
Unraveling proton-coupled electron transfer in cofactor-free oxidase- and oxygenase-catalyzed oxygen activation: a theoretical view
Qian-Qian
Wang
 a,
Yan
Qiao
*b and
Donghui
Wei
*a
a,
Yan
Qiao
*b and
Donghui
Wei
*a
aCollege of Chemistry, Zhengzhou University, 100 Science Avenue, Zhengzhou, Henan 450001, P. R. China. E-mail: donghuiwei@zzu.edu.cn
bDepartment of Pathophysiology, School of Basic Medical Sciences, Zhengzhou University, 100 Science Avenue, Zhengzhou, Henan 450001, P. R. China. E-mail: yanqiao@zzu.edu.cn
First published on 18th November 2024
Abstract
Oxygen plays a crucial role in the metabolic processes of non-anaerobic organisms. However, a detailed understanding of how triplet oxygen participates in the enzymatic oxidation of organic compounds involved in life processes is still lacking. It is noteworthy that recent studies have found that cofactor-free oxidase- and oxygenase-catalyzed oxygen activation occurs through proton-coupled electron transfer (PCET), which is significantly different from the previously proposed single electron transfer (SET) mechanism. Herein, we summarize the recent advances in the general mechanism of catalytic activation reactions of triplet oxygen by these enzymes. We believe that this review not only helps in providing a deep understanding of the processes involved in oxygen metabolism in organisms but also provides valuable theoretical reference data for designing more efficient enzyme mutants for treating diseases and handling environmental pollution in the future.
 Qian-Qian Wang | Qian-Qian Wang was born in 2000 in Henan, China. She obtained a bachelor's degree from Zhengzhou Normal University and is currently pursuing her master's degree at Zhengzhou University under the guidance of Prof. Donghui Wei. Her research interests are centered on the mechanistic studies of enzyme and organic catalyst-catalyzed reactions. |
 Yan Qiao | Yan Qiao, born in 1986 in Henan, China, received her BSc in Chemistry from Zhengzhou University in 2008 and her PhD in Physical Chemistry from the Dalian Institute of Chemical Physics in 2014. She conducted joint research at the University of Kentucky (2011–2013). Now an Associate Professor at Zhengzhou University, her research focuses on cancer chemoprevention, integrating computational biology and experimental methods to discover therapeutic targets for esophageal cancer. |
 Donghui Wei | Donghui Wei was born in 1983 in Henan, China. He graduated from Zhengzhou University (2006) and received his PhD (2012) from Zhengzhou University. While obtaining his PhD degree, he studied as a visiting scholar (2010–2012) with Prof. Chang-Guo Zhan as his advisor at the University of Kentucky, KY, USA. He joined the faculty of Zhengzhou University in 2013 and is currently a professor in the College of Chemistry. He performed a series of theoretical studies on the mechanisms of enzyme-, organocatalyst-, and transition metal-catalyzed reactions. |
1 Introduction
As we all know, the metabolic processes of non-anaerobic organisms cannot occur efficiently without the participation of oxygen (O2). Thus, the mechanistic studies of triplet O2-involving enzymatic reactions are highly valuable in the fields of chemistry, life sciences, medicine, and pharmacy.1,2 Enzymes capable of activating triplet O2 are divided into two types: oxidases that use oxygen as an oxidant to produce hydrogen peroxide or water and oxygenases that incorporate one or two oxygen atoms (monooxygenases or dioxygenases) into the substrate.3 Commonly, O2-dependent oxidase- and oxygenase-catalyzed reactions in living organisms require the participation of metal ions or organic cofactors.3–17 In the presence of these cofactors or metal ions, the activation process of triplet O2 is efficiently promoted. However, a significant number of cofactor- and metal-free enzymes can also activate triplet O2. These include cofactor-free oxidases such as uricase,18 CPO,19,20 and PqqC21,22 as well as cofactor-free monooxygenases and dioxygenases, including ActVA-Orf6,23 DpgC,24 HOD,25,26 and RluC27 (Fig. 1). At the same time, the mechanism by which triplet O2 is activated without cofactors and metal ions has attracted increasing experimental and theoretical attention.8,19,21,22,28–36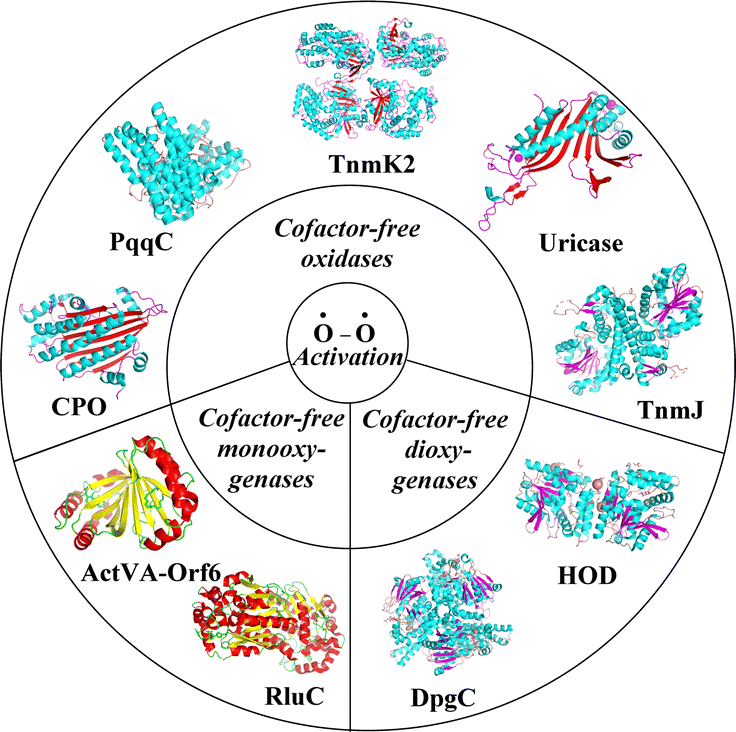 | ||
| Fig. 1 Crystal structures of selected cofactor-free oxidases and oxygenases, including uricase (PDB ID: 4N9M37), PqqC (PDB ID: 1OTW21), CPO (PDB ID: 2AEX20), ActVA-Orf6 (PDB ID: 1N5S23), RluC (PDB ID: 2PSJ27), DpgC (PDB ID: 2NP938), HOD (PDB ID: 2WJ439), TnmJ (PDB ID: 8G5S40), and TnmK2 (PDB ID: 8G5T40). | ||
For the catalytic mechanism of cofactor-free oxidases and oxygenases, the activation of triplet O2 may occur via two possible pathways, including single electron transfer (SET) and proton-coupled electron transfer (PCET), as depicted in Scheme 1A.28–35 The SET pathway involves direct transfer of a single electron from the substrate ion (Sub−) to triplet O2, resulting in the generation of ˙Sub and ˙O2− radicals. The SET mechanism has been widely investigated for the oxidase- and oxygenase-catalyzed activation of O2 in a significant number of mechanistic studies, providing a deep understanding of the metabolic processes of non-anaerobic organisms.2,3,28–31,34,41,42 On the other hand, the PCET involves the deprotonation of the substrate (Sub-H) coupled with electron transfer to O2.
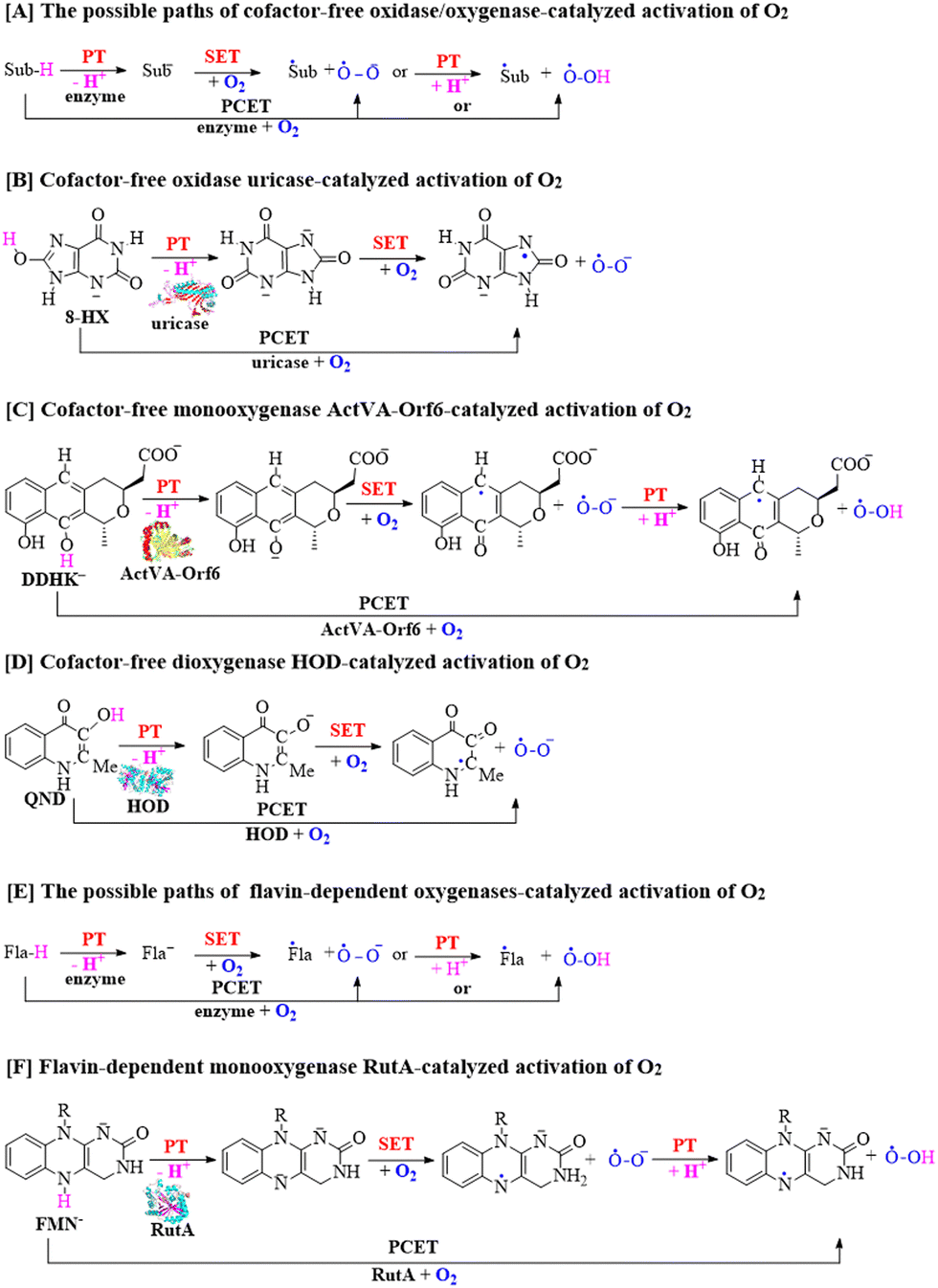 | ||
| Scheme 1 (A) Possible SET and PCET pathways of cofactor-free oxidase- and oxygenase-catalyzed activation of O2. (B)–(D) SET and PCET pathways of cofactor-free uricase-, ActVA-Orf6-, and HOD-catalyzed activation of O2. (E) Possible SET and PCET pathways of flavin-dependent oxygenase-catalyzed activation of O2. (F) SET and PCET pathways of flavin-dependent RutA-catalyzed activation of O2. Dots represent single electrons on O2, Sub-H represents substrates, and Fla-H represents flavin cofactor. | ||
It is worth mentioning that the PCET concept and mechanism have been widely studied in other reaction systems,43–57 but they have been less explored in the cofactor-free oxidase and oxygenase systems. In the previous studies, triplet O2 has always been considered and simulated to directly react with the substrate ions (Sub−) and not in the deprotonation process of substrates, causing the PCET mechanism to be rarely mentioned for a long duration. Until recently, the possible PCET pathways for the cofactor-free oxidases and oxygenases shown in Scheme 1B–D have gradually attracted increasing attention from researchers.32,33,35
Besides, regio- and stereospecific oxygen insertions by flavin-dependent oxidases and oxygenases are useful for the production of pharmaceutical ingredients and fine chemicals in industrial applications.58–67 Therefore, it is very important to study the mechanism of flavin-dependent oxidase and oxygenase-catalyzed reactions involving triplet O2. The flavin-dependent oxidase and oxygenase-promoted reactions may also include two different pathways including SET and PCET pathways to activate O2.9,68–81 As shown in Scheme 1E, the SET mechanism involves the direct transfer of a single electron from flavin ion (Fla−) to triplet O2, resulting in the generation of ˙Fla radical and ˙O2− radical. Alternatively, the PCET mechanism has been reported via the deprotonation of flavin (Fla-H) coupled with an electron transfer to O2 in the flavin-dependent oxygenase-mediated reaction, as shown in Scheme 1F.
Understanding the cofactor-free oxygenases, cofactor-free oxidases, and flavin-dependent oxygenases, catalytic mechanisms can provide valuable theoretical reference data for designing more efficient enzyme mutants to treat hyperuricemia-related diseases and address environmental pollution. Therefore, we are motivated to contribute this review on recent advances in the mechanistic studies of oxidase- and oxygenase-catalyzed reactions from a theoretical perspective. In this review, in order to better understand the metabolic processes in oxygen-dependent metabolism in organisms, we mainly summarize the latest progress on the activation of O2 by cofactor-free oxygenases, cofactor-free oxidases, and flavin-dependent oxygenases via the PCET pathway.
2 Cofactor-free oxidases and oxygenases
2.1 Cofactor-free oxidase uricase
Uric acid (UA) is the product of xanthine oxidase-catalyzed oxidation of xanthine in the human body, and it is typically maintained at a concentration of 3.6–8.3 mg dL−1 in blood plasma.82 Elevated serum UA levels predominantly lead to hyperuricemia in the human body. Persistent hyperuricemia can certainly result in gouty arthritis and renal stones, which is characterized by the deposition of monosodium UA monohydrate crystals.83–86 In the case of tumor lysis syndrome, UA levels become extremely high, resulting in the rapid loss of kidney function.87–89 Uricase regulates the concentration of uric acids in the blood of birds and reptiles, and uricase mutants could also serve as potential exogenous protein drugs for effective treatment of patients suffering from hyperuricemia disorder and related diseases.90–92 Therefore, it is necessary to understand the mechanism of cofactor-free oxidase uricase-catalyzed reactions.According to the available knowledge in the literature, the uricase-catalyzed reaction starts from 8-HX.93–97 In 2017, Zhan and co-workers reported on the PCET pathway for uricase-catalyzed activation of O2, and the QM/MM-FE calculations were performed at the [B3LYP/6-31+G(d):AMBER]//[B3LYP/6-31G(d):AMBER] level.32 In these QM/MM-FE calculations, the QM/MM reaction coordinate calculations were followed by free energy perturbation (FEP) calculations on the protein environment to account for the dynamic effects of the protein environment on the free energy barriers for the enzymatic reaction.98–102 The DFT method has been confirmed to be a good choice to study the reaction mechanism in QM part.103–108Fig. 2 depicts the uricase-catalyzed oxidation reaction pathway for 8-HX in detail. In this process, the first step involves the transfer of protons from the substrate 8-HX to the Nz atom of the residue K9 with the assistance of residue T69, while electron transfer occurs from 8-HX to the oxygen molecule to activate O2via transition state 2tst (10.0 kcal mol−1). The second reaction step involves the transfer of protons from the protonated amino group of residue K9 to the O2 atom through transition state 4tst (0.8 kcal mol−1). Subsequently, diradical recombination occurs to form a C5–O1 bond. The third reaction step is the dissociation of H2O2 molecule through transition state 7tss (13.3 kcal mol−1). Further, as reported,109 the experimentally measured rate constant (kcat) for uricase-catalyzed oxidation of UA ranges from 6.0 s−1 to 70 s−1 at T = 298.15 K for uricase. This value of calculated free energy barrier (16.2 kcal mol−1) is close to experimental measurement free energy barrier (14.9–16.4 kcal mol−1) for the rate-determining step of the uricase-catalyzed reaction. In addition, the residues K9 and T69 participate in the proton transfer process of oxygen activation catalyzed by uricase (Fig. 2).
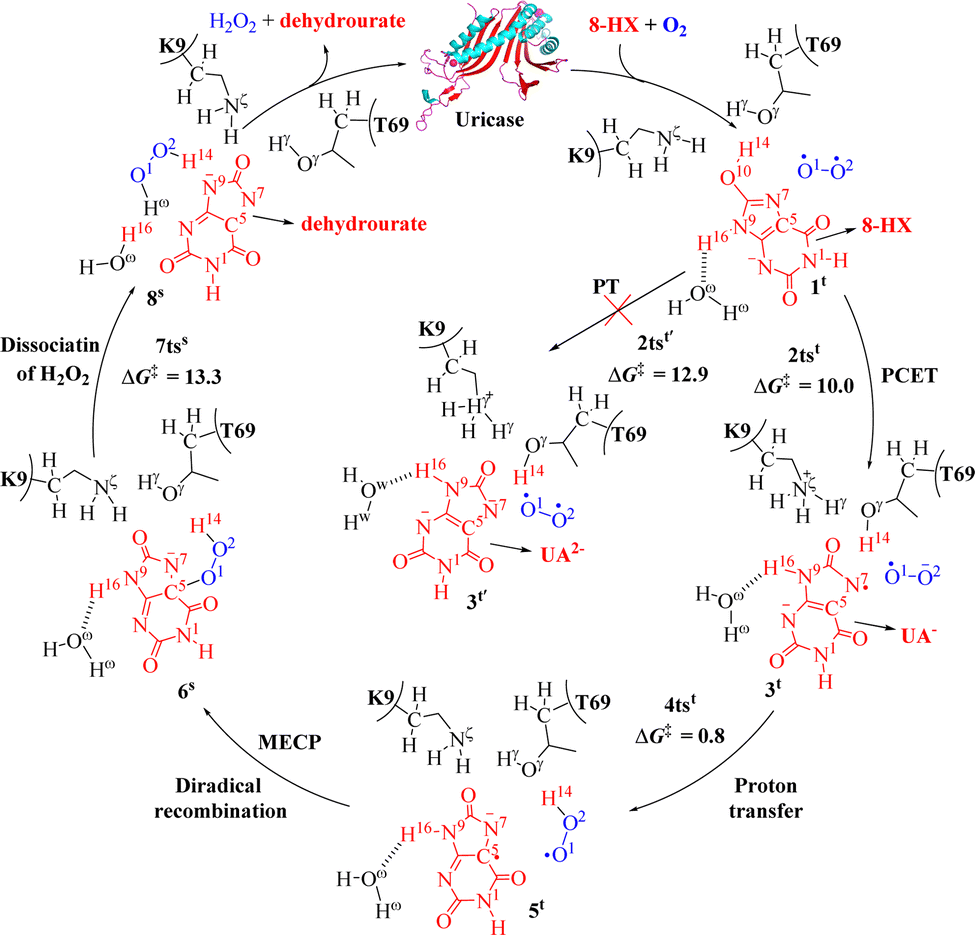 | ||
| Fig. 2 Reaction pathway for the catalytic oxidation of 8-HX by uricase and free energy profile for uricase-catalyzed oxidation of 8-HX (energy in kcal mol−1). The superscript “t” represents a triplet state, the superscript “s” represents a singlet state, the superscript “os” represents an open-shell singlet state. The meanings represented by “t”, “s”, and “os” are the same in Fig. 3 to 8 as in this figure. | ||
In almost all oxidation reactions involving molecular oxygen, changes in spin state are inevitable, involving quite complex dynamic processes that are cross correlated with various potential energy surfaces. Although the issue has not been deeply and widely explored in the cofactor-free oxidase and oxygenase-catalyzed reactions, the solutions have been mentioned in previous literature on the transformation of oxygen. For example, Bearpark et al. presented a method, which avoids the use of Lagrange multipliers, for the optimization of the lowest energy point of the intersection of two potential energy surfaces,110 and implemented in quantum chemistry packages. This method is also applied to the oxidation of flavin by O2.111
It is worth mentioning that during the activation of O2 catalyzed by uricase, protons gradually transfer from 8-HX to T69, and then to K9. Meanwhile, electron transfer occurs from 8-HX to a triplet O2 molecule, occupying the anti-π (π*) orbital of the O2 molecule. Due to this electron transfer, the spin density (SD) of the O2 molecule varies from 1.83 for 1t to 1.61 for 2tst and 1.02 for 3t; the SD value of 8-HX changes from 0.16 for 1t to 0.39 for 2tst and 0.99 for 3t (Fig. 3). The spin density value of the proton involved in the PCET process is zero, so the transfer process of hydrogen atom should not be involved in the reactions. Therefore, after the first step of the reaction, both the substrate 8-HX and O2 become free radicals, which is consistent with the experimental findings of electron spin resonance spectroscopy (ESR) that can capture substrate free radicals.112
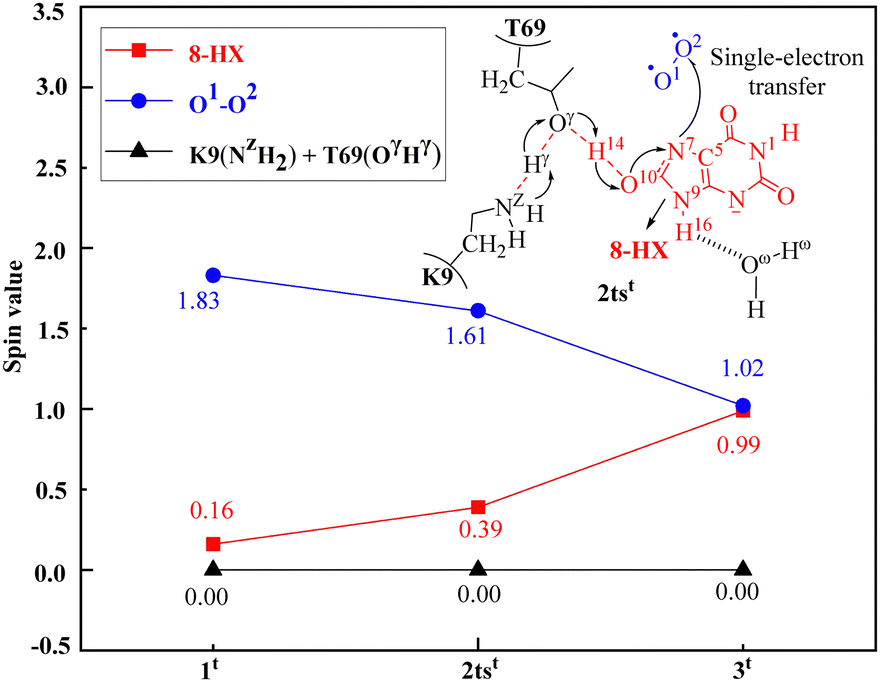 | ||
| Fig. 3 Spin densities of the substrate 8-HX, O2, the residue K9(NZH2), and the residue T69(OγHγ) in the critical transition state 2tst at the oxidation stage of the uricase-catalyzed reaction. | ||
As noted above, the intermediate 3t′ will not exist at all in the active site under aerobic conditions, because it can automatically transfer an electron to the nearby O2 molecule and activate the O2 molecule. As a result of the O2 activation, the O–O distance (RO1–O2) in the oxygen molecule changes from 1.23 Å in 1t to 1.26 Å in 2tst and to 1.34 Å in 3t. The first reaction step produces ˙UA− + ˙O2−, rather than UA2− + O2, due to the automatic electron transfer from UA2− to O2 coupled with the O2 activation. The O2 activation is associated with the increase in the O–O bond length (RO1–O2). To further test this point, Zhan and co-workers carried out the aforementioned QM/MM reaction-coordinate calculations, but with the O–O bond length (RO1–O2) frozen at that (∼1.23 Å) in the optimized 1t structure.32 The QM/MM reaction-coordinate calculations with reaction coordinate RO1–O2 frozen indeed led to UA2− and triplet O2 in the active site, but with a significantly higher energy barrier (12.9 kcal mol−1, Fig. 2). The calculation results indicate that O2 would not accept an electron from UA2− when the O–O bond length was frozen and the reaction pathway via ˙UA− + ˙O2− is the more favourable pathway. This is of great significance for understanding the metabolic process of O2 in living organisms.
It is well known that computational design and discovery of a high-activity mutant of an enzyme are extremely challenging and labour-intensive, because a truly reliable mutant design must be based on more extensive QM/MM calculations of the free energy profiles of various possible hypothetical mutants. Therefore, before carrying out extensive computational design and experiments for the oxidase and oxygenase redesign, Zhan and co-workers ensured that the key residues could affect the catalytic activity of the enzyme. For example, according to the calculation results of Zhan et al.,32 residues Asn271 and Gln299 were identified as key residues during the calculation process. In order to verify the importance of Asn271 and Gln299, they examined the effects of Asn271Leu and Gln299Ile mutations on the catalytic activity (Vmax) of Bacillus fastidiosus uricase by carrying out experimental studies including site-directed mutagenesis, protein expression, and in vitro enzyme activity assays. According to the in vitro experimental data, with the Asn271Leu mutation, the uricase activity was too low to be detected (or with Vmax < 1% of Vmax for the wild-type enzyme) within the detection limit of our activity assay. The Gln299Ile mutation also significantly decreased the Vmax value of the enzyme by ∼13-fold; the Gln299Ile mutant had ∼8% activity compared to the wild-type uricase. The experimental data are consistent with the computationally revealed roles of Asn271 and Gln299 in the enzymatic reaction process. It is worth mentioning that most of the catalytic functions of enzymes are strictly controlled by electrostatic interactions.113 The design of point mutations is a highly challenging task because the mutated residue not only changes its own (electrostatic) interactions with the reaction site, but also slightly alters the structure of the enzyme near the mutation point, thereby changing the entire pattern of electrostatic interactions with the active site. This novel PCET mechanism of uricase-catalyzed activation of oxygen provides a mechanistic base for the rational design of uricase mutants with improved catalytic activity against uric acid as an enhanced enzyme therapy.
2.2 Cofactor-free monooxygenase ActVA-Orf6
The ActVA-Orf6 protein is a homodimer quinone-forming monooxygenase with A and B subunits from Streptomyces coelicolor. It requires only molecular O2 without any assistance from the prosthetic group (i.e., metal ions and cofactors). In addition, it can catalyze the oxygenation of 6-deoxydihydrokalafungin (DDHK) to dihydrokalafungin (DHK) by releasing H2O in the actinorhodin biosynthetic pathway.23 Many efforts have been made to study the oxygenation mechanism of ActVA-Orf6 monooxygenase through spectroscopic characterization, kinetic studies, and other determination experimentally.114–118Noteworthy, Wei and co-workers have studied a PCET mechanism for O2 activation in cofactor-free monooxygenase ActVA-Orf6-catalyzed oxygenation of DDHK to form DHK and the QM/MM calculations were performed at the [B3LYP-D3/6-311++G(2df,2pd):AMBER]//[B3LYP/6-31G(d,p):AMBER] level.33 As shown in Fig. 4, the first step is a PCET process via transition state 10tst (15.5 kcal mol−1) to activate O2, which is assisted by one water molecule forming intermediate 11t. Subsequently, 11t is easy to transform into an open-shell singlet intermediate 12os, since the structures and energies are similar and almost degenerate, respectively. Notably, the spin-flipping process might be involved during the transformation process between singlet and triplet, and computations on the energy barrier of spin-flipping process should be good choice for solving this issue.119 Then the diradical complexation occurs via the transition state 13os (10.6 kcal mol−1) into a closed-shell singlet intermediate 14s with the formation of a C13–O2 bond. The final step is a dehydration process via the transition state 15tss (20.7 kcal mol−1) accompanied by the generation of monooxygenation DHK− products. It is worth mentioning that since the substrate is identified to exist in a negative deprotonated state in the active center of enzyme catalysis, an intermolecular single electron transfer (SET) process is likely to occur. The relative Gibbs free energy for the SET process is 53.3 kcal mol−1 in the QM computational modeling, indicating that the SET process is impossible to occur under experimental condition. In addition, as reported,120,121 the reaction rate constant kcat is equal to 4.17 × 10−2 s−1 at 303 K in experiments of ActVA-Orf6 monooxygenase-catalyzed oxidation of phenols to quinones. The value of the calculated energy barrier of 20.7 kcal mol−1 is very close to the energy barrier 19.7 kcal mol−1 estimated by experiments. It is worth mentioning that in the catalytic oxidation process of monooxygenase ActVA-Orf6, the residues R86 and W66 form hydrogen bonds with substrate DDHK− in the active cavity (Fig. 4).
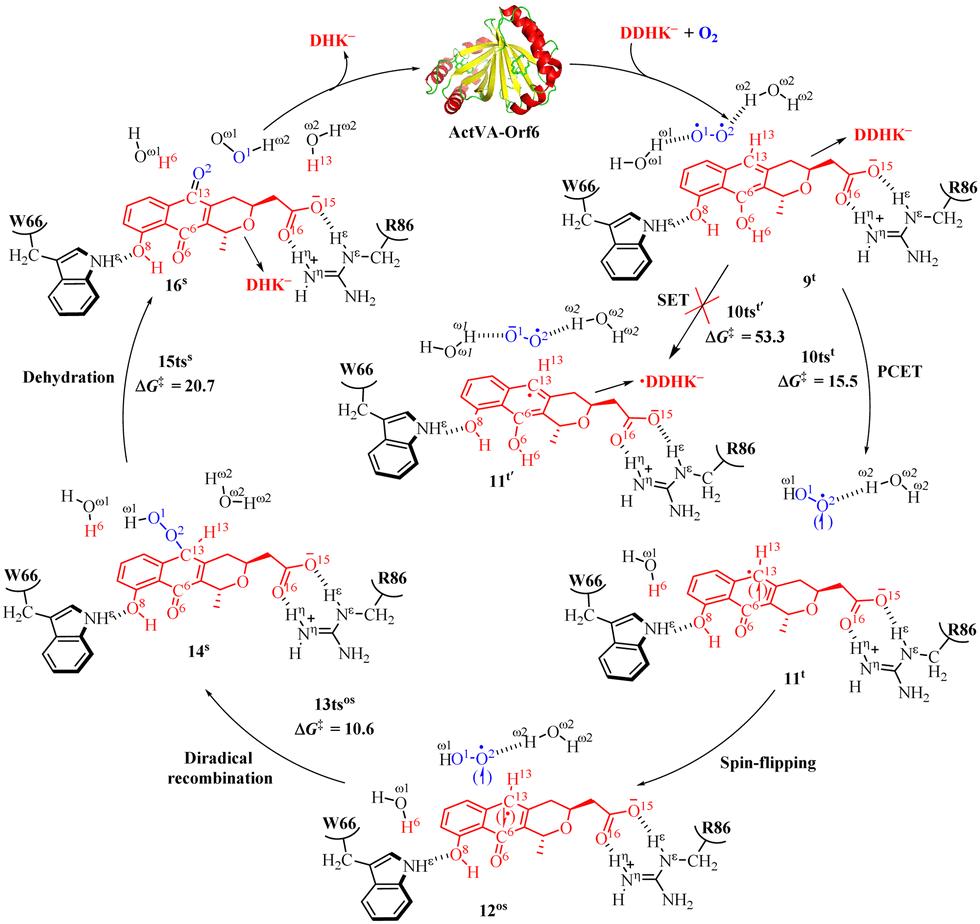 | ||
| Fig. 4 Reaction pathway of catalytic oxidation of DDHK− by monooxygenase ActVA-Orf6 and free energy profile for ActVA-Orf6-catalyzed oxidation of DDHK− (energy in kcal mol−1). | ||
In addition, the PCET process has been confirmed by analyzing the SD value changes in the first step. As shown in Fig. 5, the SD value of O2 decreases from 1.97 in 9t to 1.21 in transition state 10tst, and finally to 1.01 in 11t, while the SD value of DDHK− increases from 0.02 in 9t to 0.77 in transition state 10tst, and eventually to 0.99 in 11t. This proves that the activation of O2 occurs through the PCET process. This PCET mechanism also provides new insights into the mechanism of O2 activation by monooxygenases.
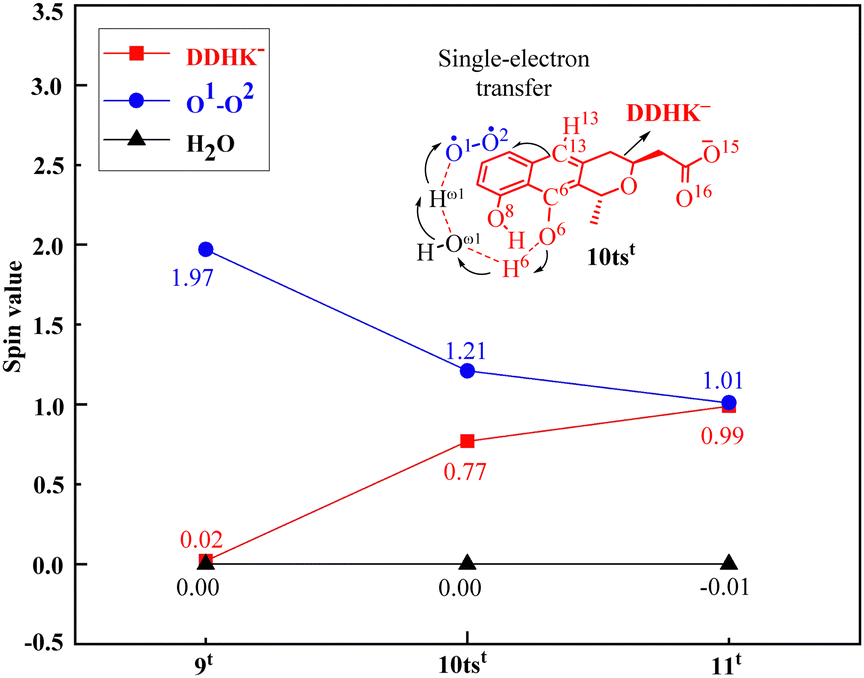 | ||
| Fig. 5 Spin densities of the substrate DDHK−, O2, and H2O via the critical transition state 10tst of the monooxygenase ActVA-Orf6-catalyzed oxidation reaction. | ||
2.3 Cofactor-free dioxygenase HOD
A prototypical cofactor-free dioxygenase is 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (HOD) from Arthrobacter nitroguajacolicus Rü61a.39 HOD can effectively promote the degradation of quinoline pollutants in the natural environment. Moreover, HOD is also useful to reduce both virulence and bacterial growth of P. aeruginosa in leaf tissues in plants.39 It catalyzes the oxygenolytic ring-opening of the N-heteroaromatic substrate (1-H-3-hydroxy-4-oxoquinaldine, QND) with concomitant release of carbon monoxide.34,122–124In the past, extensive experimental studies have been conducted on the reaction mechanism of HOD through spectroscopic characterization, kinetic studies, and various enzymatic determinations.25,125 In 2023, Wei and coworkers proposed the PCET mechanism for the O2 activation in HOD-catalyzed oxidation reaction of QND, and the QM/MM calculations were performed at the [B3LYP-D3/6-311+G(d,p):Amber]//[B3LYP/6-31G(d,p):Amber] level.35
As depicted in Fig. 6, the first step is a PCET process to activate O2 assisted by the H251/D126 dyad to form intermediate 19tvia transition state 18tst (6.7 kcal mol−1). It is followed by the recombination of diradical in 20os into a closed-shell singlet intermediate 22s with C2–O2 bond formation via transition state 21tsos (0.3 kcal mol−1). It should be noted that the structures of 19t and 20os are similar, and their energies are degenerate. After that, a ring-closure process occurs to form a bicyclic structure via transition state 23tss (4.9 kcal mol−1). The final step is the release of CO via transition state 25tss (10.5 kcal mol−1) followed by the formation of N-acetyl-anthranilate product. Further, the experimentally measured rate constant (kcat)126 for the HOD-catalyzed N-heteroaromatic ring cleavage reaction is 38.4 s−1 for HOD, and the kcat value can be used to estimate the energy barrier for the reaction. Wei and co-workers calculated the energy barrier (14.9 kcal mol−1) for the entire HOD-catalyzed reaction to be close to the experimental observation127 that the energy barrier is 15.5 kcal mol−1. In the catalytic oxidation process of dioxygenase HOD, the residues D126 and H251 form hydrogen bonds with the substrate in the active cavity and can participate in the following proton transfer process, and the residues H102 form hydrogen bonds with intermediate 22S in the active cavity (Fig. 6).
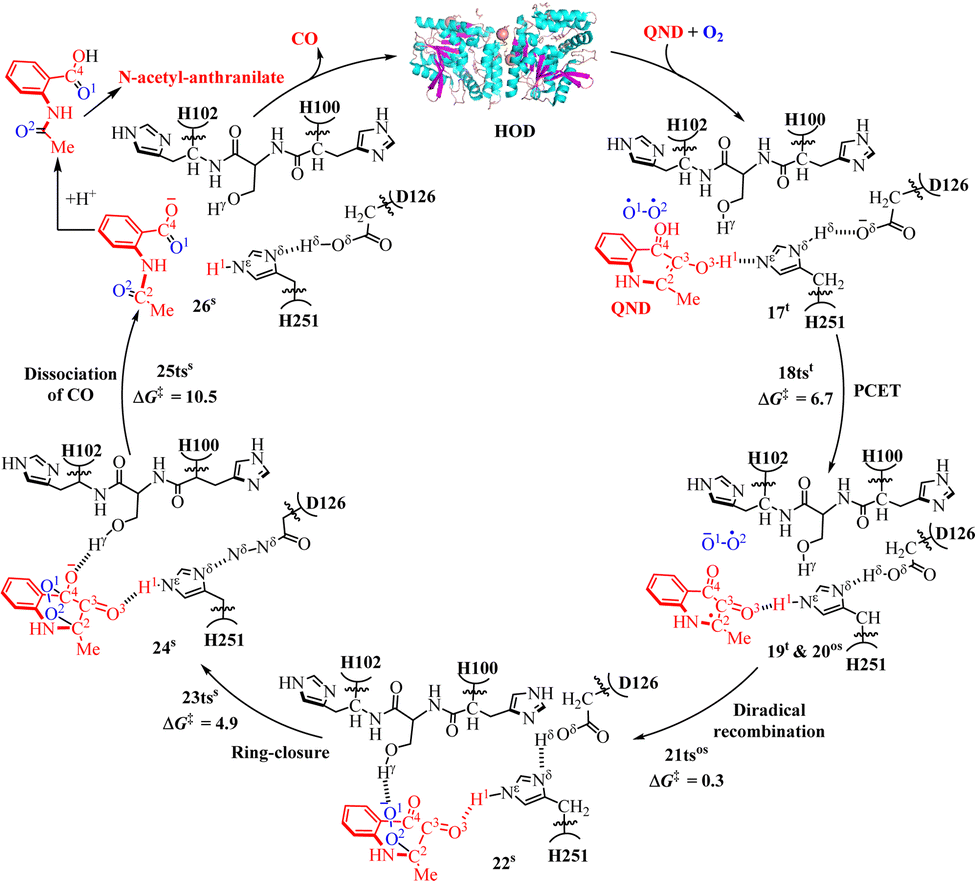 | ||
| Fig. 6 Reaction pathway for catalytic oxidation of QND by cofactor-free dioxygenase HOD and potential energy profile for the HOD-catalyzed N-heteroaromatic ring cleavage reaction (energy in kcal mol−1). | ||
It is worth mentioning that Fig. 7 clearly illustrates the PCET process in the first step of the reaction. As shown in Fig. 7, the SD value of the O2 molecule changes from 1.97 in 17t to 1.66 in 18tst and to 1.04 in 19t, while the SD value of QND changes from 0.03 in 17t to 0.34 in 18tst and to 0.96 in 19t. Therefore, it can be concluded that both the substrate QND and O2 become radicals after the first step of the reaction, and this result is consistent with the experimental observation that the existence of substrate radicals and superoxide anion radicals was detected in the electron spin resonance spectroscopy (ESR) studies. This indicates that the PCET mechanism could be a general mechanism for the cofactor-free dioxygenase-catalyzed activation of O2.
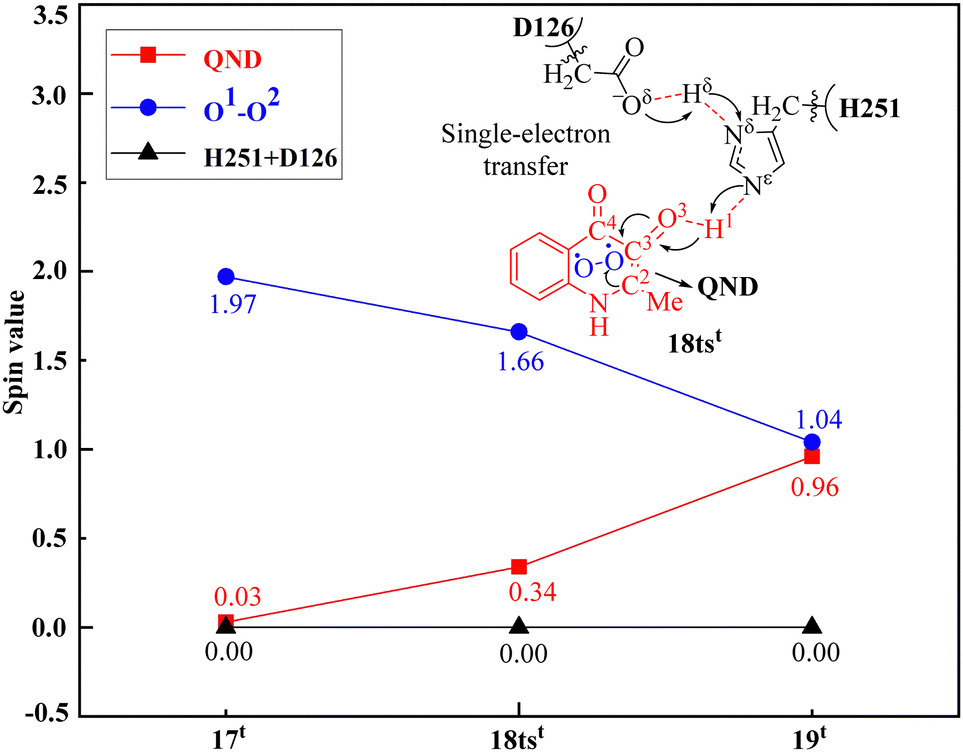 | ||
| Fig. 7 Spin densities (SDs) of the substrate QND, O2, residue H251, and residue D126 via the critical transition state 18tst in the dioxygenase HOD-catalyzed reaction. | ||
3 Flavin-dependent monooxygenase RutA
RutA belongs to a large family of flavoprotein monooxygenases (FMOs) that are widely spread across all kingdoms of life and catalyze the uracil-ring degradation to yield ureidoacrylate, allowing bacteria to utilize pyrimidine rings as the nitrogen source.60,128,129 Recent research by Wang and co-workers proposed the PCET mechanism for O2 activation in the RutA-catalyzed reaction and the QM/MM calculations were performed at the [B3LYP-D3/def2-TZVP]//[B3LYP-D3/def2-SVP] level.70 As shown in Fig. 8, the first step is the PCET process to activate O2 assisted by FMN− to form 29tvia transition state 28tst (4.9 kcal mol−1). Then the 29t intermediate flips from the triplet state to the open-shell singlet state 30os. Then it is followed by diradical recombination in 30os into a closed-shell singlet intermediate 32s with N5–O1 bond formation via transition state 31tsos (17.9 kcal mol−1). The third step is the cleavage of O1–O2 bond coupled with the formation of C4–O2 bond via transition state 34tss (1.6 kcal mol−1). The fourth step is the ring-cleavage via transition state 36tss (0.4 kcal mol−1). The final step involves a proton transfer coupled with the formation of 3-ureidoacrylate product via transition state 38tss (0.4 kcal mol−1). This indicates that the PCET mechanism could be a general mechanism for the flavin-dependent oxygenase-catalyzed activation of O2. | ||
| Fig. 8 Reaction pathway for the catalytic C–N cleavage of uracil by flavin-dependent monooxygenase RutA (free energy in kcal mol−1). | ||
Furthermore, the PCET mechanism in enzymes involving O2 activation has been supported by the recent experimental study. For example, Zhang and co-workers reported that the interaction between an electron donor and a proton donor could overcome the barrier of direct O2 activation via a proton-coupled electron transfer mechanism.130 It is worth mentioning that our group have proposed and confirmed a new mechanistic model named relayed proton-coupled electron transfer (relayed-PCET) for diradical generation in NHC organocatalysis and organic reaction systems,131–133 and Duan et al. has further confirmed the existence of relayed-PCET mechanism in the chiral phosphoric acid (CPA)-catalyzed reaction.134 As shown in Scheme 2, the relayed-PCET transition state is an effective transformation process from nonradical species to diradical species, in which the proton transfer is coupled with an electron transfer from electron donor (D) to acceptor (A). It could open a new door for understanding the diradical generation mechanism in both organic and enzymatic reactions. Therefore, exploring the new PCET mechanisms in the enzymes could be an interesting research area in theoretical and computational chemistry fields.
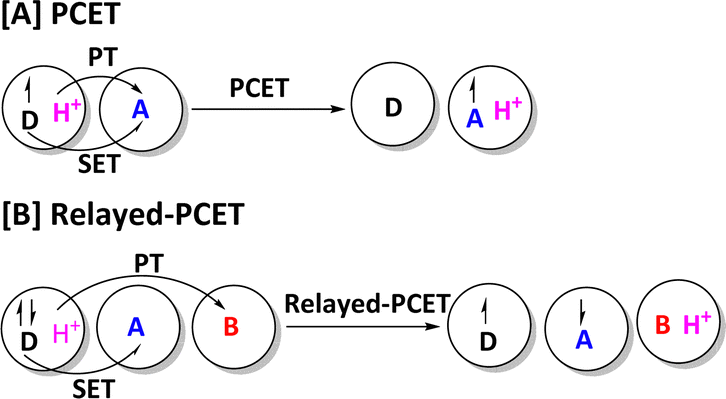 | ||
| Scheme 2 (A) and (B) Conceptual difference between standard PCET and relayed-PCET. In relayed-PCET transition state, electron donor (D), electron acceptor (A), and base (B) are all initially nonradical species. | ||
4 Conclusion
This review summarizes the general PCET mechanisms of multiple cofactor-free oxidase- and oxygenase-catalyzed activations of O2 and the universal rules of enzyme-catalyzed metabolism of organic compounds in living organisms, thereby providing a theoretical basis for designing more efficient novel enzyme mutants. In addition, this kind of PCET mechanism also exists in flavin-dependent oxygenases and biomimetic organocatalytic reactions. It is worth mentioning that the detailed mechanism of a triplet intermediate to open-shell singlet intermediate remains a challenge during the cofactor-free enzyme-catalyzed activation of O2. In the process of enzyme-catalyzed activation of O2, the PCET mechanism progresses keep in open-shell doublet system, and the remarkably different mechanism involving relayed-PCET transition states in enzyme-catalyzed reactions should be another important issue to explore in future.Author contributions
Qian-Qian Wang: conceptualization, writing – original draft, literature arrangement. Yan Qiao: discussion, literature research, writing – review & editing. Donghui Wei: conceptualization, supervision, funding acquisition, writing – review & editing.Data availability
The data supporting this review can be found in the cited reference part.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Key Project of the Joint Fund for Science and Technology Research and Development in Henan Province (no. 232301420008), the National Natural Science Fund of China (no. 22473100 and 21773214), the Basic Research and Cultivation Project for Young Teachers at Zhengzhou University (no. JC23261010), and the Training Plan for Young Key Teachers in Colleges and Universities in Henan Province (2020GGJS016).Notes and references
- E. Romero, J. R. Gómez Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Same Substrate, Many Reactions: Oxygen Activation in Flavoenzymes, Chem. Rev., 2018, 118, 1742–1769 CrossRef CAS PubMed.
- S. Fetzner, Oxygenases without requirement for cofactors or metal ions, Appl. Microbiol. Biotechnol., 2002, 60, 243–257 CrossRef CAS.
- T. D. H. Bugg, Dioxygenase enzymes: catalytic mechanisms and chemical models, Tetrahedron, 2003, 59, 7075–7101 CrossRef CAS.
- J. Münch, P. Püllmann, W. Zhang and M. J. Weissenborn, Enzymatic Hydroxylations of sp3-Carbons, ACS Catal., 2021, 11, 9168–9203 CrossRef.
- D. Troiano, V. Orsat and M.-J. Dumont, Status of Biocatalysis in the Production of 2,5-Furandicarboxylic Acid, ACS Catal., 2020, 10, 9145–9169 CrossRef CAS.
- R. Ushimaru and I. Abe, Unusual Dioxygen-Dependent Reactions Catalyzed by Nonheme Iron Enzymes in Natural Product Biosynthesis, ACS Catal., 2023, 13, 1045–1076 CrossRef CAS.
- S. H. Knauer, O. Hartl-Spiegelhauer, S. Schwarzinger, P. Hänzelmann and H. Dobbek, The Fe(II)/α-ketoglutarate-dependent taurine dioxygenases from Pseudomonas putida and Escherichia coli are tetramers, FEBS J., 2012, 279, 816–831 CrossRef CAS PubMed.
- S. Hirotsu, G. C. Chu, M. Unno, D.-S. Lee, T. Yoshida, S.-Y. Park, Y. Shiro and M. Ikeda-Saito, The crystal structures of the ferric and ferrous forms of the heme complex of HmuO, a heme oxygenase of Corynebacterium diphtheriae, J. Biol. Chem., 2004, 279, 11937–11947 CrossRef CAS PubMed.
- A. Matthews, R. Saleem-Batcha, J. N. Sanders, F. Stull, K. N. Houk and R. Teufel, Aminoperoxide adducts expand the catalytic repertoire of flavin monooxygenases, Nat. Chem. Biol., 2020, 16, 556–563 CrossRef CAS PubMed.
- R. Saleem-Batcha, F. Stull, J. N. Sanders, B. S. Moore, B. A. Palfey, K. N. Houk and R. Teufel, Enzymatic control of dioxygen binding and functionalization of the flavin cofactor, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4909–4914 CrossRef CAS.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, Proton-Coupled Electron Transfer Guidelines, Fair and Square, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function, Chem. Rev., 2018, 118, 10840–11022 CrossRef CAS PubMed.
- A. I. Cederbaum, Molecular mechanisms of the microsomal mixed function oxidases and biological and pathological implications, Redox Biol., 2015, 4, 60–73 CrossRef CAS PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, P450 enzymes: their structure, reactivity, and selectivity-modeled by QM/MM calculations, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed.
- B. Meunier, S. P. de Visser and S. Shaik, Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- S. Shaik, D. Kumar, S. P. de Visser, A. Altun and W. Thiel, Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes, Chem. Rev., 2005, 105, 2279–2328 CrossRef CAS.
- F. P. Guengerich and F. K. Yoshimoto, Formation and Cleavage of C–C Bonds by Enzymatic Oxidation-Reduction Reactions, Chem. Rev., 2018, 118, 6573–6655 CrossRef CAS PubMed.
- N. Colloc’h, M. E. Hajji, B. Bachet, G. L’Hermite, M. Schiltz, T. Prangé, B. Castro and J.-P. Mornon, Crystal Structure of the protein drug urate oxidase-inhibitor complex at 2.05 Å resolution, Nat. Struct. Mol. Biol., 1997, 4, 947–952 CrossRef.
- T. Li and J. S. Woods, Cloning, Expression, and Biochemical Properties of CPOX4, a Genetic Variant of Coproporphyrinogen Oxidase that Affects Susceptibilitly to Mercury Toxicity in Humans, Toxicol. Sci., 2009, 109, 228–236 CrossRef CAS PubMed.
- J. D. Phillips, F. G. Whitby, C. A. Warby, P. Labbe, C. Yang, J. W. Pflugrath, J. D. Ferrara, H. Robinson, J. P. Kushner and C. P. Hill, Crystal Structure of the Oxygen-dependant Coproporphyrinogen Oxidase (Hem13p) of Saccharomyces cerevisiae, J. Biol. Chem., 2004, 279, 38960–38968 CrossRef CAS PubMed.
- O. Th Magnusson, H. Toyama, M. Saeki, A. Rojas, J. C. Reed, R. C. Liddington, J. P. Klinman and R. Schwarzenbacher, Quinone biogenesis: structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7913–7918 CrossRef CAS.
- O. Th Magnusson, J. M. RoseFigura, H. Toyama, R. Schwarzenbacher and J. P. Klinman, Pyrroloquinoline Quinone Biogenesis:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Characterization of PqqC and Its H84N and H84A Active Site Variants, Biochemistry, 2007, 46, 7174–7186 CrossRef CAS PubMed.
Characterization of PqqC and Its H84N and H84A Active Site Variants, Biochemistry, 2007, 46, 7174–7186 CrossRef CAS PubMed. - G. Sciara, S. G. Kendrew, A. E. Miele, N. G. Marsh, L. Federici, F. Malatesta, G. Schimperna, C. Savino and B. Vallone, The structure of ActVA-Orf6, a novel type of monooxygenase involved in actinorhodin biosynthesis, EMBO J., 2003, 22, 205–215 CrossRef CAS.
- J. Pootoolal, M. G. Thomas, C. G. Marshall, J. M. Neu, B. K. Hubbard, C. T. Walsh and G. D. Wright, Assembling the glycopeptide antibiotic scaffold: the biosynthesis of from Streptomyces toyocaensis NRRL15009, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8962–8967 CrossRef CAS.
- I. Bauer, N. Max, S. Fetzner and F. Lingens, 2,4-Dioxygenases Catalyzing N-Heterocyclic-Ring Cleavage and Formation of Carbon Monoxide, Eur. J. Biochem., 1996, 240, 576–583 CrossRef CAS PubMed.
- R. A. Steiner, U. Frerichs-Deeken and S. Fetzner, Crystallization and preliminary X-ray analysis of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase from Arthrobacter nitroguajacolicus Rü61a: a cofactor-devoid dioxygenase of the α/β-hydrolase-fold superfamily, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2007, 63, 382–385 CrossRef CAS.
- A. M. Loening, T. D. Fenn and S. S. Gambhir, Crystal Structures of the Luciferase and Green Fluorescent Protein from Renilla reniformis, J. Mol. Biol., 2007, 374, 1017–1028 CrossRef CAS.
- K. Kahn and P. A. Tipton, Spectroscopic characterization of intermediates in the urate oxidase reaction, Biochemistry, 1998, 37, 11651–11659 CrossRef CAS PubMed.
- M. Altarsha, B. Castro and G. Monard, Intrinsic reactivity of uric acid with dioxygen: towards the elucidation of the catalytic mechanism of urate oxidase, Bioorg. Chem., 2009, 37, 111–125 CrossRef CAS.
- P. J. Silva and M. J. Ramos, A comparative density-functional study of the reaction mechanism of the O2-dependent coproporphyrinogen III oxidase, Bioorg. Med. Chem., 2008, 16, 2726–2733 CrossRef CAS.
- K. Kahn, Theoretical Study of Intermediates in the Urate Oxidase Reaction, Bioorg. Chem., 1999, 27, 351–362 CrossRef CAS.
- D. Wei, X. Huang, Y. Qiao, J. Rao, L. Wang, F. Liao and C.-G. Zhan, Catalytic Mechanisms for Cofactor-Free Oxidase-Catalyzed Reactions: Reaction Pathways of Uricase-Catalyzed Oxidation and Hydration of Uric Acid, ACS Catal., 2017, 7, 4623–4636 CrossRef CAS PubMed.
- X. Li, X. Li, Q. Zhang, P. Lv, Y. Jia and D. Wei, Cofactor-free ActVA-Orf6 monooxygenase catalysis via proton-coupled electron transfer: a QM/MM study, Org. Biomol. Chem., 2022, 20, 5525–5534 RSC.
- A. Hernández-Ortega, M. G. Quesne, S. Bui, D. J. Heyes, R. A. Steiner, N. S. Scrutton and S. P. de Visser, Catalytic mechanism of cofactor-free dioxygenases and how they circumvent spin-forbidden oxygenation of their substrates, J. Am. Chem. Soc., 2015, 137, 7474–7487 CrossRef.
- Q.-Y. Zhang, X. Li, J. Luo, X. Li, J. Song and D. Wei, Cofactor-Free Dioxygenases-Catalyzed Reaction Pathway via Proton-Coupled Electron Transfer, J. Phys. Chem. B, 2023, 127, 95–103 CrossRef CAS PubMed.
- R. C. Hart, K. E. Stempel, P. D. Boyer and M. J. Cormier, Mechanism of the enzyme-catalyzed bioluminescent oxidation of coelenterate-type luciferin, Biochem. Biophys. Res. Commun., 1978, 81, 980–986 CrossRef CAS.
- E. Oksanen, M. P. Blakeley, M. El-Hajji, U. Ryde and M. Budayova-Spano, The Neutron Structure of Urate Oxidase Resolves a Long-Standing Mechanistic Conundrum and Reveals Unexpected Changes in Protonation, PLoS One, 2014, 9, e86651 CrossRef.
- K. Li, E. N. Fielding, H. L. Condurso and S. D. Bruner, Probing the structural basis of oxygen binding in a cofactor-independent dioxygenase, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 573–580 CrossRef CAS.
- R. A. Steiner, H. J. Janssen, P. Roversi, A. J. Oakley and S. Fetzner, Structural basis for cofactor-independent dioxygenation of N-heteroaromatic compounds at the alpha/beta-hydrolase fold, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 657–662 CrossRef CAS.
- C. Gui, E. Kalkreuter, Y.-C. Liu, G. Li, A. D. Steele, D. Yang, C. Chang and B. Shen, Cofactorless oxygenases guide anthraquinone-fused enediyne biosynthesis, Nat. Chem. Biol., 2024, 20, 243–250 CrossRef CAS PubMed.
- U. Frerichs-Deeken, K. Ranguelova, R. Kappl, J. Hüttermann and S. Fetzner, Dioxygenases without Requirement for Cofactors and Their Chemical Model Reaction: Compulsory Order Ternary Complex Mechanism of 1H-3-Hydroxy-4-oxoquinaldine 2,4-Dioxygenase Involving General Base Catalysis by Histidine 251 and Single-Electron Oxidation of the Substrate Dianion, Biochemistry, 2004, 43, 14485–14499 CrossRef CAS PubMed.
- S. Bui and R. A. Steiner, New insight into cofactor-free oxygenation from combined experimental and computational approaches, Curr. Opin. Struct. Biol., 2016, 41, 109–118 CrossRef CAS PubMed.
- M. H. V. Huynh and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Theory of proton-coupled electron transfer in energy conversion processes, Acc. Chem. Res., 2009, 42, 1881–1889 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Proton-coupled electron transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Proton-Coupled Electron Transfer: Moving Together and Charging Forward, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS.
- R. E. Warburton, A. V. Soudackov and S. Hammes-Schiffer, Theoretical Modeling of Electrochemical Proton-Coupled Electron Transfer, Chem. Rev., 2022, 122, 10599–10650 CrossRef CAS PubMed.
- A. Mittal, V. K. Vashistha and D. K. Das, Recent advances in the antioxidant activity and mechanisms of chalcone derivatives: a computational review, Free Radical Res., 2022, 56, 378–397 CrossRef CAS PubMed.
- S. Hammes-Schiffer, E. Hatcher, H. Ishikita, J. H. Skone and A. V. Soudackov, Theoretical Studies of Proton-Coupled Electron Transfer: Models and Concepts Relevant to Bioenergetics, Coord. Chem. Rev., 2008, 252, 384–394 CrossRef CAS.
- J. M. Mayer, Proton-coupled electron transfer: a reaction chemist's view, Annu. Rev. Phys. Chem., 2004, 55, 363–390 CrossRef CAS.
- T. Liu, M. Guo, A. Orthaber, R. Lomoth, M. Lundberg, S. Ott and L. Hammarström, Accelerating proton-coupled electron transfer of metal hydrides in catalyst model reactions, Nat. Chem., 2018, 10, 881–887 CrossRef CAS PubMed.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Hydrogen Atom Transfer Reactions of Mononuclear Nonheme Metal–Oxygen Intermediates, Acc. Chem. Res., 2018, 51, 2014–2022 CrossRef CAS.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS.
- E. C. Gentry and R. R. Knowles, Synthetic Applications of Proton-Coupled Electron Transfer, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS.
- D. G. Nocera, Proton-Coupled Electron Transfer: The Engine of Energy Conversion and Storage, J. Am. Chem. Soc., 2022, 144, 1069–1081 CrossRef CAS.
- J. Zelenka, R. Cibulka and J. Roithová, Flavinium Catalysed Photooxidation: Detection and Characterization of Elusive Peroxyflavinium Intermediates, Angew. Chem., Int. Ed., 2019, 58, 15412–15420 CrossRef CAS.
- S. Y. Reece, J. M. Hodgkiss, J. Stubbe and D. G. Nocera, Proton-coupled electron transfer: the mechanistic underpinning for radical transport and catalysis in biology, Philos. Trans. R. Soc., B, 2006, 361, 1351–1364 CrossRef CAS.
- M. Toplak, A. Matthews and R. Teufel, The devil is in the details: the chemical basis and mechanistic versatility of flavoprotein monooxygenases, Arch. Biochem. Biophys., 2021, 698, 108732 CrossRef CAS.
- P. Chenprakhon, T. Wongnate and P. Chaiyen, Monooxygenation of aromatic compounds by flavin-dependent monooxygenases, Protein Sci., 2019, 28, 8–29 CrossRef CAS.
- C. E. Paul, D. Eggerichs, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Flavoprotein monooxygenases: versatile biocatalysts, Biotechnol. Adv., 2021, 51, 107712 CrossRef CAS.
- T. Shi, X. Sun, Q. Yuan, J. Wang and X. Shen, Exploring the role of flavin-dependent monooxygenases in the biosynthesis of aromatic compounds, Biotechnol. Biofuels, 2024, 17, 46 CrossRef CAS.
- Q. Wang, N. Liu, Y. Deng, Y. Guan, H. Xiao, T. A. Nitka, H. Yang, A. Yadav, L. Vukovic, I. I. Mathews, X. Chen and C.-Y. Kim, Triepoxide formation by a flavin-dependent monooxygenase in monensin biosynthesis, Nat. Commun., 2023, 14, 6273 CrossRef CAS PubMed.
- S. Hazra and T. P. Begley, Alkylcysteine Sulfoxide C–S Monooxygenase Uses a Flavin-Dependent Pummerer Rearrangement, J. Am. Chem. Soc., 2023, 145, 11933–11938 CrossRef CAS PubMed.
- L. Xiang, J. Shi, A. Zhu, Z. F. Xu, S. H. Liu, Y. S. Wang, Z. K. Guo, R. H. Jiao, R. X. Tan and H. M. Ge, Total Biosynthesis of Mutaxanthene Unveils a Flavoprotein Monooxygenase Catalyzing Xanthene Ring Formation, Angew. Chem., Int. Ed., 2023, 62, e202218660 CrossRef CAS PubMed.
- Y. Deng, Q. Zhou, Y. Wu, X. Chen and F. Zhong, Properties and Mechanisms of Flavin-Dependent Monooxygenases and Their Applications in Natural Product Synthesis, Int. J. Mol. Sci., 2022, 23, 2622 CrossRef CAS.
- Y. Duan, M. Toplak, A. Hou, N. L. Brock, J. S. Dickschat and R. Teufel, A Flavoprotein Dioxygenase Steers Bacterial Tropone Biosynthesis via Coenzyme A-Ester Oxygenolysis and Ring Epoxidation, J. Am. Chem. Soc., 2021, 143, 10413–10421 CrossRef CAS.
- C. Yang, L. Zhang, W. Zhang, C. Huang, Y. Zhu, X. Jiang, W. Liu, M. Zhao, B. C. De and C. Zhang, Biochemical and structural insights of multifunctional flavin-dependent monooxygenase FlsO1-catalyzed unexpected xanthone formation, Nat. Commun., 2022, 13, 5386 CrossRef CAS PubMed.
- B. Grigorenko, T. Domratcheva and A. Nemukhin, QM/MM Modeling of the Flavin Functionalization in the RutA Monooxygenase, Molecules, 2023, 28, 2405 CrossRef CAS PubMed.
- C. T. Walsh and T. A. Wencewicz, Flavoenzymes: versatile catalysts in biosynthetic pathways, Nat. Prod. Rep., 2013, 30, 175–200 RSC.
- Q. Zhang, Q. Chen, S. Shaik and B. Wang, Flavin-N5OOH Functions as both a Powerful Nucleophile and a Base in the Superfamily of Flavoenzymes, Angew. Chem., Int. Ed., 2024, 63, e202318629 CrossRef CAS PubMed.
- W. J. H. van Berkel, N. M. Kamerbeek and M. W. Fraaije, Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts, J. Biotechnol., 2006, 124, 670–689 CrossRef CAS PubMed.
- A. M. Chánique, N. Polidori, L. Sovic, D. Kracher, L. Assil-Companioni, P. Galuska, L. P. Parra, K. Gruber and R. Kourist, A Cold-Active Flavin-Dependent Monooxygenase from Janthinobacterium svalbardensis Unlocks Applications of Baeyer-Villiger Monooxygenases at Low Temperature, ACS Catal., 2023, 13, 3549–3562 CrossRef.
- A. R. Benítez, S. Tweedy, S. A. Baker Dockrey, A. L. Lukowski, T. Wymore, D. Khare, C. L. Brooks, B. A. Palfey, J. L. Smith and A. R. H. Narayan, Structural basis for selectivity in flavin-dependent monooxygenase-catalyzed oxidative dearomatization, ACS Catal., 2019, 9, 3633–3640 CrossRef PubMed.
- I. V. Polyakov, A. V. Nemukhin, T. M. Domratcheva, A. M. Kulakova and B. L. Grigorenko, Quantum-based Modeling of Protein-ligand Interaction: The Complex of RutA with Uracil and Molecular Oxygen, Mol. Inf., 2023, 42, e2200175 CrossRef PubMed.
- C.-H. Chiang, T. Wymore, A. Rodríguez Benítez, A. Hussain, J. L. Smith, C. L. Brooks and A. R. H. Narayan, Deciphering the evolution of flavin-dependent monooxygenase stereoselectivity using ancestral sequence reconstruction, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218248120 CrossRef CAS.
- J. M. Robbins and H. R. Ellis, Investigations of two-component flavin-dependent monooxygenase systems, Methods Enzymol., 2019, 620, 399–422 CAS.
- Q. Li, S. Zhang, F. Liu, H. Su and X. Sheng, Quantum chemical modeling of enantioselective sulfoxidation and epoxidation reactions by indole monooxygenase VpIndA1, Phys. Chem. Chem. Phys., 2024, 26, 16521–16528 RSC.
- T. Matsushita, S. Kishimoto, K. Hara, H. Hashimoto, H. Yamaguchi, Y. Saito and K. Watanabe, Functional Enhancement of Flavin-Containing Monooxygenase through Machine Learning Methodology, ACS Catal., 2024, 14, 6945–6951 CrossRef CAS.
- J. Kratky, D. Eggerichs, T. Heine, S. Hofmann, P. Sowa, R. H. Weiße, D. Tischler and N. Sträter, Structural and Mechanistic Studies on Substrate and Stereoselectivity of the Indole Monooxygenase VpIndA1: New Avenues for Biocatalytic Epoxidations and Sulfoxidations, Angew. Chem., Int. Ed., 2023, 62, e202300657 CrossRef CAS PubMed.
- P. Ferreira, R. P. P. Neves, F. P. Miranda, A. V. Cunha, R. W. A. Havenith, M. J. Ramos and P. A. Fernandes, DszA Catalyzes C–S Bond Cleavage through N5-Hydroperoxyl Formation, J. Chem. Inf. Model., 2024, 64, 4218–4230 CrossRef CAS PubMed.
- R. D. Barker, Y. Yu, L. De Maria, L. O. Johannissen and N. S. Scrutton, Mechanism of Action of Flavin-Dependent Halogenases, ACS Catal., 2022, 12, 15352–15360 CrossRef CAS PubMed.
- L. D. Kennedy and V. O. Ajiboye, Rasburicase for the prevention and treatment of hyperuricemia in tumor lysis syndrome, J. Oncol. Pharm. Pract., 2010, 16, 205–213 CrossRef CAS PubMed.
- Y. Zhao, L. Zhao, G. Yang, J. Tao, Y. Bu and F. Liao, Characterization of a uricase from Bacillus fastidious A.T.C.C. 26904 and its application to serum uric acid assay by a patented kinetic uricase method, Appl. Biochem., 2006, 45, 75–80 CrossRef CAS.
- Y. Zhao, X. Yang, X. Li, Y. Bu, P. Deng, C. Zhang, J. Feng, Y. Xie, S. Zhu, H. Yuan, M. Yu and F. Liao, Reversible inactivation of an intracellular uricase from Bacillus fastidiosus via dissociation of homotetramer into homodimers in solutions of low ionic strength, Biosci., Biotechnol., Biochem., 2009, 73, 2141–2144 CrossRef CAS PubMed.
- C. Zhang, X. Yang, J. Feng, Y. Yuan, X. Li, Y. Bu, Y. Xie, H. Yuan and F. Liao, Effects of modification of amino groups with poly (ethylene glycol) on a recombinant uricase from Bacillus fastidiosus, Biosci., Biotechnol., Biochem., 2010, 74, 1298–1301 CrossRef CAS PubMed.
- J. Feng, X. Li, X. Yang, C. Zhang, Y. Yuan, J. Pu, Y. Zhao, Y. Xie, H. Yuan, Y. Bu and F. Liao, A new practical system for evaluating the pharmacological properties of uricase as a potential drug for hyperuricemia, Arch. Pharmacal Res., 2010, 33, 1761–1769 CrossRef CAS.
- P. Bose and O. Qubaiah, A review of tumour lysis syndrome with targeted therapies and the role of rasburicase, J. Clin. Pharm. Ther., 2011, 36, 299–326 CrossRef CAS PubMed.
- P. Richette, C. Brière, V. Hoenen-Clavert, D. Loeuille and T. Bardin, Rasburicase for tophaceous gout not treatable with allopurinol: an exploratory study, J. Rheumatol., 2007, 34, 2093–2098 CAS.
- C.-H. Pui, Rasburicase: a potent uricolytic agent, Expert Opin. Pharmacother., 2002, 3, 433–442 CrossRef CAS.
- M. R. Sherman, M. G. P. Saifer and F. Perez-Ruiz, PEG-uricase in the management of treatment-resistant gout and hyperuricemia, Adv. Drug Delivery Rev., 2008, 60, 59–68 CrossRef CAS PubMed.
- M. S. Hershfield, L. J. Roberts, N. J. Ganson, S. J. Kelly, I. Santisteban, E. Scarlett, D. Jaggers and J. S. Sundy, Treating gout with pegloticase, a PEGylated urate oxidase, provides insight into the importance of uric acid as an antioxidant in vivo, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14351–14356 CrossRef CAS.
- M. Darmon, I. Guichard and F. Vincent, Rasburicase and tumor lysis syndrome: lower dosage, consideration of indications, and hyperhydration, J. Clin. Oncol., 2011, 29, e67–e68 CrossRef ; author reply e69.
- O. M. Pitts and D. G. Priest, Uricase reaction intermediate. Mechanism of borate and hydroxide ion catalysis, Biochemistry, 1973, 12, 1358–1363 CrossRef CAS PubMed.
- L. McGregor, T. Földes, S. Bui, M. Moulin, N. Coquelle, M. P. Blakeley, E. Rosta and R. A. Steiner, Joint neutron/X-ray crystal structure of a mechanistically relevant complex of perdeuterated urate oxidase and simulations provide insight into the hydration step of catalysis, IUCrJ, 2021, 8, 46–59 CrossRef CAS.
- Y. Dong, Y. Chi, X. Lin, L. Zheng, L. Chen and G. Chen, Nano-sized platinum as a mimic of uricase catalyzing the oxidative degradation of uric acid, Phys. Chem. Chem. Phys., 2011, 13, 6319–6324 RSC.
- L. Muñoz-Rugeles, A. Galano and J. R. Alvarez-Idaboy, The role of acid-base equilibria in formal hydrogen transfer reactions: tryptophan radical repair by uric acid as a paradigmatic case, Phys. Chem. Chem. Phys., 2017, 19, 15296–15309 RSC.
- M. Li, W. Tang and J. Gong, Unusual shape-preserved pathway of a core-shell phase transition triggered by orientational disorder, IUCrJ, 2023, 10, 38–51 CrossRef CAS PubMed.
- Y. Zhang, H. Liu and W. Yang, Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio QM/MM potential energy surface, J. Chem. Phys., 2000, 112, 3483–3492 CrossRef CAS.
- Y. Zhang, T. S. Lee and W. Yang, A pseudobond approach to combining quantum mechanical and molecular mechanical methods, J. Chem. Phys., 1999, 110, 46–54 CrossRef CAS.
- Y. Pan, D. Gao, W. Yang, H. Cho and C.-G. Zhan, Free Energy Perturbation (FEP) Simulation on the Transition-States of Cocaine Hydrolysis Catalyzed by Human Butyrylcholinesterase and Its Mutants, J. Am. Chem. Soc., 2007, 129, 13537–13543 CrossRef CAS PubMed.
- X. Huang, F. Zheng and C.-G. Zhan, Modeling differential binding of alpha4beta2 nicotinic acetylcholine receptor with agonists and antagonists, J. Am. Chem. Soc., 2008, 130, 16691–16696 CrossRef CAS PubMed.
- Y. Yao, J. Liu, F. Zheng and C.-G. Zhan, Reaction Pathway for Cocaine Hydrolase-Catalyzed Hydrolysis of (+)-Cocaine, Theor. Chem. Acc., 2016, 135, 15 Search PubMed.
- J. Wang, D. Wei, Z. Duan and F. Mathey, Cleavage of the Inert C(sp2)–Ar σ-Bond of Alkenes by a Spatial Constrained Interaction with Phosphinidene, J. Am. Chem. Soc., 2020, 142, 20973–20978 CrossRef CAS PubMed.
- S. Zhang, X. Wang, L.-L. Han, J. Li, Z. Liang, D. Wei and D. Du, Atroposelective Synthesis of Triaryl α-Pyranones with 1,2-Diaxes by N-Heterocyclic Carbene Organocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202212005 CrossRef CAS.
- G. Wang, Q.-C. Zhang, C. Wei, Y. Zhang, L. Zhang, J. Huang, D. Wei, Z. Fu and W. Huang, Asymmetric Carbene-Catalyzed Oxidation of Functionalized Aldimines as 1,4-Dipoles, Angew. Chem., Int. Ed., 2021, 60, 7913–7919 CrossRef CAS.
- Z. Hu, C. Wei, Q. Shi, X. Hong, J. Liu, X. Zhou, J. Han, W. Cao, A. K. Gupta, X. Zhang, D. Wei, Z. Fu and W. Huang, Desymmetrization of N-Cbz glutarimides through N-heterocyclic carbene organocatalysis, Nat. Commun., 2022, 13, 4042 CrossRef CAS PubMed.
- G. Wang, Q. Shi, W. Hu, T. Chen, Y. Guo, Z. Hu, M. Gong, J. Guo, D. Wei, Z. Fu and W. Huang, Organocatalytic asymmetric N-sulfonyl amide C–N bond activation to access axially chiral biaryl amino acids, Nat. Commun., 2020, 11, 946 CrossRef CAS PubMed.
- B. Dong, F. Zhao, W.-X. Lv, Y.-G. Liu, D. Wei, J. Wu and Y. R. Chi, Regio- and stereoselective access to highly substituted vinylphosphine oxides via metal-free electrophilic phosphonoiodination of alkynes, Nat. Commun., 2024, 15, 5385 CrossRef CAS.
- K. Kahn and P. A. Tipton, Spectroscopic characterization of intermediates in the urate oxidase reaction, Biochemistry, 1998, 37, 11651–11659 CrossRef CAS PubMed.
- M. J. Bearpark, M. A. Robb and H. Bernhard Schlegel, A direct method for the location of the lowest energy point on a potential surface crossing, Chem. Phys. Lett., 1994, 223, 269–274 CrossRef CAS.
- J. Stare, Oxidation of Flavin by Molecular Oxygen: Computational Insights into a Possible Radical Mechanism, ACS Omega, 2024, 9, 23431–23441 CrossRef CAS PubMed.
- L. Gabison, C. Chopard, N. Colloc’h, F. Peyrot, B. Castro, M. El Hajji, M. Altarsha, G. Monard, M. Chiadmi and T. Prangé, X-ray, ESR, and quantum mechanics studies unravel a spin well in the cofactor-less urate oxidase, Proteins, 2011, 79, 1964–1976 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Electrostatic Basis for Enzyme Catalysis, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS.
- M. Hashimoto, T. Taguchi, K. Ishikawa, R. Mori, A. Hotta, S. Watari, K. Katakawa, T. Kumamoto, S. Okamoto and K. Ichinose, Unveiling Two Consecutive Hydroxylations: Mechanisms of Aromatic Hydroxylations Catalyzed by Flavin-Dependent Monooxygenases for the Biosynthesis of Actinorhodin and Related Antibiotics, ChemBioChem, 2020, 21, 623–627 CrossRef CAS PubMed.
- T. Kumamoto, M. Kainuma, A. Takahashi, Y. Matsuo, K. Katakawa, T. Taguchi and K. Ichinose, Total Synthesis of 6-Deoxydihydrokalafungin, a Key Biosynthetic Precursor of Actinorhodin, and Its Epimer, Molecules, 2021, 26, 6397 CrossRef CAS PubMed.
- S. G. Kendrew, D. A. Hopwood and E. N. Marsh, Identification of a monooxygenase from Streptomyces coelicolor A3(2) involved in biosynthesis of actinorhodin: purification and characterization of the recombinant enzyme, J. Bacteriol., 1997, 179, 4305–4310 CrossRef CAS.
- S. Okamoto, T. Taguchi, K. Ochi and K. Ichinose, Biosynthesis of actinorhodin and related antibiotics: discovery of alternative routes for quinone formation encoded in the act gene cluster, Chem. Biol., 2009, 16, 226–236 CrossRef CAS.
- G. Pan, X. Gao, K. Fan, J. Liu, B. Meng, J. Gao, B. Wang, C. Zhang, H. Han, G. Ai, Y. Chen, D. Wu, Z.-J. Liu and K. Yang, Structure and Function of a C–C Bond Cleaving Oxygenase in Atypical Angucycline Biosynthesis, ACS Chem. Biol., 2017, 12, 142–152 CrossRef CAS.
- B. F. Minaev, Spin-orbit coupling of charge-transfer states and the mechanism for quenching singlet oxygen by amines, Theor. Exp. Chem., 1984, 20, 199–201 CrossRef.
- T. Kumamoto, M. Kainuma, A. Takahashi, Y. Matsuo, K. Katakawa, T. Taguchi and K. Ichinose, Total Synthesis of 6-Deoxydihydrokalafungin, a Key Biosynthetic Precursor of Actinorhodin, and Its Epimer, Molecules, 2021, 26, 6397 CrossRef CAS.
- S. G. Kendrew, D. A. Hopwood and E. N. Marsh, Identification of a monooxygenase from Streptomyces coelicolor A3(2) involved in biosynthesis of actinorhodin: purification and characterization of the recombinant enzyme, J. Bacteriol., 1997, 179, 4305–4310 CrossRef CAS PubMed.
- B. F. Minaev, How cofactor-free oxygenases can overcome spin prohibition in substrates oxygenation by dioxygen, Chem. Phys., 2019, 521, 61–68 CrossRef CAS.
- P. J. Silva, Refining the reaction mechanism of O2 towards its co-substrate in cofactor-free dioxygenases, PeerJ, 2016, 4, e2805 CrossRef PubMed.
- S. Bui, D. von Stetten, P. G. Jambrina, T. Prangé, N. Colloc’h, D. de Sanctis, A. Royant, E. Rosta and R. A. Steiner, Direct evidence for a peroxide intermediate and a reactive enzyme-substrate-dioxygen configuration in a cofactor-free oxidase, Angew. Chem., Int. Ed., 2014, 53, 13710–13714 CrossRef CAS PubMed.
- S. Thierbach, N. Bui, J. Zapp, S. R. Chhabra, R. Kappl and S. Fetzner, Substrate-assisted O2 activation in a cofactor-independent dioxygenase, Chem. Biol., 2014, 21, 217–225 CrossRef CAS PubMed.
- H. Eyring, The Activated Complex in Chemical Reactions, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- R. A. Steiner, H. J. Janssen, P. Roversi, A. J. Oakley and S. Fetzner, Structural basis for cofactor-independent dioxygenation of N-heteroaromatic compounds at the alpha/beta-hydrolase fold, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 657–662 CrossRef CAS.
- K. D. Loh, P. Gyaneshwar, E. Markenscoff Papadimitriou, R. Fong, K.-S. Kim, R. Parales, Z. Zhou, W. Inwood and S. Kustu, A previously undescribed pathway for pyrimidine catabolism, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5114–5119 CrossRef CAS.
- T. Mukherjee, Y. Zhang, S. Abdelwahed, S. E. Ealick and T. P. Begley, Catalysis of a flavoenzyme-mediated amide hydrolysis, J. Am. Chem. Soc., 2010, 132, 5550–5551 CrossRef CAS PubMed.
- Y.-F. Wang and M.-T. Zhang, Proton-Coupled Electron-Transfer Reduction of Dioxygen: The Importance of Precursor Complex Formation between Electron Donor and Proton Donor, J. Am. Chem. Soc., 2022, 144, 12459–12468 CrossRef CAS PubMed.
- Q. Shi, Z. Pei, J. Song, S.-J. Li, D. Wei, M. L. Coote and Y. Lan, Diradical Generation via Relayed Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2022, 144, 3137–3145 CrossRef CAS PubMed.
- Q. Shi, K. Chen, L. Guo, L. Han, D. Wei and Y. Lan, Deaminative radical reactions via relayed proton-coupled electron transfer, Org. Chem. Front., 2023, 10, 2155–2164 RSC.
- K. Chen, J. Zhang, Q. Shi, L. Han, D. Fu, D. Wei and Y. Zhu, NHC-catalyzed enantioselective radical reactions of enal and pyridinium salt: mechanism and origin of regio- and stereoselectivities, Catal. Sci. Technol., 2023, 13, 5259–5266 RSC.
- Y. Wang, W. Chen, Y. Lai and A. Duan, Activation Model and Origins of Selectivity for Chiral Phosphoric Acid Catalyzed Diradical Reactions, J. Am. Chem. Soc., 2023, 145, 23527–23532 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2025 |