
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrothermal synthesis of ZnZrOx catalysts for CO2 hydrogenation to methanol: the effect of pH on structure and activity†
Issaraporn
Rakngam‡
 a,
Gustavo A. S.
Alves‡
bc,
Nattawut
Osakoo
d,
Jatuporn
Wittayakun
a,
Thomas
Konegger
e and
Karin
Föttinger
*bc
a,
Gustavo A. S.
Alves‡
bc,
Nattawut
Osakoo
d,
Jatuporn
Wittayakun
a,
Thomas
Konegger
e and
Karin
Föttinger
*bc
aSchool of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand
bInstitute of Materials Chemistry, TU Wien, Getreidemarkt 9, 1060, Vienna, Austria. E-mail: karin.foettinger@tuwien.ac.at
cChair of Physical Chemistry, Montanuniversität Leoben, Franz-Josef-Straße 18, 8700 Leoben, Austria
dInstitute of Research and Development, Suranaree University of Technology, Thailand
eInstitute of Chemical Technologies and Analytics, TU Wien, Getreidemarkt 9, 1060, Vienna, Austria
First published on 11th October 2024
Abstract
With the growing necessity of achieving carbon neutrality in the industrial sector, the catalytic hydrogenation of carbon dioxide into methanol has been widely considered one of the key strategies for the utilization of captured CO2. For this reason, the development of alternative catalysts such as ZnZrOx has attracted considerable interest, given its superior stability and versatility in comparison to the conventional Cu-based materials. In this work, ZnZrOx has been produced by a hydrothermal synthesis method at varied synthesis pH between 7 and 10 and a positive association between pH and catalytic CO2 conversion is observed. At 2.0 MPa and 250 °C, ZnZrOx produced at pH 10 shows a methanol selectivity of 95% at a CO2 conversion of 3.4%. According to characterization, basic pH conditions enable the formation of abundant t-ZrO2 and the subsequent incorporation of Zn2+ into this phase, although the content of surface Zn does not increase between pH 8 and 10. Nevertheless, synthesis pH values can be correlated with surface oxygen content and CO2 adsorption capacity, which could be important contributors to the higher catalytic activity observed as a result of higher synthesis pH values. However, upon synthesis at pH 10, an inferior selectivity to methanol is observed above 250 °C, as a possible result of the excessive formation of ZnO. Interestingly, this secondary phase can be prevented and the selectivity can be slightly improved by utilizing NH4OH instead of NaOH in the hydrothermal method.
Sustainability spotlightOne of the key building blocks in the chemical industry, methanol, has been primarily obtained from fossil feedstocks over the last decades. With the growing necessity of developing sustainable alternatives, the production of renewable methanol from CO2 has been recently proposed as a strategy to utilize carbon derived from biomass or industrial emissions from hard-to-abate sectors. In this context, the development of more stable and versatile catalysts may facilitate the implementation of CO2 hydrogenation to methanol on a large scale. This research work addresses the following Sustainable Development Goals: Industry, Innovation and Infrastructure (SDG 9), Sustainable Consumption and Production (SDG 12) and Climate Action (SDG 13). |
Introduction
The relentless increase in atmospheric carbon dioxide (CO2) levels and the urgent need to mitigate climate change have been motivating intensive research into sustainable solutions for CO2 utilization,1 often involving its hydrogenation into carbon monoxide,2 alcohols3 and olefins.4 Among these strategies, the conversion of CO2 to methanol has been considered a promising route for the utilization of CO2 emissions due to the versatility and energy density of methanol as a liquid fuel and chemical feedstock. Typically, the industrial synthesis of methanol from CO-rich syngas at high pressure employs the Cu/ZnO/Al2O3 catalyst, which can also be applied for the direct hydrogenation of CO2.5 Nevertheless, one of the drawbacks of such copper-based materials is the deactivation due to the limited stability of the catalyst under exposure to moisture and sulfur-containing gases, which may pose a considerable obstacle to the continuous long-term operation of the methanol synthesis from industrial CO2 feedstocks.6,7 For this reason, the development of more stable and robust catalysts could benefit the yet incipient production of renewable methanol derived from a variety of CO2 sources, such as biogas,8 geothermal origins9 and steel plants.10Among the diverse catalysts explored for CO2 hydrogenation to methanol, ZnZrOx-based materials have demonstrated notable potential as next-generation catalysts, given their excellent selectivity, stability and sulfur tolerance.11,12 On the other hand, their non-metallic character engenders limited hydrogen activation, leading to lower CO2 conversion compared to commercial Cu/ZnO-based materials.13
In ZnZrOx catalysts, the synergistic interaction between Zn2+ and ZrO2 plays a vital role in promoting both catalytic activity and selectivity.11 Specifically, the Zn2+–O–Zr4+ sites from the ZnZrOx solid solution are identified as the active sites for CO2-to-methanol hydrogenation,14,15 in order that Zn species are considered responsible for dissociating H2 molecules, while Zr species facilitate the activation of CO2.15 This catalytically active Zn2+–O–Zr4+ system has been mostly observed as a result of Zn2+ ions incorporated in the tetragonal ZrO2 lattice (t-ZrO2), while the monoclinic zirconia polymorph (m-ZrO2) is considered less able to accommodate these species.16 Additionally, the recent evidence for ZnO clusters embedded in ZrO2 as a key feature of ZnZrOx catalysts indicates that ZnO/ZrO2 systems should be also taken into account as possible active sites for CO2 hydrogenation in these materials.17,18 In addition to the clear importance of Zn2+ species in ZnZrOx, recent studies have emphasized the role of lattice oxygen on H2 activation, suggesting a direct correlation between catalytic activity and surface oxygen content.19 In fact, experimental and computational studies indicate that Zn2+–O2− pairs may be responsible for the heterolytic H2 dissociation in ZnZrOx catalysts for CO2 hydrogenation to methanol.20
In several previous studies, ZnZrOx catalysts with a high content of t-ZrO2 have been typically produced by co-precipitation approaches, followed by calcination at 500 °C.11,21,22 Although t-ZrO2 is thermodynamically less stable than m-ZrO2 at such temperatures, the presence of a hydrated surface23 and small crystallites24 may promote the formation of tetragonal zirconia in these cases. Alternatively, a hydrothermal approach followed by calcination between 300 °C and 600 °C has been shown as an effective method to produce t-ZrO2-based catalysts, and the presence of Na ions has been suggested as another important factor for the stabilization of the tetragonal phase.25 Therefore, the hydrothermal synthesis method may deserve further exploration due to the possibility of obtaining nanostructured ZnZrOx catalysts with high surface area26 and suitable crystalline structure for CO2 hydrogenation to methanol. However, achieving optimal catalytic performance requires a deeper understanding of the catalyst synthesis under varying parameters, such as pH values, which are often a key factor in the nucleation, growth, and crystal size of metal-oxide particles. In Zn and Zr aqueous solutions, basic pH was shown to accelerate the crystallization of Zn and Zr oxides.27,28 Thus, exploring the catalytic improvement of ZnZrOx by systematically varying pH levels represents a promising strategy for enhancing the efficiency of CO2 conversion to methanol in this catalyst.
This work presents an investigation into the catalytic performance and material properties of ZnZrOx produced by hydrothermal synthesis. Herein, the effect of synthesis pH on structural and surface properties is investigated and the material is tested as a catalyst for CO2 hydrogenation to methanol.
Experimental
Chemicals and materials
Zirconyl chloride octahydrate (ZrOCl2·8H2O, 98%, Sigma-Aldrich), zinc chloride (ZnCl2, 98%, Fluka), sodium hydroxide (NaOH, 98%, Sigma-Aldrich), and ammonium hydroxide (NH4OH, 25%, Donau Chem) were used for the catalyst synthesis.Synthesis of ZnZrOx
A series of ZnZrOx catalysts were prepared at different pH values (7, 8, 9, and 10) using a hydrothermal treatment method based on a previously reported procedure for the preparation of t-ZrO2.25 Initially, 3.36 g of ZrOCl2·8H2O and ZnCl2 with the Zn/(Zr + Zn) mole ratio of 20% were dissolved in 20 mL of deionized water under stirring. Subsequently, a 1.0 M NaOH solution was slowly added into the mixed metal solution to achieve the desired pH value. The mixture was then transferred into an autoclave with a Teflon liner and heated at 150 °C for 18 h. After the hydrothermal treatment, the autoclave was cooled down to room temperature. The white powder was filtered, washed with deionized water until neutral pH, dried, and calcined at 500 °C for 3 h. Alternatively, a similar procedure was followed for the production of ZnZrOx at pH 10, using NH4OH as a pH adjuster instead of NaOH.Catalyst characterization
The crystalline structure of the samples was characterized by powder X-ray diffraction (XRD) on a Philips XPert diffractometer using Cu Kα radiation (λ = 1.5406 Å) at 45 kV and 40 mA operating in Bragg–Brentano reflection geometry. Transmission Electron Microscopy (TEM) and Energy-Dispersive X-ray Spectroscopy (EDX) were performed on a Thermo Scientific TALOS F200X operated at 200 kV. The morphologies of the samples were investigated by scanning electron microscopy (SEM) with a FEI Quanta 250 FEG microscope at a 5 kV voltage. N2 adsorption–desorption analysis of the samples was determined using Micromeritics ASAP 2020 at −196 °C. Before the measurement, the sample was degassed at 350 °C for 8 h under vacuum. Specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) method. Pore size distributions were determined by the Barrett–Joyner–Halenda (BJH) model.The basicity and CO2 adsorption capacity of the sample were investigated by temperature-programmed desorption of carbon dioxide (CO2-TPD) using a BELCAT-B chemisorption analyzer with a thermal conductivity detector (TCD). Prior to analysis, the sample was pretreated at 350 °C for 1 h under flowing He gas with 30 mL min−1. Then, the sample was cooled down to 50 °C and a gas mixture containing 10% CO2 in He was adsorbed on the sample surface for 1 h. The sample was purged with He and held for 30 min to remove non-adsorbed CO2. The TPD process was performed in the temperature range from 50 to 350 °C with a heating rate of 10 °C min−1 and held at 350 °C for 1 h under a He flow of 30 mL min−1.
Chemical states of surface species were identified by X-ray photoelectron spectroscopy (XPS) with a SPECS u-Focus system (AlKα source, Phoibos 150 WAL detector). XPS data evaluation was carried out using the CasaXPS software,29 considering spectra calibrated with the C 1s peak at 284.8 eV. Quantification of surface species was conducted by considering the areas of Zn 2p3/2, O 1s, Zr 3p3/2, Zr 3d and Zn 3p peaks with the respective Relative Sensitivity Factors (RSF) of 18.92, 2.93, 5.14, 7.04 and 2.83.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was carried out on a Bruker Vertex 70 spectrometer equipped with a mercury-cadmium-telluride detector cooled by liquid nitrogen. Before measurement, the sample was pretreated at 350 °C for 2 h under pure H2 flow (7.7 mL min−1) and then cooled to 250 °C under Ar flow (11.5 mL min−1). After cooling down, the background spectrum was collected from 800–4000 cm−1 with 256 scans at a resolution of 4 cm−1. H2 (7.7 mL min−1) and CO2 (2.6 mL min−1) were introduced into the reaction cell and the spectra were collected at 250 °C.
Catalytic testing for CO2 hydrogenation
The catalytic activity testing was performed in a tubular fixed-bed continuous-flow “micro effi” reactor from PID Eng&Tech. Prior to the test, 1 g of the ZnZrOx catalyst was activated under H2 flow at 350 °C for 2 h. After cooling to 250 °C, the reactant gas mixture of CO2/H2/He (20/60/20) was introduced into the reactor at a total flow of 5 mLn/min under a pressure of 2.0 MPa. The reaction was conducted in steps of 10 °C between 250 to 290 °C during 6 h in each step. Detection of products in gas phase was carried out using an Inficon Micro GC 3000 equipped with a Plot Q column. The CO2 conversion (X) and the selectivity (S) for CH3OH, CO and CH4 were calculated using the following equations: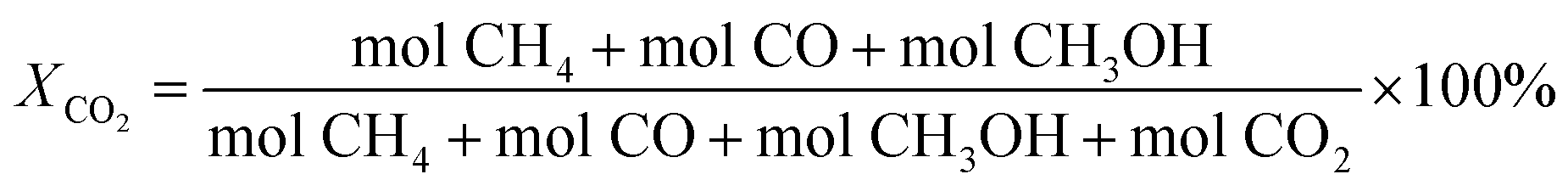 | (1) |
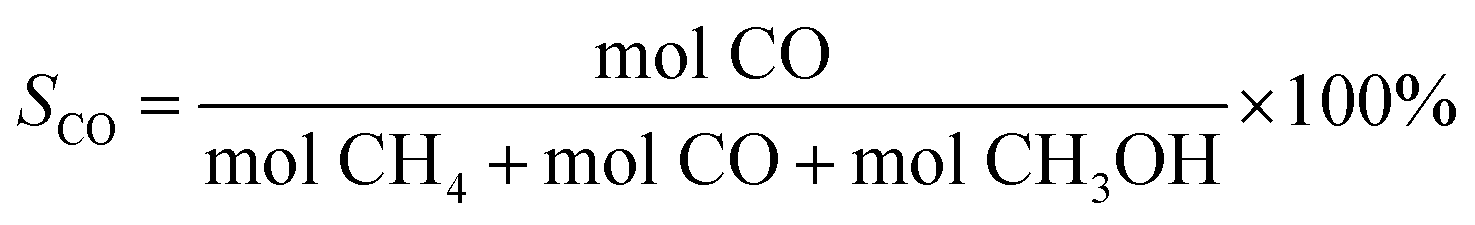 | (2) |
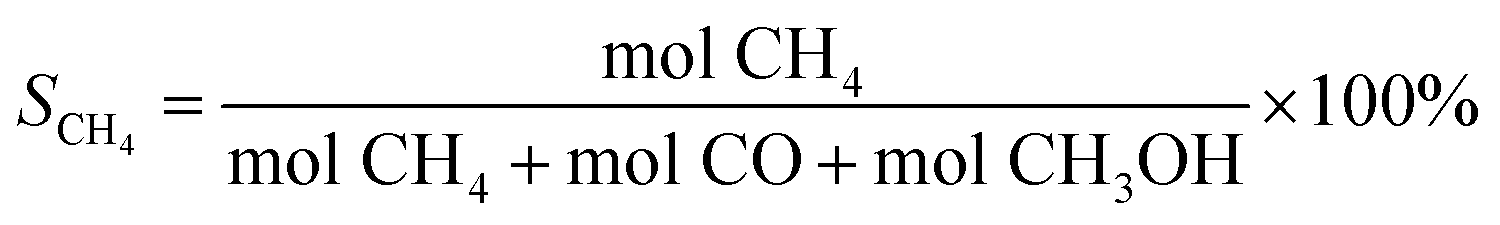 | (3) |
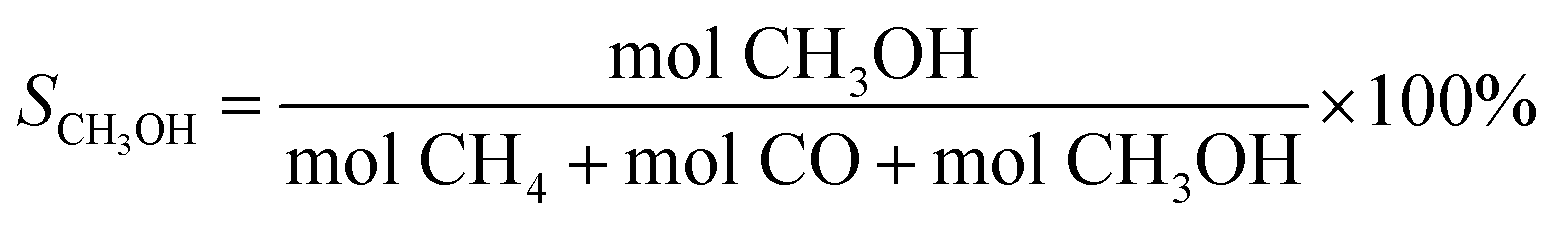 | (4) |
Results and discussion
ZnZrOx samples were produced by hydrothermal synthesis using NaOH to adjust pH values between 7 and 10. Considering that the saturation of t-ZrO2 with Zn2+ has been previously reported at 25%,16 a Zn/(Zn + Zr) atomic ratio of 20% was chosen, seeking to obtain abundant and dispersed Zn2+ sites in a ZnZrOx without the formation of segregated ZnO. In order to verify the crystalline structure of the material, XRD analysis was conducted, as shown in Fig. 1A. The XRD pattern of ZnZrOx prepared at pH 7 presents evidence for the tetragonal ZrO2 (t-ZrO2) phase (JCPDS 68-0200) mixed with a monoclinic (m-ZrO2) phase (JCPDS 65-0687).11 However, more basic pH conditions completely attenuate the pattern related to m-ZrO2, as observed for the pH 8 and pH 9 samples. Upon increasing pH during synthesis to 10, additional small features emerge at 31.9°, 34.6°, 36.4°, 47.8°, 56.8°, 63.0°, and 68.1°, which can be assigned to hexagonal ZnO phase (JCPDS 01-083-6338). This result indicates that the material produced at pH 10 presents both the t-ZrO2 phase and a secondary contribution of hexagonal ZnO. Nevertheless, in all cases the material consists primarily of tetragonal zirconia, which is observed in more detail by TEM in Fig. 1B and C, showing the characteristic (101) interplanar spacing of 0.29 nm from t-ZrO2. As shown by TEM-EDS analysis in Fig. 1D and E, this phase presents a homogeneous distribution of Zr and Zn, giving evidence for the incorporation of Zn atoms into the t-ZrO2 structure. Furthermore, as shown in the insert of Fig. 1A, the diffraction feature corresponding to t-ZrO2 shifts by approximately 0.5° towards higher angles as the pH during synthesis increases from 7 to 9, but no changes are verified between pH 9 and 10. The observed shift can be attributed to the incorporation of Zn2+ ions (ionic radius 0.74 Å) into the t-ZrO2 lattice, leading to a reduction in interplanar spacing due to the substitution of Zr4+ (ionic radius, 0.82 Å) by smaller radius size of Zn2+ ions.11,30 Therefore, these findings indicate that basic hydrothermal conditions facilitate the formation of the Zn–ZrO2 solid solution. However, above pH 9 this phase may be already saturated with Zn2+, which leads to the formation of segregated ZnO crystallites at higher pH.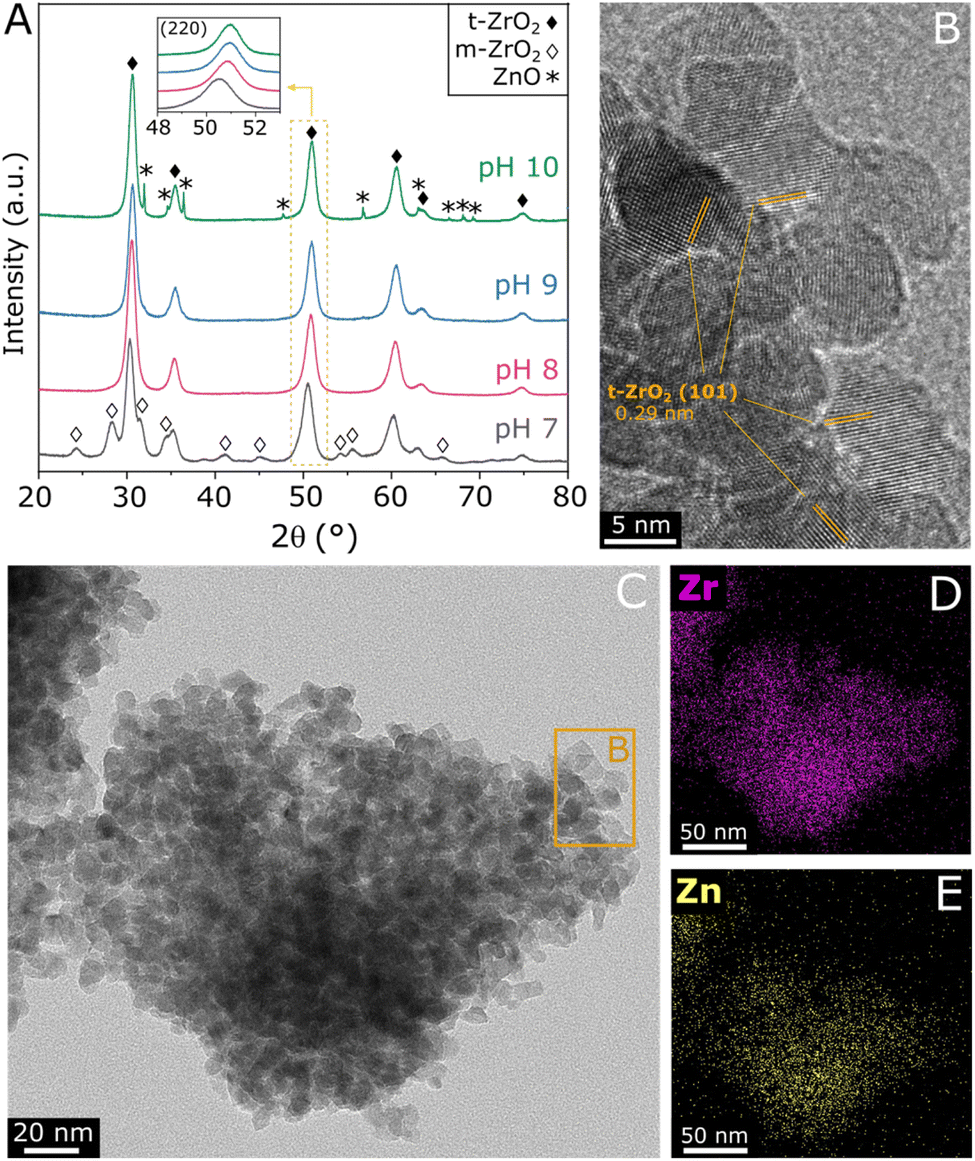 | ||
| Fig. 1 XRD patterns of ZnZrOx produced at different synthesis pH values and calcined at 500 °C (A), TEM image of ZnZrOx produced at pH 8 (B and C) and TEM-EDX mapping images showing the Zr and Zn distribution in the same region covered by C (D andE). | ||
Following the verification of clear influences of synthesis pH on the crystalline structure of ZnZrOx, surface properties of the material have been evaluated by SEM, N2 physisorption, XPS and CO2-TPD. According to the SEM micrographs shown in Fig. 2 and S1–S3,† all samples exhibit predominantly agglomerated particles with a similarly rough surface regardless of synthesis pH, which can be associated with the zirconia structure given its dominance in XRD results. As shown in Fig. 2A and B, such morphology is largely present in the pH10 sample, although here it also coexists with the characteristic rod-like ZnO particles31 shown in Fig. 2C. Fig. 2D illustrates the N2 sorption isotherms and pore size distributions of all the samples. All samples exhibit type-VI isotherms along with a type-H2 hysteresis loop. This characteristic behavior indicates the aggregation of ZrO2 particles, leading to the creation of interparticle voids within the materials.32 Elevating the pH synthesis values from 7 to 10 induced a noticeable shift in the position of the hysteresis loop towards higher relative pressures, suggesting a subtle enlargement in mesopore sizes within the material structure. Nevertheless, only limited changes in surface area are observed, as it gradually decreases from 70 m2 g−1 at pH 7 to 63 m2 g−1 at pH 10. As shown in Fig. 2E, all samples display narrow distributions of pore size, suggesting that a uniformity of pore sizes was achieved through the hydrothermal synthesis method.
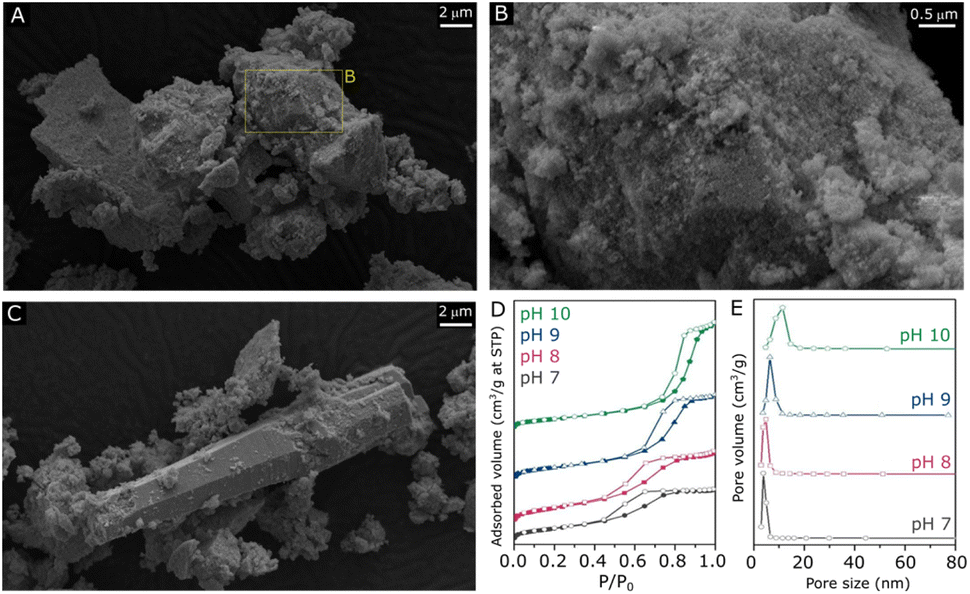 | ||
| Fig. 2 SEM micrographs of ZnZrOx produced at pH10 under different magnifications (A and B) and the region including rod-like particles (C). N2 sorption isotherms (D) with respective pore size distributions of ZnZrOx produced at pH 7, 8, 9 and 10 (E). | ||
In order to assess the surface composition of the investigated materials, XPS analysis was performed. According to high-resolution spectra in Fig. S4,† all samples present similar Zr 3d doublets with Zr 3d5/2 and Zr 3d3/2 in the region of 183.1 and 185.5 eV, corresponding to Zr4+ in tetragonal ZrO2.33 Accordingly, O 1s located at approximately 530.4 eV indicates that lattice oxygen33 is by far more abundant than adsorbed oxygen species.34 Moreover, the Zn 2p3/2 peak is verified at 1022.0 eV, as typically observed for Zn2+ species.35 Due to the severe differential charging36 experienced by the ZnZrOx samples as a result of their insulating character and surface roughness, the tailing observed in the high-resolution XPS spectra prevents fitting or precise quantification in these spectra. For this reason, quantification of Zn/Zr and O/Zr surface atomic ratios was conducted in the survey spectra, considering O 1s, the average of Zn 2p3/2 and Zn 3p, as well as the average of Zr 3d and Zr 3p3/2 for higher precision. Accordingly, Fig. 3A presents these selected regions in the survey spectra, with the respective Zn/Zr and O/Zr surface molar ratios shown in Fig. 3B. Hydrothermal synthesis under neutral pH conditions results in a low surface Zn/Zr ratio of 0.05, which can be correlated with the observation of abundant monoclinic ZrO2 by XRD, as this phase is less likely to accommodate Zn2+.16 In contrast, the pH 8 sample shows a greatly increased Zn/Zr ratio of 0.72, which is slightly decreased to 0.71 and 0.69 upon increasing pH to 9 and 10, respectively. Despite the observed differences in crystal structure upon increasing synthesis pH from 8 to 10, these results suggest that the surface has a similar content of surface Zn2+ within this basic pH range. Also in Fig. 3B, the surface O/Zr ratio presents a steady increase from 1.30 to 1.44 between pH 7 and 10, giving an indication that hydrothermal conditions with abundant OH− may provide more surface oxygen for the ZnZrOx solid solution during calcination. Furthermore, even though Na+ from NaOH has been previously suggested to stabilize the t-ZrO2 structure, surface Na was not observed by XPS, as shown in Fig. S5.† However, the Cl 2p peak at 198 eV indicates surface chlorine species in all samples, as a residue from the metal chloride precursors utilized in the hydrothermal synthesis.37 Although the effect of such species has not been deeply explored in ZnZrOx catalysts for CO2 hydrogenation to methanol, surface chlorine in Pd/ZnO was suggested to block some active sites and therefore offer a detrimental effect to catalytic activity in comparison to chlorine-free catalysts.38
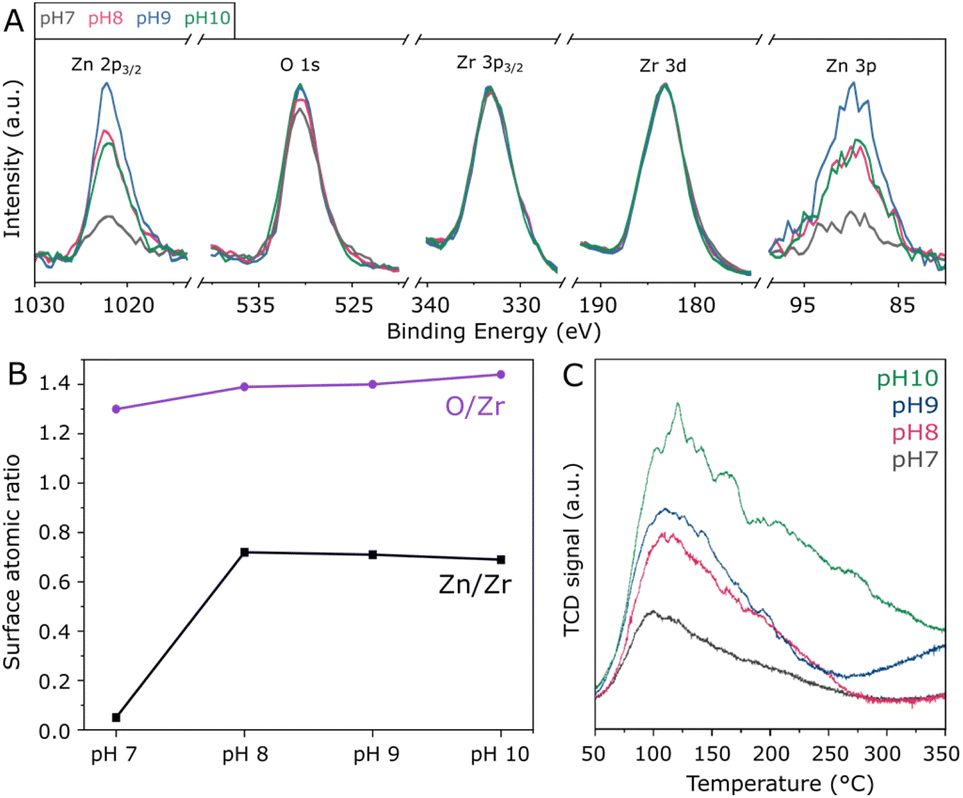 | ||
| Fig. 3 Survey XPS spectra of ZnZrOx normalized by the Zr 3d peak, showing the Zn 2p3/2, O 1 s, Zr 3p3/2, Zr 3d and Zn 3p regions (A) with the respective quantification of the Zn/Zr and O/Zr surface molar ratios (B); CO2-TPD profiles of the samples produced at pH 7 to 10 (C). | ||
Given the pivotal role of CO2 activation in its catalytic conversion, the CO2-TPD profiles of all ZnZrOx samples are investigated, as shown in Fig. 3C. In all cases, a main desorption peak is observed below 200 °C, corresponding to the desorption of CO2 from weakly basic sites.32 Interestingly, the material produced at pH 7 presents the lowest CO2 adsorption capacity despite having the highest surface area among the investigated samples. This observation cannot be directly associated with the presence of m-ZrO2, as this phase typically interacts more strongly with CO2 than t-ZrO2.39 As the synthesis pH rises from 7 to 10, this desorption peak position becomes more intense and is gradually shifted from 100 to 120 °C, indicating an increase in its basicity. Concurrently, higher synthesis pH values lead to increased desorption between 200 and 350 °C, indicating higher densities of moderately basic sites. This can be considered more relevant for CO2 hydrogenation to methanol,21 as this reaction is typically conducted around 250 °C. Such positive correlation between synthesis pH and CO2 adsorption capacity could be connected with the formation of the ZnZrOx solid solution, which may show higher CO2 adsorption with respect to pure ZrO2.40
The catalytic hydrogenation of CO2 to methanol over the ZnZrOx samples was tested in a fixed-bed reactor at various temperatures (250–290 °C), as summarized in Fig. 4. The material produced under pH 7 exhibits poor catalytic activity with CO2 conversion below 1%, as a possible result of the low surface Zn content observed by XPS, since the Zn2+–O–Zr4+ linkages have been widely recognized as the active sites in ZnZrOx catalysts. However, the pH 8 sample shows a sharply enhanced catalytic activity over the entire temperature range, as a likely effect of the improved incorporation of Zn2+ in the tetragonal zirconia phase. In this case, a CO2 conversion of 1.5% and a methanol selectivity of 84% are achieved at 250 °C, and increasing the reaction temperature to 290 °C results in a CO2 conversion of 4.5% and a methanol selectivity of 65%, as higher temperatures simultaneously favor the kinetics for CO2 hydrogenation to methanol and the endothermic production of CO via Reverse Water-Gas Shift reaction. Interestingly, increasing synthesis pH to 9 and 10 leads to progressively higher CO2 conversions. As shown in Fig. 4D, at the highest pH value a CO2 conversion of 3.4% is obtained along with a methanol selectivity of 95% at 250 °C. Such value is in a similar range to the previously reported performance at 260 °C/2.0 MPa using ZnZrOx produced by co-precipitation.11 Accordingly, an expressive increase in CO2 conversion to 8.1% is observed at 290 °C, although this coincides with a decreased methanol selectivity of 40%.
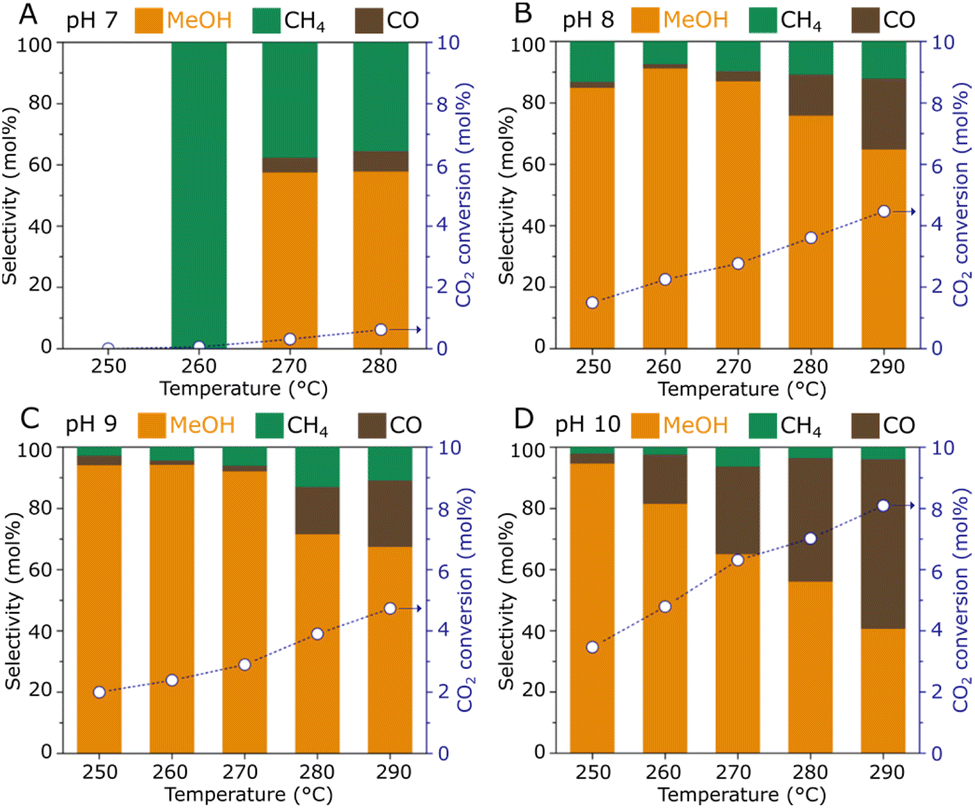 | ||
| Fig. 4 CO2 hydrogenation performance of ZnZrOx catalysts produced under synthesis pH 7 (A), 8 (B), 9 (C), and 10 (D). Reaction conditions: 0.5 g catalyst, 2.0 MPa, CO2/H2 = 1/3, 5 mL min−1. | ||
In view of the catalytic performance of ZnZrOx produced via hydrothermal synthesis at distinct pH values between 7 and 10, a clearly positive correlation between synthesis pH and CO2 conversion is verified. As pH values increase, catalytic activity at 250 °C is enhanced with high methanol selectivity, while at higher temperatures this also coexists with an enhanced production of CO. At pH 10, the higher CO production above 250 °C could be associated with the additional contribution of ZnO, similarly as previously observed when ZnZrOx is produced with an exceedingly high Zn content.11 Although the formation of abundant Zn2+/t-ZrO2 sites is important in this catalyst, surface characterization suggests that in this study, catalytic activity cannot be simply associated with surface area and the Zn surface content, as these parameters do not increase between pH 8 and 10. This is consistent with the observation that H2 activation may not simply require Zn2+ atoms but rather Zn2+–O2− pairs, as suggested by previous studies.19,20 Therefore, the enhanced CO2 conversion to methanol may be related to the stronger CO2 adsorption capacity21 and with the slightly higher lattice oxygen content at the catalyst surface, which may in turn benefit H2 dissociation.19,20 Despite the correlation of synthesis pH with catalytic activity for CO2 hydrogenation to methanol, the observed trends indicate that further increasing pH beyond 10 could lead to a ZnZrOX/ZnO system with lower selectivity due to the production of CO as a byproduct.
To obtain further insights into the hydrothermal synthesis of ZnZrOx catalysts, an analogous preparation procedure was followed using NH4OH to achieve pH 10, as an alternative to NaOH. A comparison of such materials, shown in Fig. 5A and B, shows similar catalytic activities at 250 °C, as the catalyst produced with NH4OH presents an unchanged CO2 conversion of 3.4% with a slightly lower methanol selectivity of 90%. At reaction temperatures higher than 250 °C, the material shows similar methanol production but improved selectivity due to the lower production of CO. Corresponding to such similarities, ZnZrOx produced at pH 10 with NH4OH and NaOH show similarly high O/Zr ratios of 1.47 and 1.44, calculated from the XPS spectra in Fig. 5C. Furthermore, the comparable CO2-TPD profiles in Fig. 5D indicate a similarly high density of weakly and moderately basic sites, with respect to ZnZrOx produced at lower pH. Interestingly, the XRD patterns in Fig. 5E indicate that the main contribution consists of t-ZrO2 in both catalysts, but when NH4OH is used, m-ZrO2 appears as a minor phase instead of ZnO. Given the most likely negative effect of ZnO, such differences in crystallinity suggest that the absence of ZnO may explain the higher methanol selectivity above 250 °C in the catalyst produced with NH4OH. Although this finding hints at a possible improvement by further increasing synthesis pH using NH4OH, reaching values above 10 was not feasible without drastically altering the content of the remaining reactants involved in the hydrothermal synthesis of ZnZrOx.
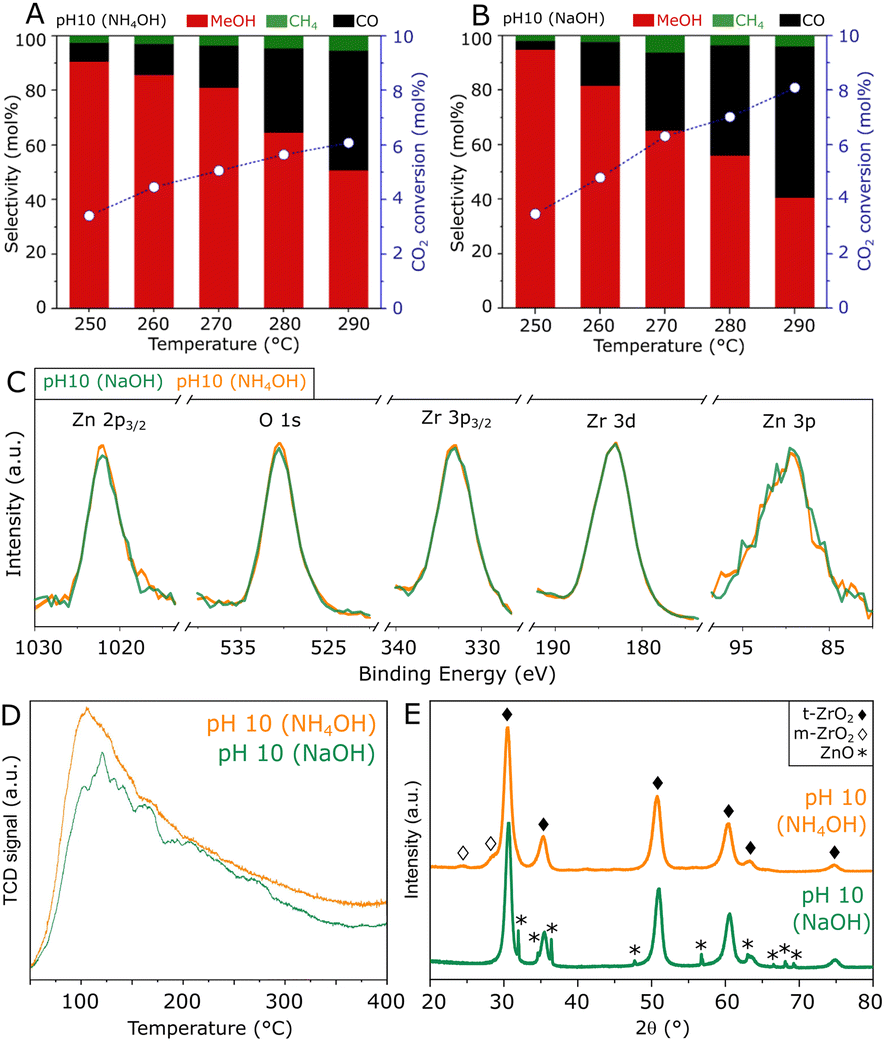 | ||
| Fig. 5 Comparison of ZnZrOx catalysts produced under synthesis pH 10 with the distinct additives NH4OH and NaOH, in terms of CO2 hydrogenation performance (A and B), Zn 2p3/2, O 1s, Zr 3p3/2, Zr 3d and Zn 3p XPS spectra (C), CO2-TPD profiles (D) and XRD patterns (E). | ||
In summary, these results emphasize that high surface oxygen content and basicity for CO2 activation are key features behind the high catalytic activity of ZnZrOx produced via hydrothermal synthesis at pH 10. As expected, these findings confirm that the Zn2+–ZrO2 solid solution is the active phase in the catalyst, and the presence of bulk ZnO may not contribute to methanol production in this case, even though the presence of ZnOx clusters within the solid solution cannot be ruled out.17,18 In fact, bulk ZnO is shown to have a detrimental effect on methanol selectivity as it promotes the formation of CO as a byproduct.
In order to obtain insights on the reaction mechanism of CO2 hydrogenation to methanol over the ZnZrOx catalyst produced at pH 10, the surface intermediates involved in the reaction were monitored by an in situ DRIFTS experiment. Spectra were collected at 250 °C under CO2/H2 flow at ambient pressure, as shown in Fig. 6A. Specifically, the peaks observed at 2978, 2881, 2737, 1385, and 1373 cm−1, which appear in the initial 2 minutes of the experiment, can be ascribed to formate species (HCOO*). Subsequently, after approximately 25 min, additional peaks corresponding to CH3O* are observed at 2935, 2823, and 1049 cm−1, with progressively increased intensity over the reaction time. These results suggest that CH3O* species are generated through the stepwise hydrogenation of HCOO* species, as part of a formate reaction pathway previously suggested in other ZnZrOx catalysts.11,22,41
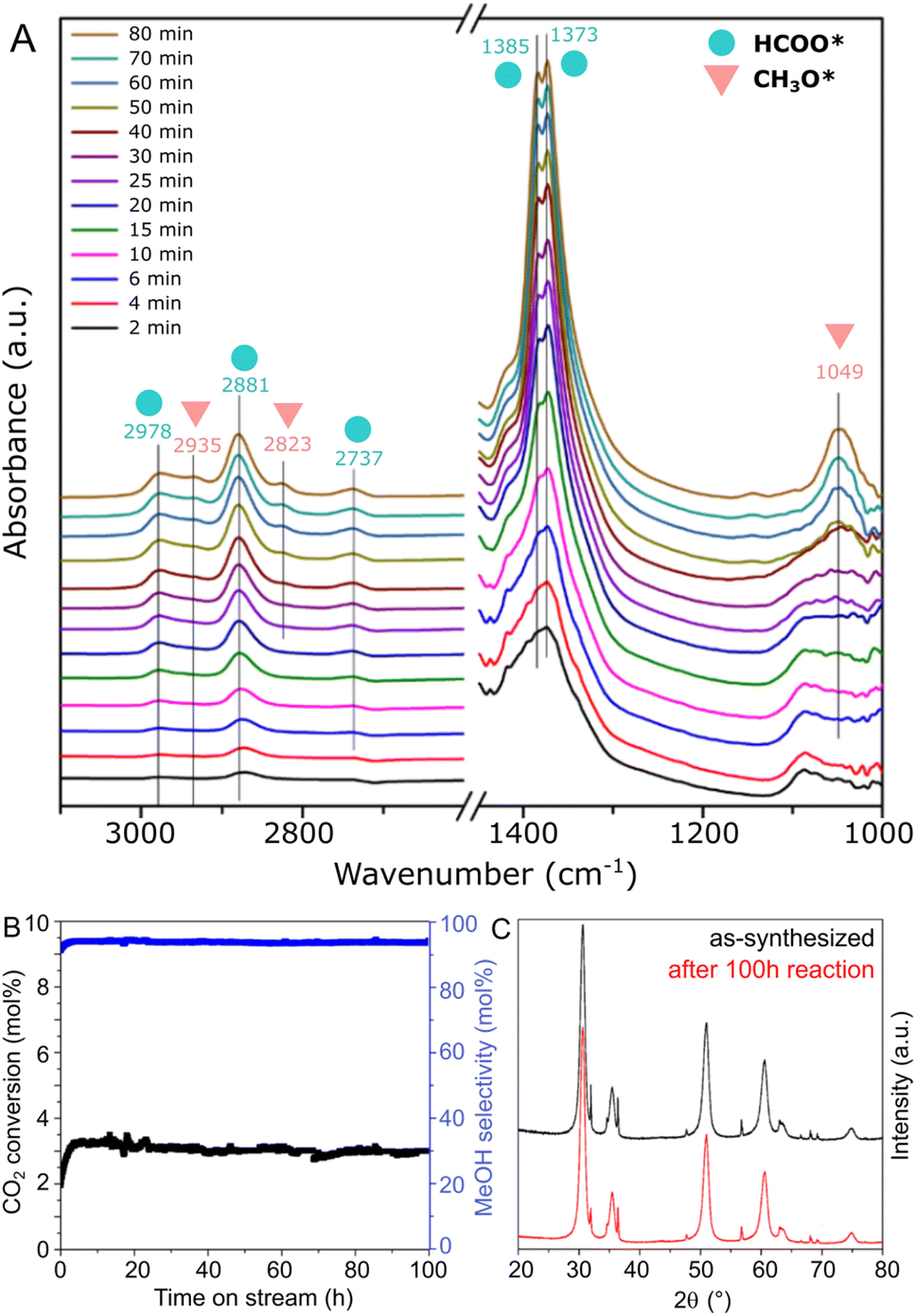 | ||
| Fig. 6 In situ DRIFTS results of CO2 hydrogenation conditions at 250 °C (A) and catalytic stability test at 250 °C using the ZnZrOx catalyst produced at pH 10 with NaOH (B), with the respective XRD patterns of as synthesized and spent material (C). | ||
In Fig. 6B, the stability of the ZnZrOx catalyst is evaluated through a 250 °C reaction carried out during 100 h. The material demonstrates remarkable stability over the test period, with no deactivation trend observed in both CO2 conversion and methanol selectivity over the 100 hours duration. Correspondingly, Fig. 6C shows that the XRD patterns of spent and fresh catalyst are highly similar, apart from a minor decrease in the peak intensities of ZnO. This change might be attributed to a slight amorphization of ZnO facilitated by the reducing reaction conditions. Nevertheless, given the key role of the Zn2+/t-ZrO2 solid solution for CO2 hydrogenation to methanol, these patterns suggest high catalyst robustness under reaction conditions.
Conclusions
In the hydrothermal synthesis of ZnZrOx catalysts for CO2 hydrogenation to methanol, altering synthesis pH significantly impacts both the structural and catalytic properties of the material. Basic pH conditions promote the formation of tetragonal ZrO2 and facilitate the incorporation of Zn2+ into this phase. As synthesis pH is increased from 8 to 10, the catalysts show a marked improvement in methanol production at 250 °C, while higher temperatures favor CO production. The correlation between hydrothermal synthesis pH and catalytic activity may be associated with the improved surface basicity verified by CO2-TPD and the slight increase in the surface oxygen content observed by XPS, given the decisive effect of Zn2+–O2− pairs on H2 activation.20 At reaction temperatures above 250 °C, the selectivity of CO2 hydrogenation to methanol can be improved by utilizing NH4OH as an alternative to NaOH in the hydrothermal synthesis at pH 10, thus suppressing the formation of bulk ZnO. In summary, these findings suggest that the hydrothermal approach is an effective and versatile method for producing ZnZrOx catalysts for CO2 hydrogenation to methanol. Nevertheless, given the inherently limited hydrogen activation in ZnZrOx, achieving catalytic activity superior to commercial Cu-based catalysts will likely require further strategies, such as the optimization of Zn2+ dispersion42 or the addition of metallic nanoparticles13,43 and promoters,22 which could offer new concepts for the hydrothermal synthesis of ZnZrOx-based catalysts.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the CO2Refinery doctoral school, the Analytical Instrumentation Center (AIC), the X-ray Center (XRC) and the University Service Facility for Transmission Electron Microscopy (USTEM) at TU Wien. This research was funded in part by the Austrian Science Fund (FWF) 10.55776/F81. I. Rakngam is supported by the Royal Golden Jubilee PhD Program (Grant No. PHD/0221/2558) from the Thailand Research Fund (TRF) and the National Research Council of Thailand (NRCT). For open access purposes, the author has applied a CC BY public copyright license to any author accepted manuscript version arising from this submission.References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS
.
- H. Dong, M. Jung, Y. Zhang, S. Wang and S. Ding, Mol. Catal., 2024, 560, 114133 CrossRef CAS
- F. Zeng, C. Mebrahtu, X. Xi, L. Liao, J. Ren, J. Xie, H. J. Heeres and R. Palkovits, Appl. Catal., B, 2021, 291, 120073 CrossRef CAS
- Z. Ma and M. D. Porosoff, ACS Catal., 2019, 9, 2639–2656 CrossRef CAS
- P. Schwiderowski, H. Ruland and M. Muhler, Curr. Opin. Green Sustainable Chem., 2022, 38, 100688 CrossRef CAS
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Ind. Eng. Chem. Res., 2019, 58, 9030–9037 CrossRef CAS
- J. T. Sun, I. S. Metcalfe and M. Sahibzada, Ind. Eng. Chem. Res., 1999, 38, 3868–3872 CrossRef CAS
- S. Ghosh, V. Uday, A. Giri and S. Srinivas, J. Cleaner Prod., 2019, 217, 615–626 CrossRef CAS
- D. S. Marlin, E. Sarron and Ó. Sigurbjörnsson, Front. Chem., 2018, 6, 446 CrossRef CAS
- S. Kleiber, A. Loder, M. Siebenhofer, A. Böhm and S. Lux, Chem. Ing. Tech., 2022, 94, 701–711 CrossRef CAS
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef
- Z. Han, C. Tang, F. Sha, S. Tang, J. Wang and C. Li, J. Catal., 2021, 396, 242–250 CrossRef CAS
- K. Lee, P. C. D. Mendes, H. Jeon, Y. Song, M. P. Dickieson, U. Anjum, L. Chen, T.-C. Yang, C.-M. Yang, M. Choi, S. M. Kozlov and N. Yan, Nat. Commun., 2023, 14, 819 CrossRef CAS
- Z. Feng, C. Tang, P. Zhang, K. Li, G. Li, J. Wang, Z. Feng and C. Li, J. Am. Chem. Soc., 2023, 145, 12663–12672 CrossRef CAS
- S. Tada, N. Ochiai, H. Kinoshita, M. Yoshida, N. Shimada, T. Joutsuka, M. Nishijima, T. Honma, N. Yamauchi, Y. Kobayashi and K. Iyoki, ACS Catal., 2022, 12, 7748–7759 CrossRef CAS
- G. Štefanić, S. Musić and M. Ivanda, J. Mol. Struct., 2009, 924, 225–234 CrossRef
- D. Salusso, E. Borfecchia and S. Bordiga, J. Phys. Chem. C, 2021, 125, 22249–22261 CrossRef CAS
- X. Zhang, G. Zhang, X. Zhou, Z. Wang, Y. Liu, J. Zhu, C. Song and X. Guo,
Ind. Eng. Chem. Res., 2023, 62, 21173–21181 CrossRef CAS
- X. Mao, Y. Zhang, Y. Xu, Y. Zhou, K. Zhuang, K. Shen and S. Ding, Catal. Sci. Technol., 2024, 14, 419–430 RSC
- K. Lee, M. P. Dickieson, M. Jung, Y. Yang and N. Yan, ACS Catal., 2024, 14, 3074–3089 CrossRef CAS
- Y. Shen, J. Yu, S. Ji, F. Hong, Q. Guo and D. Mao, Catal. Lett., 2024, 154, 3749–3758 CrossRef CAS
- F. Sha, C. Tang, S. Tang, Q. Wang, Z. Han, J. Wang and C. Li, J. Catal., 2021, 404, 383–392 CrossRef CAS
- H. Wang, G. Li, Y. Xue and L. Li, J. Solid State Chem., 2007, 180, 2790–2797 CrossRef CAS
- R. C. Garvie, J. Phys. Chem., 1965, 69, 1238–1243 CrossRef CAS
- H. Xie, J. Lu, M. Shekhar, J. W. Elam, W. N. Delgass, F. H. Ribeiro, E. Weitz and K. R. Poeppelmeier, ACS Catal., 2013, 3, 61–73 CrossRef CAS
- E. V. Dudnik, Powder Metall. Met. Ceram., 2009, 48, 238–248 CrossRef CAS
- M. Jay Chithra, M. Sathya and K. Pushpanathan, Acta Metall. Sin., 2015, 28, 394–404 CrossRef CAS
- G. Štefanić, S. Popović and S. Musić, Thermochim. Acta, 1997, 303, 31–39 CrossRef
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef
- W. Li, K. Wang, J. Huang, X. Liu, D. Fu, J. Huang, Q. Li and G. Zhan, ACS Appl. Mater. Interfaces, 2019, 11, 33263–33272 CrossRef CAS
- C. R. Chandraiahgari, G. De Bellis, P. Ballirano, S. K. Balijepalli, S. Kaciulis, L. Caneve, F. Sarto and M. S. Sarto, RSC Adv., 2015, 5, 49861–49870 RSC
- C. Temvuttirojn, Y. Poo-arporn, N. Chanlek, C. K. Cheng, C. C. Chong, J. Limtrakul and T. Witoon, Ind. Eng. Chem. Res., 2020, 59, 5525–5535 CrossRef CAS
- P. Lackner, Z. Zou, S. Mayr, U. Diebold and M. Schmid, Phys. Chem. Chem. Phys., 2019, 21, 17613–17620 RSC
- C. Morant, J. M. Sanz, L. Galán, L. Soriano and F. Rueda, Surf. Sci., 1989, 218, 331–345 CrossRef CAS
- M. Claros, M. Setka, Y. P. Jimenez and S. Vallejos, Nanomaterials, 2020, 10, 471 CrossRef CAS
- M. A. Kelly, J. Electron Spectrosc. Relat. Phenom., 2010, 176, 5–7 CrossRef CAS
- T. Nishino, M. Saruyama, Z. Li, Y. Nagatsuma, M. Nakabayashi, N. Shibata, T. Yamada, R. Takahata, S. Yamazoe, T. Hisatomi, K. Domen and T. Teranishi, Chem. Sci., 2020, 11, 6862–6867 RSC
- H. Bahruji, M. Bowker, G. Hutchings, N. Dimitratos, P. Wells, E. Gibson, W. Jones, C. Brookes, D. Morgan and G. Lalev, J. Catal., 2016, 343, 133–146 CrossRef CAS
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17, 4297–4303 CrossRef CAS
- C. Beasley, M. K. Gnanamani, M. Martinelli, K. Góra-Marek, K. Hamano, W. D. Shafer, N. Wanninayake and D. Y. Kim, ChemistrySelect, 2019, 4, 3123–3130 CrossRef CAS
- Q. Ren, K. Yang, F. Liu, M. Yao, J. Ma, S. Geng and J. Cao, Mol. Catal., 2023, 547, 113280 CrossRef CAS
- T. Zou, T. Pinheiro Araújo, M. Agrachev, X. Jin, F. Krumeich, G. Jeschke, S. Mitchell and J. Pérez-Ramírez, J. Catal., 2024, 430, 115344 CrossRef CAS
- T. Pinheiro Araújo, G. Giannakakis, J. Morales-Vidal, M. Agrachev, Z. Ruiz-Bernal, P. Preikschas, T. Zou, F. Krumeich, P. O. Willi, W. J. Stark, R. N. Grass, G. Jeschke, S. Mitchell, N. López and J. Pérez-Ramírez, Nat. Commun., 2024, 15, 3101 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00522h |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |