
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solidifying the role of hydrogen bonds in conjugated polymers for wearable electronics
Megan M.
Westwood
 *ab and
Bob C.
Schroeder
*a
*ab and
Bob C.
Schroeder
*a
aDepartment of Chemistry, University College London, London, UK
bDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Gothenburg, Sweden. E-mail: westwood@chalmers.se
First published on 11th November 2024
Abstract
Conjugated polymers are often functionalised with hydrogen bonding motifs, to impart self-healing or stretchability for wearable electronic applications. Through refinement of characterisation techniques in recent years, the effect of these supramolecular groups has become increasingly well understood. However, challenges remain to clearly decouple the effects of functionalisation with the complex nature of this polymer class. In this perspective we examine the current methods of characterisation and discuss how focus on the mechanical properties will be beneficial for setting out future design rules. Finally, we investigate the use of these techniques within the reported literature to highlight trends in the critical properties of the materials.
Introduction
In less than a century, global healthcare has been revolutionized by synthetic polymers.1 With an increasingly varied library of polymer materials and methods of processing, their application in modern medicine continues to grow.2,3 Polymers with mechanical properties analogous to human skin hold promise to realize wearable and implantable technologies that could deliver point-of-care diagnostics and rehabilitation. Their implementation promises to not only relieve strain on healthcare systems around the world but also improve the quality of treatment for patients.4,5Conjugated polymers (CPs) bring together the excellent tensile properties that are characteristic of polymers within an intrinsically semiconducting material, rendering them an ideal platform to develop wearable electronics.6,7 Over the past 20 years, research in the field of organic electronics has culminated in a thorough understanding of the intricate structure–property relationships underpinning the performance of CPs.8 Conjugated materials, composed of alternating single and double C–C bonds, allow charge carriers to move through the material due to delocalization of the π-orbitals. Microstructural order of a CP therefore dictates the efficiency of this process, where enhanced molecular orbital overlap facilitates intra- and intermolecular charge transfer along the plane of conjugation or by hopping between adjacent chains respectively.9 Accordingly, careful design and synthetic development has led to a vast range of structures that can be suited to a multitude of applications, including organic light emitting diodes (OLEDs), photovoltaics (OPVs), sensing arrays and field-effect transistors (OFETs).10–13 The active layer in these systems are typically CPs, where the alternating repeat units differ in their electronic properties to create low bandgap donor–acceptor copolymers. Additionally, the polymer chains are functionalised with solubilizing side chains to ensure adequate processability of the material. These attributes offer a high degree of versatility when functionalizing CPs, allowing tuning of the optoelectronic properties through sidechain or backbone engineering.14–16 By extension of these design principles, the focus of developing wearable electronics is to introduce skin-like properties to CPs, such that a device can conform under continuous strain whilst maintaining its functional properties and repair itself in the event of structural damage.7
An attractive strategy for enhancing intrinsic stretchability and introducing healing functionality within a CP is through addition of supramolecular interactions.17,18 Non-covalent bonds can act as energy dissipation mechanisms, to retain and restore the materials integrity, in terms of mechanical and electronic performance, when subject to strain. Hydrogen bonding has most frequently been employed as the supramolecular mechanism in this domain, because of the dynamic nature and high degree of directionality of the bonds.19,20 Although these approaches have successfully been extended to incorporate supramolecular interactions through blending with elastomeric materials, understanding the influence of crosslinkers can become increasingly complicated and herein we focus our discussion to intrinsic functionalisation routes.21,22 Introducing hydrogen bonding groups into a CP matrix is achieved through either (a) embedding the moieties within sidechains or (b) as conjugation breakers (CBs) in the polymer backbone. Understanding the effect of these interactions on the polymer structure, both on the microscale (optoelectronic properties) and macroscale (tensile properties) is a key focus for further developing these materials. However, despite systematic approaches being taken throughout the literature, disentangling the effect of hydrogen bonds on the structure–property relationships in CPs remains a challenge.23 This can be attributed to several factors, originating from variation in synthetic preparation and difficulty in accurately characterizing the materials where the effect of hydrogen bonds cannot easily be separated from other changes in physical properties. In this perspective we present a discussion on the advantages and limitations of the current characterisation techniques, in terms of spectroscopic and thermomechanical analysis, used to investigate the critical properties associated with developing skin-like conjugated polymers. As this application requires the material to maintain both good electronic properties, as well as mimic the robust mechanical properties of skin, uniting these techniques is paramount to decoupling the physical effects of the polymer and structural properties as a result of π-stacks and the introduced hydrogen bonds. We then explore the literature focused on functionalising CPs with H-bonds, with the aim of developing wearable electronics and discuss the future outlook for the design and investigation of these materials.
Characterizing hydrogen bonds
Spectroscopic analysis
As defined by the Internation Union of Pure and Applied Chemistry (IUPAC), “the hydrogen bond is an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H, and an atom or a group of atoms in the same or different molecule, in where there is evidence of bond formation”.24 The interaction between the proton donor and acceptor (Y) causes a lengthening of the X–H bond and reduction in the atomic spacing of H and Y relative to each other (depicted in Fig. 1). These attributes of hydrogen bond formation are therefore traditionally studied with Fourier-Transform Infrared spectroscopy (FTIR) and high resolution nuclear magnetic resonance (NMR) spectroscopy to monitor changes in the bond stretching frequencies and chemical environment respectively.25 As the length of the H-bond decreases, the nucleus of the binding hydrogen becomes increasingly deshielded due to loss of surrounding electronegativity.26 In 1H NMR this is observed as a downfield shift in the H-bonding proton peak and a redshifted (and potentially broadened) band in the FTIR spectrum. | ||
| Fig. 1 Schematic of hydrogen bonding between an amide (CONH) and water (H2O) molecules, where the proton donor X–H is OH or NH and the electronegative acceptor (Y) is O. | ||
Despite H-bonds holding the potential to be very strong (reaching 40 kcal mol−1 for an isolated interaction), when implemented within a polymer matrix, probing these changes can become difficult.25 Pre-existing challenges in characterising polymers by these methods are often further exaggerated in conjugated systems, due to their high side chain density and large degree of π-aggregation between neighbouring polymer chains, causing poor signal resolution for the band or peak of interest.27 Probing H-bonding interactions through 1H NMR has been employed within several studies, where a dissolved polymer is subject to temperature or concentration dependent measurements, that in the latter case can be used to measure the association constant (Ka) of the H-bond motif. Though these techniques can detect the formation of H-bonds, the environmental conditions of the measurement (e.g. solvent polarity, acidity, temperature) can not only affect the strength of the supramolecular interaction but the aggregation state of the polymer in solution itself.28 The former is of particular importance, whereby the degree of H-bonding may be suppressed through competition with solvent dipoles or bond lengthening at elevated temperatures and vice versa. By instead analysing hydrogen bonds in the solid-state (whilst under controlled atmospheric conditions), they can be probed with little deviation from the intended configuration of CPs when later implemented in flexible devices. This has been achieved in the case of FTIR measurements in recent studies, where bathochromic or hypochromic shift in the associated vibrational stretches can be observed.29–34 One limitation to this technique arises from the functional groups that are frequently incorporated in the conjugated backbone of CPs, where electron deficient carbonyl groups (e.g. amide, imide) can act as H-bond acceptors and contribute to the observed spectroscopic shifts.8,35
An alternative route to experimentally determine hydrogen bonding interactions is through means of crystallography.36,37 However, in the case of CPs, which lack long range crystalline order, these techniques become limited. For semicrystalline CPs, grazing incidence wide angle scattering (GIWAXS) is often employed to study the crystalline domains in films on the nanometre scale, as well as the π–π and lamellar stacking distances. Additional comparisons can often be made between polymers within a series, by determining the relative degree of crystallinity (rDoC) as shown in Table 1.38 Structural analysis of this type can provide insight into the effect of hydrogen bonding motifs on the crystallites, when compared with a parent polymer that incorporates analogous characteristics (i.e. molecular size and conformation) as the supramolecular motif under study with the absence of H-bonds. A promising alternative could be the employment of solid-state NMR (ssNMR) to determine the presence of intra-/intermolecular H-bonds, as has previously been demonstrated for π–π interactions in a cyclopentadithiophene (CDT) and benzothiadiazole (BT) copolymer (CDT-BT).39 Although this technique retains high sensitivity at low degrees of structural order, a multifaceted approach including computational modelling and X-ray scattering is required thus limiting accessibility.40
| Ref. | Copolymer | Sidechain or conjugation breaker | H-bonding motif | Number average molecular weight, Mn (kDa) | Weight average molecular weight, Mw (kDa) | mol% | R1 | R2 | T gα (sidechain) (°C) | T gβ (backbone) (°C) | Elastic Modulus (MPa)a | Crack-onset strain, εc (%) | μ H, average | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ε = 0% | ε = 50% | ε = 100% | |||||||||||||
| a Approximate values (denoted with ∼) are extracted from plots, in the absence of reported numerical values. | |||||||||||||||
| 41 | DPP-BT | SC | Urea | 18.8 | 59 | 0 | — | — | 0.40 | ||||||
| 28.4 | 75.5 | 3 | C5H10 | C6H13 | 1.10 | ||||||||||
| 19.8 | 46.1 | 5 | 1.60 | ||||||||||||
| 16.6 | 42.2 | 9 | 5.70 | ||||||||||||
| 29 | DPP-TVT | SC | Amide | 46.9 | 2.1 | 0 | — | — | 0.25 ± 0.02 | ||||||
| 36.5 | 2.7 | 5 | C5H10 | C6H13 | 0.58 ± 0.21 | ||||||||||
| 37.6 | 2.3 | 10 | C5H10 | C6H13 | 0.23 ± 0.35 | ||||||||||
| 9.9 | 2.4 | 20 | C5H10 | C6H13 | 0.12 ± 0.03 | ||||||||||
| 5.7 | 2.8 | 30 | C5H10 | C6H13 | 0.06 ± 0.01 | ||||||||||
| 29,42 | DPP-TVT | SC | Amide | 46.9 | 101.5 | 0 | — | — | 203 | 30 | 0.25 ± 0.02 | ||||
| 36.5 | 98.7 | 5 | C5H10 | C6H13 | 170 | 40 | 0.58 ± 0.21 | ||||||||
| 37.6 | 85.2 | 10 | 115 | 75 | 0.23 ± 0.07 | 0.016 ± 2.0 × 10−4 | 1.6 × 10−4 ± 2.0 × 10−5 | ||||||||
| 10.1 | 23.3 | 20 | 106 | 50 | 0.12 ± 0.03 | ||||||||||
| 33 | DPP-TT | SC | Urethane | 164 | 183.7 | 100 | C10H20 | CH2CH(C8H19)(C10H21) | 72 | 50 | 0.0054 | 4.0 × 10−3, 2.8 × 10−4 | 4.0 × 10−4, 3.9 × 10−4 | ||
| DPP-BT | 154 | 177.1 | 54 | 75 | 0.010 | 0.02, 0.01 | 0.01, 0.01 | ||||||||
| DPP-TVT | 171 | 188.1 | 48 | 112.5 | 0.016 | 0.02, 0.01 | 0.02, 0.01 | ||||||||
| 43 | DPP-TVT | SC | Linear | 31.4 | 131.9 | 0 | C12H25 | — | 381 ± 21.9 | 0.25 | 0.15 ± 0.04 | 0.04 ± 9 × 10−3 | |||
| Amide | 35.7 | 2.1 | 10 | C5H10 | C6H13 | 357 ± 18.5 | 0.5 | 0.20 ± 0.02 | 0.12 ± 0.05 | ||||||
| Urea | 31.3 | 1.8 | 10 | C5H10 | C6H13 | 372 ± 7.6 | 0.5 | 0.02 ± 2.0 × 10−3 | 1 × 10−3 ± 3.0 × 10−5 | ||||||
| 30 | IDT-BT | SC | — | 240 | 622 | 0 | C16H33 | 102 ± 1.0 | 1.48 ± 0.03 | ||||||
| TIPS | 217.4 | 684.4 | 5 | C16H32 | 111 ± 9.8 | ||||||||||
| 177.9 | 434.4 | 10 | 122 ± 15.6 | ||||||||||||
| 313.4 | 816.9 | 20 | 109 ± 5.4 | 1.12 ± 0.13 | 1.10 ± 0.14 | 0.75 ± 0.03 | |||||||||
| Hydroxyl | 176.3 | 525.5 | 5 | C16H32 | 119 ± 3.4 | ||||||||||
| 182.2 | 608.9 | 10 | 137 ± 18.8 | ||||||||||||
| 181.9 | 603.1 | 20 | 112 ± 4.0 | 0.47 ± 0.04 | 0.33 ± 0.02 | 0.34 ± 0.02 | |||||||||
| UPy | 144 | 405.6 | 5 | C16H32OCONHC4H8 | CH3 | 100 ± 1.8 | 0.34 ± 0.02 | 0.28 ± 0.02 | 0.23 ± 0.02 | ||||||
| 198.5 | 643.1 | 10 | 71 ± 4.7 | ||||||||||||
| 164.6 | 511.3 | 20 | 90 ± 14.5 | ||||||||||||
| 34 | DPP-TVT | SC | PDCA | 20.2 | 42.4 | 0 | — | — | −41 | 134 | 0.95 ± 0.15 | ||||
| 19.3 | 42.4 | 30 | C10H20 | C3H8 | −37 | 103 | 0.60 ± 0.25 | ||||||||
| 17.5 | 42 | 60 | C10H20 | C3H8 | −41 | 108 | 0.55 ± 0.17 | ||||||||
| CB | 20 | 40 | 10 | CH2 | CH2 | −35 | 113 | ||||||||
| 19.2 | 42 | 30 | CH2 | CH2 | −30 | 103 | |||||||||
| 10.2 | 25.2 | 60 | CH2 | CH2 | −33 | 103 | |||||||||
| 44 | DPP-TVT | CB | PDCA | 20.4 | 65.4 | 0 | — | — | 5–10 | 1.87 | |||||
| 19.7 | 61.4 | 5 | CH2 | CH2 | 50 | ∼1.64 | |||||||||
| 16.2 | 49.1 | 10 | CH2 | CH2 | 120 | 1.32 (1.28) | |||||||||
| 13.4 | 35.5 | 20 | CH2 | CH2 | 125 | 0.58 | |||||||||
| 45 | DPP-TVT | CB | Carbonate | 23.2 | 76.1 | 10 | C2H4 | C2H4 | −57.6 | 404 | 20 | 0.06 ± 0.02 | |||
| 20.2 | 61.8 | 10 | C2H4OC2H4 | C2H4OC2H4 | −53.4 | 396 | 20 | 0.15 ± 0.02 | |||||||
| Amide | 22.8 | 70.5 | 10 | C2H4 | C2H4 | −36.7 | 456 | 20 | 0.09 ± 6.3 × 10−3 | ||||||
| 27.2 | 76.8 | 10 | C2H4OC2H4 | C2H4OC2H4 | −40.5 | 440 | 30 | 0.29 ± 0.02 | |||||||
| Urethane | 21.1 | 63 | 10 | C2H4 | C2H4 | −38.6 | 336 | 20 | 0.09 ± 4.0 × 10−3 | ||||||
| 25.7 | 78.4 | 10 | C2H4OC2H4 | C2H4OC2H4 | −43.9 | 404 | 30 | 0.31 ± 0.01 | |||||||
| Urea | 23.6 | 98.9 | 10 | C2H4 | C2H4 | — | 440 | 40 | 0.10 ± 4.7 × 10−3 | ||||||
| 18.9 | 73.9 | 10 | C2H4OC2H4 | C2H4OC2H4 | −35.3 | 520 | 40 | 0.27 ± 0.02 | |||||||
| 32 | TDPP-Se | CB | UPy | 75.2 | 207.6 | 0 | — | C6H4 | 890 ± 45 | <30 | 0.82 | ||||
| 57.4 | 156.1 | 5 | (CH2)9C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH2)8 C(CH2)8 |
843 ± 62 | <30 | 0.38 | |||||||||
| 43 | 123.4 | 10 | 687 ± 53 | <30 | 0.21 | ||||||||||
| 53.9 | 141.8 | 20 | 404 ± 68 | <30 | 0.19 | ||||||||||
| 31 | DPP-TVT | CB | — | 15.2 | 19.1 | 0 | — | — | ∼880 | 50 | 0.23 | 0.20,0.02 | 8.0 × 10−3,7.0 × 10−3 | ||
| Glycine | 10.5 | 16.4 | 10 | C4H4S | C4H4S | ∼500 | 25 | 0.12 | 0.11,0.07 | 0.08,0.06 | |||||
| Valine | 13.3 | 18.9 | 10 | C4H4S | C4H4S | ∼740 | 75 | 0.15 | 0.10,0.10 | 1.0 × 10−3,1.0 × 10−3 | |||||
| Threonine | 12.2 | 14.6 | 10 | C4H4S | C4H4S | ∼800 | 100 | 0.10 | 3.0 × 10−3,0.04 | 2.0 × 10−3,3.0 × 10−3 | |||||
Similarly, characterisation of the ordered domains in CPs is routinely carried out through emission (photoluminescence) and absorption (UV-Vis) spectroscopy. The interaction of the sample with light is extremely sensitive to changes in aggregation between neighbouring polymer chains, providing insight into the optoelectronic properties of the material. By extension, measuring the UV-Vis absorption using linearly polarised light both parallel and perpendicular to the direction of crystallites in the film, the dichroic ratio can be measured.46 Tracing changes in the dichroic ratio upon applying strain to a polymer has been the subject of multiple studies, aimed at understanding the effect of supramolecular interactions on maintaining order in these domains and their recovery beyond the point of fracture.31,33,42,44 These measurements, coupled with understanding of the microstructural order through GIWAXS, are a powerful tool to probe the impact of hydrogen bonds on CP morphology. While these techniques offer valuable insights into understanding charge transport mechanisms in conducting polymers (CPs) and the impact of H-bond functionalisation, they are confined to the ordered regions of the material. This limitation makes it challenging to analyse high-performing copolymers that lack significant long-range order, within the measurement resolution, such as indacenedithiophene-co-benzothiadiazole (IDT-BT) (Fig. 2(c)).30 By extending the scope of analysis to test the physical properties of both the crystalline and amorphous regions in polymer films, a more realistic view on the applicability of these materials in flexible electronics can be achieved. Placing emphasis on characterizing H-bonds in the solid-state, in terms of the mechanical properties in addition to the discussed spectroscopic methods, provides the possibility to not only determine the presence of intermolecular hydrogen bonds but understand their effect on the structure over multiple length scales.
 | ||
| Fig. 2 (a) Schematic of comparative studies on the mechanical properties of a hydrogen bond functionalised CP compared with the unfunctionalized derivative (parent polymer) in dog-bone thin film samples. (b) Illustration of intermolecular hydrogen bonding between two ureidopyrimidinone (UPy) units. (c) structure of parent polymer IDT-BT and the H-bond functionalised derivative bearing UPy units within the sidechain.30 | ||
Thermomechanical analysis
Physical interactions between adjacent conjugated polymer chains can result in characteristic transitions (glass transition, melt and crystallisation) and therefore cause a CP to exhibit viscoelastic behaviour within a given range of parameters. Testing these properties uncovers a wealth of information that can aid the understanding of H-bonds within the CP matrix, when controlling other contributing polymer properties such as molecular weight and entanglement density. As a result, research in this field has centred around thermomechanical analysis for the determination of the glass transition (Tg), elastic modulus (E) and crack onset strain (εc) to demonstrate the improved ductility of functionalised CPs and consequently confirm the presence of H-bonds in solid-state.47–49Glass transition
Segments of a polymer chain can undergo translational and rotational movement, to adopt various conformations. These changes are initiated by temperature, allowing the polymer to transition from a rigid, glassy state to a more mobile, rubbery state. The temperature at which this occurs is the glass transition (Tg), which can be observed for both the side chains (Tα) and polymer backbone (Tβ).50 The glass transition is governed by several factors; in addition to molecular weight and polymer entanglement effects, degree of crystallization and crosslinking density also influence the polymer Tg.51 Therefore, resolving the changes in polymer relaxation is a useful tool to understanding the effects of introducing hydrogen bonds into the matrix.CPs can exhibit viscoelastic behaviour, and therefore will exhibit brittle mechanical properties below Tβ, thus the glass transition of the parent polymer should be carefully considered when designing polymers for flexible electronics. In an ideal case, the polymer would exist in a rubbery state at room temperature to allow movement of the chains in a flexible device architecture. Additionally, the formation of dynamic bonds between adjacent chains will be facilitated when a larger range of movement is available in the rubbery state above Tg.
Thermal measurements, such as differential scanning calorimetry (DSC), are often used to measure the Tg in bulk polymer samples. Alternatively, dynamic mechanical analysis (DMA) can be used to obtain Tg (including the secondary relaxation, Tα) from the complex dynamic modulus (E*).52 In addition to increased sensitivity in comparison to DSC, this technique (usually operated in tension mode), can be used to understand the tensile properties of the polymers in films. However, in both cases, the high sidechain density and rigid polymer backbone that is characteristic of CPs often limits the use of this technique due to the small heat capacity associated with backbone relaxation.50,53
Elastic modulus
The Young's modulus (E) describes how deformable a polymer is under strain. The stiffness of the material is an important physical property to consider for skin-like electronics as a device should possess mechanical compatibility with the local area of application upon or within the body. In alignment with changes to the Tg, E can be influenced by the presence of supramolecular interactions within the polymer. Monitoring how the addition of hydrogen bonding groups influences E within a series of polymers can therefore be useful to understanding their role in the matrix.Though it has been shown that little variance occurs in the mechanical properties of free-standing polymer films between 40–500 nm thickness, delicacy of the films results in inconsistencies between samples such as voids and pinholes.54,55 This, coupled with the brittle nature of soft polymeric materials, also causes difficulty in reliably loading the samples without exerting force that alters the conformation of the polymer in its as-cast form. Methods for evaluating the viscoelastic behaviour of CP thin films, such as buckling metrology, atomic force microscopy (AFM) and nanoindentation, use polymer samples supported by solid substrates.56,57 These methods offer the advantage of using >μm polymer films to determine E (and Tg in the latter case) however, the modulus extracted using these techniques is influenced by the interactions of the polymer with the support, depth of indentation and film thickness.58 Furthermore, in the case of AFM and nanoindentation, compressive forces are utilized to probe a localized area of the film, which limits the understanding of H-bonds on the bulk polymer through these techniques.
More recently, tensile testing of CPs has been carried out using DMA, through which strain can be exerted onto the solid sample with high resolution. Typically, the DMA is operated in tension mode where a polymer film sits between two parallel clamps. To evenly distribute strain upon the sample, a dog-bone sample configuration is optimal to measure films in this way. To accommodate the high E and brittle nature of CP films, supporting substrates are still regularly used to achieve pseudo free-standing films where the support Tg is outside of the expected range of the CP of interest, such as glass fibre or an elastomer (e.g. polyimide or polydimethylsiloxane (PDMS)).47 As an alternative to the latter film-on-elastomer (FOE) method for extracting E, a new derivative of tensile testing using pseudo free-standing films has been developed. Kim et al. presented a novel film-on-water (FOW) approach whereby the testing polymer is supported by water during tensile testing, owing to its high surface tension.59 To better mimic traditional tensile testing, Gu and coworkers have further developed the FOW technique for use with conjugated systems.48 Though these method developments advance the scope of DMA for analysing this class of materials, careful attention should be paid to ensuring that use of elastomers or water as supporting media do not interfere with the observed viscoelastic behaviour due to delamination or swelling respectively. This is of particular importance for materials functionalised with hydrogen bonding motifs, which can form competing H-bonds with the water acting as a support medium, as depicted in Fig. 1 for an amide functional group. Additionally, Lipomi and colleagues demonstrated how the apparent modulus determined by each of these techniques yields different values as a result of compressive or tensile forces applied to the films, and their influence on the aforementioned inconsistencies in polymer films.60 These findings highlight the difficulty in drawing cross-comparisons between studies focused on understanding the mechanical behaviour of CPs, particularly when also attempting to deduce the effect of H-bonds within the system, which are sensitive to the environment in which they are measured during FOW and FOE studies. More recently, Gu et al. developed a novel ‘SMART’ transfer method to measure free standing films.61 By instead employing water as a sacrificial layer prior to removal of the CP film using horizontal and vertical shearing film from an elastomeric support, the disruption to the viscoelastic response can be minimized.43 The thorough refinements discussed here for developing tensile testing techniques are pivotal for accessing the mechanical properties of CPs and hold potential to deepen knowledge around the effect of their functionalisation.
To gain perspective over the healing properties of a CP, the discussed tensile testing procedures (with careful sample and environmental parameter control) have potential to uncover the effect of supramolecular crosslinkers on the viscoelastic properties of a CP after deformation. This can be achieved by measuring the hysteresis of the engineering stress–strain response, to provide information on both the elasticity and ductility of the material, within the linear viscoelastic or plastic deformation regions respectively.48 The viscoelastic response depends on the sampling temperature; at T < Tg only short chains or sidechains are able to flow whereas at T > Tg longer segments can move in the amorphous region which could also lead to induced crystallization.62 The influence of H-bonds on the observed viscoelastic properties will therefore depend on their position in the polymer structure at a given temperature. If H-bonding conjugation breakers or sidechains reside in the amorphous region, they may contribute to the polymer's elasticity. Beyond the linear viscoelastic region, plastic deformation will occur which involves movement of amorphous and crystalline domains, and H-bonding groups in these regions can contribute to polymer's ductility. However, applying this methodology to deduce the direct effect of hydrogen bonds on elasticity and ductility after deformation remains a challenge, as comparative studies with parent polymers are impacted by variations in their physical properties.
Crack onset strain
As H-bonds will contribute to the mechanical properties of a CPs, determining the ductility of a polymer film through crack onset strain (COS) measurements is a useful method to identify their presence. By first mounting the film upon an elastomeric support (Fig. 3(c)), the polymer is subjected to strain up to the onset of cracking (εc) whilst using optical or atomic force microscopy to monitor crack formation.49 Though measurements of these kind offer only a qualitative view on the mechanical properties, observing trends in εc within a polymer series can provide complementary insights into the differences in physical properties when thermomechanical measurements may not be possible or inconclusive for a particular CP. However, comparisons of the results across various research studies should be made with caution as multiple sources of discrepancy occur when measuring the (εc) value, including identifying the size threshold of a crack in the film and magnification of the microscope used.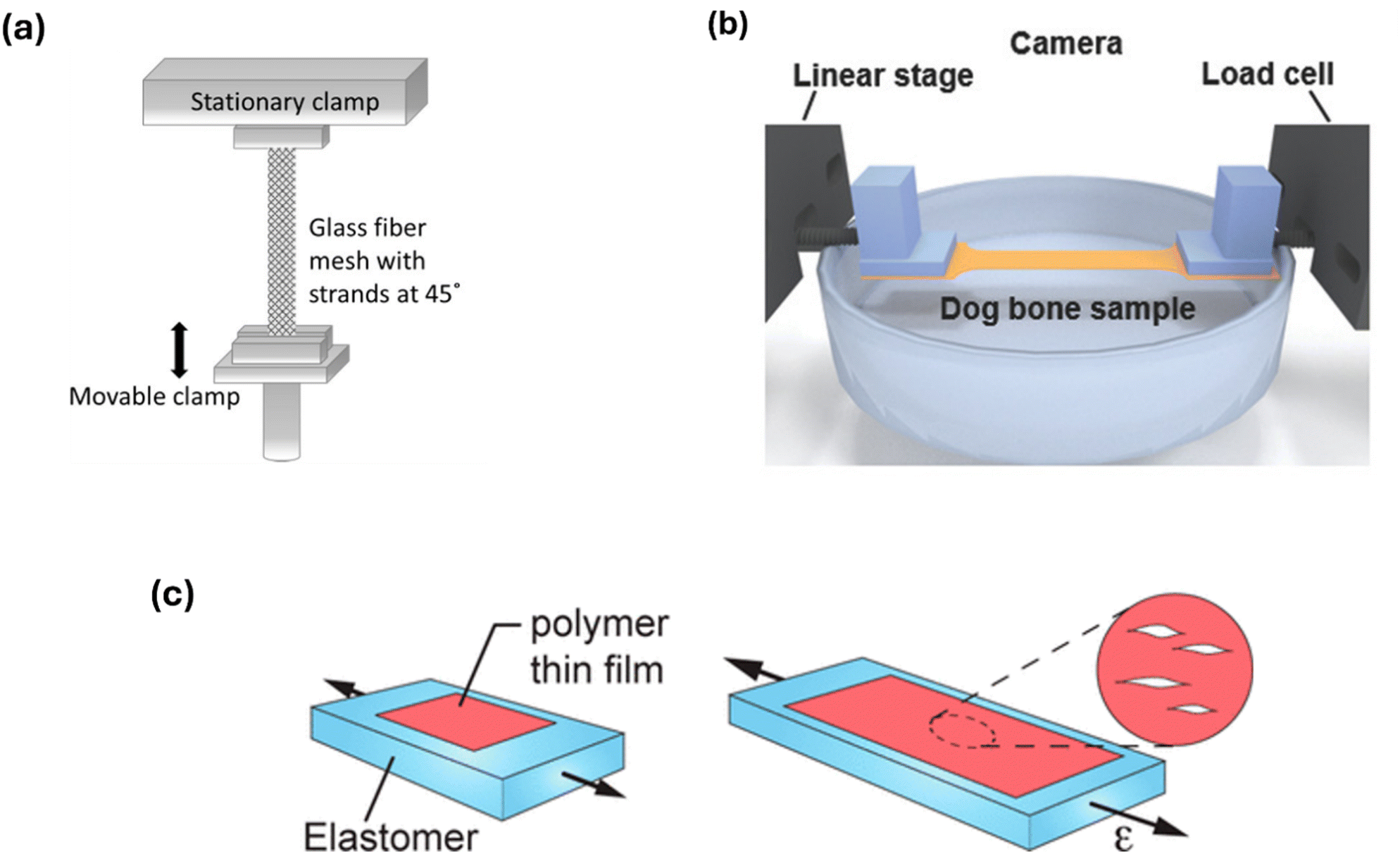 | ||
| Fig. 3 Diagrams illustrating various methods of mechanical testing for conjugated polymers. (a) Tension mode dynamic mechanical analysis (DMA), used to measure films deposited upon a glass fibre mesh support, at 45° relative to the direction of strain. Adapted with permission from A. Sharma et al., Macromolecules 2017, 50, 8, 3347–3354.47 Copyright 2017 American Chemical Society. (b) Film-on-water (FOW) technique, used for tension DMA measurements of a film supported by water. Adapted with permission from S. Zhang, et al., Macromol. Rapid Commun. 2018, 39, 1800092. Copyright 2018 John Wiley and Sons.48 (c) Crack onset strain (COS) measurement to measure the ductility of films supported on an elastomer. Adapted with permission from N. Balar & B. O’Connor, Macromolecules 2017, 50, 21, 8611–8618. Copyright 2017 American Chemical Society.49 | ||
In summary, the thorough investigation of the thin-film mechanical properties of CPs modified with intrinsic hydrogen bonding groups, in terms of the Tg, E and εc, is a powerful strategy to complement conventional spectroscopic techniques and should become a primary focus of the developmental process for flexible electronics. Thus far, practical challenges such as the typically small-scale synthesis (≤100 mg) of CPs, the difficulty in reliably developing material processing and experimental parameters for tensile measurements limits the scope for material testing. As a result, comparing the results of different studies across the research community becomes difficult. To better understand how supramolecular groups influence the properties of CPs on the micro and macrostructural level, conducting full thermomechanical analysis should be carried out. This could be achieved by the study of simplified polymer systems, where the non-functionalized polymer is already well studied in terms of its mechanical and structural properties, as has been shown for polythiophenes.48,60,63,64 In addition, the use of simpler polymer architectures that can be produced on larger scale, could offer the opportunity to optimize processing or use alternative characterisation techniques (e.g. shear rheometry) requiring larger sample quantities for mechanical analysis.64 These studies would also complement the use of solid-state FTIR and NMR, where identification of specific hydrogen bonds can be achieved in the absence of multiple competitive interactions in complex D–A backbone structures.
Hydrogen bonding in wearable electronics
Supramolecular mechanisms have often been utilized in non-conjugated polymers to introduce healing into the materials after bond dissociation. In many of these cases, this phenomenon is autonomous without requirement of an external stimulus (e.g. thermal or light activation) to restore the physical polymer properties. This has been achieved by optimizing (1) strength of the non-covalent interactions introduced to the structure and (2) maximising the polymer chain flexibility. In the case of semiconducting polymers, where the conjugation length along the planar backbone determines the ability of the material to transport charge carriers, the focus of introducing intrinsic self-healing and stretchability has mostly centred around optimizing the hydrogen bonding interactions.Sidechain functionalisation
Modification of the chemical composition of polymer sidechains is a popular technique for introducing specific structural features or functionalities within the field of organic electronics.65 Development of polymers with H-bond units hosted within or as pendant groups on sidechains has therefore gained significant attention, as it offers a pathway to improving the mechanical properties with minimal interference to the backbone conformation.Hydrogen bonding offers a strong and dynamic mechanism for self-healing in polymers and thus has gained significant attention among the flexible electronics community to induce skin-like properties. In some cases, H-bonds also contribute to improved electronic properties, as seen in a series of diketopyrrolopyrrole-bithiophene (DPP-2T) copolymers, that were investigated for their use as the donor material in an OPV device, were functionalized with urea groups set within linear alkyl chains to direct ordering of lamellae via H-bonds (visualised in Fig. 4).41 This effect was observed from GIWAXS measurements and is corroborated by the increase in mobility with loading of urea groups. To distinguish whether these differences were due to H-bonding or a lower sidechain density, the values were compared with a reference set of polymers bearing alkyl only sidechains in place of urea. It could be seen for 3 and 5 mol% of urea that the hole mobility (μh) was lower than the corresponding reference however at 9 mol% the urea functionalised polymer showed a large improvement (5.7 vs. 2.4 cm2 V−1 s−1) which was attributed to H-bonding. Though this study was not in the context of flexible electronics, it clearly demonstrates that introducing external non-covalent interactions of adequate strength and directionality can influence the ordering of a CP matrix.
 | ||
| Fig. 4 (a) Chemical structure of DPP-TVT random copolymers, as a general example to display the two routes of modification with hydrogen bonding motifs (green). H-Bonding groups in each case are bridged between linkers R1 and R2, either within sidechains or as conjugation breakers. (b) Chemical structures of the hydrogen bonding motifs employed in the discussed literature. | ||
The relationship between introducing hydrogen bonds and improved mobility is more ambiguous in other cases. Another study also employed urea, as well as amide groups, as pendant moieties at 10 mol% in DPP and (E)-2-(2-(thiophen-2-yl)-vinyl)thiophene (TVT) copolymers.43 The study compared the use of non-functionalised chains with amide and urea groups. Field-effect measurements revealed an increase in the average μh from 1.48 × 10−1 to 1.99 × 10−1 cm2 s−1 V−1 when introducing the amide H-bond motif, compared with the non-functionalised analogue. However, when increasing the number of possible hydrogen bonds per unit from one to two in urea, the mobility fell by an order of magnitude to 1.54 × 10−2 cm2 s−1 V−1. Yet, the urea derivate displayed a much higher rDoC through GIWAXS measurements. Evidence of hydrogen bonding in the materials was apparent when considering the increase in Tg with increased H-bond strength in the sidechains (culminated with a lower side chain density). And although COS measurements indicated similar low ductility in the amide and urea polymers (εc = 0.5%), reinforcing effects of the H-bonds were highlighted through an increase in modulus from 357.1 to 371.6 MPa respectively measured using the FOW technique. These values for E were lower than the non-functionalised derivative (380.7 MPa), indicating the possibility to impart higher stretchability in CPs through the introduction of supramolecular motifs.
DPP-TVT was also the subject of a healing study where amide groups were set within a linear alkyl chain at 0, 5, 10 and 20 mol%.29,42 Mechanical tests showed that elasticity was increased drastically by a near two-fold decrease in modulus from 203 to 115 MPa and εc increased from 30 to 75% upon increasing amide loading from 0 to 10 mol%. Unfortunately, no clear trend could be deduced from field-effect measurements for hole mobility although the devices (fabricated on flexible substrates) exhibited reasonable values (listed in Table 1). Electrical measurements were re-recorded for the OFET devices after tensile strength testing, showing a large decrease in μh. In agreement with the previous study, a decrease in rDoC was observed upon introducing the amide units at ε = 0%. However, increasing the content of amide up to 30 mol% resulted in continuous deterioration of rDoC, whereas previously the exchange of 10 mol% amide for urea increased the rDoC. As enhanced interdigitation of sidechains can improve the microstructural order within a material, the observed increase in rDoC with urea could be the result of not only an increased number of H-bonds but their inherent directionality, as urea is known to form “ladder” architectures.66–68
To understand the role of the backbone rigidity in a H-bond functionalised CP, a series of donor–acceptor DPP copolymers bearing urethane within branched alkyl side chains were synthesised.33 Within the copolymer, the acceptor unit was varied between thieno[3,2-b]thiophene (TT), 2,2′-bithiophene (2T) and TVT. GIWAXS measurements showed an increase in lamellar stacking for 2T and TVT derivatives compared with an alkyl only chain for pristine films. After mechanical deformation, discrepancy between d-spacing values indicated that packing of the lamellae was distributed in TT and 2T polymers at ε = 100%, this was corroborated by observed εc values at 50 and 75% respectively whereas d-spacing remained constant for TVT. In pristine films μh improved in the order TVT > 2T > TT, yet after applying 100% strain the mobility dropped in 2T and TT dramatically whereas TVT values were maintained in the parallel stretching direction and fell slightly in the perpendicular. This, as well as an increase in dichroic ratio above εc = 112.5%, would suggest that some energy dissipation occurs in both the amorphous and crystalline regions.
Another study compared DPP-TVT with broken conjugation alongside the analogous polymer that used 2,6-pyridinedicarboxamide (PDCA) (Fig. 4) as a pendant group within the sidechain.34 The use of pendant moieties caused a reduction in the hole mobility with increased loading, yet negligible difference was observed between polymers with 30 and 60 mol% PDCA (0.60 and 0.55 cm2 V−1 s−1 respectively). Yet, the rDoC and backbone melting temperature increased with PDCA loading compared to an ester derived reference polymer. Also, an intermediate glass transition was observed which scaled with the relative amount of PDCA and was absent in the case of the reference polymer. This implies that H-bonds influenced the ordering of both the amorphous and semicrystalline regions. Based on the low Mn values (∼20 kg mol−1), the lowering of μh could therefore possibly be explained by the formation of grain boundaries. DSC thermograms showed minimal differences in Tg values for polymers functionalised via the backbone or sidechains at 10–60 mol% loading. Given the previously low hole mobility at >10 mol% recorded for the CB polymers, comparable reduction of backbone rigidity with each technique highlights using hydrogen bonding pendant groups as a potential method for enhancing microstructural ordering.
The use of larger H-bonding arrays was also carried out in an IDT-BT copolymer, using ureidopyrimidinone (UPy), which hosts up to four H-bonds per dimerised unit as shown in Fig. 2.30 The study tested the influence of this bulky H-bonding unit against a triisopropylsilyl (TIPS) group and the smaller, weaker hydroxyl group. Though demonstrating high εc values, approaching or surpassing ε = 100%, COS measurements demonstrated lowering of the polymer ductility from parent IDT-BT for all types of functionalisation. Whilst they exhibit similar mechanical properties, OFET devices were constructed to understand whether a high density (20 mol%) of weaker OH units were comparable with a low density (5 mol%) of stronger UPy units in terms of the electronic performance. Though the hole mobilities were an order of magnitude lower than the parent IDT-BT (1.66 cm2 V−1 s−1) and TIPS functionalised IDT-BT polymer (ranging 1.42–1.89 cm2 V−1 s−1), the H-bonding polymers displayed similar μh values of 0.47 and 0.34 cm2 V−1 s−1 respectively. In comparison with more semicrystalline polymers, such as DPP-TVT, these results indicate that more amorphous polymers could provide scope for careful optimization of energy dissipation mechanisms within CPs for wearable electronics.
Backbone engineering
An alternative route to functionalising sidechain composition is to incorporate hydrogen bonding units within the CP backbone. By embedding the supramolecular group within a monomer unit, this type of functionalisation causes disruption to the conjugation length along the chain, and therefore they are often referred to as conjugation breakers (CBs). In many cases, this design approach has resulted in improvement of the mechanical properties of organic electronic materials.69,70 Additionally, the possibility to form intermolecular hydrogen bonds between adjacent chains can allow microstructural order to be maintained whilst also achieving improved ductility or self-healing functionality.A study using amide, urethane and urea (Fig. 4) in DPP-TVT copolymers investigated the effect of weaker H-bonding moieties as CBs.45 They also sought to better understand the effect of the local chemical environment on H-bond formation by tethering the self-healing units by either alkyl or ester chains. Tensile strength testing was carried out to determine elasticity and it was found that a lower modulus correlated with a higher hole mobility value (up to 0.31 cm2 V−1 s−1 for urethane functionalised ester derivative). Interestingly, it could be seen for the non-bonding carbonate and amide CBs that a lower E was observed when tethered to an ester chain, whereas for urethane and urea groups, the modulus was dramatically increased in ester derivatives compared with the alkyl counterparts. Although no clear trend could be observed relative to the strength of the H-bond unit, mechanical testing indicates that H-bonding is present within the sidechains in the latter two polymers. UV-Vis spectroscopy showed a higher degree of aggregation in the intramolecular charge transfer (ICT) band for ester derivatives. This was likely due to the presence of more H-bond acceptors within neighbouring chains improving alignment of the chains resulting in higher mobilities than their alkyl counterparts. GIWAXS measurements were carried out to investigate order in the microstructure, showing that with increased H-bonding strength there was an increase in the rDoC. After applying strain, the scattering profiles indicated that energy was dissipated from breaking H-bonds, realignment of crystalline domains, chain extension and realignment of amorphous regions as well as the breaking of crystallites. Only for the urea derivative tethered with ether groups was the latter mechanism not observed, however definitive trends could not be drawn for the rest of the data.
Promising results were reported in a series of DPP-TVT copolymers that were synthesised with varied loading of PDCA in the backbone (Fig. 4).44 Although an increase in CB loading saw a general decrease in hole mobility, tensile testing showed a fall in the elastic modulus and an increase onset of crack formation in films. The lowest value of E was reported for 10 mol% of PDCA which exhibited high mobility of 1.32 cm2 V−1 s−1, which encouragingly recovered to 1.00 cm2 V−1 s−1 upon release after being subjected to ε = 100%. At various strains, the 10 mol% PDCA polymer demonstrated a decrease in rDoC inferring that energy dissipation occurs by breaking of crystallites. Additionally, an increase in the dichroic ratio above the onset of crack formation (εc = 80%) indicated some energy dissipation within crystalline regions, likely due to H-bond breakage. Although these features were only partially recovered via solvothermal annealing and therefore self-healing was not observed, this work demonstrated a promising strategy to synthesising stretchable conjugated polymers.
A recent study showed that increasing the size and H-bonding ability of the CB, by using UPy in a copolymer of DPP and selenophene, caused a slight lowering of the hole mobility from 0.6 to 0.1 cm2 V−1 s−1 between 0 and 20% UPy in the backbone.32 Interestingly, this work adopted a unique approach of linking self-healing groups with alkyl linkers in a ring-like structure to keep neighbouring UPy groups in close proximity upon stretching. Mechanical measurements indicated a drastic reduction in the elastic modulus by introducing the H-bonding groups from 890 to 404 MPa between 0 and 20 mol% UPy, indicative of improved ductility through this type of functionalisation. This was also reflected in the field-effect measurements, which showed that although applying strain (ε = 100%) causes a reduction in μh, this effect was lower (85%) compared with the neutral polymer (91%).
The use of peptide linkers, glycine, valine and threonine, was also investigated as a type of CB in a DPP-TVT copolymer.31 The study showed that use of glycine, with the least steric hindrance relative to the formation of intermolecular hydrogen bonds, exhibited superior ductility through COS measurements and a reduction in modulus from ∼800 to ∼500 MPa. In line with the observed changes to hole mobility in the previous study, the glycine derivative with improved mechanical properties also showed the least reduction in μh (30% compared with 97% in the parent polymer).
Discussion and future outlook
Upon assessing the role of hydrogen bonds in CPs designed for wearable electronics, it is evident throughout the literature that intermolecular H-bonds influence both the observed ductility and microstructural ordering of crystalline domains in CPs films. Improved mechanical strength is observed in the vast majority of cases, where εc increases with H-bond strength or mol%. In contrast, although there is strong support of increased degree of structural order (through measurement of rDoC and μh in OFETs) when increasing the content or strength of the H-bond motif, within each series of analogous polymers an upper limit in improving the structural and electronic properties is often observed. This trend was more prevalent amongst literature focusing on sidechain engineering, in contrast to use of CBs, which generally saw a reduction in μh upon addition of H-bonding groups. The latter can be attributed to a reduction in effective conjugation length, where CB units reduce backbone planarization. In addition, several articles reported electronic characterisation based upon flexible OFET devices which indicated that upon application of strain, there was better retention of the mobility in polymers that incorporated the supramolecular groups within sidechains.Whilst the device fabrication to probe the electronic properties is largely consistent throughout all studies, detailed thermomechanical testing can be considered sparse as demonstrated by the lack of reported data for E and Tg in Table 1. Alternatively, the ductility of a film, which describes the ability of the material to withstand deformation before structural fracture, can be assisted by supramolecular interactions which dissipate energy under strain. COS testing to monitor ductility changes can therefore provide insights into the effect of H-bonds on the mechanical properties and is routinely performed throughout the literature (Table 1). However, this technique is limited in its accuracy and reproducibility and should be assisted with quantitative analysis under careful control of the environmental conditions (e.g. temperature, humidity).
Furthermore, the analysis can be convoluted by the difficulty in observing phase transitions for more complex chemical structures (such as those based on DPP), as demonstrated by the reporting of Tg values in only two studies (Table 1). The study of simpler polymer systems such as polythiophenes could facilitate a more detailed understanding of the effect of H-bonds on a CP structure. This approach is of further advantage when considering the wealth of knowledge already gained for these systems in terms of the thermomechanical properties in relation to electronic performance.55,71,72
Despite significant advancements in enhancing the biomimicry of organic electronics through the incorporation of hydrogen bonds, the complex characteristics of these materials are still not fully understood. To further develop these systems, more comprehensive studies of the chemical and physical mechanisms underlying the structural properties of CPs are necessary. We encourage the research community to broaden the range of characterisation methods applied to H-bonding CPs, moving beyond a selective focus on a narrow subset of properties. For example, simplifying polymer systems can facilitate the characterisation and analysis of thermomechanical properties using techniques such as DSC and DMA. Pairing these measurements with studies of optoelectronic properties would provide a more holistic understanding of how hydrogen bonds affect CP structure and function.
Currently, most research on ‘recovery’ emphasizes electronic performance or relative degree of crystallinity via UV-vis spectroscopy, focusing primarily on the more ordered polymer regions. Future efforts should prioritise the study of mechanical recovery through DMA measurements, as previously reported for CPs based on DPP. Additionally, integrating these techniques with solid-state spectroscopy methods, such as Fourier-transform infrared (FTIR) and Raman spectroscopy, would allow for the observation of changes in bands associated with hydrogen bonding when the CP is subjected to strain and subsequent recovery. We believe that this approach would contribute to a more comprehensive understanding of the impact of hydrogen bonds on CP structure and functionality and enable the development of the next generation of self-healing CPs for wearable electronics.
Conclusions
The introduction of hydrogen bonding moieties into the matrix of a conjugated polymer has critical influence on the mechanical properties, microstructural ordering and consequently the optoelectronic properties. Although an inverse relationship between ductility and structural order is often observed, to successfully further develop these systems for device implementation, a clearer understanding of the role of H-bonding units on these properties is crucial. This could be achieved through shifting focus towards the study of (1) simpler CP systems and (2) quantitative thermomechanical analysis, and therefore allow design paradigms to be established for the functionalization of CPs with supramolecular crosslinkers.Author contributions
Manuscript was written and conceptualized by M. M. W. and B. C. S. Supervision, reviewing, editing, project administration and funding acquisition by B. C. S.Data availability
No new data were collected and all data mentioned in the article are adequately referenced.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. M. W. and B. C. S. acknowledge funding from the UK Research and Innovation for Future Leaders Fellowship (MR/S031952/1). The ToC figure was created in BioRender.References
- C. Rizan, F. Mortimer, R. Stancliffe and M. F. Bhutta, J. R. Soc. Med., 2020, 113, 49–53 CrossRef PubMed.
- K. Modjarrad and S. Ebnesajjad, Handbook of Polymer Applications in Medicine and Medical Devices, Elsevier, New York, 2014 Search PubMed.
- A. Zhang, S. Zhao, J. Tyson, K. Deisseroth and Z. Bao, Nat. Synth., 2024, 3, 943–957 CrossRef.
- C. P. Price and L. J. Kricka, Clin. Chem., 2007, 53, 1665–1675 CrossRef CAS.
- J. Jagur-Grodzinski, Polym. Adv. Technol., 2006, 17, 395–418 CrossRef CAS.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- J. Y. Oh and Z. Bao, Adv. Sci., 2019, 6, 1900186 CrossRef.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- R. M. Owens and G. G. Malliaras, MRS Bull., 2010, 35, 449–456 CrossRef CAS.
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS.
- J. Y. Back, H. Yu, I. Song, I. Kang, H. Ahn, T. J. Shin, S.-K. Kwon, J. H. Oh and Y.-H. Kim, Chem. Mater., 2015, 27, 1732–1739 CrossRef CAS.
- X. Ji, H.-W. Cheng, N. J. Schuster, G. S. LeCroy, S. Zhang, Y. Wu, L. Michalek, B.-N. T. Nguyen, J. A. Chiong, M. Schrock, Y. Tomo, J. Rech, A. Salleo, S. Gam, G. H. Lee, J. B. H. Tok and Z. Bao, Chem. Mater., 2024, 36, 256–265 CrossRef CAS.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- L. Laysandra, A. Njotoprajitno, S. P. Prakoso and Y.-C. Chiu, Mater. Adv., 2022, 3, 7154–7184 RSC.
- K. Liu, Y. Jiang, Z. Bao and X. Yan, CCS Chem., 2019, 1, 431–447 CrossRef CAS.
- J. Kim, J. Fan, G. Petrossian, X. Zhou, P. Kateb, N. Gagnon-Lafrenais and F. Cicoira, Mater. Horiz., 2024, 11, 3548–3560 RSC.
- P. A. Gilhooly-Finn, M. M. Westwood and B. C. Schroeder, RSC Appl. Polym., 2024 10.1039/D4LP00163J.
- M. U. Ocheje, B. P. Charron, A. Nyayachavadi and S. Rondeau-Gagné, Flexible Printed Electron., 2017, 2, 043002 CrossRef.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CrossRef CAS.
- R. Parthasarathi and V. Subramanian, in Hydrogen Bonding—New Insights, ed. S. J. Grabowski, Springer, Netherlands, Dordrecht, 2006, pp. 1–50 DOI:10.1007/978-1-4020-4853-1_1.
- M. N. C. Zarycz and C. Fonseca Guerra, J. Phys. Chem. Lett., 2018, 9, 3720–3724 CrossRef CAS PubMed.
- K. Saalwächter, NMR Methods for Characterization of Synthetic and Natural Polymers, ed. R. Zhang, T. Miyoshi and P. Sun, The Royal Society of Chemistry, 2019 10.1039/9781788016483-00001.
- Y.-C. Xu, L. Ding, Z.-F. Yao, Y. Shao, J.-Y. Wang, W.-B. Zhang and J. Pei, J. Phys. Chem. Lett., 2023, 14, 927–939 CrossRef CAS.
- M. U. Ocheje, B. P. Charron, Y.-H. Cheng, C.-H. Chuang, A. Soldera, Y.-C. Chiu and S. Rondeau-Gagné, Macromolecules, 2018, 51, 1336–1344 CrossRef CAS.
- B. Zhao, D. Pei, Y. Shi, X. Zhang, B. Zhang, Y. Deng, Y. Han and Y. Geng, Macromolecules, 2024, 57, 528–538 CrossRef CAS.
- Y. Kwon, H. Park and J. H. Oh, Chem. Mater., 2023, 35, 10672–10683 CrossRef CAS.
- X. Wang, C. Xu, N. Xu, L. Jiang, Y. Wang, F. Ni, G. Zhang, X. Gu and L. Qiu, Macromolecules, 2023, 56, 5369–5380 CrossRef CAS.
- M. Y. Lee, S. Dharmapurikar, S. J. Lee, Y. Cho, C. Yang and J. H. Oh, Chem. Mater., 2020, 32, 1914–1924 CrossRef CAS.
- A. Gasperini, G.-J. N. Wang, F. Molina-Lopez, H.-C. Wu, J. Lopez, J. Xu, S. Luo, D. Zhou, G. Xue, J. B. H. Tok and Z. Bao, Macromolecules, 2019, 52, 2476–2486 CrossRef CAS.
- H. Zhang, W.-Y. Tung, X. Li, H. Jin, R. Deng, Y.-M. Chen, Y. Mao and Y. Zhu, Polymer, 2020, 203, 122787 CrossRef CAS.
- S. J. Grabowski, Hydrogen Bonding – New Insights, Royal Society of Chemistry (RSC), 2013 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- B. W. Boudouris, V. Ho, L. H. Jimison, M. F. Toney, A. Salleo and R. A. Segalman, Macromolecules, 2011, 44, 6653–6658 CrossRef CAS.
- D. Niedzialek, V. Lemaur, D. Dudenko, J. Shu, M. R. Hansen, J. W. Andreasen, W. Pisula, K. Müllen, J. Cornil and D. Beljonne, Adv. Mater., 2013, 25, 1939–1947 CrossRef CAS PubMed.
- M. Seifrid, G. N. M. Reddy, B. F. Chmelka and G. C. Bazan, Nat. Rev. Mater., 2020, 5, 910–930 CrossRef CAS.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed.
- M. U. Ocheje, M. Selivanova, S. Zhang, T. H. Van Nguyen, B. P. Charron, C.-H. Chuang, Y.-H. Cheng, B. Billet, S. Noori, Y.-C. Chiu, X. Gu and S. Rondeau-Gagné, Polym. Chem., 2018, 9, 5531–5542 RSC.
- L. A. Galuska, M. U. Ocheje, Z. C. Ahmad, S. Rondeau-Gagné and X. Gu, Chem. Mater., 2022, 34, 2259–2267 CrossRef CAS.
- J. Y. Oh, S. Rondeau-Gagné, Y.-C. Chiu, A. Chortos, F. Lissel, G.-J. N. Wang, B. C. Schroeder, T. Kurosawa, J. Lopez, T. Katsumata, J. Xu, C. Zhu, X. Gu, W.-G. Bae, Y. Kim, L. Jin, J. W. Chung, J. B. H. Tok and Z. Bao, Nature, 2016, 539, 411–415 CrossRef CAS PubMed.
- Y. Zheng, M. Ashizawa, S. Zhang, J. Kang, S. Nikzad, Z. Yu, Y. Ochiai, H.-C. Wu, H. Tran, J. Mun, Y.-Q. Zheng, J. B. H. Tok, X. Gu and Z. Bao, Chem. Mater., 2020, 32, 5700–5714 CrossRef CAS.
- B. O’Connor, R. J. Kline, B. R. Conrad, L. J. Richter, D. Gundlach, M. F. Toney and D. M. DeLongchamp, Adv. Funct. Mater., 2011, 21, 3697–3705 CrossRef.
- A. Sharma, X. Pan, J. A. Campbell, M. R. Andersson and D. A. Lewis, Macromolecules, 2017, 50, 3347–3354 CrossRef CAS.
- S. Zhang, M. U. Ocheje, S. Luo, D. Ehlenberg, B. Appleby, D. Weller, D. Zhou, S. Rondeau-Gagné and X. Gu, Macromol. Rapid Commun., 2018, 39, 1800092 CrossRef.
- N. Balar and B. T. O’Connor, Macromolecules, 2017, 50, 8611–8618 CrossRef CAS.
- Z. Qian, Z. Cao, L. Galuska, S. Zhang, J. Xu and X. Gu, Macromol. Chem. Phys., 2019, 220, 1900062 CrossRef.
- C. Müller, Chem. Mater., 2015, 27, 2740–2754 CrossRef.
- N. Balar, S. Siddika, S. Kashani, Z. Peng, J. J. Rech, L. Ye, W. You, H. Ade and B. T. O’Connor, Chem. Mater., 2020, 32, 6540–6549 CrossRef CAS.
- R. Xie, A. R. Weisen, Y. Lee, M. A. Aplan, A. M. Fenton, A. E. Masucci, F. Kempe, M. Sommer, C. W. Pester, R. H. Colby and E. D. Gomez, Nat. Commun., 2020, 11, 893 CrossRef CAS PubMed.
- R. Xie, R. H. Colby and E. D. Gomez, Adv. Electron. Mater., 2018, 4, 1700356 CrossRef.
- B. O’Connor, E. P. Chan, C. Chan, B. R. Conrad, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, C. L. Soles and D. M. DeLongchamp, ACS Nano, 2010, 4, 7538–7544 CrossRef.
- S. Zhang, L. A. Galuska and X. Gu, J. Polym. Sci., 2022, 60, 1108–1129 CrossRef CAS.
- C. M. Stafford, C. Harrison, K. L. Beers, A. Karim, E. J. Amis, M. R. VanLandingham, H.-C. Kim, W. Volksen, R. D. Miller and E. E. Simonyi, Nat. Mater., 2004, 3, 545–550 CrossRef CAS PubMed.
- S. H. K. Paleti, Y. Kim, J. Kimpel, M. Craighero, S. Haraguchi and C. Müller, Chem. Soc. Rev., 2024, 53, 1702–1729 RSC.
- J.-H. Kim, A. Nizami, Y. Hwangbo, B. Jang, H.-J. Lee, C.-S. Woo, S. Hyun and T.-S. Kim, Nat. Commun., 2013, 4, 2520 CrossRef.
- D. Rodriquez, J.-H. Kim, S. E. Root, Z. Fei, P. Boufflet, M. Heeney, T.-S. Kim and D. J. Lipomi, ACS Appl. Mater. Interfaces, 2017, 9, 8855–8862 CrossRef CAS.
- L. A. Galuska, E. S. Muckley, Z. Cao, D. F. Ehlenberg, Z. Qian, S. Zhang, S. Rondeau-Gagné, M. D. Phan, J. F. Ankner, I. N. Ivanov and X. Gu, Nat. Commun., 2021, 12, 2347 CrossRef CAS.
- R. Song, H. Schrickx, N. Balar, S. Siddika, N. Sheikh and B. T. O’Connor, Macromolecules, 2020, 53, 1988–1997 CrossRef CAS.
- Y. Wang, S. Zhang, G. Freychet, Z. Li, K.-L. Chen, C.-T. Liu, Z. Cao, Y.-C. Chiu, W. Xia and X. Gu, Adv. Funct. Mater., 2023, 33, 2306576 CrossRef CAS.
- R. Xie, Y. Lee, M. P. Aplan, N. J. Caggiano, C. Müller, R. H. Colby and E. D. Gomez, Macromolecules, 2017, 50, 5146–5154 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- P. J. W. Sommerville, A. H. Balzer, G. Lecroy, L. Guio, Y. Wang, J. W. Onorato, N. A. Kukhta, X. Gu, A. Salleo, N. Stingelin and C. K. Luscombe, ACS Polym. Au, 2023, 3, 59–69 CrossRef CAS.
- Y. Zhao, X. Zhao, Y. Zang, C.-A. Di, Y. Diao and J. Mei, Macromolecules, 2015, 48, 2048–2053 CrossRef CAS.
- S. Savagatrup, X. Zhao, E. Chan, J. Mei and D. J. Lipomi, Macromol. Rapid Commun., 2016, 37, 1623–1628 CrossRef CAS.
- J. Moulton and P. Smith, Polymer, 1992, 33, 2340–2347 CrossRef CAS.
- S. Savagatrup, A. S. Makaram, D. J. Burke and D. J. Lipomi, Adv. Funct. Mater., 2014, 24, 1169–1181 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |