
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of responsive MRI probes in the past and the future of molecular imaging
Ping
Yue
a,
Thavasilingam
Nagendraraj
 a,
Gaoji
Wang
b,
Ziyi
Jin
b and
Goran
Angelovski
*a
a,
Gaoji
Wang
b,
Ziyi
Jin
b and
Goran
Angelovski
*a
aLaboratory of Molecular and Cellular Neuroimaging, International Center for Primate Brain Research (ICPBR), Center for Excellence in Brain Science and Intelligence Technology (CEBSIT), Chinese Academy of Sciences (CAS), Shanghai 201602, PR China. E-mail: goran.angelovski@icpbr.ac.cn
bSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, PR China
First published on 27th November 2024
Abstract
Magnetic resonance imaging (MRI) has become an indispensable tool in biomedical research and clinical radiology today. It enables the tracking of physiological changes noninvasively and allows imaging of specific biological processes at the molecular or cellular level. To this end, bioresponsive MRI probes can greatly contribute to improving the specificity of MRI, as well as significantly expanding the scope of its application. A large number of these sensor probes has been reported in the past two decades. Importantly, their development was done hand in hand with the ongoing advances in MRI, including emerging methodologies such as chemical exchange saturation transfer (CEST) or hyperpolarised MRI. Consequently, several approaches on successfully using these probes in functional imaging studies have been reported recently, giving new momentum to the field of molecular imaging, also the chemistry of MRI probes. This Perspective summarizes the major strategies in the development of bioresponsive MRI probes, highlights the major research directions within an individual group of probes (T1- and T2-weighted, CEST, fluorinated, hyperpolarised) and discusses the practical aspects that should be considered in designing the MRI sensors, up to their intended application in vivo.
 Ping Yue | Ping Yue obtained her master's degree in the organic chemistry at the Anhui University (China) in 2018. She joined the Laboratory of Dr Angelovski at the International Center for Primate Brain Research (ICPBR), Center for Excellence in Brain Science and Intelligence Technology (CEBSIT) of the Chinese Academy of Sciences (CAS) in Shanghai, in 2022. Ping's research focuses on synthesis and characterization of bioresponsive MRI contrast agents. |
 Thavasilingam Nagendraraj | Thavasilingam Nagendraraj received his PhD in 2023 at the Madurai Kamaraj University, Madurai, Tamil Nadu, India. He then joined the ICPBR, CEBSIT, CAS in Shanghai and is currently working as a postdoctoral researcher in the laboratory of Dr Angelovski. His research interests focus on the development of bioresponsive MRI probes that are sensitive to neurotransmitters. |
 Gaoji Wang | Gaoji Wang completed his PhD in 2020 at the Max Planck Institute for Biological Cybernetics (Tübingen, Germany) under the supervision of Professor Angelovski. Currently, he is working as an Associate Professor at Jiangsu University. His research focuses on the design and synthesis of MRI contrast agents. |
 Ziyi Jin | Ziyi Jin received her Bachelor of Science degree in chemistry from Baoding University (China) in 2023 and entered Jiangsu University (China) in September of the same year to pursue a master's degree. She is now studying in the joint research group of Professors Chen and Wang and her current research focuses on the construction of multimodal MRI contrast agents. |
 Goran Angelovski | Goran Angelovski studied chemistry at the University of Belgrade (Serbia) and obtained a PhD degree in organic chemistry at the University of Dortmund (Germany). In 2005 he has started his research on the responsive MRI probes for neuroimaging at the Max Planck Institute for Biological Cybernetics (Tübingen, Germany). From 2020, Dr Angelovski heads the Laboratory of Molecular and Cellular Neuroimaging at the ICPBR, CEBSIT, CAS in Shanghai as the Senior Investigator, aiming to develop functional molecular imaging methods to study brain physiology with bioresponsive MRI probes. |
1. Introduction
Magnetic resonance imaging (MRI) has emerged as a very important tool in biomedical research and is an essential diagnostic method in clinical radiology today. It is highly regarded due to its advantageous properties that allow non-invasive imaging over large regions of tissues and organs, with high spatial and temporal resolution. Typically, MRI is performed at the 1H frequency, using the resonance of water protons in tissues to generate the signal. However, due to its ability to acquire the signal from various NMR-active nuclei, it imparts outstanding versatility for studying numerous biological phenomena. Indeed, MRI is one of the most important molecular imaging techniques, enabling the tracking of physiological changes noninvasively and allowing imaging of biological processes at the molecular or cellular level.The use of MRI probes significantly improves specificity of the measurements, especially when performing functional investigations. To this end, a great expansion in the field of (bio)responsive probes has taken place in the recent years, significantly expanding the scope of MRI applications.1 These are substances with molecular sensor features, capable of changing their NMR and MRI properties along with local environmental alterations, such as reporting on changes in the concentration of ions or molecules that reside or fluctuate in their vicinity. The presence in biological system results in their response to the ongoing physiological process, which leads to reversible or irreversible change of MRI signal they generate or affect, thus making them essential functional markers.
Several approaches of successfully using bioresponsive probes in functional imaging studies have been reported recently, giving a new momentum to the field of molecular imaging, also the chemistry of MRI probes. Importantly, their development was done hand in hand with the dynamic progress in MRI methodologies, including improvements in the hardware suitable for studies at high magnetic fields or the development of emerging techniques such as chemical exchange saturation transfer (CEST) or hyperpolarised MRI.
Due to the important role that responsive MRI probes have recently demonstrated in the development of functional MRI and molecular imaging, this perspective intends to summarize the major strategies in their development and highlight the most successful directions within individual group of probes. We also discuss the practical aspects that should be considered, from designing the MRI sensor for a particular biological target, until its anticipated application in real conditions, i.e. in vivo. In this work, we do not provide an extensive list of different responsive probes reported to date, as they were summarized in several reviews published previously.1–5 Rather, we aim to provide a summary of different approaches and types of probes suitable for diverse functional MRI methods, which can show a wide scope of potential applications of the responsive probes. Hence, the classification of following sections has been presented based on the suitability of probes for specific MRI method (T1- or T2-weighted, CEST, 19F or hyperpolarized), while highlighting their applications within each section.
2. T 1- and T2-weighted bioresponsive MRI probes
MRI contrast agents (CAs), substances that affect the T1- and T2-weighted MR signals, are the most used MRI probes in clinical practice and research.6 They alter the longitudinal (T1) and transverse (T2) relaxation times of water molecules, thereby influencing the contrast in the resulting MR image.7 The ability of MRI CAs to alter the T1 and T2 relaxation times is expressed through the longitudinal (r1) and transverse (r2) relaxivity values, respectively. These parameters are defined as a normalized value of the change in relaxation rate (R1 or R2, which is inverse of T1 or T2, respectively) per millimolar concentration of the CA. Further, more powerful CAs tend to have higher relaxivity values, whereas the responsiveness of the MRI probe, discussed in this article, can be assessed by greater differences in the relaxivity values before and after the application of the stimulus or a biological target.Typical T1 agents contain paramagnetic metal ions such as Gd(III), Fe(III), and Mn(II), and primarily enhance MRI signal intensity on T1-weighted images.8 Aforementioned metal ions are used because they have multiple unpaired electrons in their electron shells, which impact water proton relaxation and exhibit excellent magnetization effects. However, due to the toxicity of these metal ions in vivo, they must be administered in the form of complexes that have high thermodynamic stability and kinetic inertness. At a molecular level, the relaxation properties of these complexes are derived from their ability to coordinate with water and exchange quickly with bulk water. Consequently, to obtain bioresponsive T1 probes that vary their longitudinal relaxivity along with changes in a specific biomarker in their microenvironment, it is necessary to modify at least one of the important parameters that affects r1 (Fig. 1a), in specific: (i) the number of inner-sphere water molecules (hydration number, q) that are directly coordinated to the paramagnetic metal ion; (ii) the residence time of the water molecule (τm); and (iii) the rotational correlation (tumbling) time of the CA (τR). However, it should be noted that the strategies to alter r1 should consider the operating field strength: changes in q lead to r1 alterations at all magnetic fields, as well as changes in τm; however, the strategies that rely on τR change have a strong influence on r1 only at low to intermediate magnetic fields (20–60 MHz).1
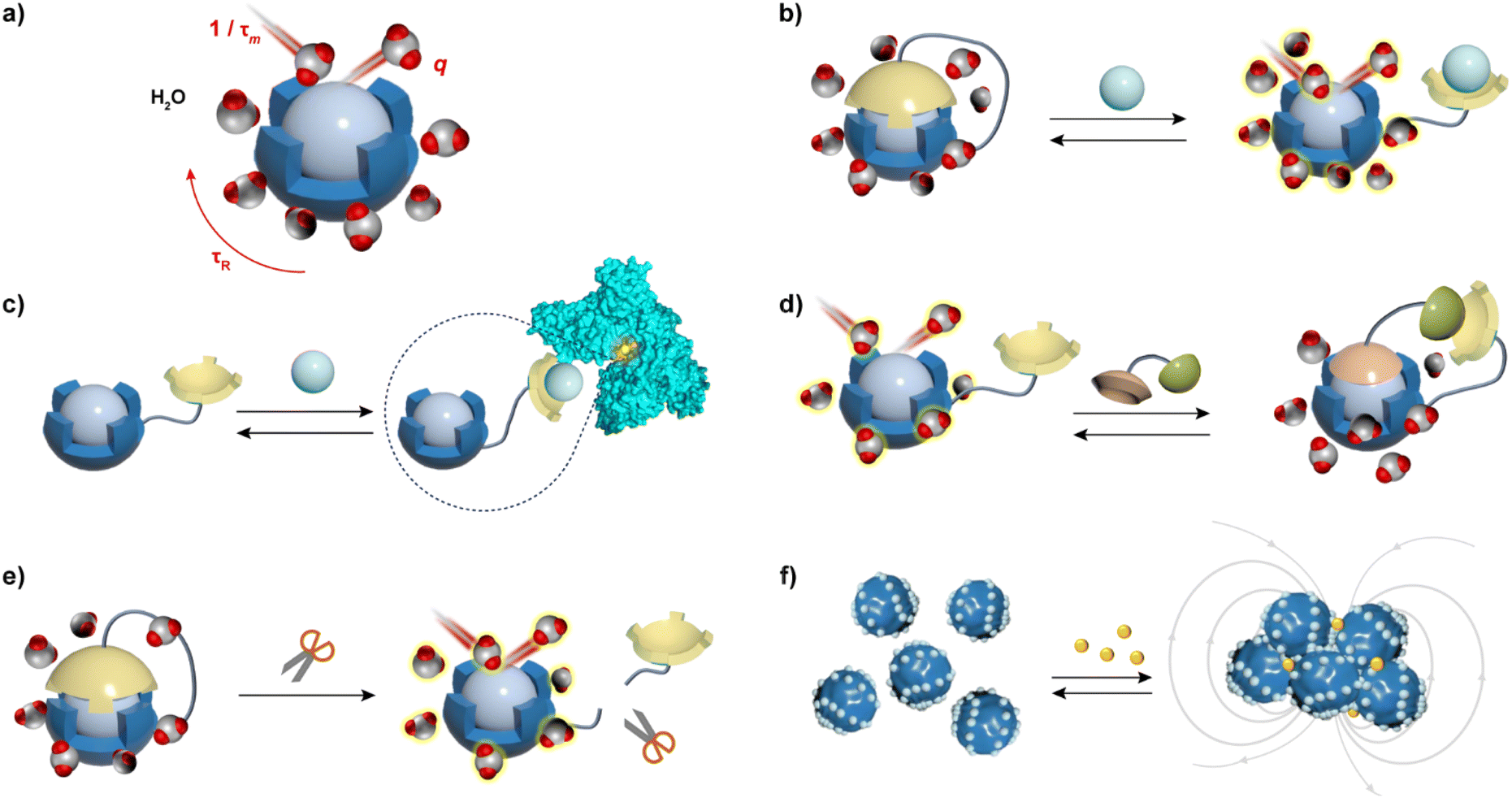 | ||
| Fig. 1 Main parameters that influence r1 and r2, and typical mechanisms that cause their changes in T1 and T2 bioresponsive MRI CAs. (a) Typical T1 agents are paramagnetic metal ion complexes and the key parameters that influence their r1 are: hydration number (q), mean residence time of bound water (τm) and rotational correlation time (τR) of the complex. (b) The recognition unit (yellow cap) of the responsive CA interacts with the target analyte (e.g. biologically relevant metal ions); the switch of recognition unit concurrently changes the q and r1 values of the CA. (c) The recognition unit on the responsive CA interacts with the target analyte, but also forms ternary complexes with large molecules, such as proteins. This leads to a decrease in τR due to formation of the high molecular weight species, which increases r1 at low-to-intermediate magnetic fields. (d) The fixed geometry and functional groups of the responsive CA interact with ditopic guests, such as zwitterionic amino acid neurotransmitters. In the absence of the analyte, water is coordinated to the paramagnetic metal ion. In the presence of the ditopic guest molecule, the formation of the ternary complex with the responsive CA decreases q, and hence the r1. (e) The enzyme activity causes the removal of functional and paramagnetic ion-coordinating group from the responsive CA, which results in increases of the q and r1 values. (f) Typical T2 CAs are composed of superparamagnetic nanoparticles with target-specific coatings on the surface. In the presence of the target analyte, the interaction between functional surfaces typically induces the aggregation of nanoparticles, which affects the τR of the agent and diffusion of water on the responsive CA surface, resulting in strong r2 changes. The processes described here lead to (b), (c), (e), (f) increase, and (d) decrease of the MRI signal. | ||
T 2 CAs cause significantly reduced signal intensity on T2-weighted MR images, most of which are based on the superparamagnetic iron oxide nanoparticles (SPION), small superparamagnetic iron oxide (SPIO) particles, and ultrasmall superparamagnetic iron oxide (USPIO) particles, while the uses of superparamagnetic particles based on Dy(III) or Tm(III) has also been reported.9,10 These CAs affect the r2 by influencing the diffusion of water molecules near their surface (Fig. 1f). The design strategy for T2 agents follows different principles than for T1 agents. The r2 changes mainly due to the local magnetic field inhomogeneities, which are mainly determined by two physicochemical properties: the agent's superparamagnetism and its tumbling time. Therefore, the size of the particles has a significant impact on the performance of CAs by influencing their tumbling time or water accessibility. These properties can further be adjusted by covering the particles with non-immunogenic coatings, to avoid their aggregation, improve biocompatibility and, in the case of bioresponsive CAs, provide the ability to be influenced by specific targets, resulting in r2 changes and hence the MRI signals.
Obviously, both types of bioresponsive T1 and T2 agents alter MRI signals indirectly, by influencing bulk water molecules through their inherent chemical properties. This effect can further be enhanced by triggering the stimulation processes through various endogenous biomarkers or changes in the CAs' microenvironment. Sources of the triggered signal may be caused by changes in the ion flux, enzyme activity, neurotransmitters, redox states, hypoxia, or other abnormal features that occur during various biological processes.
2.1. Triggering mechanisms valid for T1 and T2 MRI sensors
To develop a bioresponsive MRI probe that can alter T1 and T2 of water, one needs to be able to modulate its r1 and r2 values along with the desired stimulus, i.e. change in the concentration of the analyte or another type of change caused by the observed biochemical process (redox change, enzyme activity). The presentation of the stimulus triggers intra- and intermolecular changes of the bioresponsive MRI probe (Fig. 1), which causes its r1 and/or r2 relaxivity changes. This reflects on the MRI signal intensity, resulting in the change of the MR image contrast due to the interaction of the stimulus with MRI sensor and transduction of this recognition signal from the sensor moiety to the reporting MR unit. In turn, the MRI probe reports on the fate of the biological stimulus, through the sequence of physicochemical events that are concluded with the alteration of the MRI signal.Typical T1 responsive probes are molecules that chelate paramagnetic metals with molecular weight of usually up to ∼1.5 kDa, although they can also be replaced with proteins as chelators or can exist in form of macromolecular nanomaterials loaded with paramagnetic monomeric units. To obtain changes in relaxivity r1, changes in one or more key parameters (q, τR, τm) must be generated after the stimulus presentation, e.g. the analyte recognition. The relaxivity r1 changes are mainly achieved through the following strategies: (1) upon the analyte stimulation, the paramagnetic centre of the responsive probe switches between ‘open’ and ‘closed’ states, changing the degree of exposure of the paramagnetic centre to bulk water, which causes the (ir)reversible change in q value (Fig. 1b, d and e); (2) the binding of the CA to the analyte changes the CA conformation or forms ternary complexes with e.g. proteins or macromolecules. This increases the molecular weight of the probe and rigidifies its structure, thereby affecting its τR values, hence the r1 (Fig. 1c). (3) either of the events additionally affects the τm, which also influences the r1 value; (4) if the bioresponsive probes are based on nanoparticles (NPs), their 3D structures may significantly change upon the interaction with the stimulus, which may affect one or more key parameters that alter r1 (e.g. aggregation or disaggregation is significantly changing the access of water molecules to the paramagnetic centres).
T 2 agents respond to the target analytes differently than the T1 agents, hence the applied design strategies differ substantially. The alteration in r2 is predominantly reliant on variations in size and aggregation state of magnetic nanoparticles (NPs). Hence, the bioresponsive T2 agents have designated functional coatings on the surface of particles that exhibit excellent biocompatibility. These coatings are designed to respond to the stimulus, e.g. bind specifically to the analyte, which typically induces aggregation or an aggregate decomposition, ultimately changing the r2 value (Fig. 1f).
Finally, stimulus-induced changes in r1 and r2 generally go hand in hand with each other, although they can change with different rates depending on the size of the probe or applied triggering mechanism. Namely, small-size probes typically experience linear change in r1 and r2, in which case their T1-weighted component is dominant in MRI experiment. For NPs, the situation can be quite different – the change in r2 can be larger than that of r1, which can be exploited during the MRI acquisition. Namely, the r2/r1 ratio also changes with the stimulus in such case, opening additional possibilities for studying the target biological processes. This so-called ratiometric methodology may then enable easier analysis of results since the analyte quantification can be applied.11–14
Guided by the above principles, many types of bioresponsive probes for different targets were developed, while the number of targets (i.e. stimuli) continues to expand, as well as the use of different mechanistic principles or types of recognition or reporting moieties. The following content intends to elaborate cases of various strategies applied until now, although it will not summarize all of them due to a high diversity.
2.2. pH-based bioresponsive MRI probes
Balanced pH levels play an important role in many biological processes, which are particularly susceptible to proton concentration fluctuations and do not have a uniform pH throughout the body. In living organisms, the CO32−/HCO3− buffer primarily governs the pH range in the blood, ranging between 7.36 and 7.42.15 In this regard, several methods have been developed over the past few decades to detect pH fluctuations. Among these imaging approaches, fluorescence imaging allows investigations of pH changes at low micromolar concentration levels of probes.1,16 However, observational MRI provides a high-resolution image and deep penetration into the tissue for mapping the pH. Hence, different types of metal complexes and NPs were developed as responsive CAs for functional MRI-based techniques that study pH fluctuations.A typical example of the reversible pH-responsive probe is a Gd(III) complex with a 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid (DO3A), anchoring free terminal amine (probe 1) and the methylamine (probe 2) (Fig. 2a). In the basic medium, the amine groups from the DO3A coordinate with the Gd(III) metal centre, resulting in a lower hydration state. When pH of the medium changes to acidic, amines protonate, gaining access of water to the paramagnetic metal centre (q increases), which also increases r1, revealing the off–on response to pH.17 Further responsive CAs of this class were decorated with nitrophenol 3, sulfonamides 4–6 and alkylphosphonates 7–10, or tetraamides 11–12 and linear-length chain hydrophilic units along with DO3A to affect the q number or water exchange, respectively, and hence to achieve the pH-sensitive r1 changes.18–24
 | ||
| Fig. 2 Bioresponsive MRI probes sensitive to pH T1- and T2- weighted MRI experiments in vivo with these probes. (a) The structures of 1–16 that are responsive to pH. (b and c) The mechanism responsible for pH-dependent r1 changes of (b) MSN-NPs and (c) polymer-NP-based responsive CAs at neutral (left) and acidic (right) conditions. (d) T1 and T2 maps of NP-SSM-PEG-bearing mice (left) and corresponding R1 and R2 values (middle and right) at tumor sites at different time points. (e) T1- and T2-weighted MR images in vivo with NP-SGM-bearing VX2 tumors in rabbits. (f) Schematic illustration of the assembly and disassembly of the i-motif linker under different pH conditions switching from T2 to T1 effects (top) and corresponding in vivo small hepatocellular carcinoma diagnosis using responsive and irresponsive ultra-small iron oxide nanoclusters (bottom). Adapted with permission from (d) ref. 41 Copyright © 2022 Springer Nature, (e) ref. 42 © 2015 Elsevier, (f) ref. 45 Copyright © 2018 American Chemical Society. | ||
Other noteworthy high-paramagnetic transition metals are Mn(II/III) and Fe(III), which are abundant endogenous metals in the human body and are required for various biological functions. T1 agents based on the complexes of these metal ions have attracted much attention in recent years because of the safety concerns related to the nephrogenic systemic fibrosis disease.25 In analogy to the previously mentioned Gd(III)-based probes, a typical strategy used for the complexes 13–15 based on Fe(III) and Mn(II) was to reversibly coordinate pendant arms to the metal centres, which will de-coordinate with the change in pH, resulting in the alteration of r1.26,27 Using another principle, catechol-based Fe(III) complexes 16 showed an r1 increase of 130% and 76% in tumour and muscle regions, respectively, due to the dissociation of the agent in acidic pH. Subsequently, the probe was used to distinguish the tumour boundary in vivo in BALB/C mice.28
NPs were a frequent choice for the development of pH sensitive probes, since they exhibit features for cancer diagnosis due to their ability to accurately detect malignancies by combining single or multiple imaging components in one or multiple modalities.29 To this end, dendrimer-based NPs, combined with a macrocyclic and pH-sensitive probes were among the first NPs to be studied for this purpose. A series of studies was performed on the generation 5 poly(amidoamine) (PAMAM) dendrimer and a phosphonate-based bioresponsive monomer. Different combinations of the agent alone, with the pH-unresponsive or rhodamine-containing probes were tested to exploit the on–off responsiveness to pH.30–32
Besides dendrimers, various types of NPs were investigated as pH sensors for MRI. Mesoporous silica nanoparticles (NP-MSN) provide effective doping with Gd(III) chelates in the exterior pore channel, leading to decreased tumbling or water exchange rates while maintaining bulk water access (Fig. 2b). Recently, manganese oxide (MnO) was dispersed and coated with MSN individually showing the advantage of signal enhancement in acidic environment. It was successfully demonstrated in vivo in rabbits and mice respectively.33,34 Encouraged by these findings, the further modified monodispersed mesoporous manganese silicate-coated with silica NP (MMSSN) exhibited a 80% increase in r1 from pH 7.4 to 5.4 and a doubling of r1 upon interaction with glutathione (GSH), followed-up with an in vivo MRI testing in a mouse MDA-MB-231 xenograft model.35 Another way of incorporating manganese silicate, MnO core and Gd(III) chelates into NPs was using the polymers based on polyethylene glycol (PEG), poly(lactic-coglycolic acid) (PLGA), poly(acrylic acid) (PAA) and poly(styrene)-block-poly(acrylic acid) (PS-b-PAA) (Fig. 2c). Typically, these polymers tend to form micelle NPs when the pH changed from acidic to neutral; this process resulted in the lack of water interaction with paramagnetic ions, thereby decreasing r1. In addition, metal organic frameworks (MOF)-based Gd(III) chelates with surface block polymer showed a collapsed hydrophilic state at high pH and a prolonged hydrophilic state at low pH.36–40
Taking the alternative preparation routes, multimeric and multifunctional responsive CAs with highly beneficial properties in vitro and in vivo were developed as agents for dual use, namely T1- and T2-weighted MRI. A set of core/shell/shell models of superparamagnetic iron oxide–silicon dioxide–manganese dioxide (SPIO–SiO2–MnO2) (NP-SSM) nanosystems have been created, comprising an SPIO NP core surrounded by SiO2 and MnO2 made as shells. The PEG-attached SSM NPs are in the ‘dual quench’ condition displaying weak T1 and T2 contrasts at physiological conditions, whereas the dual activation took place at acidic condition to demonstrate the use of these NPs in differentiating between normal and diseased tissue in three different animal models (Fig. 2d).41
When a hybrid nanogel system functionalized with glycol chitosan, acrylic acid, and biotin is prepared, it can self-assemble with SPIO NPs (6 nm) and be decorated with MnOx to provide dual T1 and T2 probe for MRI. After injection of the probe in the VX2 tumour rabbit model, the results showed a significant enhancement of tumour tissue signal on the T1-weighted MR images (18%) and a darkening of the signals on the T2-weighted MR images (45%) after 50 minutes (Fig. 2e).42 In addition, SPIO-MnOx loaded graphene nanosheets and polylysine polymer coated on SPIO NPs were found to be more biocompatible and effective in vitro and in vivo in targeting the tumor acidic microenvironment.43,44
Finally, several approaches in the design of dual (T1 and T2) activated pH NP-based sensors attempted to take advantage of the ratiometric (T1/T2) response. This aimed to exploit the differences in the amplitude of T1 or T2 changes within the same probe, but also to materialize the possibility of simplifying the calculations by eliminating one of the factors that influence the MRI signal – concentration of the probe.13 To this end, a probe comprising of ultra-small iron oxide nanoclusters and i-motif DNA linkers was developed (Fig. 2f). This nanocluster dissociates from the assembly, thereby decreasing r2/r1 relaxivity ratio, resulting in prompt changes to be detected in MRI under acidic conditions. This ratiometric approach was validated in vivo to detect early hepatocellular carcinomas.45 In addition to the iron oxide NPs, incorporating ZIF-8 exhibited pH and redox-responsive T2–T1 switch in MRI signal, which can be stimulated by an acidic environment and GSH.46 Interestingly, core–shell NPs were designed from a silica NP core incorporating Gd(III) chelates and then coated with poly(MAA-co-MBAAm) polymer by a precipitation method. The shrinking and swelling of this NPs resulted in a pH-insensitive r1, but pH-sensitive r2 probe, respectively.47
2.3. Metal ion-responsive probes
Development of MRI probes sensitive to metal-ions, has attracted a lot of attention upon the great achievements in the field of fluorescent sensors and switches.48 To this end, various types of metal-responsive probes were reported meanwhile. In this section, we focus our discussion on the divalent metal ions Ca(II), Zn(II) and Cu(II), which have the highest biological relevance and physiological availability that can allow detection with MRI.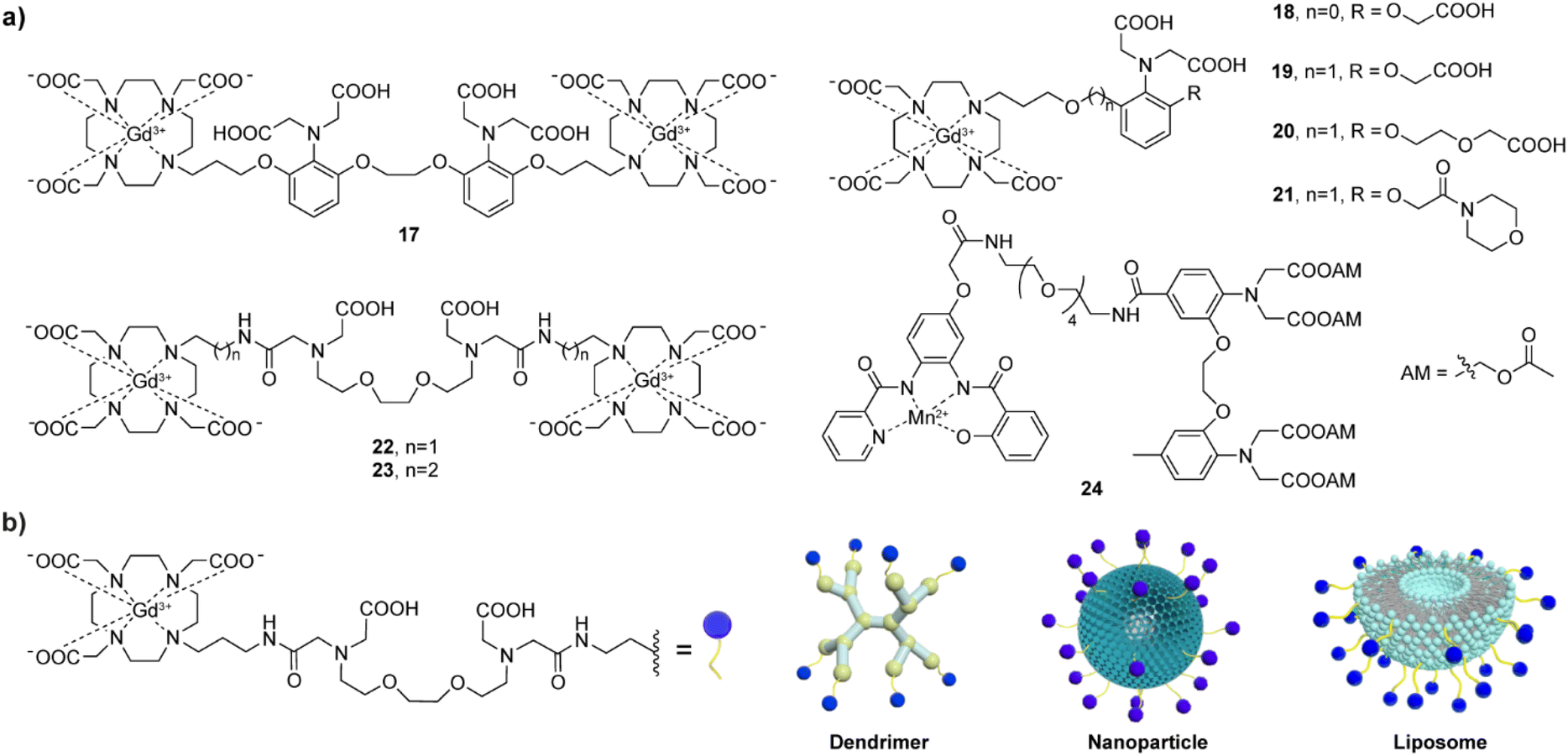 | ||
| Fig. 3 Calcium-responsive CAs. (a) Selected T1-weighted bioresponsive CA structures 17–24, which are based on paramagnetic Gd(III) (17–23) and Mn(II) (24). (b) Schematic illustrations of the Ca(II)-sensitive nanosized CAs, with a dendrimeric core, siloxane core–shell particle core, or a liposome formulation. Before coupling to the nanocarrier surface, structural modifications must be performed on the responsive CA unit (left) to allow the conjugation. | ||
In a parallel research effort, the bismacrocyclic probes 22–23, which use a chelator based on the EGTA (ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid) were explored in depth.51–53 Those probes can elicit a ‘doubled signal’, i.e. a relaxivity response from two Gd(III) concurrently, after chelating one equivalent of Ca(II). The affinity of 22–23 for Ca(II) is reduced by converting one of the carboxylates on each terminal side of the EGTA into amide groups and using them to conjugate to DO3A with appropriate linkers. The probes exhibited 69% and 83% relaxivity enhancement per equivalent of Gd(III) in the presence of Ca(II) for the ethyl and propyl linker, respectively,51 while this effective molecular structure was further functionalized using a solid-phase synthesis methodology, resulting in more biologically potent and target-specific probe.54 However, the bismacrocycle 23 was shown to have a significant potential for in vivo applications, after being used as a sensor probe to monitor the fluctuation of extracellular Ca(II) during cerebral ischemia caused by the transient middle cerebral artery occlusion (tMCAo). In this study, 23 was continuously infused into the somatosensory cortex of the rat, while the MCAo-induced ischemia and the subsequent tissue reperfusion was controlled remotely. The MRI signal experienced immediate changes that followed changes in extracellular Ca(II) concentrations, while such change was not observed in the calcium-insensitive control probe experiments (Fig. 4a).53
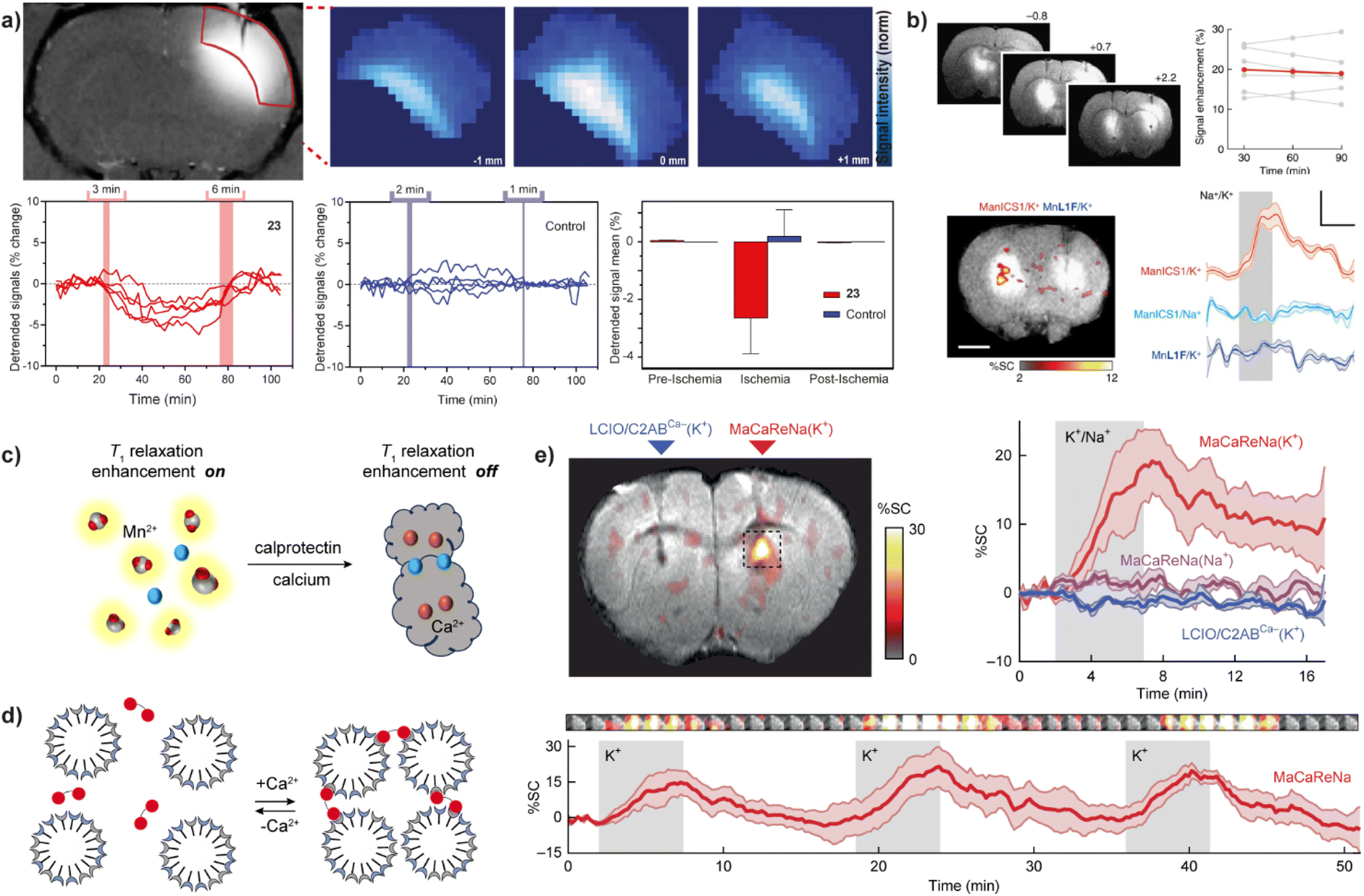 | ||
| Fig. 4 Calcium-responsive probes (second part). Functional MRI experiments in vivo with Ca(II)-sensitive CAs. (a) MRI images of probe 23 in the rat somatosensory cortex (upper left) and the regions of interest (ROIs) with signals enhanced by a paramagnetic probe <3 mm away were recorded and analyzed (upper right), while transient cerebral ischemia was induced via occlusion of the middle cerebral artery. The collected signals showed significant signal fluctuations using probe 23 (lower left) compared to the control probe that is Ca(II)-insensitive (lower middle) during the pre-ischemia, ischemia, post-ischemia periods. Average values of the detrended signals indicate that only 23 can detect changes in the Ca(II) concentration (lower right). (b) The use of probe 24 to detect neural activation in rat brain. Infusion of 24 or the control agent (left and right, respectively) into the rat striatum resulted in the signal enhancement (upper left), with an average MRI signal increase by 20 ± 2% (upper right); under K(I) stimulation, only the brain area perfused with probe 24 showed an increase in MRI signal (lower left). The time course of mean MR signals exhibited significantly greater changes during K(I) stimulation with probe 24 then both control experiments using Na(I) stimulation with probe 24 or K(I) stimulation with the control probe (lower right). (c) Mechanism of Ca(II)-triggered T1-weighted MRI change of the NP-calprotectin; in the presence of Ca(II), NP-calprotectin prevents the contact between free paramagnetic Mn(II) and water, reducing MRI signals. (d) Mechanism of Ca(II)-triggered T2-weighted MRI change of the probe NP-MaCaReNa. In the presence of Ca(II), the C2AB moieties (red) induce the aggregation of nanoparticles prepared by coating oleate-stabilized magnetic iron oxide cores with a mixture of PC and PS (grain), changing MRI signals. (e) MRI experiments with NP-MaCaReNa in living rat brain. MRI signal increases in the presence of NP-MaCaReNa but not controls (right and left, respectively)(upper left). The time courses of MR signals near areas of contrast-agent delivery show that K(I) infusion (grey shading) in the presence of NP-MaCaReNa induces clear responses (red trace), whereas controls barely affect the MRI signal (upper right). The average time courses of three consecutive K(I) stimuli (grey shading) show the reproducibility of NP-MaCaReNa responses (bottom). Adapted with permission from: (a) ref. 53 Copyright © 2019 National Academy of Sciences, (b) ref. 59 Copyright © 2019, The Author(s), (e) ref. 63 Copyright © 2018, The Author(s). | ||
A demand to improve practical features of Ca(II)-responsive MRI probes and make their studies in vivo feasible inspired further studies and development of a range of DO3A-based complexes coupled to peptides or nanosized carriers.55–57 This optimization can effectively slow down the diffusion rate of the probes and increase their retention (Fig. 3b). To this end, the effective structures of the bismacrocycle probes 22–23 have been modified by converting them to the EGTA-containing monomacrocyclic units with the aryl isothiocyanate group that can be coupled to dendrimer- or siloxane-based NPs. The effectiveness of this strategy was subsequently tested in vivo in the rat cerebral cortex, and the results showed that probes bound to dendritic molecules had much slower diffusion in vivo than small molecule probes.55 Moreover, the combination of the probe and siloxane NPs effectively detected changes in MRI signal intensity in the renal pelvis and renal parenchyma to follow the Ca(II) changes, which was the first proof-of-concept validation of Ca(II)-sensitive MRI probes in vivo.56 Using a similar modification method, one of the GdDO3A units from the bismacrocycle was replaced with lipophilic long carbon chain to allow its incorporation into a liposome formulation with good pharmacokinetics and biocompatibility. An appreciable increase of ∼420% in r1, from 7.3 mM−1 s−1 to 38.1 mM−1 s−1 at 25 °C and 21.5 MHz in the absence and presence of Ca(II), respectively, was obtained with this method.58 The relaxation change observed is a result of both the inherent q changes of small molecule probes and the τR changes caused by restricted local rotational motion after liposome binding.
Using entirely different paramagnetic centre, Mn(II)-based T1 contrast agent 24 has also been developed. In this case the bioresponsive probe exhibited excellent cell permeability, hence it was designed to detect intracellular Ca(II) fluctuations. This MRI sensor consists of an Mn(II)-based complex and a BAPTA-based calcium chelator, which is modified by acetylmethoxy (AM) ester groups to enable crossing the cell membrane. Upon cell internalization, esters are hydrolysed in vivo, activating the BAPTA and its coordination with Ca(II) to cause the MRI signal changes.59 When 24 is infused into the rat striatum and the region rich with the MRI sensor is stimulated, a substantial T1-weighted MRI signal enhancement can be recorded, which persists for over 90 minutes. The average T1-weighted signal enhances ∼20% due to probe infusion and corresponds to 19 μM calcium-free concentration in tissue (Fig. 4b).
The same type of paramagnetic metal was used to develop a calprotectin protein-based T1, T2-dual modal agent NP-calprotectin. When Ca(II) is present, their binding to calprotectin shields Mn(II), thereby blocking their impact on water and weakening MRI signals (Fig. 4c). The in vitro tests in hippocampal cell lysate reveal an increase of 18.7 ± 2.5% in T1 and 77.5 ± 1.5% in T2 with a fixed amount of calprotectin and Mn(II) at 40 μM and 30 μM, respectively.60 Although this relaxation time change is comparable to other types of CAs, the use of this bioresponsive probe to follow Ca(II) signalling in the body remains to be demonstrated. The same is valid for the generation 4 (G4) dendrimer-based nanosized probe of the previously discussed class (Fig. 3b), which can change both T1 and T2 along with changes in Ca(II) concentration. Besides the standard q change that induces T1 effect, even greater T2 effect is caused by swelling and shrinking of the dendrimeric probe in the absence and presence of Ca(II), respectively. A Ca-dependent difference in T2/T1 ratio was used to demonstrate a possibility of following Ca(II) concentration changes by employing a balanced steady-state free precession imaging protocol.61 Besides being independent of the probe's concentration (see above the discussion on ratiometric pH-sensitive probes), this protocol can also rapidly report on concentration changes, as it can record MRI signal much faster compared to the standard T1- and T2-weighted imaging protocols.
Finally, the Ca(II)-sensitive MRI probes suitable exclusively for T2-weighted MRI used magnetic NPs as materials originating from calcium-binding protein calmodulin, which was used as coating for SPIO NPs to associate and dissociate reversibly as function of Ca(II) concentration.62 More recently, its improved version based on the synaptotagmin protein was developed and reported. This highly cooperative sensing mechanism utilized a natural Ca(II)-sensitive protein that is a component of the synaptic neurotransmitter-release machinery, and phosphatidylserine-containing lipid vesicles, which drive clustering of the probe when exposed Ca(II) concentrations above 0.2 mM. Hence, the NP-MaCaReNa sensor was prepared by mixing lipid-coated iron oxide nanoparticles (LCIOs) with the fused Ca(II) sensitive domains of synaptotagmin (Fig. 4d). The transverse r2 relaxivity of NP-MaCaReNa increased from 151 ± 15 mM−1 s−1 to 261 ± 21 mM−1 s−1 when Ca(II) concentration changed from 0 mM to 1.2 mM in vitro. Subsequently, selected rat brain areas infused with NP-MaCaReNa showed an MRI signal change following dynamic Ca(II) fluctuations induced by K(I) or electrical stimuli, to allow bright perspectives for monitoring extracellular Ca(II) signaling of diverse biological phenomena (Fig. 4e).63
The first reported Zn(II)-sensitive CA, 25 (Fig. 5a), was based on the GdDTPA (Gd-diethylenetriaminepentaacetic acid) and served as the MRI reporter, tethering two bis-pyridyl-ethylamine (BPEN) units as the chelators for Zn(II).64 To acquire a more appropriate probe, a series of GdDO3A based probes 26–29 (Fig. 5a) were explored to establish an effective mechanism of Zn(II) binding and reporting of this event through the MR-active unit.65,66 The results indicated that effective r1 response can be achieved if two requirements are met. Firstly, the structure of the MRI sensor should contain at least one aminoacetate group that will coordinate with Gd(III) in absence of Zn(II), to lead to a low starting r1. However, another functional group should effectively bind with Zn(II) together with the aminoacetate, to obtain signal changes. The most prominent example of using this q-modulated mechanism is probe 30. It consists of di-(2-picolyl)amine (DPA) as the Zn(II) recognition moiety and GdDO3A as the MRI reporter, which are spaced with a modified tyrosine (Tyr) unit. This structure involves the addition of a carboxyl group to the phenolic moiety of Tyr, providing a critical collaborating assistance to DPA for Zn(II) recognition. In detail, when Zn(II) is absent, the phenoxyacetic acid group coordinates with Gd(III), preventing water from approaching Gd(III), thus producing a low initial r1 value. In the presence of Zn(II), the phenoxyacetic acid group undergoes a coordination to Zn(II) together with the DPA group, releasing the coordination space of Gd(III) for water, and resulting in a significant increase in r1. The corresponding variations in r1 reach ∼400% or ∼300%, at 25 °C or 37 °C and 7 T, respectively.67 Interestingly, an analogous probe 31 that has carboxyl group replaced for the methyl ester on the tyrosine exhibits weaker r1 relaxation changes, which may be due to the higher tendency for aggregation in the presence of Zn(II).68 Nevertheless, this platform based on the DPA with the Tyr linker provides great potential for future studies of biological role of Zn(II) in vivo.
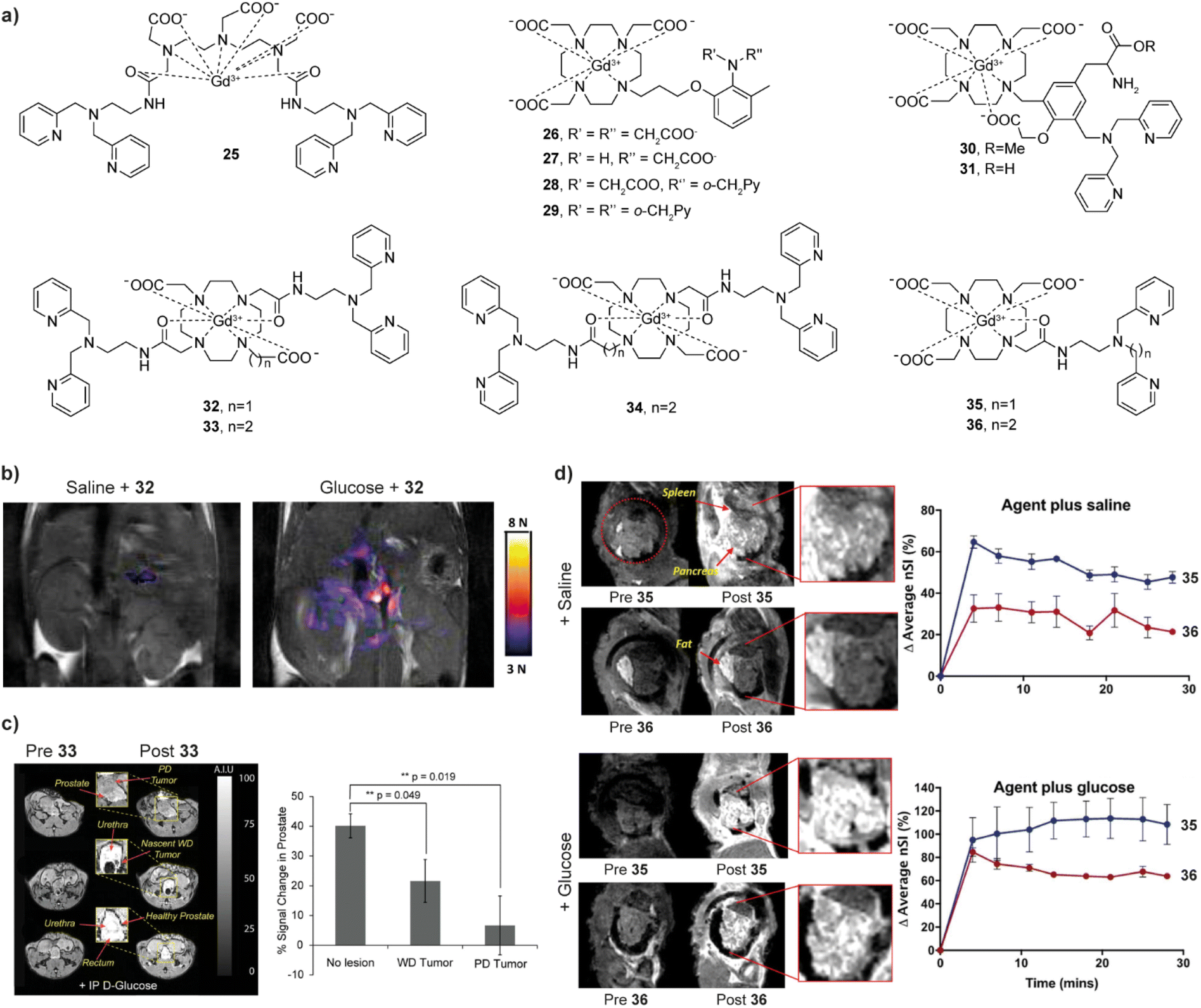 | ||
| Fig. 5 Zn(II)-sensitive CAs and functional MRI imaging experiments in vivo with these probes. (a) The structures of Zn(II)-sensitive CAs 25–36. (b) MR images of the probe 32 before (left) and 10 min after administration of glucose (right) in the pancreatic tissue of mouse, which causes the release of Zn(II). (c) MRI in vivo of the prostate during various states of tumor development before and after the administration of probe 33 (left). The average signal enhancement results indicate the lower MR signal intensities in the tumor cells, which is related to progressively lower Zn(II) secretion (right). (d) MRI in vivo of Zn(II)-sensitive CAs with different affinities. Saline was co-injected with the probes in the controls (upper panels), whereas Zn(II) was released after co-injection of glucose with the probes (lower panels). The difference between the two probes shows that the high affinity probe 35 produces a larger background signal, while the low affinity probe 36 produced higher contrast images after glucose injection. Adapted with permission from: (b) ref. 70 Copyright © 2011 National Academy of Sciences, and (c) ref. 72 Copyright © 2016 National Academy of Sciences and (d) ref. 73 Copyright © 2018 American Chemical Society. | ||
Another set of very important studies on Zn(II) sensing was made using macrocyclic Gd(III)-based systems where the r1 is changed after the probe interacts with HSA.69 In this case, the probe 32 is based on a GdDO2A motif (DO2A – 1,4,7,10-tetraazacyclododecane-1,7-diacetic acid) and uses two BPEN units to bind Zn(II) and further interact with HSA. Although the structure of the MRI probe is somewhat similar to that of 25, it exhibits moderate relaxation enhancement after binding to Zn(II). However, when the probe forms a complex with Zn(II), its affinity for binding to HSA increases, which subsequently supports formation of a ternary complex with large molecular weight, and the r1 significantly increases (165%). In vitro studies at 9.4 T have shown that the probe can detect Zn(II) concentrations as low as 30 μM, while it maintains selectivity for Zn(II) versus its physiological competitors Ca(II) and Mg(II). Further in vivo studies have been successfully applied to glucose stimulated co-release of insulin and Zn(II) in the pancreas.70 When the mice is injected with glucose, significant signal enhancement can be observed in the region corresponding to the pancreas, which is caused by the Zn(II) release and its detection by 32 (Fig. 5b). Hence the probe successfully visualised the process of insulin release from β-cells by detecting the change in Zn(II) concentration, which is of great importance for studying the pathological changes in diabetes. Based on this study, the structure of the probe was optimized, to improve the water exchange rate. The r1 value of two new sensors 33 and 34 after binding to Zn(II) and HSA increased by ∼3 times (0.47 T) if compared to the original sensor,71 allowing in vivo experiments using 33 to confirm the abnormal Zn(II) secretion levels in mice with prostate malignancy (Fig. 5c).72 Finally, the performance of two more stable and easily synthesized DO3A monoamide derivatives 35 and 36 for in vivo detection has also been studied. These probes are the structural analogues of 32, although possessing a single-sided Zn(II) BPEN recognition unit conjugated to the Gd-DO3A using different linkers to adjust the binding affinity to Zn(II). In turn, then the sensor probe extends the linker from methyl to an ethyl, the affinity for Zn(II) decreases due to the increase of distance between the Zn(II) chelating and MR reporting moieties. When used in vivo, the low affinity probe 36 displays better imaging effects due to low background signal and can be used to image “hot spot” areas of insulin secretion in the stimulated pancreas (Fig. 5d).73
Besides these most prominent examples mentioned above, a few more approaches were attempted to develop Zn(II)-sensitive probes. In the interest of presenting this topic on concisely, further examples are not going to be discussed in details but can be studied from specialized literature.74
The first-generation Cu(II) sensitive probe 37 (Fig. 6a) used iminodiacetic acid as the receptor. This hard anionic carboxylic acid donor is in a closed state of coordination with Gd(III) in the absence of Cu(II). After binding with Cu(II), it allows bulk water to enter the space of the Gd(III) inner sphere, generating 41% enhancement in r1.77 The second-generation Cu(II) sensitive probes 38–42 (Fig. 6a) introduced a thioether neutral receptor, greatly improving the probe's specificity for distinguishing Cu(I)/(II). Compared to the first generation probe, which has a moderate relaxation change and cannot report on Cu(II)-changes at 10 times excess of Zn(II), the 38–42 probes can effectively detect Cu(I), with 38 and 39 reaching a relaxation change of up to 360% at Zn(II) excess.7842 is equally sensitive to Cu(II) and Cu(I), demonstrating the ability to detect biological copper pools with a maximum triggering of 73% relaxation enhancement.
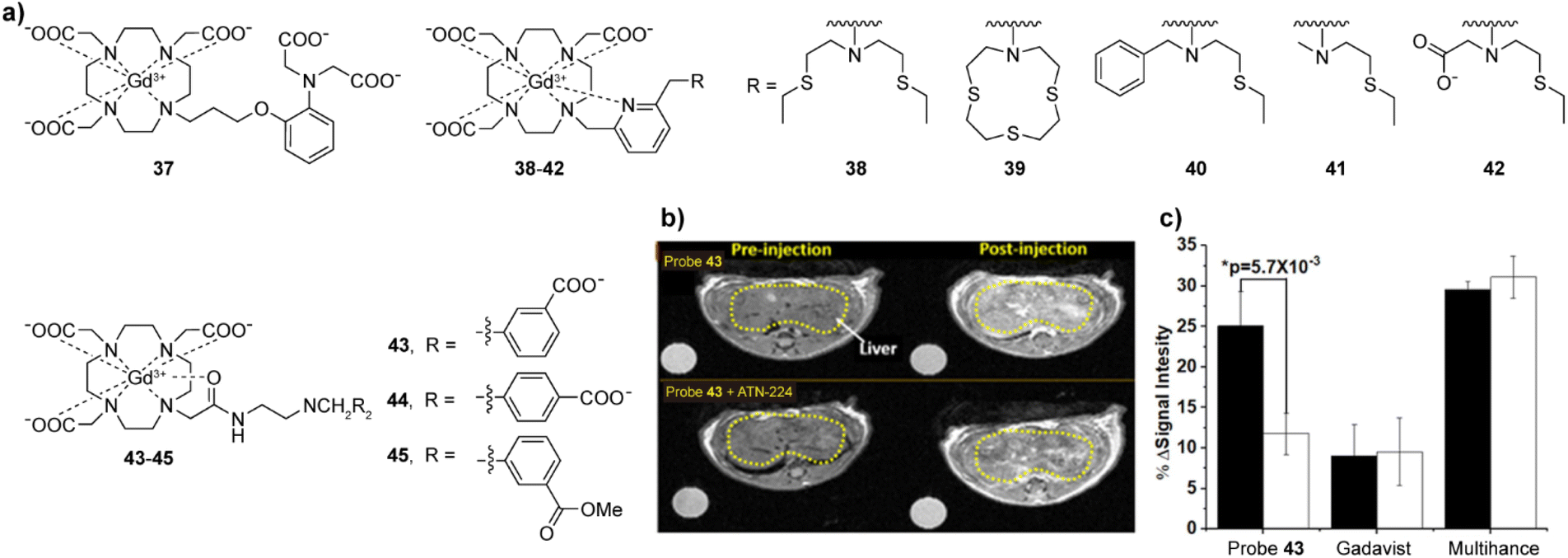 | ||
| Fig. 6 Cu(I)/(II)-sensitive CAs and functional MRI experiments in vivo with these probes. (a) The structures of probes 37–45 that are sensitive to Cu(I)/(II). (b) MRI in vivo of the wild type mouse pre- and postinjection of probe 43 without (top) or with (bottom) pretreatment with ATN-224 (the copper ion chelator). The decreased intensity reflects a reduction in freely available Cu(II) in liver. (c) The average MR signal intensity of mouse liver increases after injection of probe 43 in control mice (black bars) versus mice pretreated with ATN-224 (white bars). Mice imaged with Gadavist (an extracellular agent) and Multihance (a hepatobiliary agent) was used as control. Adapted with permission from ref. 79 Copyright © 2019 American Chemical Society. | ||
The first probe that was successfully used for the detection of Cu(II) in mice was 43. Similar to the detection of Zn(II), the combination of the probe with copper ions and the formation of a ternary complex with HSA results in a further increase in r1.79 The recognition function of 43 is achieved through the use of a bis(benzoic acid)methylamine recognition motif. In the presence of Cu(II), the r1 value increases 43%, which is speculated to be due to weakened hydrogen bonding between the inner-sphere water molecules exchanged with Gd(III) and the linker of the recognition group, affecting the exchange rate of water molecules. Nevertheless, the addition of Cu(II) in the presence of HSA increases the r1 to 270%, due to the Cu(II) binding to the N-terminal site of HSA, and formation of a stable ternary complex that leads to slower molecular rotation. Interestingly, Zn(II) can also interact in a similar manner, albeit the r1 change is twice lower than the probe's response to Cu(II). At the same time, two analogous of 43, probes 44–45, did not show significant recognition of Cu(II), indicating that positions of carboxylic acid groups in the final structure and their charges (carboxylates vs. esters) are crucial in the design of sensors. In turn, the potential of 43 for imaging extracellular Cu(II) levels in vivo has been studied. After the intravenous injection, a 25% T1 weighted image enhancement was observed in the liver area of healthy mice, which was reduced by more than 50% in the control group treated with Cu(II) at 4.7 T (Fig. 6b and c).
2.4. Redox and hypoxia-sensitive MRI probes
Changes in the concentration of biologically relevant ions and molecules such as hydrogen peroxide, GSH, cysteine (Cys) and oxygen in cells can cause an imbalance in redox state of the organism and hypoxia, an oxygen insufficiency, and hence many diseases. Maintenance of extracellular and intracellular redox processes, which are closely regulated in all organisms, plays a vital role in governing cellular redox balance, while an elevated production of reactive oxygen species (ROS) is linked to many significant cellular processes leading to proliferation, differentiation, apoptosis, and aging.80,81 Oxygen is also important to humans: low or high oxygen concentrations promote hypoxia or hyperoxia, respectively, where the former increases the production of ROS and accumulation of reduced forms of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2).82,83 To address these issues, developing a redox- or hypoxia-sensitive MRI probe appears as an adequate strategy for detecting an early-stage illness.There have been different approaches attempted to date. A typical example of a reversible redox-activated probe based on Gd(III) is GdDO3A bound to an acyclic merocyanine form 46 (Fig. 7a), which converts into the spirocyclic form upon the interaction with NADH. This causes a decoordination of the phenoxide group from the Gd(III) centre, allowing water molecules to coordinate to Gd(III), which results in an increase in r1.84 Likewise, the nicotinamide-based GdDO3A complex 47 exhibits higher q and hence r1 in its reduced form. When oxidized, the positively charged nicotinimidium attracts bicarbonates from solution, which coordinate to Gd(III) thereby reducing q and r1.85
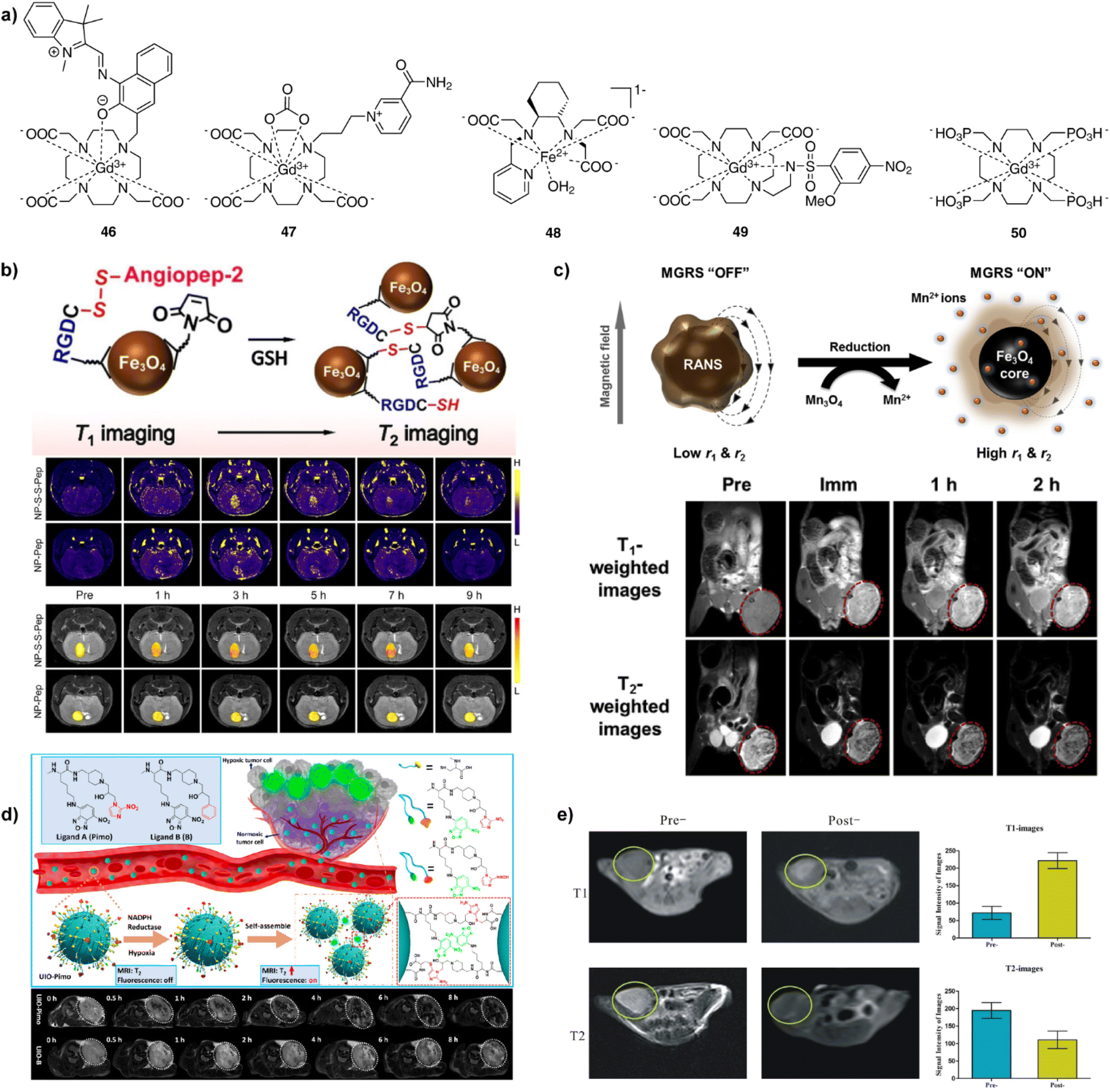 | ||
| Fig. 7 Redox- and hypoxia-sensitive CAs and MRI experiments in vivo with these probes. (a) The structures of redox- and hypoxia-sensitive MRI probes 46–50. (b) Schematic illustrations of cleavage of S–S bond, followed by the GSH-induced agglomeration of the NP-S-S-Pep probes (top). GSH levels were determined in the brains of mice bearing orthotopic U87MG glioma xenografts using NP-S-S-Pep and NP-S-Pep, administered at various time intervals before and after injection. It takes 3 h for the probes and hence the T1-weighted MR signal to be evenly dispersed throughout the brain region. In addition, the T1 signal was unevenly distributed at 9 h after injection (middle). Similarly, the T2 signal showed the same maximum intensity at 3 to 5 h post-injection and doubled signals were observed after 7 h, indicating the switch from T1 to T2 effect (bottom). (c) Schematic illustration of magnetic relaxation switch (MGRS) functions of core–shell NP-RANS with reduced intracellular targeting (top). T1- and T2-weighted MR images of MKN-45 tumor-bearing animals acquired before and after NP-RANS intravenous injection (immediate, 1 h, and 2 h) (bottom). (d) The NP-UIO self-assembly principle increases MRI and fluorescence signals generated by hypoxia (top). MRI images of 4T1 tumor-bearing mice showed a decrease in the brightness of NP-UIO-Pimo at 4 h after the intravenous NP injection as the oxygen concentration decreased, indicating hypoxia-induced self-assembly; in contrast, UIO-B did not show any similar changes with decreasing oxygen concentration (bottom). (e) Pre- and post-injection of NP-D-Fe3O4@PMn in Panc-1 tumor-bearing nude mice of T1- (upper left and middle) and T2-weighted (bottom left and middle) MRI images at 3T and corresponding signal intensity analysis (right top and bottom). Adapted with permission from: (b) ref. 92 Copyright © 2021 John Wiley and Sons, (c) ref. 94 Copyright © 2016 Elsevier, (d) ref. 97 Copyright © 2021 American Chemical Society, (e) ref. 98 Copyright © 2016, Springer Nature. | ||
Besides Gd(III) complexes, Fe(II/III) and Mn(II/III) redox-active ion pair systems have also been used as paramagnetic sources for redox responsive agents. The Fe(II) complex 48 interacts with H2O2 to undergo oxidation to Fe(III), providing a tenfold increase in r1, while the interaction with Cys reverts the structure to the original complex with the initial r1 value. This probe was successfully applied in vivo to specifically enhance the contrast in inflamed pancreatic tissue in the caerulein/LPS mouse model.86 In a similar fashion, a range of Mn(II/III) complexes interact with GSH, Cys, and H2O2 through the reversible binding mechanism, resulting in alterations redox states, and changes in r1.87–90
The research of NP-based redox sensors was focused on generating different probes able to produce changes in r1, r2, or both parameters concurrently. For instance, folic acid modified PEG was attached to extremely small iron oxide nanoparticles (ESIONPs) using a disulfide linkage. When the disulfide bond cleavage was induced by high levels of GSH, the hydrophilic PEG chains detached from the surface of ESIONPs causing the ESIONP aggregation and an increase in T2.91 Similarly, when the iron oxide NPs and angiopep-2 peptides (NP-S-Pep) were linked by disulfide bonds (NP-S-S-Pep), the GSH-induced aggregate formation and MRI changes were used to quantitatively map tumour-specific biomarkers in a mouse orthotopic brain tumour model (Fig. 7b).92 On the other hand, different kind of iron oxide NPs interact with cystamine to form nanoclusters, which provide good colloidal stability, compatibility and improved cellular uptake efficiency. Under redox conditions, this nanocluster dissociated into single NPs, to allow a ratiometric response and T2/T1-weighted dual-mode MRI of a subcutaneous tumour model in vivo.93 Finally, nanoplatforms of redox-responsive activatable nanostarshell (NP-RANS) were formed by combining superparamagnetic Fe3O4 cores and paramagnetic Mn3O4 shells (Fig. 7c). The inner Fe3O4 cores were exposed to aqueous environment, resulting in the breakdown of the Mn3O4 shells, releasing free Mn(II) ions through the GSH/Mn3O4 redox reaction. With an average hydrodynamic size of merely 21.9 nm, the fabricated RANS exhibited largest 6.8-fold and 2.8-fold increase in the r1 and r2 relaxivities, respectively. Subsequently, this sensor was used to perform T1- and T2-weighted MR imaging via effective passive tumour targeting in MKN-45 tumour-bearing mice.94
When discussing hypoxia-responsive probes, it should be noted that a fewer number of examples exists relative to the redox MRI sensors. A prototype monomacrocyclic platform is based on the ethylenearylsulfonamide side arm appended to GdDO3A complex. A typical example from this class is 2-methoxy-4-nitrobenzenesulfonamide derivative 49 (Fig. 7a); here, the hypoxic conditions promote decoordination of the sulfonamide moiety to the metal centre under, as the nitro group reduces to an aniline, which results in increased q and r1.95 Another approach was reported using 50, a Gd(III) complex with the ligand DOTP (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetra(methylenephosphonic acid)), which exhibited a significant binding affinity to oxyhemoglobin (oxy-Hb) and deoxyhemoglobin (deoxy-Hb). Due to increased rotation correlation time, the r1 relaxivity of GdDOTP bound to oxy-Hb and deoxy-Hb is almost five- and eight-fold higher than free GdDOTP. Moreover, since the difference in r1 values between Gd-DOTP-oxy-Hb and GdDOTP-deoxy-Hb is significant, the hypoxia could be evaluated in pre- and post-tumour regions of the BALB/c mice bearing murine mammary cancer.96
On the other hand, hypoxia-sensitive NP-based probes were developed using the ultrasmall iron oxides coated without and with the nitroimidazole derivatives (NP-UIO and NP-UIO-Pimo, respectively). Under the hypoxia conditions, the coating induces the NP self-assembly and formation of NP aggregates. This amplifies the T2-weighted MRI signal, revealing surprisingly deep penetration and accumulation efficiency in a mouse model bearing a breast cancer xenograft in subsequent MRI experiments in vivo (Fig. 7d).97 Similarly, a dual-modality approach was developed using an aptamer-based probe NP-D-Fe3O4@PMn that contains a hypoxia-inducible factor – 1α. This probe has the ability to generate T1-positive and T2-negative dual-mode MRI based on paramagnetic Mn(II) and superparamagnetic iron oxide, respectively. Possessing dual-mode properties, its administration in Panc-1 subcutaneous tumour-bearing nude mice in vivo yielded changes in T1- and T2-weighted MRI, with the former signal being substantially brighter and the latter giving darker MR images, respectively (Fig. 7e).98
2.5. Neurotransmitter-responsive probes
Neurotransmitters are crucial for the transmission of neural signals. They are key chemicals that aid making connections between neurons to enable their communication. Therefore, studying neurotransmitters can help to understand brain activity and neural transmission mechanisms in healthy and diseased states. Although different types of chemicals act as neurotransmitters (monoamines, amino acids, peptides, or molecules such as adenosine triphosphate and nitric oxide), the efforts to develop MRI probes for monitoring neurotransmitters were mainly focused on monoamines and amino acids.The research in this field began with on neurotransmitter-sensitive MRI probes for dopamine (DA) and serotonin (5-HT). This unique approach is based on the engineered protein technology that uses a naturally occurring paramagnetic metalloprotein from the heme domain of a P450 hydroxylase of Bacillus subtilis (BM3h). The protein probe exhibits a binding pocket with embedded paramagnetic iron, which can coordinate a water molecule. Consequently, the desired T1 signal changes take place when water-filled binding pocket is replaced with DA, which results in a decrease in q and hence the MR signal intensity. This specific recognition was obtained through a protein directed evolution method. Namely, through multiple rounds of mutations, the affinity of the binding pocket of P450-BM3 was modulated, thereby decreasing the affinity for the natural substrate arachidonic acid while increasing it for DA. The mutant protein BM3h-8C8 obtained was injected into the rat brain together with wild-type BM3h to stimulate brain cells by depolarisation at increased K(I) concentrations. The result of monitoring the DA release by T1-weighted MR imaging showed that there was a significant MR signal difference, only in the area injected with BM3h-8C8 (Fig. 8a).99 In a subsequent work, directed evolution was carried out on BM3h to obtain a series of proteins with different binding specificity and affinity.100 Among these protein-based MRI sensors, BM3h-9D7 was infused into the striatum of rats and the electrical brain stimulation was applied. Combining T1-weighted and hemodynamic functional MRI techniques, dynamics of DA was studied in a quantitative manner to successfully map DA release in the rat brain for the first time (Fig. 8b).101 Additionally, when the sensor probe was placed in the ventral striatum, the DA release in the striatum was associated with activity in more distal areas, such as the motor cortex, rather than with local changes in neuronal activity. These results demonstrated a hitherto unknown relationship between the release and clearance of neurotransmitters and large-scale activity in the brain.102
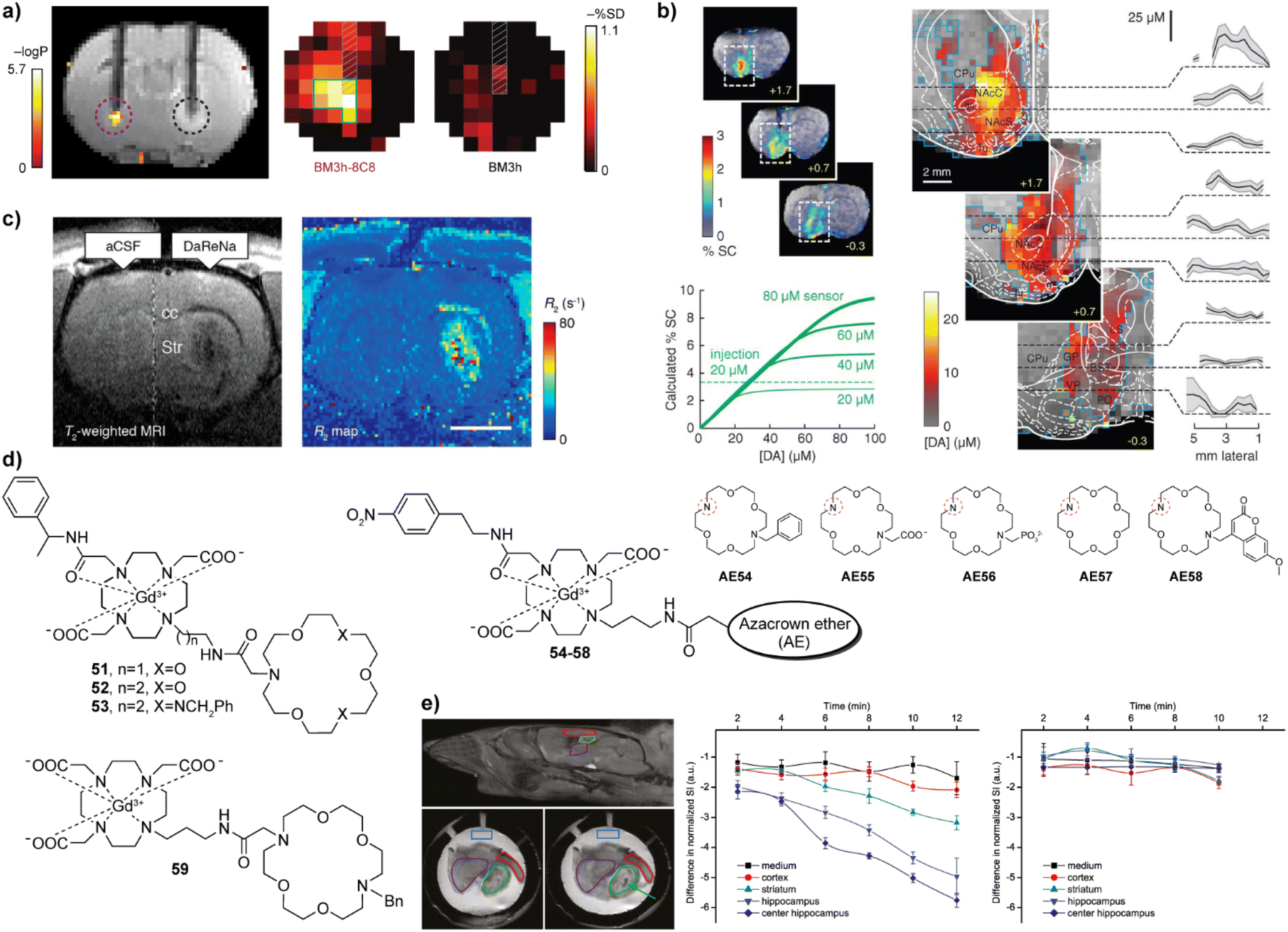 | ||
| Fig. 8 Neurotransmitter-sensitive MRI probes functional MRI experiments with these probes. (a) Dopamine-sensitive, genetically engineered protein-based probe BM3h-8C8 (purple dashed circle, left) reports significant changes compared to the wild-type protein (control probe WT BM3h, black dashed circle, right) in statistical parametric map that correlates MR intensity with low- and high-K(I) conditions (upper left). Maps of percent signal difference (SD) between high- and low-K(I) conditions observed in 2.7 mm-diameter ROIs centered around BM3h-8C8 sensor (left) and WT BM3h (right) injection sites, after spatial co-registration and averaging across multiple animals (upper right). (b) Protein-based probe BM3h-9D7 for quantitative functional imaging of DA concentration. The raw maps of average brain signal changes are significant in animals injected with BM3h-9D7 (upper left). In the unsaturated state of the probe, linearity function can be fitted between % SC and [DA], which is used as the basis for quantitative calculations. In areas that received substantial contrast agent infusion, a ratio of 8 mM DA per %SC can be used to estimate DA concentrations (lower left). Quantitative mapping of the average peak DA concentration on the infusion probe area. Plots show means (black lines) and SEMs (shading) of DA concentrations along dashed lines in respective images (right). (c) T2-weighted MRI (left) and R2 maps (right) of NP-DaReNa showing its effective diffusion in the brain. (d) The structures of ZNTs-sensitive small molecular CAs 51–59. The azacrown ethers AE54-58 are coupled to the remaining parts of the probes 54–58via the amine group indicated with the red dashed circle. (e) MRI imaging on acute brain slices with probe 53. The orientation of the investigated slices and brain structures with the ROIs (top left) and MRI of the slices in the presence of probe 53 before (lower left) and after (lower right) addition of KCl. Time courses of the normalized signal intensities in different ROIs when stimulated (membrane potential depolarization) with KCl only (middle) or KCl + tetrodotoxin (right), showing that only KCl alone caused the ZNT release and detection by the responsive CA. Adapted with permission from: (a) ref. 99 Copyright © 2010, Springer Nature America, Inc. (b) ref. 101 Copyright © 2014, The American Association for the Advancement of Science. (c) ref. 103 Copyright © 2019, American Chemical Society, (e) ref. 104 Copyright © 2015 American Chemical Society. | ||
The engineered neurotransmitter binding proteins were also used to develop the sensor probes suitable for T2-weighted imaging. Here, the above-mentioned engineered proteins were combined with SPIO nanoparticles to give DA- or 5-HT-sensitive T2-weighted probes (NP-DaReNa). The response mechanism relies on the tendency of nanoparticle conjugates to cluster in absence of the neurotransmitter, which significantly increases the r2 relaxivity and T2-weighted MR signal. After addition of neurotransmitter, cluster formation is reversibly disrupted; hence the r2 relaxivity reduces. Following this principle, the DA- or 5-HT-sensitive probes exhibited up to 20% decrease in r2, providing a new method studying monoamine neurotransmitters.103
Another approach in sensing neurotransmitters was conducted using Gd(III)-based CAs to detect zwitterionic neurotransmitters (ZNTs). This was achieved by utilising the commonality of ZNTs that their molecular framework has one negatively and one positively charged carboxylic acid and amino group, respectively. Consequently, the design principles of 51–59 (Fig. 8d) aimed to conjugate modified azacrown ethers as a recognition function with GdDO3A as a reporter unit.104–107 The recognition was hypothesized to be achieved by formation of hydrogen bonds between the probe and ZNTs, with the carboxylate group ligating to GdDO3A unit, while the amino group interacts with the crown ether. This dual binding of the ZNTs to the MRI sensor eliminates water molecules, resulting in a decrease in q and MRI signal. However, compared to the protein-based probe mentioned above, this method based on the azacrown ethers is not selective to a single ZNT molecule, but is rather suitable for exploring the overall concentration changes of several ZNTs. Overall, responsive CAs developed by this approach exhibited a response to glutamate (Glu), glycine (Gly), or γ-aminobutyric acid (GABA), with binding affinities in the mM range.104,106,107 While most NTs are typically present in pM to μM concentration in the extracellular space, the excitatory ZNT Glu can also reach low mM concentration levels.108–110 To this end, the current MRI sensors should undergo further structural optimizations to increase the affinity for ZNTs (with the exception of Glu), since the binding constants of responsive CAs should ideally match the in vivo concentrations of their targets.
The potential application of these probes to monitor the release of ZNTs was demonstrated in the MRI experiments on acute mouse brain slices (Fig. 8e). When stimulated by potassium chloride, they were able to dynamically monitor the release of ZNTs in the hippocampus.104 Lately, some structural improvements were reported, aiming to selectively detect ZNTs changes over a highly competitive metabolite in the body, bicarbonate, through the ratiometric change in r1 and r2,111 or to create the first ZNTs-sensitive nanoscale probe using the G4 PAMAM dendrimer, which should have a longer retention time in vivo.112
2.6. Enzyme responsive probes for MRI
Enzymes have a catalytic role in many physiological events in organisms and are a very important class of molecular biomarkers. To this end, development of MRI CAs that can report on the enzyme activity has been at the forefront of research in this field. Unlike ion or neurotransmitter sensitive probes, detection of the enzyme activity does not depend on the concentration of the target (enzyme), but rather on the enzyme's catalytic efficiency, i.e. the rate and amount at which the conversion of the activatable probe has been achieved. Therefore, it is important to ensure effective and specific contact between the enzyme and its substrate, as well as to design a robust mechanism that will convert chemical change in the responsive probe into the relaxivity and MR signal change.The research on enzyme-responsive MRI probes started with seminal example of the β-galactosidase (β-gal) activated agents, which consequently served as a platform to promote the bioresponsive agent technology to modern functional molecular imaging (Fig. 9a).113,114 The constructs included the paramagnetic centre suitable for T1-weighted MRI, a linker and the corresponding enzyme substrate. When the enzyme hydrolyses the substrate and the linker is severed, the paramagnetic centre is exposed to water molecules, which leads to an irreversible change in r1 (i.e. r1 increase). In case of the first-generation contrast agent 60, the conversion of low to high MRI signals was caused by the β-gal-induced hydrolysis of the galactose substrate that prevents access of water to the gadolinium complex. The magnitude of response improved when an α-methyl group was incorporated into the linker to give 61, since this modification limited the proximity of water to the paramagnetic centre and reduced the initial r1.115 The probe 61 was successfully applied to image gene expression in living Xenopus laevis embryos, to result in 45–65% higher MRI signal intensity in the mRNA-treated embryo, indicating the probe's ability to detect β-gal activity in vivo (Fig. 9b). Following these initial results, the enzyme-responsive platform technology experienced structural optimizations that aimed better exposure of the substrate unit to the enzyme, faster conversion rate and larger r1 changes. To this end, the connection between the monosaccharide (substrate) unit and gadolinium chelator was replaced with an aryl carbamate-based self-immolative linker to yield probes that respond to β-gal or β-glucuronidase activity.116–119 Consequently, the monosaccharides are distant from large volume GdDO3A unit, making them more flexible and easily accessible by enzymes. When they are hydrolysed by the enzyme, a cascade reaction is initiated inside the MRI probe, which causes a decomposition of the carbamate linker, too, exposing the paramagnetic centre to water molecules to increase q and r1. In turn, 62 and 63 are more effective than the first-generation probes 60 and 61, which was used to demonstrate their application in vivo.117,118 Based on these mechanisms, probes for detecting different enzymes have been developed, such as esterase, glutamic acid decarboxylase or tyrosinase.120–122 These and other examples were discussed in more details in previous review articles.1,2
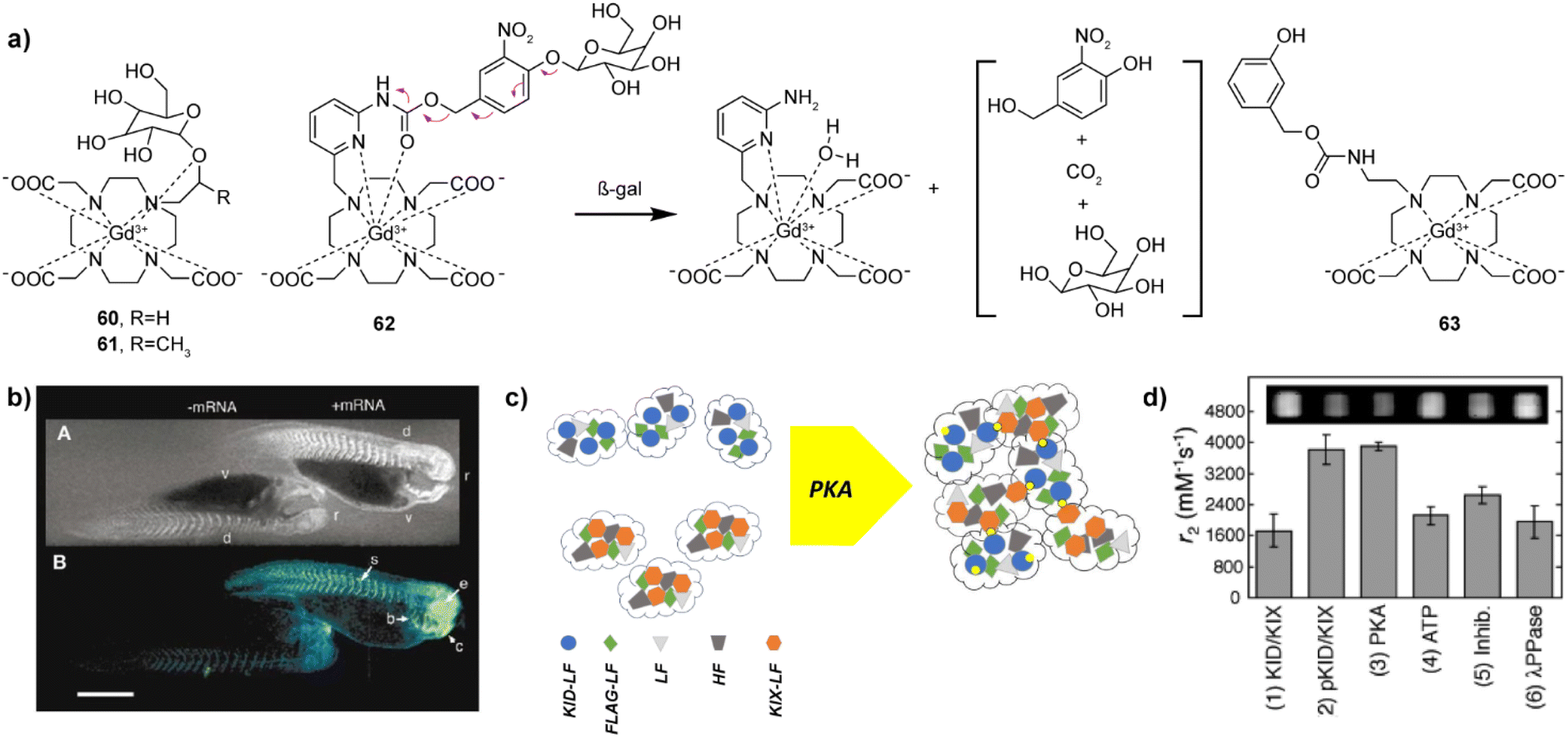 | ||
Fig. 9 Enzyme responsive MRI probes. (a) The structures of enzyme-sensitive MRI probes 60–63. (b) MR images of two embryos injected with probe 61 at the two-cell stage (top), while the embryo on the right was also injected with β-gal mRNA. Consequently, the signal strength is 45–65% greater in the embryo on the right that contains β-gal; pseudocolor rendering of the same image (top) with water made transparent (bottom). (c) Schematic illustration of the self-assembly and aggregation processes of NP-KID-FtNs in response to PKA activity. Genes encoding Ft are premixed and co-transfected into cells, where proteins are expressed and spontaneously assembled into KID-FtNs and KIX-FtNs (left). KID-LF chains are phosphorylated by PKA to form pKID-LF. Modified KID-FtNs (pKID-FtNs) containing pKID-LF form supramolecular aggregates with KIX-FtNs, which affect the MRI signal (right). (d) Measurements of the transverse relaxivity of Ft-based NP sensors in response to PKA activity. Incubation of the sensor (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of KID-FtNs with KIX-FtNs) with PKA for 2 h roughly doubled the r2 relaxivity. Adapted with permission from: (b) ref. 114 Copyright © 1969, Springer Nature, and (d) ref. 125 Copyright © 2009 American Chemical Society. :2 ratio of KID-FtNs with KIX-FtNs) with PKA for 2 h roughly doubled the r2 relaxivity. Adapted with permission from: (b) ref. 114 Copyright © 1969, Springer Nature, and (d) ref. 125 Copyright © 2009 American Chemical Society. | ||
In addition to the small molecule T1-weighted MRI responsive CAs mentioned above, iron oxide nanoparticle-based T2-weighted probes have also been developed to detect the enzyme activity.123–125 The mechanisms for their response vary, as the enzyme activity can degrade the polymer coating of the NPs to release the encapsulated magnetic cores,123 or can promote clustering of the NPs to change the T2-weighted MR signal.124 Here we highlight a variant of the latter mechanism, the probe NP-KID-FtNs, which was developed for sensing protein kinase A (PKA) activity based on protein engineering methodology.125 The probe is a nanoparticle sensor based on the iron storage protein ferritin (Ft). Ft is composed as a mixture of two polypeptides, the light chain (LF) and heavy chain (HF). Within cells, LF or HF subunits assemble into a nearly spherical protein shell together with the iron oxide core, which induces T2 relaxivity (Fig. 9c). This naturally available T2 MRI probe was used to develop a protein-engineered nanoparticle to sense the PKA activity. Namely, the N-termini of some LFs were fused with the PKA substrate known as kinase inducible domain (KID), as well as with the KID interacting domain (KIX), which binds phosphorylated KID. When genetically engineered surface-functionalized nanoprobe is exposed to PKA, the clustering of the nanoparticles is induced, resulting in r2 relaxivity changes of up to 84% (Fig. 9d). This work opened an avenue for the development of different protein engineered sensors suitable for T2-weighted MRI, with further possibility to improve the signal with more potent materials in the nanoparticle core.126,127
3. Responsive CEST MRI probes
CEST is one of the newer MR modalities, with the first work reported on this topic at the beginning of this century.128 The prerequisite for this technique is existence of exchangeable protons in the solute (CEST agent), which get saturated by a selective radiofrequency (RF) pulse.129,130 These protons then chemically exchange with bulk water protons, resulting in the partial saturation of the water pool and the decrease of the water protons signal intensity. If the proton exchange rate kex satisfies the condition of kex ≪ Δω (Δω is the difference between two shift signals), the exchangeable protons can be saturated by the applied RF pulse and exchanged chemically with water protons, giving rise to formation of CEST effect (Fig. 10a).131 Following identical principles at other frequencies, the CEST methodology can accordingly also be applied to other exchangeable species, such as 19F or 129Xe, to result in 19F CEST or 129Xe hyperCEST, respectively (see below).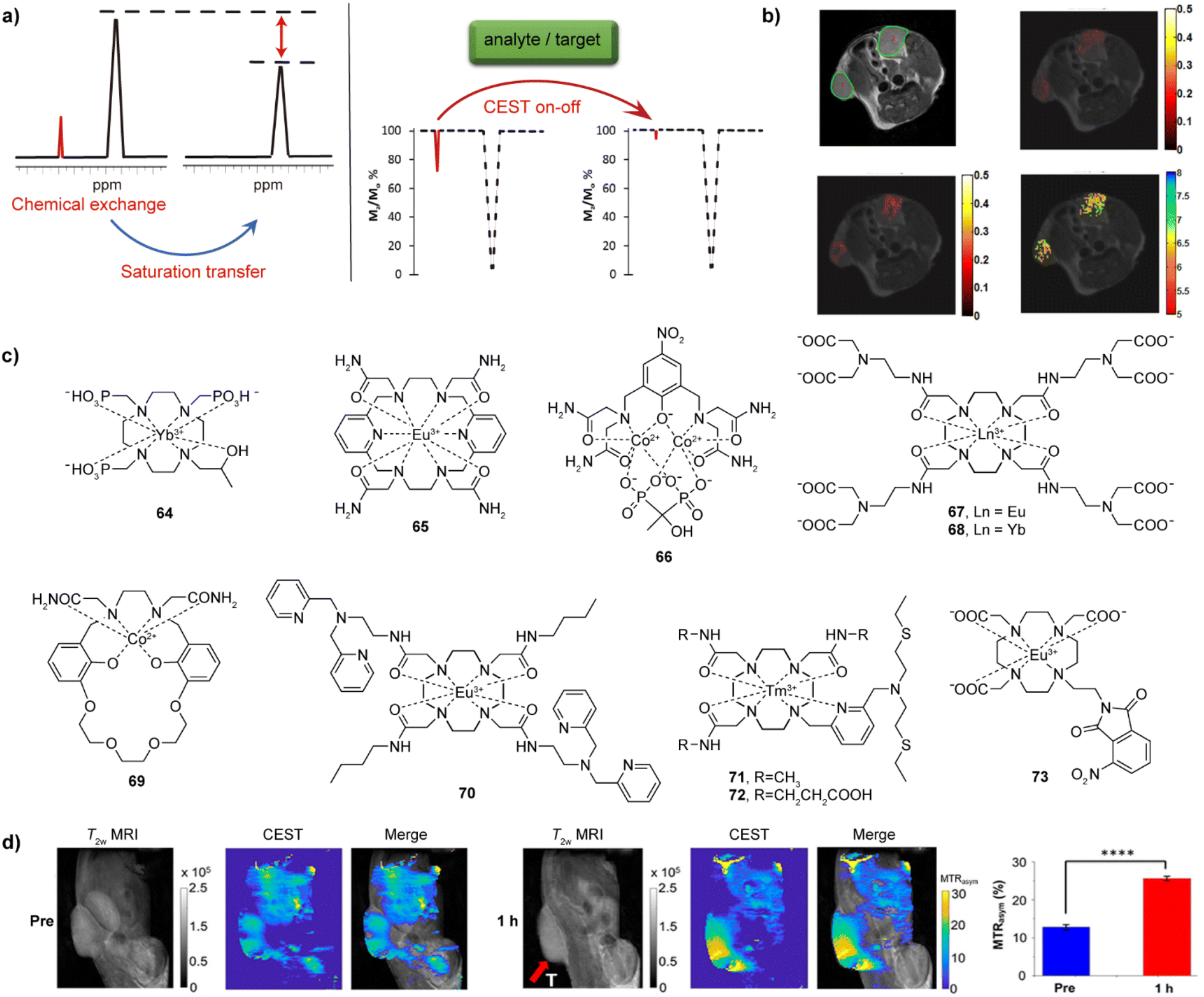 | ||
| Fig. 10 CEST effect, selected bioresponsive CEST probes and CEST MRI in vivo. (a) The illustration of CEST effect and CEST-responsive mechanism. A bioresponsive CEST probe is bearing exchangeable protons, which can be saturated by an RF pulse and can chemically exchange with bulk water protons; the chemical exchange of protons results in a partially saturated water pool, which results in a decrease of the water protons signal intensity (left). If an analyte or a target interacts with probes, the CEST signal can be affected (a turn-off CEST effect illustrated in the z-spectra) (right). (b) CEST MRI of iobitridol in the tumor-bearing mouse model: T2-weighted anatomical image of two tumour regions (top left), CEST MRI before and after injection of iobitridol at 1.5 μT (top right) and at 6 μT (bottom left), and a corresponding pH map obtained as a ratio of saturation transfer maps from these two CEST measurements (bottom right). (c) The representative CEST probes sensitive to pH (64–66), Ca(II) (67–69), Zn(II) (70), Cu(II/I) (71–72) and hypoxia (73). (d) CEST MRI of bioresponsive probe 73 in the CT26 xenograft mouse model (tumor area shown by red arrow). The panels are images obtained by recording T2-weighted and CEST MRI along with the merged image before (three left panels) and 1 h after (three right panels) intratumoral injection of probe 73, respectively. The quantitative analysis compares the mean MTRasym (%) values for the tumor region from of three measurements (right). Adapted with permission from: (b) ref. 138 Copyright © 2014, American Chemical Society. (d) Ref. 149 Copyright © 2022, American Chemical Society. | ||
As one of the advantages compared to T1- and T2-weighted agents, the CEST effect and hence the CEST-generated signal can be turned on and off by applying the selective RF pulse at a will. A wide range of external stimuli that serve as biological targets like cations pH, Ca(II) and Zn(II), anions like lactate and α-hydroxyl acids or enzymes like urokinase-type plasminogen activator (uPA) and DT-diaphorase and can have further effect on the CEST. The excess or deficiency of these analytes and targets can be related to a series of biological dysfunctions associated with chronic diseases. Therefore, different bioresponsive CEST agents have been developed in recent years, to aid the detection of these biological and disease markers.2,129
Among these agents, metal-free bioresponsive probes, termed as diaCEST agents, were frequently employed due to their advantageous properties, such as having strong CEST effect, good biocompatibility and therefore low toxicity.2,129,132,133 However, paramagnetic metal-based agents, termed as paraCEST agents, are frequently more preferred because of the comparatively lower agent required dose and generation of the larger chemical shifts.134 The latter agents are usually based on lanthanide ions such as Eu(III), Yb(III), or Tm(III), although transition metal ions such as Co(II) or Ni(II), due to their capability to shift the frequencies of CEST-active protons. The following subsections will mainly highlight examples of the bioresponsive paraCEST agents.
3.1. pH-sensitive CEST probes
The CEST probes are intrinsically pH-sensitive, since the pH alterations automatically influence their proton exchange rates and hence the CEST effect. To this end, protons of the hydroxyl, amide, primary or secondary amine units can easily be affected by pH; therefore, they were ingeniously incorporated into the structure of various chemical compounds for the construction of pH-sensitive CEST probes.135 For diaCEST agents, the employed mechanism includes the CEST process between water protons and the selectively irradiated protons on the diamagnetic probe (such as iopamidol, iobitridol or derivatives of salicylic acids), to provide a promising way for using them as pH sensors by means of CEST MRI.136,137 In turn, a diaCEST responsive probe iobitridol can be used to highlight the tumour regions in the xenografted-tumour bearing mouse, along with enabling their pH mapping in vivo (Fig. 10b).138For pH-sensitive paraCEST agents, the first example is 64 (Fig. 10c), where paramagnetic Yb(III) was chosen thanks to its ability to induce large frequency shift of exchangeable protons of over 100 ppm, as well as its high thermodynamic stability due to a chelation with a macrocyclic DO3P (1,4,7,10-tetraazacyclododecane-1,4,7-tris-(methylene phosphonic acid)) chelator.139 The pH-sensitive property is attributed to a hydroxypropyl unit of 64 and the frequency shifts change from 120 ppm to 140 ppm upon a pH change from 4 to 9 at 9.4 T and 25 °C. Although a strong CEST effect was detected in the mouse blood sample in vitro, the injection of sample into mouse tail vein did not show positive results. On the other hand, the 18-membered macrocyclic complex 65 that bears four amide groups provided a very inert environment for Eu(III). This high inertness was caused by a ten-coordinated configuration between Eu(III) and the macrocyclic ligand, which was further confirmed by a kinetic measurement; the complex remained intact in 1 M HCl over 25 days, or after a transmetalation assessment with Zn(II) in a PBS buffer at 40 °C for 3 days. The tethered amides produce two CEST effects at 8.5 and 14 ppm at 37 °C and both are pH dependent. The intensity ratio of these two peaks at 5 μT showed large changes in the pH range of 6.4–7.4, further suggesting this bioresponsive agent a desirable candidate for functional applications.140
Indeed, the labile exchangeable protons from amides and/or hydroxyl groups play a key role in producing plausible CEST effects and pH dependent properties. Using this strategy and same modality, the first-row transition metal-based complexes were developed as pH-sensitive agents. Among these, paramagnetic Fe(II), Co(II) or Ni(II) were used to form paraCEST complexes, with the high-spin Co(II) being the most preferred to construct the probes.141 Namely, this ion is well-stabilized by tri- or tetra-azamacrocyclic ligands that bear amides or hydroxypropyl motifs, while it possesses the excellent shift properties desirable for CEST applications. Due to different coordination chemistry, the commonly used ligands for these ions are derivatives of cyclam, cyclen, TACN (1,4,7-triazacyclononane), or PATA (2,2′,2′′,2′′′–((pyridine-2,6-diylbis(methylene))bis(azanetriyl))tetraacetamide). Moreover, complexes with acyclic ligands were also studied. A representative example is a di-nuclear Co(II) complex 66, which is additionally chelated by a hydroxyl-substituted bisphosphonate.142 This complex showed CEST effects as strong as ∼30% at 66 ppm and ∼25% at 106 ppm at pH 8.14 and 37 °C, deriving signals at 66 ppm or 106 ppm from carboxamide or from both carboxamide and hydroxyl protons, respectively. The ratio of CEST104ppm/CEST64ppm effects changed from 8.35 to 0.82, corresponding to the pH values from 6.50 to 7.60, which was examined and confirmed by means of MRI in vitro on tube phantoms.
3.2. Cation-responsive CEST agents
Following a similar principle of combining the cation-selective chelator with the paramagnetic reporter unit previously established for T1-weighted probes (see above), several cation-responsive paraCEST probes were also developed (Fig. 10c). The first Ca(II)-sensitive probes 67–68 involved a macrocycle based on DOTAM (tetraamide derivative of DOTA – 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate) with the iminodiacetate-bearing groups that result in the paraCEST effects on the protons of coordinated water or amide groups when Eu(III) or Yb(III) complex was use, respectively.143 Using an entirely different chelator based on a macrocyclic crown ether unit, a Co(II)-based agent 69 was developed for Ca(II) and Na(I) response. The paramagnetic metal ion was stabilized by hexadentate chelating Schiff base group and its CEST effect at 77 ppm was derived from the appended two carboxamide groups. The interaction of Na(I) with the crown ether of 69 shifted the CEST signal from 77 ppm to 69 ppm. To further investigate Ca(II) response in the presence of Na(I), 69 was employed to result in a reduction of the CEST effect at ∼69 ppm and an increase at 80 ppm. The ratio of CEST80ppm/CEST69ppm effects exhibits dependency on the amount of Ca(II), providing a possibility to quantitatively detect Ca(II) concentrations.144Previously to these works, the initial cation-responsive paraCEST probe was designed for the detection of Zn(II). This was 70, an Eu(III) complex of DOTAM, which bears two dipicolylamine units for selective detection of Zn(II). The interaction of 70 with the target metal ion affects the exchange of inner-sphere coordinated molecule, which changes the paraCEST effect.145 On the other hand, two Tm(III) complexes, positively charged 71 and charge-neutral 72, were investigated as potential paraCEST probes to respond to Zn(II) or Cu(I)/(II). These were complexes of DO3AM (triamide derivative of DO3A) appended with the chelator made from the conjugate of the bis(2-(ethylthio)ethyl)amine and 2,6-dimethylpyridine. The sensing properties were observed only in the case of 71, where amide groups exhibited the strong CEST effect. The addition of Zn(II) and Cu(I)/(II) triggers the formation of a dimmer, which enhances the protons exchange and quenches the CEST effect.146
3.3. Redox- and hypoxia- responsive CEST agents
There are a few examples of the bioresponsive probes of this class that were reported until now. To develop a redox-responsive CEST probe, EuDOTAM derivative appended with N-methylquinolinium units was prepared. The CEST-silent state of the agent was activated by the reduction of the quinolinium moiety to the dihydroquinoline derivative with NADH (nicotinamide adenine dinucleotide), which results in a large increase of CEST intensity at ∼50 ppm.147 On the other hand, several first-row transition-metal complexes were shown to be good candidates for developing on–off redox-responsive MRI agents thanks to their manifold oxidation and spin forms.148 To sense hypoxia, a Eu(III) complex of DO3A tethered with a 3-nitropthalimide unit (73, Fig. 10c) was employed. The complex can be activated using the nitroreductase (NTR) enzyme, which reduces nitro to the amino group, giving rise to the formation of CEST effect. Following affirmative in vitro experiments in buffered media and on CT26 cancer cells treated with 73, the in vivo experiments on an animal tumour model indicated necessity of applying higher probe concentrations to get higher CEST MRI signal enhancement.1493.4. Anion-responsive CEST agents
Rather than bearing a separate sensitizing unit that couples with the MR reporter, anion-sensitive paramagnetic agents are usually complexes of DOTA and DO3A derivatives with various Ln(III), where an unsaturated coordination of Ln(III) is used for interaction with the target anion (Fig. 11a). | ||
| Fig. 11 Selected bioresponsive CEST probes and CEST MRI in vivo experiments with bioresponsive CEST probes. (a) The representative CEST probes that are sensitive to lactate (74), α-hydroxyl acids (75–77) and enzymes (80–82). (b) CEST imaging of extracellular lactate in a mouse bladder by employing the probe 74. In the absence of lactate, probe 74 shows no CEST signal. Upon the injection of 1:1 mixture of probe 74 and lactate in a mouse, a strong CEST MRI signal was evident in the bladder. Adapted with permission from ref. 150 Copyright © 2017, Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The first example is a commercially available EuDO3A 74 that can be used to image extracellular lactate in vitro and in vivo, respectively. The ternary complex formation between the lactate and EuDO3A shifts the resonance of the proton of the lactate hydroxyl group, thus generating a CEST effect. The probe was used to image extracellular lactate excretion from lung cancer cells, as well as the bladder of a mouse in vivo (Fig. 11b).150 Applying the same strategy, YbDO3A(Gly)3 derivatives 75–77 with one, two and three chiral centers were employed to enantiomerically recognize α-hydroxyl acids. The ternary complexes formed via the α-hydroxyl acids bound to the Yb(III) provide different chemical shifts in R and S states. The CEST signals from R isomer are shifted further than the signal from S isomer over 20–40 ppm, and the results suggest such complexes are useful for discrimination of α-hydroxyl acids.151
3.5. Enzyme-responsive CEST agents
In analogy to other related methods, a subclass of the CEST technique that involves probes responsive to the enzyme activity is termed catalyCEST, due to the important role of enzymes wide number of biological processes as biocatalysts.152 For this purpose, different diamagnetic and paramagnetic probes can be designed to combine suitable substrates for target enzymes with the dia- or paraCEST-active moiety (Fig. 11a). The typical example of the former, diaCEST probe, is an organic compound 78. After incubation of the agent with the cathepsin B, the CEST signal at 5.0 ppm assigned to the aryl amide disappears, whereas the CEST signal from the salicylic acid at 9.2 ppm remains, allowing the pH sensitive CEST investigations.153The paraCEST probes sensitive to enzymes were the preferred choice compared to their diamagnetic analogues due to the ability to shift the resonance of CEST effect. To this end, the prototype paraCEST probe 79 sensitive to diverse enzymes was prepared by conjugating a galactopyranoside unit to YbDOTA via an amide bond. The formed linkage is self-immolative, resulting in a formation of an amine bond upon the enzyme activation, which exhibits the CEST signal.154 In a similar way, an agent 80 was developed for sulfatase recognition. In this system, the sulfatase from Helix pomatia can be sensed with the YbDO3A unit coupled to a sulphate aryl ester. The enzyme-activated, selective ester hydrolysis triggers a sequence of processes that finishes with the 1,6-elimination of the self-immolative aryl linker to form the primary amine-containing paraCEST agent with two CEST peaks at 42 and 89 ppm.155
To overcome the interference issue of the single CEST effect by the environment, addition of the second CEST effect to the same CEST agent is a plausible method. Hence the so-called self-calibrating paraCEST probe 81 as a YbDO3A-based agent is appended with two amide groups, where one serves as an internal control, while another one tethers a trimethyl lock (TML) spacer for esterase detection. The esterase-triggered cleavage of the methyl ester induces lactonization of TML moiety, leading to the conversion of 81 to an agent with newly generated amine motif that exhibits a CEST effect at +12 ppm. Combined with an unchanged amide that has a constant CEST effect at −10 ppm, ratio of these two CEST effects can be used for more accurate detection of desired enzyme-triggered stimuli.156
Despite a respectable number of the enzyme-responsive CEST probes that show activity at an in vitro level, their wider application in vivo is still not abundant. An example for this purpose describes an approach to assess an uPA (urokinase plasminogen activator) activity in vivo by using enzyme-responsive and nonresponsive agents. The former probe is Tm(III) complex 82 that has a ZGGR peptidyl unit conjugated to Tm-DOTA and displays a CEST effect at −54 ppm. Since the ZGGR is sensitive to uPA, the substrate treatment with uPA results in the cleavage of the peptidyl unit from the metal complex, making the remaining part unsuitable to generate CEST effect. In parallel, the well-known paraCEST probe Eu-DOTA-Gly4 with CEST effect at +54 ppm was used as a control probe to provide a constant reference of CEST signal. Combined, these two agents aided establishment of important MRI protocols to monitor in vivo pharmacokinetics and detection of enzyme activity using catalyCEST agents in vivo.152
4. Fluorinated responsive MRI probes
Fluorine is highly regarded for MRI applications since this is the NMR-active nucleus with high MR sensitivity, which is 83% of that existing for 1H.157 Additionally, the 19F isotope abundance of 100% in nature makes every suitable fluorinated material a potential MRI probe, while its very low abundance in human body results in the absence of any background signal and hence allows performing quantitative measurements. This is usually achieved by developing materials that possess large number of NMR equivalent fluorine atoms in the molecule to enhance the 19F signal. Such approaches may face difficulties since the water solubility of the probes may be significantly affected, usually leading to development of perfluorocarbon nanoemulsion formulations to be used as probes for 19F MRI.158 Further, fluorine has a wide NMR frequency range and is highly susceptible to large chemical shift changes. This can allow for observing signals and their changes at multiple frequencies, leading to multicolour and multiplexed imaging, i.e. observation of parallel processes simultaneously.Changing the 19F signal to provide a responsive MRI probe suitable for functional imaging can be achieved in a few ways that are analogous to 1H MRI probes. Commonly applied strategy involves a range of paramagnetic lanthanide complexes that shorten T1 and T2 relaxation times of fluorine through the paramagnetic relaxation enhancement (PRE) mechanism (Fig. 12a). Besides it, relaxation times (especially the T2) are highly sensitive on the τR of MRI probes; hence, this feature has been widely exploited in different fluorinated nanoprobe formulations to develop responsive probes suitable for 19F MRI (Fig. 12b). Finally, CEST features may also be used in for fluorinated molecules if available, especially for sensing ions (iCEST).
 | ||
| Fig. 12 The most common mechanisms used for preparation of responsive fluorinated MRI probes. (a) The design principle that uses a chelate with the paramagnetic lanthanide or transition metal ion (green ball) covalently bound to a fluorinated functional group (yellow polyhedron). The effect of paramagnetic metal ions on fluorine atoms varies depending on the distance, leading to changes in 19F MR signals: at shorter distances, T1 and T2 relaxation times are shortening (PRE) or 19F NMR frequency changes (LIS). (b) Fluorinated bioresponsive nanoprobes utilize the principle in which the low 19F MRI signal exists in assembled nanoprobe due to the significant T2 shortening (left), which, in the presence of the analyte, triggers the NP disassembly and hence an enhanced 19F MRI signal (right). (c) The structures of fluorinated bioresponsive probes 83–98, which take advantage of PRE or LIS effect. | ||
4.1. Responsive 19F MRI probes that exploit PRE effect
Using the PRE effect has been the most exploited strategy to prepare responsive fluorinated probes to date.159,160 The main reason is a possibility to use a wide range of paramagnetic lanthanide and transition metal ions in combination with 19F nucleus to result in the desired signal change, unlike a limited number of strongly paramagnetic metal ions such as gadolinium and manganese or the iron oxide for 1H. Namely, each paramagnetic lanthanide or a transition metal ion possesses unique set of properties that have different influence on the fluorine atoms in the molecule. The major effect, PRE, results in changes of T1 and T2 relaxation times. It depends on the nature of the metal ion and can further be tailored depending on the distance from the paramagnetic centre (Fig. 12a). The second effect, lanthanide induced shifts (LIS), exists in the case of lanthanide ions and combines these two parameters (paramagnetic nature and the distance) together with additional magnetic properties derived from the type of lanthanide complex and spatial positioning of fluorine to result in the change of 19F NMR frequency of the probe (Fig. 12a).The use of PRE and LIS effects can be very beneficial for developing responsive 19F MRI probes. The former shortens 19F T1 and T2, which enables faster acquisition of 19F MRI signals, thereby significantly shortening the scanning times to yield the higher signal-to-noise ratio (SNR). However, the distance and type of lanthanide ion do not shorten the relaxation times proportionally; principally, the T2/T1 ratio should remain high since significant shortening of T2 leads to loss of signal. On the other hand, LIS effect allows for events to be observed at different frequencies, which either simplify the analysis, or enable e.g. ratiometric studies, which may provide valuable quantitative information related to the monitored process.
These two powerful tools to manipulate the 19F MRI signal were widely used for the design and preparation of responsive 19F probes. For listing the most interesting examples, the same order of targets as discussed in previous sections will be followed.
To sense Ca(II), the PRE effect was used by combining a lanthanide chelate and a fluorinated group spaced with the EGTA-derived chelator. It was hypothesized that the probe is in the extended mode in Ca(II)-free environment, while interaction of the EGTA-derived moiety with Ca(II) will cause a contraction, consequently changing the distance between lanthanide ion and fluorine atoms, hence also the 19F signal. Indeed, when DO3A was used as chelator and an aromatic trifluoromethyl group as 19F MRI reporter (90), a significant decrease in 19F signal was observed on Gd(III) and Dy(III) in presence of Ca(II), due to shortening of distance between fluorine and paramagnetic centre.164 In the follow-up study, this responsive probe underwent structural modifications to improve the signal and potentially induce larger changes when interacting with Ca(II). In this modified version (91), the amount of fluorine was tripled by replacing a trifluoromethyl with perfluoro-tert-butyl group, while the AAZTA (1,4-bis(carboxymethyl)-6-[bis(carboxymethyl)]amino-6 methylperhydro-1,4-diazepine) replaced DO3A chelator.165 This time, the Ca(II)-dependent effect was clearly observed on the Dy(III) complex, while using a diamagnetic Y(III) complex as a reference. This allowed establishment of a ratiometric method for quantification of Ca(II) by means of chemical shift imaging (CSI).
Finally, a DO3A chelated with Tm(III) and conjugated to DPA group appended with the perfluoro-tert-butyl group was used for sensing Zn(II) (92). Due to fast chemical exchange of several conformational isomers in the absence of Zn(II), the 19F MRI signal cannot be observed; nevertheless, the signal recovers with the chelation of Zn(II) due to formation of more rigid conformers.166
Lanthanide redox-active ion pair system Eu(II)/Eu(III) was used to develop a fluorinated hypoxia-responsive agent. A chelate based on DOTAM ligand and several trifluoromethyl groups was prepared and studied.167 The complex 93 containing Eu(II) enhances the T1-weighted contrast in 1H MRI, while upon the oxidation to Eu(III), this system is giving rise to enhanced 19F signal due to change of paramagnetic properties of the metal ion that affirms the PRE effect. In a subsequent study, three different pairs of Eu(II)/Eu(III) complexes were employed to develop a ratiometric method that uses 19F MRI normalized with T1-weighted 1H MRI signal. This resulted in the possibility to image and quantify hypoxia, without a need to know the absolute concentration of used imaging probe.168
Further examples of redox-responsive fluorinated probes used other types of metal ions and hence suitable chelators. A Mn(II)/Mn(III) pair was used in combination with HTFBED (N,N′-bis(2-hydroxy-4-trifluoromethylbenzyl)-ethylenediamine-N,N′-diacetic acid) to give probe 94 with strong 1H, whilst weak 19F MRI signal in Mn(II)-HTFBED, due to its strong PRE effect (r1 of this oxidation form is 2.7 mM−1 s−1). When the metal ion is oxidized to Mn(III), the complex exhibits poor 1H (r1 = 0.7 mM−1 s−1), albeit strong 19F MRI signal. The oxidative and reductive responsive properties of this system were demonstrated in MRI in vitro experiments using various redox reagents.169 Another redox pair 95–96 to study cellular hypoxia is based on the Cu(I)/Cu(II) pair and the chelator ATSM (diacetyl-bis(N4-methylthiosemicarbazone)). This system switches from the paramagnetic Cu(I)-ATSM to diamagnetic Cu(II)-ATSM form under hypoxic conditions. The 19F MRI signal vanishes under normoxic conditions due to strong PRE effect, giving rise to formation of the turn-on signal response in the hypoxic environment.170 Finally, a Co(II)/Co(III) pair complexed with a TACN derivative (97) demonstrated the ability to change 19F MRI signal from low to high when Co(II) is oxidized Co(III).171 Furthermore, when a modified TACN-derived chelator is used, this process is accompanied with the 10 ppm 19F chemical shift change caused by the large magnetic anisotropy in the high spin Co(II) complex, allowing monitoring of the redox processes concurrently at two frequencies (98).172
Responsive probes for proteases such as caspase-3, a marker enzyme of apoptosis, were designed by combining a GdDOTA chelate with caspase-3 selective DEVD peptide sequence and trifluoromethyl-containing tag (Tfb) into the GdDOTA-DEVD-Tfb conjugate 99 (Fig. 13a). This probe enhances T1-weighted 1H MRI signal, concurrently cancelling 19F MRI signal due to the presence of strongly paramagnetic Gd(III) and hence significant shortening of 19F T2.173 Moreover, by replacement of the Tfb fluorinated tag with 7-amino-4-trifluoromethylcoumarin (AFC) to a Gd-DOTA-DEVD-AFC conjugate 100, the caspase-3 activity can be monitored by means of optical and 19F MR imaging.174 Similarly, the assessment of a matrix metalloprotease-2 activity was attempted with conjugate consisting of Gd-DOTA, coupled with the enzyme-sensitive peptide sequence GPLGVRGC, followed by PEGylated perfluoro-tert-butyl group.175
 | ||
| Fig. 13 Fluorinated bioresponsive MRI probes and 19F MRI in vivo studies with these probes. (a) The structures of fluorinated responsive probes 99–109, which respond to enzyme activity or are suitable for iCEST. (b) 1H and 19F MRI with pH responsive probe NP-FNPs-PEG. The 19F MRI signal in vitro at pH 7.4 is low due to the probe assembly (top left) and increases at pH 5.5 (top right), while the 1H MRI signal does not change significantly. When applied in vivo in the tumour-bearing mice, NP-FNPs-PEG delineate the boundaries of tumours from surrounding normal tissues by means of 19F MRI (bottom middle and right), when compared to 1H MRI with the same probe (bottom left and right). (c) MRI experiments with pH responsive NP-Mn-LDH@PFPE in a mouse subcutaneous MDA-MB-468 tumour model. A strong 19F MRI signal was observed at the tumour area 24 h after probe injection (left), which can also be followed over time by means of T1-weighted 1H (black) and 19F MRI (red) signal intensities (right). (d) The use of probe 103 for detecting Zn(II) secretion in a transgenic adenocarcinoma of the mouse prostate model in vivo. 19F MRI (top), iCEST MRI (middle), and iCEST spectra (bottom) of 10 week and 17 week-old mice (two left and two right panels, respectively) before and after of D-glucose injection. (e) In vivo19F iCEST maps of labile Zn(II) pools with probe 108 in the mouse brain. The probe was administered in a zinc-rich (top) and zinc-poor (bottom) regions (CA3 in hippocampus and thalamus, respectively). From left to right: schematic illustration of the setup for the delivery of 108, 1H MRI, 19F MRI at the off-resonance, 19F MRI at the on-resonance, 19F iCEST (subtracted 19F MR images overlaid on the 1H MRI). Adapted with permission from: (b) ref. 186, Copyright © 2018 Royal Society of Chemistry (c) ref. 187 Copyright © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim (d) ref. 199 Copyright © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (e) ref. 200 Copyright © 2021 The Authors. | ||
In a slightly different approach, previously mentioned visualization of gene expression through monitoring the activity of β-gal was explored using self-immolative linkers. Here the β-gal enzyme substrate, galactosidase, is coupled to the MRI modulator (GdDOTA) via the fluorine containing linker.176,177 The enzyme activity results in a cleavage of the monosaccharide unit, further followed by immolation of the fluorinated unit. Consequently, this sequential process leads to generation of high 19F signal due to the increase in distance between the fluorine atoms and paramagnetic ion in the responsive probe. Besides using Gd(III), the β-gal-responsive fluorinated probes were developed by using Ni(II) and Fe(III) metal ions as sources for generating the PRE effect. The former probe (101) used cyclam derivative as the chelator for the coordination of Ni(II), while the change in 19F MRI signal was achieved due to enzyme-induced spin-state switching of the diamagnetic low-spin (S = 0) to paramagnetic high-spin (S = 1) species.178 The latter took advantage of the in situ formed iron chelate with a product of enzyme cleavage of fluorosalicylaldehyde aroylhydrazone β-D-galactopyranoside. The chelate formation was followed by a change in 19F chemical shift, as well as strong T2 relaxation effect at 1H frequency, which aided imaging β-gal activity in vivo.179
4.2. Responsive 19F MRI nanoprobes
Given the high sensitivity of 19F relaxation times, particularly T2, on the τR change (hence the size) of MRI probes, different fluorinated nanoprobes were developed for the purpose of monitoring molecular processes by means of 19F MRI. Moreover, the use of nanoprobes allows a high fluorine load, which improves the SNR and shortens the long acquisition times that are typical for this imaging method. Different types of materials are used for the nanoprobe formulation, commonly known as perfluorocarbons (PFCs), the organic compounds in which the hydrogen atoms are replaced with fluorine. The most common molecules incorporated in these nanoformulatons are perfluoro-15-crown-5-ether (PFCE) perfluorooctyl bromide (PFOB) or perfluoropolyether (PFPE), although different fluorinated linear or branched polymers, ion liquids or nanocrystals have also been reported.4,180,181It is not necessary to use the PRE effect to design a responsive fluorinated nanoprobe probe. An assembly of individual PFC components into a nanoprobe, or its disassembly back is accompanied with significant changes in the transverse relaxation times, which typically result in the strong 19F MRI signal before, while weak to disappearance of signal after the assembly (Fig. 12b). This effect can additionally be enhanced by using paramagnetic ions as cores of the prepared nanoparticles; the current literature examples comprise of nanoprobes that solely use diamagnetic materials, but also those where paramagnetic ions were incorporated to take the additional advantage of the PRE effect. Subsequently, many types of responsive nanoprobes for 19F MRI have been developed to date and nicely summarized in a few recent reviews.4,182–184 Due to the diversity of materials used and applications, we are going to highlight a few examples, to demonstrate the common principle and purpose.
In a proof-of-concept study, ionizable diblock copolymers were prepared from different types of amines and fluorinated molecules. They self-assemble to form micelles when amines are not protonated, resulting in restricted motion of polymer chains and hence short 19F T2, which results in elimination of 19F MRI signal. At lower pH, the protonated ammonium groups induce micelle disassembly, while the 19F MRI signal recovers due to high flexibility of the polymer chains. This approach allowed the use of nanoprobes as pH indicators because the signal recovery appeared at different pH values, depending on the pKa of used amines.185
Following analogous principle, a range of probes responsive to pH or other stimuli was developed. Among these, we highlight a hybrid metal organic framework (MOF) matrix that contains 4(5)-(trifluoromethyl)imidazole as a source of 19F MRI signal. A pH dependent (dis)assembly of this NP-FNPs-PEG is resulting in same effect; hence the 19F MRI signal enhancement was recorded in tumour surrounding tissue in mice (Fig. 13b).186 As well, a PFPE conjugated on the surface of manganese-incorporated layered double hydroxide provided nanoprobes (NP-Mn-LDH@PFPE) that quench 19F MRI signal at physiological pH due to PRE effect of Mn(II). However, these ions dissolve and diffuse away at acidic pH, allowing generation of high 19F MRI signal, which can detect the presence of breast tumour tissue in the mouse tumour model (Fig. 13c).187
The fluorinated nanoprobe to detect Ca(II) is based on the amphiphiles that contain tridecafluoroheptanoyl group conjugated with three different peptides, which are used to improve the probe solubility. In presence of Ca(II), these amphiphiles self-assemble into nanoribbons, which shortens 19F T2 and eliminates the 19F MRI signal.188 Redox-activatable nanoprobes were developed using a trifluoromethyl-decorated benzene spaced via the disulfide bond to the Gd(III) chelator to take advantage of PRE effect in the self-assembled nanoparticle. Exposure of the nanoprobe to reducing environment using biothiols eliminates self-immolative linker, providing ∼30-fold stronger 19F MRI signal.189 Similarly, the fluorine accumulated silica nanoparticles for MRI contrast enhancement (FLAME) using encapsulated PFCs and Gd(III) complex attached to FLAME via the disulfide bond was used as responsive probe to detect reducing environments.190
The examples of fluorinated nanoprobes that respond to enzymes relied on the work initially describing supramolecular fluorinated nanoprobes that can detect human carbonic anhydrase I or trypsin.191 Based on this approach, a self-assembled system that can detect the legumain, a cysteine protease, was reported. The assembly of the nanoprobe is controlled by the GSH, while the subsequent disassembly is induced by the legumain to image the activity of this enzyme in tumour in zebrafish.192 Similar imaging model was used to monitor the activity of the caspase 3/7. Here also the GSH was used to aid assembly of the NPs, while the abovementioned DEVD peptide sequence integrated into the nanoprobe was used as an enzyme-specific moiety to induce the probe activation that leads to 19F MRI signal enhancement.193
Finally, worth of mentioning is also an approach using fluorinated ionic liquids capable of sensing multiple biological targets. Different fluorinated anions are loaded in hollow mesoporous silica NPs, while the pores are sealed with stimuli-responsive copolymers. After presentation of the stimulus, such as low pH, GSH or matrix metalloproteinase expression, the coating polymers will degrade to result in the ionic liquid release and recovery of 19F MRI signals.194,195
4.3. Responsive 19F CEST MRI probes
The youngest methodology that utilizes responsive fluorinated probes is based on the CEST effect; therefore, just a handful number of probes has been developed to date, for sensing metal ions only. The first probe suitable for this methodology, 5,5′-difluoro derivative of BAPTA (5F-BAPTA, 102), was reported in the early 1980's for the purpose of sensing Ca(II) by means of 19F NMR.196 Meanwhile, with the rapid development of CEST in the past two decades (see above) and a growing interest to develop responsive probes, the same molecule was utilized to demonstrate that a combination of 19F MRI and CEST can be very beneficial for sensing ions by means of CEST (iCEST). The main advantage of moving from 1H to 19F frequency and performing iCEST is a significant increase in the ratio of exchanging pools that contain fluorine. Namely, while the exchanging protons at 1H CEST usually take a small fraction of the entire proton amount in bulk water, the higher ratio of fluorine containing exchanging pools results in more efficient magnetisation transfer. Consequently, a detection of much lower concentrations is possible with iCEST.181The abovementioned first report on the iCEST methodology using the fluorinated molecule utilized the 5F-BAPTA to detect Ca(II) in presence of its competitors Zn(II) and Mg(II).197 This is possible because a formation of complex between 5F-BAPTA and Ca(II) provides 5F-BAPTA in free and Ca-bound forms in dynamic exchange, giving a CEST effect at 19F frequency. On contrary, Mg(II) does not form the complex, while Zn(II) binds strongly, disallowing exchange between free and bound 5F-BAPTA species, hence the CEST effect, too. In the subsequent study, the same principle was used with analogous probe, 5,5′,6,6′-tetrafluoro-BAPTA (TF-BAPTA, 103) to tailor the specificity of iCEST for Zn(II) and Fe(II), demonstrating the great power of this methodology for development of methods to image ions as important biomarkers.198 Consequently, the latter probe was utilized to image the changes of Zn(II) in vivo in a transgenic adenocarcinoma of the mouse prostate model by inducing the Zn(II) secretion with D-glucose injection (Fig. 13d).199
Finally, another type of fluorinated probe suitable for iCEST was developed and used to map labile Zn(II) in the hippocampus of mice. To this end, a series of fluorinated bispyridine derivatives 104–109 were prepared and examined to adjust the probe features for the desired purpose. The most suitable probe reversibly binds Zn(II) with low binding affinity and exhibits fast dissociation rate, which is beneficial for implementation of 19F-iCEST. When administered intracranially, the probe 108 was able to detect labile Zn(II) pools in hippocampus, a Zn-rich brain region, versus thalamus, a Zn-poor brain region (Fig. 13e).200 The extension of this approach may open possibilities for detection and quantification of different endogenous and biologically relevant metal ions.
5. Responsive probes for hyperpolarized MRI
Hyperpolarised MRI (HP-MRI) is an emerging technique attempting to circumvent the issues related with the low signal intensity of conventional MRI. Several hyperpolarisation methodologies, such as the dissolution dynamic nuclear polarisation (dDNP), spin-exchange optical pumping (SEOP), parahydrogen-induced polarisation (PHIP) and signal amplification by reversible exchange (SABRE) have been developed to amplify the MRI signal.201–203 Upon exposure of the MRI probe to a certain hyperpolarization procedure, the nuclear spins of the probe is largely enhanced to boost its MRI signal to a few orders of magnitude larger signal intensities.Usually, 3He, 13C, 15N, 19F, 31P or 129Xe are preferred nuclei employed as HP agents.204,205 These are highly potent for biological applications, as they: (1) contain broad chemical shift dispersion; (2) have no background signal thanks to their low or no natural existence in tissue; (3) exhibit relaxation times longer than 1H and hence permitting longer measurement time window. A high number of the nuclei prone to hyperpolarization also allows for a great variety of options to develop bioresponsive probes based on this principle.5 Such probes in the promoted state interact with their biological target, displaying an alteration in chemical shift or signal intensity. This reflects on the associated biological event by providing important information in real-time, helping quantitively map biological events or disease severity in 3D via time-resolved measurements. In turn, the magnified signals of bioresponsive and HP-MRI probes can be detected by a single MRI scan, which enables the detection of fast biological events with high, possibly subsecond temporal resolution. In the following sections we briefly highlight selected examples of HP-MRI sensor probes for pH, metal cations and ROS species. It should be noted that instead of developing such sensors for neurotransmitters, different neurotransmitter molecules (e.g. amino acids) can be hyperpolarized and studied directly.206,207 Similarly, observing the HP substrates appears to be the most suitable strategy for monitoring biological processes that involve target enzymes and metabolic processes.208,209 Selected enzymatic reactions can be followed by zero- and low-field NMR experiments, too.210
5.1. pH sensitive hyperpolarized probes
13C-labeled carboxylates are the most common functional groups used to prepare pH responsive HP probes. The typical examples are molecules 110 and 111 (Fig. 14), whose enhanced 13C signals were amplified by dDNP polarization technique and exhibited remarkable pH dependence.211,212 The carboxylic groups of these probes protonate/deprotonate at different pH values, which affects their 13C signals by changing their relaxation times and generation of a notable shift in their 13C MR signals as a function of pH. The first proof-of-principle in vivo study was performed using hyperpolarized bicarbonate (110) to map the pH values in a mouse tumour model.211 The robustness of 13C-labelled zymonic acid (111) response was initially confirmed by performing the in vitro experiments in a buffered solution and a blood sample. Further in vivo tests in rat kidney bladder, kidney and tumours demonstrated plausible properties of this probe, including non-toxicity, concentration- and temperature-independences.212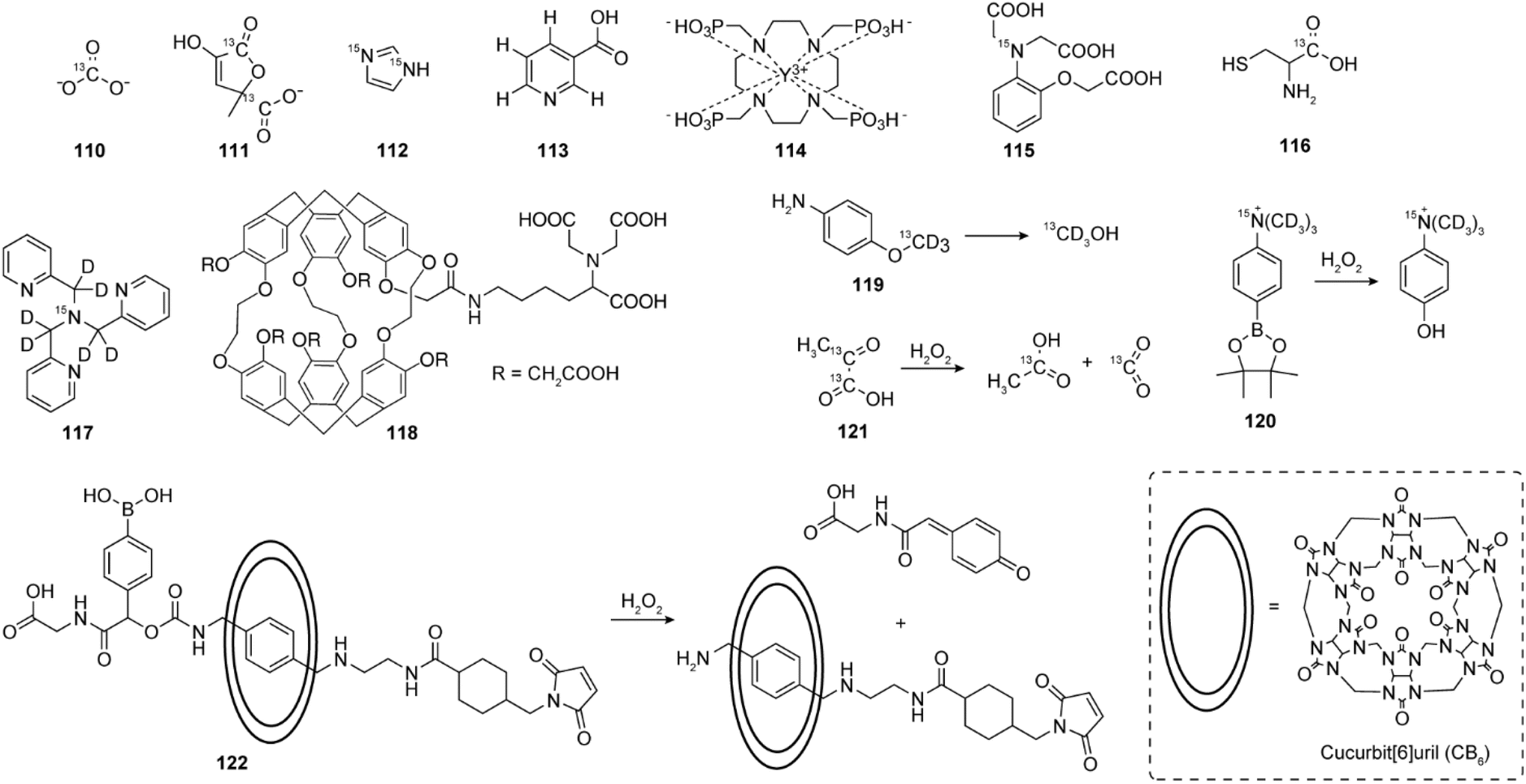 | ||
| Fig. 14 The representative HP probes that are sensitive to pH (110–114), Ca(II) (115), Zn(II) (116–118), HOCl (119) and H2O2 (120–122). These presented probes contain either 3He, 13C, 15N, 89Y or 129Xe nuclei to generate HP signal. The following techniques were employed for the signal amplification: dDNP for probes 110–111, 114–117 and 119–120; SABRE for probes 112–113 and 121; SEOP for probes 118 and 122. | ||
By employing SABRE as the hyperpolarization method, the 15N and 1H signals of probes 112 and 113, respectively, were enhanced and used for pH sensing.213,214 Probe 112 is an imidazole-15N2, a molecule enriched with two 15N nuclei, which has advantageous properties for HP-MRI. Namely, it exhibits long relaxation times from 15N signal while providing a broad chemical shift dispersion, suggesting it a suitable moiety to develop new probes for biological applications.213 Finally, metal-based complexes were also employed for pH sensing. The complex 114 chelated with 89Y(III) was investigated as a potent pH sensor.215 This probe can also provide a broader chemical shift dispersion, while having even longer relaxation times than any other nuclei mentioned in this section.
5.2. Metal-cation sensitive hyperpolarized probes
The hyperpolarized probes that sense metal cations are usually their well-known chelators, yet enriched with the appropriate NMR-active isotopes. The Ca(II)-sensor 115 is an APTRA-based and 15N-labeled probe and its signal can be enhanced by dDNP method. The complexation of 115 with Ca(II) can produce ∼5 ppm chemical shift, which helps sensing Ca(II) in a hyperpolarized state.216 In a similar fashion, chelators EDTA (ethylenediaminetetraacetic acid) and EGTA with 13C-labeled carboxylates were used to simultaneously detect several metals including Ca(II), due to their different 13C NMR shifts.217Likewise using dDNP, the amino acid-based and 13C-labeled probe [1-13C]Cys (116) was employed to quantitatively detect the concentration of Zn(II). The probe demonstrated advantageous properties such as the large changes of chemical shift, good selectivity and favourable T1.218 On the other hand, by using another well-known and 15N-labeled chelator for Zn(II), tris(2-pyridylmethyl)amine (TPA), the deuterated Zn(II)-sensor [15N]TPA-d6 (117) was prepared and its Zn(II)-binding properties were studied after being hyperpolarised with dDNP. The probe could successfully and quantitatively detect free Zn(II) contents in human prostate tissue homogenate and intact human prostate epithelial cells.219
In addition to isotope-enriched chelators of selected metal cations, structurally more complex sensors have also been reported. Suitable examples are those developed for sensing with hyperpolarised 129Xe MRI using SEOP methodology, where the polarization state is delivered from pre-polarized Rb vapor to 129Xe nuclei.201 This nucleus is preferred option for such studies due to its broad chemical shift window, chemical inertness and lower toxicity, practical solubility in aqueous solution, but also a large appetency for binding with molecular cages to provide chances for bioresponsive applications. In a notable study, probe 118 was developed by combining the cryptophane derivative as a cage for 129Xe, while the nitrilotriacetic acid served as a Zn(II) chelator. The encapsulation of xenon gives a frequency change and provides a promoted sensitivity upon the subsequently combining of Zn(II).220 Using completely different approach, a genetically encoded Zn(II) hyperCEST sensor was developed. A maltose binding protein accommodates 129Xe, which after undergoing further mutations was converted to selectively bind Zn(II). This interaction enables the control of exchange kinetics of 129Xe bound to protein and free 129Xe in solution, achieving a turn-on detection of Zn(II) at low micromolar concentration.221
5.3. ROS sensitive hyperpolarized probes
The detection of the ROS species by means of hyperpolarized methods mainly encompasses small-molecular-weight probes that are prone to chemical conversion by the redox conditions. A typical example is 119 as the probe sensitive to hypochloric acid (HOCl). This is a p-anisidine molecule with the deuterated and 13C-labeled methoxy group, which converts to 13CD3OH after the treatment with HOCl.222 Similarly, 120 is a15N-labeled probe based on a15N-labeled aryl boronate pinacol ester as a sensor for H2O2. In this molecule, boronate pinacol ester was designed as the sensing moiety, which is the appropriate substrate for H2O2. Upon polarization via dDNP method, boronate as the sensing part of this probe reacts with H2O2, leading to a cleavage of the boronate pinacol ester. This gives rise to formation of a new 15N signal present at the sensing moiety of this probe, resulting in a methodology able to indirectly detect H2O2.223 Rather than the dDNP method, SABRE/PHIP was used to polarize [1,2-13C2]pyruvate (121) with two 13C atoms by forming 13C-labeled acetic acid and carbon dioxide in response to reaction with H2O2.224 Finally, a SEOP-polarized rotaxane probe 122 that is based on the cucurbit[6]uril (CB6) host for xenon gas was used for sensing the H2O2, too. This probe can be unlocked by H2O2 to release free CB6, providing a new host molecule for the 129Xe chelation. Consequently, the 129Xe hyperCEST signal turns on to indicate the interaction of CB6 rotaxane with its target H2O2.2256. Practical challenges for using MRI responsive probes
The previous sections evidence on a huge potential that MRI responsive probes may have for the functional molecular imaging applications. However, the great abundance of the MRI sensors has not been yet reflected proportionally into their use for visualisation and assessment of various biological processes. There are multiple reasons for this state; we shall try to indicate some of them in the following discussion. In parallel, by listing these practical aspects, we would like to encourage further research on the bioresponsive MRI probes, particularly on their in vivo validation and utilisation. Many of the current examples discussed in this Perspective came from the proof-of-concept studies, whereas a long-term implementation of these probes can stimulate the unmet research directions and the future endeavours that can be highly beneficial for the research community across many disciplines.MRI is one of the least sensitive imaging techniques, especially when compared to positron emission tomography, single-photon emission computed tomography or optical imaging.226 A few consequences result from this fact, primarily that the MRI probes need to be applied in much higher amounts compared to the probes for modalities mentioned above, ranging from tens of μM for T1-weighted and T2-weighted probes to low mM concentrations for CEST or 19F probes.152,227,228 This brings important issues related to the appropriate probe delivery to the target region, but also a toxicity; consequently, imaging intracellular targets may become challenging, due to high payload of probes that need to be internalized, as well as the recognition of desired cell types.229 Due to such sensitivity of MRI, it is worth noting that selection of the biological targets needs to be carefully considered – these also need to be available in sufficient amounts to allow the detection. In turn, it is not important only to deliver detectable amount of the bioresponsive probe in the target region, but to anticipate that interaction between the probe and its target should generate sufficient and detectable MRI signal change.
Further considerations should be related to each of the MRI methods discussed above. Namely, they all display different sensitivities, which depend on the strategy used to generate the signal, also the chemistry of its MRI probes. The most abundantly used T1-weighted and T2-weighted probes are based on (super)paramagnetic species, and their detection can be achieved at the level of a few tens of μM concentrations.227 However, the use of probes based on the most potent paramagnetic ion, Gd(III), in such amounts are coupled with certain safety concerns.230 This is opening the space for development of alternative probes based on Mn(II), which then implies the use of even higher concentrations due to the lower potency of the latter type of probes. As well, water exhibits T1 and T2 relaxation even in the absence of paramagnetic probes, giving a background signal. Thus, differentiating diamagnetic from the paramagnetic signal (i.e. signals produced from bulk water from the one of the MRI probes, respectively) is often impossible, disallowing quantification of the MRI probe and other associated and essential insights, such as assessment on the concentration and the fate of the studied target analyte. To overcome this issue, the following approaches can be considered: (i) improvement of the MRI signal changes of the probe (e.g. larger r1 or r2, CEST signal or frequency shift changes) to ensure unambiguous conclusions, or (ii) design of bioresponsive probes tethered with fluorinated groups or radioisotopes that are suitable for multi-frequency and -modal investigations, to allow studies of e.g. T1-weighted bioresponsive probes through quantitative imaging methods, such as 19F MRI, positron emission tomography or single-photon emission computed tomography.231–234
Due to a different way to generate the MRI signal, CEST probes are typically applied in one to two orders of magnitude higher concentrations than T1-weighted and T2-weighted probes, which is less advantageous. Here the ability to use alternatives to lanthanide-based complexes, such as diaCEST probes, provides less concerns for CEST MRI regarding the toxicity. In addition, the use of saturation pulses is limiting the temporal resolution of the methodology, which may be crucial for observing processes with faster dynamics (a few seconds). However, designing the probes that possess two CEST effects (i.e. at two different frequencies) in a single molecule of the probe can provide advantages. In such case, the two CEST effects behave differently, with one being responsive and the other non-responsive, respectively; hence, the former CEST pool is used to sense the target biomarker, while the latter is employed as a non-responsive control, enabling the ratiometric CEST.14 Nevertheless, this approach may be challenging to apply in vivo due to the variability of line-widths. In such case, an alternative method where the frequency shifting of the responsive CEST probe is followed could be a good option to eliminate the undesirable effects.139
The use of fluorinated agents is advantageous since they provide large frequency dispersion and ability to perform quantitative studies. Unlike methods for 1H MRI where the signal essentially comes from water, high payload of 19F is needed to ensure detection.228 A couple of disadvantages are associated with this fact: first, large amounts of fluorine atoms change the solubility of 19F MRI probes, making their application into the target tissue difficult; also, acquisition times can be very long, often in the order of tens of minutes, which prevents observation of biological processes on the seconds scale. Nevertheless, despite these difficulties, the benefits of 19F MRI seen through the high specificity on the measurements or the ability to quantify the amount of the bioresponsive probe and hence the target make it the preferred method for longitudinal investigations.
Finally, HP-MRI probes have been developing to circumvent the problems related to low sensitivity of MRI measurements. Their use aims to reduce the applied concentrations of probes, shorten acquisition times, while many suitable nuclei provide a wide range of chemical platforms that can be developed as MRI sensors, thus dramatically improving specificity of the measurements. However, a significant limitation for this technique is fast relaxation of the hyperpolarised signal, which dramatically reduces the experimental time window. Therefore, these experiments need to be well planned, since the delivery of HP-MRI sensors to follow a specific biological process must be fast and efficient to ensure quick conversion of the MRI signal that is being analysed.
There are a few other practical points that influence functional MRI experiments with bioresponsive probes and go beyond any chemical nature. For instance, strength of the scanner magnetic field will influence the SNR or will make some effects be more prominent in low or high magnetic fields (e.g. r1 and r2 changes, see above). Additionally, the MRI hardware may play a very important role, since the good adjustment of electronics of the scanner and radiofrequency coils can also contribute to high SNR. Together with properly designed imaging sequencies and associated experimental protocols, these aspects altogether may be essential for improving the spatiotemporal resolution of the desired biological processes, which determines the success of the investigation.
7. Summary and conclusions
This Perspective gives an overview of the recent work done in the broad field of bioresponsive MRI probes suitable for different functional studies and MRI modalities. The richness of the approaches is evident, showing that a plethora of biological markers and processes could be studied in this manner. Indeed, a significant progress has been made in the past decade in advancing the proof-of-concept studies at the translational level, which focused on using the chemical probes to obtain functional MRI results in animal models. Achieving the next level that includes implementation of this approach into the clinical practice will be equally challenging and may be associated with further limitations on the types of sensor probes that may be used, or for which diseases. However, the use of bioresponsive probes to image biology is extremely valuable already at the current level for the biomedical sciences, as it allows collection of essential insights and knowledge. A bright future stands for this field since many avenues are still to be open, thus bringing large benefits to contemporary molecular imaging.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
G. A. conceptualized the manuscript. All authors contributed to the literature search, writing and figure making of the manuscript and were involved in revising, editing, and proofreading the initial and revised versions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Shanghai Municipal Science and Technology Major Project (Grant No. 2019SHZDZX02) Jiangsu University (Grant No. 5501310019), the Natural Science Foundation of Jiangsu Province (Grant No. BK20220528), and the National Natural Science Foundation of China (Grants No. 22174154, 22207046 and T2250710181).Notes and references
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS PubMed.
- Q. Meng, M. Wu, Z. Shang, Z. Zhang and R. Zhang, Coord. Chem. Rev., 2022, 457, 214398 CrossRef CAS.
- Y. Mo, C. Huang, C. Liu, Z. Duan, J. Liu and D. Wu, Macromol. Rapid Commun., 2023, 44, 2200744 CrossRef CAS PubMed.
- G. Angelovski, B. J. Tickner and G. Wang, Nat. Chem., 2023, 15, 755–763 CrossRef CAS PubMed.
- C. F. G. C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS PubMed.
- S. Alzola-Aldamizetxebarria, L. Fernández-Méndez, D. Padro, J. Ruíz-Cabello and P. Ramos-Cabrer, ACS Omega, 2022, 7, 36905–36917 CrossRef CAS PubMed.
- G. J. Stasiuk, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 741–770 Search PubMed.
- M. Norek and J. A. Peters, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 64–82 CrossRef CAS PubMed.
- P. Caravan, M. T. Greenfield and J. W. M. Bulte, Magn. Reson. Med., 2001, 46, 917–922 CrossRef CAS PubMed.
- N. Genicio, M. Bañobre-López, O. Gröhn and J. Gallo, Coord. Chem. Rev., 2021, 447, 214150 CrossRef CAS.
- V. Catanzaro, C. V. Gringeri, V. Menchise, S. Padovan, C. Boffa, W. Dastru, L. Chaabane, G. Digilio and S. Aime, Angew. Chem., Int. Ed., 2013, 52, 3926–3930 CrossRef CAS PubMed.
- S. Aime, F. Fedeli, A. Sanino and E. Terreno, J. Am. Chem. Soc., 2006, 128, 11326–11327 CrossRef CAS PubMed.
- L. A. Ekanger and M. J. Allen, Metallomics, 2015, 7, 405–421 CrossRef CAS PubMed.
- S. Abdella, F. Abid, S. H. Youssef, S. Kim, F. Afinjuomo, C. Malinga, Y. Song and S. Garg, Drug Discovery Today, 2023, 28, 103414 CrossRef CAS PubMed.
- Y. Liu, D. Zhang, Y. Qu, F. Tang, H. Wang, A. Ding and L. Li, Chem. Biomed. Imaging, 2024, 2, 81–97 CrossRef CAS PubMed.
- G. B. Giovenzana, R. Negri, G. A. Rolla and L. Tei, Eur. J. Inorg. Chem., 2012, 2012, 2035–2039 CrossRef CAS.
- M. Woods, G. E. Kiefer, S. Bott, A. Castillo-Muzquiz, C. Eshelbrenner, L. Michaudet, K. McMillan, S. D. K. Mudigunda, D. Grin, G. Tircso, S. R. Zhang, P. Zhao and A. D. Sherry, J. Am. Chem. Soc., 2004, 126, 9248–9256 CrossRef CAS PubMed.
- M. P. Lowe, D. Parker, O. Reany, S. Aime, M. Botta, G. Castellano, E. Gianolio and R. Pagliarin, J. Am. Chem. Soc., 2001, 123, 7601–7609 CrossRef CAS PubMed.
- I. Mamedov, A. Mishra, G. Angelovski, H. A. Mayer, L.-O. Pålsson, D. Parker and N. K. Logothetis, Dalton Trans., 2007, 5260–5267 RSC.
- S. M. Vibhute, J. Engelmann, T. Verbić, M. E. Maier, N. K. Logothetis and G. Angelovski, Org. Biomol. Chem., 2013, 11, 1294–1305 RSC.
- S. Aime, A. Barge, M. Botta, D. Parker and A. S. DeSousa, J. Am. Chem. Soc., 1997, 119, 4767–4768 CrossRef CAS.
- S. R. Zhang, K. C. Wu and A. D. Sherry, Angew. Chem., Int. Ed., 1999, 38, 3192–3194 CrossRef CAS PubMed.
- R. Hovland, C. Gløgård, A. J. Aasen and J. Klaveness, J. Chem. Soc., Perkin Trans. 2, 2001, 929–933 RSC.
- S. Lacerda, D. Ndiaye and É. Tóth, in Advances in Inorganic Chemistry, ed. C. D. Hubbard and R. van Eldik, Academic Press, 2021, vol. 78, pp. 109–142 Search PubMed.
- J. Salaam, T. Fogeron, G. Pilet, R. Bolbos, C. Bucher, L. Khrouz and J. Hasserodt, Angew. Chem., Int. Ed., 2023, 62, e202212782 CrossRef CAS PubMed.
- R. Botár, E. Molnár, G. Trencsényi, J. Kiss, F. K. Kálmán and G. Tircsó, J. Am. Chem. Soc., 2020, 142, 1662–1666 CrossRef.
- J. Han, G. Liang and D. Xing, Chem.–Eur. J., 2019, 25, 8353–8362 CrossRef CAS.
- T.-H. Shin, Y. Choi, S. Kim and J. Cheon, Chem. Soc. Rev., 2015, 44, 4501–4516 RSC.
- M. M. Ali, M. Woods, P. Caravan, A. C. L. Opina, M. Spiller, J. C. Fettinger and A. D. Sherry, Chem.–Eur. J., 2008, 14, 7250–7258 CrossRef CAS PubMed.
- M. P. I. Bhuiyan, M. P. Aryal, B. Janic, K. Karki, N. R. S. Varma, J. R. Ewing, A. S. Arbab and M. M. Ali, Contrast Media Mol. Imaging, 2015, 10, 481–486 CrossRef CAS.
- B. Janic, M. P. I. Bhuiyan, J. R. Ewing and M. M. Ali, ACS Sens., 2016, 1, 975–978 CrossRef CAS PubMed.
- Y. Chen, Q. Yin, X. Ji, S. Zhang, H. Chen, Y. Zheng, Y. Sun, H. Qu, Z. Wang, Y. Li, X. Wang, K. Zhang, L. Zhang and J. Shi, Biomaterials, 2012, 33, 7126–7137 CrossRef CAS PubMed.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon, A. Quinones-Hinojosa, J. W. M. Bulte, T. Hyeon and A. A. Gilad, J. Am. Chem. Soc., 2011, 133, 2955–2961 CrossRef CAS PubMed.
- X. Li, W. Zhao, X. Liu, K. Chen, S. Zhu, P. Shi, Y. Chen and J. Shi, Acta Biomater., 2016, 30, 378–387 CrossRef CAS PubMed.
- X. W. Li, W. R. Zhao, Y. J. Liu, X. H. Liu, P. Shi, Y. S. Li and J. L. Shi, J. Mater. Chem. B, 2016, 4, 4313–4321 RSC.
- M. F. Bennewitz, T. L. Lobo, M. K. Nkansah, G. Ulas, G. W. Brudvig and E. M. Shapiro, ACS Nano, 2011, 5, 3438–3446 CrossRef CAS PubMed.
- J. Pellico, C. M. Ellis, J. Miller and J. J. Davis, Chem. Commun., 2019, 55, 8540–8543 RSC.
- C. M. Ellis, D. Yuan, F. E. Mózes, J. J. Miller and J. J. Davis, Chem. Commun., 2023, 59, 1605–1608 RSC.
- L. Zhu, Y. Yang, K. Farquhar, J. Wang, C. Tian, J. Ranville and S. G. Boyes, ACS Appl. Mater. Interfaces, 2016, 8, 5040–5050 CrossRef CAS PubMed.
- H. Lu, A. Chen, X. Zhang, Z. Wei, R. Cao, Y. Zhu, J. Lu, Z. Wang and L. Tian, Nat. Commun., 2022, 13, 7948 CrossRef CAS PubMed.
- X. Wang, D. Niu, Q. Wu, S. Bao, T. Su, X. Liu, S. Zhang and Q. Wang, Biomaterials, 2015, 53, 349–357 CrossRef CAS PubMed.
- Y. Chen, P. Xu, Z. Shu, M. Wu, L. Wang, S. Zhang, Y. Zheng, H. Chen, J. Wang, Y. Li and J. Shi, Adv. Funct. Mater., 2014, 24, 4386–4396 CrossRef CAS.
- Y. Wei, R. Liao, A. A. Mahmood, H. Xu and Q. Zhou, Acta Biomater., 2017, 55, 194–203 CrossRef CAS PubMed.
- J. Lu, J. Sun, F. Li, J. Wang, J. Liu, D. Kim, C. Fan, T. Hyeon and D. Ling, J. Am. Chem. Soc., 2018, 140, 10071–10074 CrossRef CAS PubMed.
- J. Lin, P. Xin, L. An, Y. Xu, C. Tao, Q. Tian, Z. Zhou, B. Hu and S. Yang, Chem. Commun., 2019, 55, 478–481 RSC.
- S. Okada, S. Mizukami, T. Sakata, Y. Matsumura, Y. Yoshioka and K. Kikuchi, Adv. Mater., 2014, 26, 2989–2992 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- W. H. Li, S. E. Fraser and T. J. Meade, J. Am. Chem. Soc., 1999, 121, 1413–1414 CrossRef CAS.
- P. Yue and G. Angelovski, Analysis Sensing, 2023, 3, e202300019 CrossRef CAS.
- G. Angelovski, P. Fouskova, I. Mamedov, S. Canals, E. Toth and N. K. Logothetis, ChemBioChem, 2008, 9, 1729–1734 CrossRef CAS PubMed.
- G. Angelovski, S. Gottschalk, M. Milošević, J. Engelmann, G. E. Hagberg, P. Kadjane, P. Andjus and N. K. Logothetis, ACS Chem. Neurosci., 2014, 5, 360–369 CrossRef CAS.
- T. Savić, G. Gambino, V. S. Bokharaie, H. R. Noori, N. K. Logothetis and G. Angelovski, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 20666–20671 CrossRef.
- L. Connah, R. Joshi, S. Vibhute, G. Gambino, J. D. G. Correia and G. Angelovski, Org. Lett., 2019, 21, 5378–5382 CrossRef CAS PubMed.
- S. Gündüz, N. Nitta, S. Vibhute, S. Shibata, M. E. Maier, N. K. Logothetis, I. Aoki and G. Angelovski, Chem. Commun., 2015, 51, 2782–2785 RSC.
- A. Moussaron, S. Vibhute, A. Bianchi, S. Gündüz, S. Kotb, L. Sancey, V. Motto-Ros, S. Rizzitelli, Y. Crémillieux, F. Lux, N. K. Logothetis, O. Tillement and G. Angelovski, Small, 2015, 11, 4900–4909 CrossRef CAS.
- G. Gambino, T. Gambino, L. Connah, F. La Cava, H. Evrard and G. Angelovski, J. Med. Chem., 2021, 64, 7565–7574 CrossRef CAS PubMed.
- F. Garello, S. Vibhute, S. Gündüz, N. K. Logothetis, E. Terreno and G. Angelovski, Biomacromolecules, 2016, 17, 1303–1311 CrossRef CAS PubMed.
- A. Barandov, B. B. Bartelle, C. G. Williamson, E. S. Loucks, S. J. Lippard and A. Jasanoff, Nat. Commun., 2019, 10, 897 CrossRef PubMed.
- H. F. Ozbakir, A. D. C. Miller, K. B. Fishman, A. F. Martins, T. E. Kippin and A. Mukherjee, ACS Sens., 2021, 6, 3163–3169 CrossRef CAS PubMed.
- S. Gündüz, T. Savić, R. Pohmann, N. K. Logothetis, K. Scheffler and G. Angelovski, ACS Sens., 2016, 1, 483–487 CrossRef.
- T. Atanasijevic, M. Shusteff, P. Fam and A. Jasanoff, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 14707–14712 CrossRef CAS PubMed.
- S. Okada, B. B. Bartelle, N. Li, V. Breton-Provencher, J. J. Lee, E. Rodriguez, J. Melican, M. Sur and A. Jasanoff, Nat. Nanotechnol., 2018, 13, 473–477 CrossRef CAS PubMed.
- K. Hanaoka, K. Kikuchi, Y. Urano and T. Nagano, J. Chem. Soc., Perkin Trans. 2, 2001, 1840–1843 RSC.
- J. L. Major, G. Parigi, C. Luchinat and T. J. Meade, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 13881–13886 CrossRef CAS PubMed.
- J. L. Major, R. M. Boiteau and T. J. Meade, Inorg. Chem., 2008, 47, 10788–10795 CrossRef CAS PubMed.
- G. Wang and G. Angelovski, Angew. Chem., Int. Ed., 2021, 60, 5734–5738 CrossRef CAS PubMed.
- G. Wang, H. Martin, S. Amézqueta, C. Ràfols, C. S. Bonnet and G. Angelovski, Inorg. Chem., 2022, 61, 16256–16265 CrossRef CAS PubMed.
- A. C. Esqueda, J. A. Lopez, G. Andreu-De-Riquer, J. C. Alvarado-Monzon, J. Ratnakar, A. J. M. Lubag, A. D. Sherry and L. M. De Leon-Rodriguez, J. Am. Chem. Soc., 2009, 131, 11387–11391 CrossRef CAS PubMed.
- A. J. M. Lubag, L. M. De Leon-Rodriguez, S. C. Burgess and A. D. Sherry, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 18400–18405 CrossRef CAS.
- J. Yu, A. F. Martins, C. Preihs, V. C. Jordan, S. Chirayil, P. Y. Zhao, Y. K. Wu, K. Nasr, G. E. Kiefer and A. D. Sherry, J. Am. Chem. Soc., 2015, 137, 14173–14179 CrossRef CAS PubMed.
- M. V. Clavijo Jordan, S.-T. Lo, S. Chen, C. Preihs, S. Chirayil, S. Zhang, P. Kapur, W.-H. Li, M. De Leon-Rodriguez Luis, J. M. Lubag Angelo, M. Rofsky Neil and A. D. Sherry, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, E5464–E5471 CrossRef CAS PubMed.
- A. F. Martins, V. C. Jordan, F. Bochner, S. Chirayil, N. Paranawithana, S. R. Zhang, S. T. Lo, X. D. Wen, P. Y. Zhao, M. Neeman and A. D. Sherry, J. Am. Chem. Soc., 2018, 140, 17456–17464 CrossRef CAS PubMed.
- C. S. Bonnet, Coord. Chem. Rev., 2018, 369, 91–104 CrossRef CAS.
- Q. Xue, R. Kang, D. J. Klionsky, D. Tang, J. Liu and X. Chen, Autophagy, 2023, 19, 2175–2195 CrossRef CAS PubMed.
- K. P. Malikidogo, H. Martin and C. S. Bonnet, Pharmaceuticals, 2020, 13, 436 CrossRef CAS.
- E. L. Que and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 15942–15943 CrossRef CAS.
- E. L. Que, E. Gianolio, S. L. Baker, A. P. Wong, S. Aime and C. J. Chang, J. Am. Chem. Soc., 2009, 131, 8527–8536 CrossRef CAS.
- N. N. Paranawithana, A. F. Martins, V. Clavijo Jordan, P. Zhao, S. Chirayil, G. Meloni and A. D. Sherry, J. Am. Chem. Soc., 2019, 141, 11009–11018 CrossRef CAS PubMed.
- T. M. Hagen, Antioxid. Redox Signaling, 2003, 5, 503–506 CrossRef CAS PubMed.
- D. Trachootham, W. Lu, M. A. Ogasawara, N. R.-D. Valle and P. Huang, Antioxid. Redox Signaling, 2008, 10, 1343–1374 CrossRef CAS.
- T. L. Clanton, J. Appl. Physiol., 2007, 102, 2379–2388 CrossRef CAS PubMed.
- M. Singer, P. J. Young, J. G. Laffey, P. Asfar, F. S. Taccone, M. B. Skrifvars, C. S. Meyhoff and P. Radermacher, Crit. Care, 2021, 25, 440 CrossRef PubMed.
- C. Tu, R. Nagao and A. Y. Louie, Angew. Chem., Int. Ed., 2009, 48, 6547–6551 CrossRef CAS PubMed.
- M. Harris, J. L. Kolanowski, E. S. O'Neill, C. Henoumont, S. Laurent, T. N. Parac-Vogt and E. J. New, Chem. Commun., 2018, 54, 12986–12989 RSC.
- H. Wang, V. C. Jordan, I. A. Ramsay, M. Sojoodi, B. C. Fuchs, K. K. Tanabe, P. Caravan and E. M. Gale, J. Am. Chem. Soc., 2019, 141, 5916–5925 CrossRef CAS PubMed.
- S. M. Pinto, V. Tomé, M. J. F. Calvete, M. M. C. A. Castro, É. Tóth and C. F. G. C. Geraldes, Coord. Chem. Rev., 2019, 390, 1–31 CrossRef CAS.
- G. S. Loving, S. Mukherjee and P. Caravan, J. Am. Chem. Soc., 2013, 135, 4620–4623 CrossRef CAS PubMed.
- E. M. Gale, C. M. Jones, I. Ramsay, C. T. Farrar and P. Caravan, J. Am. Chem. Soc., 2016, 138, 15861–15864 CrossRef CAS PubMed.
- S.-S. Xue, Y. Pan, W. Pan, S. Liu, N. Li and B. Tang, Chem. Sci., 2022, 13, 9468–9484 RSC.
- Y. He, Y. Cao, Z. Mao, Y. Zhou, Y. Zhang and R. Pei, J. Mater. Chem. B, 2021, 9, 1821–1832 RSC.
- P. Zhang, J. Zeng, Y. Li, C. Yang, J. Meng, Y. Hou and M. Gao, Angew. Chem., Int. Ed., 2021, 60, 8130–8138 CrossRef CAS PubMed.
- D. Ma, M. Shi, X. Li, J. Zhang, Y. Fan, K. Sun, T. Jiang, C. Peng and X. Shi, Bioconjugate Chem., 2020, 31, 352–359 CrossRef CAS PubMed.
- M.-H. Kim, H.-Y. Son, G.-Y. Kim, K. Park, Y.-M. Huh and S. Haam, Biomaterials, 2016, 101, 121–130 CrossRef CAS PubMed.
- S. Iwaki, K. Hanaoka, W. Piao, T. Komatsu, T. Ueno, T. Terai and T. Nagano, Bioorg. Med. Chem. Lett., 2012, 22, 2798–2802 CrossRef CAS PubMed.
- E. Di Gregorio, G. Ferrauto, E. Gianolio, S. Lanzardo, C. Carrera, F. Fedeli and S. Aime, ACS Nano, 2015, 9, 8239–8248 CrossRef CAS.
- H. Zhou, M. Guo, J. Li, F. Qin, Y. Wang, T. Liu, J. Liu, Z. F. Sabet, Y. Wang, Y. Liu, Q. Huo and C. Chen, J. Am. Chem. Soc., 2021, 143, 1846–1853 CrossRef CAS PubMed.
- H. Zhu, L. Zhang, Y. Liu, Y. Zhou, K. Wang, X. Xie, L. Song, D. Wang, C. Han and Q. Chen, Sci. Rep., 2016, 6, 39245 CrossRef CAS.
- M. G. Shapiro, G. G. Westmeyer, P. A. Romero, J. O. Szablowski, B. Kuster, A. Shah, C. R. Otey, R. Langer, F. H. Arnold and A. Jasanoff, Nat. Biotechnol., 2010, 28, 264–270 CrossRef CAS PubMed.
- E. M. Brustad, V. S. Lelyveld, C. D. Snow, N. Crook, S. T. Jung, F. M. Martinez, T. J. Scholl, A. Jasanoff and F. H. Arnold, J. Mol. Biol., 2012, 422, 245–262 CrossRef CAS PubMed.
- T. Lee, L. X. Cai, V. S. Lelyveld, A. Hai and A. Jasanoff, Science, 2014, 344, 533–535 CrossRef CAS PubMed.
- N. Li and A. Jasanoff, Nature, 2020, 580, 239–244 CrossRef CAS PubMed.
- V. Hsieh, S. Okada, H. Wei, I. García-Álvarez, A. Barandov, S. R. Alvarado, R. Ohlendorf, J. Fan, A. Ortega and A. Jasanoff, J. Am. Chem. Soc., 2019, 141, 15751–15754 CrossRef CAS PubMed.
- F. Oukhatar, S. Même, W. Même, F. Szeremeta, N. K. Logothetis, G. Angelovski and É. Tóth, ACS Chem. Neurosci., 2015, 6, 219–225 CrossRef CAS PubMed.
- F. Oukhatar, H. Meudal, C. Landon, N. K. Logothetis, C. Platas-Iglesias, G. Angelovski and É. Tóth, Chem.–Eur. J., 2015, 21, 11226–11237 CrossRef CAS.
- F. Oukhatar, S. V. Eliseeva, C. S. Bonnet, M. Placidi, N. K. Logothetis, S. Petoud, G. Angelovski and É. Tóth, Inorg. Chem., 2019, 58, 13619–13630 CrossRef CAS.
- Đ. Toljić, C. Platas-Iglesias and G. Angelovski, Front. Chem., 2019, 7, 490 CrossRef.
- T. Budisantoso, H. Harada, N. Kamasawa, Y. Fukazawa, R. Shigemoto and K. Matsui, J. Physiol., 2013, 591, 219–239 CrossRef CAS PubMed.
- M. Perry, Q. Li and R. T. Kennedy, Anal. Chim. Acta, 2009, 653, 1–22 CrossRef CAS PubMed.
- S. Fliegel, I. Brand, R. Spanagel and H. R. Noori, Silico Pharmacol., 2013, 1, 7 CrossRef PubMed.
- Đ. Toljić and G. Angelovski, Chem. Commun., 2019, 55, 11924–11927 RSC.
- Đ. Toljić and G. Angelovski, ChemNanoMat, 2019, 5, 1456–1460 CrossRef.
- R. A. Moats, S. E. Fraser and T. J. Meade, Angew. Chem., Int. Ed., 1997, 36, 726–728 CrossRef CAS.
- A. Y. Louie, M. M. Huber, E. T. Ahrens, U. Rothbacher, R. Moats, R. E. Jacobs, S. E. Fraser and T. J. Meade, Nat. Biotechnol., 2000, 18, 321–325 CrossRef CAS PubMed.
- L. M. Urbanczyk-Pearson, F. J. Femia, J. Smith, G. Parigi, J. A. Duimstra, A. L. Eckermann, C. Luchinat and T. J. Meade, Inorg. Chem., 2008, 47, 56–68 CrossRef CAS PubMed.
- J. A. Duimstra, F. J. Femia and T. J. Meade, J. Am. Chem. Soc., 2005, 127, 12847–12855 CrossRef CAS PubMed.
- L. M. Lilley, S. Kamper, M. Caldwell, Z. K. Chia, D. Ballweg, L. Vistain, J. Krimmel, T. A. Mills, K. MacRenaris, P. Lee, E. A. Waters and T. J. Meade, Angew. Chem., Int. Ed., 2020, 59, 388–394 CrossRef CAS PubMed.
- T. Taghian, A. R. Batista, S. Kamper, M. Caldwell, L. Lilley, H. Li, P. Rodriguez, K. Mesa, S. Zheng, R. M. King, M. J. Gounis, S. Todeasa, A. Maguire, D. R. Martin, M. Sena-Esteves, T. J. Meade and H. L. Gray-Edwards, Mol. Ther.–Methods Clin. Dev., 2021, 23, 128–134 CrossRef CAS PubMed.
- J.-H. Tang, H. Li, C. Yuan, G. Parigi, C. Luchinat and T. J. Meade, J. Am. Chem. Soc., 2023, 145, 10045–10050 CrossRef CAS PubMed.
- M. Giardiello, M. P. Lowe and M. Botta, Chem. Commun., 2007, 4044–4046 RSC.
- R. Napolitano, G. Pariani, F. Fedeli, Z. Baranyai, M. Aswendt, S. Aime and E. Gianolio, J. Med. Chem., 2013, 56, 2466–2477 CrossRef CAS PubMed.
- H. Seo and H. A. Clark, Analyst, 2020, 145, 1169–1173 RSC.
- D. Granot and E. M. Shapiro, Magn. Reson. Med., 2011, 65, 1253–1259 CrossRef CAS PubMed.
- S. Matsumura, I. Aoki, T. Saga and K. Shiba, Mol. Pharm., 2011, 8, 1970–1974 CrossRef CAS PubMed.
- M. G. Shapiro, J. O. Szablowski, R. Langer and A. Jasanoff, J. Am. Chem. Soc., 2009, 131, 2484–2486 CrossRef CAS PubMed.
- Y. Matsumoto, R. Chen, P. Anikeeva and A. Jasanoff, Nat. Commun., 2015, 6, 8721 CrossRef CAS PubMed.
- J. Yun, M. Baldini, R. Chowdhury and A. Mukherjee, Analysis Sensing, 2022, 2, e202200019 CrossRef CAS PubMed.
- K. M. Ward, A. H. Aletras and R. S. Balaban, J. Magn. Reson., 2000, 143, 79–87 CrossRef CAS PubMed.
- M. T. McMahon, A. A. Gilad, J. W. M. Bulte and P. C. M. van Zijl, Chemical Exchange Saturation Transfer Imaging : Advances and Applications, Pan Stanford Publishing Pte. Ltd, Singapore, 2017 Search PubMed.
- P. C. M. van Zijl and N. N. Yadav, Magn. Reson. Med., 2011, 65, 927–948 CrossRef CAS PubMed.
- A. Rodríguez-Rodríguez, M. Zaiss, D. Esteban-Gómez, G. Angelovski and C. Platas-Iglesias, Int. Rev. Phys. Chem., 2021, 40, 51–79 Search PubMed.
- A. Bar-Shir, J. W. M. Bulte and A. A. Gilad, ACS Chem. Biol., 2015, 10, 1160–1170 CrossRef CAS PubMed.
- X. Yang, X. Song, Y. Li, G. Liu, S. Ray Banerjee, M. G. Pomper and M. T. McMahon, Angew. Chem., Int. Ed., 2013, 52, 8116–8119 CrossRef CAS PubMed.
- T. C. Soesbe, Y. Wu and A. Dean Sherry, NMR Biomed., 2013, 26, 829–838 CrossRef CAS PubMed.
- S. Aime and Z. Baranyai, Inorg. Chim. Acta, 2022, 532, 120730 CrossRef CAS.
- X. Yang, X. Song, S. Ray Banerjee, Y. Li, Y. Byun, G. Liu, Z. M. Bhujwalla, M. G. Pomper and M. T. McMahon, Contrast Media Mol. Imaging, 2016, 11, 304–312 CrossRef CAS PubMed.
- Z. Mohanta, S. Gori and M. T. McMahon, ACS Omega, 2024, 9, 27755–27765 CrossRef CAS PubMed.
- D. L. Longo, P. Z. Sun, L. Consolino, F. C. Michelotti, F. Uggeri and S. Aime, J. Am. Chem. Soc., 2014, 136, 14333–14336 CrossRef CAS PubMed.
- S. J. Ratnakar, S. Chirayil, A. M. Funk, S. Zhang, J. F. Queiró, C. F. G. C. Geraldes, Z. Kovacs and A. D. Sherry, Angew. Chem., Int. Ed., 2020, 59, 21671–21676 CrossRef CAS PubMed.
- T. Gambino, L. Valencia, P. Pérez-Lourido, D. Esteban-Gómez, M. Zaiss, C. Platas-Iglesias and G. Angelovski, Inorg. Chem. Front., 2020, 7, 2274–2286 RSC.
- A. Rodríguez-Rodríguez, M. Zaiss, D. Esteban-Gómez, G. Angelovski and C. Platas-Iglesias, in Metal Ions in Bio-Imaging Techniques, ed. A. Sigel, E. Freisinger and R. K. O. Sigel, De Gruyter, 2021, vol. 22, ch. 4, pp. 101–136 Search PubMed.
- A. E. Thorarinsdottir, K. Du, J. H. P. Collins and T. D. Harris, J. Am. Chem. Soc., 2017, 139, 15836–15847 CrossRef CAS PubMed.
- G. Angelovski, in Molecular Bio-Sensors and the Role of Metal Ions, ed. T. J. Meade, CRC Press, Boca Raton, 2022, vol. 23, ch. 2, pp. 23–50 Search PubMed.
- K. Du, A. E. Thorarinsdottir and T. D. Harris, J. Am. Chem. Soc., 2019, 141, 7163–7172 CrossRef CAS PubMed.
- R. Trokowski, J. M. Ren, F. K. Kalman and A. D. Sherry, Angew. Chem., Int. Ed., 2005, 44, 6920–6923 CrossRef CAS PubMed.
- K. Srivastava, G. Ferrauto, S. M. Harris, D. L. Longo, M. Botta, S. Aime and V. C. Pierre, Dalton Trans., 2018, 47, 11346–11357 RSC.
- S. J. Ratnakar, S. Viswanathan, Z. Kovacs, A. K. Jindal, K. N. Green and A. D. Sherry, J. Am. Chem. Soc., 2012, 134, 5798–5800 CrossRef CAS PubMed.
- J. R. Morrow, J. J. Raymond, M. S. I. Chowdhury and P. R. Sahoo, Inorg. Chem., 2022, 61, 14487–14499 CrossRef CAS PubMed.
- S. Karan, M. Y. Cho, H. Lee, H. S. Park, E. H. Han, Y. Song, Y. Lee, M. Kim, J.-H. Cho, J. L. Sessler and K. S. Hong, J. Med. Chem., 2022, 65, 7106–7117 CrossRef CAS PubMed.
- L. Zhang, A. F. Martins, Y. Mai, P. Zhao, A. M. Funk, M. V. Clavijo Jordan, S. Zhang, W. Chen, Y. Wu and A. D. Sherry, Chem.–Eur. J., 2017, 23, 1752–1756 CrossRef CAS PubMed.
- H. He, K. Zhao, L. Xiao, Y. Zhang, Y. Cheng, S. Wan, S. Chen, L. Zhang, X. Zhou, K. Liu and H. Zhang, Angew. Chem., Int. Ed., 2019, 58, 18286–18289 CrossRef CAS PubMed.
- B. Yoo, V. R. Sheth, C. M. Howison, M. J. K. Douglas, C. T. Pineda, E. A. Maine, A. F. Baker and M. D. Pagel, Magn. Reson. Med., 2014, 71, 1221–1230 CrossRef CAS PubMed.
- C. J. Kombala, S. D. Lokugama, A. Kotrotsou, T. Li, A. C. Pollard and M. D. Pagel, ACS Sens., 2021, 6, 4535–4544 CrossRef CAS PubMed.
- T. Chauvin, P. Durand, M. Bernier, H. Meudal, B. T. Doan, F. Noury, B. Badet, J. C. Beloeil and E. Toth, Angew. Chem., Int. Ed., 2008, 47, 4370–4372 CrossRef CAS PubMed.
- I. M. Welleman, F. Reeβing, H. H. Boersma, R. A. J. O. Dierckx, B. L. Feringa and W. Szymanski, Pharmaceuticals, 2023, 16, 1439 CrossRef CAS PubMed.
- Y. Li, V. R. Sheth, G. Liu and M. D. Pagel, Contrast Media Mol. Imaging, 2011, 6, 219–228 CrossRef CAS PubMed.
- J.-X. Yu, R. R. Hallac, S. Chiguru and R. P. Mason, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 70, 25–49 CrossRef CAS PubMed.
- J. Ruiz-Cabello, B. P. Barnett, P. A. Bottomley and J. W. M. Bulte, NMR Biomed., 2011, 24, 114–129 CrossRef CAS PubMed.
- K. L. Peterson, K. Srivastava and V. C. Pierre, Front. Chem., 2018, 6, 160 CrossRef PubMed.
- R. T. Ryan, K. M. Scott and E. L. Que, Analysis Sensing, 2023, 3, e202200041 CrossRef CAS.
- P. K. Senanayake, A. M. Kenwright, D. Parker and S. K. van der Hoorn, Chem. Commun., 2007, 2923–2925 RSC.
- A. M. Kenwright, I. Kuprov, E. De Luca, D. Parker, S. U. Pandya, P. K. Senanayake and D. G. Smith, Chem. Commun., 2008, 2514–2516 RSC.
- A. I. Gaudette, A. E. Thorarinsdottir and T. D. Harris, Chem. Commun., 2017, 53, 12962–12965 RSC.
- P. Kadjane, C. Platas-Iglesias, P. Boehm-Sturm, V. Truffault, G. E. Hagberg, M. Hoehn, N. K. Logothetis and G. Angelovski, Chem.–Eur. J., 2014, 20, 7351–7362 CrossRef CAS PubMed.
- G. Gambino, T. Gambino, R. Pohmann and G. Angelovski, Chem. Commun., 2020, 56, 3492–3495 RSC.
- M. Yu, D. Xie, R. T. Kadakia, W. Wang and E. L. Que, Chem. Commun., 2020, 56, 6257–6260 RSC.
- L. A. Basal, M. D. Bailey, J. Romero, M. M. Ali, L. Kurenbekova, J. Yustein, R. G. Pautler and M. J. Allen, Chem. Sci., 2017, 8, 8345–8350 RSC.
- S. A. A. S. Subasinghe, C. J. Ortiz, J. Romero, C. L. Ward, A. G. Sertage, L. Kurenbekova, J. T. Yustein, R. G. Pautler and M. J. Allen, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2220891120 CrossRef CAS PubMed.
- H. Chen, X. Tang, X. Gong, D. Chen, A. Li, C. Sun, H. Lin and J. Gao, Chem. Commun., 2020, 56, 4106–4109 RSC.
- D. Xie, T. L. King, A. Banerjee, V. Kohli and E. L. Que, J. Am. Chem. Soc., 2016, 138, 2937–2940 CrossRef CAS PubMed.
- M. Yu, D. Xie, K. P. Phan, J. S. Enriquez, J. J. Luci and E. L. Que, Chem. Commun., 2016, 52, 13885–13888 RSC.
- M. Yu, B. S. Bouley, D. Xie, J. S. Enriquez and E. L. Que, J. Am. Chem. Soc., 2018, 140, 10546–10552 CrossRef CAS PubMed.
- S. Mizukami, R. Takikawa, F. Sugihara, Y. Hori, H. Tochio, M. Walchli, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2008, 130, 794–795 CrossRef CAS PubMed.
- S. Mizukami, R. Takikawa, F. Sugihara, M. Shirakawa and K. Kikuchi, Angew. Chem., Int. Ed., 2009, 48, 3641–3643 CrossRef CAS PubMed.
- X. Yue, Z. Wang, L. Zhu, Y. Wang, C. Qian, Y. Ma, D. O. Kiesewetter, G. Niu and X. Chen, Mol. Pharm., 2014, 11, 4208–4217 CrossRef CAS PubMed.
- S. Mizukami, H. Matsushita, R. Takikawa, F. Sugihara, M. Shirakawa and K. Kikuchi, Chem. Sci., 2011, 2, 1151–1155 RSC.
- A. Keliris, I. Mamedov, G. E. Hagberg, N. K. Logothetis, K. Scheffler and J. Engelmann, Contrast Media Mol. Imaging, 2012, 7, 478–483 CrossRef CAS PubMed.
- D. Xie, M. Yu, Z.-L. Xie, R. T. Kadakia, C. Chung, L. E. Ohman, K. Javanmardi and E. L. Que, Angew. Chem., Int. Ed., 2020, 59, 22523–22530 CrossRef CAS PubMed.
- J.-X. Yu, V. D. Kodibagkar, R. R. Hallac, L. Liu and R. P. Mason, Bioconjugate Chem., 2012, 23, 596–603 CrossRef CAS PubMed.
- I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo, F. B. Bombelli, P. Metrangolo and G. Resnati, Chem. Rev., 2015, 115, 1106–1129 CrossRef CAS PubMed.
- A. Galisova and A. Bar-Shir, Chem. Commun., 2023, 59, 5502–5513 RSC.
- H. Lin, X. Tang, A. Li and J. Gao, Adv. Mater., 2021, 33, 2005657 CrossRef CAS PubMed.
- M. H. Cho, S. H. Shin, S. H. Park, D. K. Kadayakkara, D. Kim and Y. Choi, Bioconjugate Chem., 2019, 30, 2502–2518 CrossRef CAS PubMed.
- J. Salaam, M. Minoshima and K. Kikuchi, Analysis Sensing, 2023, 3, e202200081 CrossRef CAS.
- X. N. Huang, G. Huang, S. R. Zhang, K. Sagiyama, O. Togao, X. P. Ma, Y. G. Wang, Y. Li, T. C. Soesbe, B. D. Sumer, M. Takahashi, A. D. Sherry and J. M. Gao, Angew. Chem., Int. Ed., 2013, 52, 8074–8078 CrossRef CAS PubMed.
- C. Guo, S. Xu, A. Arshad and L. Wang, Chem. Commun., 2018, 54, 9853–9856 RSC.
- C. Zhang, L. Li, F. Y. Han, X. Yu, X. Tan, C. Fu, Z. P. Xu and A. K. Whittaker, Small, 2019, 15, 1902309 CrossRef PubMed.
- A. T. Preslar, L. M. Lilley, K. Sato, S. R. Zhang, Z. K. Chia, S. I. Stupp and T. J. Meade, ACS Appl. Mater. Interfaces, 2017, 9, 39890–39894 CrossRef CAS PubMed.
- M. Zheng, Y. Wang, H. Shi, Y. Hu, L. Feng, Z. Luo, M. Zhou, J. He, Z. Zhou, Y. Zhang and D. Ye, ACS Nano, 2016, 10, 10075–10085 CrossRef CAS PubMed.
- T. Nakamura, H. Matsushita, F. Sugihara, Y. Yoshioka, S. Mizukami and K. Kikuchi, Angew. Chem., Int. Ed., 2015, 54, 1007–1010 CrossRef CAS PubMed.
- Y. Takaoka, T. Sakamoto, S. Tsukiji, M. Narazaki, T. Matsuda, H. Tochio, M. Shirakawa and I. Hamachi, Nat. Chem., 2009, 1, 557–561 CrossRef CAS PubMed.
- Y. Yuan, S. Ge, H. Sun, X. Dong, H. Zhao, L. An, J. Zhang, J. Wang, B. Hu and G. Liang, ACS Nano, 2015, 9, 5117–5124 CrossRef CAS PubMed.
- Y. Yuan, H. Sun, S. Ge, M. Wang, H. Zhao, L. Wang, L. An, J. Zhang, H. Zhang, B. Hu, J. Wang and G. Liang, ACS Nano, 2015, 9, 761–768 CrossRef CAS PubMed.
- X. Zhu, X. Tang, H. Lin, S. Shi, H. Xiong, Q. Zhou, A. Li, Q. Wang, X. Chen and J. Gao, Chem, 2020, 6, 1134–1148 CAS.
- X. Zhu, H. Xiong, S. Wang, Y. Li, J. Chi, X. Wang, T. Li, Q. Zhou, J. Gao and S. Shi, Adv. Healthcare Mater., 2022, 11, 2102079 CrossRef CAS PubMed.
- G. A. Smith, R. T. Hesketh, J. C. Metcalfe, J. Feeney and P. G. Morris, Proc. Natl. Acad. Sci. U.S.A., 1983, 80, 7178–7182 CrossRef CAS PubMed.
- A. Bar-Shir, A. A. Gilad, K. W. Y. Chan, G. S. Liu, P. C. M. van Zijl, J. W. M. Bulte and M. T. McMahon, J. Am. Chem. Soc., 2013, 135, 12164–12167 CrossRef CAS PubMed.
- A. Bar-Shir, N. N. Yadav, A. A. Gilad, P. C. M. van Zijl, M. T. McMahon and J. W. M. Bulte, J. Am. Chem. Soc., 2015, 137, 78–81 CrossRef CAS PubMed.
- Y. Yuan, Z. Wei, C. Chu, J. Zhang, X. Song, P. Walczak and J. W. M. Bulte, Angew. Chem., Int. Ed., 2019, 58, 15512–15517 CrossRef CAS PubMed.
- N. D. Tirukoti, L. Avram, T. Haris, B. Lerner, Y. Diskin-Posner, H. Allouche-Arnon and A. Bar-Shir, J. Am. Chem. Soc., 2021, 143, 11751–11758 CrossRef CAS PubMed.
- P. Nikolaou, B. M. Goodson and E. Y. Chekmenev, Chem.–Eur. J., 2015, 21, 3156–3166 CrossRef CAS PubMed.
- K. R. Keshari and D. M. Wilson, Chem. Soc. Rev., 2014, 43, 1627–1659 RSC.
- J.-B. Hövener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Glöggler, K. V. Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier, A. Jerschow, F. Reineri, T. Theis, R. V. Shchepin, S. Wagner, P. Bhattacharya, N. M. Zacharias and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2018, 57, 11140–11162 CrossRef PubMed.
- Y. Kondo, H. Nonaka, Y. Takakusagi and S. Sando, Angew. Chem., Int. Ed., 2021, 60, 14779–14799 CrossRef CAS PubMed.
- J. Eills, D. Budker, S. Cavagnero, E. Y. Chekmenev, S. J. Elliott, S. Jannin, A. Lesage, J. Matysik, T. Meersmann, T. Prisner, J. A. Reimer, H. Yang and I. V. Koptyug, Chem. Rev., 2023, 123, 1417–1551 CrossRef CAS PubMed.
- A. N. Pravdivtsev, G. Buntkowsky, S. B. Duckett, I. V. Koptyug and J.-B. Hövener, Angew. Chem., Int. Ed., 2021, 60, 23496–23507 CrossRef CAS PubMed.
- H. Park and Q. Wang, Chem. Sci., 2022, 13, 7378–7391 RSC.
- K. M. Brindle, J. Am. Chem. Soc., 2015, 137, 6418–6427 CrossRef CAS PubMed.
- P. C. M. van Zijl, K. Brindle, H. Lu, P. B. Barker, R. Edden, N. Yadav and L. Knutsson, Curr. Opin. Chem. Biol., 2021, 63, 209–218 CrossRef CAS PubMed.
- J. Eills, R. Picazo-Frutos, O. Bondar, E. Cavallari, C. Carrera, S. J. Barker, M. Utz, A. Herrero-Gómez, I. Marco-Rius, M. C. D. Tayler, S. Aime, F. Reineri, D. Budker and J. W. Blanchard, Anal. Chem., 2023, 95, 17997–18005 CrossRef CAS PubMed.
- F. A. Gallagher, M. I. Kettunen, S. E. Day, D.-E. Hu, J. H. Ardenkjær-Larsen, R. i. t. Zandt, P. R. Jensen, M. Karlsson, K. Golman, M. H. Lerche and K. M. Brindle, Nature, 2008, 453, 940–943 CrossRef CAS PubMed.
- S. Düwel, C. Hundshammer, M. Gersch, B. Feuerecker, K. Steiger, A. Buck, A. Walch, A. Haase, S. J. Glaser, M. Schwaiger and F. Schilling, Nat. Commun., 2017, 8, 15126 CrossRef PubMed.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, T. Theis, F. Shi, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, ACS Sens., 2016, 1, 640–644 CrossRef CAS PubMed.
- A. M. Olaru, M. J. Burns, G. G. R. Green and S. B. Duckett, Chem. Sci., 2017, 8, 2257–2266 RSC.
- A. K. Jindal, M. E. Merritt, E. H. Suh, C. R. Malloy, A. D. Sherry and Z. Kovacs, J. Am. Chem. Soc., 2010, 132, 1784–1785 CrossRef CAS PubMed.
- R. Hata, H. Nonaka, Y. Takakusagi, K. Ichikawa and S. Sando, Chem. Commun., 2015, 51, 12290–12292 RSC.
- A. Mishra, G. Pariani, T. Oerther, M. Schwaiger and G. G. Westmeyer, Anal. Chem., 2016, 88, 10790–10794 CrossRef CAS PubMed.
- S. Wang, D. E. Korenchan, P. M. Perez, C. Taglang, T. R. Hayes, R. Sriram, R. Bok, A. S. Hong, Y. Wu, H. Li, Z. Wang, J. Kurhanewicz, D. M. Wilson and R. R. Flavell, Chem.–Eur. J., 2019, 25, 11842–11846 CrossRef CAS PubMed.
- E. H. Suh, J. M. Park, L. Lumata, A. D. Sherry and Z. Kovacs, Commun. Chem., 2020, 3, 185 CrossRef CAS PubMed.
- N. Kotera, N. Tassali, E. Léonce, C. Boutin, P. Berthault, T. Brotin, J.-P. Dutasta, L. Delacour, T. Traoré, D.-A. Buisson, F. Taran, S. Coudert and B. Rousseau, Angew. Chem., Int. Ed., 2012, 51, 4100–4103 CrossRef CAS PubMed.
- Z. Zhao, M. Zhou, S. D. Zemerov, R. Marmorstein and I. J. Dmochowski, Chem. Sci., 2023, 14, 3809–3815 RSC.
- T. Doura, R. Hata, H. Nonaka, K. Ichikawa and S. Sando, Angew. Chem., Int. Ed., 2012, 51, 10114–10117 CrossRef CAS PubMed.
- H. Nonaka, R. Hata, T. Doura, T. Nishihara, K. Kumagai, M. Akakabe, M. Tsuda, K. Ichikawa and S. Sando, Nat. Commun., 2013, 4, 2441 CrossRef PubMed.
- W. Iali, S. S. Roy, B. J. Tickner, F. Ahwal, A. J. Kennerley and S. B. Duckett, Angew. Chem., Int. Ed., 2019, 58, 10271–10275 CrossRef CAS PubMed.
- S. H. Klass, A. E. Truxal, T. A. Fiala, J. Kelly, D. Nguyen, J. A. Finbloom, D. E. Wemmer, A. Pines and M. B. Francis, Angew. Chem., Int. Ed., 2019, 58, 9948–9953 CrossRef CAS PubMed.
- V. Ntziachristos and D. Razansky, Chem. Rev., 2010, 110, 2783–2794 CrossRef CAS PubMed.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- M. Srinivas, P. Boehm-Sturm, C. G. Figdor, I. J. de Vries and M. Hoehn, Biomaterials, 2012, 33, 8830–8840 CrossRef CAS PubMed.
- S. M. Vithanarachchi and M. J. Allen, Curr. Mol. Imag., 2012, 1, 12–25 CrossRef CAS PubMed.
- A. D. Sherry, P. Caravan and R. E. Lenkinski, J. Magn. Reson. Imag., 2009, 30, 1240–1248 CrossRef PubMed.
- E. Gianolio, R. Napolitano, F. Fedeli, F. Arena and S. Aime, Chem. Commun., 2009, 40, 6044–6046 RSC.
- G. Gambino, T. Gambino and G. Angelovski, Chem. Commun., 2020, 56, 9433–9436 RSC.
- L. Frullano, C. Catana, T. Benner, A. D. Sherry and P. Caravan, Angew. Chem., Int. Ed., 2010, 49, 2382–2384 CrossRef CAS PubMed.
- E. Gianolio, L. Maciocco, D. Imperio, G. B. Giovenzana, F. Simonelli, K. Abbas, G. Bisi and S. Aime, Chem. Commun., 2011, 47, 1539–1541 RSC.
| This journal is © The Royal Society of Chemistry 2024 |