
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining sequential extractions with bulk and micro X-ray spectroscopy to elucidate iron and phosphorus speciation in sediments of an iron-treated peat lake†‡
Melanie A.
Münch
 *a,
Andreas
Voegelin
b,
Luis Carlos
Colocho Hurtarte
cd,
Jörg
Göttlicher
e and
Thilo
Behrends
a
*a,
Andreas
Voegelin
b,
Luis Carlos
Colocho Hurtarte
cd,
Jörg
Göttlicher
e and
Thilo
Behrends
a
aUtrecht University, Princetonlaan 8A, 3584CB Utrecht, The Netherlands. E-mail: m.a.munch@uu.nl
bEawag, Swiss Federal Institute of Aquatic Science and Technology, Überlandstrasse 133, CH-8600 Dübendorf, Switzerland
cEuropean Synchrotron Radiation Facility, Beamline ID21, Grenoble 38100, France
dDiamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK
eKarlsruhe Institute of Technology, Institute for Photon Science and Synchrotron Radiation, Hermann-von-Helmholtz Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 12th November 2024
Abstract
In shallow lakes, mobilization of legacy phosphorus (P) from the sediments can be the main cause for persisting eutrophication after reduction of external P input. In-lake remediation measures can be applied to reduce internal P loading and to achieve ecosystem recovery. The eutrophic shallow peat lake Terra Nova (The Netherlands) was treated with iron (Fe) to enhance P retention in the sediment. This treatment, however, intensified seasonal internal P loading. An earlier study suggested that Fe addition led to increased P binding by easily-reducible Fe(III) associated with organic matter (OM), which readily releases P when bottom waters turn hypoxic. In this complementary study, bulk and micro Fe K-edge and P K-edge X-ray absorption spectroscopy and micro-focused X-ray fluorescence spectroscopy were applied to characterize the P hosting Fe(III) pool. Combined with sequential extraction data, the synchrotron X-ray analyses revealed that a continuum of co-precipitates of Fe(III) with calcium, phosphate, manganese and organic carbon within the OM matrix constitutes the reducible Fe(III) pool. The complementary analyses also shed new light on the interpretation of sequential extraction results, demonstrating that pyrite was not quantitatively extracted by nitric acid (HNO3) and that most of the Fe(II) extracted by hydrochloric acid (HCl) originated from phyllosilicate minerals. Formation of an amorphous inorganic–organic co-precipitate upon Fe addition constitutes an effective P sink in the studied peaty sediments. However, the high intrinsic reactivity of this nanoscale co-precipitate and its fine distribution in the OM matrix makes it very susceptible to reductive dissolution, leading to P remobilization under reducing conditions.
Environmental significanceImpairment of surface water quality due to eutrophication is a global issue, causing ecosystem degradation and increasing greenhouse gas emissions. In many lakes, reduction of internal phosphorus loading is needed for ecosystem recovery. Iron addition offers a cost-effective remediation strategy, but little is known about its long-term effects in organic-rich systems. Our study shows that addition of Fe salts to peat lakes can induce the formation of nanoscale inorganic–organic co-precipitates embedded within the organic matter matrix that are prone to reductive dissolution. These co-precipitates can effectively bind phosphorus, but in a form highly susceptible to remobilization under reducing conditions. In organic-rich lakes, iron remediation can thus have adversary effects and potentially increase temporary internal P loading during bottom water hypoxia. |
1 Introduction
Phosphorus (P) is an essential and often limiting or co-limiting macro-nutrient in freshwater ecosystems.2,3 Increased P input due to human activities such as fertilizer use or wastewater disposal is one of the main causes for the eutrophication and deterioration of freshwater ecosystems.4 In industrialized countries, decades of anthropogenic P input have caused P to accumulate in soils and freshwater sediments.5–9 This so-called “legacy P” is a major water management challenge, since it can sustain high surface water P concentrations by internal P loading, even when external P sources have been significantly reduced, thus delaying restoration efforts for decades.10–12Methods to reduce internal P loading include iron (Fe) amendment.13,14 This method is based on the classical model of Fe and P diagenesis: the affinity of phosphate to Fe(III)(oxyhydr)oxides, which bind phosphate by adsorption or coprecipitation, closely couples the cycling of P and Fe in environmental systems.15–17 Fe amendment thus can enhance the retention of P in lake sediments by binding it to the solid phase and recycling it as a passenger of the Fe redox cycle.18,19 The natural coupling of the geochemical cycles of P and Fe makes Fe amendment less invasive than treatments with other chemicals such as e.g. aluminium (Al)-compounds or lanthanum (La)-modified bentonite. However, due to the redox sensitivity of Fe, the success of the treatment is highly dependent on the geochemistry of the sediment. In their reviews of case studies, Bakker et al.13 and Smolders et al.14 concluded that the effect and success of Fe amendment was dependent on the extent of sulfate reduction and the presence of organic matter (OM) in the sediments. Sulfate reduction produces sulfide which precipitates with Fe2+ forming Fe-sulfides (FeSx) and ultimately pyrite (FeS2), stabilizing Fe(II) and burying it into deeper sediment layers.20,21 OM influences the interaction of P with Fe by competition for adsorption sites on the Fe(III)(oxyhydr)oxide surface, hindrance of crystallization of Fe(III) minerals,22 the formation of monomeric and oligomeric Fe-OM complexes23–27 and the association with and stabilization of Fe(III)-(hydr)oxide colloids.28–30 The OM-associated Fe can bind P with Fe acting as the bridging ion between P and OM, a phenomenon which was also reported involving other metal cations such as Al, calcium (Ca), copper (Cu), zinc (Zn), magnesium (Mg) and manganese (Mn).31–33 The resulting P-metal-OM associations account for a substantial part of P in natural environments, as reported for natural waters24,34 and postulated for soils and sediments.1,29,31,32,35,36
The effects of sulfate reduction and OM on the coupling of Fe and P cycling in lake sediments is reflected by the speciation of the two elements in the sediments. Hence, knowledge on the solid phase P and Fe speciation is pivotal for the design of Fe-based remediation measures in freshwater systems. Sequential extraction procedures (SEPs), i.e., extraction over multiple sequential chemical extraction steps, are widely used to assess the binding forms of solid-phase Fe and P. Widely used and often adapted SEPs for P (P-SEP) from sediments were developed by Ruttenberg for marine sediments37 and the one by Psenner et al.38 for lake sediments and suspended matter. Popular SEPs for Fe (Fe-SEP) from sediments include the procedures developed by Claff et al.39 for acid sulfate soils, with special focus on iron sulfide minerals, and by Poulton and Canfield40 for modern and ancient sediments.
SEPs are based on consecutive extraction steps of sediment samples at increasing desorptive and/or proton-, ligand-, or reduction-promoted extraction strength, targeting solid phase Fe and P pools of increasing recalcitrance. This approach yields operationally defined Fe and P pools, which can be hypothetically assigned to distinct Fe and P phases based on the extractability of pure minerals.37–41 However, the interpretation of SEPs can be challenging, especially also in very organic-rich sediments like those of peat lakes. These sediments may contain significant amounts of P–Fe-OM phases, which are not specifically targeted in SEPs. Furthermore, the extractability of autochthonous Fe and P phases could be different in sediments of peat lakes where they form in an organic matrix.
To elucidate the nature of reactive Fe and P phases in sediments or soils, SEPs have been combined with other methods such as 31P-NMR, XRD, Mössbauer or XAS.36,42–45 To date, however, studies that systematically relate chemical fractionation to spectroscopic speciation results for P and Fe in lake sediments are scarce, in particular for organic-rich sediments. A recent study of a boreal peat lake36 combined different P-SEP approaches, with an extraction step for humic-acid-bound P46 added to the SEDEX procedure,37 and used the approach of Poulton and Canfield40 to fractionate Fe. From the combination of these SEP with 31P-NMR, 57Fe-Mössbauer and Fe K-edge and P K-edge XAS, they concluded that in spite of the anoxic conditions, a large part of the sedimentary Fe was present as Fe(III) in a continuum of ferrihydrite-like phases with a distribution of crystallinity and variable amounts of associated P and OM. They also showed that spectroscopic analyses can provide complementary information on the interpretation of SEP results. For example, SEP results indicated the presence of carbonate-associated Fe, reducible but more crystalline Fe(III)(oxyhydr)oxides and magnetite. In contrast ferrihydrite-like phases and minor amounts of nano-goethite and Fe in phyllosilicates were identified by Mössbauer spectroscopy but no magnetite or Fe-carbonates.
This study focusses on the Fe and P speciation in sediments from the shallow peat lake Terra Nova (NL), which was treated with FeCl3 in 2010 to reduce internal P loading.1 The Fe-amendment efficiently lowered the surface water P concentrations for 1.5 years before seasonal P release started again with increasing intensity, resulting in the highest P concentrations since the start of monitoring and renewed algae blooms. Our investigation of the changes in sediment geochemistry due to the Fe addition suggested that the pronounced seasonal release of P was due to the high redox turnover of Fe associated with OM.1 The interpretation in this previous paper was based on a combination of P-SEP (adapted from O'Connell et al.36 and Slomp et al.47), Fe-SEP (adapted from Poulton and Canfield,40 Raiswell et al.41 and Claff et al.48), porewater analysis and sediment incubation experiments. The P-SEP and Fe-SEP results suggested the presence of a reactive Fe-OM pool, which interacts with P. The surface sediment, in the depth interval affected by the Fe addition, was characterized by elevated contents of sodium bicarbonate extract P (P-NaHCO3) and ascorbic acid extractable Fe (Fe-Asc) with respect to the deeper sediment. Sodium bicarbonate is conceived to extract P associated to humic substances over metal cation bridges.36,46 Ascorbic acid, a weak reducing agent, targets easily reducible Fe such as in ferrihydrite.41 Although the two extractions are considered to dissolve different Fe phases and associated P, we hypothesize that the extracted Fe and P originate from the same highly reactive Fe pool. However, the nature of this postulated, highly reactive Fe pool and the bound P remained elusive.
In the present study, we therefore aim at collecting complementary information on the speciation of the corresponding Fe and P pools and their occurrence in the lake sediments. For this, we performed bulk and micro X-ray absorption spectroscopy and X-ray fluorescence and correlated the results to sediment geochemistry data, especially of Fe-SEP and P-SEP, collected in our earlier study.
2 Methods
2.1 Study site
Terra Nova is a shallow, polymictic peat lake of the Loosdrecht lake system in The Netherlands. The local water authority categorizes it as eutrophic based on the EU Water Framework Directive.49 It formed due to peat extraction in the early modern age, spanning an area of 0.85 km2 with a maximum depth of 2 m and a mean depth of 1 m. Starting in 2010 the lake was treated with 40% FeCl3 solution over a period of 1.5 years, with a total average Fe dose of 0.5 mol Fe m−2 as a remediation measure to reduce internal P loading.12.2 Sediment sampling and analysis
Undisturbed sediment cores (length 60 cm, Ø 6 cm, min. 10 cm overlying water) were collected at three positions in lake Terra Nova in June 2020 using a gravity corer (UWITEC, Mondsee, Austria). The cores were stored at bottom water temperature (15 °C) under preservation of bottom water oxygen conditions and sliced in an N2 purged glovebox within three days (sampling resolution: 1 cm for top 10 cm, 2 cm for rest of the cores). The solid phase was separated from the porewater by centrifugation (10 minutes, 3000 rpm), freeze-dried anoxically (ventilation with N2), homogenized in an N2 purged glovebox using an agate mortar and pestle, and stored in the glovebox at room temperature.1In this study we used depth-resolved solid-phase total elemental contents, sequential extraction derived Fe and P solid-phase speciation and porewater concentrations from three sediment cores, each from its respective sampling site (ESI A‡). Details on the chemical analysis of the sediment porewater and solid phase and the detailed data sets are reported in the preceding study.1 Total Fe, P, Ca, Al, S, and Mn contents of the sediment solid phase were determined by HF digestion; TOC (total organic carbon) and TON (total organic nitrogen) were determined using an NCS analyzer as reported by Helmond et al.50
Fe and the P speciation in the sediment solid phase was determined by dedicated sequential extraction procedures (SEP), conducted on samples across the entire depth of three sediment cores (one per station A to C, ESI A‡). Here, concise information regarding the SEPs is listed. Details on the used procedures and the corresponding detailed data sets are reported in Münch et al.1 The sequential Fe extraction (Fe-SEP) targeting different, operationally defined, Fe pools in sediments included ascorbic acid extractable Fe (Fe-Asc; 0.057 M ascorbic acid, carbonate buffered, pH 7.5; easily reducible Fe in ferrihydrite41), HCl-soluble Fe(II) (Fe(II)-HCl; 1 M HCl; FeS and FeCO3 (ref. 48)), HCl-soluble Fe(III) (Fe(III)-HCl; 1 M HCl; amorphous Fe(oxyhydr)oxides48), dithionite-reducible Fe (Fe-CDB; acetic acid-buffered citrate-dithionite solution, pH 4.8; crystalline Fe (oxyhydr)oxides such as lepidocrocite, goethite and hematite48), oxalate-soluble Fe (Fe-Ox; 0.37 M oxalate, pH 3.2; magnetite51,52), HNO3-soluble Fe (Fe-HNO3, conc. HNO3 at room temperature for 2 hours; pyrite48). All residual Fe (not-extr. Fe) was regarded as non-reactive Fe, mainly Fe bound in clays.40 Our earlier study suggested, that Fe-Asc also included mono- or oligomeric Fe associated with OM.1 As the extraction only yielded negligible amounts of Fe(III)-HCl and Fe-Ox, this data is not shown here. The sequential P extraction (P-SEP) provided an operational distinction of MgCl2-soluble P (P-MgCl2; 1 M MgCl2, pH 8; adsorbed P47), NaHCO3-extractable P (P-NaHCO3; 1 M NaHCO3, pH 8; P bound to OM over metal cation bridges such as Fe, Al, Ca46,53) citrate-dithionite-extractable P (P-CDB; carbonate-buffered citrate-dithionite solution, pH 7.6; P bound to Fe (oxyhydr)oxides and CaCO3 as well as Fe(II) phosphates47), HCl-soluble P (P-HCl; 1 M HCl, room temperature, 24 hours; apatite47), and P extractable by HCl after ashing (P-ashed/HCl; 550 °C for 2 hours, 1 M HCl at room temperature for 24 hours; organic P47). As the extraction only yielded negligible amounts of P-MgCl2, this data is not shown here. All solid-phase P was extracted within the 10% error margin.
2.3 X-ray absorption spectroscopy (XAS) and micro X-ray fluorescence (μ-XRF)
Bulk Fe K-edge XAS measurements were performed at the SUL-X beamline at the KIT Light Source (Karlsruhe Institute of Technology, Germany), at room temperature in transmissions mode. A Si(111) double-crystal monochromator was used for X-ray energy monochromatization. Bulk P K-edge XAS, μ-XRF and spatially resolved micro-focused XAS (μ-XAS) at the P and Fe K-edges were performed at the ID21 beamline at the European Synchrotron Radiation Facility (ESRF; Grenoble, France; storage ring energy: 6.03 GeV; Beamline design: Salomé et al., 2013;54 Beamline configuration: Werner et al., 2017 (ref. 55)), at room temperature in fluorescence mode. A Si(111) double-crystal monochromator was used for X-ray energy selection and a compact Kirkpatrick–Baez mirror system (KB), based on elliptically shaped, fixed-focus Ni coated mirrors, was used for X-ray focussing.Photon energy was calibrated based on the first maximum of the first derivative of the K-edges of reference compounds (metallic Fe: 7112 eV; tricalcium phosphate: 2153 eV), respectively.
Positions of the μ-XRF maps and μ-XAS point spectra were selected to cover the largest possible variety of Fe and P phases. Positions for lower resolution overview maps (step size 10 μm) were chosen based on differences in material morphology as assessed by optical microscopy on the mounted samples prior to X-ray beam exposure. Positions for high-resolution maps (step sizes 2 μm or 0.5 μm) were chosen based on Fe and P distributions (hotspots, occurrence of Fe and P in higher and lower intensities) and elemental correlations (Fe, P, S, Ca, Si, Al) observed in the low-resolution overview maps. Positions of μ-XAS point spectra (Fe and P K-edges) where selected per high-resolution map based on the same criteria. μ-XAS point spectra were measured right after collection of the respective high-resolution maps to guarantee position precision.
2.3.4.1 Bulk Fe K-edge XAS. For the bulk Fe K-edge XAS spectra, the pre-edge was fitted by a first-order polynomial for background removal (−120 to −20 eV before E0; with E0 set to 7123 eV) and the post edge was fitted by a third-order polynomial for normalization (50 to 500 eV after E0). The Autobk algorithm (Rbkg = 0.85; k-weight = 2; spline k-range 0–12.4 Å−1) was used to extract the EXAFS spectra, and a Kaiser–Bessel window (dk = 2) was used to calculate the Fourier-transform of the k3-weighted EXAFS spectra (k-range = 2–10 Å−1). The XANES spectra were analyzed by LCF over an energy-range of −20 to 180 eV and the EXFAS spectra over a k-range of 2 to 10 Å−. Individual fractions were constrained to values between 0 and 1, whereas the sum of all fractions was not constrained.
Preliminary LCF analyses were performed on the EXAFS and the XANES spectra separately, including an extended set of reference spectra considered relevant for the studied samples. References that were not included in significant fractions in preliminary fits or did not improve the r-factor (normalized sum of square residuals = ∑(data − fit)2/∑datai2) of the fits by at least 10%, were consecutively removed. Finally, six reference spectra that allowed to reproduce all EXAFS and XANES spectra were identified and used for LCF analysis: Fe(III)–HA, CaFeP, Fe(II)aq., smectite, biotite and pyrite (Table 1, ESI B1, and B2‡).
| Name | Material | Reference |
|---|---|---|
| Fe(III)–HA | Pahokee peat humic acid (International Humic Substances Society) loaded with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg kg−1 Fe(III) 000 mg kg−1 Fe(III) |
23 |
| CaFeP | Amorphous Ca–Fe(III)–phosphate formed by the oxidation of 0.5 mM Fe(II) in synthetic bicarbonate-buffered groundwater at pH 7.0 in presence of 4 mM Ca and 0.75 or 1 mM P, without/with 0.5 mM Si (average of four identical spectra) (CaFeP*) | 57 |
| Fe(II)aq. | Aqueous solution of 50 mM FeSO4 and 1 mM HCl | This work |
| Smectite | SWy-2 (Clay Minerals Society), Fe accounting for 15% of cations in octahedral sheet; 7% Fe(II) | 58 |
| Biotite | Natural biotite (Fe accounting for 71% of cations in octahedral sheet, 87% Fe(II)) | 58 |
| Pyrite | Natural pyrite (obtained from the mineral collection of ETH Zurich) | 59 |
| Greigite | Greigite synthesized using the method of ref. 60 according to ref. 61. Unpublished spectrum recorded in fluorescence mode at room temperature kindly provided by ed. D. Burton (Southern Cross University, Australia) | This wok |
| Mackinawite | Amorphous FeS synthesized using the method of ref. 62 | 59 |
| Hematite | Commercial synthetic hematite (Riedel-de-Haën) | This work |
| Vivianite | Synthetic vivianite, synthesized according to ref. 63, as reported in ref. 64 | 59 |
2.3.4.2 Bulk P K-edge XAS. Bulk P K-edge XAS spectra were normalized by applying linear functions for the pre-edge (−22 to −5 eV before E0, E0 = 2152 eV, post edge 28 to 45 eV above E0). The corresponding energy ranges were optimized to give consistent normalizations including spectra or references collected in transmission mode or in previous experiments. The spectra were analyzed by LCF over an energy-range of −10 to 30 eV (XANES), constraining individual fractions to values between 0 and 1 and the sum of the fractions to 1.0.
Preliminary LCF analyses were performed using an extended set of reference spectra considered relevant for the studied samples, consecutively removing references that were not used for the fits. Finally, all bulk P-XANES spectra could be described using DNA and Mg inositol hexaphosphate (Mg-IP6) as references (Table 2, ESI C‡).
| Name | Material | Reference |
|---|---|---|
| Apatite | Commercial hydroxyapatite | 65 |
| Mg-IP6 | Amorphous Mg inositol hexaphosphate (phytate) synthesized using the method of ref. 66 according to ref. 67. Unpublished spectrum kindly provided by Jörg Prietzel (Technical University of Munich, Germany) | This work |
| Fe-IP6 | Amorphous Fe(III) inositol hexaphosphate (phytate) precipitates synthesized following the procedure of:66 100 ml of 0.2 M HCl was mixed with 30 ml of 0.05 M phytic acid dodecasodium salt (C6H6O24P6Na12) – solution, to which enough Fe(III)Cl3 – solution (pH 2) was added to get a P/Fe ratio of 1. The resulting precipitate was filtered and washed three times with boiling deionized water to remove residual adsorbed Fe ions | 67 |
| DNA | Commercial deoxyribonucleic acid sodium salt | 68 |
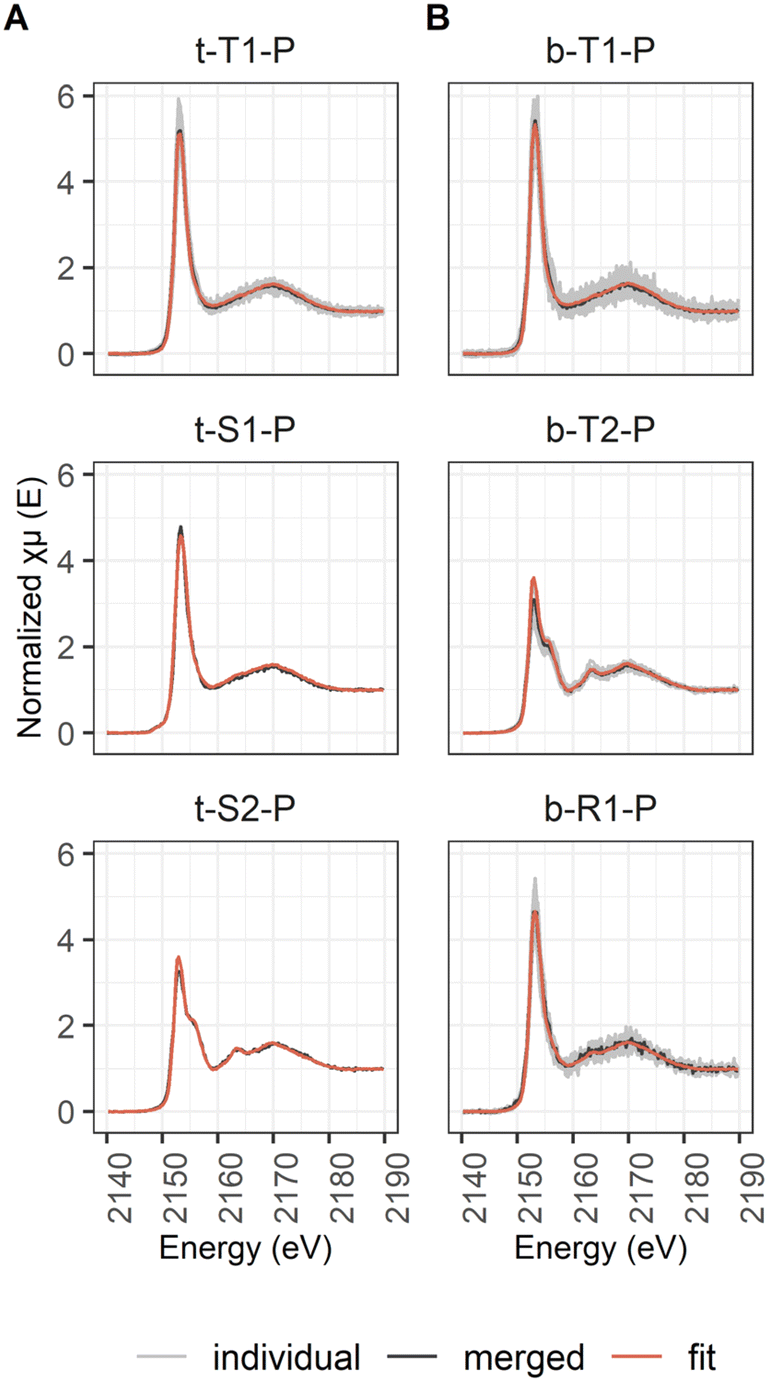
2.3.4.3 Fe and P K-edge μ-XAS. For the Fe K-edge point spectra, the pre-edge was fitted by a first order polynomial for background removal (−100 to −10 eV before the edge) and the post edge was fitted by a third order polynomial for normalization (50 to 800 eV, edge step at 7120 eV). Per spot one spectrum was collected, as the first measurements with multiple scans per spot showed indications of beam damage (ESI D1‡).
The P K-edge μ-XAS spectra were extracted and normalized as were the bulk P K-edge XAS spectra (Section 2.3.4.2). At each spot, three scans were collected.
A total of 190 and 90 viable μ-XANES spectra were recorded at the Fe and P K-edges, respectively. The spectra collected at the respective K-edges were compared by eye after extraction and normalization. All spectra that were identical within the range of noise (variations within the small high frequency features of the spectra) were merged using I0 normalized spectra to yield concentration weighed average spectra. Average spectra of point spectra measured on different maps from the same sample were classified to represent an abundant type (Type 1 to n). Average spectra of point spectra all located on the same map (distance larger than beam size) were classified as regions (Region 1 to n). Unique single spectra were classified as spots (Spot 1 to n). Numbering of the classified spectra was done according to abundance with the number 1 of one class being the one comprised of most individual spectra. The classified average μ-XANES spectra are listed in Table 3 for Fe and in Table 4 for P, including information on how many spectra were merged and on how many μ-XRF maps those spectra were located. The identity of the individual spectra are listed in ESI Table D2(1).‡
| Class | Top: 2 cm depth | Bottom: 40 cm depth | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Merged spectra | Number of maps | Number of spectra | % of spectra | Merged spectra | Number of maps | Number of spectra | % of spectra | |||
| Name | Abbreviation | Name | Abbreviation | |||||||
| Abundant type | Type 1 | t-T1-Fe | 5 | 41 | 43 | Type 1 | b-T1-Fe | 4 | 20 | 21 |
| Type 2 | t-T2-Fe | 4 | 25 | 26 | Type 2 | b-T2-Fe | 3 | 19 | 20 | |
| Type 3 | t-T3-Fe | 4 | 8 | 8 | Type 3 | b-T3-Fe | 2 | 7 | 7 | |
| Type 4 | b-T4-Fe | 3 | 6 | 6 | ||||||
| Region | Region 1 | t-R1-Fe | 1 | 9 | 9 | Region 1 | b-R1-Fe | 1 | 9 | 9 |
| Region 2 | t-R2-Fe | 1 | 7 | 7 | Region 2 | b-R2-Fe | 1 | 8 | 8 | |
| Region 3 | t-R3-Fe | 1 | 2 | 2 | Region 3 | b-R3-Fe | 1 | 6 | 6 | |
| Region 4 | t-R4-Fe | 1 | 3 | 3 | Region 4 | b-R4-Fe | 1 | 6 | 6 | |
| Region 5 | b-R5-Fe | 1 | 5 | 5 | ||||||
| Region 6 | b-R6-Fe | 1 | 3 | 3 | ||||||
| Region 7 | b-R7-Fe | 1 | 3 | 3 | ||||||
| Region 8 | b-R8-Fe | 1 | 2 | 2 | ||||||
| Spot | Spot 1 | b-S1-Fe | 1 | 1 | 1 | |||||
| Total | 95 | 95 | ||||||||
| Class | Top: 2 cm depth | Bottom: 40 cm depth | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Merged spectra | Number of maps | Number of spectra | % of spectra | Merged spectra | Number of maps | Number of spectra | % of spectra | |||
| Name | Abbreviation | Name | Abbreviation | |||||||
| a Two scans showed compressed spectra, which were not included in the LCF. Thus, 12 spectra were used for LCF. | ||||||||||
| Abundant type | Type 1 | t-T1-P | 4 | 33 | 85 | Type 1 | b-T1-P | 3 | 24 | 47 |
| Type 2 | b-T2-P | 3 | 18a | 35 | ||||||
| Region | Region 1 | b-R1-P | 1 | 9 | 18 | |||||
| Spot | Spot 1 | t-S1-P | 1 | 3 | 8 | |||||
| Spot 2 | t-S2-P | 1 | 3 | 8 | ||||||
| Total | 39 | 51 | ||||||||
Average Fe μ-XANES spectra were analyzed by LCF over an energy-range of −20 to 80 eV (XANES), constraining individual fractions to values between 0 and 1 and leaving the sum of the fractions unconstrained. As a first fit, the same six references were used as for LCF of the bulk Fe XAS spectra (see Section 2.3.4.1). If the fit was not satisfying, additional references were included to find the optimal fit. The quality of a fit was assessed according to its r-factor and the visual match between sample and LCF spectra. All fits with an r-factor within 20% of the lowest r-factor were considered as statistically viable fits. All references used are listed in Table 1 and shown in ESI Fig. B1(1).‡ The LCF data is listed in ESI Table B4(1).‡
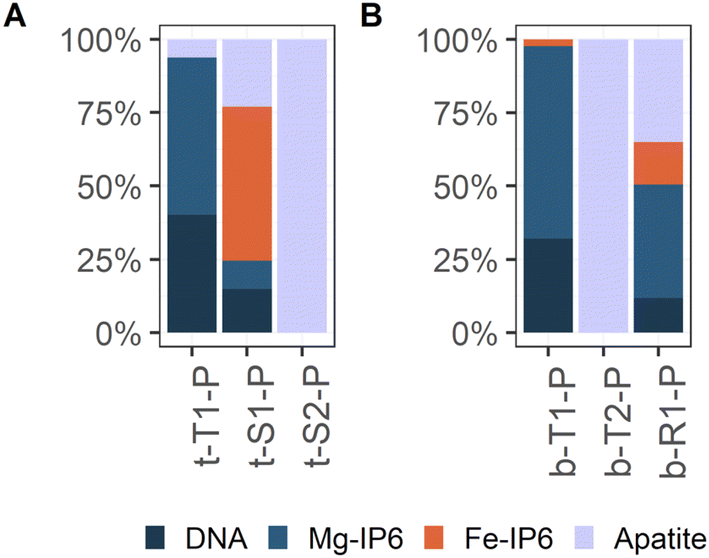
Average P μ-XANES spectra were analyzed by LCF in analogy to the bulk P XANES spectra (Section 2.3.4.2). As for LCF of the bulk P K-edge XANES spectra, preliminary fits with the extended set of reference spectra and consecutive removal of non-used references were performed. Finally, all P μ-XANES spectra could be described using DNA, Mg inositol hexaphosphate (Mg-IP6), Fe inositol hexaphosphate (Fe-IP6) and hydroxyapatite (apatite) as references. All references used are listed in Table 2 and shown in ESI Fig. C1(1).‡ The LCF data is listed in ESI Table C4(1).‡
3 Results
3.1 Total elemental contents
Fig. 1 shows the averaged depth profiles of elemental contents obtained from sampling the solid phase of three sediment cores (one per station A to C, ESI A‡). The enrichment of Fe in the top 5 cm with respect to the deeper sediment, reflected in increased Fe/Al ratios, can be attributed to the Fe treatment.1 In the same depth range, the contents of Ca and Mn were higher compared to the deeper sediment. Sulfur (S) contents were maximum at 20 cm depth with ca. 0.8 mmol per g dw. The Al contents were around 0.95 mmol per g dw in the upper 18 cm, below which they increased to 1.1 mmol per g dw. At the sediment surface, the P content was ca. 80 μmol per g dw and decreased with depth to ca. 25 μmol per g dw. Total organic carbon (TOC) contents were constant with depth at ca. 37 wt%, corresponding to a total organic matter content of approximately 75%.73 Total organic nitrogen (TON) was at ca. 3 wt% in the top 10 cm of the core and decreased with depth to 2 wt%.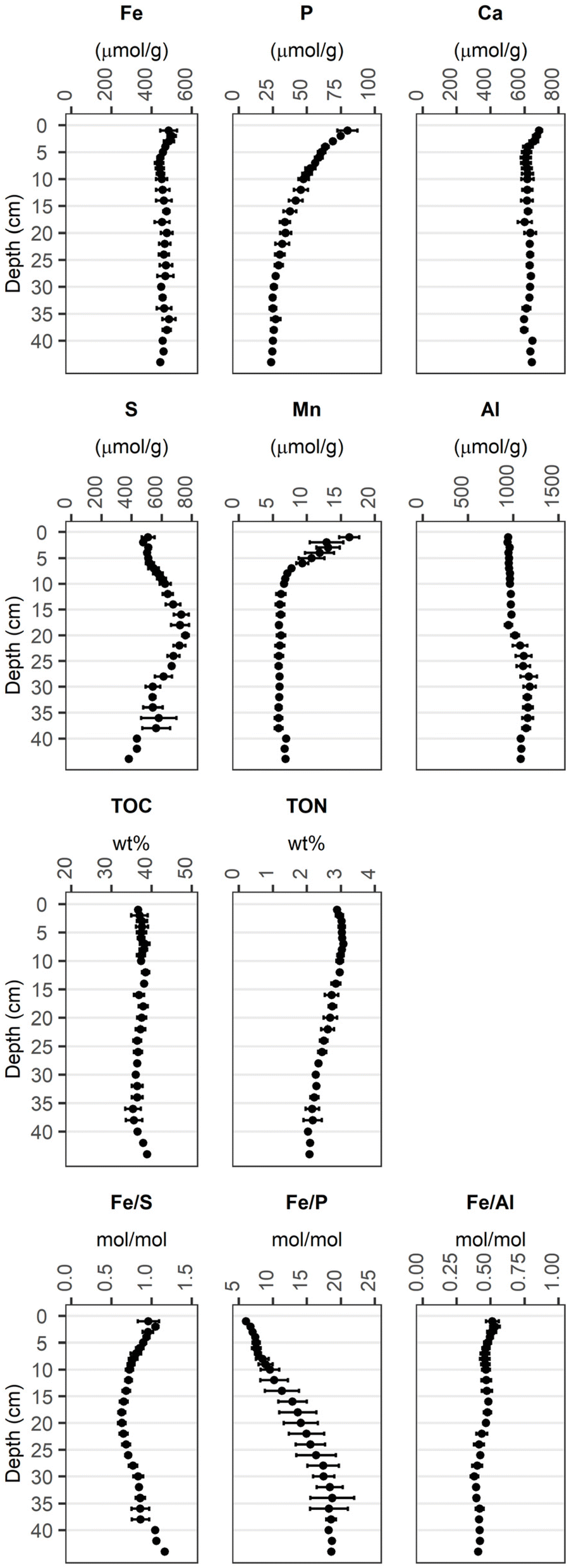 | ||
| Fig. 1 Average total elemental contents of the sediment solid phase (1–36 cm: n = 3, 38 cm: n = 2, 40–44 cm: n = 1). Error bars denote the standard deviation of the values obtained from 3 sediment cores for the corresponding depth. Individual measurements are reported in Münch et al.1 | ||
3.2 Fe and P fractionation according to sequential extraction (SEP) of the sediment solid phase
According to Fe-SEP (Fig. 2A and B) the largest extracted Fe pool throughout the investigated sediment depth was Fe(II) extracted by HCl (Fe(II)-HCl). On average, this step extracted ∼32% of the total Fe from the top 10 cm of sediment. Below 10 cm, Fe(II)-HCl decreased to a minimum of 19% of total Fe between 18 and 28 cm depth and increased again to ∼36% towards the lowest probed sediment depth. The second largest Fe pool was ascorbic acid extractable Fe (Fe-Asc) with up to ∼28% in the top 12 cm and up to ∼26% of total Fe below 28 cm depth. Between 12 and 28 cm depth, Fe-Asc was the smallest extracted Fe pool, while Fe-HNO3 was second largest pool with up to ∼8% of total Fe at 16 cm depth. In the top 6 cm Fe-HNO3 was the smallest extracted Fe pool, accounting for a constant ∼4% of total Fe, as it did below 40 cm depth. Fe-CDB did not show a clear trend with depth, with slightly higher fractions of ∼9% of total Fe in the top 6 cm and oscillating around ∼6% of total Fe in the deeper sediment. While in the surface and the deeper sediment more than 50% of Fe was extracted, between 14 and 28 cm depth over 50% and up to ∼64% of the present Fe was not extracted by the SEP.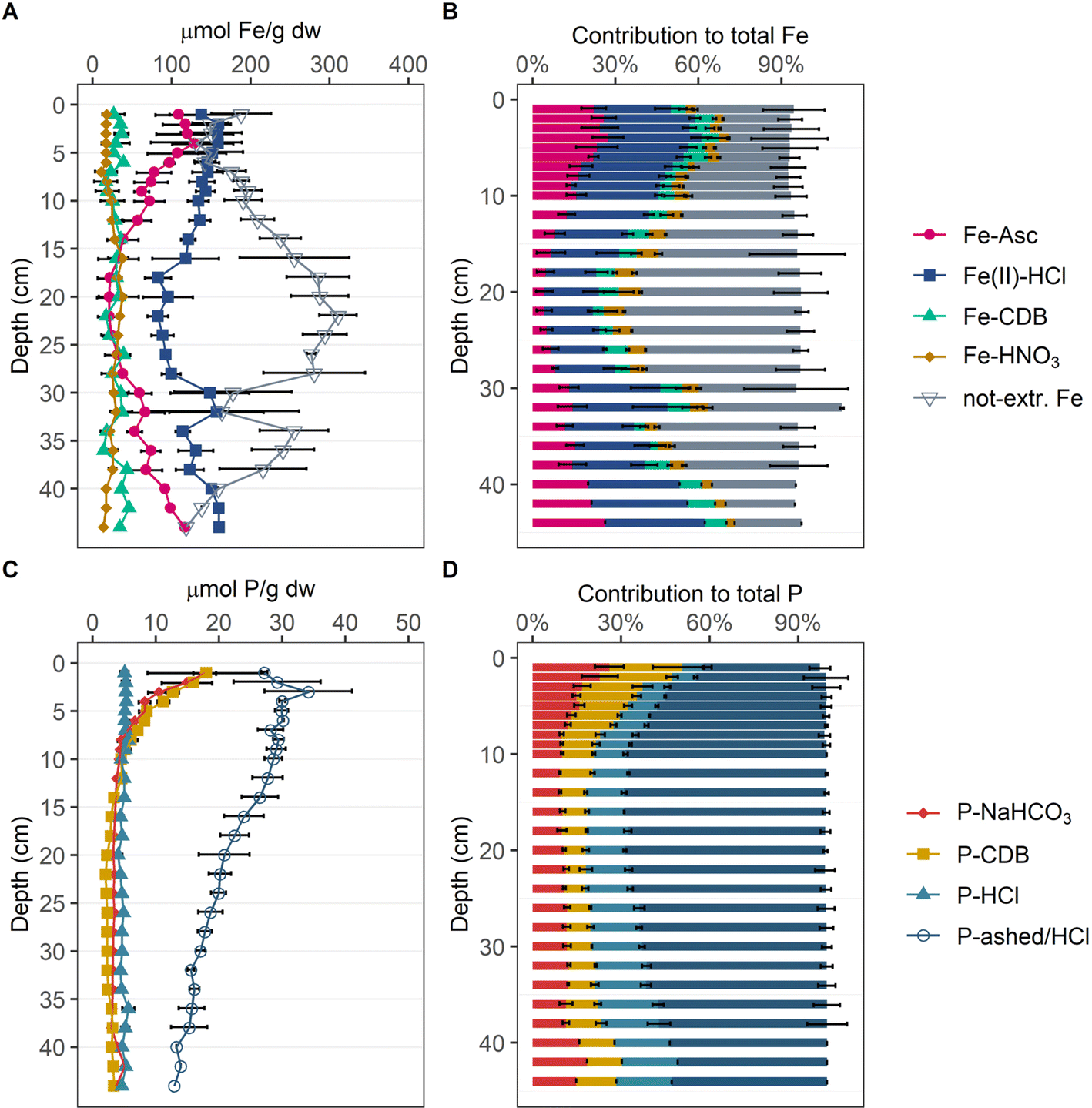 | ||
| Fig. 2 Depth profiles of the Fe and P pools determined by SEP. Respective Fe content of the four extracted and the not extracted Fe pools in μmolFe per g dw (A). Contribution of determined Fe pools to total Fe in averaged percentages (B). Respective P content of four extracted P pools in μmolP per g dw (C). Contribution of determined P pools to total P in averaged percentages (D). For contents individual measurements at each depth were averaged and error bars denote their standard deviation. For the contribution to total contents, percentages of individual measurements were averaged, and error bars denote their standard deviation (1–36 cm: n = 3; 38 cm: n = 2; 40–44 cm: n = 1). Individual measurements are reported in Münch et al.1 | ||
According to the P-SEP (Fig. 2C and D), organic P (P-ashed/HCl) is the dominant P phase over the entire depth accounting for up to ∼69% of total P, except in the top 2 cm, where it accounts for only ∼40% of total P. In the top 6 cm the two Fe-associated P pools P-NaHCO3 and P-DCB are enriched with respect to the deeper sediment, accounting for up to ∼27% and ∼26% of total P, respectively. Apatite (P-HCl) is constant at ∼16% of total P over the entire investigated sediment depth, but nevertheless represents the second largest pool below 10 cm depth.
3.3 Bulk XAS analyses
The LCF analysis of the EXAFS spectra returned consistently higher CaFeP and lower Fe(III)–HA, smectite, and pyrite fractions than the LCF analysis of the corresponding XANES spectra (data listed in ESI Table B3-1‡), but the deviation for individual phases was typically below 10%. For further interpretation of the XAS-derived Fe speciation, the results of the LCF analysis of the EXAFS and XANES range were averaged to avoid bias towards the similarity in local coordination environment or similarity in redox state and coordination geometry (data listed in ESI Table B3–2‡).
The LCF-derived fractions of Fe phases for sediment samples from 1–36 cm depth are displayed in % of total Fe in Fig. 3A and as absolute Fe contents (fractions times total Fe contents) in Fig. 3B. Fe(III)–HA contributed ∼14% to total Fe in the top 3 cm and decreased to below 1% of total Fe below 10 cm depth. CaFeP represented ∼29% of total Fe in the top 7 cm and decreased to ∼17% below 14 cm depth. Fe(II)aq. contribution to total Fe was approximately constant at, on average, ∼6% of total Fe across the investigated sediment depth. The fraction of the spectra from the clay minerals smectite and biotite steadily increased from ∼20% and ∼11%, respectively, at 1 cm depth, to ∼27% and ∼14%, respectively, at 36 cm depth. The contribution of pyrite was lowest in the top 5 cm with a minimum of ∼20% of total Fe at 2 cm depth, constant at around ∼25% of total Fe at 5–7 cm and increased to a maximum of ∼46% of total Fe at 20 cm depth. Below that, pyrite content decreased again, but never contributed less than ∼30% until the bottom of the core. In the top 5 cm of sediment CaFeP contributed most to total Fe, below 5 cm pyrite was the most abundant Fe phase.
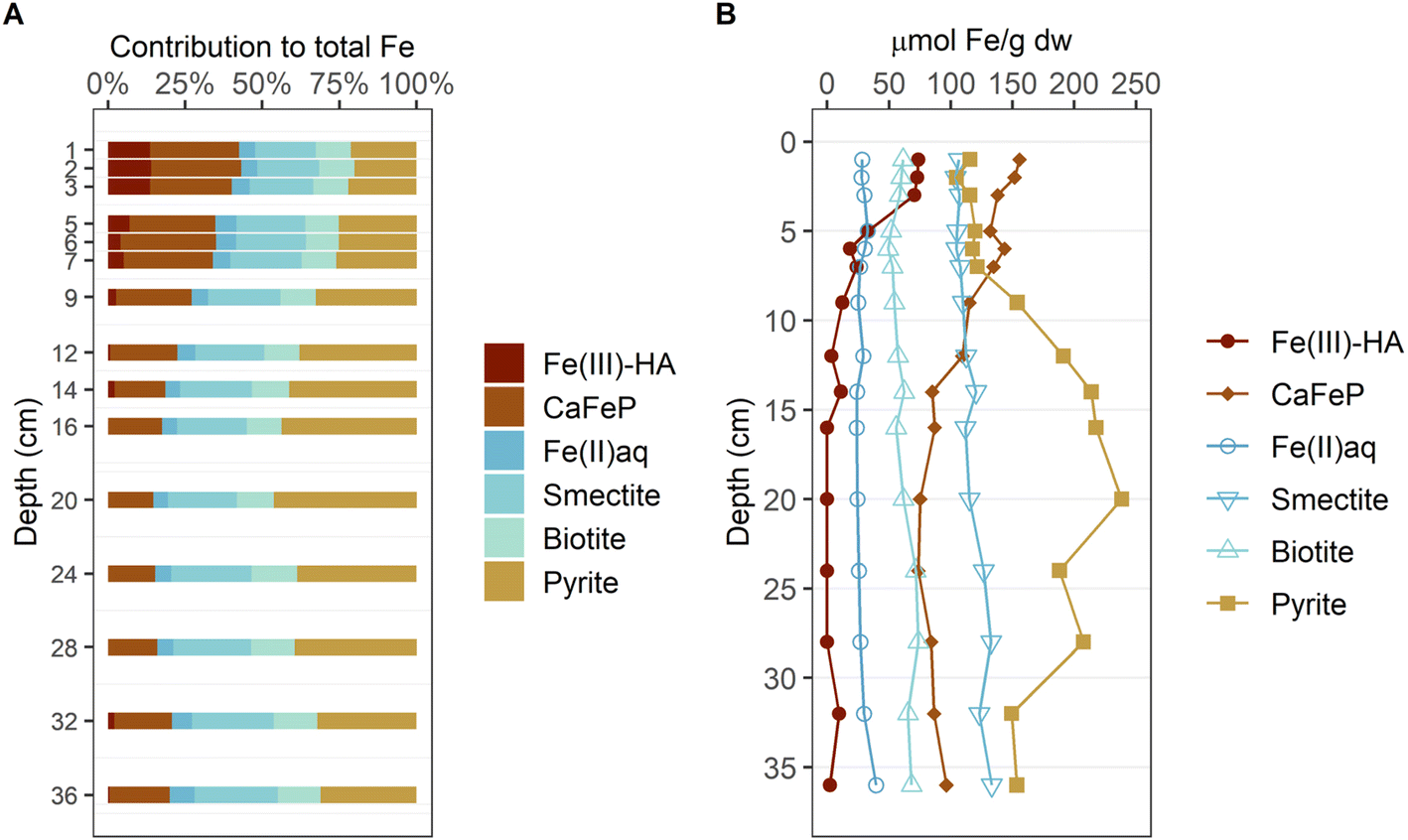 | ||
| Fig. 3 Depth-resolved contribution of Fe reference phase to total Fe as determined by linear combination fitting (LCF) of the bulk Fe K-edge XAS. The given values represent averaged LCF of XANES and EXAFS (A). Concentration of each component as inferred from total Fe content and component contribution from LCF (B). The used references are grouped by similar coordination environment: highly amorphous mono- or oligomeric Fe(III) (red colors), Fe in oxygen-coordinated environments with varying degree of organization and little Fe–Fe coordination (blue colors) and pyrite (yellow). The LCF data is available in Tables ESI B3(1) and B3(2).‡ | ||
3.4 Scanning X-ray microscopy
The ground sediment from both, 2 cm (Fig. 4A) and 40 cm (Fig. 4B), depths, consisted of large fibrous aggregates, hundreds of micrometers in size, with uniform distribution of Fe, Ca and P. Al was also ubiquitously present in the aggregates at both depths but showed more variation in intensity compared to the other three elements (Fig. 4D, H, 5D, E(2), and F(2)). Within or on these aggregates, spherical particles with higher densities of Fe and S occurred, having a size of a few to a few tens of micrometers (Fig. 4B, and F). These particles were identified as framboidal pyrite based on morphology and elemental content in earlier scanning electron microscopy analyses.1 At both depths, also particles were found with a size of tens of micrometers and containing predominately Fe, among the detectable elements (Fig. 4B–D, and F–H). At both depths, hotspots with S as the only detectable element, with sizes ranging from below ten to tens of micrometers (Fig. 4G, F, 5B, C, G, and K), suggested the presence of elementary or organic material locally enriched with S. Spherical particles with high Ca content with a size of tens of micrometers were encountered in material from 2 cm depth, while those particles were less abundant at 40 cm depth and Ca predominately occurred as part of the large aggregates (Fig. 4C, G, and 5). Next to the occurrence in the large aggregates, Si-rich particles with blurred contours, variable shapes and sizes ranging from tens to hundreds of micrometers were found at both sediment depths (Fig. 4D, H, 5D, E(2), and F(2)). | ||
| Fig. 4 μ-XRF maps collected on samples from 2 cm depth (A–D) and 40 cm depth (E–H), at 7200 eV for Fe (A–C and E–G) and at 2400 eV for P (D and H), with a spatial resolution of 10 μm (A and E), and 2 μm (B–D and F–H). The identities of the maps are listed in ESI Table D2(2).‡ | ||
 | ||
| Fig. 5 μ-XRF maps collected on samples from 2 cm depth (A–E) and 40 cm depth (F–L), at 7200 eV for Fe (A–D, F(1), G(1), H, I, K, L, M, and N) and at 2400 eV for P (E, F(2), G(2), and J). The identities of the maps are listed in ESI Table D2(2).‡ Markers and labels refer to positions of μ-XANES spectra (listed in Tables 3 (Fe) and 4 (P), identity in ESI Table D2(1),‡ spectra in Fig. 6 (Fe) and 8 (P), composition according to LCF in Fig. 7 (Fe) and 9 (P)). | ||
Based on the elemental maps Fe K-edge and P K-edge μ-XANES spectra were collected at spots representing the different features described above. A selection of positions of μ-XANES spectra are shown in Fig. 5.
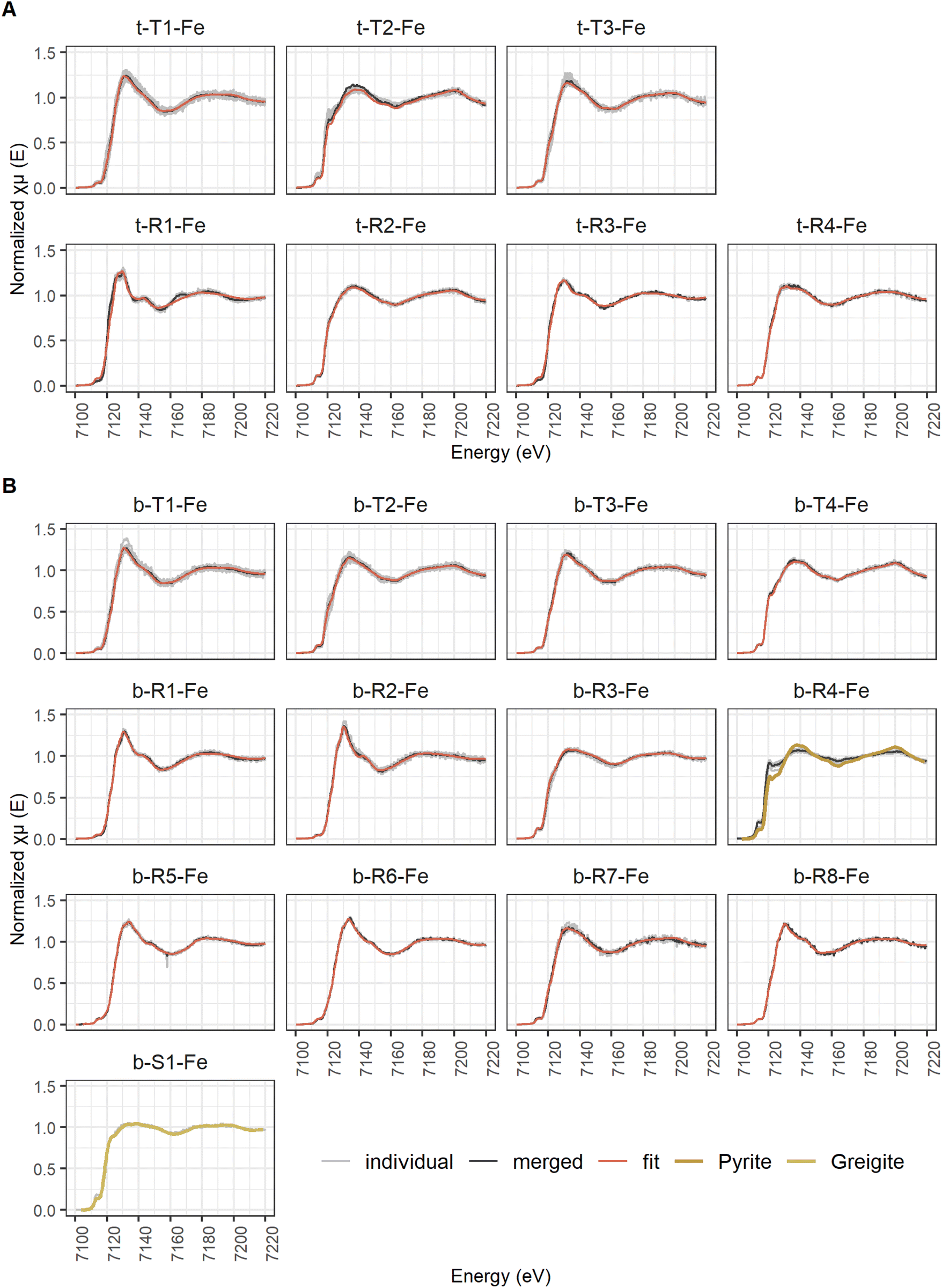 | ||
| Fig. 6 Classified Fe μ-XANES spectra showing individual spectra (individual), the merged spectra (merged) and the LCF spectra (fit) for the samples from 2 cm (A) and 40 cm (B) sediment depth. For the sample at 40 cm depth, the spectra of Region 4 (b-R4-Fe) are compressed, but their shape clearly indicates pyrite and the spectrum of Spot 1 (b-S1-Fe) is almost identical to the greigite reference spectrum. | ||
 | ||
| Fig. 7 Normalized LCF results for aggregated μ-XANES type, region and spot spectra collected on sediment samples from 2 cm (A) and 40 cm (B) depth. The derivation and occurrence of the aggregated spectra are listed in Table 3. Complete LCF results are listed in ESI B3.‡ | ||
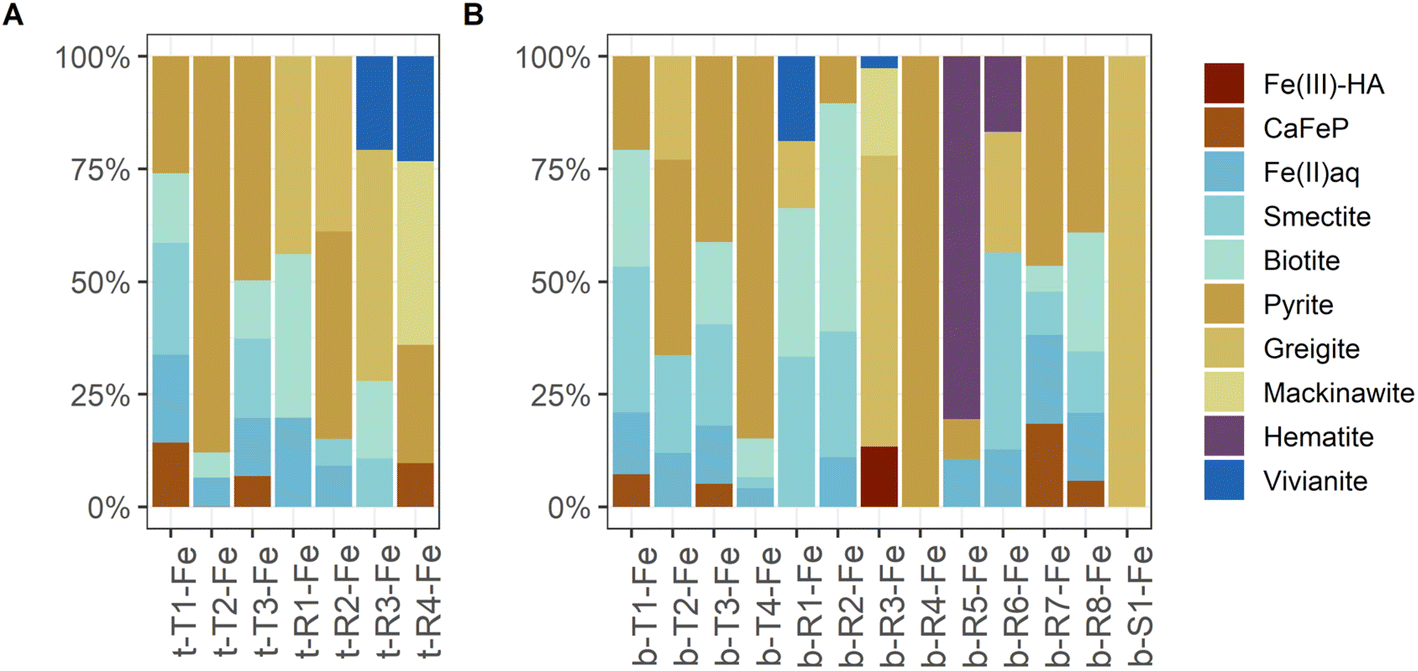
Although the same number of Fe μ-XANES spectra were recorded at both depths, the diversity of spectra was larger at 40 cm depth (Table 3b, spectra in Fig. 6B, LCF results in Fig. 7B and ESI Table B4(1)‡). Four types of Fe spectra were frequently encountered on multiple maps (b-T1-Fe to b-T4-Fe). Type 1 (b-T1-Fe) and Type 2 (b-T2-Fe) were most abundant, both accounting for ∼20% of the measured spectra and found on 4 and 3 maps respectively. Spectrum b-T1-Fe was, as t-T1-Fe at 2 cm depth, mainly found as part of the large aggregates where no element showed pronounced relative densities (Fig. 5G(1), H, and I), b-T2-Fe was both found as part of the aggregates and in areas with slightly higher S abundance (Fig. 5G(1) and K). Type 3 (b-T3-Fe) and Type 4 (b-T4-Fe) were less abundant, accounting for 7% and 6% of the measured spectra and found on 2 and 3 maps, respectively. While b-T3-Fe was mainly part of the matrix of large aggregates (Fig. 5G(1)), b-T4-Fe was next to the large aggregates (Fig. 5G(1)) also obtained from Fe and S hotspots (Fig. 5H, and K). Based on LCF, the composition of the frequently encountered Fe spectra is similar at 40 cm depth and at 2 cm depth. The most abundant types of Fe spectra at both depths (t-T1-Fe and b-T1-Fe) had the highest abundances of CaFeP, next to Fe(II)aq., smectite, biotite and pyrite. b-T1-Fe, however, had higher clay content with in total 72% clays and Fe(II)aq. and slightly less pyrite than t-T1-Fe. At both depths, the LCF -results of Type 3 (t-T3-Fe and b-T3-Fe) were similar to Type 1 (t-T1-Fe and b-T1-Fe), but showed higher pyrite content, mainly on the expense of CaFeP. At both depths, one of the frequently encountered types of Fe spectra essentially represented pyrite (t-T2-Fe and b-T4-Fe). At 2 cm depth, this was the second most abundant type, which was not the case at 40 cm. At 40 cm depth the second most abundant type, b-T2-Fe, was described by ∼66% S-bound Fe, of which ∼23% were greigite, next to pyrite, Fe(II)aq. and smectite. At 40 cm depth, there was a high diversity of Fe spectra encountered on just one map (b-R1-Fe to b-R8-Fe) and one unique Fe spectrum (b-S1-Fe). Region 7 (b-R7-Fe) and Region 8 (b-R8-Fe) were variations of b-T1-Fe, both containing CaFeP, but with considerably more pyrite according to LCF. b-R8-Fe was indeed located on an area of elevated relative densities of Fe and Ca (Fig. 5M). But b-R7-Fe, even though having the highest CaFeP contribution (∼18%) measured by focused beam on both samples, was located on an area with an agglomeration of ∼10 micrometer-sized Fe and S hotspots (Fig. 5F(1)). This agglomeration resembled clusters of euhedral pyrite observed by SEM on the same sample in an earlier study.1 Indeed, the other two groups of Fe μ-XANES spectra located on this area of Fe and S agglomerations (Fig. 5F(1)), Region 3 (b-R3-Fe) and Spot 1 (b-S1-Fe), were characterized by high abundance of iron sulfides according to LCF. b-R3-Fe closely resembled the greigite reference spectrum, but also required mackinawite, Fe(III)–HA and vivianite for a satisfying reproduction by LCF. b-S1-Fe was identical to the greigite reference spectrum. Region 1 (b-R1-Fe) and Region 2 (b-R2-Fe) showed the characteristic pre and post white line shoulders of biotite. Accordingly, LCF indicated that most Fe in those regions was associated to phyllosilicate minerals. Nevertheless, LCF of b-R1-Fe yielded the highest vivianite contribution of all spectra measured at 40 cm depth. While b-R1-Fe was located on a rod-shaped particle of increased relative Fe density (Fig. 5N), b-R2-Fe was part of the matrix of a large aggregate (Fig. 5M). LCF of Region 5 (b-R5-Fe) and Region 6 (b-R6-Fe) indicated the presence of hematite, with b-R5-Fe being composed of ∼80% hematite and located on Fe hotspot without occurrence of other detectable elements (Fig. 5I). The spectrum at Region 4 (b-R4-Fe) showed the features of the pyrite reference spectrum but the features were damped. b-R4-Fe was located on a rod shaped area of high relative densities of Fe and S, indicating that the features are damped due to self-absorption on a spot with high Fe concentration (Fig. 5L).
 | ||
| Fig. 8 Classified P μ-XANES spectra showing individual spectra (light grey), the merged spectra (dark grey) and the LCF spectra (orange) for the samples from 2 cm (A) and 40 cm (B) sediment depth. | ||
3.5 Correlations of total element contents, results from sequential extractions and X-ray spectroscopy
Correlation matrices between elemental contents and SEP results for sediment samples above 10 cm show distinct differences to those below 10 cm. In contrast to deeper sediment layers, Ca, Mn, Fe and P are stronger correlated with each other and to SEP-derived contents in the top 10 cm of the sediment. In the top 10 cm, Fe-Asc and Fe(II)-HCl are strongly correlated with total Fe contents, indicating that the variability in Fe content is connected to variations in these two SEP fractions. Furthermore, the total Fe contents are correlated to Ca, Mn and P, which is not the case for the latter two elements in the deeper layers. Thus, the strong correlation between Mn and P contents is an exclusive feature of the top 10 cm of the sediment. Another remarkable and differencing feature are the correlations of P-NaHCO3 and P-CDB with other variables in the top 10 cm: P-NaHCO3 and P-CDB are strongly correlated with the total P content, and they are stronger correlated with the Fe, Ca and Mn contents compared to the deeper sediments. In the deeper sediment layers, next to Ca, Fe is correlated with S and the Fe-SEP fractions Fe-HNO3 and not extractable Fe. The stronger correlation between S with Fe-HNO3 and not extractable Fe distinguishes the lower sediment layers from the top 10 cm. For P, the strong correlation with TON and the P-ashed/HCl fraction is only observed in the lower sediment layer.The correlation of LCF-based Fe species with element contents as well as SEP-based Fe and P fractions can be found in Fig. 10C. Of all LCF-based Fe species, Fe(III)–HA and CaFeP showed the most positive correlations with other variables and they are also strongly correlated with each other. In particular, Fe(III)–HA and CaFeP strongly correlate with P, Mn and Fe-Asc. Also all SEP-based P fractions are positively correlated with Fe(III)–HA and CaFeP, but correlation is strongest with P-NaHCO3 and P-CDB. The LCF-derived pyrite-content is strongly correlated with the S contents and shows strong and medium correlations with the HNO3 and not-extractable Fe pool, respectively. The contents of smectite and biotite, obtained by LCF, are strongly correlated with Al as well as to each other, supporting their interpretation as proxies for phyllosilicate-Fe in the sediment.
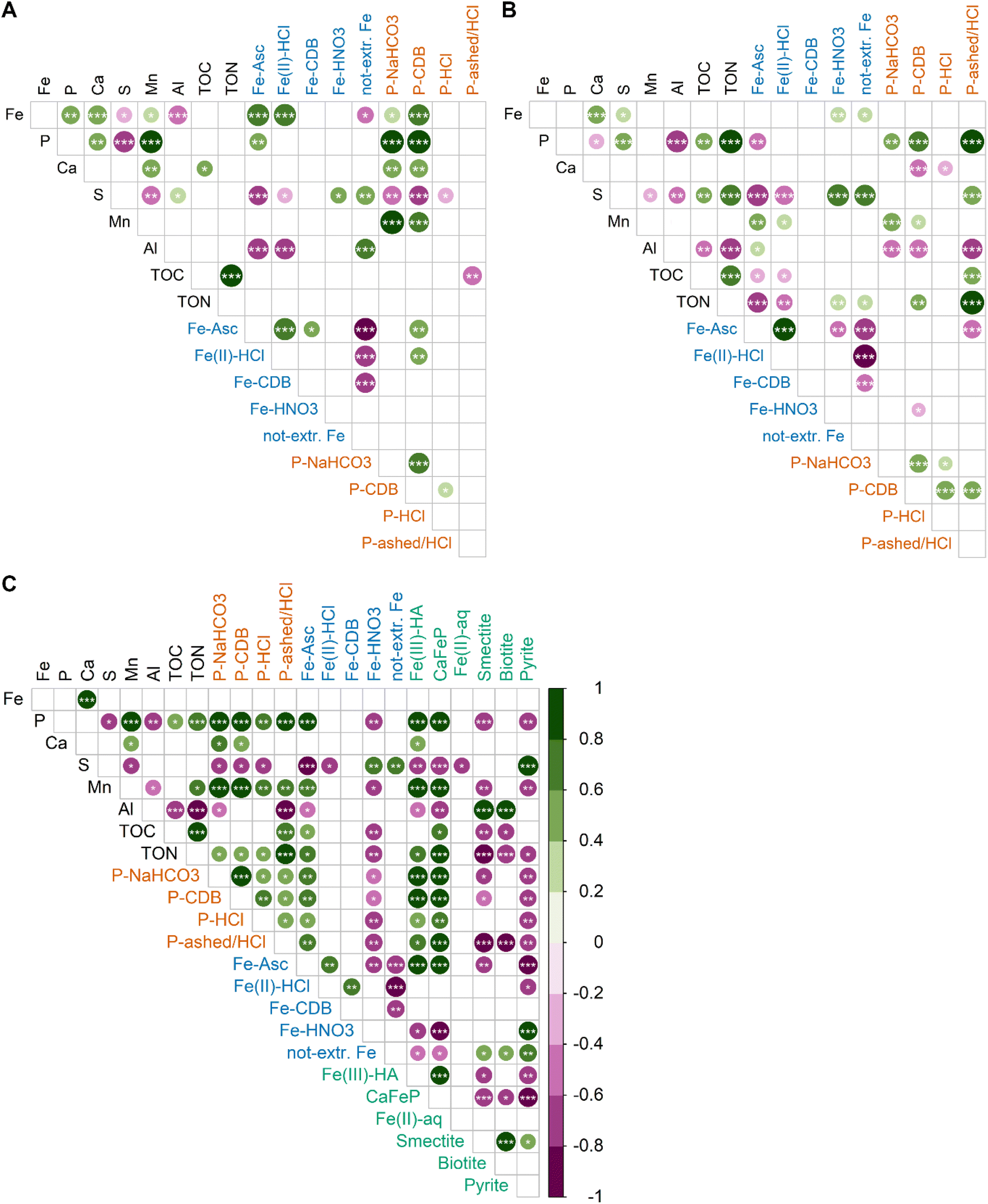 | ||
| Fig. 10 (A, and B) Pearson correlations of total elemental contents (black labels), Fe pools according to Fe-SEP (blue labels), P pools according to P-SEP (orange labels) in (A) the top 10 cm of sediment (n = 30 samples) and (B) below 10 cm depth (n = 44 samples). (C) Pearson correlations including the Fe species according to Fe K-edge bulk XAS LCF (green labels) across the entire investigated depth (n = 15 samples). Correlations were calculated using contents in μmol per g dw and are significant with p-values < 0.001 (***), < 0.01 (**) and < 0.05 (*). Insignificant correlations (p ≥ 0.05) are not shown. | ||
4 Discussion
4.1 Reducible Fe(III) phases and associated P
The subject of this study is the shallow peat lake Terra Nova in which Fe treatment induced seasonal peaks of internal P loading. An earlier investigation of the system suggested that Fe addition led to the build-up of a highly redox sensitive OM-associated Fe pool, which can bind considerable amounts of P but tends to release this P during periods of low O2 concentrations in the bottom water. This reducible Fe pool, mainly extracted by Fe-Asc is pivotal for the surface water quality of Terra Nova,1 and elucidating its nature was the main objective of this study.Correlations between Fe-Asc and CaFeP as well as with Ca, Fe, P contents indicate that oxidation of Fe2+ in the top sediment results in the co-precipitation of these elements. The correlation of Ca, Fe and P is reflected in concurrently decreasing contents with depth in the top 10 cm of sediment. Presence of dissolved Ca and Si and P/Fe ratios >0.5 in the porewaters in the corresponding depth interval (ESI Fig. E(1)‡) resemble solution compositions that lead to the precipitation of CaFeP.57 This implies that Fe redox cycling in these sediments does not lead to the precipitation of pure Fe (oxyhydr)oxides such as ferrihydrite or lepidocrocite but to the formation of an amorphous co-precipitate. Precipitation of Fe together with Ca and P in aquatic sediments has been previously reported by Hyacinthe and van Cappellen76 in a heavily bioturbated freshwater site in the Scheldt estuary (Belgium/The Netherlands). They found that on average 77% of Fe(III) in the top 14 cm of sediment was present in an authigenic amorphous Fe–Ca–Mn–P phase, which was extractable by the same ascorbic acid extraction we used in the Fe-SEP. This finding is in line with the correlation of Ca, Fe, P as well as Fe-Asc and CaFeP with Mn contents in the top 10 cm of the sediments from lake Terra Nova (Fig. 10A). Hyacinthe and van Cappellen76 further reported that Mn-contents decreased from freshly formed Fe–Ca–Mn–P precipitates to older precipitates at larger depth. Our results are in line with those reported by Hyacinthe and van Cappellen76 and hence support the presence of authigenic CaFeP co-precipitates in the surface sediment of Terra Nova, which become Mn depleted when buried deeper in the sediment: in sediments for Terra Nova the Fe and Mn contents were correlated in sections of the fibrous aggregates without pronounced enrichment of pyrite particles and the Mn/Fe ratio was larger in 2 cm depth compared to 40 cm depth (ESI F‡). Pearson correlation further showed that Fe(III)–HA fractions were closely correlated with CaFeP (LCF), Fe and P fractions mobilizable by reduction (SEPs), and total Ca, Mn, and P contents across the entire sediment depth (Fig. 10C). This offers additional evidence for the presence of a Fe–Ca–Mn–P phase, but also indicates that part of the reducible Fe(III) is coordinated with functional groups of the OM. Formation of complexes between Fe and functional groups of OM is key regarding the influence of OM on the formation of Fe(III)–precipitates. Organic matter can limit Fe(III) polymerization to monomeric or oligomeric Fe-OM complexes or ferrihydrite, depending on the molar OC/Fe ratio and pH.27,77–79 In laboratory experiments on the oxidation and precipitation of Fe in the presence of Leonardite humic acid at pH 6.5 and at OC/Fe from ∼10 to 50, Beauvois et al.80 observed the formation of oligomeric Fe(III) and ferrihydrite-like Fe(III) nanoparticles embedded in OM, with a decreasing share of ferrihydrite at increasing OC/Fe. For OC/Fe from 50 to 500, at neutral to acidic pH, Karlsson and Persson27 reported a shift to predominantly monomeric Fe(III) bound in chelate complexes. At OC/Ca ratios <3.8, Beauvois et al.80 found Ca to act as a coagulant promoting the formation of large, branched organic networks in which Fe(III) nano-aggregates of ∼6 nm size were distributed. At higher OC/Ca, on the other hand, they observed the development of smaller Fe-OM-Ca aggregates. Considering contents of ∼30 mol kg−1 TOC, 0.5 mol kg−1 Fe, and 0.6 mol kg−1 Ca in the sediments from Lake Nova, an overall OC/Ca ratio of ∼50 and an overall OC/Fe ratio of ∼60 can be estimated. Taking into account that CaFeP and Fe(III)–HA accounted for ∼1/3 of the total Fe, an OC/Fe ratio of ∼180, can be estimated for the Fe(III) represented by these two LCF references. Based on all aforementioned considerations, we therefore conclude that Fe(III)–HA and CaFeP identified by LCF do not constitute two distinctive Fe phases in our peat lake samples, but that Fe(III)–HA and CaFeP together describe a co-precipitate of Ca, P, and mono- and oligomeric Fe(III) that is also coordinated with OC. This mixed inorganic–organic co-precipitate is finely distributed within the OM matrix of the peat sediment.
Indeed, X-ray micro-microscopy supports the presence of Fe–Ca–P oligomers and nanoparticles associated with the OM matrix. At both investigated depths, the majority of the measured μ-XAS spectra (t-T1-Fe and b-T1-Fe) contained CaFeP (Fig. 7), all of them found on large aggregates with uniform distribution of Fe, Ca and P (Fig. 4 and 5). These spots exhibited relatively low densities of these elements, in contrast to spots with high local densities for which LCF pointed to high abundance of Fe sulfides or apatite, respectively. That is, the inorganic–organic precipitates occur finely dispersed within the OM matrix. Consequently, it was not possible to discern and characterize individual particles when analyzing the material with a spatial resolution of several hundred nm. Notably, LCF of most μ-XANES spectra returned higher fraction of Fe(II)aq. than LCF analyses of bulk XANES and EXAFS spectra, but did not include the Fe(III)–HA reference. This could be due to lower sensitivity in the microanalyses constrained to the XANES region, but could indicate that the orders of magnitude higher flux density of the focused X-ray beam induced photoreduction of organically complexed Fe(III).
In contrast to what Fe K-edge XAS results suggest, the P K-edge XAS data indicate that the majority of P in the Terra Nova sediment is present as organic P esters. The bulk P K-edge XAS is exclusively fitted by inositol phosphate and DNA in the LCF with no indication of considerable contributions of an inorganic P phase (ESI C2 and C3‡). Organic P esters thus constitute an important P pool in the Terra Nova sediment. While according to P-SEP the majority of sediment P is indeed P structurally bound in OM, extracted as P-ashed/HCl (Fig. 2C), this pool accounts for at most 70% of the total P (Fig. 2D). In the surface sediment, up to 51% of sediment P is extracted as P-NaHCO3 and P-CDB. According to the used P-SEP, P-NaHCO3 represents P bound to OM over metal cation bridges46,53 and P-CDB represents P bound to Fe (oxyhydr)oxides, CaCO3 and Fe-phosphates.45,47,81 Indeed, correlation analysis suggests that NaHCO3 and CDB extract P bound to the Fe–Ca–P-OM co-precipitates. P-NaHCO3 and P-CDB show almost identical depth profiles (Fig. 2C), correlate to each other (Fig. 10A and B) and to Fe(III)–HA, CaFeP and Fe-Asc (Fig. 10C) over the entire investigated sediment depth. Further, P-concentrations in the SEP-extracts where measured by colorimetry,1 which is exclusively sensitive to ortho-phosphate. These findings suggest that bulk P K-edge XAS underestimates inorganic P content. P K-edge XANES analysis has previously been reported to have its limitations for determining the P speciation in peat soils. Kruse and Leinweber82 found that P-XANES spectra did not show a pronounced change upon extractions targeting different P phases. Successive removal of P pools was not reflected in distinct spectral changes as all spectra were dominated by organic P, which lacks marked pre- or post-edge features and obscures the signal from other phases. Even distinguishing organic and inorganic phosphate groups complexed to the same solid phases is difficult by P K-edge,67 as the corresponding XANES spectra differ only marginally. Even if inorganic PO4 features a higher white line intensity than inositol phosphate,67 the intensity of the white line is particularly sensitive to self-absorption and, consequently, the contribution of inorganic P can be underestimated at the expense of organic P when P XANES spectra are collected in fluorescence mode. These methodological constraints can explain why inorganic P was not included in the LCF of bulk sample spectra. Nevertheless, μ-XAS analysis revealed local variations in P speciation. The characteristic pre-edge feature of Fe-bound P was detected on several spots (Fig. 5 and 8), and was reproduced by the Fe-IP6 reference in the LCF (Fig. 9, ESI C3‡). Further, apatite particles were unequivocally detected (Fig. 5, 8 and 9), in line with 14% (on average) HCl-extractable P across the entire investigated depth (Fig. 2C and D) that is interpreted as Ca–phosphate P. Despite the limitations of distinguishing organic and inorganic phosphate as delineated above, the results of P K-edge XAS thus further point towards the presence of Fe–Ca–P-OM co-precipitates, and also suggest that organic phosphate groups in addition to carboxyl or phenolic groups might contribute to Fe(III) binding by OM.
4.2 Other Fe pools
High abundance of organic matter and the embedding of pyrite into the OM matrix might retard or prevent pyrite dissolution in HNO3. Most of the μ-XRF maps at both investigated depths show areas of high density collocated Fe and S, often also in the characteristic spherical shape of framboidal pyrite (Fig. 10), which is confirmed by the dominance of pyrite in the LCF of the corresponding spectra (t-T2-Fe, b-R4-Fe, Fig. 6, 5C, L, and 7). However, also the LCF of two most abundant types of Fe μ-XANES spectra, t-T1-Fe and b-T1-Fe (Table 3, and Fig. 6) collected from OM aggregates with low Fe density (Fig. 5), indicated up to 25% pyrite (Fig. 7). This suggests that an abundant part of pyrite is embedded in the organic matrix in finely dispersed form. Oxidative dissolution by HNO3 can be conceived as an electrochemical reaction83 whereby the cathodic reduction of nitrate could be slowed down when pyrite is encapsulated by OM, in analogy to the retarding effect of the formation of a passivating layer.84 As the oxidation of pyrite is autocatalytic and formation of Fe3+ ion accelerates the oxidation of pyrite85 scavenging of Fe3+ ions by organic matter via reduction or complexation can impede the progress of pyrite dissolution. That is, two hours of HNO3 extraction might have been too short to completely extract pyrite from the OM-rich sediments studied here.
Next to pyrite, mackinawite (FeS) and greigite (Fe3S4) were detected in the sediment samples by μ-XANES analyses. The three sulfide phases occurred in vicinity of each other, from a mix of mainly mackinawite and greigite (b-R3-Fe), to pure greigite (b-S1-Fe), to ∼50% pyrite (b-R7-Fe) (spectra: Fig. 6B, LCF results: Fig. 7B, ESI B4‡) in the sample from 40 cm depth. The spectra were collected from an area with elevated Fe and S densities extending over tens of micrometers and looking like an assembly of euhedral pyrite (Fig. 5F(1)). Also in the sample from 2 cm depth, mackinawite and greigite were occasionally detected but only in small areas with the size of few micrometers (region t-R4-Fe containing ∼40% mackinawite and ∼25% pyrite, region t-R2-Fe containing ∼40% greigite and ∼50% pyrite, spectra: Fig. 6A, LCF results: Fig. 7A, ESI B4‡). Mackinawite and greigite can be conceived as intermediates in the formation of pyrite upon the sulfidation of Fe(III) phases. That is, initially FeS is formed as the primary product of the reaction of dissolved sulfide with Fe(III) (oxyhydr)oxides86 and greigite can be the intermediate on the polysulfide pathway of pyrite formation from mackinawite.87 Hence, the presence of these two sulfides might bear witness to the active formation of pyrite in the whole sediment of Terra Nova, which is most pronounced in layers below 10 cm depth, where pyrite contents increase and pyrite becomes the dominant phase based on bulk LCF analysis (Fig. 3).
4.3 Implications for sediment P dynamics
Combining results from sequential extractions with elemental and spectroscopic analyses leads to the conclusion that the addition of Fe salts to lake Terra Nova has resulted in the formation of multi-elemental co-precipitates containing Fe, Ca, P and minor amounts of Mn, which form a continuum of oligomers and nanoparticles embedded in the OM matrix and involving inner-sphere complexes of Fe with functional groups of the OM. These precipitates are likely authigenic and have been formed as a consequence of redox cycling of Fe in the sediments. During dosing of FeCl3, Fe3+ hydrolysis caused the formation of large flocs in the water column, which settled on top of the sediment in the form of Fe hydroxides.93 The transformation of these primary products of Fe addition into multi-elemental precipitates upon redox cycling in the sediment has consequences for the P binding and release in the sediment.The formation of co-precipitates can enhance the efficiency of Fe addition for P binding in the sediment. Ratios between P and Fe in the corresponding Fe pools obtained by sequential extractions reached values up to about 0.3 in the top sediments of Terra Nova.1 Fe(III)-OM complexes and CaFeP can have P/Fe ratios of up to 1.31,57 Hence, the formation of Fe–Ca–P-OM co-precipitates can constitute an efficient sink for P in sediments of Fe treated lakes. However, a threshold P/Fe ratio of 0.12 was reported, at which, in aquatic sediments, the retention of P via adsorption is insufficient to prevent a significant increase in benthic P fluxes during temporal bottom water anoxia.94–96 Consequently, the P/Fe ratios in Terra Nova sediment indicate that the amount of P bound in Fe(III)-OM complexes and CaFeP could further increase in the future. Nevertheless, the current ratio already exceeds the threshold value beyond which release of P into porewater upon Fe(III) reduction under summer low oxygen conditions cannot be attenuated by adsorption to remaining Fe(III) phases. Further, analyses of P XANES spectra suggest that part of the Fe(III) is coordinated with organic P groups. As organically-bound P, mostly in the form of phosphate mono- and diesters, constitutes the largest P pool in the sediment, further research would be necessary to inquire possible competition of organic P for inorganic P adsorption and complexation with Fe in these sediments.
Although formation of co-precipitates is an efficient pathway for P binding, these precipitates might be particularly sensitive to P mobilization upon changes in redox conditions when bottom water hypoxia emerge. Freshly precipitated Fe(III) phosphates exhibit a high reactivity towards microbial reduction and the similarity of kinetic parameters with those obtained in estuarine sediments containing Fe(III) phosphate precipitates suggests, that those are the preferential electron acceptor for Fe reducing bacteria in these sediments.97 Furthermore, linkage of Fe(III) (oxyhydroxides) with OM enhances microbial and chemical Fe(III) reduction98,99 and, in general, the small particle size and its wide distribution is expected to increase the reactivity of the Fe(III) containing co-precipitates in the sediment of Lake Terra Nova. In conclusion, formation of co-precipitates with Fe and Ca in the presence of OM presents an effective P sink in the sediments with respect to P/Fe ratio, but the product is eminently prone to P release under reducing conditions.
5 Conclusion
In organic-rich dynamic systems such as peat sediments, the classical model of Fe and P diagenesis, based on oxidative precipitation and reductive dissolution of Fe (oxyhydr)oxides does not apply. Addition of Fe and its redox cycling in the peat sediment of lake Terra Nova does not result in the formation of distinctive Fe(III) (oxyhydr)oxides in the top part of the sediment. Instead, the presence of OM and Ca inhibits polymerization and particle growth leading to finely dispersed solids intimately linked to the OM matter. Fe(III) produced by Fe(II) oxidation forms a continuum of highly amorphous Fe–Ca–(Mn–)P-OM co-precipitates, which are highly redox sensitive and responsible for the rapid release of P upon emerging bottom water hypoxia.Methodologically, this study shows that X-ray spectroscopy provides significant additional information next to SEPs, with scanning X-ray microscopy as a valuable asset for qualitative insights into the presence and spatial distribution of individual species. Our results suggest that Fe-Asc represents a good estimate of the most redox-sensitive amorphous Fe(III) pool in organic-rich sediments and that P-NaHCO3 and P-CDB together yield an estimate of P associated with nanoscale inorganic–organic Ca–Fe–P-OM co-precipitates. On the other hand, the study demonstrates that concentrated HNO3 does not quantitatively extract pyrite in the assigned reaction time and may therefore lead to its underestimation. Furthermore, Fe release from phyllosilicate minerals contributes to the HCl-extractable Fe, and this fraction should therefore not be interpreted exclusively as Fe(II) incorporated in authigenic phases. SEPs can thus deliver an indication of Fe and P pool reactivities in organic rich systems, but should be interpreted as such and not as discrete phases.
In conclusion, our study shows that organic-rich systems should not be approached using models and methods developed for organic poor systems, and that there is a need for new models and more fundamental research on OM-associated Fe phases to be able to reliably predict Fe and P dynamics in organic rich sediments.
Data availability
Data collected for this article including results of chemical analysis, correlation calculations and XAS spectra are available at Zenodo at https://doi.org/10.5281/zenodo.12610105. Data supporting this article including linear combination fit results and map and spectra identities are listed in the ESI.‡ Data of XAS reference spectra are available via the respective publication, or with the person who collected them, as listed in the ESI.‡ XAS raw data will be available from 2025 on under https://doi.esrf.fr/10.15151/ESRF-ES-845384380.Author contributions
Melanie Münch: conceptualization, investigation, formal analysis, visualization, writing – original draft, writing – review & editing. Andreas Voegelin: conceptualization, methodology, investigation, formal analysis, supervision, writing – review & editing. Luis Carlos Colocho Hurtarte: methodology, investigation, formal analysis, resources, Jörg Göttlicher: investigation, formal analysis, resources. Thilo Behrends: conceptualization, funding acquisition, project administration, methodology, supervision, investigation, formal analysis, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was part of the P-Trap project funded by the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 813438. We acknowledge the European Radiation Facility (ESRF) for provision of synchrotron radiation facilities under proposal number ES-1169 and the staff of beamline ID21 for their great support. We would like to thank Dr Edward D. Burton (Southern Cross University, Australia) and Prof. Jörg Prietzel (Technical University of Munich, Germany) for providing XAS reference spectra, and MSc Niccolò Pesenti for helping with the XAS measurements. We further thank three anonymous reviewers for their valuable comments which improved the manuscript.References
- M. A. Münch, R. van Kaam, K. As, S. Peiffer, G. T. Heerdt and C. P. Slomp, et al., Impact of iron addition on phosphorus dynamics in sediments of a shallow peat lake 10 years after treatment, Water Res., 2023, 120844 Search PubMed , available from, https://www.sciencedirect.com/science/article/pii/S0043135423012848.
- R. W. Sterner, On the phosphorus limitation paradigm for lakes, Int. Rev. Hydrobiol., 2008, 93(4-5), 433–445 CrossRef CAS.
- D. W. Schindler, R. E. Hecky, D. L. Findlay, M. P. Stainton, B. R. Parker and M. J. Paterson, et al., Eutrophication of lakes cannot be controlled by reducing nitrogen input: Results of a 37-year whole-ecosystem experiment, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(32), 11254–11258 CrossRef CAS PubMed.
- W. J. Brownlie, M. A. Sutton, D. S. Reay, K. V. Heal, L. Hermann and C. Kabbe, et al., Global actions for a sustainable phosphorus future, Nat. Food, 2021, 2(2), 71–74 CrossRef CAS PubMed.
- M. Shore, P. Jordan, P. E. Mellander, M. Kelly-Quinn, K. Daly and J. T. Sims, et al., Characterisation of agricultural drainage ditch sediments along the phosphorus transfer continuum in two contrasting headwater catchments, J. Soils Sediments, 2016, 16(5), 1643–1654, DOI:10.1007/s11368-015-1330-0.
- P. Geranmayeh, K. M. Johannesson, B. Ulén and K. S. Tonderski, Particle deposition, resuspension and phosphorus accumulation in small constructed wetlands, Ambio, 2018, 47(1), 134–145, DOI:10.1007/s13280-017-0992-9.
- P. Kleinman, A. Sharpley, A. Buda, R. McDowell and A. Allen, Soil controls of phosphorus in runoff: Management barriers and opportunities, Can. J. Soil Sci., 2011, 91(3), 329–338, DOI:10.4141/cjss09106.
- P. J. A. Withers and H. P. Jarvie, Delivery and cycling of phosphorus in rivers: A review, Sci. Total Environ., 2008, 400(1), 379–395 CrossRef CAS , available from, https://www.sciencedirect.com/science/article/pii/S0048969708008139.
- M. Søndergaard, J. P. Jensen and E. Jeppesen, Role of sediment and internal loading of phosphorus in shallow lakes, Hydrobiologia, 2003, 506(1), 135–145 CrossRef.
- A. D. Steinman, and B. M. Spears, Internal Phosphorus Loading in Lakes: Causes, Case Studies, and Management, J. Ross Publishing, 2020 Search PubMed.
- B. M. Spears, D. P. Hamilton, Y. Pan, C. Zhaosheng and L. May, Lake management: is prevention better than cure?, Inland Waters, 2022, 12(1), 173–186 CrossRef.
- M. Søndergaard, R. Bjerring and E. Jeppesen, Persistent internal phosphorus loading during summer in shallow eutrophic lakes, Hydrobiologia, 2013, 710(1), 95–107 CrossRef.
- E. S. Bakker, E. Van Donk and A. K. Immers, Lake restoration by in-lake iron addition: a synopsis of iron impact on aquatic organisms and shallow lake ecosystems, Aquat. Ecol., 2016, 50(1), 121–135 CrossRef.
- A. J. P. Smolders, L. P. M. Lamers, L. Echet, G. Van Der Velde and J. G. M. Roelofs, Internal eutrophication: How it works and what to do about it – A review, Chem. Ecol., 2006, 22(2), 93–111 CrossRef CAS.
- W. Einsele, Über die Beziehungen des Eisenkreislaufs zum Phosphatkreislauf im eutrophen See, Arch. Hydrobiol., 1936, 29, 664–686 CAS.
- C. H. Mortimer, The Exchange of Dissolved Substances Between Mud and Water in Lakes, J. Ecol., 1941, 29(2), 280–329 CrossRef CAS , https://www.jstor.org/stable/2256395.
- P. Campbell and T. Torgersen, Maintenance of Iron Meromixis by Iron Redeposition in a Rapidly Flushed Monimolimnion, Can. J. Fish. Aquat. Sci., 1980, 37(8), 1303–1313 CrossRef CAS.
- A. Kleeberg, C. Herzog and M. Hupfer, Redox sensitivity of iron in phosphorus binding does not impede lake restoration, Water Res., 2013, 47(3), 1491–1502 CrossRef CAS.
- A. Kleeberg, A. Köhler and M. Hupfer, How effectively does a single or continuous iron supply affect the phosphorus budget of aerated lakes?, J. Soils Sediments, 2012, 12(10), 1593–1603 CrossRef CAS.
- R. A. Berner, Sedimentary pyrite formation, Am. J. Sci., 1970, 268, 1–23 CrossRef CAS.
- P. Kraal, E. D. Burton and R. T. Bush, Iron monosulfide accumulation and pyrite formation in eutrophic estuarine sediments, Geochim. Cosmochim. Acta, 2013, 122, 75–88 CrossRef CAS.
- J. Gerke, Phosphate adsorption by humic/Fe-oxide mixtures aged at pH 4 and 7 and by poorly ordered Fe-oxide, Geoderma, 1993, 59(1–4), 279–288 CrossRef CAS.
- C. Mikutta and R. Kretzschmar, Spectroscopic evidence for ternary complex formation between arsenate and ferric iron complexes of humic substances, Environ. Sci. Technol., 2011, 45(22), 9550–9557 CrossRef CAS.
- A. Sundman, T. Karlsson, S. Sjöberg and P. Persson, Impact of iron–organic matter complexes on aqueous phosphate concentrations, Chem. Geol., 2016, 426, 109–117 CrossRef CAS.
- K. Lalonde, A. Mucci, A. Ouellet and Y. Gélinas, Preservation of organic matter in sediments promoted by iron, Nature, 2012, 483(7388), 198–200, DOI:10.1038/nature10855.
- T. Karlsson and P. Persson, Coordination chemistry and hydrolysis of Fe(III) in a peat humic acid studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Acta, 2010, 74(1), 30–40 CrossRef CAS.
- T. Karlsson and P. Persson, Complexes with aquatic organic matter suppress hydrolysis and precipitation of Fe(III), Chem. Geol., 2012, 322–323, 19–27 CrossRef CAS.
- L. K. Thomasarrigo, J. M. Byrne, A. Kappler and R. Kretzschmar, Impact of Organic Matter on Iron(II)-Catalyzed Mineral Transformations in Ferrihydrite-Organic Matter Coprecipitates, Environ. Sci. Technol., 2018, 52(21), 12316–12326, DOI:10.1021/acs.est.8b03206.
- J. Dolfing, W. Chardon and J. Japenga, Assiciation between colloidal iron, aluminum, phosphorus and humic acids, Soil Sci., 1999, 164(3), 171–179 CrossRef.
- C. Moens and E. Smolders, Suwannee River Natural Organic Matter concentrations affect the size and phosphate uptake of colloids formed by iron oxidation, Geochim. Cosmochim. Acta, 2021, 312, 375–391 CrossRef CAS.
- J. Gerke, Humic (organic matter)-Al(Fe)-phosphate complexes: An underestimated phosphate form in soils and source of plant-available phosphate, Soil Sci., 2010, 175(9), 417–425 CrossRef CAS.
- I. Guardado, O. Urrutia and J. M. Garcia-Mina, Size Distribution , Complexing Capacity , and Stability of Phosphate–Metal–Humic Complexes, J. Agric. Food Chem., 2007, 55, 408–413 CrossRef CAS PubMed.
- J. Riggle and R. von Wandruszka, Binding of inorganic phosphate to dissolved metal humates, Talanta, 2005, 66(2), 372–375 CrossRef CAS , available from, https://www.sciencedirect.com/science/article/pii/S0039914004006605.
- C. Yu, J. J. Virtasalo, T. Karlsson, P. Peltola, P. Österholm and E. D. Burton, et al., Iron behavior in a northern estuary: large pools of non-sulfidized Fe (II) associated with organic matter, Chem. Geol., 2015, 413, 73–85 CrossRef CAS.
- I. Guardado, O. Urrutia and J. M. Garcia-Mina, Some structural and electronic features of the interaction of phosphate with metal–humic complexes, J. Agric. Food Chem., 2008, 56(3), 1035–1042 CrossRef CAS.
- D. W. O’Connell, N. Ansems, R. K. Kukkadapu, D. Jaisi, D. M. Orihel and B. J. Cade-Menun, et al., Changes in Sedimentary Phosphorus Burial Following Artificial Eutrophication of Lake 227, Experimental Lakes Area, Ontario, Canada, J. Geophys. Res.: Biogeosci., 2020, 125(8), e2020JG005713 CrossRef.
- K. C. Ruttenberg, Development of a sequential extraction method for different forms of phosphorus in marine sediments, Limnol. Oceanogr., 1992, 37(7), 1460–1482 CrossRef CAS.
- R. Psenner and R. Puckso, Phosphorus fractionation: Advantages and limits of the method for the study of sediment P origins and interactions, Arch. Hydrobiol., 1988, 30, 43–59 CAS.
- S. R. Claff, L. A. Sullivan, E. D. Burton and R. T. Bush, A sequential extraction procedure for acid sulfate soils: Partitioning of iron, Geoderma, 2010, 155(3–4), 224–230, DOI:10.1016/j.geoderma.2009.12.002.
- S. W. Poulton and D. E. Canfield, Development of a sequential extraction procedure for iron: Implications for iron partitioning in continentally derived particulates, Chem. Geol., 2005, 214(3–4), 209–221 CrossRef CAS.
- R. Raiswell, H. P. Vu, L. Brinza and L. G. Benning, The determination of labile Fe in ferrihydrite by ascorbic acid extraction : Methodology , dissolution kinetics and loss of solubility with age and de-watering, Chem. Geol., 2010, 278(1–2), 70–79, DOI:10.1016/j.chemgeo.2010.09.002.
- I. C. Regelink, A. Voegelin, L. Weng, G. F. Koopmans and R. N. J. Comans, Characterization of Colloidal Fe from Soils Using Field-Flow Fractionation and Fe K - Edge X - ray Absorption Spectroscopy, Environ. Sci. Technol., 2014, 48(8), 4307–4316 CrossRef CAS PubMed.
- L. L. Baker, D. G. Strawn, K. L. Vaughan and P. A. McDaniel, XAS study of Fe mineralogy in a chronosequence of soil clays formed in basaltic cinders, Clays Clay Miner., 2010, 58, 772–782 CrossRef CAS.
- W. Li, S. R. Joshi, G. Hou, D. J. Burdige, D. L. Sparks and D. P. Jaisi, Characterizing phosphorus speciation of Chesapeake Bay sediments using chemical extraction, 31P NMR, and X-ray absorption fine structure spectroscopy, Environ. Sci. Technol., 2015, 49(1), 203–211 CrossRef CAS.
- P. Kraal, N. Dijkstra, T. Behrends and C. P. Slomp, Phosphorus burial in sediments of the sulfidic deep Black Sea: Key roles for adsorption by calcium carbonate and apatite authigenesis, Geochim. Cosmochim. Acta, 2017, 204, 140–158, DOI:10.1016/j.gca.2017.01.042.
- D. S. Baldwin, The phosphorus composition of a diverse series of australian sediments, Hydrobiologia, 1996, 335(1), 63–73 CrossRef CAS.
- C. P. Slomp, E. H. G. Epping, W. Helder and W. Van Raaphorst, A key role for iron-bound phosphorus in authigenic apatite formation in North Atlantic continental platform sediments, J. Mar. Res., 1996, 54(6), 1179–1205 CrossRef CAS.
- S. R. Claff, E. D. Burton, L. A. Sullivan and R. T. Bush, Effect of sample pretreatment on the fractionation of Fe, Cr, Ni, Cu, Mn, and Zn in acid sulfate soil materials, Geoderma, 2010, 159(1–2), 156–164, DOI:10.1016/j.geoderma.2010.07.007.
- J. van Dijk, M. Amesz, M. Ouboter, L. Moria, G. ter Heerdt, and T. van der Wijngaart, Aktualisatie KRW-Waterlichamen AGV: Maatregelenprogramma 2022-2027, Amsterdam, 2020 Search PubMed.
- N. A. G. M. van Helmond, T. Jilbert and C. P. Slomp, Hypoxia in the Holocene Baltic Sea: Comparing modern versus past intervals using sedimentary trace metals, Chem. Geol., 2018, 493(June), 478–490, DOI:10.1016/j.chemgeo.2018.06.028.
- D. R. Lovley and E. J. P. Phillips, Rapid Assay for Microbially Reducible Ferric Iron in Aquatic Sediments, Appl. Environ. Microbiol., 1987, 53(7), 1536–1540 CrossRef CAS PubMed.
- J. A. McKeague and J. H. Day, Dithionite and oxalate-extractable Fe and Al as aids in differentiating various classes of soils, Can. J. Soil Sci., 1966, 46(1), 13–22, DOI:10.4141/cjss66-003.
- C. T. Parsons, F. Rezanezhad, D. W. O'Connell and P. Van Cappellen, Sediment phosphorus speciation and mobility under dynamic redox conditions, Biogeosciences, 2017, 14(14), 3585–3602 CrossRef CAS.
- M. Salomé, M. Cotte, R. Baker, R. Barrett, N. Benseny-Cases and G. Berruyer, et al., The ID21 Scanning X-ray Microscope at ESRF, J. Phys.: Conf. Ser., 2013, 425(18), 182004, DOI:10.1088/1742-6596/425/18/182004.
- F. Werner, C. W. Mueller, J. Thieme, A. Gianoncelli, C. Rivard and C. Höschen, Micro-scale heterogeneity of soil phosphorus depends on soil substrate and depth, Sci. Rep., 2017, 7(1), 3203 CrossRef PubMed.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 537–541, DOI:10.1107/FS0909049505012719&partnerID=40&md5=450ffc75c60d60335c23ce51c2dd2635.
- A. C. Senn, R. Kaegi, S. J. Hug, J. G. Hering, S. Mangold and A. Voegelin, Composition and structure of Fe(III)-precipitates formed by Fe(II) oxidation in water at near-neutral pH: Interdependent effects of phosphate, silicate and Ca, Geochim. Cosmochim. Acta, 2015, 162, 220–246 CrossRef CAS.
- J. Frommer, A. Voegelin, J. Dittmar, M. A. Marcus and R. Kretzschmar, Biogeochemical processes and arsenic enrichment around rice roots in paddy soil: results from micro-focused X-ray spectroscopy, Eurasian J. Soil Sci., 2011, 62(2), 305–317 CrossRef.
- M. Ma, P. Overvest, A. Hijlkema, S. Mangold, C. McCammon and A. Voegelin, et al., Phosphate burial in aquatic sediments: Rates and mechanisms of vivianite formation from mackinawite, Chem. Eng. J. Adv., 2023, 16, 100565 CrossRef CAS.
- X. Chen, X. Zhang, J. Wan, Z. Wang and Y. Qian, Selective fabrication of metastable greigite (Fe3S4) nanocrystallites and its magnetic properties through a simple solution-based route, Chem. Phys. Lett., 2005, 403(4–6), 396–399 CrossRef CAS.
- E. D. Burton, R. T. Bush, L. A. Sullivan, R. K. Hocking, D. R. G. Mitchell and S. G. Johnston, et al., Iron-Monosulfide Oxidation in Natural Sediments: Resolving Microbially Mediated S Transformations Using XANES, Electron Microscopy, and Selective Extractions, Environ. Sci. Technol., 2009, 43(9), 3128–3134, DOI:10.1021/es8036548.
- M. Wolthers, S. J. Van Der Gaast, and D. Rickard. The structure of disordered mackinawite. 200388(11–12):2007–2015. Available from: doi: DOI:10.2138/am-2003-11-1245.
- A. Al-Borno and M. B. Tomson, The temperature dependence of the solubility product constant of vivianite, Geochim. Cosmochim. Acta, 1994, 58(24), 5373–5378 CrossRef CAS.
- J. Wielinski, J. Jimenez-Martinez, J. Göttlicher, R. Steininger, S. Mangold and S. J. Hug, et al., Spatiotemporal Mineral Phase Evolution and Arsenic Retention in Microfluidic Models of Zerovalent Iron-Based Water Treatment, Environ. Sci. Technol., 2022, 56(19), 13696–13708, DOI:10.1021/acs.est.2c02189.
- L. C. Colocho Hurtarte, L. F. Souza-Filho, W. Oliveira Santos, L. Vergütz, J. Prietzel and D. Hesterberg, Optimization of Data Processing Minimizes Impact of Self-Absorption on Phosphorus Speciation Results by P K-Edge XANES, Soil Syst., 2019, 3(3), 61 CrossRef.
- Z. He, C. W. Honeycutt, T. Zhang and P. M. Bertsch, Preparation and FT–IR characterization of metal phytate compounds, J. Environ. Qual., 2006, 35(4), 1319–1328 CrossRef CAS PubMed.
- J. Prietzel, G. Harrington, W. Hausler, K. Heister, F. Werner and W. Klysubun, Reference spectra of important adsorbed organic and inorganic phosphate binding forms for soil P speciation using synchrotron-based K-edge XANES spectroscopy, J. Synchrotron Radiat., 2016, 23(2), 532–544, DOI:10.1107/S1600577515023085.
- H. L. C. Colocho, H. C. Santana Amorim, J. Kruse, J. Criginski Cezar, W. Klysubun and J. Prietzel, A Novel Approach for the Quantification of Different Inorganic and Organic Phosphorus Compounds in Environmental Samples by P L2,3-Edge X-ray Absorption Near-Edge Structure (XANES) Spectroscopy, Environ. Sci. Technol., 2020, 54(5), 2812–2820, DOI:10.1021/acs.est.9b07018.
- V. A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra, Spectrochim. Acta, Part B, 2007, 62(1), 63–68 CrossRef.
- J. Schindelin, C. T. Rueden, M. C. Hiner and K. W. Eliceiri, The ImageJ ecosystem: An open platform for biomedical image analysis, Mol. Reprod. Dev., 2015, 82(7–8), 518–529 CrossRef CAS.
- L. K. Huang and M. J. J. Wang, Image thresholding by minimizing the measures of fuzziness, Pattern Recogn., 1995, 28(1), 41–51 CrossRef.
- F. E. Harrell Jr, Harrell Jr M. F. E. Package ‘hmisc’. CRAN2018, 2019, pp. 235–236 Search PubMed.
- J. Babej, R. Červenka, J. Kuta, K. Brabec and Z. Máčka, Organic and inorganic carbon storage in riparian zones of Central European rivers–A comparison of methods for their determination, Quat. Int., 2023, 681, 44–51 CrossRef.
- L. Celi, and E. Barberis, Abiotic reactions of inositol phosphates in soil, 2007 Search PubMed.
- B. L. Turner, M. J. Papházy, P. M. Haygarth and I. D. Mckelvie, Inositol phosphates in the environment, Philos. Trans. R. Soc. London, Ser. B, 2002, 357(1420), 449–469 CrossRef CAS.
- C. Hyacinthe and P. van Cappellen, An authigenic iron phosphate phase in estuarine sediments: composition, formation and chemical reactivity, Mar. Chem., 2004, 91(1–4), 227–251 CrossRef CAS.
- A. Vilgé-Ritter, J. Rose, A. Masion, J. Y. Bottero and J. M. Lainé, Chemistry and structure of aggregates formed with Fe-salts and natural organic matter, Colloids Surf., A, 1999, 147(3), 297–308 CrossRef.
- H. Guénet, M. Davranche, D. Vantelon, J. Gigault, S. Prévost and O. Taché, et al., Characterization of iron–organic matter nano-aggregate networks through a combination of SAXS/SANS and XAS analyses: impact on As binding, Environ. Sci.: Nano, 2017, 4(4), 938–954 RSC.
- D. Vantelon, M. Davranche, R. Marsac, C. La Fontaine, H. Guénet and J. Jestin, et al., Iron speciation in iron–organic matter nanoaggregates: a kinetic approach coupling Quick-EXAFS and MCR-ALS chemometrics, Environ. Sci.: Nano, 2019, 6(8), 2641–2651 RSC.
- A. Beauvois, D. Vantelon, J. Jestin, C. Rivard, M. Bouhnik-Le Coz and A. Dupont, et al., How does calcium drive the structural organization of iron–organic matter aggregates? A multiscale investigation, Environ. Sci.: Nano, 2020, 7(9), 2833–2849 RSC.
- N. Dijkstra, C. P. Slomp and T. Behrends, Vivianite is a key sink for phosphorus in sediments of the Landsort Deep, an intermittently anoxic deep basin in the Baltic Sea, Chem. Geol., 2016, 438, 58–72, DOI:10.1016/j.chemgeo.2016.05.025.
- J. Kruse and P. Leinweber, Phosphorus in sequentially extracted fen peat soils: AK-edge X-ray absorption near-edge structure (XANES) spectroscopy study, J. Plant Nutr. Soil Sci., 2008, 171(4), 613–620 CrossRef CAS.
- J. R. Flatt and R. Woods, A voltammetric investigation of the oxidation of pyrite in nitric acid solutions: relation to treatment of refractory gold ores, J. Appl. Electrochem., 1995, 25(9), 852–856 CrossRef CAS.
- H. Long and D. G. Dixon, Pressure oxidation of pyrite in sulfuric acid media: a kinetic study, Hydrometallurgy, 2004, 73(3–4), 335–349 CrossRef CAS.
- C. O. Moses, D. K. Nordstrom, J. S. Herman and A. L. Mills, Aqueous pyrite oxidation by dissolved oxygen and by ferric iron, Geochim. Cosmochim. Acta, 1987, 51(6), 1561–1571 CrossRef CAS.
- D. T. Rickard, Kinetics and mechanism of the sulfidation of goethite, Am. J. Sci., 1974, 274(8), 941–952 CrossRef CAS.
- S. Hunger and L. G. Benning, Greigite: a true intermediate on the polysulfide pathway to pyrite, Geochem. Trans., 2007, 8, 1–20 CrossRef PubMed.
- J. W. Stucki, D. C. Golden and C. B. Roth, Effects of Reduction and Reoxidation of Structural Iron on the Surface Charge and Dissolution of Dioctahedral Smectites, Clays Clay Miner., 1984, 32(5), 350–356, DOI:10.1346/CCMN.1984.0320502.
- A. Neumann, S. Petit and T. B. Hofstetter, Evaluation of redox-active iron sites in smectites using middle and near infrared spectroscopy, Geochim. Cosmochim. Acta, 2011, 75(9), 2336–2355 CrossRef CAS , available from, https://www.sciencedirect.com/science/article/pii/S0016703711000883.
- M. A. Arshad, R. J. St. Arnaud and P. M. Huang, Dissolution of trioctahedral layer silicates by ammonium oxalate, sodium dithionite-citrate-bicarbonate, and potassium pyrophosphate, Can. J. Soil Sci., 1972, 52(1), 19–26, DOI:10.4141/cjss72-002.
- M. E. Tuccillo, I. M. Cozzarelli and J. S. Herman, Iron reduction in the sediments of a hydrocarbon-contaminated aquifer, Appl. Geochem., 1999, 14(5), 655–667 CrossRef CAS , available from, https://www.sciencedirect.com/science/article/pii/S0883292798000894.
- T. S. Peretyazhko, J. M. Zachara, R. K. Kukkadapu, S. M. Heald, I. V. Kutnyakov and C. T. Resch, et al., Pertechnetate (TcO4−) reduction by reactive ferrous iron forms in naturally anoxic, redox transition zone sediments from the Hanford Site, USA, Geochim. Cosmochim. Acta, 2012, 92, 48–66 CrossRef CAS , available from, https://www.sciencedirect.com/science/article/pii/S0016703712003511.
- G. ter Heerdt, Ijzersuppletie in Laagveenplassen – Rapport 2012-43, STOWA, STOWA, Amersfoort, Nederlands, 2012, available from, https://www.stowa.nl/ Search PubMed.
- I. Hilbrandt, V. Lehmann, F. Zietzschmann, A. S. Ruhl and M. Jekel, Quantification and isotherm modelling of competitive phosphate and silicate adsorption onto micro-sized granular ferric hydroxide, RSC Adv., 2019, 9(41), 23642–23651, 10.1039/C9RA04865K.
- E. Smolders, E. Baetens, M. Verbeeck, S. Nawara, J. Diels, M. Verdievel, B. Peeters, W. De Cooman and S. Baken, Internal Loading and Redox Cycling of Sediment Iron Explain Reactive Phosphorus Concentrations in Lowland Rivers, Environ. Sci. Technol., 2017, 51(5), 2584–2592 CrossRef CAS PubMed.
- L. Xia, The Application of Iron-Rich Sorbents to Mitigate Phosphorus Release from Sediments in Lowland Rivers, KU Leuven, Leuven, 2022 Search PubMed.
- C. Hyacinthe, S. Bonneville and P. Van Cappellen, Reactive iron(III) in sediments: chemical versus microbial extractions, Geochim. Cosmochim. Acta, 2006, 70(16), 4166–4180 CrossRef CAS.
- J. Chen, B. Gu, R. A. Royer and W. D. Burgos, The roles of natural organic matter in chemical and microbial reduction of ferric iron, Sci. Total Environ., 2003, 307(1–3), 167–178 CrossRef CAS.
- A. Fritzsche, J. Bosch, M. Sander, C. Schroder, J. M. Byrne and T. Ritschel, et al., Organic matter from redoximorphic soils accelerates and sustains microbial Fe(III) reduction, Environ. Sci. Technol., 2021, 55(15), 10821–10831 CrossRef CAS PubMed.
Footnotes |
| † Preceding study: M. A. Münch, R. V. Kaam, K. As, S. Peiffer, G. T. Heerdt, C. P. Slomp and T. Behrends, Impact of iron addition on phosphorus dynamics in sediments of a shallow peat lake 10 years after treatment, Water Res., 2023, 248, 120844, https://doi.org/10.1016/j.watres.2023.120844. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00402g |
| This journal is © The Royal Society of Chemistry 2025 |