
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous integration of poly(dimethylsiloxane) elastomer in polymer donor and dimer acceptor enables strain-induced power enhancement in intrinsically-stretchable organic photovoltaics†
Jin-Woo
Lee‡
a,
Trieu Hoang-Quan
Nguyen‡
 a,
Won Jung
Kang
b,
Soodeok
Seo
a,
Seungbok
Lee
c,
Seungjin
Lee
d,
Jaeyoung
Choi
a,
Jimin
Park
a,
Jung-Yong
Lee
c,
Taek-Soo
Kim
b and
Bumjoon J.
Kim
*a
a,
Won Jung
Kang
b,
Soodeok
Seo
a,
Seungbok
Lee
c,
Seungjin
Lee
d,
Jaeyoung
Choi
a,
Jimin
Park
a,
Jung-Yong
Lee
c,
Taek-Soo
Kim
b and
Bumjoon J.
Kim
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: bumjoonkim@kaist.ac.kr
bDepartment of Mechanical Engineering, KAIST, Daejeon 34141, Republic of Korea
cSchool of Electrical Engineering, KAIST, Daejeon 34141, Republic of Korea
dPhotoenergy Research Center, Korea Research Institute of Chemical Technology (KRICT), Daejeon, 34114, Republic of Korea
First published on 28th February 2025
Abstract
Intrinsically stretchable organic solar cells (IS-OSCs) are an emerging class of wearable power sources owing to their ability to stretch in multiple directions. However, their current stretchability remains insufficient to meet the demands of wearable electronics. In this study, we develop a poly(dimethylsiloxane) (PDMS)-incorporated dimer acceptor (DYPDMS) and a PDMS integrated block-copolymer donor (PM6-b-PDMS) to achieve IS-OSCs with a high power conversion efficiency (PCE = 12.7%) and remarkable mechanical stretchability, maintaining over 80% of their initial PCE under 40% strain. Notably, we demonstrate the critical role of simultaneously integrating PDMS into both the polymer donor (PD) and acceptor materials to achieve superior photovoltaic and mechanical performance in IS-OSCs. The dual incorporation of PDMS significantly enhances the blend morphology by improving the thermodynamic compatibility between the PM6-b-PDMS PD and the dimer acceptors while effectively suppressing macrophase separation of PDMS elastomers from the photoactive materials. Consequently, IS-OSCs based on the PM6-b-PDMS:DYBT:DYPDMS system achieve significantly higher PCE and stretchability compared to systems using PM6-b-PDMS:DYBT (without PDMS in dimer acceptors) or PM6-b-PDMS:DYBT:PDMS (with PDMS physically mixed). Importantly, these IS-OSCs exhibit an increase in overall power output under stretching up to 35% strain, demonstrating a successful example of IS-OSCs with strain-induced power enhancement.
Broader contextIntrinsically stretchable organic solar cells (IS-OSCs) are promising power sources for wearable electronics due to their multidirectional stretchability, but the current level of stretchability is insufficient. This study develops a PDMS-incorporated dimer acceptor (DYPDMS) and block-copolymer donor (PM6-b-PDMS), achieving IS-OSCs with a high power conversion efficiency (PCE = 12.7%) and excellent device stretchability (over 80% PCE retention under 40% strain). Incorporation of PDMS elastomers into both polymer donor and acceptor materials enhances blend morphology by improving thermodynamic compatibility and suppressing macrophase separation, resulting in superior photovoltaic and mechanical performance. Importantly, these IS-OSCs increase their overall power output under stretching up to 35% strain, demonstrating one of the first examples of strain-induced power output enhancement in OSCs. |
Introduction
The growing demand for wearable electronics drives the need for advanced portable power solutions. Organic solar cells (OSCs) have emerged as a promising candidate due to their lightweight nature, solution processability, and mechanical flexibility.1–4 For wearable applications, OSCs must possess a high degree of stretchability to adapt to the complex, multi-directional movements of the human body.5–9 Unlike conventional flexible or stretchable OSCs that rely on structural engineering strategies and are therefore limited in their stretchable direction and extent, intrinsically stretchable OSCs (IS-OSCs) offer a more robust solution for wearable applications.10–15 By integrating polymer- and liquid metal-based electrodes and buffer layers, IS-OSCs enable stretching in multiple directions.A unique advantage of IS-OSCs is their ability to expand the photoactive area during stretching, which provides the potential for strain-induced power enhancement.16,17 For example, when the expansion in the photoactive area compensates for any decline in power conversion efficiency (PCE) of the IS-OSCs, their total power output (PCE × photoactive area) can improve. This capability introduces a novel approach to enhancing performance and broadens the potential applications of IS-OSCs in wearable technologies.
However, achieving reliable strain-induced power enhancement remains a significant challenge due to the mechanical limitations of current IS-OSCs. Although recent advancements in polymer donors (PDs) and small molecule acceptors (SMAs) have driven OSC PCEs to nearly 20% in rigid devices and around 15% in IS-OSCs,18–35 many high-performance IS-OSCs still suffer from rapid degradation of PCE and power output under strain. This issue is particularly crucial for wearable applications. For example, human skin and joints experience repeated tensile and shear strains often exceeding 50%, while most IS-OSCs retain less than 80% of their initial PCE at 50% strain (strain at PCE80% < 50%). This leads to a continuous decline in power output under such strain conditions, significantly restricting the practical usability of IS-OSCs. The primary reason for this mechanical fragility is the brittleness of the photoactive layer, which typically has a low crack-onset strain (COS < 10%).36–40 Most high-performance photoactive materials are designed with rigid molecular structures to promote strong π–π interactions, which are crucial for superior optoelectronic performance. However, these structures also promote the formation of large, hard crystalline domains, making the layers prone to cracking under mechanical stress.31,36–42
To address these limitations, recent research efforts have focused on developing photoactive layers with enhanced mechanical robustness. Strategies include forming entangled polymer networks or increasing the proportion of amorphous domains within the active layers to dissipate mechanical stress more effectively. A straightforward approach involves incorporating highly stretchable elastomers, such as polydimethylsiloxane (PDMS) or polystyrene-block-poly(ethylene-ran-butylene)-block-polystyrene (SEBS), into the photoactive layers as a third component. For instance, the Shao group demonstrated that adding 5 wt% PDMS into PTB7-Th:IEICO-4F active layers significantly increased the free volume and amorphous regions, resulting in a fourfold improvement in stretchability (COS from 5 to 20%).43 Similarly, the Ye group incorporated SEBS into high-performance PM6:N3 systems, achieving a twofold increase in COS (from 6.9 to 13.0%) with 30 wt% SEBS incorporation.37 Despite these successes, the physical incorporation of elastomers results in severe phase separation of elastomers from the photoactive materials due to their strong thermodynamic incompatibility, causing a significant decrease in the PCE of OSCs.
Chemically binding elastomers to photoactive materials can be a promising solution for enhancing their mechanical properties. This approach effectively suppresses macrophase separation of elastomers and promotes the formation of amorphous regions within the photoactive materials, thereby enhancing mechanical robustness while minimally compromising the photovoltaic performance, as demonstrated in the recent example of elastomer-integrated PD materials.44,45 However, this strategy has not yet been applied to acceptor materials. This is an urgent and important task, as the overall mechanical performance of photoactive layers is primarily determined by SMAs, which inherently exhibit much lower mechanical properties compared to PD. Dimerized SMAs provide an ideal model platform for chemically linking elastomers to acceptor molecules for the following reasons: (1) the larger molecular sizes of dimer acceptors significantly reduce molecular diffusion kinetics, enhancing the long-term stability of the resulting OSCs compared to monomeric SMAs.46–50 (2) The linker positions connecting the two SMA units in dimer acceptors provide an accessible and versatile site for chemical modification, enabling efficient elastomer incorporation. (3) Linker engineering minimizes disruptions to the molecular packing of the main backbone and end-groups within dimer acceptors, ensuring the retention of their optoelectronic properties.51–54 Thus, we propose that the design of elastomer-linked dimer acceptors, in combination with elastomer-incorporated PDs, can improve compatibility and intermixing between donor and acceptor materials, thereby enhancing both the photovoltaic and mechanical properties of the photoactive layers. Importantly, we anticipate that such a high-performance and mechanically robust photoactive system would enable IS-OSCs to deliver not only a high initial power output but also enhanced power output under strain, ensuring a continuous and reliable power supply for wearable electronics.
Here, we design and synthesize a new elastomer-incorporated dimer acceptor (DYPDMS) by connecting two Y-based SMA units with PDMS linkers. By integrating DYPDMS with a PDMS-incorporated block-copolymer donor (PM6-b-PDMS), we develop high-performance, mechanically robust photoactive layers, achieving high-performance IS-OSCs that maintain 80% of their initial PCE under 41% strain (strain at PCE80% = 41%). Importantly, we demonstrate the critical role of simultaneously incorporating the same PDMS elastomers into both PD and acceptor materials to achieve IS-OSCs with superior PCE and mechanical robustness. Specifically, photoactive systems (PM6-b-PDMS:DYBT:DYPDMS) incorporating PDMS in both components exhibit significantly higher PCE and mechanical robustness compared to systems with PDMS in only one (PM6-b-PDMS:DYBT system) or neither component (PM6:DYBT system). Morphological analyses reveal that the use of PDMS-incorporated active materials enhances the entanglement density and increases the number and size of amorphous domains within the photoactive films. In addition, the dual PDMS incorporation into the PD and dimer acceptor improves their thermodynamic compatibility, suppressing phase separation and strengthening donor–acceptor interfaces. These improvements facilitate efficient charge generation while enhancing the dissipation of mechanical stress in OSCs. As a result, IS-OSCs based on the PM6-b-PDMS:DYBT:DYPDMS system deliver a high PCE (12.7%) and an impressive stretchability (strain at PCE80% = 41%). Importantly, these devices demonstrate a strain-induced power output enhancement, in contrast to other IS-OSC systems that experience continuous power decreases under strain.
Results and discussion
Materials design and synthesis
The chemical structures of the conventional PD (poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)], PM6) and dimer acceptor (2,2′-((2Z,2′Z)-(((((2Z,2′Z)-[2,2′-bithiophene]-5,5′-diylbis(1-(dicyanomethylene)-3-oxo-1,3-dihydro-2H-indene-5-yl-2-ylidene))bis(methaneylylidene))bis(3,9-bis(2-butyloctyl)-12,13-bis(2-octyldodecyl)-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-10,2-diyl))bis(methaneylylidene))bis(5,6-dichloro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile, DYBT), as well as the elastomer-incorporated PD (poly(dimethylsiloxanes)-block-poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)]-block-poly(dimethylsiloxanes)), PM6-b-PDMS) and dimer acceptor ((((poly(dimethylsiloxane)bis(propane-3,1-diyl))bis(oxy))bis(ethane-2,1-diyl)bis(5′-((Z)-2-((3,9-bis(2-butyloctyl)-10-(((Z)-5,6-dichloro-1-(dicyanomethylene)-3-oxo-1,3-dihydro-2H-inden-2-ylidene)methyl)-12,13-bis(2-octyldodecyl)-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5] pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indol-2-yl)methylene)-1-(dicyanomethylene)-3-oxo-2,3-dihydro-1H-inden-5-yl)-[2,2′-bithiophene]-5-carboxylate), DYPDMS) used in this study, are presented in Fig. 1a and b. Conventional high-performance photoactive materials such as PM6 and DYBT, have rigid molecular structures consisting of multiple fused rings and exhibit brittle mechanical properties in films due to the formation of large and hard crystalline domains.55,56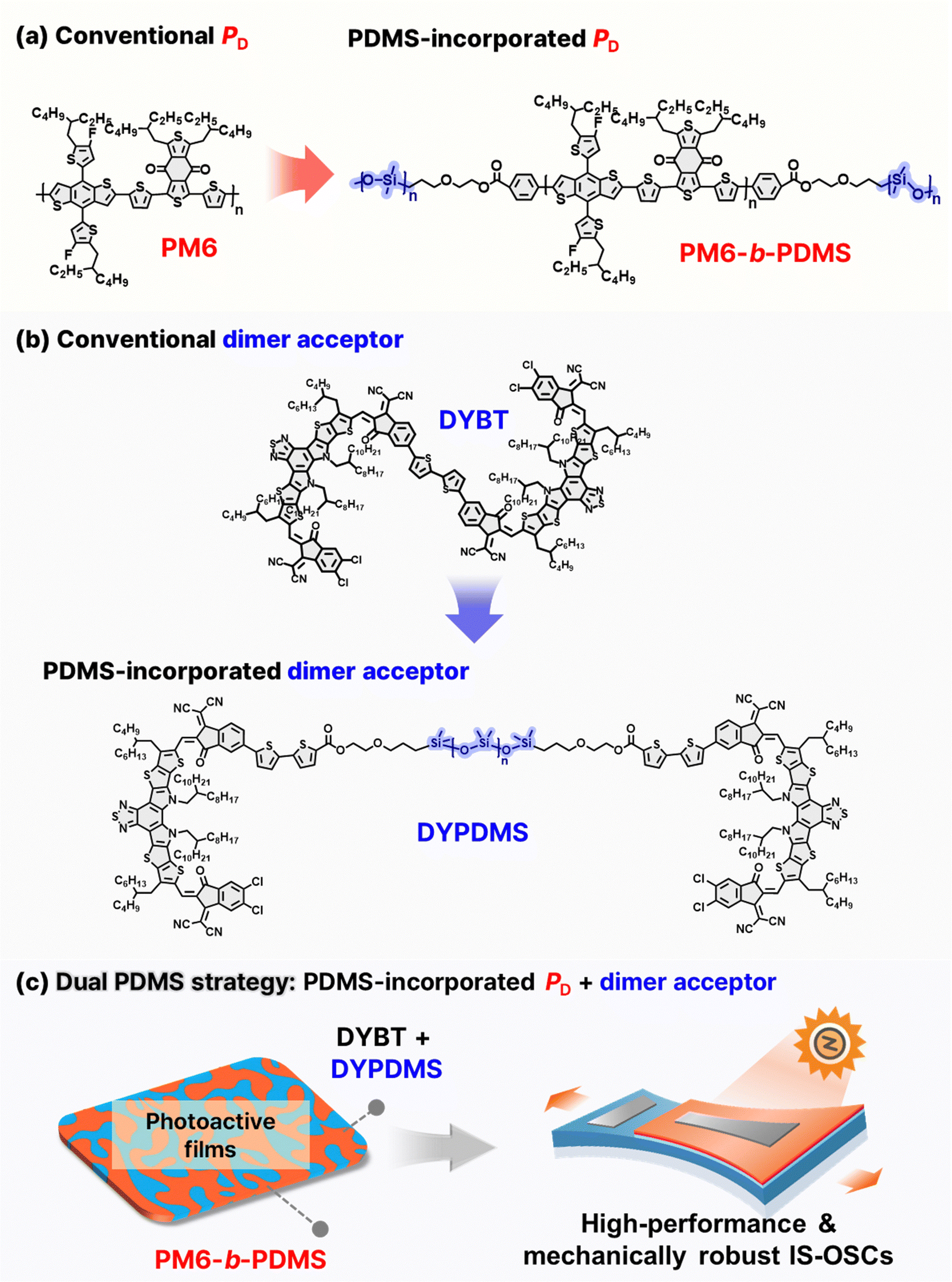 | ||
| Fig. 1 (a) Chemical structures of conventioanl (PM6) and PDMS-incorporated PDs (PM6-b-PDMS). (b) Chemical structures of conventional (DYBT) and PDMS-incorporated dimer acceptors (DYPDMS). (c) Conceptual illustration of the dual PDMS incorporation strategy to achieve enhanced mechanical robustness and photovoltaic performance in IS-OSCs. | ||
To address this limitation, we developed PDMS-incorporated PD (PM6-b-PDMS) and dimer acceptor (DYPDMS) (Fig. 1a and b). The design strategy for incorporating PDMS units into both donor and acceptor materials offers key advantages: (1) enhanced mechanical robustness: covalently linking PDMS chains with a very low glass-transition temperature (Tg < −50 °C)57 to conjugated backbones imparts sufficient chain flexibility and deformability to the photoactive materials. (2) The strong covalent bonds between PDMS and photoactive materials restrict the aggregation of PDMS chains and prevent phase separation between PDMS and the photoactive components.44,45 (3) Importantly, dual incorporation of PDMS units into both donor and acceptor materials improves their thermodynamic compatibility, promoting the development of intermixed domains and optimizing the blend morphology. This results in the formation of large and broad donor–acceptor interfaces, which not only facilitate efficient charge generation but also help the effective dissipation of mechanical stress during the stretching. To the best of our knowledge, PDMS-incorporated acceptor materials have not been reported previously. Therefore, the combined use of elastomer-integrated PD and acceptor materials in the OSCs has not been explored. In particular, we designed a dimer acceptor with a PDMS linker. Dimer acceptors are known to exhibit superior long-term stability of the OSCs compared to SMAs due to their reduced molecular diffusion kinetics. Also, the linker position between the acceptor cores in dimer acceptors provides a convenient handle for effective integration of PDMS with minimal influence on the molecular packing between the acceptor cores and electrical properties.
The conventional PM6 and DYBT materials, which do not contain PDMS, were synthesized following previously reported procedures.55,58 Detailed synthetic procedures are provided in the ESI† (Schemes S1 and S2). The successful synthesis of PM6 was confirmed using nuclear magnetic resonance (NMR) spectroscopy and size exclusion chromatography (SEC) (Fig. S1, S2 and Tables S1, S2, ESI†), while the synthesis of DYBT was validated using both NMR and matrix-assisted laser desorption ionization-time of flight (MALDI-ToF) mass spectrometry (Fig. S3 and S4, ESI†).
For the synthesis of DYPDMS, we initially prepared an asymmetric Y-series-based compound featuring a dichlorinated 1,1-dicyanomethylene-3-indanone (IC) group at one end and an in-isomeric brominated IC group at the other end (compound 2, Scheme S1, ESI†). Then, the monobrominated sites in compound 2 were functionalized with Sn-terminated thiophene groups (compound 3) to enable dimerization with dibrominated linkers (Scheme S3, ESI†). For the linker unit, brominated thiophene-ended PDMS was synthesized by brominating carbinol-terminated PDMS polymers (number-average molecular weight (Mn) = 1 kg mol−1) with 5-bromo-2-thiophenecarboxylic acid (compound 4, Scheme S4, ESI†). We note that compared to the PDMS block (Mn = 19 kg mol−1) used in PM6-b-PDMS, a shorter PDMS unit was chosen to yield DYPDMS to ensure a sufficient volume fraction of Y-SMA units relative to PDMS within each molecule. The monostannylated Y-SMAs (compound 3) were subsequently dimerized with the brominated PDMS linkers through Stille coupling to produce the final dimer acceptor, DYPDMS (Scheme 1a and Scheme S5, ESI†). Detailed synthesis and purification protocols are provided in the ESI.†
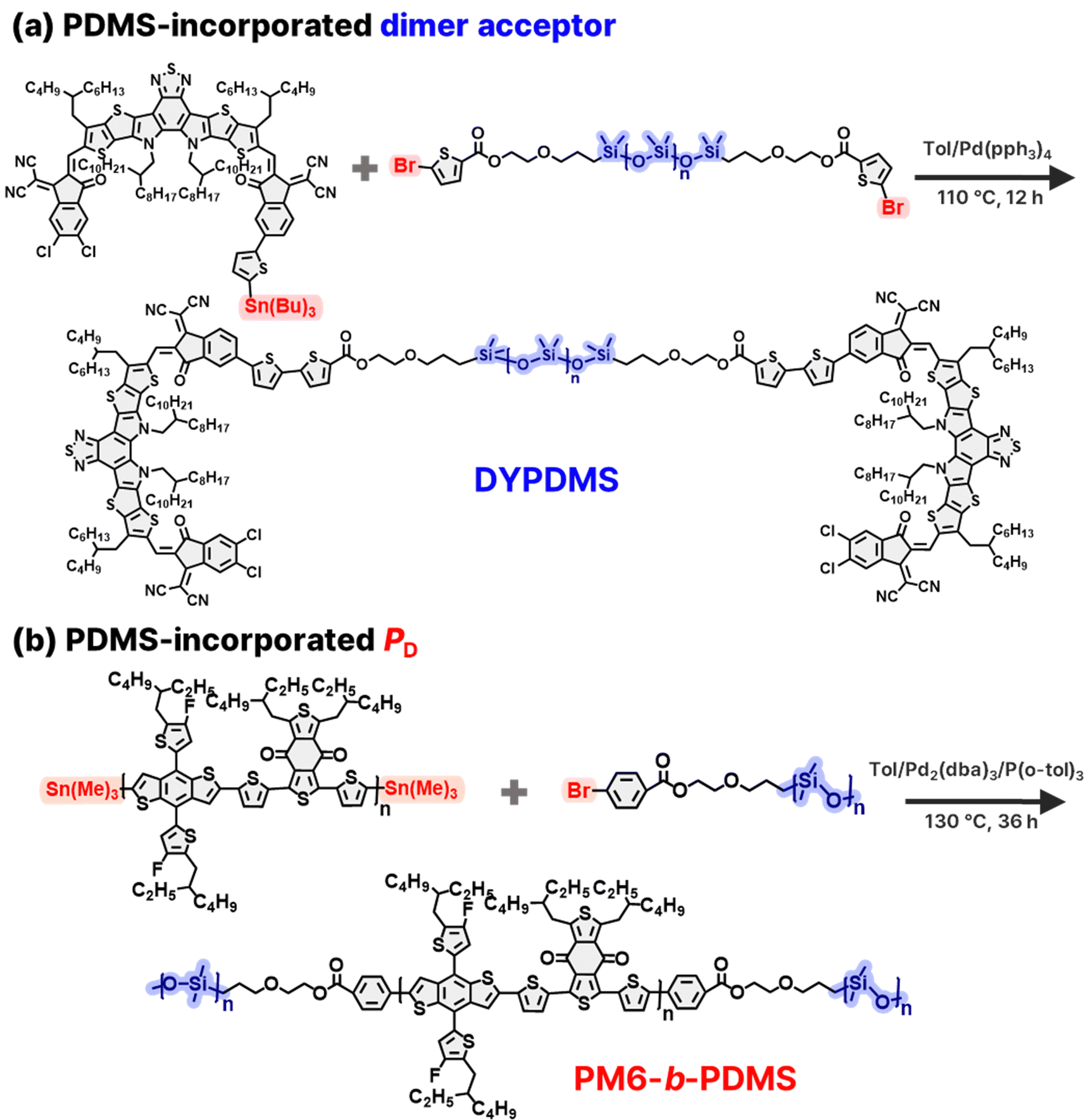 | ||
| Scheme 1 Synthetic schemes of (a) PM6-b-PDMS and (b) DYPDMS. | ||
The successful synthesis of the intermediates and final product, DYPDMS, was confirmed using a combination of SEC, NMR, and thermogravimetric analysis (TGA) measurements (Fig. S5–S11 and Table S3, ESI†). The SEC results showed that the PDMS linkers had an Mn of 1.0 kg mol−1, corresponding to 10.3 repeating dimethylsiloxane units, while DYPDMS exhibited an Mn of 5.2 kg mol−1 (Table S3, ESI†). In the NMR spectrum of DYPDMS (Fig. S8, ESI†), characteristic proton peaks of dimethylsiloxane were observed between 0.060 and 0.084 ppm. These were compared to the peaks at 8.998–9.267 ppm, assigned to the vinylene groups in the two acceptor blocks, yielding a proton ratio of 4.0 to 63.5, respectively. Using this ratio, we estimated that DYPDMS contains 10.6 repeating dimethylsiloxane units, closely matching the 10.3 units determined from SEC analysis of pristine PDMS. This confirms the successful integration of PDMS linkers within DYPDMS.
TGA analysis was performed to confirm the synthesis of DYPDMS and its composition (Fig. S10, ESI†). TGA measurements were conducted in an oxygen atmosphere from ambient temperature to 900 °C. Under these conditions, organic compounds decompose via thermo-oxidative reactions, releasing gaseous products such as CO2, H2O, SOx, and NOx, while silicon reacts with oxygen to form solid SiO2 (Fig. S11, ESI†). Consequently, the residual weight observed at 900 °C reflects the silicon content in the material. The dimethylsiloxane content in DYPDMS, estimated from the TGA results, corresponded to 9.4 repeating units, which aligns closely with that from the NMR measurement. In contrast, the asymmetric Y-core acceptor (compound 2), which lacks silicon, showed no residue after TGA analysis. These results validate the successful synthesis and structural composition of DYPDMS.
The PM6-b-PDMS block copolymer PD was synthesized via a two-step Stille coupling reaction (Scheme 1b and Scheme S6, S7, Fig. S12, ESI†).44 First, Sn-terminated PM6 blocks were prepared by the Stille coupling reaction between the monomers (4,8-bis(5-(2-ethylhexyl)-4-fluorothiophen-2-yl)benzo[1,2-b:4,5-b′′]dithiophene-2,6-diyl)bis(trimethylstannane) (1.1 equivalent) and 1,3-bis(5-bromothiophen-2-yl)-5,7-bis(2-ethylhexyl)benzo[1,2-c:4,5-c′′]dithiophene-4,8-dione (1 equivalent). In the second step, the Sn-terminated PM6 (1 equivalent) was reacted with an excess amount of Br-terminated PDMS with Mn of 19 kg mol−1 through a sequential Stille coupling reaction (Scheme 1b). The PDMS length was chosen to exceed its entanglement molecular weight (Me ∼ 12 kg mol−1)59 to provide sufficient mechanical stretchability of the resulting PM6-b-PDMS. Residual unreacted PDMS units were thoroughly removed by Soxhlet extraction using hexane as the solvent, yielding the desired PM6-b-PDMS PD.
The NMR spectra of PM6 and PM6-b-PDMS PDs are presented in Fig. S1 (ESI†). The NMR spectrum of PM6-b-PDMS showed distinct proton peaks at 0.2 ppm, corresponding to the PDMS blocks. In contrast, no such PDMS peaks were observed in the spectrum of PM6, confirming the successful incorporation of PDMS into the PM6-b-PDMS structure. In addition, the SEC profiles of the PM6 and PM6-b-PDMS indicated Mn values of 134 and 164 kg mol−1, respectively (Fig. S2 and Table S1, ESI†). In addition, the SEC profile of PM6-b-PDMS showed a single peak, confirming that complete removal of unreacted PDMS units after purification. By combining the SEC and NMR results, the average number of PDMS blocks attached to each PM6 block in PM6-b-PDMS was estimated to be 1.5 (Table S2, ESI†), suggesting that the PM6-b-PDMS consists of approximately equal proportions of di-block (i.e., PM6-PDMS) and tri-block (i.e., PDMS-PM6-PDMS) structures.
Basic material properties
The electrochemical and optical properties of the PDs and acceptors used in this study were investigated using cyclic voltammetry (CV) and ultraviolet-visible (UV-Vis) absorption spectroscopy measurements (Fig. 2a–c and Fig. S13, ESI†). Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the photoactive materials were estimated using CV measurements (Fig. S13, ESI†). The HOMO and LUMO energy levels of PM6 and PM6-b-PDMS PDs were well aligned with those of the acceptors (DYBT and DYPDMS), with sufficient energy offset (>0.1 eV) to afford efficient charge generation and transfer in OSCs (Fig. 2a). The optical properties of the photoactive materials in chlorobenzene (CB) solution and thin films were assessed using UV-Vis absorption spectroscopy (Fig. 2b and c). The PDs (PM6 and PM6-b-PDMS) and the acceptors (DYBT and DYPDMS) showed complementary absorption profiles in both solution and film states, which is advantageous for efficient light harvesting across a broad absorption wavelength range (300–900 nm). PM6-b-PDMS PD exhibited a slightly red-shifted maximum absorbance wavelength in the film (λfilmmax) of 617 nm compared to that of PM6 (λfilmmax = 614 nm) (Table 1). In addition, the intensity ratio of the (0–0) transition peak to the (0–1) transition peak for PM6-b-PDMS was 1.12, which was higher than that for PM6 (1.06), indicating a higher degree of aggregation in PM6-b-PDMS film. Interestingly, the UV-Vis absorption spectra of the DYBT and DYPDMS acceptors revealed a different trend in aggregation depending on the PDMS linker. DYPDMS exhibited a blue-shifted λfilmmax of 803 nm compared to DYBT (λfilmmax = 810 nm), indicating that DYBT without PDMS has stronger aggregation than DYPDMS (Table 1). | ||
| Fig. 2 (a) Frontier orbital energy level alignments, (b) and (c) UV-Vis absorption spectra in (b) CB solution and (c) thin film, and (d) GIXS 2D-images of photoactive materials. | ||
| Material | λ filmmax (nm)a | E LUMO (eV)b | E HOMO (eV)b | r-DoC(010)c |
|---|---|---|---|---|
| a Wavelength of maximum UV-Vis absorbance (λmax) in film. b Measured by CV. c Estimated from (010) peaks in GIXS scattering profiles. | ||||
| PM6 | 614 | −3.55 | −5.42 | 0.45 |
| PM6-b-PDMS | 617 | −3.51 | −5.40 | 0.68 |
| DYBT | 810 | −3.72 | −5.57 | 1.00 |
| DYPDMS | 803 | −3.71 | −5.54 | 0.32 |
The crystalline structures of the materials in thin films were analyzed using grazing-incidence wide-angle X-ray scattering (GIXS) measurements (Fig. 2d and Fig. S14, S15, ESI†). Both PM6 and PM6-b-PDMS PDs showed a preferential face-on orientation, characterized by distinct (100) and (010) scattering peaks in the in-plane (IP) and out-of-plane (OOP) directions, respectively. The relative crystal sizes corresponding to these peaks were estimated by calculating coherence lengths (Lcs) using Scherrer equation.60 PM6-b-PDMS film showed higher Lc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (100)IP of 12.6 nm and Lc(010)OOP of 3.4 nm compared to PM6 (Lc(100)IP = 7.6 nm and Lc(010)OOP = 2.5 nm), indicating that PM6-b-PDMS film contains larger crystalline domains than PM6 film (Table S4, ESI†). Furthermore, the relative degree of crystallinity (r-DoC) for the OOP (010) peak of PM6-b-PDMS film was 0.68, which was higher than that of PM6 film (0.45) (Table 1). The enhanced crystallinity of PM6-b-PDMS film is mainly attributed to the improved ordering of PM6 blocks, induced by the confinement effect of the adjacent PDMS domains.44,61 However, the dimer acceptors displayed a different trend. DYBT film exhibited face-on preferential packing, as evidenced by distinct IP (100) and OOP (010) scattering peaks. In comparison, DYPDMS film exhibited ring-shaped (100) and (010) peaks, indicating more amorphous-like, isotropic molecular packing structures. The r-DoC(010) of DYBT was 1.00, which was significantly higher than that of DYPDMS (0.32), demonstrating that the intermolecular assembly of DYPDMS was weakened by the PDMS linkers. In comparison, neat PDMS polymers (Mn = 1 kg mol−1) showed amorphous peaks at q = 0.8 Å−1 in the GIXS 2D-images (Fig. 2d).
(100)IP of 12.6 nm and Lc(010)OOP of 3.4 nm compared to PM6 (Lc(100)IP = 7.6 nm and Lc(010)OOP = 2.5 nm), indicating that PM6-b-PDMS film contains larger crystalline domains than PM6 film (Table S4, ESI†). Furthermore, the relative degree of crystallinity (r-DoC) for the OOP (010) peak of PM6-b-PDMS film was 0.68, which was higher than that of PM6 film (0.45) (Table 1). The enhanced crystallinity of PM6-b-PDMS film is mainly attributed to the improved ordering of PM6 blocks, induced by the confinement effect of the adjacent PDMS domains.44,61 However, the dimer acceptors displayed a different trend. DYBT film exhibited face-on preferential packing, as evidenced by distinct IP (100) and OOP (010) scattering peaks. In comparison, DYPDMS film exhibited ring-shaped (100) and (010) peaks, indicating more amorphous-like, isotropic molecular packing structures. The r-DoC(010) of DYBT was 1.00, which was significantly higher than that of DYPDMS (0.32), demonstrating that the intermolecular assembly of DYPDMS was weakened by the PDMS linkers. In comparison, neat PDMS polymers (Mn = 1 kg mol−1) showed amorphous peaks at q = 0.8 Å−1 in the GIXS 2D-images (Fig. 2d).
The crystallinity of the materials in bulk states was further investigated using differential scanning calorimetry (DSC) measurement (Fig. S16, ESI†). PM6 and PM6-b-PDMS did not exhibit any melting transition peaks in the 2nd heating profiles between 100–300 °C, which is consistent with the typical behavior of benzodithiophene-based PDs. In contrast, DYBT and DYPDMS showed melting transition peaks at 268 and 263 °C, respectively, with corresponding melting enthalpies (ΔHm) of 13.0 and 2.8 J g−1. The higher melting temperature (Tm) and ΔHm of DYBT indicate stronger crystalline behavior compared to DYPDMS, consistent with UV-Vis absorption spectra and GIXS results. The optimized molecular structure of DYPDMS calculated via density functional theory simulation, reveals helical structure of PDMS linkers, which support amorphous packing structures and reduced crystallinity in DYPDMS (Fig. S17, ESI†).
Photovoltaic properties
The photovoltaic properties of the materials were evaluated by fabricating OSCs with a normal device structure (Fig. 3a–d and Table 2). Detailed information on the device structures and fabrication procedures is provided in the ESI.† To elucidate the influence of incorporated PDMS in PM6-b-PDMS and DYPDMS on photovoltaic performance, we systematically compared six different PD:acceptor pairs. The three different acceptors were used: (1) DYBT, (2) DYBT:PDMS (a physical mixture of DYBT and PDMS), and (3) DYBT:DYPDMS (PDMS chemical bonded to DYBT). Each paired with two different PDs: (1) PM6 and (2) PM6-b-PDMS. We anticipate that this comparison will elucidate the impact of PDMS incorporation into PD and dimer acceptors via physical mixing versus chemical bonding on the performance of OSCs. It is noted that for a fair comparison, the PDMS Mn (1 kg mol−1) in the ternary PD:DYBT:PDMS- and PD:DYBT:DYPDMS-based devices was kept consistent across all experiments. In addition, since DYPDMS contains two Y-SMA cores, the weight ratio of DYBT:PDMS in the device was set to 4:1 w/w, which is lower than the ratio of DYBT:DYPDMS (3:2 w/w). The detailed photovoltaic performance of PD:DYBT:PDMS and PD:DYBT:DYPDMS-based OSCs, as functions of the acceptor types and their ratio, is presented in Fig. S18, S19 and Tables S5–S8 (ESI†).
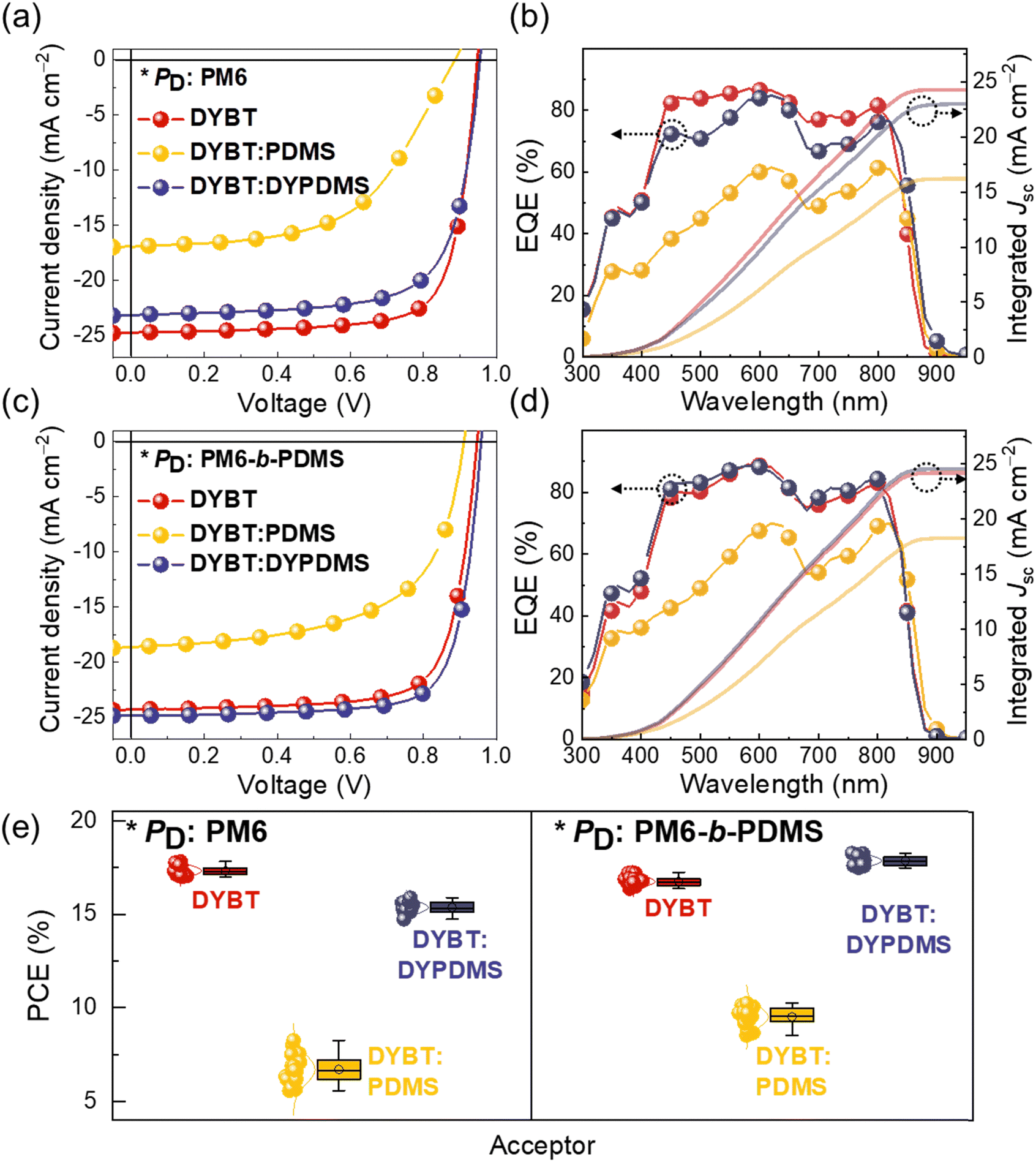 | ||
| Fig. 3 (a) J–V curves and (b) EQE spectra of PM6:acceptors-based OSCs. (c) J–V curves and (d) EQE spectra of PM6-b-PDMS:acceptors-based OSCs. (e) Gaussian function fitted PCE distributions of the OSCs. | ||
| P D | Acceptor | V oc (V) | J sc (mA cm−2) | Cal. Jsc (mA cm−2)a | FF | PCEmax(avg)b (%) |
|---|---|---|---|---|---|---|
| a Calculated from EQE spectra. b Average values obtained from 10 independent devices. | ||||||
| PM6 | DYBT | 0.95 (0.95 ± 0.01) | 24.73 (24.25 ± 0.14) | 24.26 | 0.76 (0.75 ± 0.01) | 17.83 (17.33 ± 0.24) |
| DYBT:PDMS | 0.89 (0.89 ± 0.01) | 16.42 (15.02 ± 0.81) | 16.17 | 0.55 (0.54 ± 0.01) | 8.25 (7.02 ± 0.54) | |
| DYBT:DYPDMS | 0.95 (0.95 ± 0.01) | 23.13 (22.56 ± 0.18) | 22.96 | 0.72 (0.72 ± 0.01) | 15.89 (17.33 ± 0.28) | |
| PM6-b-PDMS | DYBT | 0.95 (0.95 ± 0.00) | 24.23 (23.50 ± 0.20) | 24.16 | 0.75 (0.75 ± 0.01) | 17.26 (16.76 ± 0.25) |
| DYBT:PDMS | 0.91 (0.91 ± 0.01) | 18.62 (17.38 ± 0.73) | 18.23 | 0.60 (0.59 ± 0.01) | 10.24 (9.50 ± 0.52) | |
| DYBT:DYPDMS | 0.96 (0.96 ± 0.01) | 24.84 (24.38 ± 0.16) | 24.50 | 0.77 (0.76 ± 0.01) | 18.28 (17.81 ± 0.23) | |
The current density–voltage (J–V) curves of the OSCs are presented in Fig. 3a and c. PM6:DYBT-based OSC exhibited a high PCE of 17.83%, with an open-circuit voltage (Voc) of 0.95, short-circuit current density (Jsc) of 24.73 mA cm−2, and fill-factor (FF) of 0.76. When DYBT:PDMS and DYBT:DYPDMS acceptors were used with the same PM6 donor, the PCEs decreased to 8.25% and 15.89%, respectively. This performance decline upon PDMS incorporation was mainly due to reductions in both Jsc and FF. Notably, the DYBT:DYPDMS-based OSCs (PCE = 15.89%) exhibited a higher PCE than the DYBT:PDMS-based OSCs (PCE = 8.25%), owing to higher Voc (0.95 vs. 0.89 V), Jsc (23.13 vs. 16.42 mA cm−2), and FF (0.72 vs. 0.55), suggesting the importance of chemical linking of PDMS.
Interestingly, the use of PM6-b-PDMS PD resulted in different trend of OSC performance depending on the acceptors. While PM6-b-PDMS:DYBT-based OSC exhibited a slightly lower PCE (17.26%) compared to that of PM6:DYBT-based OSC, the combination of PM6-b-PDMS PD with DYBT:PDMS and DYBT:DYPDMS acceptors led to notable increase in the PCEs. PM6-b-PDMS:DYBT:PDMS- and PM6-b-PDMS:DYBT:DYPDMS-based OSCs exhibited the PCE values of 10.24% and 18.28%, respectively, which significantly outperform those of PM6:DYBT:PDMS- and PM6:DYBT:DYPDMS-based OSCs (PCE = 8.25% and 15.89%, respectively). The improved PCE values of PM6-b-PDMS-based OSCs with these acceptors were mainly attributed to higher Jsc and FF compared to the corresponding PM6-based OSCs. These results underscore the importance of consistent PDMS incorporation in both the PD and acceptor materials to enhance OSC performance. Additionally, the superior performance of DYBT:DYPDMS-based OSCs compared to DYBT:PDMS OSCs, regardless of the PD type, highlights the advantage of chemically bonding PDMS to acceptors over physical mixing of PDMS in OSCs. Notably, the PCE of PM6-b-PDMS:DYBT:DYPDMS-based OSCs (18.28%) also exceeded that of PM6-b-PDMS:DYBT-based OSCs (17.26%). This enhancement in PCE for the PM6-b-PDMS:DYBT:DYPDMS system is attributed to improvements in both Jsc (24.84 mA cm−2) and FF (0.77).
The external quantum efficiency (EQE) spectra of the PM6:acceptors- and PM6-b-PDMS:acceptors-based OSCs are presented in Fig. 3b and d, respectively. All OSCs demonstrated efficient charge generation across a broad wavelength range from 300 to 900 nm, which is attributed to the complementary absorption of the PDs and acceptors. The integrated photocurrent densities (Jcal.) from the EQE spectra closely matched the measured device Jsc values, with deviations of less than 3%. The Gaussian-function fitted PCE distributions of the OSCs are shown in Fig. 3e. These distributions reveal small deviations in PCE values, confirming the reproducibility of the fabricated OSC devices.
To elucidate the origins of different photovoltaic properties of the OSCs depending on the PD:acceptor pairs, we investigated their charge transport, generation, and recombination characteristics. The charge transport properties of the OSCs were evaluated using space-charge limited current (SCLC) charge mobility measurements (Tables S9 and S10, ESI†).62 For the pristine constituent films, the PM6-b-PDMS film exhibited a hole mobility (μh) comparable to that of PM6, while the DYPDMS film showed a slightly lower electron mobility (μe) than DYBT (Table S9, ESI†). These trends are consistent with the observed differences in aggregation and crystallinity for the PDs and dimer acceptors (Fig. 2). In the blend films, both μh (6.1–8.7 × 10−5 cm2 V−1 s−1) and μe (2.8–4.4 × 10−5 cm2 V−1 s−1) values of PD:DYBT:PDMS systems were an order of magnitude lower compared to those of the PD:DYBT (μh = 4.0–4.6 × 10−4 cm2 V−1 s−1 and μe = 3.1–3.6 × 10−4 cm2 V−1 s−1) or PD:DYBT:DYPDMS systems (μh = 3.1–4.4 × 10−4 cm2 V−1 s−1 and μe = 1.2–2.8 × 10−4 cm2 V−1 s−1), regardless of the PD type (Table S10, ESI†). This observation supports the importance of chemically bonding PDMS to the photoactive materials. The higher charge mobilities of DYBT and DYBT:DYPDMS systems account for their higher Jsc values in OSCs compared to the DYBT:PDMS system.
Charge generation properties were assessed by measuring the photocurrent density (Jph) under different effective voltages (Veff) (Fig. S20, ESI†). Regardless of the donor type, exciton dissociation probabilities (P(E,T)s) of the DYBT (88–90%) and DYBT:DYPDMS systems (87–93%) were significantly higher compared to those of the DYBT:PDMS systems (70–79%). Notably, the P(E,T) of PM6-b-PDMS:DYBT:DYPDMS (93%) was significantly higher than those of PM6:DYBT:DYPDMS (87%) and PM6-b-PDMS:DYBT (88%). These findings suggest that dual PDMS incorporation enhances charge generation properties, consistent with the highest Jsc observed for PM6-b-PDMS:DYBT:DYPDMS OSCs among the systems studied.
The charge recombination properties assessed through light intensity (P)-dependent Voc measurements showed similar trends (Fig. S21, ESI†). The slopes (S) of the Vocvs. P plots for DYBT:PDMS systems (1.48–1.55 kT q−1) were significantly higher than those for DYBT- (1.17–1.21 kT q−1) and DYBT:DYPDMS-based systems (1.07–1.29 kT q−1). Furthermore, PM6-b-PDMS:DYBT:DYPDMS, with dual incorporation of PDMS into both the PD and acceptor, exhibited the lowest S (1.07 kT q−1) among all systems studied, indicating minimal monomolecular/trap-assisted recombination and explaining superior Jsc and FF values observed in the OSC device.
Next, we investigated the long-term thermal stability of PM6-b-PDMS:DYBT:PDMS- and PM6-b-PDMS:DYBT:DYPDMS-based OSCs under continuous heating at 80 °C in an inert atmosphere (Fig. S22, ESI†), and observed the critical role of using DYPDMS in achieving superior thermal stability. PM6-b-PDMS:DYBT:DYPDMS-based OSCs demonstrated a high thermal stability, retaining over 80% of their initial PCE after 500 h of heating. In contrast, PM6-b-PDMS:DYBT:PDMS-based OSCs exhibited rapid degradation, retaining only 63% of their initial PCE after just 120 h of heating. This stark difference is mainly attributed to variations in morphological stability. In the PM6-b-PDMS:DYBT:PDMS system, the rapid diffusion of physically-mixed PDMS molecules caused significant changes in the blend morphology under thermal stress. In contrast, in the PM6-b-PDMS:DYBT:DYPDMS system, the morphology remained highly stable, with the molecular diffusion of PDMS being suppressed by its chemical bonding to DYPDMS as well as high molecular compatibility between DYPDMS and other materials.
Mechanical properties of photoactive films
To evaluate the suitability of the developed blend systems for IS-OSC applications, we investigated the mechanical properties of the blend films using pseudo free-standing tensile tests (Fig. 4 and Table 3).63,64 The stress–strain (S–S) curves from these tests are presented in Fig. 4a and b. First, we compared the tensile properties of the blend films depending on the PD types, while keeping the acceptor fixed as DYBT (Fig. 4a). The PM6:DYBT blend film exhibited brittle mechanical behavior, as evidenced by a very low COS of 3%. In contrast, the PM6-b-PDMS:DYBT blend film demonstrated significantly improved mechanical stretchability, with a COS of 20%. This enhancement in stretchability resulted in a substantial 6.5-fold increase in toughness for the PM6-b-PDMS:DYBT blend film (toughness = 4.6 MJ m−3) compared to the PM6:DYBT blend film (toughness = 0.7 MJ m−3). This result highlights the effectiveness of chemically bonding PDMS chains to the PD on enhancing mechanical robustness. We also attribute this improvement to the sufficient length of the PDMS chains incorporated into the PD. Specifically, the PDMS block in PM6-b-PDMS has a Mn of 19 kg mol−1 that exceeds the entanglement molecular weight (Me ∼ 12 kg mol−1) of PDMS.44,59 Thus, the PDMS chains are expected to contribute to the stretchability of the resulting blend films by increasing the entanglement density.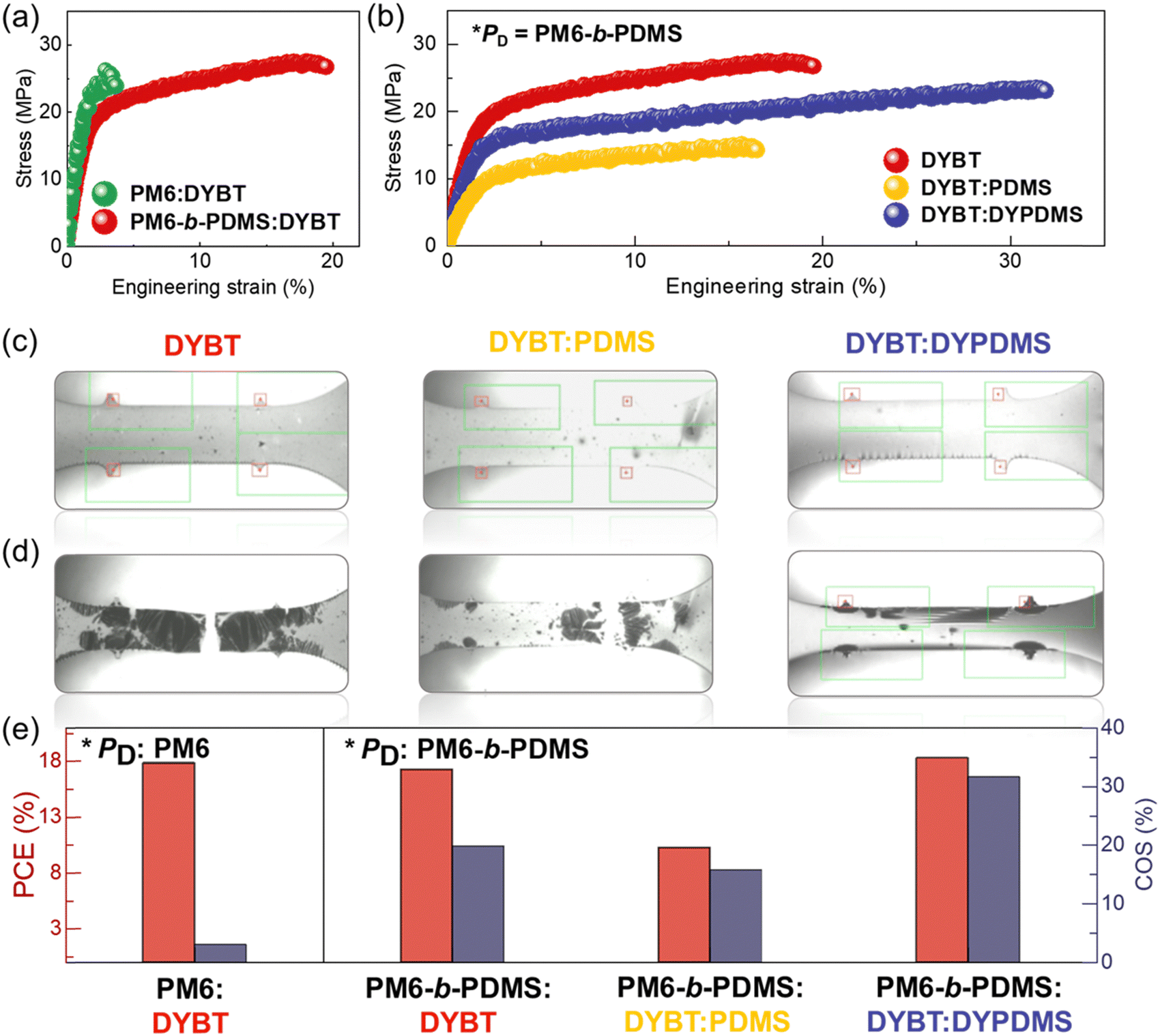 | ||
| Fig. 4 (a) and (b) S–S curves of blend films for (a) PD:DYBT and (b) PM6-b-PDMS:acceptors, (c) and (d) OM images of the of PM6-b-PDMS:acceptors-based blend films at (c) 0% and (d) 20% strain. (e) comparison of PCE and COS values among the photoactive systems studied. | ||
Next, we investigated the tensile properties of the blend films as a function of acceptor type, with PM6-b-PDMS PD (Fig. 4b). Interestingly, blend films containing a physical mixture of PDMS polymers (PM6-b-PDMS:DYBT:PDMS) exhibited a lower COS (16%) compared to the PM6-b-PDMS:DYBT binary blend (COS = 20%). This unexpected reduction in stretchability is attributed to severe aggregation of PDMS and phase separation with photoactive materials due to thermodynamic immiscibility. Notably, the PM6-b-PDMS:DYBT:DYPDMS blend films demonstrated significantly higher stretchability, with a COS of 32%, compared to the other two blends. These results indicate that the chemical bonding of PDMS in acceptors (DYBT:DYPDMS) is more effective than the physical mixture of PDMS (DYBT:PDMS) for increasing the stretchability of the blend films. Considering the short PDMS length in DYPDMS (Mn = 1 kg mol−1), we speculate that the improvement in stretchability of DYPDMS-based blend films is not attributed to an increase in entanglement density, as observed for PM6-b-PDMS. Instead, the incorporation of PDMS into DYPDMS reduces aggregation and crystallinity, increasing the number and size of amorphous domains within the blend films.65 The influence of DYPDMS on blend morphology will be explored in detail in the next section.
Optical microscopy (OM) images of the blend films at 0 and 20% strain are compared in Fig. 4c and d, respectively. The PM6-b-PDMS:DYBT and PM6-b-PDMS:DYBT:PDMS blend films exhibited sharp crack tips and stress concentrations at defect sites after 20% strain (Fig. 4d). In contrast, PM6-b-PDMS:DYBT:DYPDMS film did not show any crack at the same strain. Consequently, the PM6-b-PDMS:DYBT:DYPDMS photoactive system achieved an excellent combination of photovoltaic performance (PCE = 18.3%) and stretchability (COS = 32%). This performance is distinct not only from the other photoactive systems examined in this study but also from many previously reported systems, which typically face a trade-off between photovoltaic efficiency and mechanical robustness (Fig. 4e).
Morphological properties of photoactive films
To analyze origins of different photovoltaic and mechanical properties, we investigated morphology of the PM6-b-PDMS:acceptor blend films using atomic force microscopy (AFM), transmission electron microscopy (TEM), resonant soft X-ray scattering (RSoXS), and GIXS measurements (Fig. 5 and Table 4). The surface morphology of the blend films was examined using AFM (Fig. 5a). The PM6-b-PDMS:DYBT:PDMS blend films, containing physically mixed PDMS, displayed macrophase separation of PDMS domains with much higher surface roughness (root-mean-square averaged roughness, Rq = 14.8 nm) compared to PM6-b-PDMS:DYBT (Rq = 2.3 nm). In contrast, PM6-b-PDMS:DYBT:DYPDMS (1.4 nm) blend films, incorporating chemically linked DYPDMS, exhibited much smoother surfaces and more intermixed morphologies compared to PM6-b-PDMS:DYBT.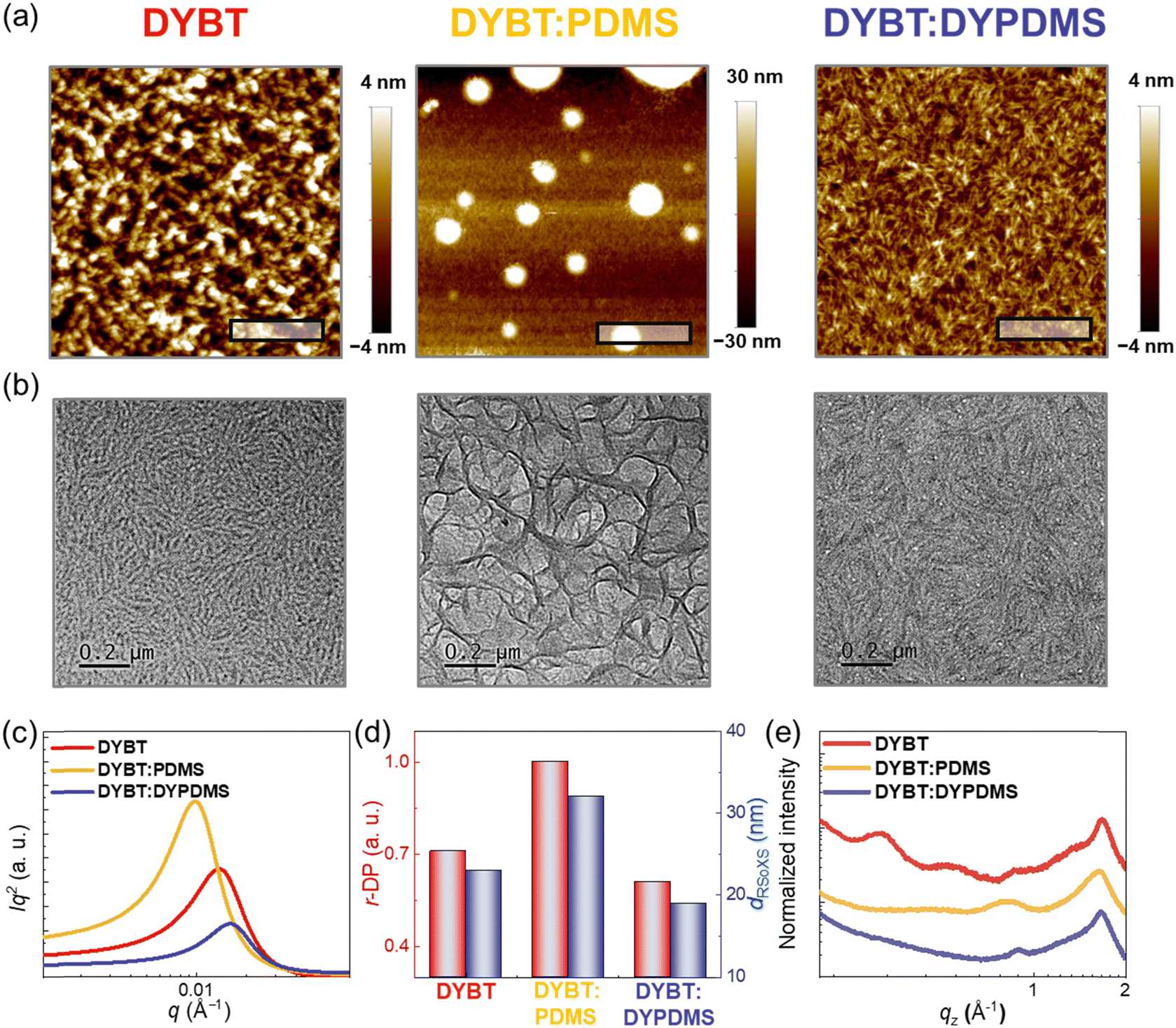 | ||
| Fig. 5 (a) AFM height images (scale bars are 200nm), (b) TEM images (scale bars are 200nm), (c) RSoXS profiles, (d) r-DP and dRSoXS values obtained from the RSoXS measurements, and (e) GIXS line-cut profile in the OOP direction of the PM6-b-PDMS:acceptors blend films. | ||
TEM results revealed that PM6-b-PDMS:DYBT blend films exhibited distinct and fine nano-fibrillar structures, attributed to the high crystallinity of DYBT (Fig. 5b). In contrast, PM6-b-PDMS:DYBT:PDMS blend films displayed severely phase-separated morphology with large grain boundaries and collapsed fibrillar structures. Notably, PM6-b-PDMS:DYBT:DYPDMS blend films retained the fibrillar structures originating from DYBT, but featured finer fibrils and a more intermixed morphology compared to PM6-b-PDMS:DYBT. Domain properties, including size and purity, analyzed through RSoXS experiments, supported the findings from AFM and TEM measurements (Fig. 5c). For example, PM6-b-PDMS:DYBT:PDMS blend films exhibited higher relative domain purity (r-DP = 1.00) and domain spacing (dRSoXS = 64 nm) compared to PM6-b-PDMS:DYBT (r-DP = 0.74 and dRSoXS = 46 nm). In contrast, PM6-b-PDMS:DYBT:DYPDMS blend films exhibited lower r-DP of 0.61 and a smaller dRSoXS of 38 nm compared to PM6-b-PDMS:DYBT blend films (Fig. 5d).
To elucidate how the incorporation of PDMS into PDs and/or acceptors affects photovoltaic properties, we compared the morphology of four blend films—PM6:DYBT, PM6-b-PDMS:DYBT, PM6:DYBT:DYPDMS, and PM6-b-PDMS:DYBT:DYPDMS using RSoXS measurements (Fig. S23 and Table S11, ESI†). When DYBT (without PDMS) was used as the acceptor, the PM6:DYBT blend films exhibited lower relative domain purity (r-DP = 0.68) and smaller domain spacing (dRSoXS = 39 nm) compared to PM6-b-PDMS:DYBT (r-DP = 0.74 and dRSoXS = 46 nm). In contrast, when DYBT:DYPDMS (with PDMS) was used as the acceptor, the PM6-b-PDMS:DYBT:DYPDMS films (r-DP = 0.61 and dRSoXS = 38 nm) showed less pronounced phase separation compared to PM6:DYBT:DYPDMS (r-DP = 0.64 and dRSoXS = 43 nm). These results suggest that blend morphologies are more intermixed when both the PD and acceptor either lack PDMS or include PDMS, compared to when PDMS is present in only one component. This enhanced intermixing in PM6:DYBT and PM6-b-PDMS:DYBT:DYPDMS blends can improve charge generation and suppress charge recombination in OSCs, resulting in superior device Jsc and PCE compared to PM6-b-PDMS:DYBT and PM6:DYBT:DYPDMS, respectively.
To further understand different morphologies depending on the presence of PDMS in PDs and acceptors, we measured contact angles of water and glycerol droplets on pristine films of PM6, PM6-b-PDMS, DYBT, and DYPDMS (Fig. S24 and Table S12, ESI†). From these contact angles, we calculated the surface tension of each component and the interfacial tension (γ) between donor–acceptor pairs using the Wu model.66 The interfacial tension between PM6 and DYBT (γPM6:DYBT = 0.57) was lower than that with PM6-b-PDMS and DYBT (γPM6-b-PDMS:DYBT = 1.65). Conversely, the interfacial tension between PM6-b-PDMS and DYPDMS (γPM6-b-PDMS:DYPDMS = 0.41) was significantly lower than that between PM6 and DYPDMS (γPM6:DYPDMS = 1.67). These results highlight that non-PDMS acceptors are thermodynamically more compatible with non-PDMS donors, while PDMS-containing acceptors are more compatible with PDMS-incorporated donors.
Crystalline structures in the blend films were analyzed using GIXS (Fig. 5e and Fig. S25–S27, ESI†). The relative crystal sizes of the (010) scattering peaks in the OOP direction, associated with the π–π stacking of the materials, were determined by calculating Lc values using the Scherrer equation. The Lc(010)OOP values for PM6-b-PDMS:DYBT (3.8 nm) and PM6-b-PDMS:DYBT:DYPDMS (3.3 nm) were significantly larger than that of PM6-b-PDMS:DYBT:PDMS (2.2 nm), indicating better-developed crystalline networks in the former two blend films. The r-DoC of the (010) scattering peaks showed a similar trend. For example, the r-DoC values for PM6-b-PDMS:DYBT (1.00) and PM6-b-PDMS:DYBT:DYPDMS (0.91) were higher than that for PM6-b-PDMS:DYBT:PDMS (0.79).
The morphological characteristics of the blend films strongly correlate with their photovoltaic and mechanical properties. The PM6-b-PDMS:DYBT system exhibited well-defined fibrillar structures due to the high crystallinity of DYBT, facilitating efficient charge transport and a high PCE (17.3%). However, the sharp donor–acceptor interfaces created by rigid DYBT molecules acted as defect sites under elongation, resulting in moderate stretchability (COS = 20%). In contrast, the physical mixing of PDMS (PM6-b-PDMS:DYBT:PDMS) led to severe phase separation and a collapse of crystalline structures due to the strong immiscibility of PDMS with the active components, resulting in the poor PCE and low COS value. However, the use of PDMS-incorporated acceptor (DYPDMS) significantly improved the blend morphology through two mechanisms. (1) Suppression of phase separation via chemical bonding: Unlike the physically added PDMS in the DYBT:PDMS system, which undergoes severe phase separation during film formation due to immiscibility, PDMS in DYPDMS is chemically bonded to the conjugated backbone. This structural integration prevents macro-phase separation, leading to a more uniform morphology. (2) Enhanced molecular compatibility and stabilized donor–acceptor interfaces: The introduction of PDMS blocks in both the PD (PM6-b-PDMS) and the dimer acceptor (DYPDMS) improves their chemical compatibility, reducing thermodynamic barriers to mixing. This suppression of demixing strengthens donor–acceptor interactions, resulting in a more stable and well-intermixed film morphology. As a result, PM6-b-PDMS:DYBT:DYPDMS retained the high crystallinity and fibrillar networks of DYBT, while the combined use of PM6-b-PDMS and DYPDMS significantly increased the interfacial areas between the donor–acceptor interfaces. The preserved fibrillar networks enabled efficient charge transport, while the increased interfaces facilitated more effective charge generation and mechanical stress dissipation. These synergistic effects led to the optimized OSC system to achieve both high PCE (18.3%) and improved mechanical stretchability (COS = 32%).
Photovoltaic and mechanical performances of IS-OSCs
To demonstrate the feasibility of the developed photoactive systems for wearable applications, IS-OSC devices were fabricated (Fig. 6a).67–72 The photovoltaic and mechanical properties of IS-OSCs based on four blend films of (1) PM6:DYBT, (2) PM6-b-PDMS:DYBT, (3) PM6-b-PDMS:DYBT:PDMS, and (4) PM6-b-PDMS:DYBT:DYPDMS were thoroughly investigated. The J–V curves of the IS-OSCs are shown in Fig. 6b, and the corresponding photovoltaic parameters are summarized in Table 5. The initial PCEs of the IS-OSCs followed the same trend as the rigid OSC devices. Among the systems, PDMS:DYBT:DYPDMS-based IS-OSCs exhibited the highest initial PCE of 12.74%, surpassing devices based on PM6:DYBT- (PCE = 12.08%), PM6-b-PDMS:DYBT (PCE = 11.58%), and PM6-b-PDMS:DYBT:PDMS (PCE = 7.02%). | ||
| Fig. 6 (a) Schematic illustration of the IS-OSC device structure. (b) J–V curves of the IS-OSCs. (c) Normalized PCE of the IS-OSCs vs. engineering strain. (d) Changes in the area of the devices in terms of engineering strain. The plot is used to estimate the υ value by quadratic function fitting. (e) Strain-induced power output changes in IS-OSCs: normalized power output vs. engineering strain curves. | ||
| P D | Acceptor | V oc (V) | J sc (mA cm−2) | FF | PCEmax(avg)a (%) | Strain at PCE80% (%) |
|---|---|---|---|---|---|---|
| a Average values obtained from 5 independent devices. | ||||||
| PM6 | DYBT | 0.92 (0.92 ± 0.01) | 19.99 (19.18 ± 0.40) | 0.66 (0.65 ± 0.01) | 12.08 (11.14 ± 0.36) | 12 |
| PM6-b-PDMS | DYBT | 0.92 (0.91 ± 0.01) | 19.52 (18.84 ± 0.32) | 0.65 (0.64 ± 0.01) | 11.58 (11.02 ± 0.31) | 27 |
| DYBT:PDMS | 0.90 (0.89 ± 0.02) | 14.71 (13.84 ± 0.67) | 0.53 (0.52 ± 0.01) | 7.02 (6.48 ± 0.65) | 21 | |
| DYBT:DYPDMS | 0.93 (0.93 ± 0.01) | 20.22 (19.30 ± 0.41) | 0.68 (0.67 ± 0.01) | 12.74 (12.23 ± 0.44) | 41 | |
The stretchability of the IS-OSCs also significantly varied depending on the photoactive material systems (Fig. 6c). For PD:DYBT systems, PM6-b-PDMS:DYBT-based IS-OSCs exhibited a strain at PCE80% of 27%, more than double that of PM6:DYBT-based devices (12%). Among PM6-b-PDMS:acceptor systems, PM6-b-PDMS:DYBT:DYPDMS-based IS-OSCs demonstrated the highest strain at PCE80% (41%), significantly outperforming both PM6-b-PDMS:DYBT (strain at PCE80% = 27%) and PM6-b-PDMS:DYBT:PDMS-based devices (strain at PCE80% = 21%). In addition, the PM6-b-PDMS:DYBT:DYPDMS-based IS-OSCs exhibited significantly higher cyclic durability compared to those based on PM6-b-PDMS:DYBT, PM6-b-PDMS:DYBT:PDMS, and PM6:DYBT systems (Fig. S28 and S29, ESI†). For example, the normalized PCE values for the IS-OSCs based on these blend films after 100 stretching cycles at 10% strain were 80, 67, 53, and 31%, respectively. These findings align with the mechanical and morphological properties of different photoactive layers, underscoring the importance of designing mechanically robust photoactive systems to achieve high stretchability in IS-OSCs (Table S13, ESI†).
Unlike rigid OSCs, where their photoactive area remains constant, the photoactive area of IS-OSCs can expand during stretching.16,17 Considering that IS-OSCs with a Poisson's ratio of υ under a strain (ε), the length increases by (1 + ε), and the width decreases by (1 − υε). Thus, the total area as a function of ε follows the quadratic form: −υε2 + (1 − υ)ε + 1. In this case, the total power output (PCE × photoactive area) of IS-OSCs may increase with strain due to the expanding photoactive area. To assess the area expansion of IS-OSCs under stretching, we measured changes in the top electrode area across various strain levels. The observed area changes were fitted to the quadratic equation: −υε2 + (1 − υ)ε + 1, resulting in a calculated ν value of 0.354 (Fig. 6d).17 Using this ν value, the potential for power output enhancement in IS-OSCs was analyzed by plotting the normalized power output (normalized PCE × area) as a function of strain (Fig. 6e). Notably, PM6-b-PDMS:DYBT:DYPDMS-based IS-OSCs exhibited a continuous power output increase during the stretching in the ε range from 0 to 35%. This represents one of the first examples showing strain-induced power enhancement in OSCs. In contrast, all other IS-OSCs including PM6:DYBT, PM6-b-PDMS:DYBT, and PM6-b-PDMS:DYBT:PDMS showed decreases in power output with increasing strain. Therefore, this result highlights the potential of the dual elastomer incorporated photoactive systems for developing high-performance IS-OSCs with high mechanical stretchability suitable for wearable applications.
Conclusions
We designed new DYPDMS and developed efficient and highly-stretchable IS-OSCs capable of increasing power output under strain. Importantly, we demonstrated the critical role of simultaneously incorporating PDMS into both the PD and the acceptor materials to achieve both high photovoltaic and mechanical performances in IS-OSCs. The combined use of the PDMS-incorporated PD (PM6-b-PDMS) and PDMS-incorporated dimer acceptor (DYPDMS) led to photoactive films with enhanced entanglement density and an increased fraction of amorphous domains. Additionally, dual PDMS incorporation significantly improved molecular compatibility between the PD and acceptor molecules, reducing phase separation and strengthen donor–acceptor interfaces. As a result, IS-OSCs based on the PM6-b-PDMS:DYBT:DYPDMS system exhibited a high PCE of 12.7% and outstanding stretchability (strain at PCE80% = 41%), which outperformed those based on PM6-b-PDMS:DYBT (PCE = 11.6% and strain at PCE80% = 27%) and PM6-b-PDMS:DYBT:PDMS system (PCE = 7.0% and strain at PCE80% = 21%). Importantly, PM6-b-PDMS:DYBT:DYPDMS-based IS-OSCs successfully demonstrated strain-induced power enhancement even at a high strain of 35%. Our results underscore the importance of (1) elastomer-incorporated acceptor design and (2) simultaneous inclusion of elastomers in active components in achieving IS-OSCs with high performance and mechanical robustness.Experimental
Supplementary data (e.g., cyclic voltammogram, UV-Vis spectra, DSC, and additional GIXS and RSoXS analyses) and experimental procedures are included in ESI.†Author contributions
J.-W. Lee fabricated the solar cells, and wrote the original draft. Q. Nguyen synthesized the dimer acceptor materials and performed synthetic characterizations. W. J. Kang and T.-S. Kim conducted the tensile tests. S. Seo synthesized the polymer donor materials and assisted synthetic characterizations. J. Choi assisted TEM analysis. S. Lee and J-Y. Lee assisted IS-OSC fabrication. S. Lee and J. Park reviewed and revised the manuscript. B. J. Kim supervised the study and revised the manuscript. All authors commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (2022R1A2B5B03001761, RS-2023-00217884, and RS-2024-00358817). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.References
- Y. Li, G. Xu, C. Cui and Y. Li, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- D. J. Lipomi, Joule, 2018, 2, 195–198 CrossRef.
- E. Dauzon, X. Sallenave, C. Plesse, F. Goubard, A. Amassian and T. D. Anthopoulos, Adv. Mater., 2021, 33, 2101469 CrossRef CAS PubMed.
- S. Dong, T. Jia, K. Zhang, J. Jing and F. Huang, Joule, 2020, 4, 2004–2016 CrossRef CAS.
- Y. T. Hsieh, J. Y. Chen, S. Fukuta, P. C. Lin, T. Higashihara, C. C. Chueh and W. C. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 21712–21720 CrossRef CAS PubMed.
- Z. Jiang, K. Fukuda, W. C. Huang, S. Park, R. Nur, M. O. G. Nayeem, K. Yu, D. Inoue, M. Saito, H. Kimura, T. Yokota, S. Umezu, D. Hashizume, I. Osaka, K. Takimiya and T. Someya, Adv. Funct. Mater., 2019, 29, 1808378 CrossRef.
- J.-W. Lee, G. U. Kim, D. J. Kim, Y. Jeon, S. Li, T. S. Kim, J. Y. Lee and B. J. Kim, Adv. Energy Mater., 2022, 12, 2200887 CrossRef CAS.
- J. M. Huang, Z. Lu, J. Q. He, H. Hu, Q. Liang, K. Liu, Z. W. Ren, Y. K. Zhang, H. Y. Yu, Z. J. Zheng and G. Li, Energy Environ. Sci., 2023, 16, 1251–1263 RSC.
- J.-W. Lee, S. Seo, S.-W. Lee, G.-U. Kim, S. Han, T. N.-L. Phan, S. Lee, S. Li, T.-S. Kim, J.-Y. Lee and B. J. Kim, Adv. Mater., 2022, 34, 2207544 CrossRef CAS PubMed.
- Z. Y. Wang, M. C. Xu, Z. L. Li, Y. R. Gao, L. Yang, D. Zhang and M. Shao, Adv. Funct. Mater., 2021, 31, 2103534 CrossRef CAS.
- Y. T. Hsieh, J. Y. Chen, S. Fukuta, P. C. Lin, T. Higashihara, C. C. Chueh and W. C. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 21712–21720 CrossRef CAS PubMed.
- J. Noh, G. U. Kim, S. Han, S. J. Oh, Y. Jeon, D. Jeong, S. W. Kim, T. S. Kim, B. J. Kim and J. Y. Lee, ACS Energy Lett., 2021, 6, 2512–2518 CrossRef CAS.
- D. J. Lipomi, B. C. K. Tee, M. Vosgueritchian and Z. N. Bao, Adv. Mater., 2011, 23, 1771–1775 CrossRef CAS PubMed.
- Z. Wang, D. Zhang, M. Xu, J. Liu, J. He, L. Yang, Z. Li, Y. Gao and M. Shao, Small, 2022, 18, 2201589 CrossRef CAS PubMed.
- J. Wang, Y. Ochiai, N. Wu, K. Adachi, D. Inoue, D. Hashizume, D. Kong, N. Matsuhisa, T. Yokota, Q. Wu, W. Ma, L. Sun, S. Xiong, B. Du, W. Wang, C.-J. Shih, K. Tajima, T. Aida, K. Fukuda and T. Someya, Nat. Commun., 2024, 15, 4902 CrossRef CAS PubMed.
- K. Zhou, D. Han, K. Xian, S. Li, M. Gao, K. Zhang, B. Zhao, X. Li, Y. Chen, Y. Geng and L. Ye, Energy Environ. Sci., 2024, 17, 5950–5961 RSC.
- J.-W. Lee, E. S. Oh, S. Lee, T. N.-L. Phan, T.-S. Kim, J.-Y. Lee, J. R. Reynolds and B. J. Kim, Joule, 2025, 9, 101792 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- F. Liu, L. Zhou, W. Liu, Z. Zhou, Q. Yue, W. Zheng, R. Sun, W. Liu, S. Xu, H. Fan, L. Feng, Y. Yi, W. Zhang and X. Zhu, Adv. Mater., 2021, 33, 2100830 CrossRef CAS PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- H. Lu, W. Liu, G. Ran, Z. Liang, H. Li, N. Wei, H. Wu, Z. Ma, Y. Liu, W. Zhang, X. Xu and Z. Bo, Angew. Chem., Int. Ed., 2024, e202314420 Search PubMed.
- S. Luo, C. Li, J. Zhang, X. Zou, H. Zhao, K. Ding, H. Huang, J. Song, J. Yi, H. Yu, K. S. Wong, G. Zhang, H. Ade, W. Ma, H. Hu, Y. Sun and H. Yan, Nat. Commun., 2023, 14, 6964 CrossRef CAS PubMed.
- C. Sun, J.-W. Lee, Z. Tan, T. N.-L. Phan, D. Han, H.-G. Lee, S. Lee, S.-K. Kwon, B. J. Kim and Y.-H. Kim, Adv. Energy Mater., 2023, 13, 2301283 CrossRef CAS.
- Z. Gan, L. Wang, J. Cai, C. Guo, C. Chen, D. Li, Y. Fu, B. Zhou, Y. Sun, C. Liu, J. Zhou, D. Liu, W. Li and T. Wang, Nat. Commun., 2023, 14, 6297 CrossRef CAS PubMed.
- J.-W. Lee, C. Sun, J. Lee, D. J. Kim, W. J. Kang, S. Lee, D. Kim, J. Park, T. N.-L. Phan, Z. Tan, F. S. Kim, J.-Y. Lee, X. Bao, T.-S. Kim, Y.-H. Kim and B. J. Kim, Adv. Energy Mater., 2024, 14, 2303872 CrossRef CAS.
- B. Liu, W. Xu, R. Ma, J.-W. Lee, T. A. Dela Peña, W. Yang, B. Li, M. Li, J. Wu, Y. Wang, C. Zhang, J. Yang, J. Wang, S. Ning, Z. Wang, J. Li, H. Wang, G. Li, B. J. Kim, L. Niu, X. Guo and H. Sun, Adv. Mater., 2023, 35, 2308334 CrossRef CAS PubMed.
- S. H. Chen, S. T. Zhu, L. Hong, W. Y. Deng, Y. Zhang, Y. Fu, Z. Y. Zhong, M. H. Dong, C. C. Liu, X. H. Lu, K. Zhang and F. Huang, Angew. Chem., Int. Ed., 2024, e202318756 CAS.
- X. Wu, X. Zheng, T. Chen, S. Zhang, Y. Zhou, M. Wang, T. Chen, Y. Wang, Z. Bi, W. Fu, M. Du, W. Ma, L. Zuo and H. Chen, Adv. Mater., 2024, 36, 2406879 CrossRef CAS PubMed.
- J.-W. Lee, H.-G. Lee, E. S. Oh, S.-W. Lee, T. N.-L. Phan, S. Li, T.-S. Kim and B. J. Kim, Joule, 2024, 8, 204–223 CrossRef CAS.
- X. Li, H. Ke, S. Li, M. Gao, S. Li, J. Yu, H. Xie, K. Zhou, K. Zhang and L. Ye, Adv. Funct. Mater., 2024, 34, 2400702 CrossRef CAS.
- M. Wang, T. Chen, Y. Li, G. Ding, Z. Chen, J. Li, C. Xu, A. Wupur, C. Xu, Y. Fu, J. Xue, W. Fu, W. Qiu, X. Yang, D. Wang, W. Ma, X. Lu, H. Zhu, X. Chen, X. Wang, H. Chen and L. Zuo, Energy Environ. Sci., 2024, 17, 2598–2609 RSC.
- Y.-J. Xue, Z.-Y. Lai, H.-C. Lu, J.-C. Hong, C.-L. Tsai, C.-L. Huang, K.-H. Huang, C.-F. Lu, Y.-Y. Lai, C.-S. Hsu, J.-M. Lin, J.-W. Chang, S.-Y. Chien, G.-H. Lee, U. S. Jeng and Y.-J. Cheng, J. Am. Chem. Soc., 2024, 146, 833–848 CrossRef CAS PubMed.
- H. Wang, Z. Zhong, S. Gámez-Valenzuela, J.-W. Lee, B. Li, C. Xu, J. Yang, H. Sun, B. J. Kim, B. Liu and X. Guo, Adv. Funct. Mater., 2024, 2418805, DOI:10.1002/adfm.202418805.
- X. Li, H. Ke, S. Li, M. Gao, S. Li, J. Yu, H. Xie, K. Zhou, K. Zhang and L. Ye, Adv. Funct. Mater., 2024, 2400702, DOI:10.1002/adfm.202400702.
- Z. Peng, K. Xian, Y. Cui, Q. Qi, J. Liu, Y. Xu, Y. Chai, C. Yang, J. Hou, Y. Geng and L. Ye, Adv. Mater., 2021, 33, 2106732 CrossRef CAS PubMed.
- W. Tang, Z. Ding, Y. Su, Q. Weng, Y. Zhang, R. Li, W. Huang, Z. Wang, Y. Wu, Y. Han, K. Zhao, Z. Yang, X. Wang and S. Liu, Adv. Funct. Mater., 2024, 34, 2312289 CrossRef CAS.
- A. A. Shafe, H. M. Schrickx, K. Ding, H. Ade and B. T. O’Connor, ACS Energy Lett., 2023, 8, 3720–3726 CrossRef CAS.
- J. Han, F. Bao, D. Huang, X. Wang, C. Yang, R. Yang, X. Jian, J. Wang, X. Bao and J. Chu, Adv. Funct. Mater., 2020, 30, 2003654 CrossRef CAS.
- Y.-C. Tseng, Q. Fan, C.-Y. Tsai, J.-F. Chang, M.-H. Yu, H.-Z. Tseng, H.-L. Yip, F. R. Lin, A. K.-Y. Jen and C.-C. Chueh, Adv. Funct. Mater., 2024, 34, 2408993 CrossRef CAS.
- Q. Wan, H. Jeon, S. Seo, E. S. Oh, J.-W. Lee, C. Wang, T.-S. Kim, B. J. Kim and B. C. Thompson, Chem. Mater., 2023, 35, 10476–10486 CrossRef CAS.
- Z. Wang, M. Xu, Z. Li, Y. Gao, L. Yang, D. Zhang and M. Shao, Adv. Funct. Mater., 2021, 31, 2103534 CrossRef CAS.
- S. Seo, J.-W. Lee, D. J. Kim, D. Lee, T. N.-L. Phan, J. Park, Z. Tan, S. Cho, T.-S. Kim and B. J. Kim, Adv. Mater., 2023, 35, 2300230 CrossRef CAS PubMed.
- H.-G. Lee, J.-W. Lee, E. S. Oh, M. J. Lee, T.-S. Kim, C. Lee and B. J. Kim, J. Mater. Chem. A, 2024, 12, 19039–19051 RSC.
- J.-W. Lee, J. S. Park, H. Jeon, S. Lee, D. Jeong, C. Lee, Y.-H. Kim and B. J. Kim, Chem. Soc. Rev., 2024, 53, 4674–4706 RSC.
- S. Li, R. Zhang, M. Zhang, J. Yao, Z. Peng, Q. Chen, C. Zhang, B. Chang, Y. Bai, H. Fu, Y. Ouyang, C. Zhang, J. A. Steele, T. Alshahrani, M. B. J. Roeffaers, E. Solano, L. Meng, F. Gao, Y. Li and Z.-G. Zhang, Adv. Mater., 2023, 35, 2206563 CrossRef CAS PubMed.
- H. Wang, C. Cao, H. Chen, H. Lai, C. Ke, Y. Zhu, H. Li and F. He, Angew. Chem., Int. Ed., 2022, 61, e202201844 CrossRef CAS PubMed.
- Y. Liang, D. Zhang, Z. Wu, T. Jia, L. Lüer, H. Tang, L. Hong, J. Zhang, K. Zhang, C. J. Brabec, N. Li and F. Huang, Nat. Energy, 2022, 7, 1180–1190 CrossRef CAS.
- C. Sun, J.-W. Lee, C. Lee, D. Lee, S. Cho, S.-K. Kwon, B. J. Kim and Y.-H. Kim, Joule, 2023, 7, 416–430 CrossRef CAS.
- Y. Li, L. Mei, Z. Ge, C. Liu, J. Song, Y. Man, J. Gao, J. Zhang, Z. Tang, X.-K. Chen and Y. Sun, Adv. Mater., 2024, 36, 2403890 CrossRef CAS PubMed.
- Z. Zhang, S. Yuan, T. Chen, J. Wang, Y.-Q.-Q. Yi, B. Zhao, M. Li, Z. Yao, C. Li, X. Wan, G. Long, B. Kan and Y. Chen, Energy Environ. Sci., 2024, 17, 5719–5729 RSC.
- H. Fu, Q. Wang, Q. Chen, Y. Zhang, S. Meng, L. Xue, C. Zhang, Y. Yi and Z.-G. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202403005 CrossRef CAS PubMed.
- J.-W. Lee, C. Sun, H. Jeon, T. H.-Q. Nguyen, T. N.-L. Phan, X. Bao, Y.-H. Kim and B. J. Kim, Adv. Funct. Mater., 2024, 34, 2404569 CrossRef CAS.
- J.-W. Lee, C. Sun, S. Lee, D. J. Kim, E. S. Oh, T. N.-L. Phan, T. H.-Q. Nguyen, S. Seo, Z. Tan, M. J. Lee, J.-Y. Lee, X. Bao, T.-S. Kim, C. Lee, Y.-H. Kim and B. J. Kim, Nano Energy, 2024, 125, 109541 CrossRef CAS.
- H. You, Austin L. Jones, B. S. Ma, G.-U. Kim, S. Lee, J.-W. Lee, H. Kang, T.-S. Kim, J. R. Reynolds and B. J. Kim, J. Mater. Chem. A, 2021, 9, 2775–2783 RSC.
- S. J. Clarson, K. Dodgson and J. A. Semlyen, Polymer, 1985, 26, 930–934 CrossRef CAS.
- J.-W. Lee, D. Jeong, D. J. Kim, T. N.-L. Phan, J. S. Park, T.-S. Kim and B. J. Kim, Energy Environ. Sci., 2021, 14, 4067–4076 RSC.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639–4647 CrossRef CAS.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- J. Xu, S. Wang, G.-J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y. H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B. H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59–64 CrossRef CAS PubMed.
- Z. Chiguvare and V. Dyakonov, Phys. Rev. B:Condens. Matter Mater. Phys., 2004, 70, 235207 CrossRef.
- J. H. Kim, A. Nizami, Y. Hwangbo, B. Jang, H. J. Lee, C. S. Woo, S. Hyun and T. S. Kim, Nat. Commun., 2013, 4, 2520 CrossRef PubMed.
- J.-W. Lee, T. H.-Q. Nguyen, E. S. Oh, S. Lee, J. Choi, H. S. Kwon, C. Wang, S. Lee, J.-Y. Lee, T.-S. Kim and B. J. Kim, Adv. Energy Mater., 2024, 14, 2401191 CrossRef CAS.
- J.-W. Lee, S.-W. Lee, J. Kim, Y. H. Ha, C. Sun, T. Ngoc-Lan Phan, S. Lee, C. Wang, T.-S. Kim, Y.-H. Kim and B. J. Kim, J. Mater. Chem. A, 2022, 10, 20312–20322 RSC.
- S. Wu, J. Polym. Sci., Polym. Symp., 1971, 34, 19–30 CrossRef.
- X. Zheng, X. Wu, Q. Wu, Y. Han, G. Ding, Y. Wang, Y. Kong, T. Chen, M. Wang, Y. Zhang, J. Xue, W. Fu, Q. Luo, C. Ma, W. Ma, L. Zuo, M. Shi and H. Chen, Adv. Mater., 2024, 36, 2307280 CrossRef CAS PubMed.
- J. S. Park, G. U. Kim, S. Lee, J.-W. Lee, S. Li, J. Y. Lee and B. J. Kim, Adv. Mater., 2022, 34, 2201623 CrossRef CAS PubMed.
- W. Yang, X. Luo, M. Li, C. Shi, Z. Wang, Z. Yang, J. Wu, X. Zhang, W. Huang, D. Ma, C. Wang, W. Zhong and L. Ying, Adv. Energy Mater., 2024, 2403259, DOI:10.1002/aenm.202403259.
- J.-W. Lee, C. Lim, S. W. Lee, Y. Jeon, S. Lee, T. S. Kim, J. Y. Lee and B. J. Kim, Adv. Energy Mater., 2022, 12, 2202224 CrossRef CAS.
- W. Song, Q. Ye, Z. Chen, J. Ge, L. Xie and Z. Ge, Adv. Mater., 2024, 36, 2311170 CrossRef CAS PubMed.
- J.-W. Lee, T. N.-L. Phan, E. S. Oh, H.-G. Lee, T.-S. Kim and B. J. Kim, Adv. Funct. Mater., 2023, 33, 2305851 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00002e |
| ‡ J.-W. Lee and Q. Nguyen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |