2024, 36, 2307290 CrossRef CAS.
- A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS.
- C.-Y. Shi, Q. Zhang, B.-S. Wang, D.-D. He, H. Tian and D.-H. Qu, CCS Chem., 2023, 5, 1422–1432 CrossRef CAS.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gándara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS.
- J. Sautaux, F. Marx, I. Gunkel, C. Weder and S. Schrettl, Nat. Commun., 2022, 13, 356 CrossRef CAS PubMed.
- Y. Deng, Q. Zhang, B. L. Feringa, H. Tian and D. Qu, Angew. Chem., Int. Ed., 2020, 59, 5278–5283 CrossRef CAS PubMed.
- O. Ikkala and G. Ten Brinke, Science, 2002, 295, 2407–2409 CrossRef CAS PubMed.
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198–201 CrossRef CAS PubMed.
- J. Wang, Q. Xiao, H. Zhou, P. Sun, Z. Yuan, B. Li, D. Ding, A.-C. Shi and T. Chen, Adv. Mater., 2006, 18, 3284–3288 CrossRef CAS.
- B. Hasenknopf, J.-M. Lehn, N. Boumediene, E. Leize and A. Van Dorsselaer, Angew. Chem., Int. Ed., 1998, 37, 3265–3268 CrossRef CAS.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- M. W. Schulze, L. D. McIntosh, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2014, 14, 122–126 CrossRef CAS PubMed.
- M. Seo and M. A. Hillmyer, Science, 2012, 336, 1422–1425 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- R. Motokawa, Y. Iida, Y. Zhao, T. Hashimoto and S. Koizumi, Polym. J., 2007, 39, 1312–1318 CrossRef CAS.
- R. Motokawa, T. Taniguchi, T. Kumada, Y. Iida, S. Aoyagi, Y. Sasaki, M. Kohri and K. Kishikawa, Macromolecules, 2016, 49, 6041–6049 CrossRef CAS.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 8839–8850 CrossRef CAS PubMed.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 17954–17963 CrossRef CAS PubMed.
- R. J. Gaymans, Prog. Polym. Sci., 2011, 36, 713–748 CrossRef CAS.
- V. A. Bobrin, K. Lee, J. Zhang, N. Corrigan and C. Boyer, Adv. Mater., 2022, 34, 2107643 CrossRef CAS.
- V. A. Bobrin, Y. Yao, X. Shi, Y. Xiu, J. Zhang, N. Corrigan and C. Boyer, Nat. Commun., 2022, 13, 3577 CrossRef CAS PubMed.
- X. Shi, V. A. Bobrin, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2022, 61, e202206272 CrossRef CAS PubMed.
- S. Toki, T. Fujimaki and M. Okuyama, Polymer, 2000, 41, 5423–5429 CrossRef CAS.
- N. Candau, R. Laghmach, L. Chazeau, J.-M. Chenal, C. Gauthier, T. Biben and E. Munch, Macromolecules, 2014, 47, 5815–5824 CrossRef CAS.
- I. J. Rao and K. R. Rajagopal, Int. J. Solids Struct., 2001, 38, 1149–1167 CrossRef.
- L. Song, T. Zhu, L. Yuan, J. Zhou, Y. Zhang, Z. Wang and C. Tang, Nat. Commun., 2019, 10, 1315 CrossRef PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 18, 1740–1741 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Wu, Z. Zhang, Z. Wu, D. Liu, X. Yang, Y. Wang, X. Jia, X. Xu, P. Jiang and X. Wang, Adv. Funct. Mater., 2023, 33, 2210395 CrossRef CAS.
- T. Xie, Polymer, 2011, 52, 4985–5000 CrossRef CAS.
- C. B. Cooper, S. Nikzad, H. Yan, Y. Ochiai, J.-C. Lai, Z. Yu, G. Chen, J. Kang and Z. Bao, ACS Cent. Sci., 2021, 7, 1657–1667 CrossRef CAS PubMed.
- K. Ariga, J. P. Hill and Q. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319–2340 RSC.
- Z. Yu, F. Tantakitti, T. Yu, L. C. Palmer, G. C. Schatz and S. I. Stupp, Science, 2016, 351, 497–502 CrossRef CAS.
- T. P. Bigioni, X.-M. Lin, T. T. Nguyen, E. I. Corwin, T. A. Witten and H. M. Jaeger, Nat. Mater., 2006, 5, 265–270 CrossRef CAS PubMed.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. de, A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- T. Wen, J.-Y. Lee, M.-C. Li, J.-C. Tsai and R.-M. Ho, Chem. Mater., 2017, 29, 4493–4501 CrossRef CAS.
- H. Yamagishi, H. Sato, A. Hori, Y. Sato, R. Matsuda, K. Kato and T. Aida, Science, 2018, 361, 1242–1246 CrossRef CAS PubMed.
- S. Bera, S. Mondal, B. Xue, L. J. W. Shimon, Y. Cao and E. Gazit, Nat. Mater., 2019, 18, 503–509 CrossRef CAS PubMed.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed.
- Q. Zhang, Y.-X. Deng, H.-X. Luo, C.-Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D.-H. Qu, J. Am. Chem. Soc., 2019, 141, 12804–12814 CrossRef CAS PubMed.
- Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2018, 4, eaat8192 CrossRef CAS PubMed.
- C.-Y. Shi, Q. Zhang, B.-S. Wang, M. Chen and D.-H. Qu, ACS Appl. Mater. Interfaces, 2021, 13, 44860–44867 CrossRef CAS.
- Y. Deng, Q. Zhang, D. Qu, H. Tian and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202209100 CrossRef CAS.
- T. Ikami, Y. Watanabe, H. Ogawa, M. Takenaka, N. L. Yamada, M. Ouchi, H. Aoki and T. Terashima, ACS Macro Lett., 2021, 10, 1524–1528 CrossRef CAS PubMed.
- W. Lei, S. Qi, Q. Rong, J. Huang, Y. Xu, R. Fang, K. Liu, L. Jiang and M. Liu, Adv. Mater., 2019, 31, 1808217 CrossRef.
- A. Galeski, Prog. Polym. Sci., 2003, 28, 1643–1699 CrossRef CAS.
- T. Nicolai, F. Prochazka and D. Durand, Phys. Rev. Lett., 1999, 82, 863–866 CrossRef CAS.
- T. Nakajima, Polym. J., 2017, 49, 477–485 CrossRef CAS.
- L. Chen, C. Zhao, J. Huang, J. Zhou and M. Liu, Nat. Commun., 2022, 13, 6821 CrossRef CAS.
- H. Wan, B. Wu, L. Hou and P. Wu, Adv. Mater., 2023, 2307290 Search PubMed.
- M. Hua, S. Wu, Y. Ma, Y. Zhao, Z. Chen, I. Frenkel, J. Strzalka, H. Zhou, X. Zhu and X. He, Nature, 2021, 590, 594–599 CrossRef CAS PubMed.
- C. Shi, Q. Zhang, C. Yu, S. Rao, S. Yang, H. Tian and D. Qu, Adv. Mater., 2020, 32, 2000345 CrossRef CAS.
- C. N. Maganaris and J. P. Paul, J. Physiol., 1999, 521, 307–313 CrossRef CAS PubMed.
- J. Zhao, Z. Zhang, C. Wang and X. Yan, CCS Chem., 2024, 6, 41–56 CrossRef CAS.
- Z. Jiang, A. Bhaskaran, H. M. Aitken, I. C. G. Shackleford and L. A. Connal, Macromol. Rapid Commun., 2019, 40, 1900038 CrossRef.
- V. Nagarajan, A. K. Mohanty and M. Misra, ACS Sustain. Chem. Eng., 2016, 4, 2899–2916 CrossRef CAS.
- Z. Li, C. Chen, R. Mi, W. Gan, J. Dai, M. Jiao, H. Xie, Y. Yao, S. Xiao and L. Hu, Adv. Mater., 2020, 32, 1906308 CrossRef CAS PubMed.
- X. Wang, S. Zhan, Z. Lu, J. Li, X. Yang, Y. Qiao, Y. Men and J. Sun, Adv. Mater., 2020, 32, 2005759 CrossRef CAS PubMed.
- Z. Guo, X. Lu, X. Wang, X. Li, J. Li and J. Sun, Adv. Mater., 2023, 35, 2300286 CrossRef CAS PubMed.
- J. Zhu, G. Y. Chen, L. Yu, H. Xu, X. Liu and J. Sun, CCS Chem., 2020, 2, 280–292 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- M. Liu, P. Liu, G. Lu, Z. Xu and X. Yao, Angew. Chem., Int. Ed., 2018, 57, 11242–11246 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS.
- T. Dayyoub, A. V. Maksimkin, O. V. Filippova, V. V. Tcherdyntsev and D. V. Telyshev, Polymers, 2022, 14, 3511 CrossRef CAS.
- M. Zare, M. P. Prabhakaran, N. Parvin and S. Ramakrishna, Chem. Eng. J., 2019, 374, 706–720 CrossRef CAS.
- T. Chung, A. Romo-Uribe and P. T. Mather, Macromolecules, 2008, 41, 184–192 CrossRef CAS.
- Q. Zhao, W. Zou, Y. Luo and T. Xie, Sci. Adv., 2016, 2, e1501297 CrossRef.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed.
- H. Song, Z. Fang, B. Jin, P. Pan, Q. Zhao and T. Xie, ACS Macro Lett., 2019, 8, 682–686 CrossRef CAS PubMed.
- C. Keplinger, J.-Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed.
- N. Sata, K. Eberman, K. Eberl and J. Maier, Nature, 2000, 408, 946–949 CrossRef CAS.
- X. C. Chen, R. L. Sacci, N. C. Osti, M. Tyagi, Y. Wang, J. K. Keum and N. J. Dudney, Front. Chem., 2021, 8, 592604 CrossRef PubMed.
- Q. Zeng, Y. Lu, P. Chen, Z. Li, X. Wen, W. Wen, Y. Liu, S. Zhang, H. Zhao, H. Zhou, Z. Wang and L. Zhang, J. Energy Chem., 2022, 67, 157–167 CrossRef CAS.
- J. Chen, Y. Gao, L. Shi, W. Yu, Z. Sun, Y. Zhou, S. Liu, H. Mao, D. Zhang, T. Lu, Q. Chen, D. Yu and S. Ding, Nat. Commun., 2022, 13, 4868 CrossRef CAS PubMed.
- M. Zhang, R. Yu, X. Tao, Y. He, X. Li, F. Tian, X. Chen and W. Huang, Adv. Funct. Mater., 2023, 33, 2208083 CrossRef CAS.
- E. B. Trigg, T. W. Gaines, M. Maréchal, D. E. Moed, P. Rannou, K. B. Wagener, M. J. Stevens and K. I. Winey, Nat. Mater., 2018, 17, 725–731 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |