
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge-assisted hydrogen bonding in a bicyclic amide cage: an effective approach to anion recognition and catalysis in water†
Chengkai
Xu
 ,
Quy Gia
Tran
,
Dexin
Liu
,
Canjia
Zhai
,
Lukasz
Wojtas
and
Wenqi
Liu
*
,
Quy Gia
Tran
,
Dexin
Liu
,
Canjia
Zhai
,
Lukasz
Wojtas
and
Wenqi
Liu
*
Department of Chemistry, University of South Florida, 4202 E. Fowler Ave, Tampa, FL 33620, USA. E-mail: wenqi@usf.edu
First published on 18th September 2024
Abstract
Hydrogen bonding is prevalent in biological systems, dictating a myriad of life-sustaining functions in aqueous environments. Leveraging hydrogen bonding for molecular recognition in water encounters significant challenges in synthetic receptors on account of the hydration of their functional groups. Herein, we introduce a water-soluble hydrogen bonding cage, synthesized via a dynamic approach, exhibiting remarkable affinities and selectivities for strongly hydrated anions, including sulfate and oxalate, in water. We illustrate the use of charge-assisted hydrogen bonding in amide-type synthetic receptors, offering a general molecular design principle that applies to a wide range of amide receptors for molecular recognition in water. This strategy not only revalidates the functions of hydrogen bonding but also facilitates the effective recognition of hydrophilic anions in water. We further demonstrate an unconventional catalytic mechanism through the encapsulation of the anionic oxalate substrate by the cationic cage, which effectively inverts the charges associated with the substrate and overcomes electrostatic repulsions to facilitate its oxidation by the anionic MnO4−. Technical applications using this receptor are envisioned across various technical applications, including anion sensing, separation, catalysis, medical interventions, and molecular nanotechnology.
Introduction
Hydrogen bonding plays a crucial role in nature, enabling biological receptors to recognize hydrated substrates. This mechanism underpins numerous vital functions in aqueous environments.1–5 However, replicating this process with synthetic analogs poses significant challenges, particularly in using hydrogen bonds for substrate recognition in water.6–9 One major hurdle is the strong hydration of both receptor and substrate, creating high energetic barriers due to the need for desolvation during the binding process. Additionally, most organic building blocks used to synthesize hydrogen bonding receptors are poorly soluble in water, complicating the design and synthesis of water-soluble receptors. The pursuit of water-soluble hydrogen bonding receptors capable of effectively recognizing strongly hydrated substrates marks a cutting-edge frontier in supramolecular chemistry. Achieving this functionality could have profound impacts across various fields, including catalytic chemical transformations, biology, healthcare, environmental science, and molecular nanotechnology.10–16Current approaches to revalidating hydrogen bonding in water mainly focus on two strategies: hydrophobicity-assisted and charge-assisted hydrogen bonding. Hydrophobicity-assisted hydrogen bonding, in particular, involves the design of receptors that mimic the structure of protein-binding pockets.17–20 These receptors feature hydrogen bonding residues buried within a hydrophobic binding pocket. This design leverages the hydrophobic effect to offset the energy cost of dehydration, thus serving as the primary driving force. At the same time, hydrogen bonding ensures the selectivity in the binding process. This approach has successfully been applied in a variety of hydrogen bonding receptors, such as molecular temples,21–27 tetralactam macrocycles,19,20,28–31 naphthotubes,32–40 and cyclopeptides,41–43 demonstrating its viability and effectiveness.
Charge-assisted hydrogen bonding is another critical strategy for the binding of anionic substrates in water, leveraging electrostatic interactions to offset hydration energy. However, this strategy is predominantly confined to polyammonium and guanidinium receptors. Polyammonium receptors exhibit44,45 (Fig. 1a, left) high effectiveness in acidic conditions but lose their anion-binding efficiency and water solubility under neutral or basic pH, thereby restricting their applications. Guanidinium-based receptors can maintain8,9,46–48 (Fig. 1a, right) their charge in neutral environments but face challenges in conformational flexibility and strong solvation. For instance, a single guanidinium ion's binding with carbonate or phosphate in water is negligible, with a binding constant (Ka) of less than 5 M−1.9 This limitation narrows the charge-assisted hydrogen bonding application in guanidinium receptors to specific molecular frameworks that either incorporate constrained amino imidazoline, bicyclic guanidinium residues, or use multiple guanidinium groups for anion binding.11,46–50 This restriction significantly limits the diversity of molecular scaffolds available for receptor design. Consequently, despite the effectiveness of leveraging electrostatic interactions in enhancing hydrogen bonds in water, there is a pressing need for a broader molecular design strategy to fully exploit the power of charge-assisted hydrogen bonding.
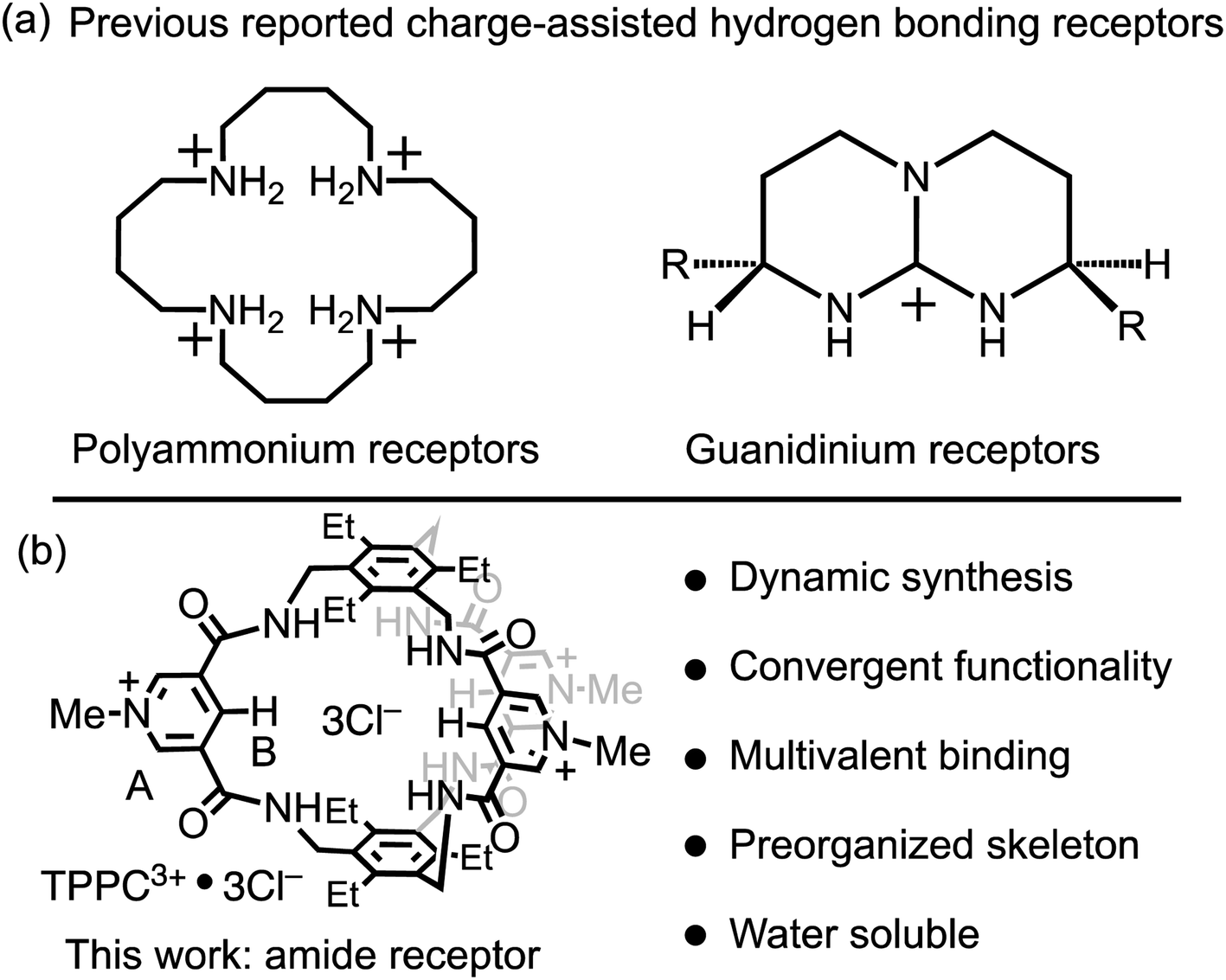 | ||
| Fig. 1 (a) Representative anion receptors with charge-assisted hydrogen bonding functionalities. (b) The structural formula of TPPC3+·3Cl− and a summary of its structural features. | ||
Hydrogen-bonding receptors, especially those utilizing amide functionalities, have been the subject of extensive research in the field of anion recognition. However, the majority of this research has focused51–67 on anion binding in organic solvents. Given the significant number of hydrogen bonding receptors that either cannot bind anions in water due to solubility issues or lose their effectiveness because of strong solvation, there is a crucial need for a universal molecular design strategy that revalidates the hydrogen bonding capability of these receptors through the principle of charge-assisted hydrogen bonding. Such a strategy promises to have a far-reaching impact on the field of anion recognition in water.
In this research, we showcase a general approach to revalidate the hydrogen bonding capability of amide receptors for anion recognition in water. We demonstrate a water-soluble tripodal tricationic cage-type receptor TPPC3+·3Cl− under the principle of charge-assisted hydrogen bonding. The receptor utilizes (Fig. 1b) pyridinium moieties both as charge carriers and for water solubilization groups.68–71 The introduction of positive charges creates electrostatic attractions with anions, which serve as the driving force to overcome their dehydration energy barrier, while hydrogen bonding patterns within the binding pocket provide substrate selectivity. This receptor is synthesized through a dynamic process, beginning with an imine condensation reaction, followed by a crucial imine-to-amide oxidation step to complete the cyclization. TPPC3+·3Cl− exhibits remarkable affinities and selectivities for hydrophilic anions such as C2O42−, SO42−, and NO3−. The binding to these hydrophilic anions is primarily driven by a favorable entropic effect resulting from the anion desolvation upon their associations by the cage. Additionally, the encapsulation of HC2O4− by TPPC3+ inverts its overall charge, forming the [HC2O4−⊂TPPC3+]2+ complex. This charge inversion overcomes the electrostatic repulsion and increases the reactivity between HC2O4− and MnO4−. As a result, TPPC3+·3Cl− shows a catalyst activity for the oxidation of HC2O4− by MnO4−, illustrating its practical application in catalytic chemical transformation in water.
Results and discussion
Molecular design and synthesis
The 3,5-dicarboxamide pyridinium building block has been extensively explored by the Beer group in mechanically interlocked structures, leveraging the charge and hydrogen bonds for anion recognition in organic solvents.72–79 As a proof of principle for charge-assisted hydrogen bonding in amide-based receptors, we have introduced (Fig. 1b) this pyridinium moiety as the charge carrier into a rigid, preorganized bicyclic molecular cage developed by Anslyn and Mastalerz.59,80–82 The trimethyl pinwheel-shaped pyridinium cage, TPPC3+·3Cl−, is equipped with six hydrogen bond donors converging towards its binding cavity. The C–H bonds at the para position of the pyridinium rings are polarized, creating three additional hydrogen bond donors pointing toward the cavity.69,71 Additionally, the three pyridinium residues serve as charge carriers, assisting the hydrogen bonding by providing electrostatic attractions with anions in water. When associated with the hydrophilic counter-anion Cl−, the receptor maintains good water solubility.83 This molecular design strategy exemplifies a general approach that could be extended to other amide-based hydrogen bonding receptors for binding hydrophilic substrates in water.54,55The TPPC3+·3Cl− cage can be synthesized (Fig. 2) through two distinct methods. The first method involves a traditional approach, where acyl chloride 2 is reacted with the pinwheel-shaped triamine 1 under high dilution conditions. This process results in the critical intermediate, amide cage 5, at a low yield of 9%, which is common for irreversible macrocyclization reactions not relying on the use of a template.84–88 An alternative and more effective method employs68,81,82,89 dynamic imine chemistry followed by the Pinnick oxidation reaction. The triamine 1 is condensed with bisaldehyde 3, leading to a quantitative formation of the bicyclic imine skeleton. The resulting imine cage 4 can then be converted into the key intermediate amide cage 5 through Pinnick oxidation, achieving a much higher yield of 61%. The final steps involve alkylating the pyridine moiety to form a pyridinium salt, followed by standard anion exchange with a yield of 76%. These three steps produce the water-soluble amide cage TPPC3+·3Cl− with an overall yield of 46% from easily accessible starting materials.
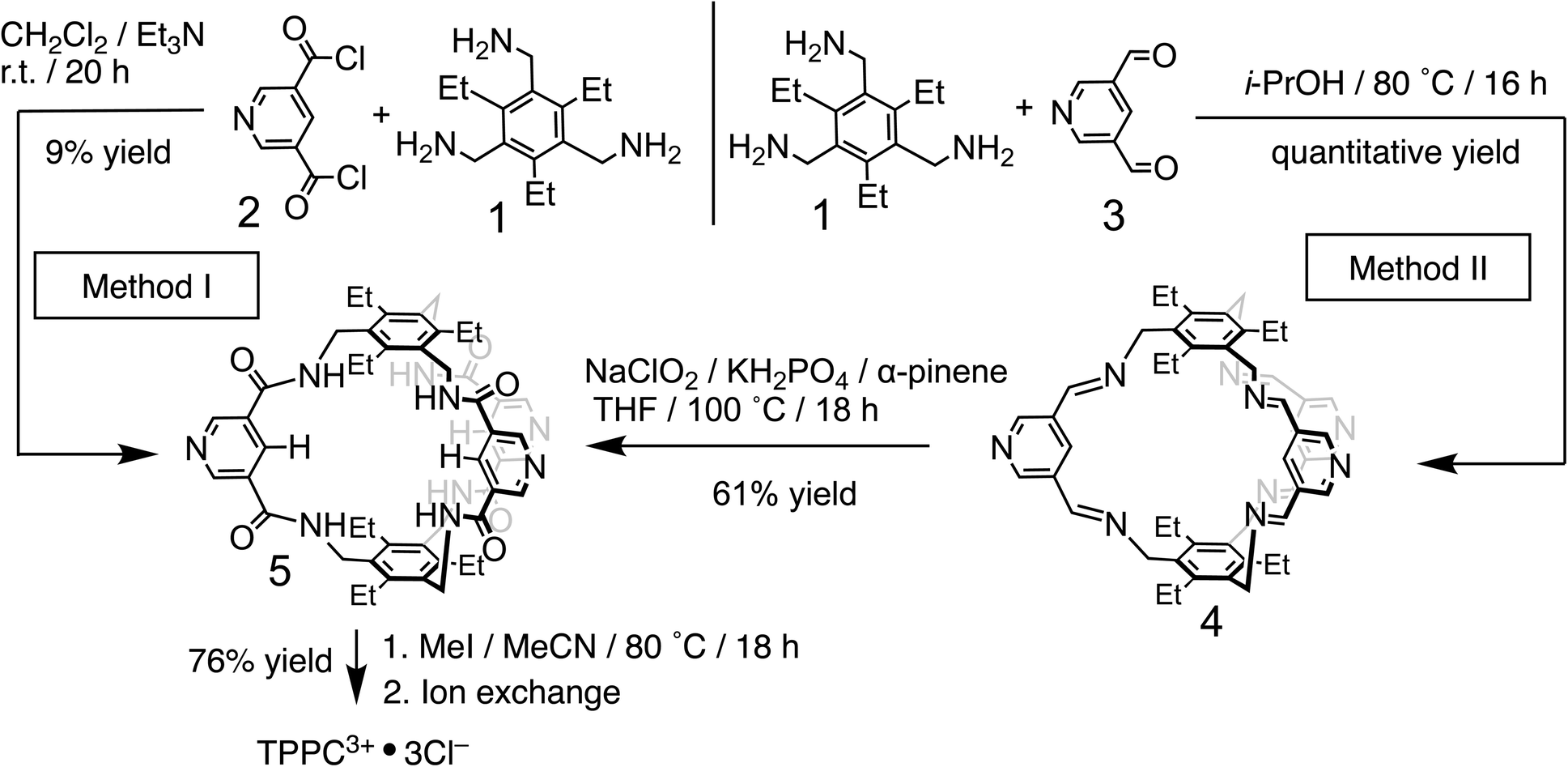 | ||
| Fig. 2 Synthesis of TPPC3+·3Cl− using a conventional high dilution approach (method I) and a dynamic approach (method II). | ||
Anions binding in water
The anion-binding properties of TPPC3+·3Cl− in water were analyzed through 1H NMR titration experiments. Our findings (Fig. S45†) show that the chemical shift of the inward-facing C–H proton B depends on the concentration of TPPC3+·3Cl−, indicating that the cage is partially occupied by Cl− counter anions and exists as a mixture of TPPC3+ and Cl−⊂TPPC3+. Therefore, the anion binding affinities measured should be considered as apparent binding affinities, reflecting the displacement of chloride ions from the binding cavity by the target anions. To avoid D/H exchange between the protons from the cage and D2O and to obtain direct evidence of the hydrogen bonding between the anions and TPPC3+·3Cl−, we performed the experiments in a solution of 10% D2O and 90% H2O. The broad peak around 8.84 pm represents the NH proton signals. Upon the addition of Na2SO4, this peak showed dramatic downfield shifts (Δδ = +0.69 ppm), suggesting the hydrogen bonds formation between SO42− and the NH protons from TPPC3+·3Cl− in water. The inward-facing C–H protons, labeled (Fig. 1b) as proton B, are polarized, making them effective hydrogen bond donors. The binding of SO42− by TPPC3+·3Cl− results (Fig. 3a) in a notable upfield shift (Δδ = −0.15 ppm) of proton B. This observed shift in proton B indicates a change in the hydrogen bonding state of these protons, likely transitioning from bonding with H2O or Cl− to SO42−. Nonlinear fitting of the chemical shift changes of proton B against the concentration of SO42−, based on a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model, yielded an apparent binding constant (Ka) of (1.7 ± 0.1) × 104 M−1. In a separate experiment, where the titration was performed in D2O, the same trend in the changes of the chemical shifts was observed (Fig. S37†). The nonlinear fitting of the changes in the chemical shift of proton B in D2O yielded (Fig. S38†) a similar apparent binding affinity of (2.5 ± 0.3) × 104 M−1.
:1 binding model, yielded an apparent binding constant (Ka) of (1.7 ± 0.1) × 104 M−1. In a separate experiment, where the titration was performed in D2O, the same trend in the changes of the chemical shifts was observed (Fig. S37†). The nonlinear fitting of the changes in the chemical shift of proton B in D2O yielded (Fig. S38†) a similar apparent binding affinity of (2.5 ± 0.3) × 104 M−1.
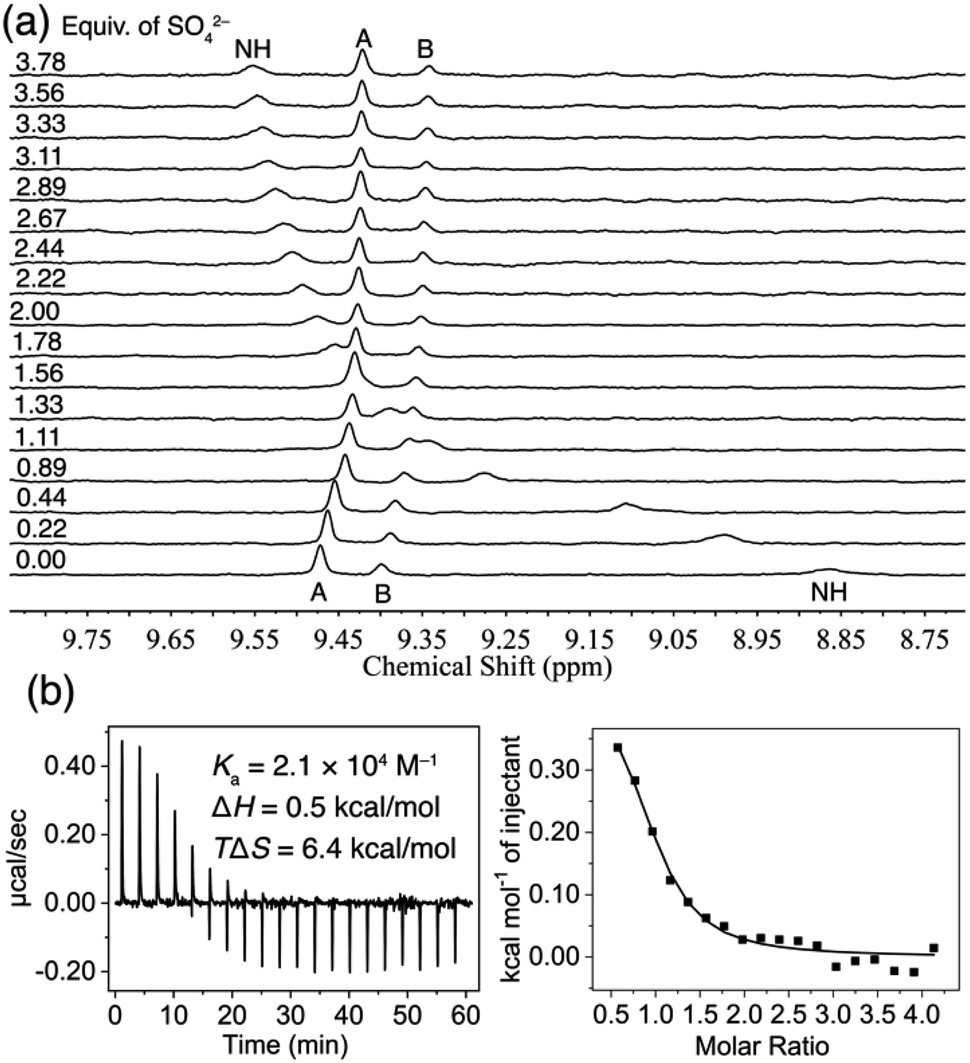 | ||
| Fig. 3 (a) 1H NMR spectra (400 MHz, 10% D2O + 90% H2O) of TPPC3+·3Cl− (0.1 mM) titrated with Na2SO4. (b) ITC profile of TPPC3+·3Cl− (0.5 mM) titrated with Na2SO4 in H2O. | ||
The binding of SO42− by TPPC3+·3Cl− was further confirmed using isothermal titration calorimetry (ITC). A solution of Na2SO4 in H2O was incrementally added to a 0.5 mM solution of TPPC3+·3Cl−, and the heat change in the system was monitored over time. The ITC data revealed (Fig. 3b) an endothermic binding process mainly assigned to the interaction between SO42− and TPPC3+. Analyzing the binding isotherm with a 1:1 binding model yielded an apparent binding affinity similar to our previous findings at a Ka of (2.0 ± 0.6) × 104 M−1. The ITC results indicated that the binding is primarily driven by favorable entropy (−TΔS = −6.35 kcal mol−1), with only a minor enthalpic penalty (ΔH = +0.48 kcal mol−1). This outcome aligns with our hypothesis that the dehydration of the highly hydrophilic SO42− demands significant enthalpic energy. The combined hydrogen bonding and electrostatic interactions between SO42− and TPPC3+·3Cl− do not completely offset this high enthalpic requirement, resulting in a small net enthalpic penalty of +0.48 kcal mol−1. At the same time, the dehydration process releases water molecules from both the hydration shell of SO42− and the binding cavity of the cage into the bulk solution, providing a substantial entropic driving force for the molecular recognition of this hydrophilic anion. Such an entropically driven binding mechanism, commonly observed in the association of hydrophobic molecules in water (known as the hydrophobic effect), is rare for hydrophilic substrates as it typically requires significant enthalpic compensation for dehydration.8,90 In this case, charge-assisted hydrogen bonding proves to be a unique and effective strategy to facilitate the dehydration of hydrophilic anions in water.
The binding pattern of C2O42− anion with TPPC3+·3Cl− presents distinct characteristics compared to that of SO42−. The 1H NMR spectra of TPPC3+·3Cl− obtained in a mixture of 10% D2O and 90% H2O show (Fig. 4a) notable downfield shifts for both the NH protons (Δδ = + 0.65 ppm) and protons B (Δδ = +0.35 ppm) with an increasing concentration of Na2C2O4. This result serves as direct evidence of hydrogen bonding formation between TPPC3+ and C2O42− in water. Nonlinear fitting of changes in the chemical shift of proton B yielded a Ka higher than 106 M−1. This strong apparent binding affinity was independently corroborated by ITC, which indicated (Fig. 4b) a Ka of (2.0 ± 0.9) × 106 M−1. The ITC experiments revealed an exothermic process for this binding, with a favorable enthalpic contribution (ΔH) of −3.3 kcal mol−1. Additionally, a substantial favorable entropic contribution (−TΔS = −5.3 kcal mol−1) was identified as the primary driving force for the interaction between C2O42− and TPPC3+. These results suggest that the charge-assisted hydrogen bond not only overcomes the dehydration barrier of C2O42− but also provides additional binding enthalpy, serving as an additional driving force for the recognition of hydrophilic C2O42− anion in water. More importantly, the anion binding facilitated by charge-assisted hydrogen bonding significantly benefits from the release of constrained water molecules from both the hydration shell of the anions and the binding pocket of the cage. This entropic effect serves as the primary driving force behind the effective anion binding in water.
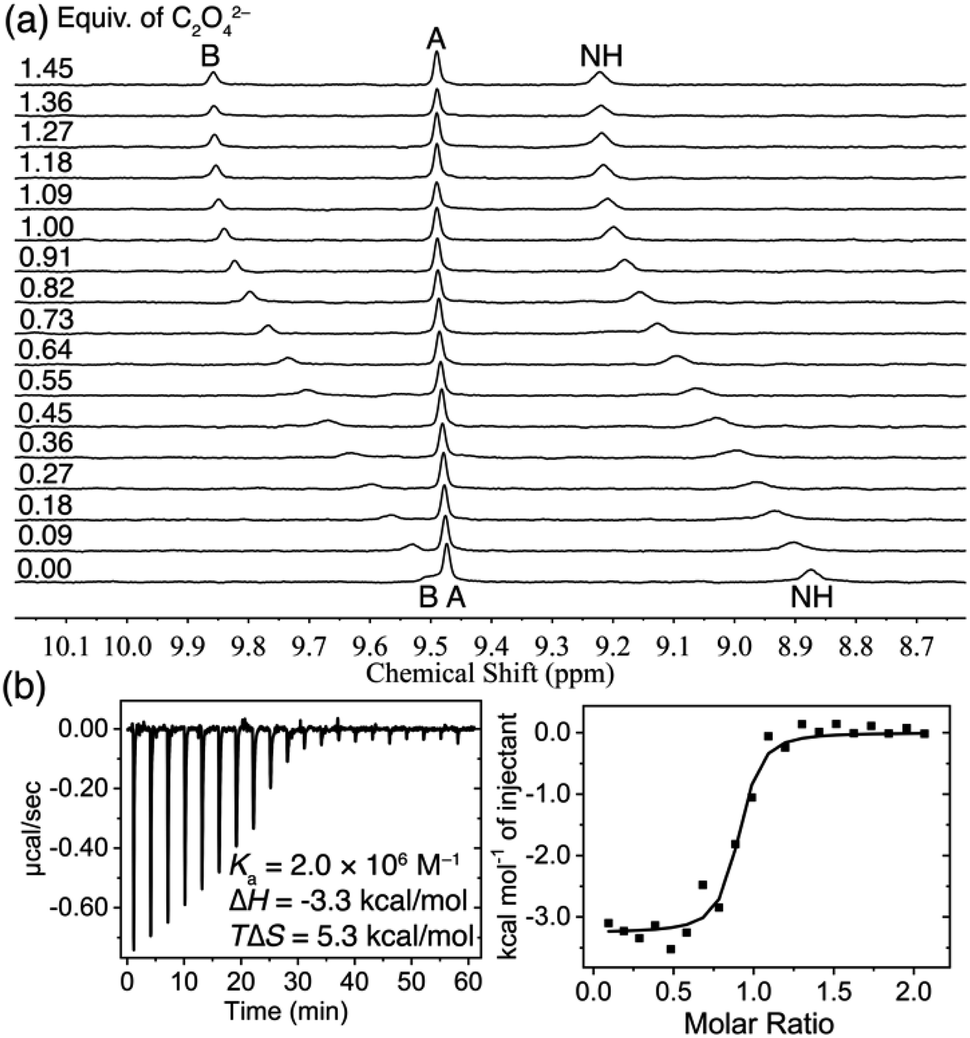 | ||
| Fig. 4 (a) 1H NMR spectra (400 MHz, 10% D2O + 90% H2O) of TPPC3+·3Cl− (0.2 mM) titrated with Na2C2O4. (b) ITC profile of TPPC3+·3Cl− (0.1 mM) titrated with Na2C2O4 in H2O. | ||
The binding affinities of TPPC3+·3Cl− with various anions in water are detailed in Table 1. TPPC3+ shows a strong affinity for I− and NO3−, with similar binding strengths in the order of 104 M−1. AcO− and NO2− exhibit comparable affinities, each around 1500 M−1, while Br− and Cl− are both around 600 M−1. In terms of selectivity for halide anions,91 we observed a trend of I− > Br− ≈ Cl−. The higher affinity for I− compared to Br− and Cl− suggests that the hydrophobic effect is a predominant driving force in anion binding in water. Although Br− is more hydrophobic than Cl−, Br− has a lower charge density, resulting in similar apparent binding affinities for both anions. This observation indicates that electrostatic interactions may also play a significant role in binding. Generally, TPPC3+ demonstrates higher affinities for anions with more negative charges, as evident in the observed trend: C2O42− > SO42− > NO3−. Additionally, TPPC3+ shows notable shape selectivity in anion binding. More binding sites correlate with higher affinity, and anions with similar structures exhibit comparable affinities, as seen in the trend NO3− > AcO− ≈ NO2−. Further thermodynamic analysis indicates that the anion binding by TPPC3+ is consistently associated with a positive entropy change, which acts as a major driving force in the recognition process. Overall, TPPC3+·3Cl− demonstrates effective selectivity in anion binding in water. This selectivity is influenced by a combination of factors, including electrostatic attraction, shape complementarity, and hydrophobicity.
| Anionsa | K a/M−1 (NMR)b | K a/M−1 (ITC)c | ΔH/kcal mol−1 | TΔS/kcal mol−1 |
|---|---|---|---|---|
| a All anions come with sodium as the counter cation. b The titration experiments were performed in D2O. c The titration experiments were performed in H2O. d Apparent binding affinity is too high to be accurately determined by direct 1H NMR titration. e Apparent binding affinity is too low to be accurately determined by ITC. | ||||
| C2O42− | >106d | (2.0 ± 0.9) × 106 | −3.3 | 5.3 |
| SO42− | (2.5 ± 0.3) × 104 | (2.0 ± 0.6) × 104 | 0.5 | 6.4 |
| I− | (1.6 ± 0.2) × 104 | (0.9 ± 0.1) × 104 | −1.4 | 4.0 |
| NO3− | (1.4 ± 0.1) × 104 | (1.1 ± 0.4) × 104 | −1.6 | 3.9 |
| AcO− | 1614 ± 11 | N.A.e | N.A.e | N.A.e |
| NO2− | 1439 ± 33 | N.A.e | N.A.e | N.A.e |
| Br− | 643 ± 38 | N.A.e | N.A.e | N.A.e |
| Cl− | 589 ± 25 | N.A.e | N.A.e | N.A.e |
Structural analysis by X-ray single crystallography and DFT calculations
Three distinct single crystal structures of TPPC3+ were obtained (Fig. 5a–c), corresponding to its association with Cl−, I−, and CF3CO2− as counter anions. In all these structures, TPPC3+ maintains a similar conformation, with all NH bonds converging toward the binding cavity. In the Cl− complex (Fig. 5a), two Cl− anions and one water molecule were found within the TPPC3+ binding pocket. Each Cl− forms two [NH⋯O] and one [CH⋯O] hydrogen bonds with one of the three pyridinium panels. Additionally, a water molecule located in the cavity aligns its hydrogens toward the two Cl− guests, forming [OH⋯Cl] hydrogen bonds. The I− complex (Fig. 5b) is analogous to the Cl− complex, with each TPPC3+ binding two I− ions and one water molecule through hydrogen bonds in its binding pocket. In the case of the CF3CO2− complex (Fig. 5c), three CF3CO2− anions are associated with each TPPC3+. Each carboxylate group interacts with two of the three pyridinium units, with each oxygen in the carboxylate group forming one [NH⋯O] and one [CH⋯O] hydrogen bond with the pyridinium unit.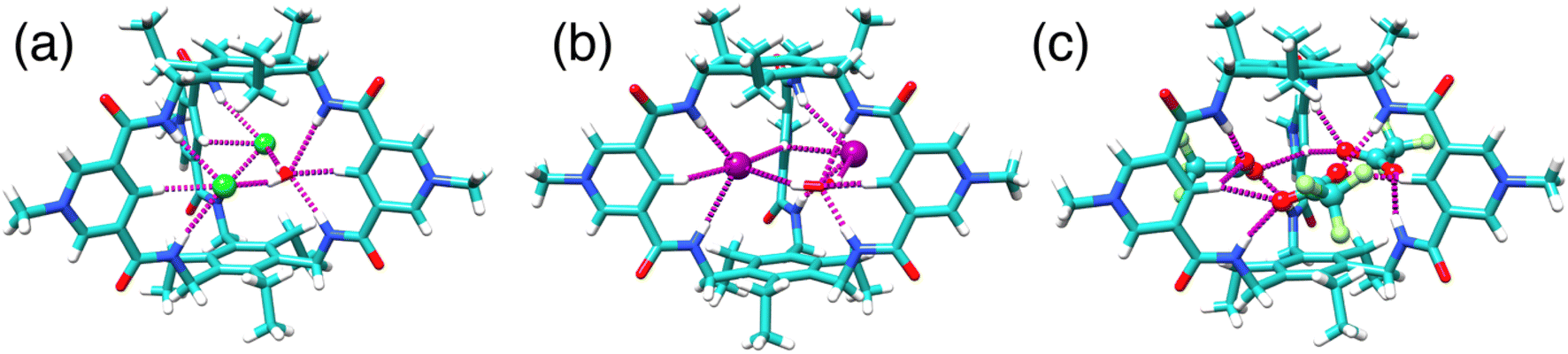 | ||
| Fig. 5 X-ray single crystal structures of (a) 2Cl−·H2O⊂TPPC3+, (b) 2I−·H2O⊂TPPC3+, (c) 3CF3CO2−⊂TPPC3+. | ||
Notably, the solid-state single-crystal structures were obtained from organic solvents and do not fully represent the binding states observed in water. One key difference lies in the binding stoichiometry. For example, a 2:1 binding stoichiometry is observed for binding I− in the solid state. In contrast, a 1:1 binding stoichiometry between I− and TPPC3+ was determined in solution using 1H NMR titration, where the shift of the inward-facing C–H proton B stopped after adding 1.0 equivalent of I−. Complementary results supporting the 1:1 binding stoichiometry are also found in high-resolution mass spectrometry (HRMS), where the m/z peaks for the 1:1 complexes were observed as the major peaks for all anions investigated, indicating that the 1:1 complex is the primary species in solution. The absence of m/z peaks for 2:1 complexes suggests these species are much less stable and should exist minimally compared to the 1:1 complex. In water, anions primarily form 1:1 complexes with TPPC3+. Forming higher-order anion complexes would require dehydrating a second anion with weaker electrostatic interactions and overcoming strong electrostatic repulsion, making such binding unlikely. In contrast, TPPC3+ tends to bind multiple anions in solid-state structures to balance its charges within the crystal lattice. Despite this difference, these crystal structures are valuable as they reveal the conformation of TPPC3+ with its convergent functionalities and demonstrate its interactions with anions through hydrogen bonds.
To visualize the potential binding modes of TPPC3+ with anions in solution, we carried out structural optimization by DFT calculations at the BLYP-D3/SVP level in implicit water solvent for complexes with Cl−, Br−, I−, NO3−, SO42−, and C2O42−. All halide anions form (Fig. 6a–c and S72–S74†) hydrogen bonds with two of the three pyridinium units, establishing four [NH⋯O] and two [CH⋯O] hydrogen bonds. The NO3− anion associates (Fig. 6d and S76†) with two pyridinium units of TPPC3+ through two [NH⋯O] and two [CH⋯O] hydrogen bonds. The SO42− anion can interact (Fig. 6e and S77†) with all three pyridinium units of TPPC3+. Of its four oxygen atoms of the SO42− anion, three of them engage in a total of three [NH⋯O] and three [CH⋯O] hydrogen bonds, while the fourth oxygen forms one additional [NH⋯O] bond. This binding pattern results in a total of seven hydrogen bonds stabilizing the SO42−⊂TPPC3+ complex. The distances between the oxygen atoms on SO42− and the pyridinium nitrogens on TPPC3+ range from 5.4 to 6.0 Å, indicating strong electrostatic interactions that stabilize the complex. The larger C2O42− anion fits (Fig. 6f and S78†) better within the TPPC3+ cavity. Two of its oxygen atoms form three [NH⋯O] and three [CH⋯O] hydrogen bonds with two of the pyridinium units, while the remaining two oxygens form one [NH⋯O] and two [CH⋯O] hydrogen bonds with the third unit. This binding pattern results in a total of nine hydrogen bonds, stabilizing the C2O42−⊂TPPC3+ complex. The distances between the oxygen atoms on C2O42− and the pyridinium nitrogens on TPPC3+ are measured at 5.8 to 5.9 Å, suggesting strong electrostatic interactions that stabilize the C2O42−⊂TPPC3+ complex.
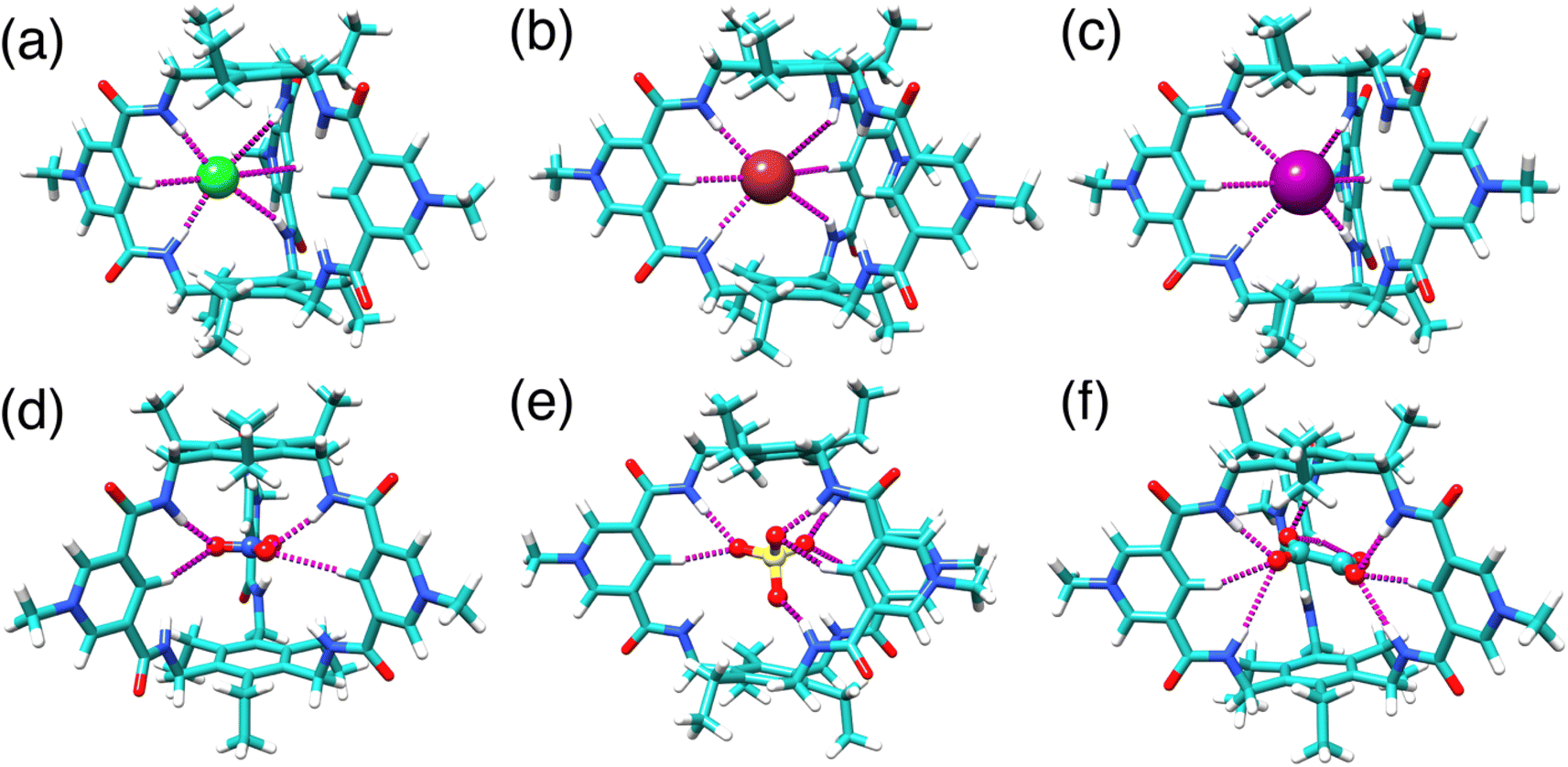 | ||
| Fig. 6 DFT optimized structure of (a) Cl−⊂TPPC3+, (b) Br−⊂TPPC3+, (c) I−⊂TPPC3+, (d) NO3−⊂TPPC3+, (e) SO42−⊂TPPC3+, and (f) C2O42−⊂TPPC3+. | ||
Catalysis for oxalate oxidation
We next explored how oxalate encapsulation in water with TPPC3+·3Cl− affects its reactivity in the oxidation process by the permanganate anion. This reaction between permanganate and oxalate is a well-known example of redox reactions exhibiting autocatalytic kinetics.92–96 The reaction involves (Fig. 7a) C2O42− or HC2O4− (depending on the pH) and MnO4−, yielding Mn2+ and CO2. Initially, the electrostatic repulsion between HC2O4− and MnO4− results in a sluggish reaction rate. The reaction product, Mn2+, acts as a catalyst by forming a neutral complex with C2O42− as MnC2O4, which can be more efficiently oxidized by MnO4− to produce CO2 and more Mn2+. This newly formed Mn2+ further catalyzes the reaction, leading to a characteristic reaction kinetics with an initial induction period needed to generate sufficient Mn2+ catalyst, followed by a rapid reaction rate acceleration.94 We hypothesize (Fig. 7b) that TPPC3+·3Cl− can act as a catalyst to bypass the initial electrostatic repulsion between HC2O4− and MnO4−, thereby speeding up the reaction by eliminating the induction period.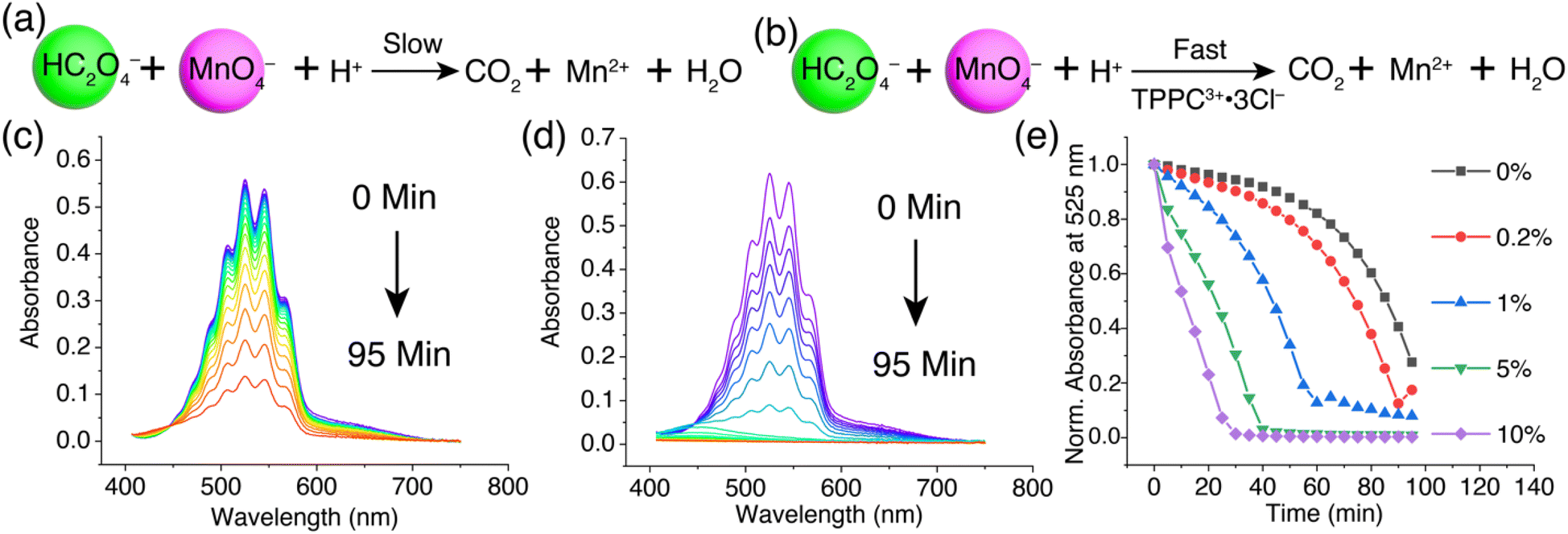 | ||
| Fig. 7 Reaction formula showing (a) the slow oxidation of oxalate by MnO4− and (b) its rate acceleration by TPPC3+·3Cl−. (c) Changes of the UV-vis spectra in a KMnO4 solution (0.2 mM) over 95 minutes after adding H2C2O4 (1 mM). (d) Changes of the UV-vis spectra in a KMnO4 (0.2 mM) solution in the presence of 5% TPPC3+·3Cl− (0.01 mM) over 95 minutes following H2C2O4 addition (1 mM). (e) The changes in absorbance at 525 nm for a KMnO4 solution (0.2 mM) over 95 minutes, demonstrating the effects of different loadings of TPPC3+·3Cl− as a catalyst on the reaction rate post H2C2O4 (1 mM) addition. | ||
To test our hypothesis, we mixed a solution of KMnO4 (0.2 mM) with H2C2O4 (1 mM) and monitored (Fig. 7c) the reaction by observing the decrease in the KMnO4 absorbance band at 400–700 nm. Initially, the reaction progressed slowly, with only 20% of the KMnO4 reduced (Fig. 7e) by H2C2O4 in the first 60 minutes, marking the induction period. From 60 to 95 minutes, the reaction rate significantly increased, resulting in an 80% overall reduction of KMnO4. Introducing 5% TPPC3+·3Cl− (0.01 mM) into the KMnO4 and H2C2O4 mixture drastically accelerated (Fig. 7d) the reaction, with the KMnO4 absorbance band dropping (Fig. 7e) to zero within 30 minutes, indicating a remarkable catalytic effect of TPPC3+·3Cl−. Monitoring the absorbance at 525 nm over time showed the absence of an induction period with the addition of 5% TPPC3+·3Cl−. The catalysis of oxalate oxidation by KMnO4 with TPPC3+·3Cl− is efficient even at a catalyst load (Fig. 7e) as low as 1%, attributed to the strong affinity between HC2O4− and TPPC3+ in water.97 With 10% TPPC3+·3Cl− as the catalyst, the reaction achieved an 80% MnO4− reduction in just 20 minutes, which is 4.8 times faster than the autocatalytic reaction without TPPC3+·3Cl− as the catalyst. The catalytic turnover number was estimated at 70 when 1% of the TPPC3+ was used as the catalyst.
The catalytic mechanism facilitated by TPPC3+·3Cl− is illustrated in Fig. 8 and is relatively straightforward. The encapsulation of HC2O4− by TPPC3+ forms a complex (HC2O4−⊂TPPC3+) carrying a net charge of +2. This electrostatic attraction between the encapsulated complex and MnO4− enhances the oxidation of HC2O4− into CO2, subsequently releasing the free TPPC3+ cage to capture another HC2O4− molecule, thereby perpetuating the catalytic cycle. To validate the effect of molecular recognition between TPPC3+ and HC2O4− on the observed catalytic effect, we synthesized (Scheme S2†) a TPy3+·3Cl− molecule as a control compound. This control compound carries identical charges to TPPC3+ but shows no binding with Na2C2O4 in water (Fig. S44†). When we introduced 10% TPy3+·3Cl− as a catalyst, we observed a slight rate acceleration for the oxidation of HC2O4− by KMnO4, indicating that charge neutralization from electrostatic interactions is a critical factor for the rate acceleration. In comparison, we achieved an even faster rate of acceleration using only 1% of TPPC3+·3Cl−, suggesting that molecular recognition mediated by charge-assisted hydrogen bonding enhances the catalytic effect by more than tenfold. To further confirm that the rate acceleration of the reaction was directly attributable to the binding between HC2O4− and TPPC3+, we performed (Fig. S63†) an additional control experiment by adding Na2SO4 (10 mM) as a competitive binding agent into a reaction mixture of TPPC3+·3Cl− (0.01 mM), KMnO4 (0.2 mM) and H2C2O4 (1.0 mM). The consequent oxidation rate of HC2O4− decreased by 30% as a result of the competitive binding of SO42− with TPPC3+, serving as compelling evidence that the specific interaction between HC2O4− and TPPC3+ is a pivotal factor in accelerating the reaction. To rule out the possibility of encapsulation of the anionic MnO4− by TPPC3+, we examined a 1:1 mixture of KMnO4 and TPPC3+·3Cl− by 1H NMR spectroscopy and observed (Fig. S43†) no change in chemical shift of TPPC3+·3Cl−, suggesting a lack of appreciable binding between MnO4− and TPPC3+. HRMS analysis of a mixture of KMnO4 and TPPC3+·3Cl− confirmed (Fig. S11†) the absence of binding, as it only displayed peaks corresponding to the free TPPC3+. Conversely, the HRMS spectra of a mixture containing KMnO4, H2C2O4, and TPPC3+·3Cl− showed (Fig. S11†) a distinct peak at m/z 512.7495, pointing to the selective formation of the HC2O4−⊂TPPC3+ complex in the presence of the MnO4− anion.
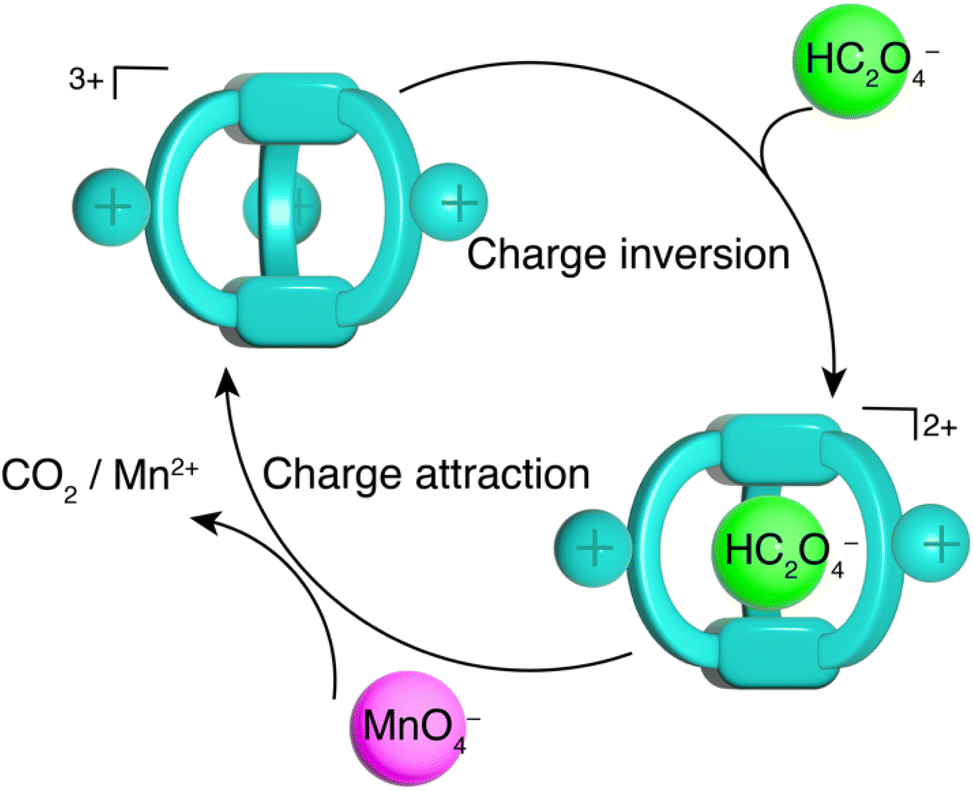 | ||
| Fig. 8 Schematic illustration of the catalytic cycle of oxalate oxidation catalyzed by TPPC3+. | ||
Conclusions
In conclusion, a tangible outcome of this study is the development of the water-soluble amide cage, TPPC3+·3Cl−, which can be readily synthesized by a dynamic approach. This cage demonstrates strong affinities and excellent selectivities for hydrophilic anions in water. Notably, TPPC3+·3Cl− exhibits remarkable binding affinities towards divalent anions such as C2O42− and SO42−, which play crucial roles in conditions like kidney stone formation98,99 and the management of radioactive nuclear waste,100 respectively. These findings are poised to advance efforts in oxalate removal during hemodialysis and enhance sulfate separation processes in the treatment of nuclear waste. Moreover, this research introduces the encapsulation of an anionic substrate by a cationic cage as a new mechanism for catalysis in water. This molecular recognition approach effectively inverts the charges of the substrate and facilitates reactions by overcoming electrostatic repulsions, showcasing an innovative strategy for developing catalysts in water.From a fundamental standpoint, our work broadened the scope of charge-assisted hydrogen bonding by illustrating its utility in an amide-type synthetic receptor, offering a general molecular design principle that applies to a wide range of amide receptors. This principle not only revalidates their hydrogen bonding capabilities but also opens avenues for the efficient molecular recognition of hydrophilic substrates in water. These findings will pave the way for the development of synthetic receptors for medical intervention,101–105 anion sensing,106–112 separation and sequestration,10,100,113–115 and catalysis in biologically and environmentally relevant aqueous media.116–120
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
C. X. and W. L. conceived and designed the project and wrote the manuscript. X. C., Q. T., D. L., C. Z. and W. L. performed the experiments and analyzed the results. L. W. solved the crystal structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Science Foundation (CHE-2337419) and the University of South Florida start-up funding. This research made use of the XRAY and CPAS Core facilities at the University of South Florida. The research was supported in part by the computational resources provided by the CIRCE research cluster facility at the University of South Florida.Notes and references
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer Berlin Heidelberg, Berlin, Heidelberg, 1991 Search PubMed.
- J. C.-H. Chen, B. L. Hanson, S. Z. Fisher, P. Langan and A. Y. Kovalevsky, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 15301–15306 CrossRef CAS PubMed.
- H. Luecke and F. A. Quiocho, Nature, 1990, 347, 402–406 CrossRef CAS PubMed.
- J. W. Pflugrath and F. A. Quiocho, Nature, 1985, 314, 257–260 CrossRef CAS PubMed.
- A. R. Fersht, J.-P. Shi, J. Knill-Jones, D. M. Lowe, A. J. Wilkinson, D. M. Blow, P. Brick, P. Carter, M. M. Y. Waye and G. Winter, Nature, 1985, 314, 235–238 CrossRef CAS PubMed.
- J. Dong and A. P. Davis, Angew. Chem., Int. Ed., 2021, 60, 8035–8048 CrossRef CAS PubMed.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- S. Kubik, ChemistryOpen, 2022, 11, e202200028 CrossRef CAS PubMed.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648 RSC.
- J. Samanta, M. Tang, M. Zhang, R. P. Hughes, R. J. Staples and C. Ke, J. Am. Chem. Soc., 2023, 145, 21723–21728 CrossRef CAS PubMed.
- A. Metzger and E. V. Anslyn, Angew. Chem., Int. Ed., 1998, 37, 649–652 CrossRef CAS PubMed.
- J. Krämer, R. Kang, L. M. Grimm, L. De Cola, P. Picchetti and F. Biedermann, Chem. Rev., 2022, 122, 3459–3636 CrossRef.
- C.-L. Deng, S. L. Murkli and L. D. Isaacs, Chem. Soc. Rev., 2020, 49, 7516–7532 RSC.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed.
- I. V. Kolesnichenko and E. V. Anslyn, Chem. Soc. Rev., 2017, 46, 2385–2390 RSC.
- G. T. Williams, C. J. E. Haynes, M. Fares, C. Caltagirone, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2021, 50, 2737–2763 RSC.
- D. R. Carcanague, C. B. Knobler and F. Diederich, J. Am. Chem. Soc., 1992, 114, 1515–1517 CrossRef CAS.
- F. Diederich and D. R. Carcanague, Helv. Chim. Acta, 1994, 77, 800–818 CrossRef CAS.
- E. M. Peck, W. Liu, G. T. Spence, S. K. Shaw, A. P. Davis, H. Destecroix and B. D. Smith, J. Am. Chem. Soc., 2015, 137, 8668–8671 CrossRef CAS PubMed.
- W. Liu, A. Johnson and B. D. Smith, J. Am. Chem. Soc., 2018, 140, 3361–3370 CrossRef CAS PubMed.
- A. P. Davis, Chem. Soc. Rev., 2020, 49, 2531–2545 RSC.
- P. Stewart, C. M. Renney, T. J. Mooibroek, S. Ferheen and A. P. Davis, Chem. Commun., 2018, 54, 8649–8652 RSC.
- P. Ríos, T. J. Mooibroek, T. S. Carter, C. Williams, M. R. Wilson, M. P. Crump and A. P. Davis, Chem. Sci., 2017, 8, 4056–4061 RSC.
- P. Rios, T. S. Carter, T. J. Mooibroek, M. P. Crump, M. Lisbjerg, M. Pittelkow, N. T. Supekar, G. J. Boons and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 3387–3392 CrossRef CAS PubMed.
- T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. M. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2016, 8, 69–74 CrossRef CAS PubMed.
- T. S. Carter, T. J. Mooibroek, P. F. N. Stewart, M. P. Crump, M. C. Galan and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 9311–9315 CrossRef CAS PubMed.
- C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718–723 CrossRef CAS PubMed.
- D. H. Li and B. D. Smith, Beilstein J. Org. Chem., 2019, 15, 1086–1095 CrossRef CAS PubMed.
- W. Liu, C. F. A. Gómez-Durán and B. D. Smith, J. Am. Chem. Soc., 2017, 139, 6390–6395 CrossRef CAS PubMed.
- W. Liu, E. M. Peck, K. D. Hendzel and B. D. Smith, Org. Lett., 2015, 17, 5268–5271 CrossRef CAS PubMed.
- S. K. Shaw, W. Liu, S. P. Brennan, M. de Lourdes Betancourt-Mendiola and B. D. Smith, Chem.–Eur. J., 2017, 23, 12646–12654 CrossRef CAS PubMed.
- Y. F. Wang, H. Yao, L. P. Yang, M. Quan and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202211853 CrossRef CAS PubMed.
- X. Wang, M. Quan, H. Yao, X. Y. Pang, H. Ke and W. Jiang, Nat. Commun., 2022, 13, 2291 CrossRef CAS PubMed.
- M. Li, Y. Dong, M. Quan and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202208508 CrossRef CAS PubMed.
- X. Huang, X. Wang, M. Quan, H. Yao, H. Ke and W. Jiang, Angew. Chem., Int. Ed., 2021, 60, 1929–1935 CrossRef CAS PubMed.
- L. P. Yang, X. Wang, H. Yao and W. Jiang, Acc. Chem. Res., 2020, 53, 198–208 CrossRef CAS PubMed.
- H. Ke, L. P. Yang, M. Xie, Z. Chen, H. Yao and W. Jiang, Nat. Chem., 2019, 11, 470–477 CrossRef CAS PubMed.
- H. Yao, H. Ke, X. Zhang, S. J. Pan, M. S. Li, L. P. Yang, G. Schreckenbach and W. Jiang, J. Am. Chem. Soc., 2018, 140, 13466–13477 CrossRef CAS PubMed.
- L.-L. Wang, Z. Chen, W.-E. Liu, H. Ke, S.-H. Wang and W. Jiang, J. Am. Chem. Soc., 2017, 139, 8436–8439 CrossRef CAS PubMed.
- G. B. Huang, S. H. Wang, H. Ke, L. P. Yang and W. Jiang, J. Am. Chem. Soc., 2016, 138, 14550–14553 CrossRef CAS PubMed.
- S. Kubik, R. Kirchner, D. Nolting and J. Seidel, J. Am. Chem. Soc., 2002, 124, 12752–12760 CrossRef CAS PubMed.
- S. Otto and S. Kubik, J. Am. Chem. Soc., 2003, 125, 7804–7805 CrossRef CAS PubMed.
- Z. Rodriguez-Docampo, S. I. Pascu, S. Kubik and S. Otto, J. Am. Chem. Soc., 2006, 128, 11206–11210 CrossRef CAS PubMed.
- J. M. Llinares, D. Powell and K. Bowman-James, Coord. Chem. Rev., 2003, 240, 57–75 CrossRef CAS.
- E. García-España, P. Díaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952–2986 CrossRef.
- P. Blondeau, M. Segura, R. Pérez-Fernández and J. De Mendoza, Chem. Soc. Rev., 2007, 36, 198–210 RSC.
- M. Berger and F. P. Schmidtchen, Angew. Chem., Int. Ed., 1998, 37, 2694–2696 CrossRef CAS PubMed.
- W. Peschke, P. Schiessl, F. P. Schmidtchen, P. Bissinger and A. Schier, J. Org. Chem., 1995, 60, 1039–1043 CrossRef CAS.
- A. Metzger, V. M. Lynch and E. V. Anslyn, Angew. Chem., Int. Ed., 1997, 36, 862–865 CrossRef CAS.
- M. D. Best, S. L. Tobey and E. V. Anslyn, Coord. Chem. Rev., 2003, 240, 3–15 CrossRef CAS.
- Q. Q. Wang, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2012, 51, 2119–2123 CrossRef CAS PubMed.
- S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882–7894 CrossRef CAS PubMed.
- K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed.
- L. Chen, S. N. Berry, X. Wu, E. N. W. Howe and P. A. Gale, Chem, 2020, 6, 61–141 CAS.
- P. A. Gale, Acc. Chem. Res., 2006, 39, 465–475 CrossRef CAS PubMed.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- W. A. Harrell, M. L. Bergmeyer, P. Y. Zavalij and J. T. Davis, Chem. Commun., 2010, 46, 3950–3952 RSC.
- J. T. Davis, P. A. Gale, O. A. Okunola, P. Prados, J. C. Iglesias-Sánchez, T. Torroba and R. Quesada, Nat. Chem., 2009, 1, 138–144 CrossRef CAS PubMed.
- A. P. Bisson, V. M. Lynch, M.-K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed., 1997, 36, 2340–2342 CrossRef CAS.
- L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- V. E. Zwicker, K. K. Y. Yuen, D. G. Smith, J. Ho, L. Qin, P. Turner and K. A. Jolliffe, Chem.–Eur. J., 2018, 24, 1140–1150 CrossRef CAS PubMed.
- S. N. Berry, L. Qin, W. Lewis and K. A. Jolliffe, Chem. Sci., 2020, 11, 7015–7022 RSC.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS PubMed.
- S. Peng, Q. He, G. I. Vargas-Zúñiga, L. Qin, I. Hwang, S. K. Kim, N. J. Heo, C. H. Lee, R. Dutta and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 865–907 RSC.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CrossRef CAS PubMed.
- H. Wang, S. Fang, G. Wu, Y. Lei, Q. Chen, H. Wang, Y. Wu, C. Lin, X. Hong, S. K. Kim, J. L. Sessler and H. Li, J. Am. Chem. Soc., 2020, 142, 20182–20190 CrossRef CAS PubMed.
- C. Zhai, C. Xu, Y. Cui, L. Wojtas and W. Liu, Chem.–Eur. J., 2023, 29, e202300524 CrossRef CAS PubMed.
- W. Liu, S. Bobbala, C. L. Stern, J. E. Hornick, Y. Liu, A. E. Enciso, E. A. Scott and J. Fraser Stoddart, J. Am. Chem. Soc., 2020, 142, 3165–3173 CrossRef CAS PubMed.
- W. Liu, C. Lin, J. A. Weber, C. L. Stern, R. M. Young, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 8938–8945 CrossRef CAS PubMed.
- W. Liu, Y. Tan, L. O. Jones, B. Song, Q. H. Guo, L. Zhang, Y. Qiu, Y. Feng, X. Y. Chen, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 15688–15700 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- J. Lehr, T. Lang, O. A. Blackburn, T. A. Barendt, S. Faulkner, J. J. Davis and P. D. Beer, Chem.–Eur. J., 2013, 19, 15898–15906 CrossRef CAS PubMed.
- M. J. Langton and P. D. Beer, Chem.–Eur. J., 2012, 18, 14406–14412 CrossRef CAS PubMed.
- N. H. Evans, H. Rahman, A. V. Leontiev, N. D. Greenham, G. A. Orlowski, Q. Zeng, R. M. J. Jacobs, C. J. Serpell, N. L. Kilah, J. J. Davis and P. D. Beer, Chem. Sci., 2012, 3, 1080 RSC.
- A. Brown and P. D. Beer, Dalton Trans., 2012, 41, 118–129 RSC.
- N. H. Evans, C. J. Serpell, N. G. White and P. D. Beer, Chem.–Eur. J., 2011, 17, 12347–12354 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, Org. Biomol. Chem., 2011, 9, 92–100 RSC.
- S. R. Bayly, T. M. Gray, M. J. Chmielewski, J. J. Davis and P. D. Beer, Chem. Commun., 2007, 2234 RSC.
- G. Hennrich and E. V. Anslyn, Chem.–Eur. J., 2002, 8, 2218 CrossRef CAS PubMed.
- J. C. Lauer, A. S. Bhat, C. Barwig, N. Fritz, T. Kirschbaum, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2022, 28, e202201527 CrossRef CAS PubMed.
- A. S. Bhat, S. M. Elbert, W. Zhang, F. Rominger, M. Dieckmann, R. R. Schröder and M. Mastalerz, Angew. Chem., Int. Ed., 2019, 131, 8911–8915 CrossRef.
- TPPC3+ showed a poor water solubility when associated with hydrophobic counter anions such as BF4− and PF6−, making it impractical to evaluate its anion binding properties in water.
- R. S. Das, D. Maiti, S. Kar, T. Bera, A. Mukherjee, P. C. Saha, A. Mondal and S. Guha, J. Am. Chem. Soc., 2023, 145, 20451–20461 CrossRef CAS PubMed.
- A. Szumna and J. Jurczak, Eur. J. Org Chem., 2001, 4031–4039 CrossRef CAS.
- M. J. Chmielewski and J. Jurczak, Chem.–Eur. J., 2005, 11, 6080–6094 CrossRef CAS PubMed.
- M. J. Chmielewski and J. Jurczak, Chem.–Eur. J., 2006, 12, 7652–7667 CrossRef CAS PubMed.
- A. Martinez-Cuezva, L. V. Rodrigues, C. Navarro, F. Carro-Guillen, L. Buriol, C. P. Frizzo, M. A. P. Martins, M. Alajarin and J. Berna, J. Org. Chem., 2015, 80, 10049–10059 CrossRef CAS PubMed.
- V. W. Liyana Gunawardana, C. Ward, H. Wang, J. H. Holbrook, E. R. Sekera, H. Cui, A. B. Hummon and J. D. Badjić, Angew. Chem., Int. Ed., 2023, 62, e202306722 CrossRef CAS PubMed.
- F. Biedermann, W. M. Nau and H. J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171 CrossRef CAS PubMed.
- A downfield shift of proton B was observed during the titration with Cl−, while an upfield shift occurred when titrated with Br− and I−. The downfield shift of proton B indicates Cl− binding to the partially filled cavity of the cage. The upfield shift with Br− and I− can be attributed to the displacement of Cl− by these anions, which have lower charge densities and thus lead to a weaker ion-dipole interaction with proton B, resulting in its upfield shift.
- C. M. Hindson, Z. M. Smith, N. W. Barnett, G. R. Hanson, K. F. Lim and P. S. Francis, J. Phys. Chem. A, 2013, 117, 3918–3924 CrossRef CAS PubMed.
- M. A. Kelland, J. Chem. Educ., 2011, 88, 276–278 CrossRef CAS.
- K. A. Kovács, P. Gróf, L. Burai and M. Riedel, J. Phys. Chem. A, 2004, 108, 11026–11031 CrossRef.
- V. Pimienta, D. Lavabre, G. Levy and J. C. Micheau, J. Phys. Chem., 1995, 99, 14365–14371 CrossRef CAS.
- H. F. Launer, J. Am. Chem. Soc., 1932, 54, 2597–2610 CrossRef CAS.
- 1H NMR titration experiment between H2C2O4 and TPPC3+·3Cl− revealed (Fig. S41 and S42†) a binding constant of 3.8 × 105 M−1 in water.
- T. Ermer, C. Kopp, J. R. Asplin, I. Granja, M. A. Perazella, M. Reichel, T. D. Nolin, K.-U. Eckardt, P. S. Aronson, F. O. Finkelstein and F. Knauf, Kidney International Reports, 2017, 2, 1050–1058 CrossRef PubMed.
- C. F. M. Franssen, Nephrol., Dial., Transplant., 2005, 20, 1916–1921 CrossRef CAS PubMed.
- C. J. Fowler, T. J. Haverlock, B. A. Moyer, J. A. Shriver, D. E. Gross, M. Marquez, J. L. Sessler, M. A. Hossain and K. Bowman-James, J. Am. Chem. Soc., 2008, 130, 14386–14387 CrossRef CAS PubMed.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- E. S. Williams, H. Gneid, S. R. Marshall, M. J. González, J. A. Mandelbaum and N. Busschaert, Org. Biomol. Chem., 2022, 20, 5958–5966 RSC.
- S. R. Herschede, R. Salam, H. Gneid and N. Busschaert, Supramol. Chem., 2022, 34, 26–33 CrossRef CAS.
- R. Salam, S. M. Chowdhury, S. R. Marshall, H. Gneid and N. Busschaert, Chem. Commun., 2021, 57, 13122–13125 RSC.
- R. Cao, R. B. Rossdeutcher, Y. Zhong, Y. Shen, D. P. Miller, T. A. Sobiech, X. Wu, L. S. Buitrago, K. Ramcharan, M. I. Gutay, M. F. Figueira, P. Luthra, E. Zurek, T. Szyperski, B. Button, Z. Shao and B. Gong, Nat. Chem., 2023, 15, 1559–1568 CrossRef CAS PubMed.
- R. Hein, P. D. Beer and J. J. Davis, Chem. Rev., 2020, 120, 1888–1935 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed.
- K. M. Bak, S. C. Patrick, X. Li, P. D. Beer and J. J. Davis, Angew. Chem., Int. Ed., 2023, 62, e202300867 CrossRef CAS PubMed.
- R. Hein, A. Docker, J. J. Davis and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 8827–8836 CrossRef CAS PubMed.
- R. Hein and P. D. Beer, Chem. Sci., 2022, 13, 7098–7125 RSC.
- S. C. Patrick, R. Hein, P. D. Beer and J. J. Davis, J. Am. Chem. Soc., 2021, 143, 19199–19206 CrossRef CAS PubMed.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS PubMed.
- C. R. Benson, L. Kacenauskaite, K. L. VanDenburgh, W. Zhao, B. Qiao, T. Sadhukhan, M. Pink, J. Chen, S. Borgi, C. H. Chen, B. J. Davis, Y. C. Simon, K. Raghavachari, B. W. Laursen and A. H. Flood, Chem, 2020, 6, 1978–1997 CAS.
- A. Aydogan, D. J. Coady, S. K. Kim, A. Akar, C. W. Bielawski, M. Marquez and J. L. Sessler, Angew. Chem., Int. Ed., 2008, 47, 9648–9652 CrossRef CAS PubMed.
- S. K. Kim, J. Lee, N. J. Williams, V. M. Lynch, B. P. Hay, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 15079–15085 CrossRef CAS PubMed.
- J. M. Ovian, P. Vojáčková and E. N. Jacobsen, Nature, 2023, 616, 84–89 CrossRef CAS PubMed.
- Q. Li, S. M. Levi, C. C. Wagen, A. E. Wendlandt and E. N. Jacobsen, Nature, 2022, 608, 74–79 CrossRef CAS PubMed.
- S. M. Banik, A. Levina, A. M. Hyde and E. N. Jacobsen, Science, 2017, 358, 761–764 CrossRef CAS PubMed.
- M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed.
- T. Du, B. Shen, J. Dai, M. Zhang, X. Chen, P. Yu and Y. Liu, J. Am. Chem. Soc., 2023, 145, 27788–27799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2334098 and 2334101–2334103. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05236f |
| This journal is © The Royal Society of Chemistry 2024 |