
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiphosphaenones: beyond the phosphorus analogue of enones†
Jieli
Lin
,
Shihua
Liu
,
Shunlin
Zheng
,
Hansjörg
Grützmacher
 ,
Cheng-Yong
Su
and
Zhongshu
Li
*
,
Cheng-Yong
Su
and
Zhongshu
Li
*
LIFM, IGCME, School of Chemistry, Sun Yat-Sen University, Guangzhou 510006, China. E-mail: lizhsh6@mail.sysu.edu.cn
First published on 11th November 2024
Abstract
Phosphaenones, like their carbon analogue enones (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CO), are promising building blocks for synthetic chemistry and materials science. However, in contrast to the α- and β-phosphaenones, structurally and spectroscopically well-defined diphosphaenones (DPEs) are rare. In this study, we disclose the isolation and spectroscopic characterization of N-heterocyclic vinyl (NHV) substituted acyclic DPEs 3a,b [NHV–PP–C(O)–NHV]. X-ray diffraction methods allowed determination of the structures, which show a central planar trans PP–CO configuration. Compound 3a behaves like classical enones and shows 1,4-addition across the PP–CO unit, which proceeds in a stepwise manner. In contrast, 3a exhibits also 1,2-addition across the PP but not the CO double bond, which differentiates it from enones.
C–CO), are promising building blocks for synthetic chemistry and materials science. However, in contrast to the α- and β-phosphaenones, structurally and spectroscopically well-defined diphosphaenones (DPEs) are rare. In this study, we disclose the isolation and spectroscopic characterization of N-heterocyclic vinyl (NHV) substituted acyclic DPEs 3a,b [NHV–PP–C(O)–NHV]. X-ray diffraction methods allowed determination of the structures, which show a central planar trans PP–CO configuration. Compound 3a behaves like classical enones and shows 1,4-addition across the PP–CO unit, which proceeds in a stepwise manner. In contrast, 3a exhibits also 1,2-addition across the PP but not the CO double bond, which differentiates it from enones.
Introduction
The diagonal relationship between carbon and phosphorus has not only inspired the synthesis of organophosphorus compounds but also expanded our understanding of molecular properties on one side and the limits of concepts based on analogies on the other.1 Phosphabenzenes, phosphaalkenes, phosphaalkynes, diphosphenes, and other related compounds closely mimic their carbon analogues while also exhibiting distinctive electronic properties due to the incorporation of heavier elements.2 These compounds became valuable building blocks in synthetic chemistry and materials science and like their carbon counterparts allow to synthesize organophosphorus compounds in an especially efficient and frequently atom-economic way.3 This is also true for enones [R2CCR–C(O)R], which represent a widely investigated class of compounds in classical organic syntheses.4 By replacing one or two CR groups of enones with P atoms, α-phosphaenones [α-PEs, R2CP–C(O)R], β-phosphaenones [β-PEs, RPCR–C(O)R], and diphosphaenones [DPEs, RPP–C(O)R] can be engineered (Fig. 1a).
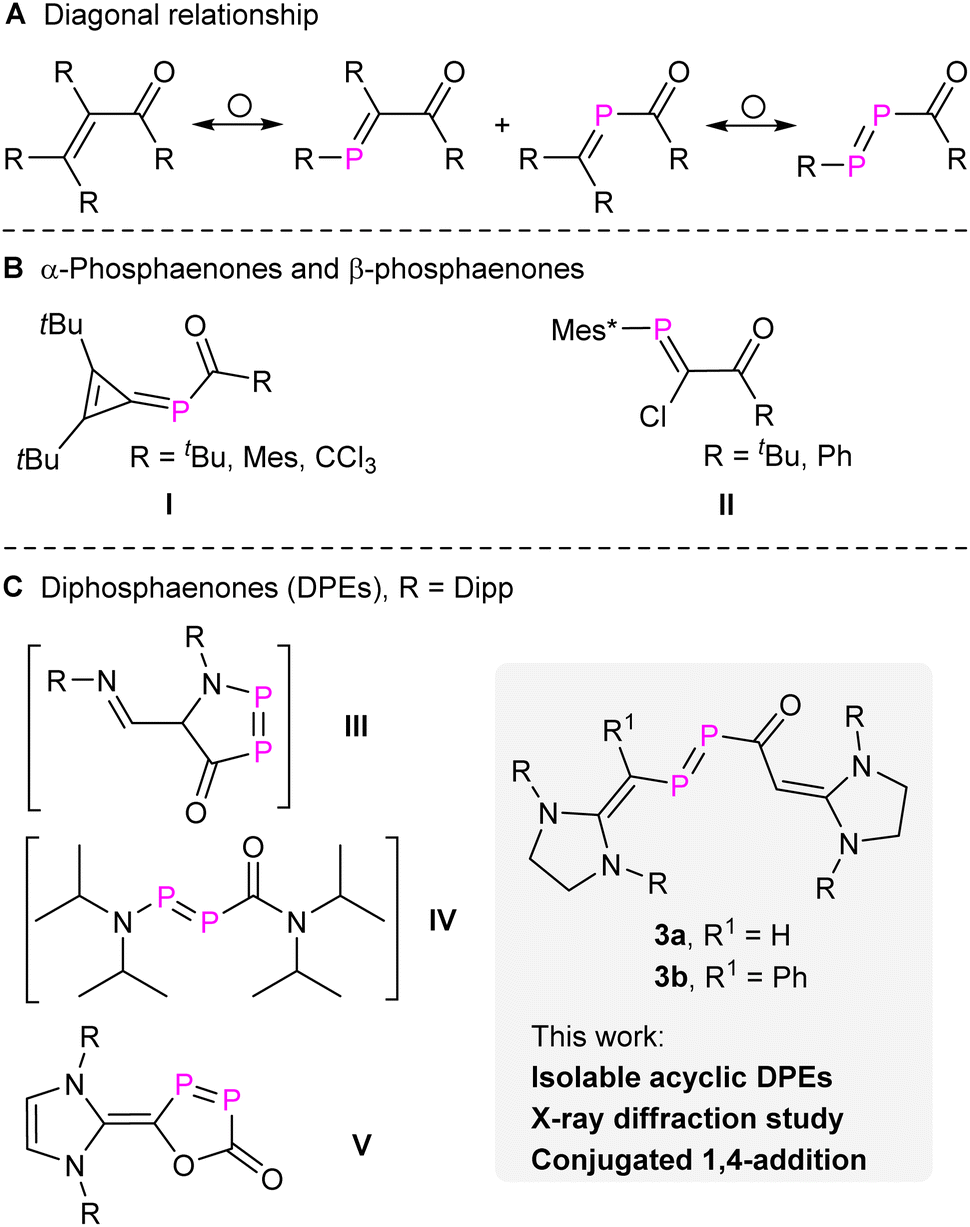 | ||
| Fig. 1 (A) Diagonal relationship between enones and their phosphorus analogues. (B and C) Selected examples of α-phosphaenones, β-phosphaenones, and diphosphaenones. Mes, 2,4,6-tri-methylphenyl; Mes*, 2,4,6-tri-tertbutylphenyl; Dipp, 2,6-di-isopropylphenyl. | ||
The synthesis of stable α-PEs (I, Fig. 1) was initially achieved by M. Regitz and co-workers through the nucleophilic attack from P-silyl-substituted phosphaalkene [R2CP–SiMe3] to acyl chlorides.5 The first stable β-PEs (II, Fig. 1) were independently reported by the groups of F. Bickelhaupt6 and M. Yoshifuji7 through nucleophilic attack of a carbenoid phosphanylidene [Mes*PC(Cl)Li] to acyl chlorides. Other stable β-PEs were also successfully prepared using cyclic rigid frameworks.8 However, the synthesis and isolation of DPEs remains challenging. Previously, we proposed the presence of two transient DPEs, the first cyclic (III, Fig. 1)9 and acyclic DPEs (IV, Fig. 1),10 which were confirmed through trapping experiments via the [2 + 4] or [2 + 2] cycloaddition across the PP double bond. Seminal work from the group of Protasiewicz revealed the first stable cyclic DPE (V, Fig. 1)11 through the treatment of [(NHC–Cl)Cl] (NHCN-heterocyclic carbene) with two equivalents of sodium phosphaethynolate [Na(OCP)].12 However, stable acyclic DPEs remain elusive to date.
In the present work, we report the synthesis of N-heterocyclic vinyl (NHV)13 substituted compounds 3a,b [NHV–PP–C(O)–NHV] (Fig. 1). The characterization of 3a,b as DPEs is strongly supported by X-ray crystal diffraction (XRD) studies, multinuclear NMR spectra analyses, and quantum chemical calculations. In addition, 3a behaves like enones showing 1,4-additions in a stepwise manner, which differentiates it from classical diphosphenes. To date only one example among PEs, a cyclic cis β-PE, has been reported to undergo likewise a conjugated 1,4-addition.8e The replacement of both CR with P atoms in enones alters more profoundly the electronic structure and indeed, 3a exhibits also 1,2-addition across the PP double bond, but not the CO double bond.
Results and discussion
Synthesis and characterization of DPEs 3a,b
The NHV substituted14 phosphenium chloride 1a (RH) and 1b (RPh) ([LC(R)–P–CHL)+Cl−], L = SIPr = (1,3-bis-(2,6-diisopropylphenyl)imidazolidin-2-ylidine) were prepared as orange powders [1a: δ(31P) = 331.7 ppm]; 1b: δ(31P) = 323.3 ppm; see ESI† for further details). Treatment of 1a,b with an equimolar amount of Na(OCP) exclusively afforded 3a,b (LC(R)–PP–C(O)–C(H)L, 3a: RH; 3b: RPh) as red powders in more than 74% yield under the elimination of sodium chloride (Fig. 2A). The formation of 3a,b is likely to occur through the intramolecular rearrangement of transient phosphanyl phosphaketenes 2a,b ([NHV–P(PCO)–NHV]).10 Both 3a,b are stable in the solid state under a nitrogen atmosphere for weeks without noticeable decomposition, but they are highly sensitive to air and moisture. Additionally, no apparent decomposition or isomerization products were detected after irradiating a toluene solution of 3a,b with LED light at 455 or 520 nm at 0 °C for 30 minutes.14d But 3a slowly dimerizes in solution via the [2 + 2] cycloaddition across the PP double bond.15 Similar dimerization process was not observed for 3b, likely due to the presence of additional phenyl substituents. After storing the saturated hexane solution of 3a at room temperature under an inert atmosphere via slow evaporation for one-week, yellow crystalline product as the dimer of 3a precipitated in 61% yield. The multinuclear NMR spectra analyses (Fig. S14–18†) and XRD study (Fig. S120†) confirmed that the dimerization proceeds in a highly regioselective manner affording thermodynamically favorable head to head dimer (Fig. S129†).15
 | ||
| Fig. 2 (A) Synthesis of 3–5. (B) Solid-state structure of 3a and 3b (ellipsoids are set to 50% probability; H atoms are omitted for clarity). Selected distances (Å): 3a: P1–P2 2.0609(7), P2–C1 1.874(19), C1–O1 1.242(2), C1–C2 1.416(3), C2–C3 1.385(3), P1–C4 1.749(2), C4–C5 1.389(3); 3b: P1–P2 2.0657(19), P2–C1 1.861(6), C1–O1 1.220(6), C1–C2 1.454(7), C2–C3 1.376(7), P1–C4 1.762(6), C4–C5 1.391(7), C4–C6 1.500(7). | ||
Red single crystals of 3a,b were grown from saturated hexane solutions at −30 °C and studied by XRD analyses (Fig. 2B). In the case of 3a, the central PP–CO fragment is almost planar [∠P1P2C1O1 = 173.26(13)°], and coplanar with the two terminal NHV moieties [∠C5C4P1P2 = 170.1(2)°, ∠P2C1C2C3 = 174.03(16)°]. This indicates the presence of electron delocalization across the NHV–P–P–C(O)–NHV skeleton. The delocalized double bonds C5C4 [1.389(3) Å], P1P2 [2.0609(7) Å], C1O1 [1.242(2) Å], and C2C3 [1.385(3) Å] are connected via single bonds C4–P1 [1.749(2) Å], P2–C1 [1.874(19) Å], and C1–C2 [1.416(3) Å] [Σrcov(P–C) = 1.86 Å, Σrcov(PC) = 1.69 Å, Σrcov(C–C) = 1.50 Å, Σrcov(CC) = 1.34 Å, Σrcov(C–O) = 1.38 Å, Σrcov(CO) = 1.24 Å],16 respectively. The P2–C1 bond is longer than that of the C4–P1 bond, which is likely due to hyperconjugation n(O1) → σ*(P2–C1).173b exhibits similar structural metrics to those of 3a, but with an additional phenyl substituent at C4 (C4–C6 [1.500(7) Å]).
Natural bond orbital (NBO) calculations18 allow a more detailed insight into the bonding scenario of a simplified model 3aM, where the NHC SIPr was replaced with SIMe (1,3-dimethyl-imidazoline-2-ylidine) (Fig. S121†). An inspection of the Wiberg bond indices (WBIs) of 3aM suggested that the WBIs of alternating double [C5C4 (1.51), P1P2 (1.75), C1O1 (1.64), and C2C3 (1.40)] and single [C4–P1 (1.10) and C1–C2 (1.20)] bonds are substantially smaller or larger than expected, respectively. Only single bond P2–C1 (0.93) for which the bond order turned to be even smaller. Second-order perturbation theory analyses revealed the donor–acceptor energies E(2) for the delocalization of the lone pair localized on the O1 to σ*(P2–C1) is 29.86 kcal mol−1, which is larger than the donation into σ*(C1–C2) (22.60 kcal mol−1). The strength of the π-type interaction, including both electron-donation and acception between the central double bonds of 3aM, follows the order: 49.75 kcal mol−1 (C1O1 ↔ C2C3) > 23.63 kcal mol−1 (P1P2 ↔ C4C5) > 10.65 kcal mol−1 (C1O1 ↔ P1P2). Thus, NBO calculations support the presence of significant hyperconjugation n(O1) → σ*(P2–C1) and π-type electron delocalization within the PP–CO moiety, which aligns with the relatively long P2–C1 bond and nearly planar PP–CO configuration (see XRD analyses).
The proton-decoupled 31P NMR spectra of 3a and 3b show two characteristic doublets at δ(31P) = 487.5 (P1) and 385.0 ppm (P2, 1JPP = 529.5 Hz), and δ(31P) = 525.9 (P1) and 376.7 ppm (P2, 1JPP = 515.9 Hz), respectively. The chemical shift assignment was made by comparison with the calculated chemical shifts [3aM: 511.3 (P1) and 413.9 ppm (P2), Table S2†].19 The large 1JPP coupling constants are indicative of the presence of a PP double bond in both 3a,b.3h,15 In the 1H NMR spectra, a broad singlet at 5.77 ppm due to the small and unresolved 2JPH coupling, and a sharp singlet at 4.77 ppm were attributed to the protons of the C4–H and C2–H at the NHV moieties for 3a. A sharp singlet at 4.54 ppm is assigned to the proton of C2–H and characterizes 3b, which shows no further resonances between 4.0–6.5 ppm. The proton-decoupled 13C NMR spectra displayed high-frequency shifted multiplets assigned to the carbonyl group at 198.9 ppm and 198.3 ppm for 3a and 3b, respectively. Solutions of 3a and 3b in toluene are red and exhibit strong absorptions at λmax = 488 nm (5.0 × 104 M−1 cm−1) and 496 nm (2.0 × 104 M−1 cm−1), respectively. According to time-dependent density functional theory (TD-DFT) calculations, these absorptions arise from electronic transitions of the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) (π–π*, Fig. S90 and S91†).
Chlorophosphine 4 [δ(31P) = 96.3 ppm] with NHV and 2,4,6-tri-methylphenyl substituents was prepared as a white powder in 90% yield (see ESI† for details). Treatment of 4 with an equimolar amount of Na(OCP) provided exclusively the phosphanyl phosphaketene 5 [δ(31P) = −41.0 and −248.6 ppm, 1JPP = 170.9 Hz] as white powder in 55% yield (Fig. 2).9,20 In solution at room temperature, 5 did not convert to the DPE but decomposed into an unidentified mixture (Fig. S39†).
To gain a deeper understanding of a possible mechanism leading to 3a,b, DFT calculations were carried out for the rearrangement of the simplified model 2bM at the M062X/Def2TZVP-SMD(toluene)//M062X/Def2SVP level of theory.21 The minimum energy reaction pathways (MERPs) for two possible rearrangements are shown in Fig. 3A. The rearrangement starts with a nucleophilic attack of the parent or phenyl substituted NHV on the electron-deficient carbon of the PCO moiety via the activated complex TS1-b (14.2 kcal mol−1) or TS1-c (17.1 kcal mol−1). Subsequently, the four-membered heterocycles IN1-b (10.5 kcal mol−1) or IN1-c (6.9 kcal mol−1) are formed in an endothermic reaction. A subsequent P–C cleavage is easily achieved viaTS2-b (9.0 kcal mol−1) or TS2-c (6.4 kcal mol−1) to generate kinetically and thermodynamically favourable products 3bM (−7.9 kcal mol−1) or 3cM (−12.9 kcal mol−1). Natural population analyses (NPA) charges were obtained from a NBO analysis18 and indicate that the carbon of the PCO moiety is positively charged (+0.31e), whereas the vinyl carbons from the two NHV substituents are negatively charged. Specifically, the vinyl carbon (−0.80e) in the parent NHV possesses more electron density than the one with phenyl substituted NHV moiety (−0.59e; Fig. 3B). The exergonic rearrangement from 2bM to 3bM is kinetically favoured by 2.9 kcal mol−1 over the formation of 3cM, which is in consistent with our experimentally observation that only 3b was formed.
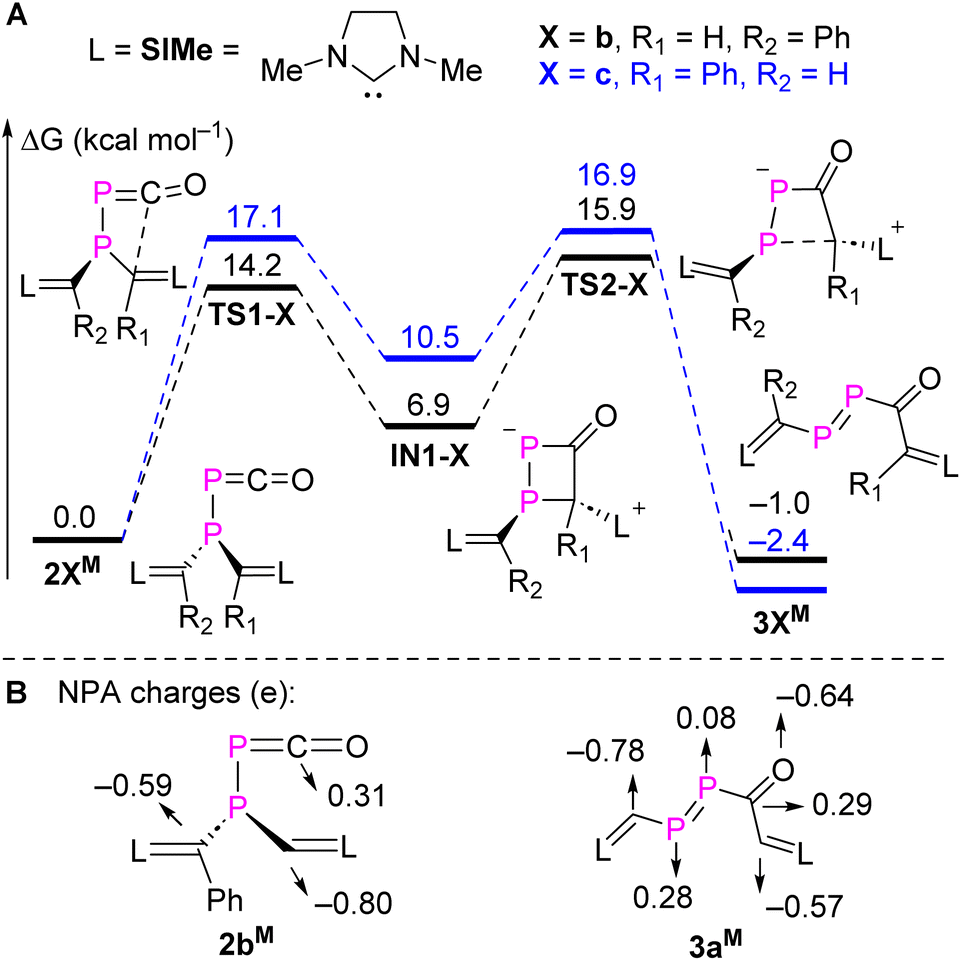 | ||
| Fig. 3 (A) MERPs of the selective intramolecular rearrangement of 2bM, and (B) selected NPA charges of 2bM and 3aM at M062X/Def2-TZVP-SMD(toluene)//M062X/Def2SVP level of theory. | ||
A detailed MO analysis at the M062X/Def2SVP level of theory provided insights into the electronic structure between 3aM and its hypothetical carbon analogue B, parent DPE A and parent enone C (Fig. 4). For the parent DPE A, the LUMO and HOMO-1 are primarily the π*-type and π-type orbitals of the PP unit. The HOMO corresponds mainly the lone-pair type orbital derived from phosphorus and oxygen atoms. The HOMO and HOMO-1 of the parent enone C are also mainly combinations of the lone-pair type orbital from oxygen and π-type orbitals of the CC unit. But the LUMO of C is the π*-type orbitals of the conjugated CC–CO. These frontier orbitals in A and C are seen in the NHV substituted derivatives 3aM and B as LUMO, HOMO, and HOMO-2. The strong electron releasing substituent NHV raises the energy levels of all orbitals discussed so far. The energy levels of the π-type orbitals (HOMO) are even higher than that of the combination of lone-pair type orbitals at O and both P (HOMO-2) in 3aM and B due to the strong π-electron delocalization over the NHV groups. Both HOMO-1 of 3aM and B are mainly attributed to the π-type bonding of one NHV group with a minor polarization towards the carbonyl group. Consequently, both 3aM and B are best viewed as NHV substituted DPE and enone.
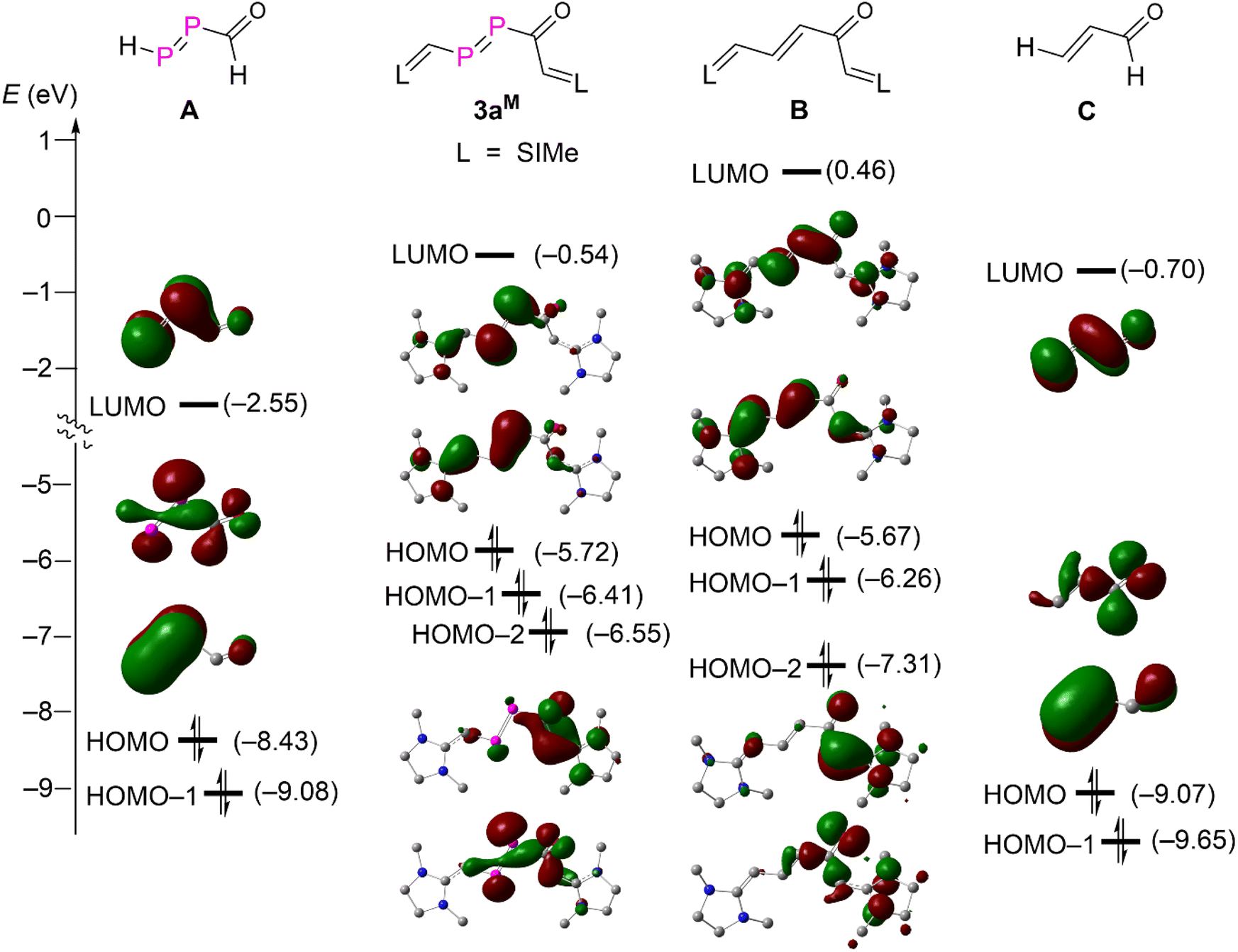 | ||
| Fig. 4 Energy diagram for the frontier Kohn–Sham orbitals (isovalue = 0.04) of compound 3aM and A–C. | ||
Stepwise nucleophilic and electrophilic 1,4-addition (P,O) of 3a
As the phosphorus analogues of enones, DPEs are expected to exhibit also 1,4-addition reactivity over the PP–CO moiety. To probe this, we first treated 3a with relatively weak nucleophiles such as an equivalent of either potassium bis(trimethylsilyl)amide [KN(TMS)2] or potassium tert-butoxide [KOtBu] in tetrahydrofuran as solvent at room temperature. This resulted in nearly quantitative conversion to 6a or 6b, which were obtained as yellow powders in over 90% yield (Fig. 5A). The proton-decoupled 31P NMR spectra of 6a or 6b displayed two doublets at δ(31P) = 61.6 and 29.7 ppm (1JPP = 276.8 Hz) or δ(31P) = 104.9 and 82.1 ppm (1JPP = 301.8 Hz), suggesting the conversion of the PP π-bond into single bonds during the addition process. Reactions of 3a with smaller or stronger nucleophiles, such as sodium methoxide, phenylmagnesium bromide or different hydride sources, led to rapid conversion of the 3a but mixtures of unidentified products were obtained.
 | ||
| Fig. 5 (A) Synthesis of 6–8. (B) Plots of the molecular structure of 6a, 7b, and 8a (ellipsoids are set to 50% probability; H atoms and solvents are omitted for clarity). Selected distances (Å): 6a: P1–K1 3.1275(6), O1–K1 2.5557(11), P1–N1 1.7716(13), P1–P2 2.1923(6), P2–C1 1.8058(17), C1–O1 1.2705(19), C1–C2 1.469(2), C2–C3 1.360(2), P1–C4 1.7957(17), C4–C5 1.352(2); 7b: P1–O2 1.6834(13), O1–Si1 1.6593(13), P1–P2 2.2243(7), P2–C1 1.7203(19), C1–O1 1.390(2), C1–C2 1.430(3), C2–C3 1.370(3), P1–C4 1.7903(19), C4–C5 1.353(3); 8a: P1–N1 1.739(3), P2–C6 1.862(3), P1–P2 2.2367(10), P2–C1 1.891(3), C1–O1 1.229(4), C1–C2 1.424(4), C2–C3 1.384(4), P1–C4 1.797(3), C4–C5 1.363(4). | ||
Single crystals of 6a suitable for XRD analysis were obtained from a fluorobenzene solution layered with hexane and stored at −30 °C. The XRD study of 6a unambiguously confirms the formation of a new P1–N1 single bond [1.7716(13) Å; Σrcov(P–N) = 1.82 Å, Σrcov(PN) = 1.62 Å],16 and the presence of a potassium cation coordinating to P1 [P1–K1 3.1275(6) Å] and O1 [O1–K1 2.5557(11) Å] (Fig. 5B). The nucleophilic attack to the β-P atom of 3a aligns with the inspection of the individual atom contributions to the frontier MOs in 3aM (Fig. 4). The negative charge is delocalized over the P–C–O fragment as indicated by the lengths of the P2–C1 bond [1.8058(17) Å] and C1–O1 bond [1.2705(19) Å], which are in between the PC and C–O bonds in 7b [1.7203(19) Å; 1.390(2) Å] and P–C and CO bonds in 8a [1.891(3) Å; 1.229(4) Å]. The P1–P2 [2.1923(6) Å] bond in 6a is significantly longer than that in 3a [2.0609(7) Å]. The P1–C4 [1.7957(17)] and C1–C2 [1.469(2)] bonds are elongated, while the C2C3 [1.360(2)] and C4C5 [1.352(2)] bonds are shortened as compared to those in 3a. These data indicate a less delocalized π-electron system in 6a.
The oxygen atom of the carbonyl group of a α-PE shows a higher affinity for a silyl group than the phosphorus atom.5e In order to investigate whether this accounts also for DPEs, 6a or 6b were treated with trimethylsilyl triflate (TMS-OTf), and the products 7a or 7b were isolated as yellow powders in 36% or orange powders in 52% yield (spectroscopic yield >90%, see Fig. S47 and S68†), respectively (Fig. 5A). Single crystals of 7b suitable for XRD analysis were obtained from a saturated acetonitrile solution stored at −30 °C. XRD studies revealed that the TMS group is bound to the O1 atom [O1–Si1 1.6593(13) Å]. The C1–O1 [1.390(2) Å] bond in 7b corresponds to a typical single bond length. The P2C1 [1.7203(19) Å] bond is shorter than the one observed in 3a [1.874(19) Å] and 6a [1.8058(17) Å], and is best regarded as a double bond. Thus, 7a,b are the products of the stepwise nucleophilic and electrophilic 1,4-addition reactions.
Alternatively, 6a or 6b can be reacted with methyl triflate (Me-OTf) to afford yellow powders 8a or 8b in 49% or 57% yield (spectroscopic yield >70%, see Fig. S53 and S75†). Multinuclear NMR spectra analyses and XRD studies confirmed that the methyl group is bonded to the P2 rather than the O1, resulting in 8a,b as the 1,2-addition product (Fig. 4B) Note that the reaction of 6a with triflic acid (H-OTf) yielded a mixture of unidentified products. But we could observe the formation of a protonated product with a P2–H group in solution after reaction with triethylamine hydrochloride (Et3NHCl), which decomposed upon attempted isolation (Fig. S60 and S61†). Thus, 3a exhibits also the reactivity of typical diphosphenes,3h,15b and the nucleophilic 1,2-addition reaction across the carbonyl group was not observed.
Stepwise electrophilic and nucleophilic 1,4-addition (P,O) of 3a
To probe the electrophilic 1,4-addition reactivity of 3a, we reacted 3a with Me-OTf and TMS-OTf yielding product 9a,b as purple powders in over 90% yield (Fig. 6A), respectively. The proton-decoupled 31P NMR spectra of 9a displayed two doublets at δ(31P) = 471.3 and 229.7 ppm (1JPP = 500.6 Hz). The 1H and 13C NMR data as well as the HSQC spectrum (Fig. S84†) of 9a confirmed the presence of one methoxyl group [δ(1H) = 2.82 ppm (s) and δ(13C) = 59.6 ppm (d, 3JPC = 24.4 Hz)], and two CH moieties from two NHV groups [δ(1H) = 4.85 ppm (s), δ(13C) = 87.9 ppm (d, 2JPC = 15.8 Hz); and δ(1H) = 6.11 ppm (t, 2JPH ≈ 3JPH = 14.1 Hz), δ(13C) = 103.9 ppm (t, 1JPH ≈ 2JPH = 54.3 Hz)]. The HMBC experiment of 9a displayed correlations from the protons of the methoxyl group to the carbon of the PPCO fragment, but not to the carbon nuclei of both CH moieties from the NHV groups (Fig. 6B). This assignment is in agreement with the calculated chemical shifts (Table S3†). The solution of 9a in acetonitrile is purple and exhibits a strong absorption at λmax = 569 nm (4.1 × 104 M−1 cm−1). According to TD-DFT calculations this absorption corresponds to the electronic transition from the HOMO to the LUMO of 9a (546 nm). Compound 9b exhibits similar spectroscopic feature to those observed for 9a. The reaction of 3a and H-OTf also led to a product [δ(31P) = 504.3 and 250.3 ppm (1JPP = 492.8 Hz)], which was too unstable to be isolated (Fig. S100†). Thus, 3a reacts with electrophiles under attack of the carbonyl oxygen center in line with the predicted NPA charges of the PP–CO fragment of 3aM, where only the oxygen atom is negatively charged (−0.64e, Fig. 3B).
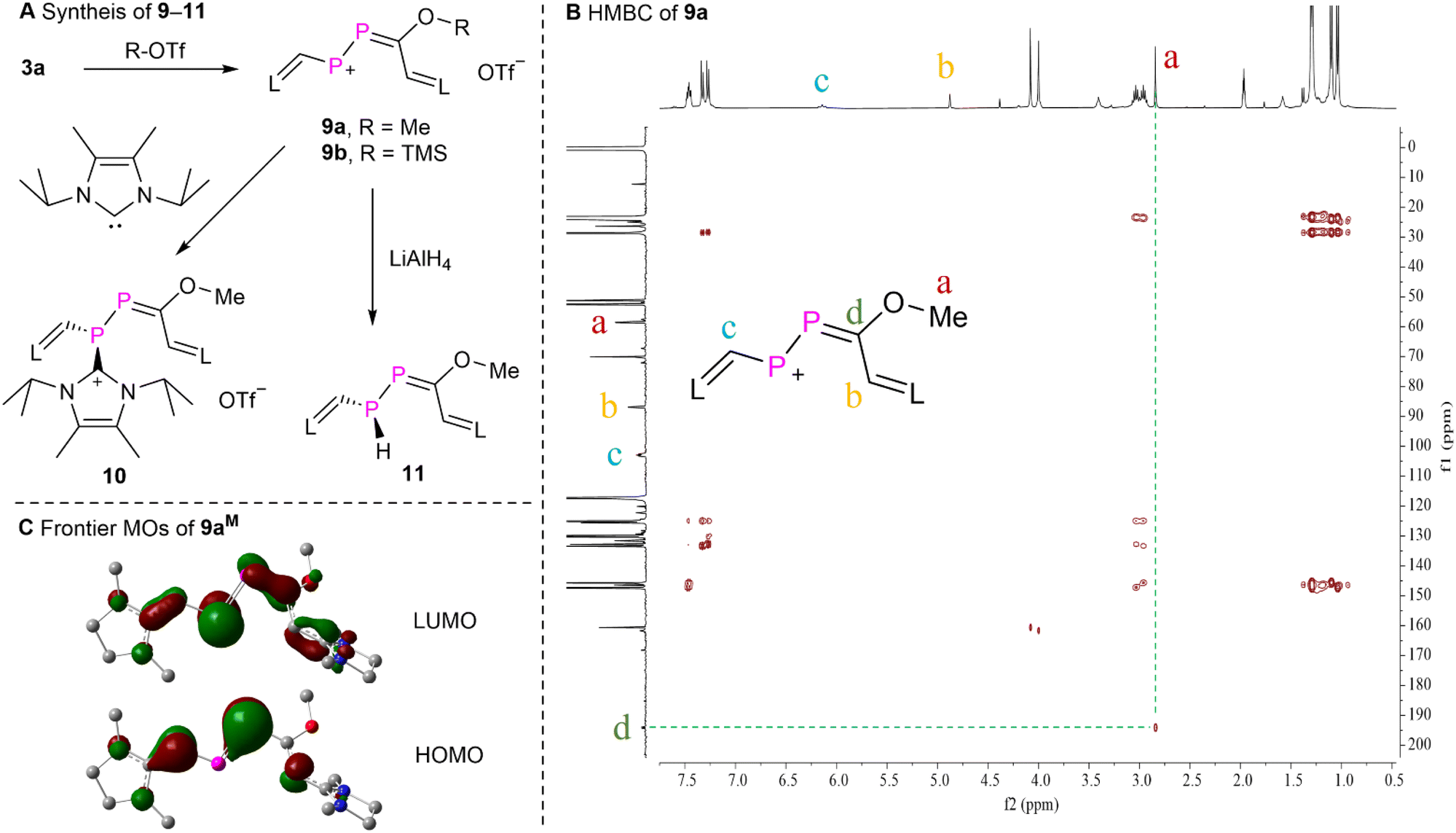 | ||
| Fig. 6 (A) Synthesis of 9–11, (B) the HMBC spectrum of 9a, and (C) the frontier Kohn–Sham orbitals (isovalue = 0.05) of compound 9aM. | ||
The MOs of 9aM were calculated at the M062X/Def2SVP level of theory and reveal that the HOMO is primarily the π-type lone-pair localized on one of the phosphorus atoms adjacent to the methoxyl group with a minor contribution from two terminal NHV substituents. The LUMO is mainly the π*-type long-pair centered at the other phosphorus atom with small contributions from π*-type orbitals of the PC bond and two NHV groups (Fig. 6C). And the NPA charges of the central P–PC moiety of 9aM are +0.43e, +0.03e, and +0.15e, respectively. These MO analyses and NBO calculations unveil that the phosphorus atom adjacent to one of the NHV groups is more susceptible to nucleophilic attack. Consequently, 9a was reacted with nucleophiles such as 4,5-dimethyl-1,3-diisopropylimidazol-2-ylidene22 and lithium aluminium hydride (LiAlH4). These reactions afforded products 10 and 11 as dark red powder or yellow oil, respectively, in over 60% yield. The relatively small JPP coupling constants of 10 [δ(31P) = 35.0 and −54.1 ppm (1JPP = 265.2 Hz)] and 11 [δ(31P) = 49.6 (d) and −105.9 ppm (dd, 1JPP = 221.2 Hz, 1JPH = 180.7 Hz)] indicates the diminished double bond character between two phosphorus atoms. According to calculated chemical shifts (Table S3†), the phosphorus atom (predicted to be −100.0 ppm) with a P–H bond is situated in β-position of the C–OMe groups of 11. Further purification of the oil 11 was not possible in our hands because of its high air sensitivity. The 13C NMR spectrum of 10 displayed a characteristic doublet at δ(13C) = 152.8 ppm (d, 1JPC = 80.8 Hz), indicating the presence of a P–C bond in which the C2 nucleus of an NHC participates.23 These results suggest that 10 and 11 are the products of the electrophilic conjugated 1,4-addition reaction (Fig. 6).
To differentiate the electrophilic reactivity of 3a from diphosphenes, the diphosphene 12,14d that has the same framework of 3a except the carbonyl group, was selected to react with Me-OTf. As result product 14 was obtained as yellow powder in 91% yield (Fig. 7). Multiple NMR spectroscopic analyses and XRD study unambiguously confirmed that 14 is a three-membered heterocyclic diphosphirane. The formation of 14 proceeds likely via an intramolecular rearrangement via the hypothetical intermediate 13, which is formed by initial electrophilic attack of a methyl cation to one of the phosphorus atoms of 12. In contrast, the cyclic alkyl amino carbene (CAAC) substituted diphosphene showed different reactivity with Me-OTf.24
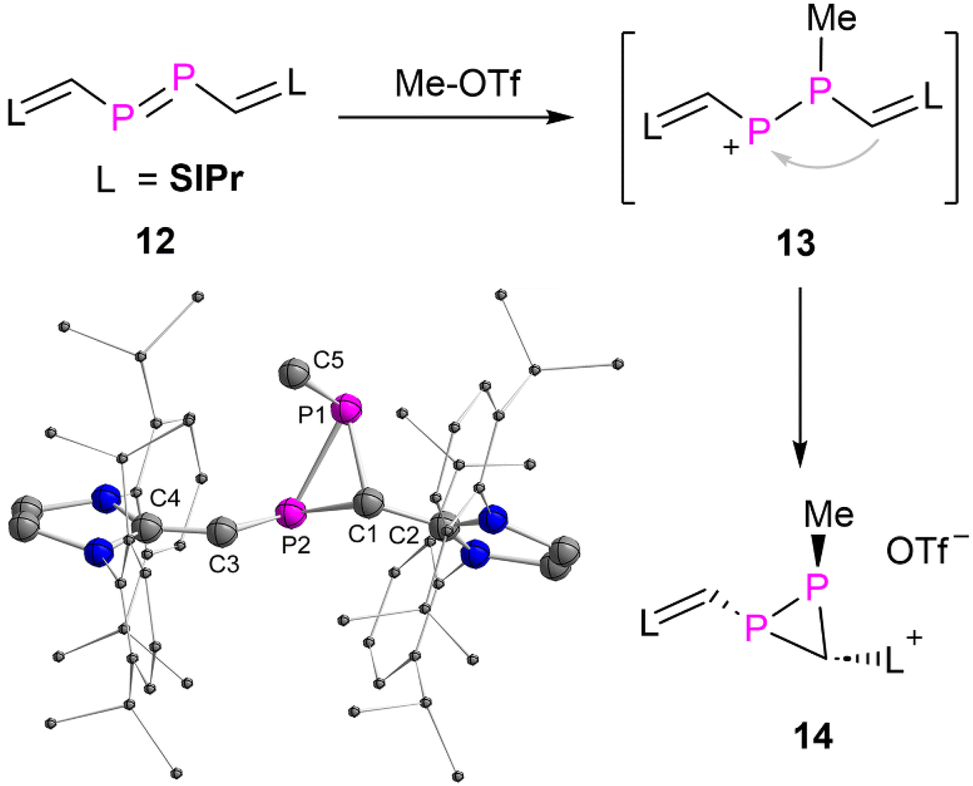 | ||
| Fig. 7 Synthesis of 14 and the plot of its molecular structure (ellipsoids are set to 50% probability; H atoms, anion and solvents are omitted for clarity). Selected distances (Å): P1–P2 2.2463(7), P1–C1 1.900(2), P1–C5 1.845(2), P2–C1 1.8625(19), P2–C3 1.777(2), C3–C4 1.373(3), C1–C2 1.455(2). | ||
Conclusions
Over nearly three decades after the first syntheses and isolation of α-PE5 and β-Pes,6,7 this work demonstrates that the acyclic DPEs 3a,b [NHV–PP–C(O)–NHV] can be obtained and isolated. These novel DPEs show structural features analogous to those observed in enones. However, the replacement of both CR moieties by P atoms in enones also alter profoundly their electronic structures. Experimentally, 3a exhibits not only 1,4-addition, but also nucleophilic 1,2-addition across the PP bond instead of the CO double bond. We show here that our previously established synthetic method for the preparation of transient DPEs can be generalized for the synthesis of stable DPEs via an intramolecular rearrangement reaction from transient phosphanyl phosphaketenes.10 Given the variety of synthetically accessible N-heterocyclic olefines14 and carbenes,25 the synthetic method applied here may intrigue the preparation of previously inaccessible DPEs and other related derivatives.
Data availability
All the characterization data are available from the electronic ESI.†Author contributions
J. Lin carried out most of the experimental work. S. Liu and S. Zheng assisted with the NMR spectra and X-ray single crystallographic diffraction measurements. Z. Li carried out the computational studies. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22271315, 22171291, 21821003, 21890380), Guangdong Basic and Applied Basic Research Foundation (2021A1515012028, 2023A1515010092), Science and Technology Planning Project of Guangzhou (202102080189), National Key Research and Development Program of China (2021YFA1500401). We thank Dr Long Jiang from the Instrumental Analysis & Research Center of Sun Yat-Sen University for assistance with the SC-XRD analyses.Notes and references
- (a) J. Li, Z. Lu and L. L. Liu, J. Am. Chem. Soc., 2022, 144, 23691–23697 CrossRef CAS; (b) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578–1604 CrossRef CAS PubMed; (c) S. Wang, J. D. Sears, C. E. Moore, A. L. Rheingold, M. L. Neidig and J. S. Figueroa, Science, 2022, 375, 1393–1397 CrossRef CAS PubMed; (d) G. Rayner-Canham, Found. Chem., 2011, 13, 121–129 CrossRef CAS; (e) E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2024, 146, 6326–6335 CrossRef CAS; (f) E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2023, 145, 7107–7112 CrossRef CAS PubMed; (g) K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy: From Organophosphorus to Phospha-organic Chemistry, Wiley, 1998 Search PubMed.
- (a) G. Märkl, Angew. Chem., Int. Ed. Engl., 1966, 5, 846–847 CrossRef; (b) G. Recker, Z. Anorg. Allg. Chem., 1976, 423, 242–254 CrossRef; (c) G. Becker, G. Gresser and W. Uhl, Z. Naturforsch., B, 1981, 36, 16–19 CrossRef; (d) M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS; (e) K. B. Dillon, V. C. Gibson and L. J. Sequeira, J. Chem. Soc., Chem. Commun., 1995, 2429–2430 RSC.
- (a) S. P, N. Gautam, S. Maji, K. Bhattacharyya and S. K. Mandal, J. Am. Chem. Soc., 2024, 146, 16743–16752 CrossRef CAS; (b) Q. Luo, T. Liu, L. Huang, C. Yang and W. Lu, Angew. Chem., Int. Ed., 2024, e202405122 CAS; (c) L. L. Liu, J. Zhou, R. Andrews and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 7466–7470 CrossRef CAS PubMed; (d) T. G. Saint-Denis, T. A. Wheeler, Q. Chen, G. Balázs, N. S. Settineri, M. Scheer and T. D. Tilley, J. Am. Chem. Soc., 2024, 146, 4369–4374 CrossRef CAS; (e) Y. Chen, P. Su, D. Wang, Z. Ke and G. Tan, Nat. Commun., 2024, 15, 4579 CrossRef CAS; (f) N. T. Coles, A. Sofie Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 433, 213729 CrossRef CAS; (g) T. Görlich, P. Coburger, E. S. Yang, J. M. Goicoechea, H. Grützmacher and C. Müller, Angew. Chem., Int. Ed., 2023, 62, e202217749 CrossRef; (h) L. Weber, F. Ebeler and R. S. Ghadwal, Coord. Chem. Rev., 2022, 461, 214499 CrossRef CAS; (i) A. Chirila, R. Wolf, J. Chris Slootweg and K. Lammertsma, Coord. Chem. Rev., 2014, 270–271, 57–74 CrossRef CAS; (j) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- (a) L. V. R. Reddy, V. Kumar, R. Sagar and A. K. Shaw, Chem. Rev., 2013, 113, 3605–3631 CrossRef CAS PubMed; (b) C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338–14349 CrossRef CAS; (c) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2018, 118, 2080–2248 CrossRef CAS; (d) S. Zhang, H. Neumann and M. Beller, Chem. Soc. Rev., 2020, 49, 3187–3210 RSC.
- (a) E. P. O. Fuchs, H. Heydt, M. Regitz, W. W. Schoeller and T. Busch, Tetrahedron Lett., 1989, 30, 5111–5114 CrossRef CAS; (b) E. Fuchs, B. Breit, H. Heydt, M. Regitz, W. Schoeller, T. Busch, C. Krüger and P. Betz, Chem. Ber., 1991, 124, 2843–2855 CrossRef CAS; (c) J. Grobe, D. L. Van and G. Lange, Z. Naturforsch., B, 1993, 48, 58–67 CrossRef CAS; (d) L. Weber, S. Uthmann, H. Bögge, A. Müller, H.-G. Stammler and B. Neumann, Organometallics, 1998, 17, 3593–3598 CrossRef CAS; (e) L. Weber, S. Uthmann, H.-G. Stammler, B. Neumann, W. W. Schoeller, R. Boese and D. Bläser, Eur. J. Inorg. Chem., 1999, 1999, 2369–2381 CrossRef; (f) L. Weber, Eur. J. Inorg. Chem., 2000, 2000, 2425–2441 CrossRef; (g) L. Weber, G. Noveski, S. Uthmann, H.-G. Stammler and B. Neumann, Eur. J. Inorg. Chem., 2007, 2007, 4011–4016 CrossRef; (h) A. S. Ionkin, Y. Wang, W. J. Marshall and V. A. Petrov, J. Organomet. Chem., 2007, 692, 4809–4827 CrossRef CAS; (i) G. Becker, M. Schmidt, W. Schwarz and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 608, 33–42 CrossRef CAS; (j) G. Becker, M. Birkhahn, W. Massa and W. Uhl, Angew. Chem., Int. Ed. Engl., 1980, 19, 741–742 CrossRef.
- (a) M. V. D. Sluis, F. Bickelhaupt, N. Veldman, H. Kooijman, A. L. Spek, W. Eisfeld and M. Regitz, Chem. Ber., 1995, 128, 465–476 CrossRef; (b) M. Van Der Sluis, J. B. M. Wit and F. Bickelhaupt, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 109, 585–588 CrossRef.
- (a) M. Yoshifuji, S. Ito, K. Toyota and M. Yasunami, Bull. Chem. Soc. Jpn., 1995, 68, 1206–1212 CrossRef CAS; (b) D. Ghereg, E. André, H. Gornitzka, J. Escudié, F. Ouhsaine, N. Saffon, K. Miqueu and J.-M. Sotiropoulos, Chem.–Eur. J., 2011, 17, 12763–12772 CrossRef CAS PubMed; (c) M. Yoshifuji, Y. Ichikawa and K. Toyota, Tetrahedron Lett., 1997, 38, 1585–1588 CrossRef CAS; (d) M. Helena, A. Benvenutti, P. B. Hitchcock, J. L. Kiplinger, J. F. Nixon and T. G. Richmond, Chem. Commun., 1997, 1539–1540 Search PubMed; (e) M. Scheer and J. Krug, Z. Anorg. Allg. Chem., 1998, 624, 399–405 CrossRef CAS.
- (a) M. Ghalib, B. Niaz, P. G. Jones and J. W. Heinicke, Tetrahedron Lett., 2012, 53, 5012–5014 CrossRef CAS; (b) B. R. Aluri, M. K. Kindermann, P. G. Jones and J. Heinicke, Chem.–Eur. J., 2008, 14, 4328–4335 CrossRef CAS; (c) N. Gupta, C. B. Jain, J. Heinicke, N. Bharatiya, R. K. Bansal and P. G. Jones, Heteroat. Chem., 1998, 9, 333–339 CrossRef CAS; (d) X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef CAS PubMed; (e) X. Chen, W. Chen, T. Ren and J. D. Protasiewicz, Tetrahedron Lett., 2005, 46, 5941–5944 CrossRef CAS.
- Z. Li, X. Chen, M. Bergeler, M. Reiher, C.-Y. Su and H. Grützmacher, Dalton Trans., 2015, 44, 6431–6438 RSC.
- J. E. Borger, G. Le Corre, Y. Mei, R. Suter, E. Schrader and H. Grützmacher, Chem.–Eur. J., 2019, 25, 3957–3962 CrossRef CAS.
- R. J. Gilliard, R. Suter, E. Schrader, Z. Benkő, A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Chem. Commun., 2017, 53, 12325–12328 RSC.
- (a) G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS; (b) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS; (c) A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed; (d) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 831–840 RSC; (e) I. Krummenacher and C. C. Cummins, Polyhedron, 2012, 32, 10–13 CrossRef CAS.
- (a) M. M. D. Roy and E. Rivard, Acc. Chem. Res., 2017, 50, 2017–2025 CrossRef CAS PubMed; (b) R. S. Ghadwal, Acc. Chem. Res., 2022, 55, 457–470 CrossRef CAS; (c) C. E. I. Knappke, J. M. Neudörfl and A. J. von Wangelin, Org. Biomol. Chem., 2010, 8, 1695–1705 RSC.
- (a) I. C. Watson, A. Schumann, H. Yu, E. C. Davy, R. McDonald, M. J. Ferguson, C. Hering-Junghans and E. Rivard, Chem.–Eur. J., 2019, 25, 9678–9690 CrossRef CAS; (b) C. Hering-Junghans, P. Andreiuk, M. J. Ferguson, R. McDonald and E. Rivard, Angew. Chem., Int. Ed., 2017, 56, 6272–6275 CrossRef CAS; (c) D. Rottschäfer, M. K. Sharma, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem.–Eur. J., 2019, 25, 8127–8134 CrossRef; (d) J. Lin, S. Liu, J. Zhang, H. Grützmacher, C.-Y. Su and Z. Li, Chem. Sci., 2023, 14, 10944–10952 RSC; (e) O. Back, B. Donnadieu, M. von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858–861 RSC; (f) F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS; (g) A. J. Arduengo, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 119, 12742–12749 CrossRef CAS; (h) H. Klöcker, M. Layh, A. Hepp and W. Uhl, Dalton Trans., 2016, 45, 2031–2043 RSC.
- (a) M. Yoshifuji, Eur. J. Inorg. Chem., 2016, 2016, 607–615 CrossRef CAS; (b) L. Weber, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS.
- (a) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed; (b) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- (a) Q. Shen, J. Xu and X. Chen, Dalton Trans., 2022, 51, 10240–10248 RSC; (b) A. C. Tsipis, Organometallics, 2006, 25, 2774–2781 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018 Search PubMed.
- S. Patchkovskii and T. Ziegler, J. Phys. Chem. A, 2002, 106, 1088–1099 CrossRef CAS.
- (a) M. M. Hansmann, D. A. Ruiz, L. L. Liu, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 3720–3725 RSC; (b) Z. Li, X. Chen, Z. Benkő, L. L. Liu, D. A. Ruiz, J. L. Peltier, G. Bertrand, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 6018–6022 CrossRef CAS PubMed; (c) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CrossRef CAS; (d) J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS; (e) D. W. N. Wilson, J. Feld and J. M. Goicoechea, Angew. Chem., Int. Ed., 2020, 59, 20914–20918 CrossRef CAS.
- (a) M. J. Frisch and et al., Gaussian 16, Revision C.01, 2016 Search PubMed; (b) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- N. Kuhn and T. Kratz, Synthesis, 1993, 1993, 561–562 CrossRef.
- (a) Z. Li, X. Chen, Y. Li, C.-Y. Su and H. Grützmacher, Chem. Commun., 2016, 52, 11343–11346 RSC; (b) D. Dhara, P. Kalita, S. Mondal, R. S. Narayanan, K. R. Mote, V. Huch, M. Zimmer, C. B. Yildiz, D. Scheschkewitz, V. Chandrasekhar and A. Jana, Chem. Sci., 2018, 9, 4235–4243 RSC; (c) M. Balmer, H. Gottschling and C. von Hänisch, Chem. Commun., 2018, 54, 2659–2661 RSC; (d) H. Chen, Y. Chen, T. Li, D. Wang, L. Xu and G. Tan, Inorg. Chem., 2023, 62, 20906–20912 CrossRef CAS; (e) M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS; (f) J. E. Walley, L. S. Warring, E. Kertész, G. Wang, D. A. Dickie, Z. Benkő and R. J. Gilliard Jr, Inorg. Chem., 2021, 60, 4733–4743 CrossRef CAS; (g) A. Doddi, D. Bockfeld, T. Bannenberg, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2014, 53, 13568–13572 CrossRef CAS PubMed; (h) A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC; (i) M. K. Sharma, S. Chabbra, C. Wölper, H. M. Weinert, E. J. Reijerse, A. Schnegg and S. Schulz, Chem. Sci., 2022, 12643–12650 RSC.
- L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273–277 CrossRef CAS PubMed.
- (a) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (b) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS; (d) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem, 2021, 5, 711–725 CrossRef CAS; (e) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS; (f) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (g) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS; (h) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, NMR spectra, crystallographic, and computational details. CCDC 2343829, 2343830, 2343833–2343835, 2346470, and 2385608. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06462c |
| This journal is © The Royal Society of Chemistry 2024 |