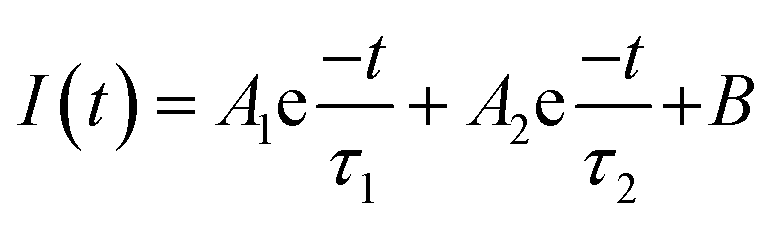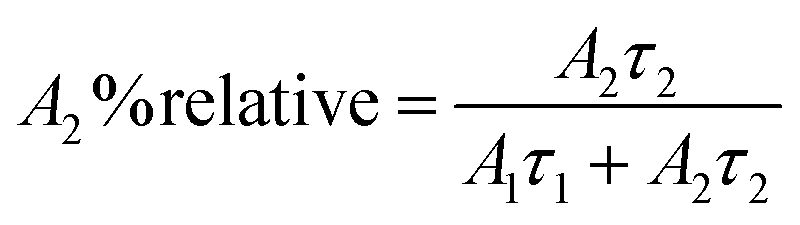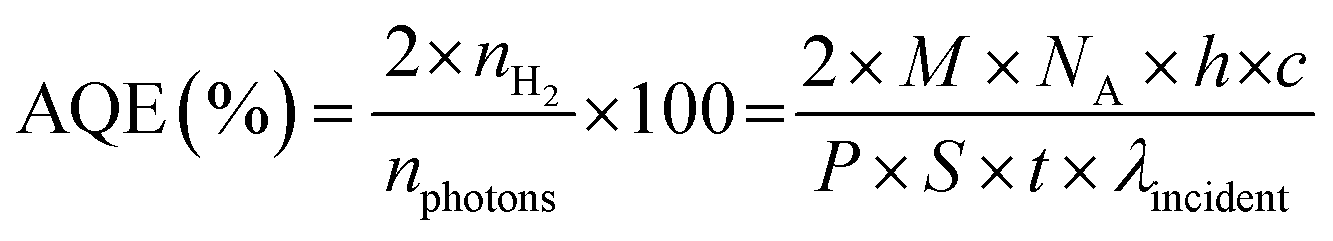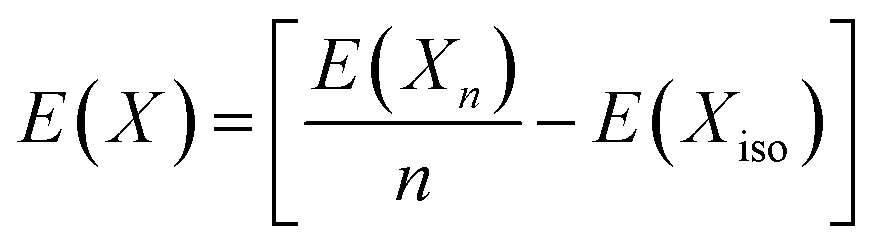Boosted photoredox capability of visible light-active P-doped C3N4 with efficient harvesting of electron–hole pairs†
Received
21st June 2024
, Accepted 17th August 2024
First published on 18th September 2024
Abstract
Photocatalytic production of solar fuels and high-value chemicals by photogenerated carriers has been at the forefront as one of the promising sustainable approaches. However, most of the studies focus only on one of the half reactions, either photoreduction or photooxidation, leading to underutilization of the potentiality of photocatalysis due to inefficient harvesting of electron–hole pairs. Herein, the efficient utilization of photogenerated electron–hole pairs was demonstrated by employing phosphorous-doped graphitic carbon nitride (P-doped g-C3N4) as a visible light-active photocatalyst that is capable of simultaneously producing hydrogen and benzaldehyde from benzyl alcohol. P-doping into g-C3N4 was achieved using an eco-friendly P source. P doping induced changes in the light-harvesting capacity of g-C3N4, and its consequence on the dual-functional photocatalytic activity of P-doped g-C3N4 was systematically investigated using various characterization techniques. P-doped g-C3N4 exhibited an ≈3-fold increase in photocatalytic activity in the production of H2 and benzaldehyde as compared to that of pristine g-C3N4. Density functional theory (DFT) studies reveal that the P-dopant preferentially replaces the corner C-site as compared to the N site of the tri-s-triazine ring of g-C3N4, which results in the creation of mid-gap states that enable the enhanced visible light absorption of P-doped g-C3N4. Mechanistic investigation studies suggest that photogenerated holes drive the selective oxidation of benzyl alcohol to benzaldehyde while photogenerated electrons drive H2 evolution, leading to concomitant production of H2 and benzaldehyde by P-doped g-C3N4. The selective conversion of benzyl alcohol proceeds through a carbon-centred radical mechanism, according to experimental and DFT studies. This work elucidates the importance of P-doping in g-C3N4 for the simultaneous production of solar fuels (such as H2) and high-value chemicals (such as benzaldehyde).
Introduction
The global demand for energy is rising significantly, leading to the development of sustainable alternate energy sources due to the depletion of non-renewable fossil fuels.1 The efficient conversion of abundant renewable solar energy into clean and storable chemical fuels has long been recognised as a viable and effective solution to the world's rising energy needs. In recent years, photocatalytic hydrogen (H2) production via water splitting using visible light-active semiconductors has been considered a promising strategy for addressing the energy crisis.2,3 In semiconductor-based photocatalytic H2 evolution from pure water, photoexcited electrons reduce protons and generate H2, whereas photogenerated holes oxidize water to evolve oxygen (O2). The efficiency of photocatalytic H2 evolution from pure water is severely constrained by the recombination of photoexcited electron–hole pairs, sluggish water-oxidation kinetics, and high-water oxidation overpotential.4 To increase the efficiency of H2 generation via photocatalytic means, studies are often conducted in the presence of sacrificial electron donors (SEDs) such as lactic acid, triethanolamine (TEOA), and methanol. However, associated costs and waste production pose additional challenges, which may impede the practical implementation. Additionally, the oxidation power of photogenerated holes is under-utilized as they participate in unwanted oxidation reactions. Therefore, it would be beneficial if one uses accessible feedstock or feedstock-derived molecules as SEDs in order to improve H2 evolution and profitability, as the oxidation of feedstock may generate high-value chemicals at the same time.5,6 Thus, benzyl alcohol (BA), a biomass-derived molecule,5 can be an interesting SED as the selective photocatalytic oxidation of BA yields benzaldehyde (BADH) in an eco-friendly manner as compared to traditional methods that involve partial oxidation of toluene or hydrolysis of benzylidene chloride.7 BADH is an important industrial solvent and a key intermediate for different perfumes, dyes, etc.7 The simultaneous production of H2 and oxidation of BA to BADH using photogenerated electrons and holes, respectively, can be a novel approach to harvest holes and electrons effectively.8 In order to realize this, the selection of a suitable bifunctional semiconductor photocatalyst is an important step.8 Among the various photocatalysts, graphitic carbon nitride (g-C3N4) has gained immense interest due to its good thermal and chemical stability, visible-light response, and tunable electronic structure.9 In 2009, Wang et al. have first described the use of g-C3N4 as a promising visible-light-responsive photocatalyst for H2 production that triggered the research on g-C3N4 for various photocatalytic applications.10 However, it should be noted that poor separation of charge carriers, and fast recombination rate provide opportunities to further explore this material.11,12 In recent years, various modification approaches have been implemented to enhance the photocatalytic activity of g-C3N4 such as thermal exfoliation of bulk g-C3N4 into two-dimensional (2D) nanosheets, constructing heterojunctions, heteroatom doping and band gap engineering. 9,12–21 Non-metallic heteroatom doping-assisted tunable C3N4 has attracted immense attention to attain high activity by creating effective catalytic sites and band edges. The heteroatom is known to create localised states between the valence band (VB) and conduction band (CB) via atomic orbital hybridisation, which is beneficial to achieve wide visible absorption and to produce more active sites.13 Additionally, the dopants play a crucial role in lowering the recombination rates of charge carriers and thereby increasing the effective utilization of photocarriers that participate in surface reactions.14 Among various heteroatoms such as C, S, P, and O-doped g-C3N4, P-dopants have received considerable attention in the recent past because they may create P–N linkages that act as new migration channels for the charge carriers.15 Notably, it has been reported that the delocalized orbitals (HOMO and LUMO) around P and neighbouring N atoms help in achieving facile charge separation.14,15 For instance, Shi et al. reported the synthesis of P-doped C3N4 using hexachlorocyclotriphosphazene as a P precursor and reported 2.9-fold increment in H2 evolution activity as compared to undoped C3N4.22 Qiao et al. have used 2-aminoethylphosphonic acid as a P-doping precursor to synthesize P-doped C3N4 nanosheets that exhibited 3.6 times higher photocatalytic H2 evolution activity than that of bulk carbon nitride.16 Mu et al. have reported the synthesis of P-doped C3N4 for dye degradation study using 1-butyl-3-methylimi-imidazolium hexafluorophosphate (BmimPF6) as the P-precursor.23 Jia et al. have synthesized P-doped C3N4 using phosphoric acid as the P-source, which shows a H2 activity rate of 256.4 μmol h−1, which is almost 24 times higher than that of pristine C3N4.24 Yuan et al. have reported that P-doped C3N4, synthesized using melamine and (hydroxyethylidene)diphosphonic acid (HEDP) precursors, showed a H2 evolution activity of 104.1 μmol h−1, which is 9.2 times higher than that of undoped C3N4.25 Other phosphorous precursors reported to synthesize P-doped C3N4 are ammonium hexafluorophosphate, diammoniumhydrogen phosphate, phosphoric acid, diphosphonic acid, red P etc.26 It is to be noted that the majority of the earlier studies on P-doped C3N4 are related to photocatalytic H2 evolution in the presence of SEDs such as TEOA and methanol, thus under-utilizing the efficacy of the photogenerated holes. Thus, the improved lifetime of photocarriers upon doping was not harvested effectively as the studies focus only on reductive half-reactions. Additionally, unlike most of the earlier reports, the present study focuses on the use of eco-friendly inositol hexaphosphate (IP6), commonly known as phytic acid as a P precursor. Owing to its high abundance, biocompatibility and most importantly the environmental-friendly aspects, IP6 is believed to be a suitable candidate in comparison to majority of the P-precursors reported in the literature to prepare P-doped C3N4. Due to the presence of six phosphate groups and twelve ionizable protons of various acidic strengths, IP6 is capable of interacting with C3N4-precursors such as dicyandiamide (DCDA) quite effectively. There exist few reports on the use of phytic acid as the P-precursor for the synthesis of P-doped C3N4; however, the photocatalytic studies are limited in exploring reductive half reactions, i.e., photocatalytic H2 evolution activity, in addition to the use of multiple time-consuming steps to yield P-doped C3N4.13,27 The development of dual-functional semiconductors for effective utilization has recently attracted attention.8 Yu et al. have demonstrated the simultaneous production of H2 and BADH from BA oxidation using CN/BP@Ni heterostructure nanosheets with the activity of H2 and BADH as 0.928 μmol h−1 and 0.93 μmol h−1 respectively.28 Zhang et al. reported dual-functional, S or P-doped C3N4 using tri-thiocyanuric acid or melamine polyphosphate, respectively.29 Chen et al. have synthesized P-doped ZnxCd1−xS for hydroxyl methylfurfural (HMF) oxidation and H2 production simultaneously with the activity of H2 production increased to 786 μmol h−1 g−1 with 2,5-diformylfuran (DFF) as a value-added oxidation product.30
In the present study, we report the doping of P-atom in the g-C3N4 matrix using environmentally friendly inositol hexaphosphate (IP6), commonly known as phytic acid. The enhanced visible-light photocatalytic activity and efficient harvesting of hole–electron pairs of P-doped C3N4 were studied for the simultaneous production of H2 and BADH.
Results and discussion
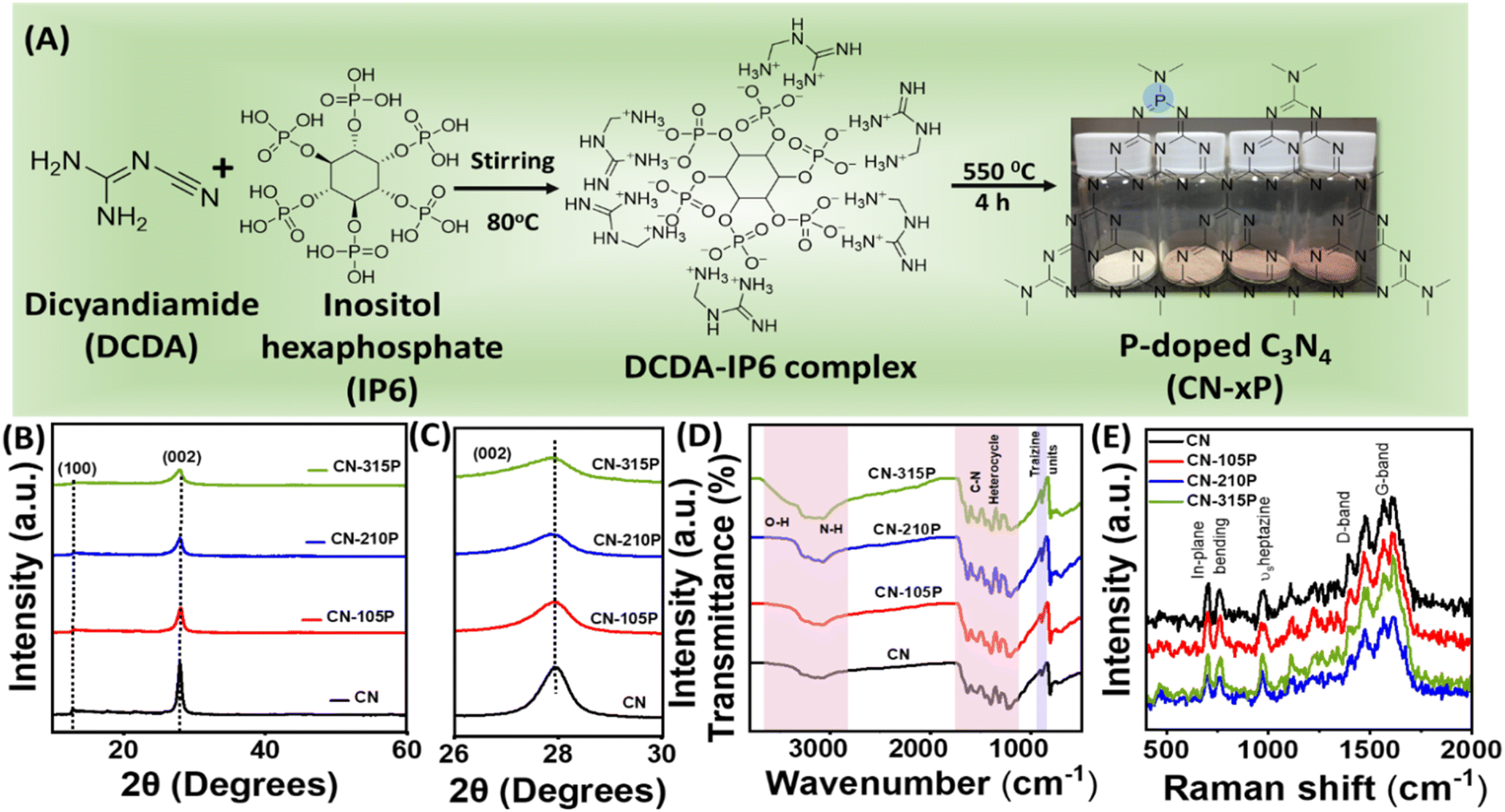
P-doped graphitic carbon nitride (CN-xP) samples with various amounts (x) of phosphorous (P) dopants were prepared via the thermal condensation of dicyandiamide (DCDA) and inositol hexaphosphate (IP6) that acted as the source of P. Various amounts of P-dopants were introduced into the C3N4 lattice by tuning the amount of IP6 in the precursor mixture. It is noted that an intermediate complex is formed when DCDA reacts with IP6, due to electrostatic interactions. The obtained powders after solvent evaporation were heated at 550 °C to obtain CN-xP samples (Fig. 1A). The as-obtained powders of various CN samples (doped and undoped) were analysed by powder X-ray diffraction (PXRD) technique to investigate the crystal structure. As shown in Fig. 1B, the PXRD pattern of CN shows two distinct diffraction peaks at 2θ values of 27.9° and 12.9°, which are indexed to the (002) and (100) planes, respectively (JCPDF no. 87-1526).15,16 The intense (002) diffraction peak observed at around 27.9° represents the inter-planar graphitic stacking and the diffraction peak observed at 12.9° corresponds to the (100) plane due to the in-plane tri-s-triazine packing motif.13 A noticeable shift in the diffraction peak at 27.9° towards the lower 2θ values is observed for CN-xP samples in comparison to undoped CN, indicating an increase in interlayer distance by lattice expansion along the c-axis direction due to the presence of bulkier P dopants (100 pm), which replace relatively smaller C atoms (70 pm) (Fig. 1C).16 In addition, the broadening of diffraction peaks with decreased intensity observed in CN-xP suggests a disordered structure due to the incorporation of bulkier P in the C3N4 matrix. The peak at 12.9° shifts to a lower 2θ value, which indicates an increase in planar nitride pores.13 The FTIR spectra of CN and CN-xP are shown in Fig. 1D. The band located at 803 cm−1 is assigned to the out-of-plane breathing vibrations of tri-s-triazine units. The observed bands ranging from 3000 cm−1 to 3500 cm−1 appear due to water molecules adsorbed on samples and uncondensed amine groups. The bands observed between 1200 cm−1 and 1600 cm−1 are assigned to the stretching vibrational modes of CN heterocycles.17 Further, the band located at around 801 cm−1 for CN shits to a higher wavenumber in the case of CN-xP samples. Moreover, the decrease in the intensity of the bands observed in the range of 3000 cm−1 to 3300 cm−1 as a function of P-amount suggests successful incorporation of P into the CN matrix, 17 which is in agreement with the XRD data. Peaks related to P-containing species are not observed, probably due to the low phosphorus contents present in the C3N4 lattice.17 The formation of carbon nitride was further confirmed by Raman spectroscopy. As shown in Fig. 1E, all characteristic Raman bands corresponding to C3N4 are intact in CN-xP, suggesting that the basic structure of C3N4 is unaltered even after P doping. The bands observed at 980 cm−1 and 690 cm−1 originate due to the symmetric N-breathing mode and in-plane bending modes of heptazine units respectively. The bands located between 1200 and 1700 cm−1 are ascribed to the disordered graphitic carbon nitrogen vibrations.18 The band located at around 1570 cm−1 is due to the G band of C3N4, which is related to the degree of graphitization while the D band located at around 1398 cm−1 is related to the structural defects and partially distorted structures of C-sp2. The ratio of the intensity of D-band (ID) to that of G-band (IG) is known to provide information about the structural distortion of CN. It is observed that the value of ID/IG is high for P-doped samples as compared to the undoped sample, suggesting a relatively more structural disorder in case of P-doped samples (Table S1†).31 The morphology of CN and CN-210P samples was characterized by field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) techniques. The FESEM images of CN and CN-210P indicate the presence of layered morphology (Fig. 2A and B). As evident from the images, CN-210P consists of loosely bound thin layers as compared to that of CN, where it consists of bulk aggregated particles. The FESEM images of CN-105P and CN0315P are given in Fig. S1.† The TEM images further confirm layered morphology composed of thin nanosheets (Fig. 2C and D). The high-resolution TEM (HRTEM) of CN-210P shows well-resolved lattice fringes that correspond to the (002) plane of C3N4 as the spacing between the fringes is found to be ≈0.32 nm (Fig. 2E). Further, the ring pattern observed in the SAED pattern suggests the poly crystallinity of CN-210P and the diffraction ring can be indexed to the (002) reflection (Fig. 2F), which is consistent with HRTEM and XRD data. The presence and homogeneous distribution of P-dopants across CN-210P nanosheets are clearly evident from the elemental mapping obtained using STEM-EDS (Fig. 2G–J).
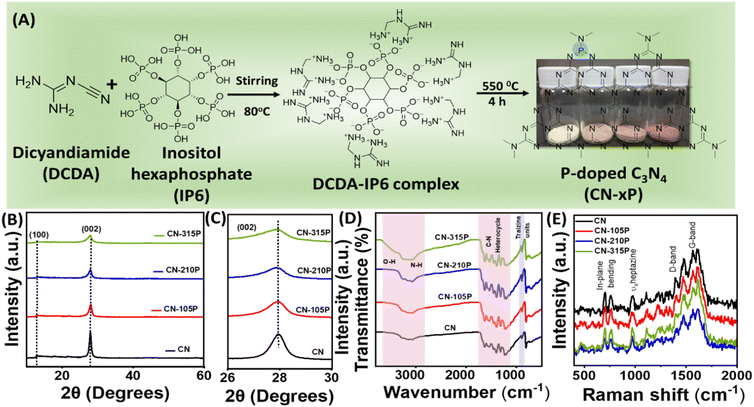 |
| | Fig. 1 (A) Schematic representation of the preparation of P-doped carbon nitride, (B and C) XRD patterns, (D) FTIR and (E) Raman spectra of pristine and P-doped carbon nitride. Digital pictures of CN-xP powders are also shown in (A). | |
 |
| | Fig. 2 FESEM images of (A) CN and (B) CN-210P. (C and D) TEM images of CN-210P at different magnifications. (E) HRTEM, (F) SAED, (G) STEM-HAADF images of CN-210P. (H–J) STEM elemental mapping corresponding to C, N and P, scale bar 500 nm. | |
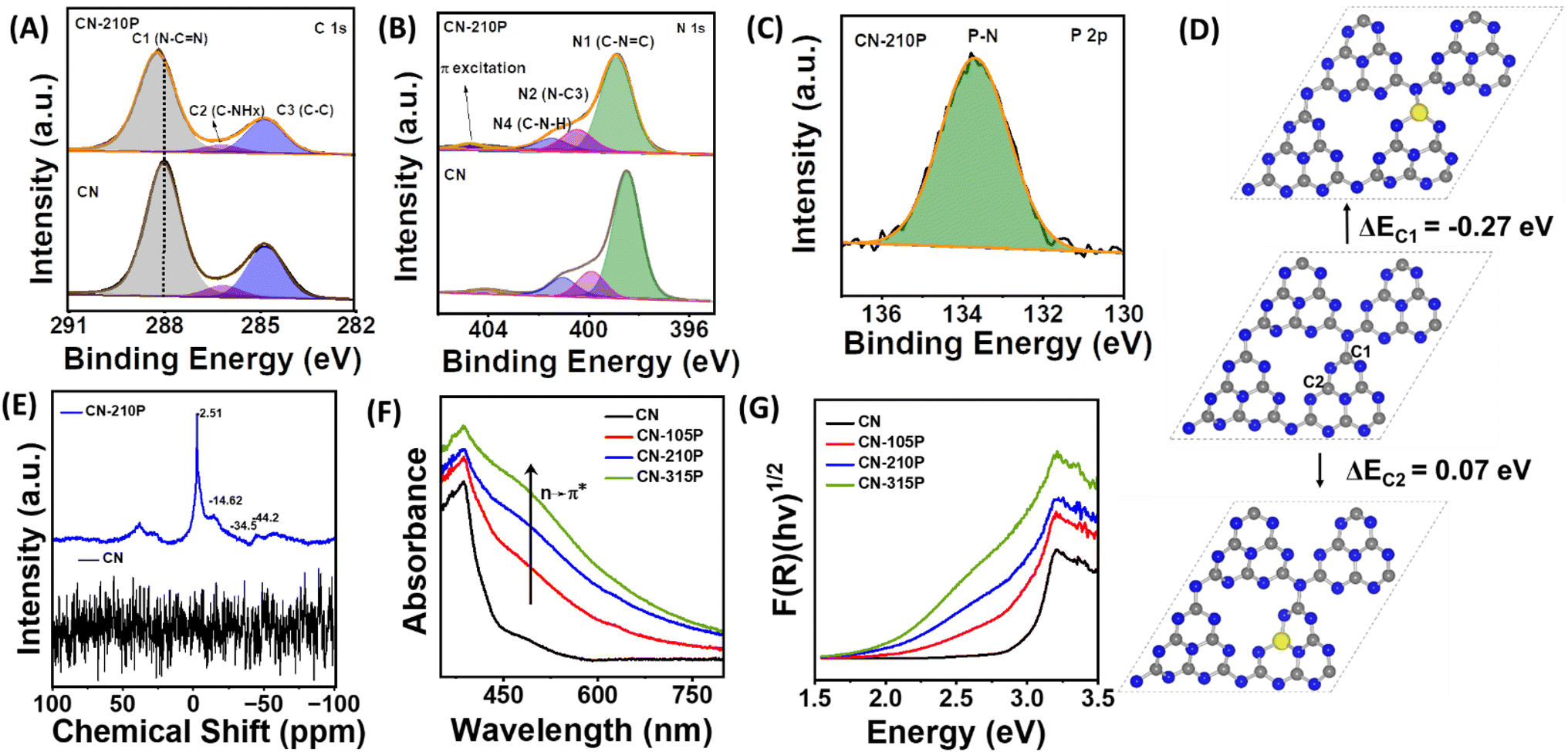
The survey spectra of the samples reveal the presence of corresponding elements (Fig. S2†). The C 1s core level XPS data of CN-210P can be deconvoluted into three peaks (Fig. 3A) namely, C1 observed at a binding energy (BE) of 288.0 eV, C2 at 286.2 eV and C3 at 284.8 eV, which are attributed to the carbon atom of the heptazine (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–(N)2 group, C–NHx and graphitic carbon (C–C) respectively.27 The N 1s XPS spectra of the CN and CN-210P samples (Fig. 3B) can be fitted into four distinct peaks, which are attributed to C–NC (N1, 398.6 eV), N–C3 (N2, 400.5 eV), C–NH2 (N3 401.5 eV amino functional group) and for charging effects or π-excitations in heterocycles (N4, 404.7 eV).27 It is noteworthy that the peak positions observed for CN-210P are slightly shifted to higher BE values than those of CN, indicating that P-doping modifies the electronic structure C3N4.27 The P 2p core level spectra of the CN-210P sample show a characteristic peak at 133.6 eV, which corresponds to the P–N linkage 13,27 (Fig. 3C). Additionally, the P 2p XPS data of CN-210P do not exhibit any peak corresponding to the P–C bonding, which usually appears at 1-2 eV lower than P–N bonding, suggesting that the P-dopant most probably replaces the C atom in the C–N framework of g-C3N4, thus forming P–N bonds.13 The O 1s spectrum of CN-210P shows two peaks located at BE values of 532.6 eV and 533.8 eV, which corresponds to adsorbed water and O–P linkages, respectively (Fig. S3†).32 The undoped and P-doped samples were analysed using solid-state nuclear magnetic resonance (SSNMR) measurements. As shown in Fig. S4A,† the 13C NMR spectra of CN and CN-210P show two prominent peaks at 153 ppm and 162.3 ppm, originating due to the carbon atoms of N–CN (C3N) and –C–NHx groups (C2N-NHx), respectively.33 The absence of additional peaks confirm that carbon atoms are not directly linked to P-dopants. In other words, 13C NMR spectra reveal the non-existence of P–C linkages in P-doped samples, supplementing XPS results. The ratio of intensity of peaks due to C3N and C2N-NHx (IC3N/IC2N-NHx)) may provide insights into the doping of P in heptazine rings. It is noted that the value of IC3N/IC2N-NHx) is found to be 0.37 for CN, while it is 0.43 for CN-210P, suggesting that P-doping is attained by preferential replacement of the C-site of C2N-NHx. Further, the shifts in the peak positions in the case of CN-210P as compared to CN may be due to the presence of P-dopants, which may change the electron density and local chemical environment.34 The 15N NMR spectrum (Fig. S4B†) of CN-210P and CN shows four signals at 191.3, 152.3, 132.5 and 112.4 ppm corresponding to CN–C(N1), N3C (N2), C–NH–C (N3) and NH2 (N4) respectively.33 The incorporation of P-dopants into g-C3N4 can be clearly seen in the 31P NMR spectrum that show four signals between 0 and -50 ppm (Fig. 3E), attributed to four possible different positions in the framework structure. Thus, it is envisaged that P-dopants can have four possibilities to replace either bay or corner C atoms.35 Among bay and corner carbon atoms, the P-atom prefers to replace the corner C atom rather than the bay C atom, as replacing the former is reported to be relatively easy than the latter.13,35 The substitution of corner C atoms in the tri-s-triazine unit by the P atom is known to result in the elongation of in-planar tri-s-triazine motifs, complementing XRD data, as shown in Fig. 1B. In order to validate this, DFT calculations were performed. The single-layer g-C3N4 structure was optimized and its cell parameter was found to be 6.93 Å. A 2 × 2 × 1 super cell consisting of 32 N and 24 C atoms was considered to model the P doped system with a vacuum layer of 15 Å, and the corresponding optimized geometry is reported in Fig. S5.† Substitutional doping of P at different N (N1, N2 and N3) and C (C1 and C2) sites, as depicted in Fig. 3D and S5,† was considered and their optimized structures along with the formation energies are reported in Fig. 3D and S5.† In the case of P doped at the N-site of g-C3N4, the doping at the N1 site which is connecting three heptazine units is found to be the minimum energy configuration while for the doping at the C-site, the corner C (C1)-site is the minimum energy one, which is consistent with the earlier reports.13,14 From the minimum energy structures, P-doping energy at C1 and N1 sites were calculated to be −0.27 eV and 1.70 eV respectively, clearly indicating that the P-doping at C1-site is exothermic while it is endothermic at all the three N-sites, in consistence with the experimental observations. In the optimized structure of P-dopants at the C1-site, the three P–N bond distances were measured to be 1.613 Å, 1.624 Å and 1.722 Å. The CHNS elemental analysis data of doped and undoped samples indicate that the molar ratio of C/N for all samples was found to be around 0.66 (Table S2†), similar to the values reported earlier.33 The surface area and pore size distribution of the samples were measured using the Brunauer–Emmett–Teller (BET) measurements. As evident from Fig. S6,† all the prepared materials exhibit type-IV adsorption.33 The specific surface areas of CN-xP samples are slightly lower than those of the pristine CN. It has been reported that doping usually modifies the electronic structure and the structural distortion caused by doping may result in a reduction in the surface area, as reported earlier.36 The pore size of CN-210P (46.2 nm) was found to be higher than that of CN (43.3 nm). It is anticipated that larger pores of the mesoporous structure are beneficial for facile diffusion of reactants and efficient light capture.37 Parameters such as specific surface area, average pore volume and total pore volume of all the catalysts obtained from BET data are tabulated in Table S3.† Further, the effect of doping on the absorption characteristics of C3N4 was studied by UV-vis diffuse reflectance spectroscopy (UV-vis DRS) measurements. The samples showed apparent visual color change from light yellow to brown upon increasing the P content in the P-doped samples (Fig. 1A). As displayed in Fig. 3F, extended visible light absorption is noted in the case of P-doped samples as compared to that of the undoped sample, indicating enhanced light-harvesting ability of CN-xP samples. A gradual shift in the absorption edge to higher wavelengths is noted with gradual increase in the P content present in C3N4, suggesting bandgap engineering upon P-doping. The optical bandgap (Eg) of all the samples was measured from the Tauc plot, as shown in Fig. 3G and S7.† The values of Eg for CN, CN-105P, CN-210P, and CN-315P were found to be 2.98 eV, 2.82 eV, 2.61 eV and 2.45 eV, respectively. The observed Eg values portray the narrowing of Eg due to the P-doping of C3N4. Moreover, a faint shoulder around 500 nm observed in the absorption spectra of doped samples is known to originate due to the relatively disordered C3N4 framework wherein n → π* excitation of lone pair electrons of edge N becomes allowed unlike the case of planar C3N4 where it is forbidden. This assists in the absorption of photon energy in the visible region.38 Additionally, the P-dopant results in the creation of sub-band gap states (ET), as indicated in Fig. S7 and S8.†13 The P-dopant-induced changes in the band structure of C3N4 were analysed by locating the positions of the VB and CB of P-doped and undoped CN samples using UV-vis DRS data in combination with either XPS VB spectra or Mott–Schottky (M–S) measurements. Using eqn (1),33 the position of the VB vs. normal hydrogen electrode (NHE) was estimated from the valence band maxima (VBM), which was
determined from the XPS-VB spectra (Fig. 4A).
C–(N)2 group, C–NHx and graphitic carbon (C–C) respectively.27 The N 1s XPS spectra of the CN and CN-210P samples (Fig. 3B) can be fitted into four distinct peaks, which are attributed to C–NC (N1, 398.6 eV), N–C3 (N2, 400.5 eV), C–NH2 (N3 401.5 eV amino functional group) and for charging effects or π-excitations in heterocycles (N4, 404.7 eV).27 It is noteworthy that the peak positions observed for CN-210P are slightly shifted to higher BE values than those of CN, indicating that P-doping modifies the electronic structure C3N4.27 The P 2p core level spectra of the CN-210P sample show a characteristic peak at 133.6 eV, which corresponds to the P–N linkage 13,27 (Fig. 3C). Additionally, the P 2p XPS data of CN-210P do not exhibit any peak corresponding to the P–C bonding, which usually appears at 1-2 eV lower than P–N bonding, suggesting that the P-dopant most probably replaces the C atom in the C–N framework of g-C3N4, thus forming P–N bonds.13 The O 1s spectrum of CN-210P shows two peaks located at BE values of 532.6 eV and 533.8 eV, which corresponds to adsorbed water and O–P linkages, respectively (Fig. S3†).32 The undoped and P-doped samples were analysed using solid-state nuclear magnetic resonance (SSNMR) measurements. As shown in Fig. S4A,† the 13C NMR spectra of CN and CN-210P show two prominent peaks at 153 ppm and 162.3 ppm, originating due to the carbon atoms of N–CN (C3N) and –C–NHx groups (C2N-NHx), respectively.33 The absence of additional peaks confirm that carbon atoms are not directly linked to P-dopants. In other words, 13C NMR spectra reveal the non-existence of P–C linkages in P-doped samples, supplementing XPS results. The ratio of intensity of peaks due to C3N and C2N-NHx (IC3N/IC2N-NHx)) may provide insights into the doping of P in heptazine rings. It is noted that the value of IC3N/IC2N-NHx) is found to be 0.37 for CN, while it is 0.43 for CN-210P, suggesting that P-doping is attained by preferential replacement of the C-site of C2N-NHx. Further, the shifts in the peak positions in the case of CN-210P as compared to CN may be due to the presence of P-dopants, which may change the electron density and local chemical environment.34 The 15N NMR spectrum (Fig. S4B†) of CN-210P and CN shows four signals at 191.3, 152.3, 132.5 and 112.4 ppm corresponding to CN–C(N1), N3C (N2), C–NH–C (N3) and NH2 (N4) respectively.33 The incorporation of P-dopants into g-C3N4 can be clearly seen in the 31P NMR spectrum that show four signals between 0 and -50 ppm (Fig. 3E), attributed to four possible different positions in the framework structure. Thus, it is envisaged that P-dopants can have four possibilities to replace either bay or corner C atoms.35 Among bay and corner carbon atoms, the P-atom prefers to replace the corner C atom rather than the bay C atom, as replacing the former is reported to be relatively easy than the latter.13,35 The substitution of corner C atoms in the tri-s-triazine unit by the P atom is known to result in the elongation of in-planar tri-s-triazine motifs, complementing XRD data, as shown in Fig. 1B. In order to validate this, DFT calculations were performed. The single-layer g-C3N4 structure was optimized and its cell parameter was found to be 6.93 Å. A 2 × 2 × 1 super cell consisting of 32 N and 24 C atoms was considered to model the P doped system with a vacuum layer of 15 Å, and the corresponding optimized geometry is reported in Fig. S5.† Substitutional doping of P at different N (N1, N2 and N3) and C (C1 and C2) sites, as depicted in Fig. 3D and S5,† was considered and their optimized structures along with the formation energies are reported in Fig. 3D and S5.† In the case of P doped at the N-site of g-C3N4, the doping at the N1 site which is connecting three heptazine units is found to be the minimum energy configuration while for the doping at the C-site, the corner C (C1)-site is the minimum energy one, which is consistent with the earlier reports.13,14 From the minimum energy structures, P-doping energy at C1 and N1 sites were calculated to be −0.27 eV and 1.70 eV respectively, clearly indicating that the P-doping at C1-site is exothermic while it is endothermic at all the three N-sites, in consistence with the experimental observations. In the optimized structure of P-dopants at the C1-site, the three P–N bond distances were measured to be 1.613 Å, 1.624 Å and 1.722 Å. The CHNS elemental analysis data of doped and undoped samples indicate that the molar ratio of C/N for all samples was found to be around 0.66 (Table S2†), similar to the values reported earlier.33 The surface area and pore size distribution of the samples were measured using the Brunauer–Emmett–Teller (BET) measurements. As evident from Fig. S6,† all the prepared materials exhibit type-IV adsorption.33 The specific surface areas of CN-xP samples are slightly lower than those of the pristine CN. It has been reported that doping usually modifies the electronic structure and the structural distortion caused by doping may result in a reduction in the surface area, as reported earlier.36 The pore size of CN-210P (46.2 nm) was found to be higher than that of CN (43.3 nm). It is anticipated that larger pores of the mesoporous structure are beneficial for facile diffusion of reactants and efficient light capture.37 Parameters such as specific surface area, average pore volume and total pore volume of all the catalysts obtained from BET data are tabulated in Table S3.† Further, the effect of doping on the absorption characteristics of C3N4 was studied by UV-vis diffuse reflectance spectroscopy (UV-vis DRS) measurements. The samples showed apparent visual color change from light yellow to brown upon increasing the P content in the P-doped samples (Fig. 1A). As displayed in Fig. 3F, extended visible light absorption is noted in the case of P-doped samples as compared to that of the undoped sample, indicating enhanced light-harvesting ability of CN-xP samples. A gradual shift in the absorption edge to higher wavelengths is noted with gradual increase in the P content present in C3N4, suggesting bandgap engineering upon P-doping. The optical bandgap (Eg) of all the samples was measured from the Tauc plot, as shown in Fig. 3G and S7.† The values of Eg for CN, CN-105P, CN-210P, and CN-315P were found to be 2.98 eV, 2.82 eV, 2.61 eV and 2.45 eV, respectively. The observed Eg values portray the narrowing of Eg due to the P-doping of C3N4. Moreover, a faint shoulder around 500 nm observed in the absorption spectra of doped samples is known to originate due to the relatively disordered C3N4 framework wherein n → π* excitation of lone pair electrons of edge N becomes allowed unlike the case of planar C3N4 where it is forbidden. This assists in the absorption of photon energy in the visible region.38 Additionally, the P-dopant results in the creation of sub-band gap states (ET), as indicated in Fig. S7 and S8.†13 The P-dopant-induced changes in the band structure of C3N4 were analysed by locating the positions of the VB and CB of P-doped and undoped CN samples using UV-vis DRS data in combination with either XPS VB spectra or Mott–Schottky (M–S) measurements. Using eqn (1),33 the position of the VB vs. normal hydrogen electrode (NHE) was estimated from the valence band maxima (VBM), which was
determined from the XPS-VB spectra (Fig. 4A).
| | | EVB-NHE = φ + EVB-XPS − 4.44 | (1) |
where
EVB-NHE is the VB potential
vs. NHE,
φ is the electron work function of the XPS analyser and the value is 4.21 eV in the present case, and
EVB-XPS is the VBM value obtained from the XPS-VB spectra. From the as-obtained VB values and
Eg values determined from UV-vis DRS data (
Fig. 3G), the positions of the CB of CN and CN-
xP were estimated (
ECB =
EVB −
Eg) and are schematically shown in Fig. S8.
†33 Similarly, the VB and CB positions determined using Mott–Schottky measurements (Fig. S9
†) are comparable to the values obtained from the VB-XPS spectra (see ESI for more details, Tables S4 and S5
†).
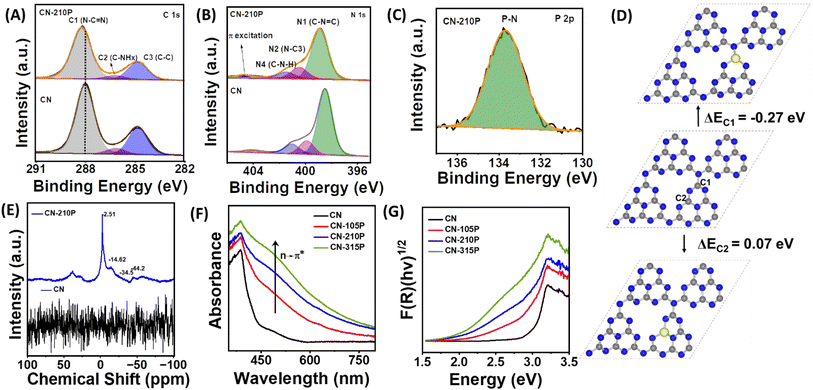 |
| | Fig. 3 XPS core-level spectra corresponding to (A) C 1s, (B) N 1s, and (C) P2p of pristine and CN-210P. (D) Optimized structures of the 2 × 2 × 1 super cell of pristine C3N4 and P-doped systems at different possible C-sites with the corresponding formation energies. (E) SS-31P NMR spectra of CN and CN-210P. (F) UV-vis DRS and (G) Tauc plot of CN and CN-xP. | |
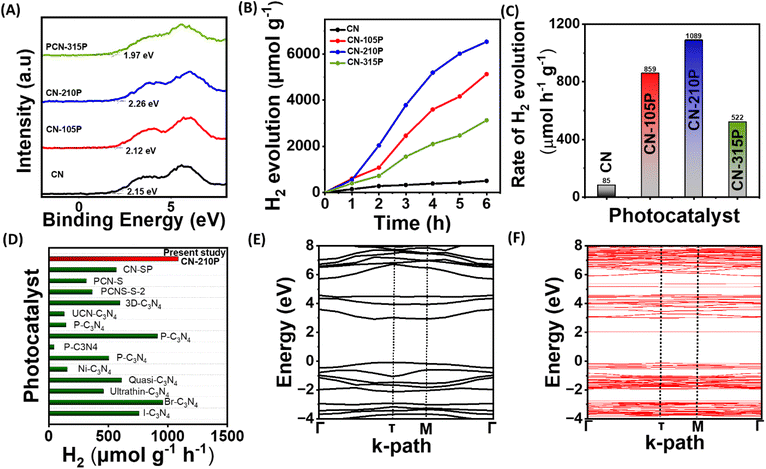 |
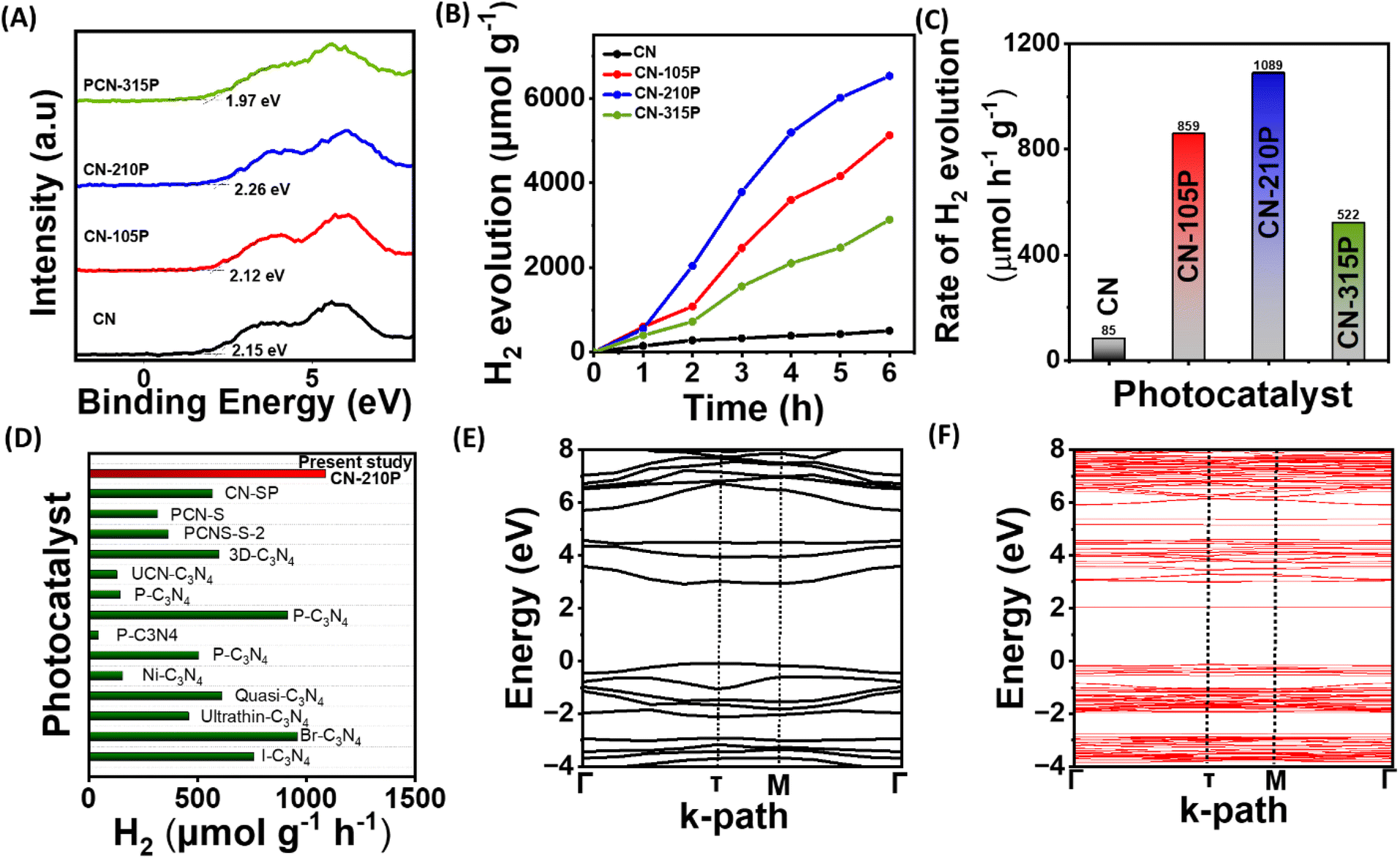
| | Fig. 4 (A) XPS VB spectra. (B) Photocatalytic hydrogen evolution as a function of time using TEOA as the SED. (C) Hydrogen evolution rate obtained using the CN and CN-xP sample. (D) Comparison plot depicting the H2 evolution activity of CN-210P with some of the reported systems. Electronic band structure of (E) g-C3N4 and (F) P-doped C3N4 from the HSE06 functional. | |
Impressed by the enhanced light absorption and suitable VB and CB positions of CN-xP, photocatalytic activity studies were performed using a custom-built white LED light source. The spectral output of the LED used in the present study is given in Fig. S10.† Prior to dual functional photocatalytic activity studies, H2 evolution studies for all the photocatalysts (doped and undoped) with photodeposited Pt co-catalysts were carried out in the presence of triethanolamine (TEOA) as the SED. It is to be noted that the reflections corresponding to Pt are not seen in PXRD data, indicating high dispersibility of small Pt particles on the CN-210P surface. The TEM data also support high dispersibility of Pt particles on CN-210P (Fig. S11†). Fig. 4B depicts the photocatalytic H2 evolution performance of undoped and P-doped CN consisting of different amounts of P. As observed from Fig. 4C, around 6 to 12.8 times increase in the H2 production rate was observed in the case of P-doped samples depending on the P content, as compared to undoped C3N4. For instance, CN-210P exhibits a H2 production rate of around 1089 μmol h−1 g−1, 12.8 times higher than that of undoped CN (85 μmol h−1 g−1), suggesting better charge separation ability and availability of more reactive sites in P-doped samples unlike pristine CN. It is noteworthy that the activity is tunable with the P-content present in the C3N4 lattice. The photocatalytic activity increases with the increase in the P-content; however, beyond a certain threshold (Fig. 4B and C), the activity decreases due to the presence of excess P dopants, which may themselves act as recombination centres and thus inhibit the charge carrier separation.27 Among all the P-doped catalysts, CN-210P with a P-dopant quantity of 0.66 at% (from XPS) appears to be the optimum quantity and shows the highest rate of H2 production among other studied catalysts. The enhanced visible light absorption and associated narrow band gap are responsible for the observed higher activity of CN-xP than that of CN. Table S6† and Fig. 4D show the comparison of the photocatalytic activity of CN-210P with other reported materials. These results indicate the appreciative effect of P that induces electronic structure modifications, which assist in increasing photocatalytic activity. The P atoms that replace C-sites in g-C3N4 form P–N bonding with the adjacent N atoms, leading to disorder in the heptazine skeleton. It has been reported that these P–N bonds in P-doped C3N4 are known to break the localized state around the highly symmetric heptazine units of undoped C3N4, leading to the creation of charge carrier-rich regions that facilitates charge transfer. The extra non-bonding electron of P dopants will be delocalized in the tri-s-triazine ring, creating P+ centres, and thus enhanced electron-rich tri-s-triazine moieties present in P-doped C3N4.39 Notably, the HOMO and LUMO localized around P atoms and neighbouring N atoms, which facilitates the movement of photogenerated carriers under visible light between neighbouring heptazine units through the N–P–N–C channel.14
Further, DFT calculations were performed to understand the electronic structure and its manifestation on P-doping in g-C3N4. The electronic band structure and density of states (DOS) of both pure and P-doped C3N4 were calculated using PBE and HSE06 functionals. The band structure plots shown in Fig. S12† indicate that the electronic band gap of C3N4 has been significantly underestimated to 1.86 eV from the PBE functional, while the corresponding band gap determined from HSE06 is found to be 3.01 eV, which is close to the experimental value (Fig. 4E). From the projected DOS (PDOS) reported in Fig. S13,† it is observed that the valence band maxima (VBM) is mostly contributed by the N 2p states while the conduction band minima (CBM) is coming from C 2p and N 2p states, which can also be seen from the band decomposed charge density iso-surface plots reported in Fig. S14.† The band structure of P-doped C3N4 shows an isolated mid-gap state around 1 eV below the CB resulting in a reduction in the band gap from 3.01 eV in the pure system to 2.13 eV (Fig. 4F), which is consistent with the observed improved photoactivity after P-doping. From the PDOS reported in Fig. S13B† and the band decomposed charge density iso-surface shown in Fig. S15C,† it can be observed that the mid-gap state arises from the P 2p, N 2p and C 2p states. From the charge density iso-surface plots, it can also be observed that both the highest single electron occupied band (Fig. S15B†) and the mid-gap band (Fig. S15C†) are majorly contributed by the P and the N atoms bound to P. It is also clear from these band decomposed charged densities that in the case of pure systems, the VBM and CBM are almost uniformly distributed over the cell while in the P-doped system, better spatial separation of charge carries can be expected.
The apparent quantum efficiency (AQE) of the optimized catalyst was determined (See ESI). The AQE was found to be 1.6%, 1.01% and 0.16% at 452, 517 and 636 nm respectively. As shown in Fig. S16,† the wavelength-dependent AQE values determined for CN-210P are consistent with the absorption spectrum of CN-210P. The AQE values obtained in the present study is comparable to many of the reported photocatalysts (Fig. S16, Table S7†).
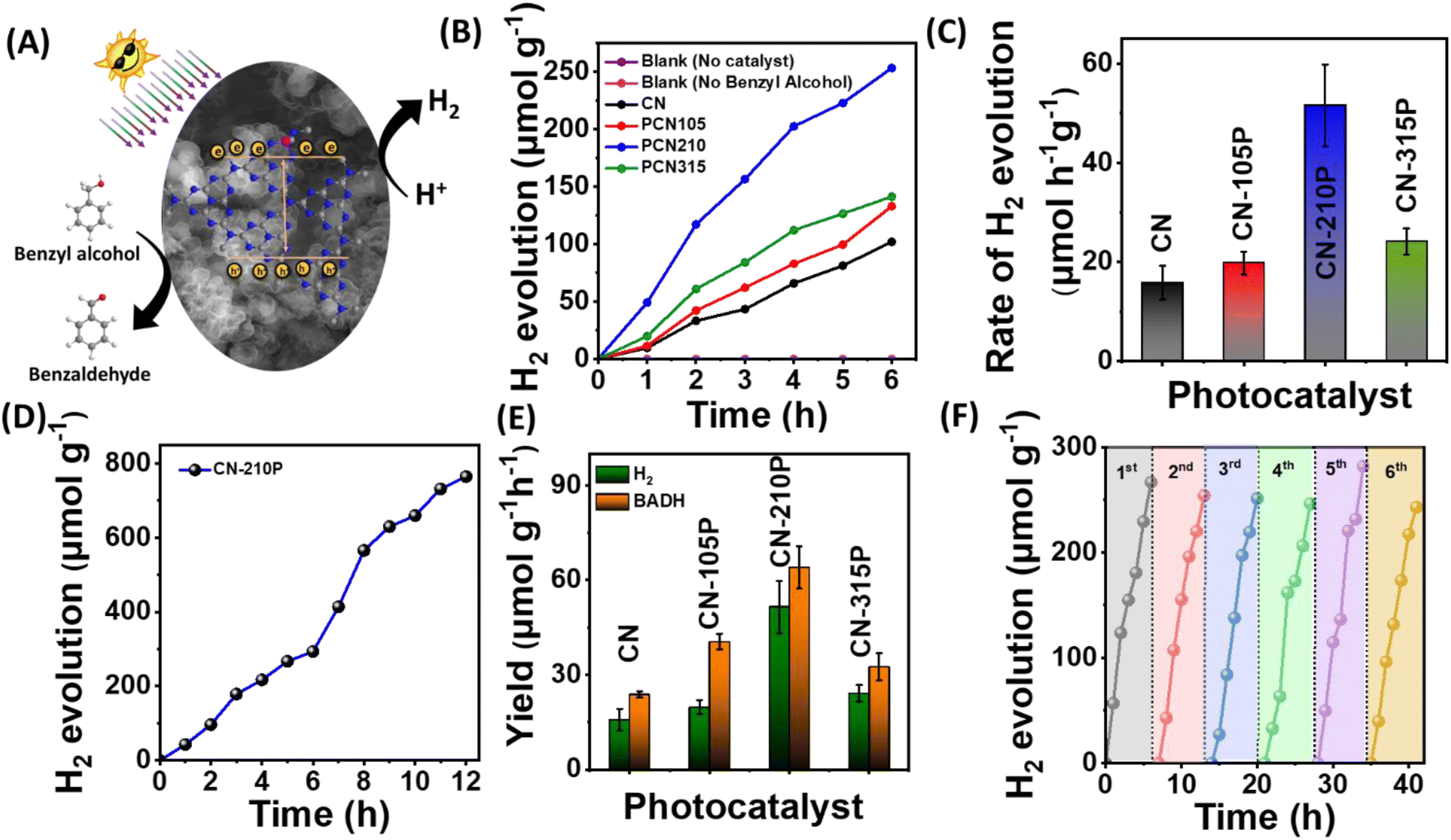
The suitability of CN-xP as a dual-functional catalyst was demonstrated by carrying out photocatalytic studies that produce H2 and benzaldehyde, simultaneously. In other words, we utilized photogenerated electrons to produce H2 and photogenerated holes to produce BADH, an approach wherein the charge carriers are harvested effectively without wasting the oxidizing ability of holes (Fig. 5A). The H2 production from photocatalytic studies conducted in the presence of BA obtained using CN-xP is shown in Fig. 5B. The P-doped C3N4 (CN-210P) shows 3 times higher rate of H2 production than that of pristine CN, confirming the determining role of P in dictating the photocatalytic activity. Among all the CN-xP samples, CN-210P exhibited better activity (Fig. 5B and C). On prolonged illumination for 12 h, CN-210P yields appreciable amounts of H2 (Fig. 5D). Further, the products formed during the photocatalytic oxidation of BA were monitored using various techniques such as 1H NMR, liquid chromatography (LC), and gas chromatography-mass spectrometry (GC-MS). Fig. S17† shows the 1H NMR spectra recorded for the reaction mixture obtained using CN and CN-xP samples collected after continuous illumination of light for 7 h. As shown in the figure, the peaks related to BADH are clearly evident, confirming the selective oxidation of BA. It should be noted that the yield of BADH using CN-210P was found to be 57.3 μmol h−1 g−1, which is 2.8 times higher than that of CN (Fig. 5E). Similarly, LC and GC-MS data also revealed the formation of BADH as the only oxidation product formed during the photocatalytic reaction, indicating good selectivity (Fig. S18 and S19†). Overall, the CN-210P sample demonstrates excellent dual-functional photocatalytic activity for the simultaneous production of H2 and BADH. It is to be noted that CN-210P is capable of producing H2 and BADH even in the presence of natural sunlight, indicating the applicability of this material in practical applications (Fig. S20†). Further, the positions of VB and CB are appropriately located with respect to proton reduction and BA oxidation to realize proton reduction by photogenerated electrons and BA oxidation by photogenerated holes.40 Additionally, the sub-band states originated due to P-doping located near the Fermi level also contributes to the enhanced activity in case of P-doped C3N4.41 To gain further insights, a range of control experiments were conducted. The experiments conducted in the absence of BA do not result in the production of H2 and BADH. Similar observation has been noted when CN-210P was used as a photocatalyst under dark conditions, co-catalyst free conditions, as well as photocatalyst-free conditions, suggesting the essential role of light and photocatalysts. Notably, experiments were conducted to probe the reactive species that participated in photocatalysis, by adding tetrachloromethane (CCl4) and TEOA to the reaction mixture, as these are known to act as scavengers for electrons (e−) and holes (h+) respectively. A remarkable decrease in H2 evolution rate was noted, when CCl4 or K2S2O8 was used in the reaction mixture, indicating that the proton reduction process was driven by photogenerated electrons (Fig. S21A†). With the addition of AO or TEOA, the yield of BADH decreased remarkably, suggesting that photogenerated holes drive oxidation of BA to BADH (Fig. S21B†). Thus, it can be concluded that CN-xP harvests electron–hole pairs effectively in concomitant production of H2 and BADH. Further, when the measurements were carried out in the presence of isopropyl alcohol (IPA), which is known to acts as a scavenger for ˙OH radicals, the yields of H2 and BADH remain unchanged, suggesting that ˙OH radicals may not be involved in the conversion of BA to BADH (Fig. S21A and B†). Notably, when experiments were carried out in a pure acetonitrile medium (water-free conditions), no H2 was observed (minor trace), indicating that water plays a key role in H2 production. In addition, increased amounts of H2 evolution are noted when the concentration of BA is increased. These measurements indicate that water and BA mutually promote the photocatalytic H2 production. Additionally, electron spin resonance (ESR) measurements were carried out to further understand the conversion of BA into BADH (Fig. S22†). As shown in Fig. S22,† the ESR spectrum does not show any signals when the reaction is carried out in the darkness, whereas evident ESR signals are noticed when the reaction is carried out in the presence of light. Based on these observations, it is envisaged that the conversion of BA to BADH proceeds via a carbon-centred radical route, as proposed in Fig. S23.† The DFT calculations also support the experimental observations (see ESI†).
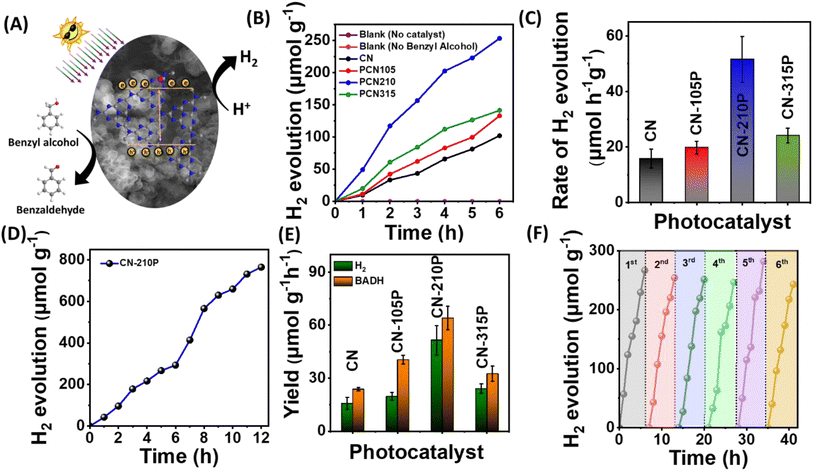 |
| | Fig. 5 (A) Schematic depicting the dual functionality of CN-xP for the simultaneous production of H2 and benzaldehyde. (B) Hydrogen evolution studies in the presence of 20 mM benzylalcohol. (C) Rate of hydrogen evolution using CN and CN-xP. (D) Hydrogen evolution observed using CN-210P as the photocatalyst for longer durations. (E) Yields of H2 and benzaldehyde obtained using CN and CN-xP. (F) Cyclic runs of H2 evolution over CN-210P. | |
The robustness of CN-210P was examined by performing photocatalytic measurements for repeated cycles and long hours of continuous illumination. As shown in Fig. 5D, CN-210P exhibited excellent stability when the measurements continued for 12 h of illumination. Notably, the activity of CN-210P towards H2 generation remains unaltered for six cycles, that is, for 36 h (Fig. 5F). In order to confirm the robustness of CN-210P, the catalyst after the photocatalytic studies was collected and characterized by FTIR spectroscopy, XRD, XPS and Raman spectroscopy measurements. As shown in Fig. S26–S28,† insignificant changes are noted even after repeated use of the photocatalyst in photocatalytic reactions, suggesting the stability of CN-210P.
To understand the observed enhanced photocatalytic activity of P-doped samples, steady-state photoluminescence (PL) and time-resolved PL (TRPL) measurements were carried out. As shown in Fig. 6A, the PL spectra of undoped CN recorded at an excitation wavelength of 325 nm exhibit a broad peak at around 490 nm, which shifts to a higher wavelength in the case of P-doped CN, suggesting the narrowing of bandgap upon P-doping.42 Additionally, the data indicate that the PL intensity is quenched in P-doped samples as compared to the pristine CN, suggesting suppressed recombination of the photo-induced charge carriers in P-doped samples. Besides, the lifetime of photogenerated charge carriers was determined from the TRPL spectra, as shown in Fig. 6B. The PL decay curves recorded at 296 nm of doped and undoped CN samples were fitted using the biexponential decay function (eqn (2)):
| | 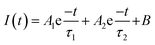 | (2) |
where
A1 and
A2 are the relative amplitudes,
I(
t) is the intensity of TRPL decay, and
τ1 and
τ1 are the short and long fluorescence lifetimes respectively.
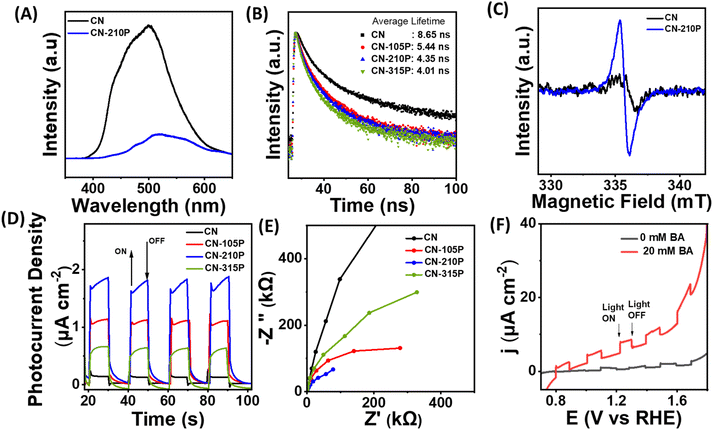 |
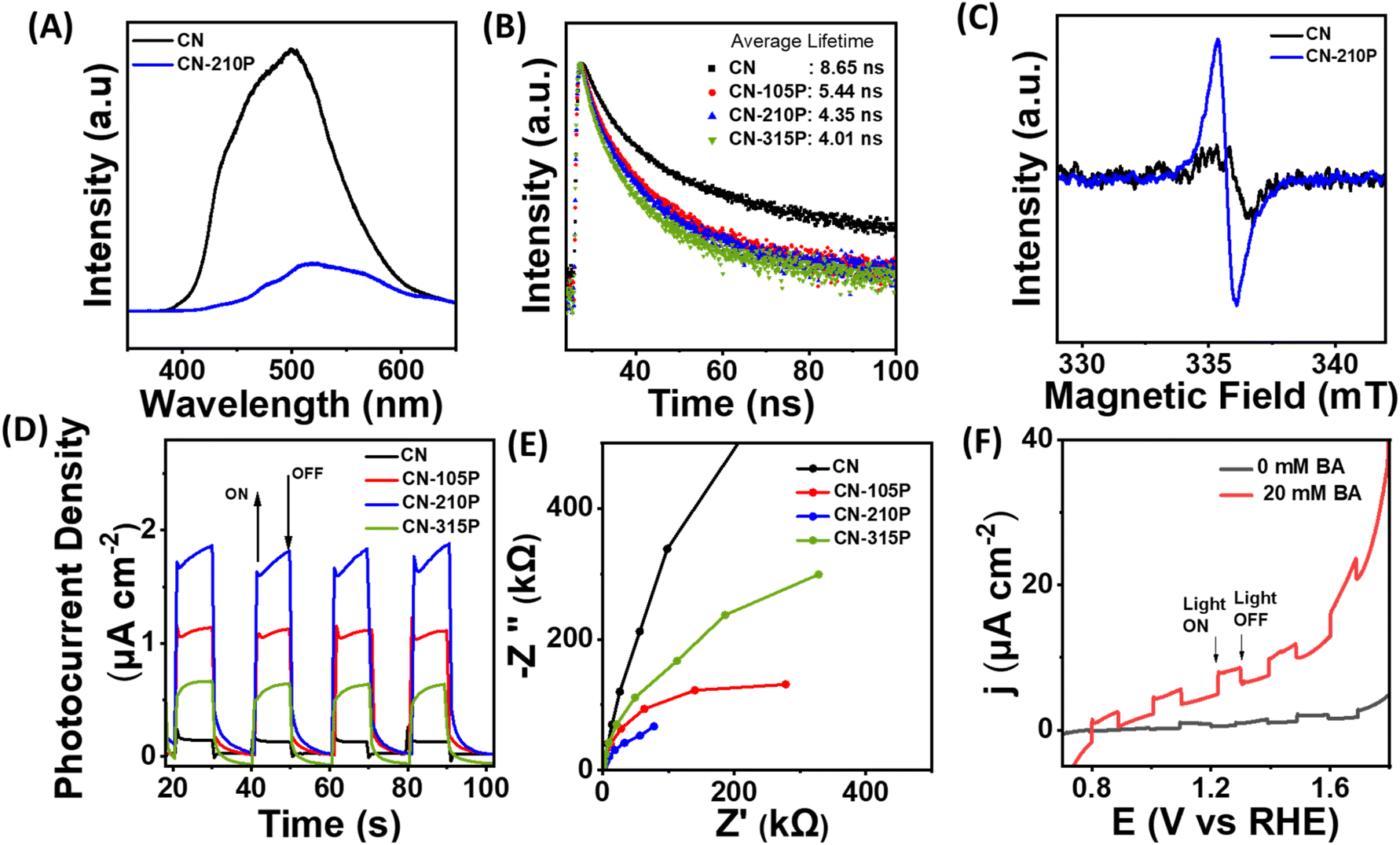
| | Fig. 6 (A) Steady-state photoluminescence of CN and CN-210P. (B) Time-resolved photoluminescence of CN-xP. (C) ESR spectra of CN and CN-210P solid powders, recorded at room temperature. (D) Photoelectrochemical measurements depicting photocurrent recorded at 1.23 V vs. RHE. (E) EIS data obtained at 1.23 V vs. RHE. (F) LSVs recorded in the presence and absence of BA. | |
The relative% of the pre-exponential factor was calculated using eqn (3) and (4):
| | 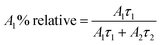 | (3) |
| | 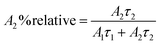 | (4) |
The average PL lifetime (τ) was calculated using eqn (5):
| | 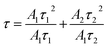 | (5) |
The average decay lifetime of P-doped samples (CN-xP) is found to be smaller than that of pristine CN, reaffirming the enhanced charge separation efficiency and rapid charge carrier migration in CN-xP than CN. It is anticipated that the additional migration channels arising due to P–N linkages in CN-xP samples enable faster charge transfer.15,27 Table S8† summarizes the average decay life time of CN-xP and CN samples determined from the TRPL data. Further, CN and CN-210P powder samples were characterized using ESR recorded at room temperature. As shown in Fig. 6C, the samples exhibited a single Lorentzian line, originating due to the presence of unpaired electrons of π-conjugated aromatic rings of g-C3N4. Notably, the intensity of the ESR signal of CN-210P is enhanced as compared to that of CN, suggesting high concentration of delocalized electrons on π-conjugated aromatic rings of CN-210P. Thus, the P-doping in C3N4 leads to the formation of electron-rich C3N4. The delocalized electrons on π-conjugated aromatic rings are known to facilitate the charge transfer, and enhanced photocatalytic activity can be expected in P-doped samples. To further validate the improved life time of photogenerated charge carriers, photoelectrochemical (PEC) measurements were carried out. The photocurrent responses were recorded at 1.23 V vs. RHE by chopping the light at regular intervals of time. As shown in Fig. 6D, around 1.6 times increase in photocurrent density is noted with CN-210P in comparison to CN, indicating the relatively improved electron–hole separation. It is expected that the dopants (P) can act as trap centres and minimize the recombination of photogenerated charge carriers.27 The electrochemical impedance spectroscopy (EIS) studies indicate smaller charge transfer resistance (Rct) of P-doped samples, suggesting their good charge transfer ability at the electrode/electrolyte interface (Fig. 6E). Further, as shown in Fig. 6F, the higher photocurrents observed from the voltammograms recorded in the presence of BA clearly indicate the oxidative ability of CN-210P towards BA.43a Additionally, the electrochemical hydrogen evolution reaction (HER) polarization curves recorded in the darkness suggest that the overpotential to reach 0.1 mA cm−2 is 0.47 V vs. RHE for CN-210P, while it is 0.54 V vs. RHE for CN (Fig. S29). This again confirms the beneficial role of the presence of P-dopants in achieving high HER activity.
Conclusions
In summary, P-doped carbon nitride with different amounts of P dopants has been prepared using a mixture of DCDA and eco-friendly inositol hexaphosphate (IP6) as the P-source. Enhanced light harvesting due to the narrowing of the band gap of C3N4 upon P-doping was demonstrated. The systematic variations in the band structure of C3N4 with dopant (P) quantity were studied. DFT studies revealed that the doping of P into C3N4 occurs preferentially through the replacement of the corner carbon atom. Among all the P-doped CN samples, CN-210P exhibited high photocatalytic activity for H2 evolution as compared to undoped CN. Around 12.8 times higher H2 production rate was observed in the case of CN-210P than that of pristine CN when TEOA was the SED. The appreciable enhancement in photocatalytic activity in CN-210P is due to the result of P-doping, which induced modifications in the electronic structure that enable wide visible light absorption and provide more active sites, as suggested by DFT calculations. The presence of P–N linkages acts as additional migration channels and facilitates efficient charge transfer. Most importantly, it is confirmed that CN-210P can act as a dual functional catalyst for producing H2 and BADH simultaneously. The yield of H2 and BADH was found to be 51.6 μmol h−1 g−1 and 64.2 μmol h−1 g−1 respectively. ESR measurements and DFT calculations suggested that the photocatalytic oxidation proceeds via a carbon-centred radical route to selectively yield BADH. The study provides opportunities to explore the ability of CN-xP for oxidizing other biomass-derived platform molecules as SEDs to simultaneously produce H2 and high-value products.
Experimental
Chemicals
All chemical reagents were purchased from commercial suppliers and used directly as received without further purification. The chemicals used are as follows: dicyandiamide (DCDA) (Loba Chemie, 98%), inositol hexaphosphate (50% in water, TCI Chemicals), benzyl alcohol (Loba Chemie, 99%), benzaldehyde (Sigma-Aldrich, > 99%), triethanolamine (Loba Chemie), methanol (Molychem 99%), H2PtCl6. 6H2O (Sigma-Aldrich, 37.50% Pt basis), sodium sulfate (Molychem, 99%), and maleic Acid (Loba Chemie, 99%).
Preparation of P-doped carbon nitride (CN-xP)
A precursor mixture containing a known amount (5 g) of DCDA and a certain amount of inositol hexaphosphate (IP6) was added to 4.55 mL water, followed by evaporation of the solvent by heating at 80 °C. Subsequently, the white solid obtained was transferred into a cylindrical crucible of volume 30 mL covered with a lid, kept in a muffle furnace, and heated at 550° for 4 h at a ramping rate of 5 °C min−1. After natural cooling to room temperature, the products were obtained and hand-milled before use. CN with different amounts of P-dopants (CN-xP) was obtained by varying the amounts of IP6 in the precursor mixture, where x represents the amount (in μL) of IP6 taken in the precursor mixture.
Preparation of bulk g-C3N4 (CN)
A conventional calcination method was used to prepare pristine g-C3N4 (CN). A known amount (5 g) of DCDA was taken in a 30 mL cylindrical crucible covered with a lid, transferred into a muffle furnace and heated at 550° for 4 hours at a ramping rate of 5 °C min−1. After cooling to room temperature naturally, the product was milled into a fine powder, washed with water and dried overnight to obtain the final CN.
Characterization
The powder XRD patterns were recorded using a Bruker D8 Advance with Cu Kα radiation (λ = 1.5406 Å). The FESEM images were recorded using a Quanta FEG 250. The samples for SEM were prepared by drop-casting the dilute dispersions of samples onto a pre-cleaned Si substrate. The Fourier transform infrared (FTIR) spectra were recorded using a PerkinElmer spectrophotometer (Model: spectrum two) in the ATR mode. The surface area measurements were carried out using a quanta chrome NOVA 1000e BET surface area analyser. The X-ray photoelectron spectroscopy (XPS) characterization was performed using a K-alpha Thermo Fisher Scientific Instrument with an Al Kα X-ray source (1486.6 eV). The UV-visible diffuse reflectance spectra (UV-vis DRS) were recorded using a UV-visible Jasco V770 spectrometer with an integrated sphere setup and BaSO4 as a reference. Raman and PL spectra were recorded using a LABRAM HR, Horiba France at an excitation wavelength of 325 nm. The photoelectrochemical measurements were performed using an Iviumstat/Biologic SP150e electrochemical workstation in a three-electrode electrochemical cell in a de-aerated 0.1 M Na2SO4 solution (pH = 6.8), where Ag/AgCl (sat. KCl) was used as the reference electrode, a graphite rod was used as the auxiliary electrode, and the photocatalyst-modified fluorine-doped-tin-oxide (FTO) glass obtained by electrophoretic deposition was used as the working electrode. The transient photocurrent responses were measured at 1.23 V vs. RHE with chopping light illumination at a constant interval of 10 s. The electrochemical impedance spectra were recorded at 1.23 V vs. RHE in the frequency range of 1000 Hz–0.01 Hz with an amplitude of 0.05 V. Mott–Schottky (M–S) measurements were performed in 0.1 M Na2SO4 at a frequency of 1000 Hz. TRPL measurements were carried out using a Horiba DeltaPro Time-Correlated Single Photon Counting (TCSPC) Lifetime Fluorimeter with a 296 nm delta diode as the excitation source. The electron spin resonance (ESR) spectra were recorded using a JEOL with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a spin-trapping agent to detect the carbon-centred free radicals. A microwave frequency of 9.44 GHz and a microwave power of 0.995 mW were used. The sample was collected after 20 min to record the ESR spectra. For the solid samples, the spectra were recorded at room temperature.
Photocatalytic studies
Photocatalytic H2 evolution.
The photocatalytic tests were carried out in custom-build reactors using a custom-designed white LED as the light source. The temperature of the reactor was maintained using a water bath and the reactors was cooled with fans to avoid heating of the reactor. Prior to hydrogen evolution studies, CN and CN-xP catalysts were modified with Pt via photodeposition. Briefly, a known amount of photocatalyst was dispersed in 15 mL of water consisting of 10 vol% of methanol. To this, a required amount of H2PtCl6, corresponding to 1.5 wt% of Pt, was added and the mixture was sonicated for 20 min and de-aerated by purging with high pure Ar for 30 min. Subsequently, the mixture was illuminated for 1 h to photoreduce Pt-ions to Pt. After that, the sample was centrifuged, washed and kept for drying at 60 °C for 12 h before further use. Hydrogen evolution studies were carried out by suspending known amounts (10 mg) of CN and CN-xP in water containing 10 vol% TEOA that acts as the SED. Prior to the measurements, the reactor was deaerated by purging high-purity Ar for 30 min. The amount of H2 produced was quantified by collecting 0.2 mL of gas from the headspace of the reactor at regular intervals of time using air tight Hamilton syringes. The collected samples were injected into a gas chromatograph (Agilent 8890) equipped with a TCD detector. The carrier gas used was Ar at a flow rate of 0.5 mL min−1. A MolSieve 5A 80/100 SS (9Ft 2 mm) packed column was used. The temperature of the column was maintained at 40 °C, and the inlet and detector temperatures were kept at 150 °C. The apparent quantum efficiency (AQE) was determined in the presence of TEOA using LEDs that emit 452, 517, and 636 nm of light (Fig. S16†). The intensity of light was measured using a spectroradiometer. The AQE was determined using the following equation:| | 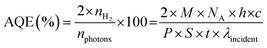 | (6) |
where nH2 is the number of electrons transferred in H2 evolution, nphotons is the total number of incident photons, M is the amount of H2 in moles, NA is the Avogadro number, h is Planck's constant, c is the speed of light, S is the irradiation area, P is the intensity of irradiation light, t is the photoreaction time and λ is the wavelength of incident light.
Photocatalytic co-production of H2 and benzaldehyde
Studies on the co-production of H2 and benzaldehyde were conducted in deaerated aqueous solutions of 20 mM BA. The amount of H2 produced was measured by GC as mentioned above. The oxidation product formed during photocatalysis was characterized by 1H NMR (Bruker Avance Neo 500 MHz). The NMR sample consists of 350 μL of the reaction mixture collected after a certain time of illumination from the photocatalyst, 100 μL D2O and 100 μL of 1 mM maleic acid (internal standard). The NMR signal integrals were obtained using the Bruker Topsin software. The concentration of the species was calculated using the following formula:43b| | 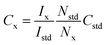 | (7) |
where Ix is the integral of the species, namely BADH (δ 9.8 ppm) or BA (δ 7.4–7.2 ppm), Istd is the integral of the internal standard (δ 6.2 ppm), Nstd is the number of protons of internal standard, Nx is the number of protons of BA or BADH and Cstd is the concentration of the internal standard.
The number of moles of the species was calculated using eqn (3):
| |  | (8) |
where
Cx is the concentration of the species determined from
eqn (2),
VNMR tube is the total volume (sample + internal standard + D
2O) present in the tube,
VR is the total volume of the reaction mixture taken in reactor (10 mL in the present case), and
Vsample is the volume of the sample aliquot from the reactor taken for analysis. For mass spectral studies, the reaction mixture collected after illumination of a known period of time was separated from the photocatalyst and was extracted in CH
2Cl
2. Briefly, the catalyst was separated from the reaction mixture by filtering it through a 0.22 μm Nylon syringe filter and extracted with CH
2Cl
2. The mixture extracted in CH
2Cl
2 is injected into GC equipped with an MS detector (HP-5 fused silica capillary column, 0.25 m × 0.25 mm × 30 m). The liquid chromatography (LC) measurements were carried out using an LC 1290 infinity (Model 6460 Triple Quadrupole) with a Zorbax Eclipse plus C18 rapid resolution column HD: 2.1 × 50 mm, 1.8 microns. The mobile phase used for LC measurements was 70
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
:
30, that is 0.1% formic acid in water and acetonitrile. The column temperature was kept at 30 °C.
DFT calculations
Periodic density functional theory (DFT) studies were conducted using the planewave based ab initio code, Vienna Ab initio Simulation Package (VASP).44–46 The interactions between the core and valence electrons were treated using the projector augmented wave (PAW) potentials.47 The exchange-correlation energy density functional of Perdew–Burke–Ernzerhof along with Grimme's D3 semi-empirical method (PBE-D3) was used.48,49 Since the electronic band gaps from pure DFT functionals are known to be underestimated, a more accurate hybrid DFT functional by Heyd–Scuseria–Ernzerhof (HSE06)50 was used to calculate the electronic band structure. Plane-wave basis-sets with a kinetic energy cut-off of 520 eV were used to expand the Kohn–Sham wave functions of the valence electrons. The graphical software VESTA was used to generate all the reported structures.51 The adsorption energies (ΔE) of different reaction intermediates over the surface were calculated as follows:| | | ΔE(int) = E(sur–int) − [E(sur) + E(int)] | (9) |
where E(sur–int), E(sur) and E(int) represent the energies of intermediate (int) adsorbed surface, pure surface and reaction intermediate respectively. The energies of all the molecular systems were calculated in a cubic cell of 20 Å length. The formation energy of different P-doped systems at both the N- and C-sites in g-C3N4 was calculated as follows:| | | ΔEF = E(P@X-C3N4) − E(C3N4) − E(X) | (10) |
where E(P@X-C3N4), E(C3N4), and E(X) represent the energies of P doped at the X-site in g-C3N4, pure g-C3N4 and element X respectively. The energies of C, N and P were calculated from their standard states (graphite for C, N2 gas for N and black phosphorus for P) as follows:| | 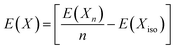 | (11) |
where E(Xn) and E(Xiso) represent the energy of the molecule/material with n atoms in its standard state and the energy of a single atom in a large cubic cell respectively.
Author contributions
ADG carried out experimental work, data interpretation, write-up; SP assisted in synthesis and data interpretation; SK performed DFT calculations and write-up related to DFT; KV conceived the idea, overall supervision, manuscript writing and editing. All the authors have given approval for the final version.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
KV is indebted to SERB-DST (SRG/2020/000719) for financial support. ADG and SP are grateful to SERB-DST and BITS Pilani, K Birla Goa campus for research fellowship respectively. We gratefully acknowledge CSIF, BITS Pilani, K K Birla Goa campus for providing XRD, FESEM, Raman, NMR and LC-MS facilities. Central analytical laboratory, BITS Pilani, Hyderabad is acknowledged for XPS facility. Horiba India Technical Centre (HITC), Indian Institute of Science (IISc) is acknowledged for helping us with TRPL measurements. SAIF of IIT Bombay is acknowledged for ESR (ESR-JEOL) facility. We thank Mr Laxman Raikar and Prof. Halan Prakash, Department of Chemistry, BITS Pilani, K K Birla Goa campus for assisting us in measuring the intensity of LEDs used in the present study.
Notes and references
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- X. B. Li, C. H. Tung and L. Z. Wu, Nat. Rev. Chem, 2018, 2, 160–173 CrossRef CAS.
- M. Y. Qi, M. Conte, M. Anpo, Z. R. Tang and Y. J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed.
- L. I. Granone, F. Sieland, N. Zheng, R. Dillert and D. W. Bahnemann, Green Chem., 2018, 20, 11691192 RSC.
- K. A. Davis, S. Yoo, E. W. Shuler, B. D. Sherman, S. Lee and G. Leem, Nano Convergence, 2021, 8, 6 CrossRef CAS PubMed.
- R. Marotta, I. Di Somma, D. Spasiano, R. Andreozzi and V. Caprio, Chem. Eng. J., 2011, 172, 243–249 CrossRef CAS.
- S. Kampouri and K. C. Stylianou, ACS Catal., 2019, 9, 4247–4270 CrossRef CAS.
- J. Barrio, M. Volokh and M. Shalom, J. Mater. Chem. A, 2020, 8, 11075 RSC.
-
(a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed;
(b) O. Savateev and J. Zhuang, ChemPhotoChem, 2024, 8, e202300306 CrossRef CAS;
(c) N. Karjule, R. S. Phatake, S. Barzilai, B. Mondal, A. Azoulay, A. I. Shames, M. Volokh, J. Albero, H. García and M. Shalom, J. Mater. Chem. A, 2022, 10, 16585–16594 RSC;
(d) Q. Chen, C. Ning, J. Fang, B. Ping, G. Li, L. Kong, J. Chen, Z. Sun, J. Wang, Q. Ruan, X. Niu and L. Tao, Chem. Eng. J., 2024, 487, 150581 CrossRef CAS;
(e) M. Thangamuthu, Q. Ruan, P. O. Ohemeng, B. Luo, D. Jing, R. Godin and J. Tang, Chem. Rev., 2022, 122, 11778–11829 CrossRef CAS PubMed;
(f) Q. Ruan, M. K. Bayazit, V. Kiran, J. Xie, Y. Wang and J. Tang, Chem. Commun., 2019, 55, 7191–7194 RSC;
(g) R. C. Sahoo, H. Lu, D. Garg, Z. Yin and H. S. S. R. Matte, Carbon, 2022, 192, 101–108 CrossRef CAS.
- Y. Li, X. Li, H. Zhang and Q. Xiang, Nanoscale Horiz., 2020, 5, 765 RSC.
-
(a) L. Zhou, H. Zhang, H. Sun, S. Liu, M. O. Tade, S. Wang and W. Jin, Catal. Sci. Technol., 2016, 6, 7002–7023 RSC;
(b) Y. Wang, M. K. Bayazit, S. J. A. Moniz, Q. Ruan, C. C. Lau, N. Martsinovich and J. Tang, Energy Environ. Sci., 2017, 10, 1643–1651 RSC.
- H. Bin Fang, X. H. Zhang, J. Wu, N. Li, Y. Z. Zheng and X. Tao, Appl. Catal., B, 2018, 225, 397–405 CrossRef.
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CrossRef CAS.
- W. Wang, L. Du, R. Xia, R. Liang, T. Zhou, H. K. Lee, Z. Yan, H. Luo, C. Shang, D. L. Phillips and Z. Guo, Energy Environ. Sci., 2023, 16, 460–472 RSC.
- J. Ran, T. Y. Ma, G. Gao, X. W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 RSC.
- B. Li, Y. Si, Q. Fang, Y. Shi, W. Q. Huang, W. Hu, A. Pan, X. Fan and G. F. Huang, Nano-Micro Lett., 2020, 12, 52 CrossRef CAS PubMed.
- Y. Wang, F. Silveri, M. K. Bayazit, Q. Ruan, Y. Li, J. Xie, C. R. A. Catlow and J. Tang, Adv. Energy Mater., 2018, 24, 1801084 CrossRef.
- Q. Zhang, P. Chen, L. Chen, M. Wu, X. Dai, P. Xing, H. Lin, L. Zhao and Y. He, J. Colloid Interface Sci., 2020, 568, 117–129 CrossRef CAS PubMed.
- J. Zhang, W. Zhang, L. Yue, X. Hu, H. Lin, L. Zhao and Y. He, Appl. Surf. Sci., 2022, 592, 153337 CrossRef CAS.
- P. Chen, L. Chen, S. Ge, W. Zhang, M. Wu, P. Xing, T. B. Rotamond, H. Lin, Y. Wu and Y. He, Int. J. Hydrogen Energy, 2020, 45, 14354–14367 CrossRef CAS.
- Y. Zhou, L. Zhang, J. Liu, X. Fan, B. Wang, M. Wang, W. Ren, J. Wang, M. Li and J. Shi, J. Mater. Chem. A, 2015, 3, 3862–3867 RSC.
- L. Zhang, X. Chen, J. Guan, Y. Jiang, T. Hou and X. Mu, Mater. Res. Bull., 2013, 48, 3485–3491 CrossRef CAS.
- H. Yang, Y. Zhou, Y. Wang, S. Hu, B. Wang, Q. Liao, H. Li, J. Bao, G. Ge and S. Jia, J. Mater. Chem. A, 2018, 6, 16485–16494 RSC.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 16850–16856 CrossRef CAS PubMed.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- M. Wu, J. Zhang, B. bei He, H. wen Wang, R. Wang and Y. sheng Gong, Appl. Catal., B, 2019, 241, 159–166 CrossRef CAS.
- M. Wen, N. Yang, J. Wang, D. Liu, W. Zhang, S. Bian, H. Huang, X. He, X. Wang, S. Ramakrishna, P. K. Chu, S. Yang and X. F. Yu, ACS Appl. Mater.
Interfaces, 2021, 13, 50988–50995 CrossRef CAS PubMed.
- F. Zhang, J. Li, H. Wang, Y. Li, Y. Liu, Q. Qian, X. Jin, X. Wang, J. Zhang and G. Zhang, Appl. Catal., B, 2020, 269, 118772 CrossRef CAS.
- H. F. Ye, R. Shi, X. Yang, W. F. Fu and Y. Chen, Appl. Catal., B, 2018, 233, 70–79 CrossRef CAS.
- H. Wang, X. Zhang, J. Xie, J. Zhang, P. Ma, B. Pan and Y. Xie, Nanoscale, 2015, 7, 5152–5156 RSC.
- J. Zou, Y. Yu, K. Qiao, S. Wu, W. Yan, S. Cheng, N. Jiang and J. Wang, J. Mater. Sci., 2020, 55, 13618–13633 CrossRef CAS.
- C. Yang, S. Zhang, Y. Huang, K. Lv, S. Fang, X. Wu, Q. Li and J. Fan, Appl. Surf. Sci., 2020, 505, 144654 CrossRef CAS.
- S. Zhao, Y. Liu, Y. Wang, J. Fang, Y. Qi, Y. Zhou, X. Bu and S. Zhuo, J. Colloid Interface Sci., 2022, 616, 152–162 CrossRef CAS.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- M. Alejandra Quintana, R. R. Solís, M. Ángeles Martín-Lara, G. Blázquez, F. Mónica Calero and M. J. Muñoz-Batista, Sep. Purif. Technol., 2022, 298, 121613 CrossRef CAS.
- G. Li, Y. Wu, M. Wang, W. Zhou, X. Liu, Z. Zhu, X. Song and P. Huo, ACS Appl. Nano Mater., 2023, 6, 14513–14526 CrossRef CAS.
- G. Zhang, G. Li, Z. A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- X. Chi, S. Tan, J. Song, F. Liu, Y. Tian, H. Yuan and R. Guan, Catal. Lett., 2021, 151, 3592–3602 CrossRef CAS.
- J. Wang, L. Guan, S. Yuan, J. Zhang, C. Zhao, X. Hu, B. Teng, Y. Wu and Y. He, Sep. Purif. Technol., 2023, 314, 123554 CrossRef CAS.
- J. Wang, H. Liang, C. Zhang, B. Jin and Y. Men, Appl. Catal., B, 2019, 256, 117874 CrossRef CAS.
- X. Xiang Fang, L. B. Ma, K. Liang, S. J. Zhao, Y. F. Jiang, C. Ling, T. Zhao, T. Y. Cheang and A. W. Xu, J. Mater. Chem. A, 2019, 7, 11506 RSC.
-
(a) C. Pulignani, C. A. Mesa, S. A. J. Hillman, T. Uekert, S. Gimenez, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2022, 134, 202211587 CrossRef;
(b) M. Abdinejad, S. Subramanian, M. K. Motlagh, M. Noroozifar, S. Duangdangchote, I. Neporozhnii, D. Ripepi, D. Pinto, M. Li, K. Tang, J. Middelkoop, A. Urakawa, O. Voznyy, H. Kraatz and T. Burdyny, Adv. Energy Mater., 2023, 2300402 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shraddha
Paniya
a,
Shraddha
Paniya