
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect photochemical intramolecular [4 + 2] cycloadditions of dehydrosecodine-type substrates for the synthesis of the iboga-type scaffold and divergent [2 + 2] cycloadditions employing micro-flow system†
Gavin
Tay‡
,
Soushi
Nishimura‡
and
Hiroki
Oguri
 *
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: hirokioguri@g.ecc.u-tokyo.ac.jp
First published on 25th September 2024
Abstract
Photocyclisation reactions offer a convenient and versatile method for constructing complex polycyclic scaffolds, particularly in the synthesis of natural products. While the [2 + 2] photocycloaddition reaction is well-established and extensively reported, the [4 + 2] counterpart via direct photochemical means remains challenging and relatively unexplored. In this work, we devised the rapid assembly of the iboga-type scaffold through photochemical intramolecular Diels–Alder reaction using a common biomimetic dehydrosecodine-type intermediate having vinyl indole and dihydropyridine (DHP) sub-units. Exploiting a micro-flow system, the medicinally important iboga-type scaffold was obtained up to 77% yield under mild, neutral conditions at room temperature. This study demonstrated the site-selective activation of the DHP moiety by direct UV-LED irradiation, eliminating the need for external photocatalysts or photosensitisers and showing good tolerance to a wide range of stabilised dehydrosecodine-type substrates. By adjusting the spatial arrangement of the DHP ring and the vinyl indole group, this versatile photochemical approach efficiently facilitates both [4 + 2] and [2 + 2] cyclisations, assembling architecturally complex multicyclic scaffolds. Precise photoactivation of the DHP subunit, generating short-lived biradical species, enabled the new way of harnessing the hidden but innately pre-encoded reactivity of the polyunsaturated dehydrosecodine-type intermediate. These photo-mediated [4 + 2] cyclisation and divergent [2 + 2] cycloadditions are distinct from biosynthetic processes, which are mainly mediated through concerted thermal cycloadditions.
Introduction
Biologically important natural products frequently contain architecturally complex three-dimensional ring systems as intricate components of their core skeletal structure.1 In particular, the vast majority of these compounds possesses elaborately fused and bridged frameworks, adding an extra layer of complexity to their retrosynthetic analyses and the design of appropriate intermediates for streamlined chemical assembly. Towards this goal, the development of straightforward and flexible synthetic methodologies has become a fundamental objective in organic chemistry. To construct polycyclic rings having a carbon-rich backbone, the classical Diels–Alder (DA) reaction has been extensively employed as a canonical strategy, wherein two unsaturated sub-units, namely the diene and dienophile, undergo a concerted pericyclic annulation to form a new six-membered ring (Fig. 1a).2 According to the molecular orbital symmetry rules devised by Woodward and Hoffmann in the 1960s,3 it is well understood that the concerted [4 + 2] cycloaddition process is thermally-allowed in the ground state. While the DA reaction has been proven to be a reliable process, its efficiency and reactivity are greatly dependent on the electronic compatibility between the dienes and dienophiles, constituting a limiting factor in the design of suitable substrates. When this electronic demand is not met, high temperatures are often required to surpass the large activation barrier.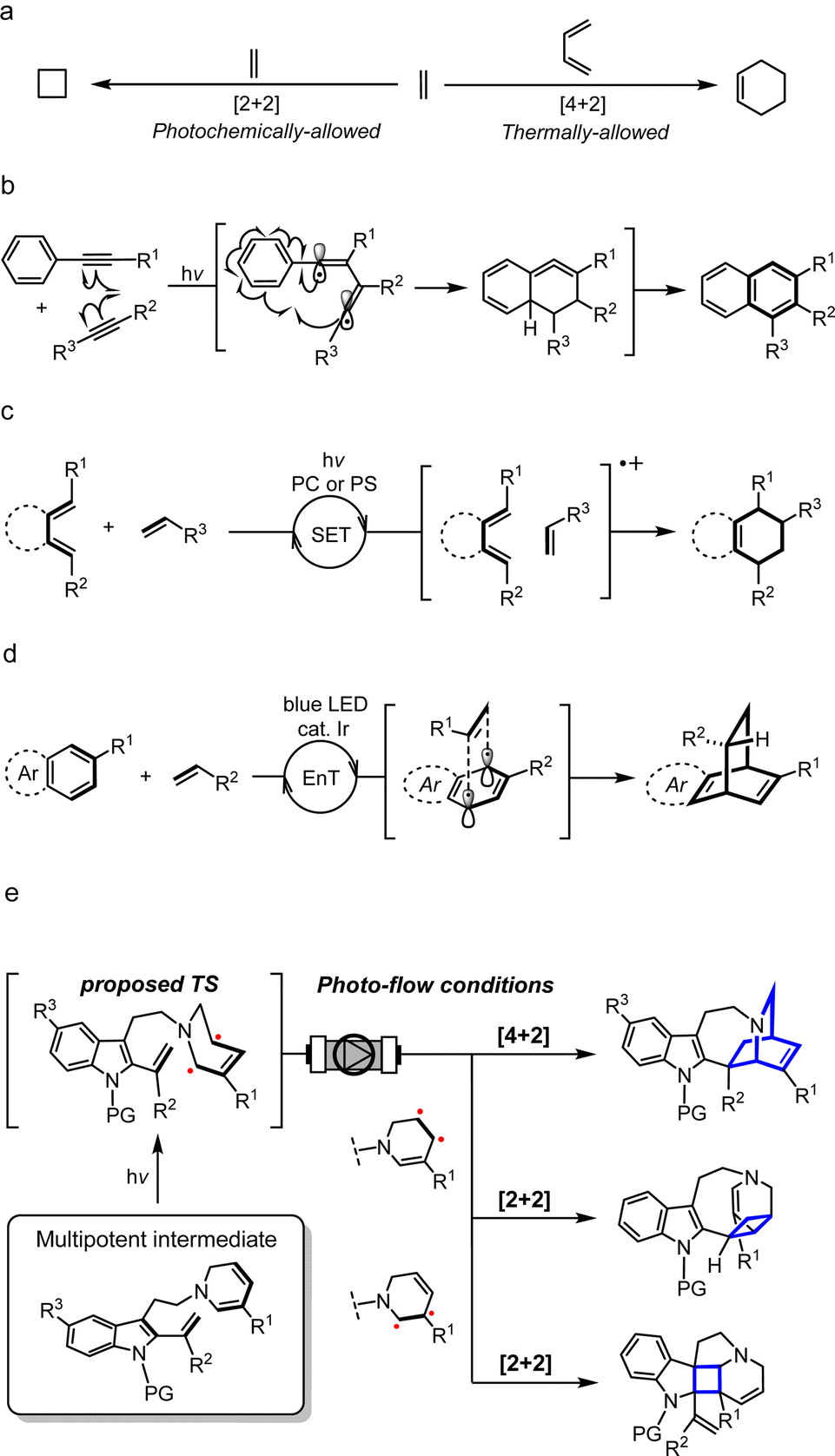 | ||
| Fig. 1 Scope and chemical space of [4 + 2] cycloaddition reactions. PC: photocatalyst; PS: photosensitizer; SET: single-electron transfer; EnT: energy transfer; TS: transition state. | ||
An alternative means by which molecules in the ground state can be activated is with light. Over the last few decades, the application of photochemistry in organic synthesis has grown exponentially and has witnessed remarkable progress in its chemical toolbox.4 When energy in the form of photons is absorbed, the molecule is elevated to a high-energy excited state. Initiation of the chemical reaction from this vantage point not only facilitates overcoming high kinetic barriers but can also unveil new and previously inconceivable reaction pathways which are otherwise hidden under thermal conditions. The [2 + 2] cycloaddition to construct a cyclobutane ring is the most ubiquitous and well-known photocycloaddition reaction,5 and along with numerous other light-driven cycloaddition reactions have provided a straightforward and versatile strategy in organic chemistry for achieving challenging synthetic transformations, particularly the synthesis of natural products.6
Since its advent, the scope and chemical space of classical DA reactions have greatly expanded and rapidly advanced into the realm of photochemistry. Early on, it was discovered that the dearomative [4 + 2] cycloaddition of anthracene and maleic anhydride could proceed both thermally and photochemically.7 Fascinated by the unique reactivities that govern regio- and stereoselectivity, such dearomative DA reactions have been a subject of intense mechanistic investigation. Under thermal conditions, a concerted pathway is widely postulated although some studies have provided evidence for the involvement of biradicals or charge-transfer species depending on the electronic nature of the diene and dienophile.7a,b On the contrary, the photochemically-driven process is believed to proceed via a biradical mechanism in a stepwise manner, maintaining the orbital symmetry requirements of the Woodward–Hoffmann theory.
Similar to the dearomative DA reaction, dienes in the form of an aryl-alkyne were also found to react under both heat and light.8 The photochemical process, formerly referred to as the photo-dehydro-Diels–Alder (PDDA) reaction, involves the activation of aryl-alkynes in a triplet excited state, leading to the formation of a C–C single bond with an alkyne as a dienophile, resulting in the generation of a 1,2-butadiene-1,4-diyl biradical intermediate (Fig. 1b). A second C–C bond formation with an aromatic ring generates a strained cyclic allene intermediate, followed by transposition of double bond, which affords a naphthalene ring. Wessig and coworkers recently reported the use of triplet photosensitisers (PSs) such as xanthone to enhance the efficiency of PDDA compared to direct photoirradiation.8c
By far the most common strategy for photochemical DA reactions to date is using external photocatalysts (PCs) and PSs, which has seen significant progress since the 1980s (Fig. 1c).9 Organic photoinduced-electron-transfer (PET) PCs such as pyrylium salts and cyanoaromatic compounds were extensively employed to generate a radical cation as an excited-state intermediate.9c,d Triplet ketone PSs including benzoylthiophenes can also induce this transformation via triplet state energy-transfer (EnT).9e,f On the other hand, Yoon achieved the visible light photocatalysed DA reaction using transition-metal complexes, including Ru and Pt/TiO2 heterogeneous catalyst, to promote the generation of the radical cation intermediate via a photoredox process.9g–j More recently, Glorius and Ma developed the metal-catalysed EnT dearomative [4 + 2] cycloaddition of azarenes, predominantly using Ir catalyst under blue LED irradiation (Fig. 1d).10 On the other hand, several total synthesis routes, including that of estrones11 and hamigerans,12 utilised the photoenolisation Diels–Alder (PEDA) reaction as a key strategy to construct the benzannulated cores.13 UV light activation of the aromatic carbonyl unit triggers o-quinodimethane diene formation in situ,14 followed by sequential DA trapping of this intermediate by a dienophile, although the cyclisation step itself is not a photochemically-dependent process.
Nonetheless, DA reactions by direct excitation, particularly towards the biomimetic synthesis of natural products, are still very rare. Herein, we report the photo-induced intramolecular [4 + 2] cycloaddition reaction for the rapid assembly of the bridged [2.2.2]-bicyclic isoquinuclidine ring system of the pharmaceutically intriguing iboga-type scaffold from a common dehydrosecodine-type intermediate having 1,6-DHP and vinyl indole moieties as the diene and dienophile, respectively (Fig. 1e). Iboga-type mono-terpene indole alkaloids (MIAs) are a significant class of bioactive compounds that possess promising anti-addiction and psychoactive properties.15 Fascination with this medicinally important family of alkaloids has inspired the development of numerous synthetic routes, especially towards the construction of the characteristic isoquinuclidine core.15a In this novel approach, we utilise direct UV light irradiation to site-selectively photoactivate the 1,6-DHP unit to efficiently promote the formal [4 + 2] cycloaddition. Contrary to common photochemical DA reactions, our strategy does not require the addition of any PCs or PSs. This cyclisation is presumably achieved through stepwise C–C bond formation of transient biradical intermediates. Importantly, the application of a micro-flow system facilitated streamlined assembly of the pentacyclic scaffold of iboga-type MIAs in modest to high yields under mild reaction conditions at room temperature, with short irradiation times through enhancing the photoirradiation efficiency while minimising decomposition. Furthermore, removal of the methyl ester moiety at the vinyl indole unit induced subtle but significant changes in the pre-organised conformation of the stabilised dehydrosecodine-type intermediate, paving the way for the divergent photo-mediated [2 + 2] cyclisations. These intricate and topologically complex multicyclic scaffolds are inaccessible through the enzymatic conversions and biomimetic synthesis relying on the thermal concerted cycloadditions.
Results and discussion
Synthesis of intermediate and its biomimetic thermal DA reaction leading to iboga-type scaffold
Azepinoindole intermediate 1a was prepared from tryptamine hydrochloride through the modular assembly of three building blocks in four steps, which included a Pictet–Spengler cyclisation, ring expansion, reduction, and N-propargylation (Fig. 2). In a previous study, we reported the biogenetically-inspired synthesis of iboga/aspidosperma-type alkaloidal scaffolds, utilising a common multipotent intermediate 3a that possesses both vinyl indole and 1,6-DHP moieties.16 This intermediate 3a, designed to mimic and preserve the structural features of dehydrosecodine 4, only differs by installing an electron-withdrawing ester group at the C3 position of the labile DHP ring in place of the ethyl substituent for improved stability through modulation of its electron density. Under microwave-assisted heating at 60 °C, Cu(I)-catalysed in situ 6-endo cyclisation of the 1,6-DHP ring (2a → 3a)17 and subsequent intramolecular biomimetic DA cycloaddition proceeded to give the iboga-type scaffold 5a in 48% yield (two steps).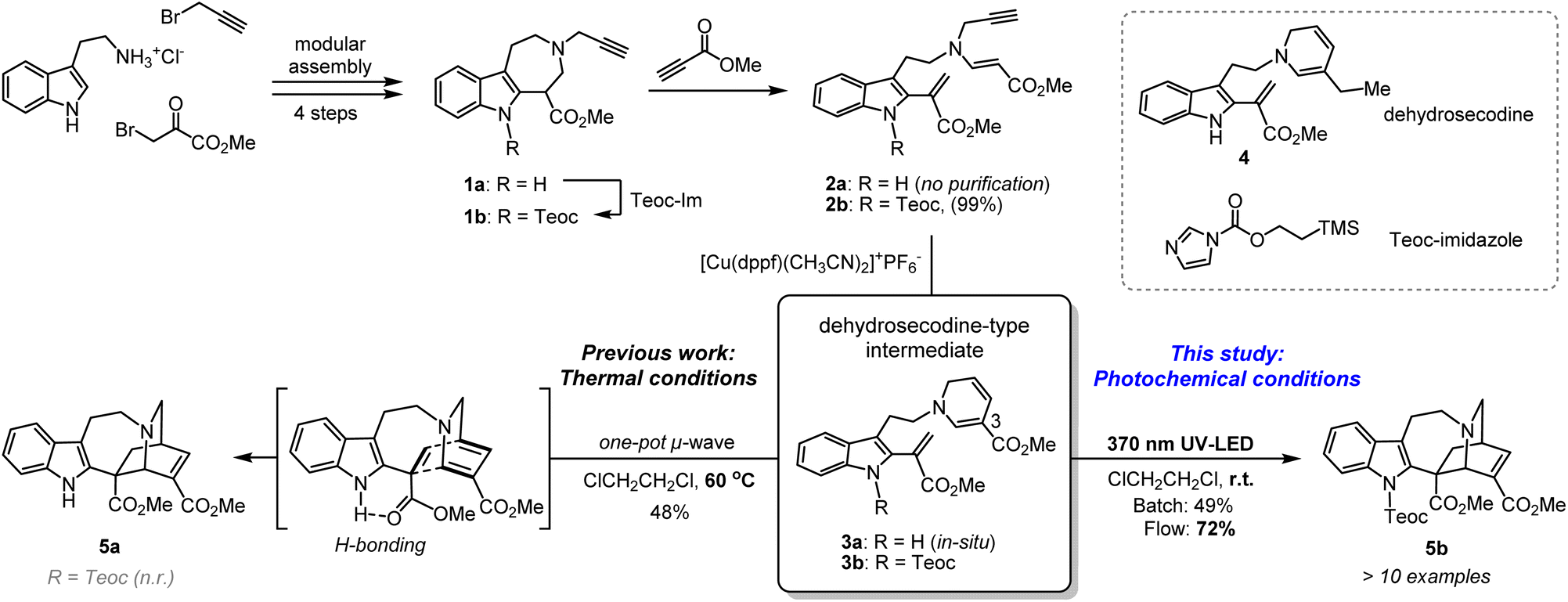 | ||
| Fig. 2 Synthesis of common biomimetic dehydrosecodine-type intermediates 3 and iboga-type scaffolds 5via thermal and photochemical pathways. | ||
A notable drawback of our previously designed synthetic process has been the instability of the indole-free ene-yne precursor 2a, without protection of the indole, which is prone to gradual decomposition by intra- and intermolecular Michael additions.16 To stabilise this precursor, and ease its handling, we installed an electron-withdrawing 2-(trimethylsilyl)ethoxycarbonyl (Teoc) carbamate protecting group at the indole N1 position of 1a to give 1b. Then, treatment with methyl propiolate triggered a tandem one-pot transformation involving hetero-Michael addition and Hofmann elimination to afford the stable indole-protected ene-yne 2b in almost quantitative yield. Cu(I)-catalysed conversion of 2b generated the dehydrosecodine-type intermediate 3b with efficient formation of the 1,6-DHP ring. However, subsequent one-pot DA cycloaddition with microwave-assisted heating to yield the corresponding iboga-product 5b did not proceed at all. Raising the temperature to 80 °C only led to gradual isomerisation of the 1,6-DHP unit in 3b to the 1,4-DHP. Regarding the biomimetic dehydrosecodine-type intermediate 3a, it is believed that the contribution of hydrogen bonding interactions between the free indole NH and neighbouring methyl ester unit plays a crucial role in increasing the electrophilicity of the dienophile, as well as achieving precise conformational pre-organisation suitable for the concerted DA reaction.16
Photochemical [4 + 2] cycloaddition to assemble iboga-type scaffold
With this in mind, we instead envisioned the construction of the isoquinuclidine [2.2.2]-bicyclic bridged ring system of the iboga-type skeleton by photochemical means. Preliminary UV-Vis analysis of the dehydrosecodine-type intermediate 3b showed a strong UV absorption peak at 356 nm (ε = 6.16 × 103 M−1 cm−1) in CHCl3 (Fig. S9†). Based on the absorption wavelength of the intermediate 3b, we attempted the photoreaction using a 370 nm UV-LED lamp (30 W). Cu(I)-catalysed 6-endo cyclisation of the Teoc-protected ene-yne precursor 2b in degassed 1,2-dichloroethane formed the 1,6-DHP ring. To our delight, subsequent irradiation for 4 h successfully furnished the corresponding iboga-product 5b with formation of a quaternary carbon centre, albeit in low yield of 30% over two steps (see Table S1†). Taking this finding as a starting point, we commenced the systematic optimisation of photoreaction conditions under batch conditions (Table 1). Upon photo-irradiation of 2b in 1,2-dichloroethane as the solvent, we found that 90 min was the optimal reaction time. This condition afforded the product 5b in 42% yield (41% isolated yield) based on NMR analysis using an internal standard (entry 1). Prolonged exposure of the reaction solution to UV light irradiation potentially led to the decomposition of the product, although such byproducts could not be isolated or identified. A survey of various solvents suggested that toluene was the ideal choice (entries 1–4), and irradiation at 370 nm gave the best results (entries 5–7), consistent with the UV-vis spectrum (Fig. S9†). When we changed the bidentate phosphine ligand for the Cu(I) catalyst from 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) (6) to 1,1′-bis(diphenylphosphino)ferrocene (DPPF) (7), the yield for the two-step sequential conversion (2b → 3b → 5b) improved to 55% (49% isolated yield) (entry 8).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst | Wavelength | Yield of 5ba |
| a Yield of 5b was determined as a two-step yield by 1H NMR spectroscopic analyses using triphenylmethane as an internal standard. b Reaction mixture was irradiated for 3 h. c Isolated yield over two steps. Xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene; DPPF = 1,1′-bis(diphenylphosphino)ferrocene. | ||||
| 1 | ClCH2CH2Cl | A | 370 nm | 42% (41%)c |
| 2 | MeCN | A | 370 nm | 27% |
| 3 | Toluene | A | 370 nm | 43% |
| 4 | Benzene | A | 370 nm | 41% |
| 5b | Toluene | A | 280 nm | 4% |
| 6 | Toluene | A | 427 nm | 30% |
| 7 | Toluene | A | 456 nm | Trace |
| 8 | Toluene | B | 370 nm | 55% (49%) |
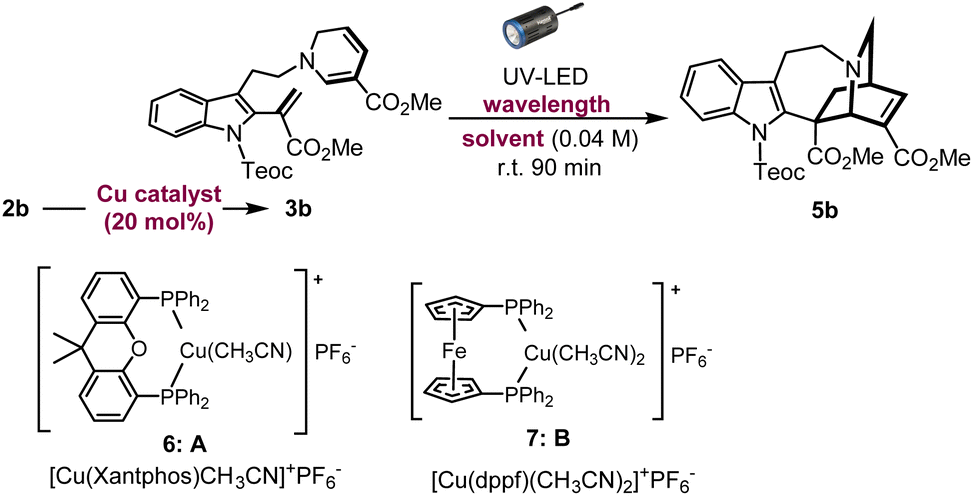
Substantial improvement using micro-flow system
In an effort to enhance the completion of the photoreaction, we conceived of employing the micro-flow system to simultaneously increase the photoirradiation efficiency and control the exposure time of the reaction solution under UV light, thereby suppressing the overreaction of the resulting iboga-product 5b. Recently, the application of photo-flow technology in synthetic organic chemistry, both in the laboratory and industrial settings, is rapidly expanding due to its improved reaction efficiency, ease of scalability, as well as practical cost and environmental benefits.18 Unlike thermal batch conditions, whereby the temperature within the reaction vessel remains uniform, photochemical reactions are highly sensitive to distance between the light source and reaction media owing to the rapid attenuation of light intensity. By employing the micro-flow chamber as the reaction vessel, we can achieve a more uniform irradiation and significantly reduce the distance that light needs to penetrate through the solvent media to enhance the overall irradiation efficiency. Under flow conditions, diluting the reaction concentration from 0.04 M to 0.01 M led to a substantial increase in yield, reaching 61% (51% isolated yield) with an optimal reaction time of just 25 min (Table 2: entries 1–6). However, the slight insolubility of intermediate 3b in toluene, the ideal solvent under batch conditions, caused gradual precipitation inside the flow chamber. Consequently, changing the solvent to 1,2-dichloroethane considerably improved the yield, affording the iboga-product in 74% (72% isolated yield) from 2b over two steps (entry 7). When the reaction scale was increased by six-fold, the iboga-product 5b (213 mg, see Page S28† for details) was still obtained in 68% yield.|
|
|||
|---|---|---|---|
| Entry | Deviation from batch | Residence time | Yield of 5ba |
| a Yield of 5b was determined as a two-step yield by 1H NMR spectroscopic analyses using triphenylmethane as an internal standard. b Isolated yield over two steps. Cu cat. B = [Cu(dppf)(CH3CN2)]+PF6−. | |||
| 1 | None | 15 min | 24% |
| 2 | None | 20 min | 41% |
| 3 | None | 25 min | 35% |
| 4 | 0.01 M | 15 min | 53% |
| 5 | 0.01 M | 20 min | 55% |
| 6 | 0.01 M | 25 min | 61% (51%)b |
| 7 | 0.01 M ClCH2CH2Cl | 25 min | 74% (72%)b |
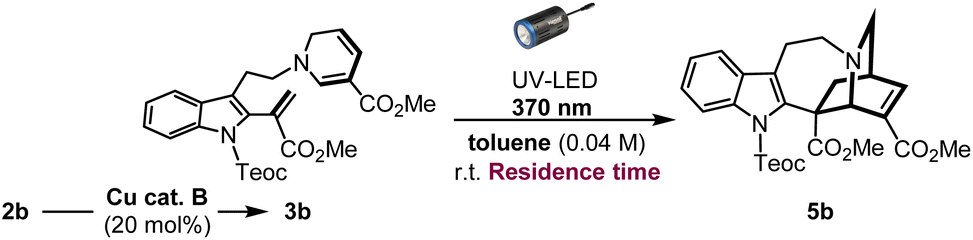
Substrate scope and limitation
Using the optimised photoreaction conditions, we aimed to explore the general applicability of the formal [4 + 2] cycloaddition through substrate scope expansion (Fig. 3). Firstly, all tested substituents on the indole nitrogen, including carbamate, methyl, and benzyl groups, were well tolerated for this photo-[4 + 2]-cycloaddition. Among the five substrates, 3b–3f, protection with an electron-withdrawing carbamate group at the indole N1 led to efficient conversions, and the best result was achieved with the cycloaddition of 3d with a methyl carbamate protection to furnish corresponding iboga-type scaffold 5d in high yield of 77%. Meanwhile, substrates 3e and 3f having an electron-donating substituent at the indole N1 resulted in slower reactions, potentially due to an electronic mismatch between the reacting sub-units, although the photoreaction still proceeded modestly. Next, the substrate scope for the DHP ring was examined. Installation of electron-withdrawing groups in the substrates (3g–h, and 3m) at the C3 position of the DHP ring yielded mixed results. Substrates 3g and 3h, bearing sulfonyl and ketone moiety, respectively, provided good yields of the corresponding [4 + 2] cycloadducts, 5g and 5h,19 comparable to their methyl ester counterpart 5b. However, substrate 3i with an aldehyde group instead resulted in a significantly slower reaction, presumably attributed to its excessively strong electron-withdrawing nature. Meanwhile, an attempted conversion of substrate 3j bearing an amide group did not yield the corresponding product 5j at all. We presume that the weakly electron-withdrawing amide group was insufficient to adequately stabilise the oxidation-labile 1,6-DHP unit, potentially causing an undesired hydride shift from the C6 position and rapid decomposition into the pyridinium species. The negligible differences in both the molar absorption coefficient and spectral profiles among the UV-vis spectra in CHCl3 for 3b (ε = 6.16 × 103 M−1 cm−1), 3e (ε = 6.32 × 103 M−1 cm−1) (Fig. S10†), and 3i (ε = 6.31 × 103 M−1 cm−1) (Fig. S11†) strongly suggest that there is essentially no decrease in the light absorption efficiency between the substrates, despite the lower yields observed for iboga-type products such as 5e and 5i.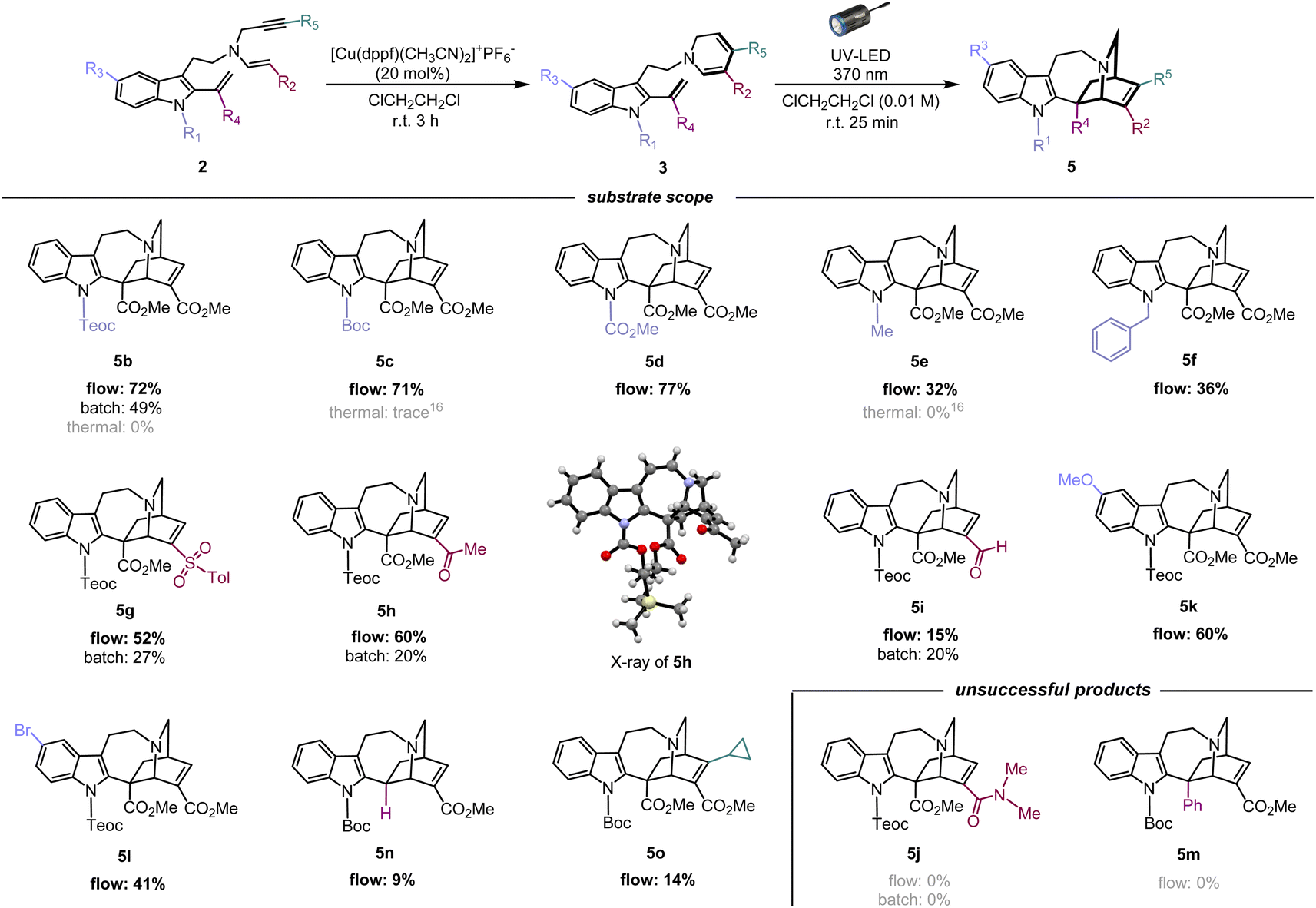 | ||
| Fig. 3 Substrate scope and limitations of [4 + 2] photocycloaddition for assembly of the iboga-type product. | ||
Subsequently, to assess the necessity of the ester group at the vinyl indole position, we attempted to replace it with a phenyl group via Au(I)-catalysed alkenylation20 of the indole C2 position, with substrate 3m having a N-Boc carbamate protecting group at the indole (for synthesis scheme, see Pages S13–S16†). However, the resulting photo-mediated [4 + 2] cyclisation of 3m to 5m failed to proceed, likely due to the steric hindrance near the phenyl group.
Divergent [2 + 2] cycloadditions
As an attempt to reduce the steric hindrance in 3m, we then prepared substrate 3n with removal of the phenyl group at the vinyl indole unit (for synthesis scheme, see Pages S16–S19†). The photoirradiation of 3n under micro-flow conditions led to distinct and notable results (Fig. 4a and see Page S30† for details). Unlike the attempt with substrate 3c bearing a methyl ester moiety, the formation of the iboga-type scaffold was turned to be substantially diminished and resulted in 5n in a minute amount with the yield of 9%. More importantly, we unexpectedly encountered divergent [2 + 2] cyclisations to two distinct scaffolds in combined yield of 87%. The subtle structural change, the removal of the methyl ester group in precursor 3n, induced a drastic alteration of the intramolecular cyclisation mode. The major product 8n was assembled in 49% yield via a [2 + 2] cycloaddition between the vinyl indole and the C4–C5 olefinic double bond of the 1,6-DHP.19 In addition, a dearomative [2 + 2] cycloaddition between the indole C2–C3 double bond and an olefin at the C2–C3 position of the 1,6-DHP also proceeded to form pentacyclic spiroindoline scaffold 9n in 38% yield. This photo-mediated dearomative [2 + 2] cyclisation enabled the diastereo-controlled formation of the three consecutive quaternary carbon centres within the central cyclobutane framework in 9n.19 In fact, the relevant intra- and intermolecular [2 + 2] cycloaddition reactions with the indole ring have been reported using blue-LED or visible light in the presence of PCs.21 We postulate that the absence of any substituent at the vinyl indole unit induces a slight conformational shift in the spatial arrangement of the reacting sub-units, sufficient to favour the [2 + 2] cycloaddition while severely inhibiting the [4 + 2] cyclisation reaction pathway.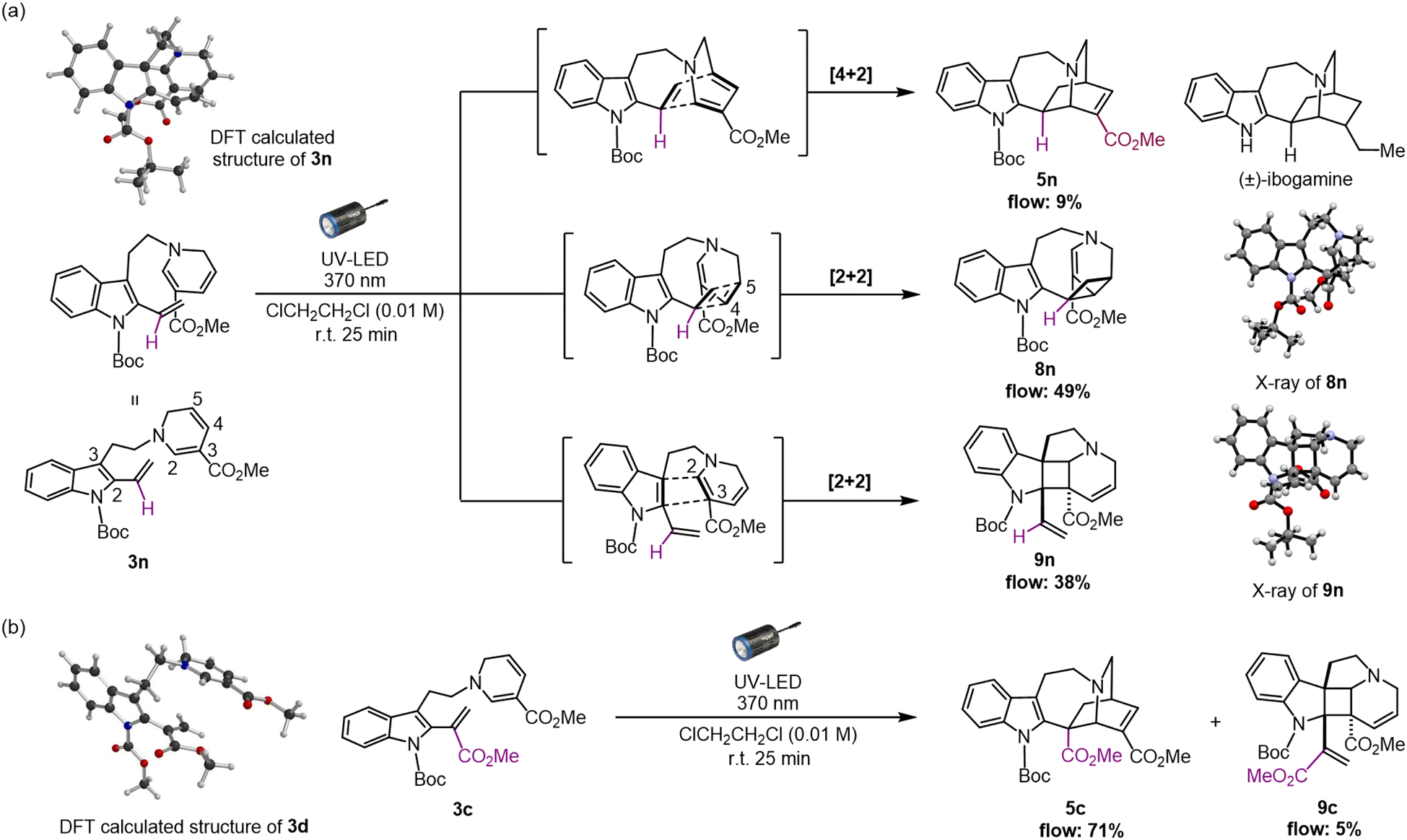 | ||
| Fig. 4 (a) Unexpected divergent [4 + 2] and [2 + 2] cycloadditions of 3n to form iboga-type scaffold 5n, and novel indole alkaloidal scaffolds 8n and 9n, respectively. (b) Re-analysis of photoreaction from 3c. DFT modelling of 3d and 3n were performed using the B3LYP-D3 (6-31+G*) level of theory. | ||
To confirm the possibility that the [2 + 2] cycloadditions compete with the photo-mediated [4 + 2] cyclisation, even in trace amounts, we carefully re-analysed the photochemical reaction using 3c as a representative substrate for the high-yield formation of the iboga-type scaffold 5c (Fig. 4b). Indeed, the formation of 9c through the corresponding dearomative [2 + 2] cycloaddition was observed, albeit with a very low yield of 5%. Meanwhile, the occurrence of the other [2 + 2] cycloaddition leading to scaffold 8c was not detected at all.
In our recent study, we successfully elucidated the X-ray crystallographic structure of unstable dehydrosecodine-type intermediate 3 through host-guest encapsulation within a self-assembled coordination cage.22 The resulting X-ray analysis of guest molecule 3d revealed the unique stacking of both reacting sub-units, the 1,6-DHP ring and vinyl indole moiety, conformationally primed to undergo the [4 + 2] cycloaddition. This finding was consistent with DFT computational modelling (Fig. S7†).23 Notably, the energy minimised structure of 3n (Fig. S8†) revealed an adopted conformation with the folding of the DHP ring towards the indole ring and evidently aligning the four reacting olefins, representing a pre-organised spatial arrangement that favours the [2 + 2] cycloadditions over the corresponding [4 + 2] annulation. Thus, the methyl ester group in the vinyl indole moiety of 3c appears to possess the “Goldilocks effect” as a precise steric handle that selectively facilitates the assembly of the iboga-type scaffold 5c. To the best of our knowledge, the [2 + 2] cycloadducts 8 and 9 are novel, bis-nitrogen containing, densely functionalised sp3-rich scaffolds relevant to MIAs. These unnatural intricate alkaloidal scaffolds are inaccessible under previous thermal conditions or by chemoenzymatic means.
Mechanistic studies
We then turned our attention to elucidating the photoreaction mechanism (Fig. 5, see the ESI† for details). To probe the origin of the characteristic UV absorption at approximately 356 nm for 3b, we first conducted regio-selective hydrogenation of the C4–C5 double bond of the 1,6-DHP unit to generate the stable tetrahydropyridine (THP) product 10 (Fig. 5a).16 After reduction, no UV absorption was observed at this wavelength, indicating that the 356 nm absorption is derived from the conjugated system of the 1,6-DHP ring. This also strongly suggests that direct irradiation induces site-selective activation of the DHP moiety to trigger the [4 + 2] cyclisation. | ||
| Fig. 5 Mechanistic studies of [4 + 2] cycloaddition. (a) UV-vis spectral analysis of DHP intermediate 3b and THP 10 in CHCl3. (b) UV-vis spectral analysis of EDA complex via model substrates 11 and 12 in CHCI. (c) Photoreaction under Cu(I)-free conditions. (d) Radical trapping experiments. (e) Radical clock experiment. | ||
To investigate whether an electron donor–acceptor (EDA) pair could form between the electron-rich DHP and electron-deficient vinyl indole moieties upon photoexcitation,24 we prepared a pair of model substrates, vinyl indole 11 and 1,6-DHP 12,16,22,25 and verified the possibility of their complexation through UV-vis analysis in CHCl3 (Fig. 5b). However, no significant shifts in the absorption spectrum were observed, indicating that an EDA pair is not likely to be involved in this system. The absence of solvent-dependent spectral shifts of the cyclisation precursor 3b, as the stabilised dehydrosecodine-type substrate, also supports this result (Fig. S15†).
We then wondered whether the Cu(I) catalyst, utilised in the initial step to form the 1,6-DHP ring (2b → 3b), could act as a PC to facilitate the photoreaction. DA reactions promoted by copper salts have been extensively reported, and are particularly useful for hetero-DA reactions, as well as for enantioselective synthesis by employing chiral copper complexes.26 After cyclisation of the DHP ring, commercially available metal scavenger, SiliaMetS Diamine (10.0 equiv. to Cu) was added and then stirred for 1 h (Fig. 5c). Successful complexation of the scavenger with Cu(I) was confirmed by a visible change to a blue colour of the silica support, which was subsequently removed by filtration through a short pad of silica. When the resulting solution was subjected to photoreaction under standard flow conditions, the iboga-scaffold 5b was still obtained in 65% yield. The slight decrease in yield could be attributed to the additional manipulation of the labile intermediate 3b. Furthermore, the Cu(I) catalyst [Cu(dppf)(MeCN)2]+PF6− (7) itself does not show UV absorption at around 370 nm (Fig. S16†). These experimental results have confirmed that the light-mediated [4 + 2] intramolecular cyclisations proceeds without the need for the copper complex to act as a photocatalyst.
At this point, we anticipated that the photo-mediated [4 + 2] cycloaddition of 3b proceeds via a series of transient biradical intermediates upon photoactivation of the 1,6-DHP unit. To attest our hypothesis, we attempted radical trapping under flow conditions using (2,2,6,6-tetramethyl-1-piperidinyloxyl) (TEMPO) radical, 1,1-diphenylethylene, and butylated hydroxytoluene (BHT) (Fig. 5d). In the presence of the radical trapping agents (10 equiv.), the yield of iboga-product 5b decreased by approximately half (Table 3). LCMS-IT-TOF analysis indeed revealed formation of the corresponding adduct bearing two additional hydrogens donated from BHT (Fig. S17†) as well as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct with 1,1-diphenylethylene (Fig. S18†), although isolation of these products to deduce their exact structures was not possible. It is likely that the competing intramolecular cyclisation occurred very rapidly, therefore the formation of the iboga-product 5b could not be sufficiently prevented even when excessive quantities of the radical trapping agents were added.
:1 adduct with 1,1-diphenylethylene (Fig. S18†), although isolation of these products to deduce their exact structures was not possible. It is likely that the competing intramolecular cyclisation occurred very rapidly, therefore the formation of the iboga-product 5b could not be sufficiently prevented even when excessive quantities of the radical trapping agents were added.
| Entry | Radical trapping agent | Yield of 5b |
|---|---|---|
| 1 | 2,2,6,6-Tetramethylpiperidinyloxyl (TEMPO) radical | 38% |
| 2 | Butylated hydroxytoluene (BHT) | 34% |
| 3 | 1,1′-Diphenylethylene | 40% |
Next, we designed and synthesised radical clock substrate 3o, by incorporating a cyclopropane moiety at the C4 position of the DHP ring (Fig. 5e). To attain 3o, the cyclopropane unit was pre-installed to the propargyl bromide fragment prior to N-propargylation (for synthesis scheme, see Pages S19 and S20†). Upon the photo-mediated generation of biradicals from the radical clock substrate 3o, we anticipated a sequential radical-mediated cyclopropane ring opening to generate intermediate Int-1, followed by the formation of a five-membered ring leading to 13. However, contrary to our expectations, products like 13 were not observed while the iboga-scaffold 5o still formed, albeit in a low yield of 15%. Unexpectedly, the formation of a nitrogen-containing bicyclo[2.2.0] ring system, presumably through Int-2, predominantly occurred to produce 14 in 32% yield as the major product. These experimental findings are consistent with our previous results, where photoexcitation of the 1,6-DHP ring leads to the generation of biradical species. The 4π-electrocyclic ring-contraction of relatively electron-rich DHPs, pyridones, and other conjugated cyclic systems has been reported to occur under UV light irradiation via the singlet excited state.27 The electron density of the DHP moiety likely increased due to inductive effects from the introduction of the electron-donating cyclopropane moiety into the electron-deficient DHP ring bearing the methyl ester substituent. This change in electronic properties probably facilitated the 4π-electrocyclisation of 3o to 14, a reaction otherwise not observed in the photochemical cycloadditions of substrates 3b–n without the cyclopropane substituent on the DHP ring.
A plausible reaction mechanism
Based on a series of spectral analyses, experimental outcomes, as well as accumulated circumstantial evidence, we propose a stepwise biradical mechanism for the [4 + 2] cyclisation reaction (Fig. 6). Upon UV light irradiation, the 1,6-DHP moiety of 3 undergoes photochemical excitation to reach a high energy excited state, leading to the formation of interconvertible biradical intermediates denoted as Int-A. Among the three biradicals in equilibrium, Int-A2, having the biradicals at the DHP C2 and C5 positions, is expected to have the highest spin density, as evidenced by the formation of the bicyclo[2.2.0] ring (3o → 14). From Int-A2, two distinct radical reaction pathways to form a C–C bond can be conceived. It is likely that pathway B is favoured over pathway A, since pathway A generates a biradical intermediate Int-B which contains a highly unstable primary radical. In contrast, pathway B forms a corresponding intermediate Int-C capable of stabilising the resulting radical through conjugation with the indole ring on the left. Subsequent radical coupling from this stabilised intermediate Int-C would furnish iboga-scaffold 5.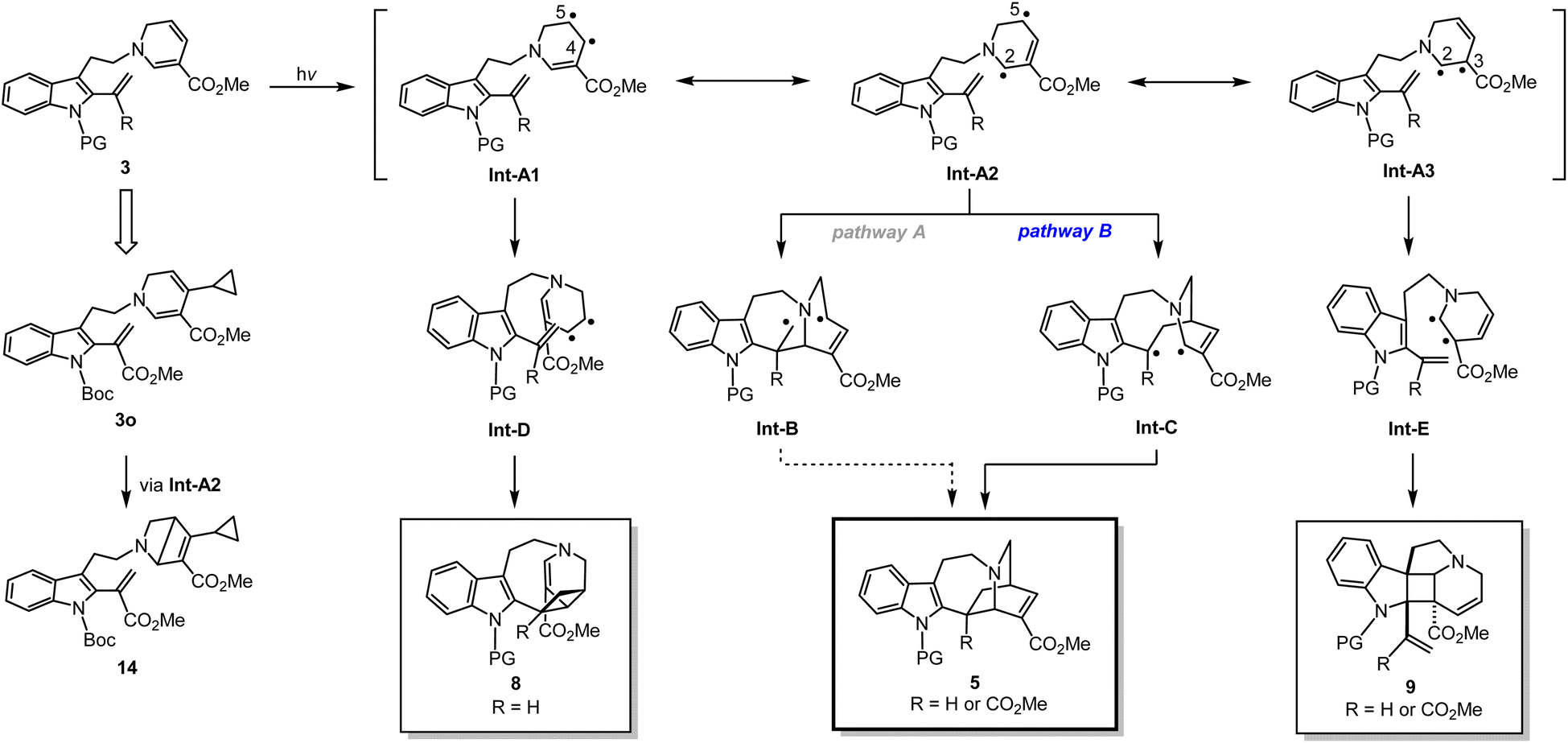 | ||
| Fig. 6 Proposed reaction mechanism of formal [4 + 2] and [2 + 2] cycloadditions via a biradical pathway. | ||
In a similar manner, we postulate the divergent intramolecular [2 + 2] annulations of 3 to assemble 8 and 9 could also proceed from the same excited state intermediate Int-A (Fig. 6). UV-vis analysis (Fig. S12†) revealed that there is negligible red-shift in the absorption spectrum of the conjugated vinyl indole moiety in substrate 3n compared to 3b. These experimental findings strongly suggest that the [2 + 2] cycloadditions are also initiated through the direct photoactivation of the 1,6-DHP ring rather than the vinyl indole. Thus, Int-A1 and Int-A3 could directly and rapidly give rise to scaffolds 8 and 9, respectively, with exquisite control of diastereoselectivity. The retro-[2 + 2] reactions of the cycloadducts 8 and 9 do not occur at all under 370 nm light irradiation at room temperature, strongly indicating that there is no photo-reversible interconversion among the three scaffolds 8, 9, and 5 under the reaction conditions. Importantly, the ability to induce C–C bond formation at all four olefinic centres on the 1,6-DHP ring (C2 to C5) also confirms the presence of transient resonance stabilised biradical species Int-A1 to Int-A3. It is worth noting that unlike previously reported [2 + 2] cycloadditions with the indole C2–C3 double bond, our photochemical cyclisation does not rely on the use of external PCs or PSs to generate the cyclobutane-fused indoline-type products 9.21 As revealed by DFT computational modelling of 3n, the challenging dearomative transformation (3 → 9) could be facilitated by the inherent pre-organisation of the two reacting olefinic double bonds. The novelty of this finding thus holds considerable significance as a direct and feasible approach to access such fused heterocyclic spiroindoline scaffolds.
Through our investigations, we have showcased the efficiency and adaptability of a biogenetically-inspired approach that flexibly accesses diverse indole alkaloidal scaffolds from the common stabilised multipotent intermediates 3, mimicking dehydrosecodine (4), which is biosynthetically derived from tryptamine and secologanin (Fig. 7).16 The synthetically tractable indole-free analogue 3a has paved the way to the biomimetic assembly of iboga-type alkaloid 5a, aided by hydrogen-bonding-mediated conformational pre-organisation and heating. Yet, because of its high reactivity, the utility of 3a has been severely restricted to an in situ generated species. Such labile intermediates bearing multiple sensitive structural components are highly susceptible to unwanted side-reactions under heat due to unpredictable conformational changes and uncontrollable activation of reactive sites. Moreover, efforts to further stabilise 3avia protection of the indole N1 position was ultimately met with a complete loss of desirable reactivity, even under harsher thermal conditions. Using this newly developed photo-mediated strategy, we were able to successfully circumvent these synthetic complications and shortcomings. Despite the dormancy of indole-protected substrates like 3b towards thermal-mediated DA cyclisations, its inherent reactivity can be efficiently triggered by site-selective photochemical excitation of the DHP unit. The simple modification of the substituent on the vinyl indole moiety in the substrate 3n revealed the remarkable efficiency of the [2 + 2] cycloaddition reactions. This enables the divergent synthesis of the pentacyclic scaffolds 8n and 9n with high cumulative yield and excellent regio- and diastereoselectivity in the formation of the densely functionalised central cyclobutane ring. Yet, by leveraging the exquisite conformational control afforded by the methyl ester substituent on the vinyl indole moiety, we circumvented these rapid and photochemically-intrinsic [2 + 2] cycloadditions. Instead, we successfully engaged in the more challenging and less common photo-mediated [4 + 2] cyclisation, leading to the iboga-type scaffold through a stepwise reaction pathway involving a series of reactive biradical intermediates. This unique feature of our study is distinctly defined by the precise light activation and the preservation of the innate proximity of the reaction sites with minimal conformational changes, as demonstrated with substrates 3c and 3n. Hence, we have unveiled a novel synthetic utility for multipotent intermediates 3b–n, which possesses the dehydrosecodine-type polyunsaturated system. These versatile yet stabilised and manipulable intermediates enable the efficient and precise construction of intricately fused and densely functionalised scaffolds under mild, neutral photochemical conditions otherwise unattainable by thermal processes.
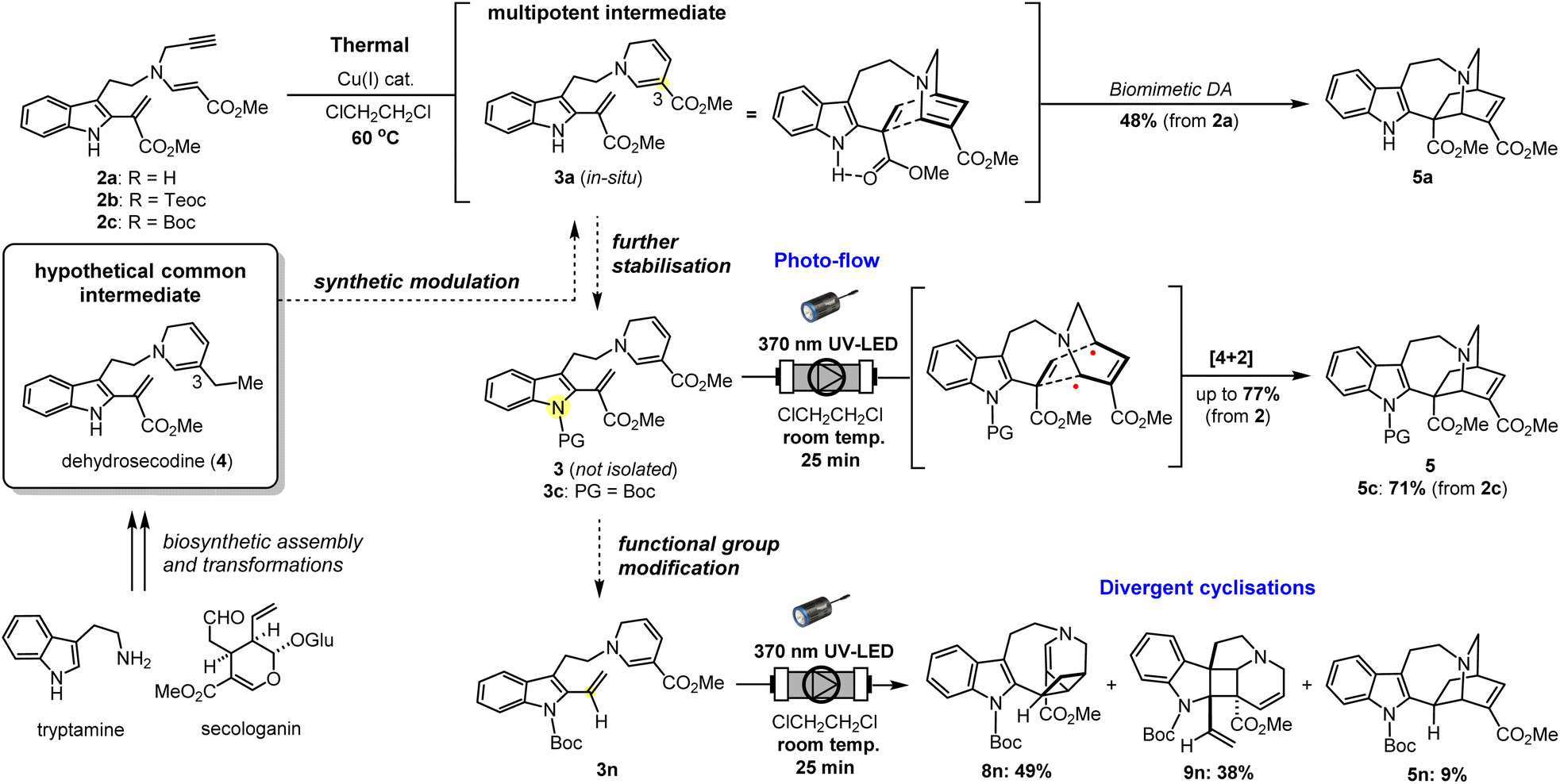 | ||
| Fig. 7 Biosynthesis of dehydrosecodine (4) initiates from tryptamine and secologanin as building blocks. The thermal biomimetic [4 + 2] cycloaddition proceeds via a dehydrosecodine-type multipotent intermediate (3a → 5a) facilitated by both heating and an intramolecular hydrogen-bonding. The new photo-flow approach features site-selective activation of DHP moiety using stabilised substrates 3 with indole N1 protection. A biradical reaction pathway enabled the divergent [4 + 2] and [2 + 2] cyclisations with minimal conformational changes in the pre-organised structures of 3c and 3n, respectively. | ||
Conclusions
In conclusion, we achieved the direct and additive-free photochemical [4 + 2] cycloaddition reaction to efficiently assemble the bridged [2.2.2]-bicyclic isoquinuclidine ring system of the iboga-type scaffold. The introduction of an indole N1 protecting group substantially stabilised the ene-yne precursors 3 for ease of handling and storage. Simultaneous implementation of micro-flow conditions to our reaction system was crucial to substantially enhance the photoirradiation efficiency while taming unwanted overreactions arising from overexposure to UV light under traditional batch methods. Compared to previously reported thermal conditions, our facile and scalable photochemical approach showed excellent tolerance to a diverse range of substrates in modest to high yields. Mechanistic studies strongly suggest the generation of biradical species upon photoactivation of the highly reactive 1,6-DHP moiety, followed by a stepwise pathway to furnish the iboga-type product 5. Moreover, we were able to harness the biradical excited state of the DHP moiety, together with appropriate conformational pre-organisation, to generate two novel unnatural scaffolds 8 and 9via divergent [2 + 2] cycloadditions, including construction of a pentacyclic spiroindoline-type product via the dearomatisation of the indole ring. Simple modification of the substituent at the vinyl indole position allowed precise control of both the mode-of-cyclisation and diastereoselectivity, enabling divergent assembly of a total of three distinct indole alkaloidal fused scaffolds. We believe that the reactive 1,6-DHP ring, capable of undergoing thermal and photo-mediate transformations, holds untapped synthetic potential as a versatile intermediate to feasibly construct difficult-to-access molecular frameworks.Data availability
The ESI† includes all experimental details, including optimisation of photoreaction conditions, synthesis and characterisation of compounds reported in this study, and mechanistic studies. Crystallographic data for 5h, 8n, and 9n have been deposited under CCDC accession numbers 2333565, 2375152, and 2375172, respectively. The NMR spectra of all products, UV-vis spectra, crystallographic data, and computational modelling details are also included.Author contributions
G. T. and S. N. contributed equally to this work. G. T., S. N., and H. O. conceived the projects and analysed the experimental results. G. T. and H. O. wrote the manuscript with feedback from S. N.Conflicts of interest
A patent application for these photo-mediated divergent cyclisations of the multipotent intermediates will be filed by The University of Tokyo.Acknowledgements
The authors thank Dr Haruki Mizoguchi, Hiroki Kubota, and Shun Oyadomari for preliminary synthetic investigations. We also appreciate Prof. Masanobu Uchiyama for discussions regarding radical trapping experiments. This work was supported in part by JSPS KAKENHI (JP19H02847, JP22H00346, and JP22H05127), the Naito Foundation, and the Asahi Glass Foundation (H. O.). G. Tay is grateful to the Global Science Graduate Course (GSGC) supported by The University of Tokyo. This work was also inspired by the international and interdisciplinary environments of the JSPS Asian CORE Program, “Asian Chemical Biology Initiative” as well as the JSPS A3 Foresight Program.References
- For selected papers, see: (a) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (b) E. K. Davison and J. Sperry, J. Nat. Prod., 2017, 80, 3060–3079 CrossRef CAS PubMed; (c) C. Zhao, Z. Ye, Z. Ma, S. A. Wildman, S. A. Blaszczyk, L. Hu, I. A. Guizei and W. Tang, Nat. Commun., 2019, 10, 4015 CrossRef PubMed; (d) Y. Chen, C. Rosenkranz, S. Hirte and J. Kirchmair, Nat. Prod. Rep., 2022, 39, 1544–1556 RSC.
- For selected papers, see: (a) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; (c) A. A. Sara, U. Um-e-Farwa, A. Saeed and M. Kalesse, Synthesis, 2022, 54, 975–998 CrossRef CAS.
- (a) R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395–397 CrossRef CAS; (b) R. B. Woodward and R. Hoffmann, Angew Chem. Int. Ed. Engl., 1969, 8, 781–932 CrossRef CAS.
- For selected papers, see: (a) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS; (b) B. Beeler, Chem. Rev., 2016, 116, 9629–9630 CrossRef.
- For selected papers, see: (a) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed; (b) D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org Chem., 2020, 2020, 1310–1326 CrossRef CAS.
- For selected papers, see: (a) J. Iriondo-Alberdi and M. F. Greaney, Eur. J. Org. Chem., 2007, 4801–4815 CrossRef CAS; (b) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed.
- For selected papers, see: (a) J. Sauer and R. Sustmann, Angew Chem. Int. Ed. Engl., 1980, 19, 779–807 CrossRef; (b) S. Jones and J. C. C. Atherton, Tetrahedron: Asymmetry, 2001, 12, 1117–1119 CrossRef CAS; (c) J. C. C. Atherton and S. Jones, Tetrahedron, 2003, 59, 9039–9057 CrossRef CAS.
- For selected papers, see: (a) P. Wessig, A. Matthes and C. Pick, Org. Biomol. Chem., 2011, 9, 7599–7605 RSC; (b) J. Zhuang, S. Zhang, H. Hao and L. Jiang, J. Photochem. Photobiol., A, 2013, 270, 14–18 CrossRef CAS; (c) P. Wessig, D. Badetko and M. Koebe, ChemistrySelect, 2022, 7, e202202648 CrossRef CAS.
- For selected papers, see: (a) B. Harirchian and N. L. Bauld, J. Am. Chem. Soc., 1989, 111, 1826–1828 CrossRef CAS; (b) A. Gieseler, E. Steckhan, O. Wiest and F. Knoch, J. Org. Chem., 1991, 56, 1405–1411 CrossRef CAS; (c) M. Martiny, E. Steckhan and T. Esch, Chem. Ber., 1993, 126, 1671–1682 CrossRef CAS; (d) M. A. Miranda and H. García, Chem. Rev., 1994, 94, 1063–1089 CrossRef CAS; (e) J. Pérez-Prieto, S. E. Stiriba, M. González-Béjar, L. R. Domingo and M. A. Miranda, Org. Lett., 2004, 6, 3905–3908 CrossRef; (f) M. González-Béjar, S. E. Stiriba, L. R. Domingo, J. Pérez-Prieto and M. A. Miranda, J. Org. Chem., 2006, 71, 6932–6941 CrossRef; (g) S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS; (h) S. Lin, C. E. Padilla, M. A. Ischay and T. P. Yoon, Tetrahedron Lett., 2012, 53, 3073–3076 CrossRef CAS PubMed; (i) S. P. Pitre, J. C. Scaiano and T. P. Yoon, ACS Catal., 2017, 7, 6440–6444 CrossRef CAS PubMed; (j) K. Nakayama, N. Maeta, G. Horiguchi, H. Kamiya and Y. Okada, Org. Lett., 2019, 21, 2246–2250 CrossRef CAS.
- For selected papers, see: (a) J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown, K. N. Houk and F. Glorius, Science, 2021, 371, 1338–1345 CrossRef CAS; (b) P. Rai, K. Maji, S. K. Jana and B. Maji, Chem. Sci., 2022, 13, 12503–12510 RSC; (c) R. Guo, S. Adak, P. Bellotti, X. Gao, W. W. Smith, S. N. Le, J. Ma, K. N. Houk, F. Glorius, S. Chen and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 17680–17691 CrossRef CAS PubMed.
- G. Quinkert, W.-D. Weber, U. Schwartz and G. Dürner, Angew Chem. Int. Ed. Engl., 1980, 19, 1027–1029 CrossRef.
- K. C. Nicolaou, D. Gray and J. Tae, Angew. Chem., Int. Ed., 2001, 40, 3675–3678 CrossRef CAS.
- N. C. Yang and C. Rivas, J. Am. Chem. Soc., 1961, 83, 2213 CrossRef CAS.
- J. L. Sugura and N. Martín, Chem. Rev., 1999, 99, 3199–3246 CrossRef PubMed.
- For selected papers, see: (a) R. N. Iyer, D. Favela, G. Zhang and D. E. Olsen, Nat. Prod. Rep., 2021, 38, 307–329 RSC; (b) B. M. M. Michele and A. A. Sophie, Pharmacol. Res. Nat. Prod., 2023, 1, 100006 Search PubMed.
- H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 CrossRef CAS PubMed.
- H. Mizoguchi, R. Watanabe, S. Minami, H. Oikawa and H. Oguri, Org. Biomol. Chem., 2015, 13, 5955–5963 RSC.
- For selected papers, see: (a) T. H. Rehm, Chem. Eur. J., 2020, 26, 16952–16974 CrossRef CAS PubMed; (b) T. Fukuyama, T. Kasakado, M. Hyodo and I. Ryu, Photochem. Photobiol. Sci., 2022, 21, 761–775 CrossRef CAS; (c) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed; (d) S. D. A. Zondag, D. Mazzarella and T. Noël, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 283–300 CrossRef CAS PubMed; (e) A. Slattery, Z. Wen, P. Tenblad, J. S. J. Orduna, D. Pintossi, T. D. Hartog and T. Noël, Science, 2024, 383, 382 CrossRef PubMed.
- Crystallographic data for 5h, 8n, and 9n have been deposited under accession numbers 2333565, 2375152, and 2375172, respectively.
- E. B. McLean, F. M. Cutolo, O. J. Cassidy, D. J. Burns and A.-L. Lee, Org. Lett., 2020, 22, 6977–6981 CrossRef CAS.
- For selected papers, see: (a) M. Zhu, C. Zheng, X. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2636–2644 CrossRef CAS PubMed; (b) Z. Zhang, D. Yi, M. Zhang, J. Wei, J. Lu, L. Yang, J. Wang, N. Hao, X. Pan, S. Zhang, S. Wei and Q. Fu, ACS Catal., 2020, 10, 10149–10156 CrossRef CAS; (c) M. Zhu, X. Zhang, C. Zheng and S.-L. You, Acc. Chem. Res., 2022, 55, 2510–2525 CrossRef CAS PubMed.
- G. Tay, T. Wayama, H. Takezawa, S. Yoshida, S. Sato, M. Fujita and H. Oguri, Angew. Chem., Int. Ed., 2023, 62, e202305122 CrossRef CAS.
- (a) B. J. Deppmeier, A. J. Driessen, W. J. Hehre, T. S. Hehre, J. A. Johnson, S. Ohlinger and P. E. Klunzinger, Spartan 20, Wavefunction Inc., Irvine CA, 2020 Search PubMed; (b) Y. Shao, et al. , Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- For selected papers, see: (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS; (b) Y. Yuan, S. Majumder, M. Yang and S. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS.
- T. Wayama, Y. Arai and H. Oguri, J. Org. Chem., 2022, 87, 5938–5951 CrossRef CAS PubMed.
- For selected papers, see: (a) Y. Li, Y. Hu, S. Zhang, J. Sun, L. Li, Z. Zha and Z. Wang, J. Org. Chem., 2016, 81, 2993–2999 CrossRef CAS; (b) S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359–5406 CrossRef CAS PubMed.
- For selected papers, see: (a) F. W. Fowler, J. Org. Chem., 1972, 37, 1321–1323 CrossRef; (b) P. Beeken, J. N. Bonfiglio, I. Hasan, J. J. Piwinski, B. Weinstein, K. A. Zollo and F. W. Fowler, J. Am. Chem. Soc., 1979, 101, 6677–6682 CrossRef CAS; (c) S. M. N. Sieburth, CRC Handbook of Organic Photochemistry and Photobiology, CRC Press, 2004, 2nd edn, p. 103 Search PubMed; (d) S. C. Coote, Eur. J. Org Chem., 2020, 1405–1423 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, spectroscopy, crystallography, computational and characterisation details (PDF). CCDC 2333565, 2375152 and 2357172. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02597k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |